

Abstract
We report the crystal structures of a translesion DNA polymerase, Dpo4, complexed with a matched or mismatched incoming nucleotide and with a pyrophosphate product after misincorporation. These structures suggest two mechanisms by which Dpo4 may reject a wrong incoming nucleotide with its preformed and open active site. First, a mismatched replicating base pair leads to poor base stacking and alignment of the metal ions and as a consequence, inhibits incorporation. By replacing Mg2+ with Mn2+, which has a relaxed coordination requirement and tolerates misalignment, the catalytic efficiency of misincorporation increases dramatically. Mn2+ also enhances translesion synthesis by Dpo4. Subtle conformational changes that lead to the proper metal ion coordination may, therefore, be a key step in catalysis. Second, the slow release of pyrophosphate may increase the fidelity of Dpo4 by stalling mispaired primer extension and promoting pyrophosphorolysis that reverses the polymerization reaction. Indeed, Dpo4 has robust pyrophosphorolysis activity and degrades the primer strand in the presence of pyrophosphate. The correct incoming nucleotide allows DNA synthesis to overcome pyrophosphorolysis, but an incorrect incoming nucleotide does not.
Keywords: base stacking, fidelity, metal ion, mismatch, pyrophosphorolysis
Introduction
Replicative DNA polymerases share the common features of high fidelity and high processivity, which allow genetic material to be copied faithfully and promptly (Bebenek and Kunkel, 2004). While highly accurate and efficient in replicating normal DNA, replicative polymerases are often impeded by a variety of DNA lesions where bases are lost or modified. In contrast, the recently identified Y-family DNA polymerases (Ohmori et al, 2001), which exhibit low fidelity and low processivity in replicating normal bases, are capable of traversing these lesions and can substitute for replicative polymerases when a lesion is encountered.
The replicative and Y-family polymerases share a similar catalytic core, which consists of thumb, palm and finger domains arranged like a right hand, and a conserved active site composed of three conserved carboxylates (Yang, 2003). The difference between the two classes of polymerases is the formation of their catalytic center. Replicative polymerases undergo a so-called ‘induced-fit' conformational change upon binding of a deoxynucleoside triphosphate that perfectly matches the templating base (Doublie et al, 1999). As a result, the polymerase surface is seamlessly complementary to the replicating base pair, particularly in the minor groove where all four Watson–Crick base pairs have a similar shape and hydrogen bonding potential (Kool, 2002). The active site thus formed is secluded from bulk solvent and juxtaposes the 3′-OH of the primer strand and the α phosphate of the incoming nucleotide for nucleotidyl transfer (Huang et al, 1998; Li et al, 1998; Doublie et al, 1999; Franklin et al, 2001; Johnson and Beese, 2004). In contrast, the Y-family polymerases, for example, archaeal Dpo4 from Sulfolobus solfataricus and Dbh from Sulfolobus acidocaldarius, human Pol κ and ι, each possesses a preformed and solvent-accessible active site (Ling et al, 2001; Silvian et al, 2001; Zhou et al, 2001; Nair et al, 2004; Uljon et al, 2004). As the crystal structures of Dpo4 complexed with a cyclobutane pyrimidine dimer (CPD), benzo[a]pyrene diol epoxide (BPDE) adduct and abasic (Ab) lesions have shown (Ling et al, 2003, 2004a, 2004b), these polymerases appear able to accept a variety of modified base pairs in their large and nondiscriminatory active site.
Replicative polymerases often contain an intrinsic 3′ to 5′ exonuclease activity that removes a newly misincorporated nucleotide to improve replication fidelity (Kunkel and Bebenek, 2000). None of Y-family polymerases possess such proofreading exonuclease activity. Instead, each Y-family polymerase contains a little-finger (LF) domain, which binds a primer–template duplex in the major groove and influences both processivity and fidelity of the polymerases (Boudsoçq et al, 2004).
It is estimated that if solely determined by the shapes and hydrogen bonds of base pairs, the fidelity of a polymerase would be in the range of 10−1–10−2 (Friedberg et al, 1995). The replicative polymerases attain an error rate of 10−5–10−6 owing to the ‘induced-fit' mechanism in base selection and the proofreading function of the 3′ → 5′ exonuclease. Interestingly, despite a preformed and open active site and the lack of proofreading exonuclease, the fidelity of several Y-family polymerases has been reported to be better than merely dictated by base pairing (Bebenek and Kunkel, 2004). For example, Dpo4 exhibits an error rate of 10−2–10−3 when replicating normal DNA (Boudsoçq et al, 2001, 2004), 10 times better than warranted by base pairing alone. Pre-steady-state kinetic studies suggest that Dpo4 undergoes conformational changes upon binding of a correct incoming nucleotide (Fiala and Suo, 2004), but structural analyses show no evidence of discernable domain movement around the binding pocket of a replicating base pair (Silvian et al, 2001; Ling et al, 2004a). Could a much subtler conformational change be the rate-limiting step of DNA synthesis by Dpo4?
To determine what factors govern the fidelity of a Y-family polymerase, we have first determined the crystal structure of Dpo4 when dATP is incorporated opposite a template dT. This structure supersedes the previously reported Dpo4 ternary complex with a degraded incoming nucleotide (ddADP) (Ling et al, 2001). We then obtained the crystal structures of Dpo4 poised to incorporate dGTP opposite a template dT and immediately after misincorporation of dGTP, where pyrophosphate (PPi) has yet to be released. To corroborate the crystallographic results, we have carried out steady-state kinetic studies of misincorporation and mispaired primer extension by Dpo4. We have also examined the metal ion usage in terms of the active site formation and the role of pyrophosphorolysis in DNA synthesis by Dpo4. Finally, all our results are analyzed and interpreted in comparison with replicative polymerases.
Results
Crystal structures of Dpo4 incorporating correct and incorrect nucleotides
The original crystal structure of the Dpo4 ternary complexes (type I) was obtained from a 2′,3′-dideoxy primer and dideoxy incoming nucleotide (ddATP) (Ling et al, 2001). The ddATP was hydrolyzed to ddADP, and the remaining α and β phosphates adopted an unusual conformation in the crystal. Only one metal ion was located in the active site. Since other Dpo4 structures determined with dNTP retain the intact triphosphate moiety, we replaced ddATP with dATP as the incoming nucleotide. The new crystals of Dpo4 with dATP opposite dT (T/dATP) diffracted X-rays to 2.1 Å resolution (Table I). After model fitting and refinement (Jones et al, 1991; Brünger et al, 1998), the structure is nearly isomorphous to that of type I and contains an intact dATP and two metal ions in the active site (Figure 1A and Table I).
Table 1.
Summary of crystallographic data
| Crystal (space group) | T/dATP (P21212) | T/dGTP (P212121) | T/G (P21212) |
|---|---|---|---|
| Unit cell (a,b,c) (Å) | 98.1, 103.0, 52.4 | 97.9, 101.5, 105.6 | 99.0, 102.0, 53.0 |
| Complexes per AUa | 1 | 2 | 1 |
| Non-hydrogen atoms | 3548 (165 H2O) | 7505 (737 H2O) | 3542 (165 H2O) |
| Resolution range (Å)b | 30.0–2.1 (2.14–2.10) | 30–2.9 (3.0–2.9) | 30–2.85 (2.9–2.85) |
| Rmergeb,c | 0.060 (0.405) | 0.064 (0.354) | 0.048 (0.418) |
| Unique reflections | 29 072 | 22 411 | 12 652 |
| Completeness (%)b | 95.1 (87.6) | 93.5 (91.5) | 96.6 (98.8) |
| R-valued | 0.231 | 0.207 | 0.220 |
| Rfreee | 0.250 (1430) | 0.264 (1121) | 0.263 (1053) |
| R.m.s.d. bond length (Å) | 0.007 | 0.009 | 0.009 |
| R.m.s.d. bond angle (deg) | 1.22 | 1.50 | 1.67 |
| Ave. B (Wilson) (Å2) | 44.6 (33.8) | 62.3 (78.8) | 60.6 (89.2) |
| a AU means asymmetric unit. | |||
| b Data in the highest resolution shell are in parentheses. | |||
| c Rmerge=∑h∑i ∣Ihi−〈Ih〉∣/∑〈Ih〉, where Ihi is the intensity of the ith observation of reflection h and 〈Ih〉 is the average intensity of redundant measurements of the h reflections. | |||
| d R-value=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣, where Fo and Fc are the observed and calculated structure factor amplitudes. | |||
| e Rfree is monitored with the number of reflections shown in parentheses excluded from refinement. | |||
Figure 1.
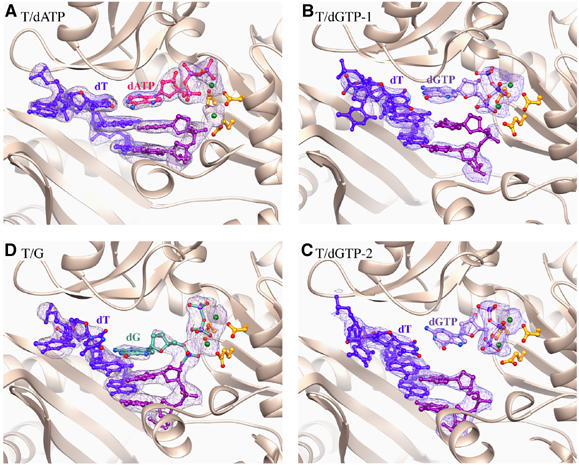
Ribbon diagrams of the T/dATP (A), T/dGTP-1 (B) and T/dGTP-2 (C) and T/G (D) structures around the active site. Dpo4 is shown as ribbons. The three conserved carboxylates in the active site, the last two base pairs of the primer/template and the replicating base pair are shown as ball-and-stick models. The template strand is shown in blue and the primer strand in purple. The incoming nucleotide is shown in different colors for each crystal structure. The metal ions are shown as green spheres. The 2Fo−Fc electron density maps are contoured at 1σ level and superimposed onto the nucleic acid portion.
Crystals of Dpo4 bound to a mismatched replicating base pair were obtained with dGTP opposite a template dT. The primer terminus is dideoxy as in the T/dATP structure. To avoid template misalignment (slippage) observed in the type II crystal structure of Dpo4 (Ling et al, 2001), we used a template containing three consecutive dTs immediately beyond the template–primer junction. Crystals (T/dGTP) were obtained in a different space group from that of the T/dATP crystals and diffracted X-rays to 2.9 Å resolution (Table I). Two Dpo4 complexes were found in each asymmetric unit (T/dGTP-1 and T/dGTP-2), and the structure was refined to an R and Rfree of 20.7 and 26.4%, respectively. In the T/dGTP-1 complex, the dGTP clearly forms a wobble base pair with the templating dT (Figure 2A), but its base-stacking partner at the 3′-end of the primer strand has very weak electron density, especially the base and sugar (Figure 1B). In the T/dGTP-2 complex, the 3′ nucleotide of the primer strand is well stacked with the rest of the DNA duplex, but the guanine base of the dGTP is 5.3 Å instead of 3.4 Å above it and is not coplanar with the templating base (Figure 1C). It appears that when the incoming nucleotide is incorrect, the bases of either the incoming nucleotide or the 3′ nucleotide of the primer strand are disordered, indicating destabilized base stacking.
Figure 2.
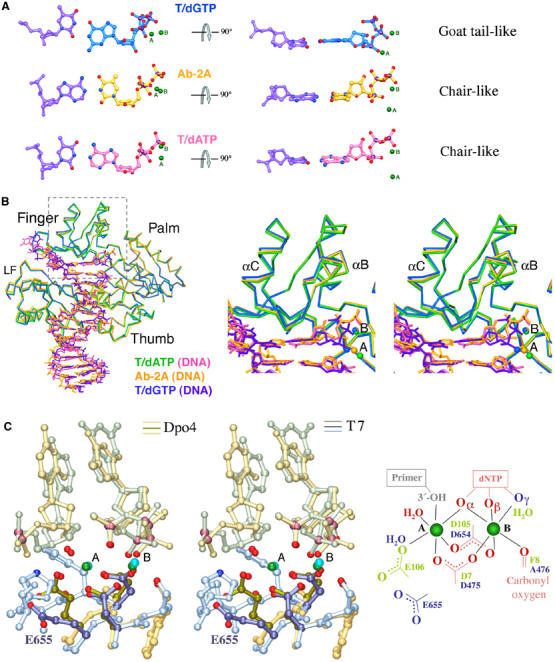
Structural comparison of Dpo4 and T7 DNA polymerase. (A) The replicating base pairs in three Dpo4 structures (T/dGTP, T/dATP and Ab-2A) are shown as ball-and-stick models. The two metal ions (A and B) are shown as green spheres. The A-metal ion position differs in each structure. The conformation of the triphosphate is denoted as ‘chair-like' and ‘goat tail-like'. (B) Superposition of T/dATP, T/dGTP and Ab-2A structures. The Cα traces, DNA and nucleotide substrate are shown in stick models. A zoom-in stereo view of the finger domain with the replicating base pair and metal ions (outlined in gray) is shown on the right. The colors representing each structure are indicated. (C) Superposition of the metal ion coordination in Dpo4 (Ab-2A, yellow and brown colors) and T7 DNA polymerase (PDB: 1T7P, blue and green colors) in a stereo view. The oxygen atoms of the three conserved carboxylates and those involved in metal ion coordination are highlighted in red. The metal ion coordination is schematically drawn on the right. Red indicates ligands conserved in both polymerases, light green in Dpo4 only and blue in T7 only. The hypothesized 3′-OH of the primer strand is shown in gray.
The alternate conformations of dNTP and metal ion displacement
Besides weakened base stacking in the T/dGTP structure compared to that of T/dATP, we find that the triphosphate moiety of the incoming nucleotide assumes different conformations, either ‘chair-like' ( ) in T/dATP or ‘goat tail-like' (
) in T/dATP or ‘goat tail-like' ( ) in T/dGTP (Figure 2A). In addition, the catalytic divalent cation in the A site, which should coordinate and activate the 3′-OH of the primer strand for the nucleophilic attack, is shifted by ∼3 Å between the two structures in the absence of the 3′-OH (Figure 2A and B).
) in T/dGTP (Figure 2A). In addition, the catalytic divalent cation in the A site, which should coordinate and activate the 3′-OH of the primer strand for the nucleophilic attack, is shifted by ∼3 Å between the two structures in the absence of the 3′-OH (Figure 2A and B).
The crystal structure of a misincorporation product
When the 2′,3′ dideoxyribonucleotide at the 3′-end of the primer strand was replaced by a normal deoxyribonucleotide, crystals of Dpo4, template/primer DNA and dGTP opposite a template dT were obtained after a short incubation with Mg2+. The crystals were isomorphous to type I and T/dATP crystals and contain one protein–DNA complex in each asymmetric unit. After refinement to 2.85 Å resolution (Table I), we found that dGTP was incorporated into the primer strand and that the product PPi remains bound to Dpo4 and occupies the same space as the triphosphate moiety of dNTP (Figure 1D). Primer or terminal extension by Dpo4 during crystallization occurred previously (Ling et al, 2001, 2004a, 2004b), but this is the first time that PPi is retained in the crystal structure, suggesting a slow release of PPi because of misincorporation. The structure represents the state after incorporation of a wrong nucleotide by Dpo4, and we name this crystal structure ‘T/G' for the mismatch. Interestingly, the T/G mismatched base pair is not coplanar. The dG at the primer end is translocated about half way toward the usual position in the preinsertion state, but its base-pairing partner dT remains at the templating position (Figure 1D). In this product complex, the two metal ions are still present and coordinated by the PPi and the three conserved carboxylates in the active site.
Between the substrate (T/dGTP) and product (T/G) complexes, changes in the Dpo4 structure are subtle and mainly in the LF domain in spite of the large shift of DNA, particularly the primer strand. In contrast, comparison of the matched (T/dATP) and mismatched (T/dGTP) structures reveals a noticeable shift in the finger domain, which interacts most extensively with the replicating base pair (Figure 2B).
Kinetic analyses of Dpo4-catalyzed nucleotide incorporation
To assess the catalytic efficiencies of incorporating correct versus incorrect incoming nucleotides, we determined steady-state kinetic parameters of primer extension by Dpo4 using the same DNA template sequence context as in the mismatch structures (Materials and methods). To emulate the native environment of S. solfataricus, we carried out standing-start kinetic assays at 60°C with a 44-mer template and 20-mer primer (Table II). Depending on the terminal base pair at the template–primer junction (G/C versus T/A), the KM of incorporating dATP opposite dT varies by 15-fold and the catalytic efficiencies differ by eight-fold (Table II). With the same sequence context and a correct incoming nucleotide, whether it is dATP opposite dT or dGTP opposite dC, the kcat and KM are similar (Table II). When the incoming nucleotide is incorrect, for example, dGTP opposite dT, the kcat is similar to that of the matched base pair, but the KM is increased by 25-fold (Table II). The requirement for high concentrations of an incorrect incoming nucleotide to facilitate polymerization agrees well with the unstable base stacking observed in the T/dGTP structures.
Table 2.
Steady-state kinetic analysis of nucleotide insertion by Dpo4
| DNA template | dNTP | Metal ion | kcat (min−1) | Km (μM) | kcat/Km (μM−1 min−1) | fmis | fext |
|---|---|---|---|---|---|---|---|
| 5′–C↓3′–GC– | G | Mg | 133±17 | 1.09±0.21 | 146±27 | N/A | N/A |
| 5′–C↓3′–GT– | A | Mg | 157±9 | 1.25±0.09 | 128±7 | 1 | N/A |
| 5′–C↓3′–GT– | G | Mg | 154±22 | 31.7±4.5 | 5.3±0.9 | 4.1 × 10−2 | N/A |
| 5′–C↓3′–GT– | A | Mn | 212±15 | 0.49±0.07 | 451±45 | 1 | N/A |
| 5′–C↓3′–GT– | G | Mn | 215±8 | 3.37±1.00 | 83±10 | 1.75 × 10−1 | N/A |
| 5′–CA↓3′–GTT– | A | Mg | 81±3.9 | 0.085±0.008 | 998±137 | N/A | 1 |
| 5′–CG↓3′–GTT– | A | Mg | 31.8±3.8 | 1.5±0.3 | 24.2±4.9 | N/A | 2.4 × 10−2 |
| Standing-start kinetic assays were performed at 60°C for 2 min in reaction mixtures containing 0.4 nM Dpo4 and 10 nM primer–templates (see Materials and methods). To determine the efficiency of nucleotide misincorporation opposite the template dT, dGTP concentrations ranged from 1.25 to 50 μM. For templates with mismatched primer/template termini, the concentrations of correct dATP ranged from 0.25 to 10 μM. The kinetic parameters were determined as described in Materials and methods. The misinsertion frequency (fmis) was determined as the ratio of insertion efficiencies (kcat/Km) for incorrect and correct nucleotides. The frequency of mispair extension (fext) was determined as the ratio of extension efficiencies (kcat/Km) for incorrectly and correctly paired primer–template termini. Incorrect nucleotide is indicated in bold. N/A: not applicable. | |||||||
We also examined the kinetics of primer extension with a preformed mismatch at the template/primer junction. The kcat of primer extension was reduced by three-fold in the presence of a T/G mismatch, compared with a correct T/A pair, and the KM was increased by 16-fold (Table II). The overall catalytic efficiencies of misinsertion (fmis) versus primer extension of a mismatched base pair (fext), however, are surprisingly similar. Making a mismatch may be slightly more efficient (1.7-fold) than extending it (Table II).
Effects of Mg2+ and Mn2+ on misincorporation
The displacement of the A-site metal ion observed in the T/dATP and T/dGTP structures suggests that in Dpo4, the preformed active site is insufficient for catalysis to take place, and DNA synthesis may require precise coordination of the metal ions by the conserved carboxylates, 3′-OH of the primer strand and an incoming nucleotide. Because of the open catalytic center, if one of the coordination ligands is missing or incorrect, the metal ions may not be perfectly situated to engage in catalysis. To test whether metal ion coordination plays a role in the fidelity of DNA synthesis by Dpo4, we compared the kinetic parameters of nucleotide incorporation by Dpo4 in the presence of Mn2+ versus Mg2+. Mn2+ is known to be more tolerant of distortions in substrate and mutations in enzymes than Mg2+ (Vermote et al, 1992; Junop and Haniford, 1996; Vipond et al, 1996; Villani et al, 2002). In the case of Dpo4, Mn2+ increases the kcat and decreases the KM for both correct and incorrect nucleotide incorporation (Table II), but Mn2+ improves the efficiency of primer extension with an incorrect nucleotide significantly more than with a correct incoming nucleotide (Figure 3A and Table II). Even with the least affected nucleotide, dGTP (Figure 3A), the misincorporation frequency increased by ∼4.3-fold when Mg2+ was substituted by Mn2+ (Table II). Interestingly, Mn2+ fails to improve the efficiency of mispaired primer extension (Figure 3B). This may be because of the abnormal structure of a mismatched template/primer junction, where a T/G mismatch was shown to form an unusual reverse-wobble base pair (Trincao et al, 2004). We suspect that Mn2+ decreases the fidelity of Dpo4 only when the substrate configuration in the active site is near-native.
Figure 3.
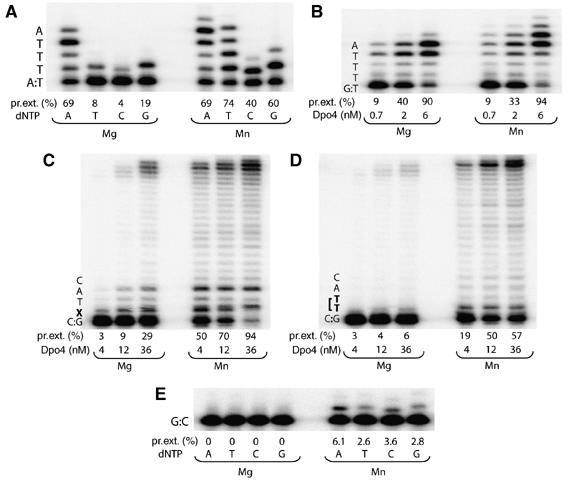
Effect of divalent metal ion on the fidelity and lesion bypass ability of Dpo4. (A) Specificity of nucleotide incorporation. (B) Mispaired primer extension. Bypass of (C) a synthetic abasic site and (D) a cis-syn CPD. (E) Terminal nucleotidyl transferase activity of Dpo4. Reactions were performed in the presence of 5 mM Mg2+ or Mn2+. The local template sequence is shown to the left of each gel. Dpo4 concentration in each reaction was 2 nM, or as indicated at the bottom of the gels. Primer elongations were calculated as the percent of total primer termini, and are indicated at the bottom of the gels.
When tested with an abasic or CPD lesions, the effects of Mn2+ on relaxing base-pairing specificity and increasing tolerance of non-native substrate are spectacular (Figure 3C and D). The improvement of bypass efficiencies on either lesion is more than 10-fold. In addition, Mn2+ enhances the template-independent terminal nucleotidyl transferase activity of Dpo4 (Figure 3E).
Pyrophosphorolysis by Dpo4 requires a matched template/primer junction
It is well known that PPi can promote the reversal of polymerization by degrading the primer strand and regenerating deoxynucleotide triphosphate, a process known as pyrophosphorolysis (Deutscher and Kornberg, 1969). The slow release of PPi after incorporating an incorrect nucleotide by Dpo4, as indicated in the T/G structure, suggests that reversal of polymerization (degradation) may occur and contribute to the low efficiency of misincorporation. We have thus examined the influence of exogenous PPi on primer extension catalyzed by Dpo4. Indeed, Dpo4 catalyzes pyrophosphorolysis in the presence of 0.125–0.5 mM PPi and can efficiently shorten the primer strand from a 20-mer to 15-mer (Figure 4A, panels I and V). Addition of a correct dNTP effectively arrests pyrophosphorolysis and allows Dpo4 to carry out the normal forward polymerization reaction in the presence of up to 2 mM concentrations of PPi (Figure 4A, panels II and IV). However, if a dNTP does not match the templating base, primer extension is greatly reduced and pyrophosphorolysis overtakes polymerization in the presence of 0.5 mM PPi (Figure 4A, panels III and V). When the PPi concentration is above 2 mM, both the forward and reverse reactions are inhibited because of the depletion of Mg2+ (Figure 4; Supplementary data).
Figure 4.
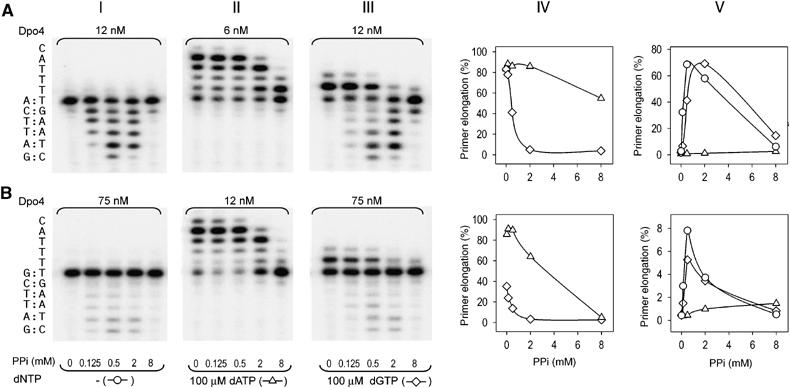
Pyrophosphorolytic activity of Dpo4. Comparison of polymerization and pyrophosphorolytic activities of Dpo4 with correctly paired (A) or mispaired (B) primer–template termini. Effect of increasing PPi concentrations on primer extension and degradation was studied in the absence of added dNTP (panel I), in the presence of the next correct dATP (panel II) or in the presence of incorrect dGTP nucleotide (panel III). The local template sequence is shown to the left of the gel. PPi-dependent inhibition of primer elongation (panel IV) or primer degradation (panel V) in reactions shown in panels I, II and III was calculated as percent of total primer termini. Open triangles (▵) represent the presence of dATP, open diamonds (◊) the presence of dGTP and open circles (○) the absence of dNTP.
To examine whether pyrophosphorolysis plays a bona fide proofreading role, we analyzed the effect of PPi on a preformed mismatch at the template–primer junction. The primer with a G/T mismatch at the template/primer junction was readily extended by Dpo4 in the presence of a correct incoming nucleotide (Figure 4B, panel II). In contrast, extension with a wrong incoming nucleotide, for instance dGTP, required excessive amounts of Dpo4 (Figure 4B, panel III). The mismatched template–primer pair also greatly inhibits pyrophosphorolysis (Figure 4B, panel I). Even with an eight-fold excess of Dpo4 over the mispaired template/primer, pyrophosphorolysis was reduced by more than 10-fold whether in the presence or absence of an incorrect dNTP (Figure 4). Apparently, Dpo4 is very inefficient in removing a preformed mismatch by pyrophosphorolysis.
Pyrophosphorolysis of mispaired primer may require de novo synthesis
The T/G crystal structure suggests that removal of an incorrect dNMP by pyrophosphorolysis may occur immediately after misincorporation and before PPi release. To check whether the endogenous PPi that results from misincorporation plays a role in removing the wrongly incorporated dNMP or preventing dNTP binding and hence primer extension, we compared the efficiencies of extending a primer containing a mismatch either preformed at the template/primer junction or acquired through ongoing polymerization. Even in the absence of exogenous PPi, Dpo4 is less efficient in making a mismatch (dG or dC opposite a template dT) (Figure 5A) than extending a preformed mismatch (Figure 5B). Interestingly, Dpo4 appears to utilize mismatches made de novo more efficiently than a preformed mispair (Figure 5). The differences are not large, but are reproducible. A possible explanation is that the mismatch made by Dpo4 is either extended if the next incoming nucleotide succeeded in replacing the PPi product (97%) or removed by pyrophosphorolysis, so that only 77% of the primers were extended (Figure 5A). The presence of 0.5–2 mM PPi precipitously reduced the misincorporation (from 64 to 4%; Figure 5A) but had only mild effects on extending a mismatched template/primer that is either preformed (from 86 to 29%; Figure 5B) or made by Dpo4 (from 95 to 47%; Figure 5A). These observations suggest that pyrophosphorolysis may increase the fidelity of Dpo4 by preventing misincorporation.
Figure 5.
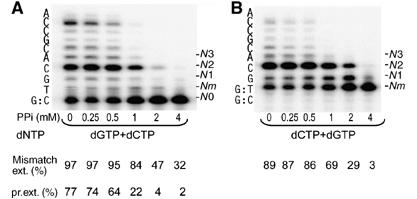
Comparison of extension of a preformed primer/template terminal mismatch and mismatch formed as a result of nucleotide misincorporation. (A) PPi-dependent inhibition of primer extension in the presence of 100 μM incorrect dGTP and 10 μM of the next correct dCTP. (B) PPi-dependent inhibition of G:T mismatch extension in the presence of 10 μM correct dCTP and dGTP. Reactions were performed in the presence of 8 nM Dpo4. The local template sequence is shown to the left of the gels. Mismatch extension was calculated as 100 × (N1+N2+⋯+Nn)/(Nm+N1+N2+⋯+Nn). Primer elongation was calculated as 100 × (Nm+N1+N2+⋯+Nn)/(N0+Nm+N1+N2+⋯+Nn).
It should be noted that the absence of pyrophosphorolysis in Figure 5B is a result of the preformed template/primer mismatch, as shown in Figure 4B. Pyrophosphorolysis of the matched template and primer (Figure 5A) may occur before or after misincorporation of dGMP. But the presence of dGTP, which complements the templating dC at the template/primer junction, promotes polymerization and prevents the primer from further degradation.
The role of the little finger domain in pyrophosphorolysis
We examined the pyrophosphorolysis activity of other members of the Y-family polymerases and found that none of the human Y-family polymerases (Pol η, ι and κ) perform pyrophosphorolysis (data not shown). Moreover, we were unable to detect PPi-induced primer degradation by Dbh, a close relative of Dpo4 from S. acidocaldarius (Figure 6), although inhibition of the correct nucleotide incorporation occurs at lower PPi concentrations than those required to inhibit Dpo4 (Figure 6A, panels I and III). Similar to DNA synthesis (Boudsoçq et al, 2004), the pyrophosphorolysis activities of Dpo4 and Dbh are strongly influenced by the LF domain. Replacing the LF domain of Dpo4 with that from Dbh completely abolishes pyrophosphorolytic activity (Figure 6B, panel IV), and Dbh with the LF from Dpo4 becomes capable of PPi-induced primer degradation (Figure 6B, panel II).
Figure 6.
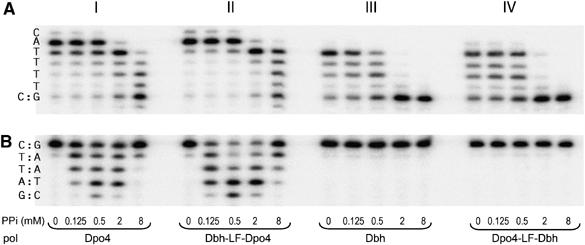
Comparison of pyrophosphorolytic activity of Dpo4, Dbh, Dpo4-LF-Dbh and Dbh-LF-Dpo4. (A) PPi-dependent inhibition of dATP (100 μM) incorporation and (B) pyrophosphorolysis catalyzed by 2.5 nM Dpo4 (panel I), 5 nM Dbh-LF-Dpo4 (panel II), 10 nM Dbh (panel III) or 25 nM Dpo4-LF-Dbh (panel IV). The local template sequence is shown to the left of each set of reactions.
Discussion
Mismatched base pair destabilizes base stacking and inhibits misincorporation
Mismatched base pairs are well known to form wobble hydrogen bonds and distort the width of a DNA duplex. In this study, we show that a mismatched base pair also leads to severely weakened base stacking, so that it hinders Dpo4 from binding and incorporating an incorrect incoming nucleotide (Figure 1 and Table II). A close examination of the kinetic data reveals that base stacking may play an important role even when Dpo4 makes Watson–Crick duplexes. The catalytic efficiency of Dpo4 is ∼7 times better if an incoming nucleotide and the 3′-end of the primer are both A's, than if they are A and C or G and C (Table II). The effect of base stacking may be considerable for Dpo4 because of the limited interactions between the protein and the replicating base pair (Figure 1). The destabilized base stacking probably leads to the slight structural alterations in the finger domain (Figure 2B), which in turn may underlie the distorted conformation of the triphosphate moiety of the incoming nucleotide (Figure 2A).
Poor base stacking as a result of a wobble base pair may also prevent the ‘induced-fit' movement of the finger domain in a replicative polymerase, more so than the altered hydrogen-bond pattern per se. The crystal structures, however, may not reveal the destabilized base stacking because a mismatched replicating base pair is sandwiched between the finger domain and the template/primer base pair (Johnson and Beese, 2004). In fact, weakened base stacking is the common feature among varieties of mismatched base pairs that MutS-like proteins recognize and rely upon to kink a mismatch-containing DNA (Obmolova et al, 2000; Natrajan et al, 2003).
Mg2+ enhances fidelity, and metal coordination may be the rate-limiting step
The metal ion displacement observed in the T/dATP and T/dGTP structures prompts us to examine its cause. All Dpo4 ternary complex crystals were grown in the presence of 100 mM CaCl2. Could the abnormal metal ion coordination be due to the Ca2+ replacement of Mg2+ in the active site? Among the previously determined Dpo4 structures, the two Ca2+ ions in the active site of the Ab-2A structure (Ling et al, 2004a), where an abasic lesion upstream from the template/primer junction is looped out and the DNA duplex and the replicating base pair are all Watson–Crick type, are superimposable with the two Mg2+ ions found in the active site of the ternary complex structure of T7 DNA polymerase (Doublie et al, 1998) (Figure 2C). The similarity indicates that Ca2+ ions are not the cause for the metal ion displacement. The A-site metal in T/dATP and T/dGTP is shifted by 1–2 Å relative to that in Ab-2A (Figure 2A). The movement of the A-metal ion in the T/dATP and T/dGTP structures is correlated with the absence of a 3′-OH in the primer strand and the wobble base pair in T/dGTP. In addition to the dideoxy primer end, modified substrates, including CPD and BPDE lesions, appear to also instigate displacement of the A-site metal (Table III). It is believed that the preformed and open active site enables the Y-family polymerases to accommodate lesions and mismatched base pairs, thereby allowing them to make more errors than replicative polymerase. But the large and solvent-accessible active site also has difficulty in trapping the metal ions. Thus, even after a replicating base pair is formed, Dpo4 may remain uncommitted to catalysis.
Table 3.
Summary of Dpo4 structures
| Struct. name | Ref.a | Description | Template/primer junctionb | Base stacking | Triphosphatec | Metal ions |
|---|---|---|---|---|---|---|
| Type I | 2001 | Normal DNA | dT/ddATP, ddA(p) | Normal, WC | Permutated | 1 Ca2+ at B |
| Type II | 2001 | Intended mismatch | dC/ddGTP, dA(p) | Misaligned, WC | Goat tail-like | 1 Ca2+ at B |
| TT-1 | 2003 | 3′ CPD templating | CPD/ddATP, dC(p) | Normal, WC | Goat tail-like | 2 Ca2+, shifted A |
| TT-2 | 2003 | 5′ CPD templating | CPD/ddATP, dT(p) | OK, Hoogsteen | Goat tail-like | 2 Ca2+, shifted A |
| Ab-2A | 2004a | Abasic lesion (at −1), looped out | dA/dTTP, dC(p) | Normal, WC, | Chair-like | 2 Ca2+, T7-like |
| BP-1 | 2004b | BPDE adduct (at −1), intercalated | dT/dATP, dT(p) | Stacked with intercal. BPDE | Permutated | 2 Ca2+, shifted A |
| BP-2 | 2004b | BPDE adduct (at −1), extruded | dT/dATP, dT(p) | OK, Hoogsteen | Chair-like | 2 Ca2+, shifted A |
| T/dATP | Normal DNA | dT/dATP, ddC(p) | Normal, WC | Chair-like | 2 Ca2+, shifted A | |
| T/dGTP | Mismatch | dT/dGTP, ddC(p) | Unstable | Goat tail-like | 2 Ca2+, shifted A | |
| a References are Ling et al., and year of publication are as listed in References. | ||||||
| b Template/primer junction includes the nature of the replicating base pair and the deoxy d(p) or dideoxy dd(p) state of the primer strand. | ||||||
c Permutated means the α phosphate is shifted to the β position, the β phosphate to the γ position and the γ phosphate to the α position; chair-like is ( ) when looked at the base of dNTP edge-on (Figure 2A); and goat-tail like is ( ) when looked at the base of dNTP edge-on (Figure 2A); and goat-tail like is (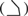 ). ). | ||||||
The formation of an active catalytic center in Dpo4 requires a 3′-OH at the primer terminus, a matched template base and the incoming nucleotide, the three catalytic carboxylates and properly coordinated metal ions (Figure 2C). The metal ions, particularly at the A-site, can ‘sense' the presence of a suboptimal substrate and fail to assume a correct position, thus preventing the chemical reaction from occurring. Replacing the highly stringent Mg2+ ion with Mn2+ for its relaxed metal coordination requirement increases the catalytic efficiency of Dpo4 bypassing otherwise unfavorable substrates (Figure 3C and D). Mn2+ also reduces the fidelity of Dpo4 by increasing the efficiency of misincorporation (Figure 3A and Table II). Nature probably exploits the stringent coordination requirement of Mg2+ ions to enhance substrate specificity of polymerases and to increase the fidelity to levels higher than can be achieved by base pairing alone.
The reliance on Mg2+ ions for ‘sensing' the alignment of the substrates in the polymerization reaction and for improvement of fidelity has also been reported for the A-family and X-family DNA-repair polymerases (Kunkel and Loeb, 1979; Shah et al, 2001; Yang et al, 2004). In the ternary complexes of T7 DNA polymerase, the A-site metal ion assumes a defined position once the finger domain is closed by the ‘induced-fit' mechanism, even without a 3′-OH group on the primer strand or with lesions in the template strand (Doublie et al, 1998; Brieba et al, 2004; Dutta et al, 2004; Li et al, 2004). Although the metal ions remain in the same position, subtle adjustment of the active site is still essential because the third catalytic carboxylate of T7 polymerase, E655, is not engaged in metal coordination in any T7 structure reported to date (Figure 2C). In Dpo4, although the catalytic carboxylates do not undergo conformational changes upon substrate binding, the arrangement of substrates and positions of the metal ion require fine-tuning before the chemical reaction takes place. Pulse-quench and pulse-chase experiments, which also indicate the existence of a Dpo4 ternary complex intermediate and a rate-limiting step as a result of conformational change, support this assessment (Fiala and Suo, 2004). The subtle conformational changes that lead to the proper metal ion coordination may be the key and rate-limiting step of catalysis by all polymerases.
Side reactions catalyzed by DNA polymerases
In the original type I crystals of Dpo4 (Ling et al, 2001), because of the absence of 3′-OH groups on the primer strand and the incoming nucleotide, the triphosphate moiety was permutationally rearranged (i.e., the α phosphate occupies the β position, the β the γ position and the γ the α position) such that instead of primer extension, the incoming ddATP was degraded to ddADP. The degradation of ddATP to ddADP by Dpo4 is barely detectable without DNA substrate and is enhanced by the presence of the template and primer strands. Such a reaction has not been observed in other misaligned ternary complexes of Dpo4 and is prevented by the 3′-OH group on the incoming nucleotide, as shown in the T/dATP structure. These observations suggest that when the substrate is distorted in a certain way, the active site of Dpo4 may carry out side reactions other than DNA synthesis.
Related side reactions, for example degradation of an incoming nucleotide triphosphate (dNTP → dNMP+PPi) and PPi exchange (PPi+dNTP* → PPi*+dNTP or d(d)XMP+dNTP → d(d)XTP+dNMP), have been reported for both repair (Pol β in the X-family) and replicative polymerases (Lecomte et al, 1986; Rozovskaia et al, 1989; Dahlberg and Benkovic, 1991). Excising chain terminators by HIV reverse transcriptase (RT) using PPi or ATP is the most thoroughly investigated phenomenon (Arion et al, 1998; Meyer et al, 1999). Certain HIV strains become resistant to drugs, such as AZT, which acts as a chain terminator during reverse transcription, and the resistance was because of mutations in the RT resulting in enhanced pyrophosphorolysis activity. Mutations in the finger and palm domain, which facilitate the HIV RT to use either PPi or ATP to remove chain terminators, are selected under the pressure of anti-AIDS drug treatment.
Can pyrophosphorolysis play a role in proofreading?
Pyrophosphorolysis was first established by Deutscher and Kornberg (1969), and a role in performing proofreading and improving fidelity by removing misincorporated nucleotides has been proposed (Lecomte et al, 1986). However, such an activity has long been dismissed, since replicative polymerases are highly processive and contain an intrinsic proofreading exonuclease activity. On the contrary, the Y-family polymerases have no intrinsic proofreading exonuclease activity and limited processivity. It is therefore possible that pyrophosphorolysis plays a role in improving polymerase fidelity.
Dpo4 is active in pyrophosphorolysis in the absence of any incoming nucleotide (Figure 4A, panel I). Pyrophosphorolysis can be inhibited and eliminated by the presence of a correct incoming nucleotide (Figure 4A, panel II). For example, 10 μM of dATP is enough to overcome primer degradation by 0.5 mM PPi (SI). However, pyrophosphorolysis persists if a dNTP does not match the templating base (Figure 4, panel III). Although Dpo4 does not proofread a preformed mismatch, a newly incorporated ‘wrong' nucleotide may be removed by the pyrophosphorolytic activity of Dpo4 because of the slow release of PPi. In our mixed dNTP experiment (Figure 5A) that monitors the active proofreading by Dpo4, the absence of accumulated mismatched template/primer junction as a result of misincorporation hints at the possibility of removal of de novo-synthesized mismatches by pyrophosphorolysis.
The slow release of PPi after misincorporation (Figure 1D) may have two other possible consequences: namely, preventing incorporation of the next dNTP and impeding translocation of the DNA substrate. Both lead to reduction of mispaired primer extension. The LF domain swapping experiments (Figure 6) indicate that pyrophosphorolysis depends on prolonged association between the polymerase and DNA substrate. Dbh with weak DNA association and poor processivity has no detectable pyrophosphorolysis. Intriguingly, even without pyrophosphorolysis, the chimeric Dpo4-LF-Dbh (Dpo4 catalytic core plus LF of Dbh) was reported to have a lower rate of base substitution (misincorporation) than Dpo4 (Boudsoçq et al, 2004). We suspect that the weak DNA binding by the LF of Dbh results in the increased dissociation of polymerase after misincorporation and the reduced amount of complete product containing mismatches (Boudsoçq et al, 2004).
Fidelity check in DNA synthesis: domain movement and metal ion coordination
The fidelity of replicative polymerases is achieved by an induced-fit mechanism, which includes both global movement of the finger domain upon substrate binding and subtle changes in the active site for metal ion coordination, prior to the chemical reaction. The active site of replicative polymerases has evolved to form a similar binding surface that seamlessly complements a Watson–Crick base pair and a conserved active site excluded from bulk solvent. Unlike the replicative polymerases, Y-family polymerases contain significant alterations around the conserved active site. For example, sequence alignment has revealed a variable loop in the finger domain that contacts the replicating base pair. Between the T/dATP and T/dGTP structures of Dpo4, this loop moves depending on whether the replicating base pair is a match or mismatch (Figure 2B). Mutation of a Met to Gly in this loop in human Pol η makes the enzyme much more efficient at bypassing CPDs (Glick et al, 2003). Other changes surrounding this loop may make the active site of yeast Pol η more solvent secluded (Ling et al, 2003). Alterations are also observed in the thumb domain, which interacts with the primer strand. These differences probably allow each Y-family polymerase to favor one or several particular lesion types over others. In a sense, the replicative polymerases are made in ‘one size that fits all' Watson–Crick base pairs, and they verify the base-pair authenticity by large domain movement followed by subtle adjustment of metal coordination. In contrast, the Y-family polymerases are each made for a specific type of lesion. Only when the substrate and preformed active site match, are the two metal ions settled into the right position for catalysis.
Materials and methods
Crystallization and structure determination
Dpo4 was purified as described (Ling et al, 2001). For the T/dATP crystals, the oligos (5′-TTC ATG AGT CCT GTA GCC-3′ and 5′-GGC TAC AGG ACT ddC-3′) were purchased from the Keck Facility at Yale University. For the T/dGTP and T/G crystals, a 17-nt template (5′-TTT TGA ATC CTT CCC CC-3′) and a complementary 13-nt primer with either a dC or ddC at the 3′-end (5′ GGG GGA AGG ATT C*-3′) were purchased from Oligos Etc. Inc. Appropriate pairs of template and primer were annealed and mixed with Dpo4 in a 1.2:1 molar ratio. Crystal growth and data collection were performed as described (Ling et al, 2001) with slight variations of PEG concentrations. Streak seeding was often applied. Diffraction data of the three crystals were collected at −178°C using an R-axis IPII detector mounted on a Rigaku RU 200 generator. The data were processed using DENZO and SCALEPACK (Otwinowski and Minor, 1997). All three structures were modeled based on previously solved Dpo4 ternary complex structures (Ling et al, 2004a) and refined using routine procedures for rigid-body and positional refinement (Jones et al, 1991; Brünger et al, 1998). Non-crystallographic symmetry (NCS) restraints were applied to the refinement of the T/dGTP structure that contains two molecules in the asymmetric unit. The data collection and refinement statistics are reported in Table I, and none of the residues is in the disallowed region of a Ramachandran plot.
DNA templates
The sequence of the 44-mer oligonucleotide templates used in most experiments is 5′-CTC TCA CAA GCA CGA CAT TTN1 GAA TCC TTC CCC CGC GGC GCC GC-3′, where N1 is either C or T. For insertion assays, the primer was 20-mer complementary to the 4–23 positions at the 3′-end of the 44-mer template (5′-GCG CCG CGG GGG AAG GAT TC-3′). For extension assays, the primers were 21-mers (5-GCG CCG CGG GGG AAG GAT TCN2-3) in which the 3′ base (N2) is either A or G, and pairs with the T at the position 24 from the 3′-end of the template. The sequences of the 50-mer templates used for the translesion synthesis studies were 5′-CTC TCA CAA GCA GCC AGG CAT-TCT CCG CAC TCG TCT CTA CAC CGC TCC GC-3′, where T-T at positions 21–22 (shown in bold) are either undamaged or cis-syn dimer-containing thymidines, and 5′-CTC TCA CAA GCA GCC AGG CAT XCT CCG CAC TCG TCT CTA CAC CGC TCC GC-3′, where X is an abasic analog, dSpacer. These three templates were primed with a 23-mer oligonucleotide with the following sequence: 5′-GCG GTG TAG AGA CGA GTG CGG AG-3′. Most of the oligonucleotides including abasic-containing ones were synthesized by Loftstrand Laboratories (Gaithersburg, MD) and gel-purified before use. The cis-syn CPD-containing oligonucleotide was synthesized and purified by Phoenix Biotechnologies (Huntsville, AL).
The primers were 5′-end-labeled using T4 polynucleotide kinase and [γ-32P]ATP. DNA substrates were prepared by annealing templates with labeled primers at a 1.5:1 molar ratio in 50 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 50 μg/ml bovine serum albumin (BSA) and 1.42 mM 2-mercaptoethanol for 10 min at 100°C, followed by slow cooling. Annealing efficiencies were >95%, judged by nondenaturing polyacrylamide gels.
Kinetic analysis
Steady-state kinetic parameters Km and Vmax for dNTP incorporation were measured in standing-start reactions as described previously (Vaisman et al, 2001). The efficiencies of nucleotide incorporation were determined after 2 min incubation. DNA substrates (10 nM) were replicated by Dpo4 (Boudsoçq et al, 2004) at 60°C in reaction mixtures containing 40 mM Tris–HCl (pH 8.0), 5 mM MgCl2 or MnCl2, 10 mM dithiothreitol (DTT), 250 μg/ml BSA, 2.5% glycerol, 0.4 nM Dpo4 and variable concentrations of dNTP. Reactions were stopped by the addition of an equivalent amount of gel loading buffer containing 50 mM EDTA, 0.1% xylene cyanol and 0.1% bromphenol blue in 90% formamide. Reaction products were separated in a 20% polyacrylamide gel containing 8 M urea and then visualized and quantified using a Molecular Dynamics PhosphorImager and ImageQuant software (Molecular Dynamics, CA). The velocity of dNTP incorporation was determined by dividing the percent of reaction product by the respective time of the reaction. The apparent Vmax and Km were determined from a Hanes–Woolf plot by linear least squares fit using the Sigma Plot software. Apparent kcat values were calculated based on the assumption that Dpo4 was fully active. The kcat values were obtained by dividing Vmax (in nM primer extended per min) by the enzyme concentration (nM). The efficiency of nucleotide insertion by polymerase was calculated as kcat/Km. Results are means±standard errors from four to five measurements. The misinsertion frequency (fmis) was determined as the ratio of insertion efficiencies (kcat/Km) for incorrect and correct nucleotides. The frequency of mispair extension (fext) was determined as the ratio of extension efficiencies (kcat/Km) for incorrectly and correctly paired primer–template termini.
Dpo4-catalyzed pyrophosphorolysis
DNA template (10 nM) was preincubated at room temperature for 5 min with various amounts of Dpo4 in a reaction mixture containing 40 mM Tris (pH 8.0), 5 mM MgCl2, 10 mM DTT, 250 μg/ml BSA and 2.5% glycerol to allow the formation of the polymerase/DNA binary complex. Pyrophosphorolysis was then initiated by the addition of various concentrations of PPi with or without dNTPs. After 2 min at 60°C, the reactions were stopped and analyzed on a 12% polyacrylamide/8 M urea sequencing gel. The resolved products were visualized and quantified using Molecular Dynamics PhosphorImager and ImageQuant software. Primer elongation and/or degradation were calculated as percent of total primer termini.
Primer extension assays
DNA template (10 nM) was incubated at 60°C for 2 min with different amounts of Dpo4 and 1–500 μM of either all four dNTPs or each dNTP individually in the reaction buffer containing 40 mM Tris (pH 8.0), 5 mM MgCl2 or MnCl2, 10 mM DTT, 250 μg/ml BSA and 2.5% glycerol. Reactions were terminated and products were resolved by denaturing polyacrylamide gel electrophoresis (12% acrylamide, 8 M urea, 2 h at 2000 V), visualized and quantified using Molecular Dynamics PhosphorImager and ImageQuant software. Primer elongation was calculated as percent of total primer termini.
Coordinates
The Protein Data Bank (PDB) accession code for the atomic coordinates and structure factors of T/dATP is 2AGQ, for T/dGTP is 2AGO and for T/G is 2AGP.
Supplementary Material
Supplementary Information
Supplementary data
Acknowledgments
We thank the referees for constructive and insightful comments and suggestions. This research was supported by the Intramural Research Program of the NIH, NIDDK and NICHD.
References
- Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA (1998) Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 37: 15908–15917 [DOI] [PubMed] [Google Scholar]
- Bebenek K, Kunkel TA (2004) Functions of DNA polymerases. Adv Protein Chem 69: 137–165 [DOI] [PubMed] [Google Scholar]
- Boudsoçq F, Iwai S, Hanaoka F, Woodgate R (2001) Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res 29: 4607–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudsoçq F, Kokoska RJ, Plosky BS, Vaisman A, Ling H, Kunkel TA, Yang W, Woodgate R (2004) Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J Biol Chem 279: 32932–32940 [DOI] [PubMed] [Google Scholar]
- Brieba LG, Eichman BF, Kokoska RJ, Doublie S, Kunkel TA, Ellenberger T (2004) Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J 23: 3452–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Dahlberg ME, Benkovic SJ (1991) Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry 30: 4835–4843 [DOI] [PubMed] [Google Scholar]
- Deutscher MP, Kornberg A (1969) Enzymatic synthesis of deoxyribonucleic acid. 28. The pyrophosphate exchange and pyrophosphorolysis reactions of deoxyribonucleic acid polymerase. J Biol Chem 244: 3019–3028 [PubMed] [Google Scholar]
- Doublie S, Sawaya MR, Ellenberger T (1999) An open and closed case for all polymerases. Struct Fold Des 7: R31–R35 [DOI] [PubMed] [Google Scholar]
- Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391: 251–258 [DOI] [PubMed] [Google Scholar]
- Dutta S, Li Y, Johnson D, Dzantiev L, Richardson CC, Romano LJ, Ellenberger T (2004) Crystal structures of 2-acetylaminofluorene and 2-aminofluorene in complex with T7 DNA polymerase reveal mechanisms of mutagenesis. Proc Natl Acad Sci USA 101: 16186–16191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala KA, Suo Z (2004) Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry 43: 2116–2125 [DOI] [PubMed] [Google Scholar]
- Franklin MC, Wang J, Steitz TA (2001) Structure of the replicating complex of a pol α family DNA polymerase. Cell 105: 657–667 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W (1995) DNA Repair and Mutagenesis. Washington, DC: ASM Press [Google Scholar]
- Glick E, Chau JS, Vigna KL, McCulloch SD, Adman ET, Kunkel TA, Loeb LA (2003) Amino acid substitutions at conserved tyrosine 52 alter fidelity and bypass efficiency of human DNA polymerase η. J Biol Chem 278: 19341–19346 [DOI] [PubMed] [Google Scholar]
- Huang H, Chopra R, Verdine GL, Harrison SC (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282: 1669–1675 [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Beese LS (2004) Structures of mismatch replication errors observed in a DNA polymerase. Cell 116: 803–816 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW (1991) Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Junop MS, Haniford DB (1996) Multiple roles for divalent metal ions in DNA transposition: distinct stages of Tn10 transposition have different Mg2+ requirements. EMBO J 15: 2547–2555 [PMC free article] [PubMed] [Google Scholar]
- Kool ET (2002) Active site tightness and substrate fit in DNA replication. Annu Rev Biochem 71: 191–219 [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K (2000) DNA replication fidelity. Annu Rev Biochem 69: 497–529 [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Loeb LA (1979) On the fidelity of DNA replication. Effect of divalent metal ion activators and deoxyrionucleoside triphosphate pools on in vitro mutagenesis. J Biol Chem 254: 5718–5725 [PubMed] [Google Scholar]
- Lecomte P, Doubleday OP, Radman M (1986) Evidence for an intermediate in DNA synthesis involving pyrophosphate exchange. A possible role in fidelity. J Mol Biol 189: 643–652 [DOI] [PubMed] [Google Scholar]
- Li Y, Dutta S, Doublie S, Bdour HM, Taylor JS, Ellenberger T (2004) Nucleotide insertion opposite a cis-syn thymine dimer by a replicative DNA polymerase from bacteriophage T7. Nat Struct Mol Biol 11: 784–790 [DOI] [PubMed] [Google Scholar]
- Li Y, Korolev S, Waksman G (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J 17: 7514–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W (2003) Replication of a cis-syn thymine dimer at atomic resolution. Nature 424: 1083–1087 [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W (2001) Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 107: 91–102 [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W (2004a) Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol Cell 13: 751–762 [DOI] [PubMed] [Google Scholar]
- Ling H, Sayer JM, Plosky BS, Yagi H, Boudsocq F, Woodgate R, Jerina DM, Yang W (2004b) Crystal structure of a benzo[a]pyrene diol epoxide adduct in a ternary complex with a DNA polymerase. Proc Natl Acad Sci USA 101: 2265–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer PR, Matsuura SE, Mian AM, So AG, Scott WA (1999) A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol Cell 4: 35–43 [DOI] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK (2004) Replication by human DNA polymerase-ι occurs by Hoogsteen base-pairing. Nature 430: 377–380 [DOI] [PubMed] [Google Scholar]
- Natrajan G, Lamers MH, Enzlin JH, Winterwerp HH, Perrakis A, Sixma TK (2003) Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res 31: 4814–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obmolova G, Ban C, Hsieh P, Yang W (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407: 703–710 [DOI] [PubMed] [Google Scholar]
- Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R (2001) The Y-family of DNA polymerases. Mol Cell 8: 7–8 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Rozovskaia TA, Tarusova NB, Minasian S, Atrazhev AM, Kukhanova MK (1989) [Analogs of pyrophosphate in a pyrophosphorolysis reaction catalyzed by DNA polymerases]. Mol Biol (Mosk) 23: 862–871 [PubMed] [Google Scholar]
- Shah AM, Li SX, Anderson KS, Sweasy JB (2001) Y265H mutator mutant of DNA polymerase β. Proper teometric alignment is critical for fidelity. J Biol Chem 276: 10824–10831 [DOI] [PubMed] [Google Scholar]
- Silvian LF, Toth EA, Pham P, Goodman MF, Ellenberger T (2001) Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nat Struct Biol 8: 984–989 [DOI] [PubMed] [Google Scholar]
- Trincao J, Johnson RE, Wolfle WT, Escalante CR, Prakash S, Prakash L, Aggarwal AK (2004) Dpo4 is hindered in extending a G.T mismatch by a reverse wobble. Nat Struct Mol Biol 11: 457–462 [DOI] [PubMed] [Google Scholar]
- Uljon SN, Johnson RE, Edwards TA, Prakash S, Prakash L, Aggarwal AK (2004) Crystal structure of the catalytic core of human DNA polymerase κ. Structure (Camb) 12: 1395–1404 [DOI] [PubMed] [Google Scholar]
- Vaisman A, Tissier A, Frank EG, Goodman MF, Woodgate R (2001) Human DNA polymerase ι promiscuous mismatch extension. J Biol Chem 276: 30615–30622 [DOI] [PubMed] [Google Scholar]
- Vermote CL, Vipond IB, Halford SE (1992) EcoRV restriction endonuclease: communication between DNA recognition and catalysis. Biochemistry 31: 6089–6097 [DOI] [PubMed] [Google Scholar]
- Villani G, Tanguy Le Gac N, Wasungu L, Burnouf D, Fuchs RP, Boehmer PE (2002) Effect of manganese on in vitro replication of damaged DNA catalyzed by the herpes simplex virus type-1 DNA polymerase. Nucleic Acids Res 30: 3323–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vipond IB, Moon BJ, Halford SE (1996) An isoleucine to leucine mutation that switches the cofactor requirement of the EcoRV restriction endonuclease from magnesium to manganese. Biochemistry 35: 1712–1721 [DOI] [PubMed] [Google Scholar]
- Yang L, Arora K, Beard WA, Wilson SH, Schlick T (2004) Critical role of magnesium ions in DNA polymerase β's closing and active site assembly. J Am Chem Soc 126: 8441–8453 [DOI] [PubMed] [Google Scholar]
- Yang W (2003) Damage repair DNA polymerases Y. Curr Opin Struct Biol 13: 23–30 [DOI] [PubMed] [Google Scholar]
- Zhou BL, Pata JD, Steitz TA (2001) Crystal structure of a DinB lesion bypass DNA polymerase catalytic fragment reveals a classic polymerase catalytic domain. Mol Cell 8: 427–437 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary data