

ABSTRACT
TIGIT (T cell immunoreceptor with immunoglobulin and tyrosine‐based inhibitory motif (ITIM) domain), Vstm3, and VSIG9, are newly recognized immunological checkpoints. They are prominently expressed on CD4+ and CD8+ T cells, tumor‐infiltrating lymphocytes (TILs), natural killer (NK) cells, and regulatory T cells (Tregs). The TIGIT (TIGIT) protein is crucial for immune modulation since it diminishes NK cell populations and hinders T cell activity in cancer patients and experimental models. CD155, the principal ligand of TIGIT in humans, has been recognized as a pivotal target for immunotherapy owing to its interaction with TIGIT. CD155 is linked to the efficacy of anti‐programmed cell death protein 1 (PD‐1) therapy, even without TIGIT expression, underscoring its importance in immune checkpoint suppression. Anti‐TIGIT medicines, either independently or in conjunction with anti‐PD‐1 treatments, have demonstrated potential in augmenting immune responses to malignancies. This review examines the structural and functional characteristics of the TIGIT protein, new developments in anti‐TIGIT drugs, and their prospective use in cancer immunotherapy.
Keywords: cancer, immunotherapy, PD‐1, resistance, TIGIT

1. Introduction
TIGIT, also known as T cell immunoreceptor with immunoglobulin and tyrosine‐based inhibitory motif (ITIM) domain (TIGIT), V‐set and immunoglobulin domain‐containing protein 9 (VSIG9), and V‐set and transmembrane domain‐containing protein 3 (VSTM3), is a co‐inhibitory protein that was first identified in 2009 and is a part of the immunoglobulin (Ig) superfamily [1, 2, 3, 4, 5]. It is composed of an immunoglobulin tail tyrosine (ITT)‐like motif and an immunoreceptor tyrosine‐based inhibitory motif (ITIM), as well as an extracellular immunoglobulin variable (IgV) domain and type I transmembrane domain [6, 7]. NK cells, T cells, and regulatory T cells (Tregs) are the only cell types on which TIGIT is expressed [8, 9].
The expression of TIGIT on regulatory B cells (Bregs) inhibits immunological responses by enhancing anti‐inflammatory cytokines such as IL‐10, hence fostering immune tolerance and aiding tumor immune evasion inside the tumor microenvironment (TME) [10].
1.1. Function and Mechanism
TIGIT family receptors regulate innate and adaptive immunity by transferring inhibitory signals to immune cells through intricate interactions with ligands [11, 12]. TIGIT, on the one hand, naturally suppresses T cell activation. First, TIGIT attaches itself to CD155 to send intracellular inhibitory signals that essentially block the production and signaling of the T cell receptor (TCR). When TIGIT is engaged, TCR‐induced p‐ERK signaling and interferon‐γ (IFNγ) production in CD8+ T cells are reduced, as well as the TCRα chain and other TCR complex constituents are down‐regulated [13, 14]. Second, TIGIT competes with its costimulatory counterpart CD226 with a stronger affinity for CD155 (Figure 1), which impairs T cell activity by either directly disrupting CD226 homodimerization or by reducing T‐bet expression and IFNγ production [16, 17].
FIGURE 1.
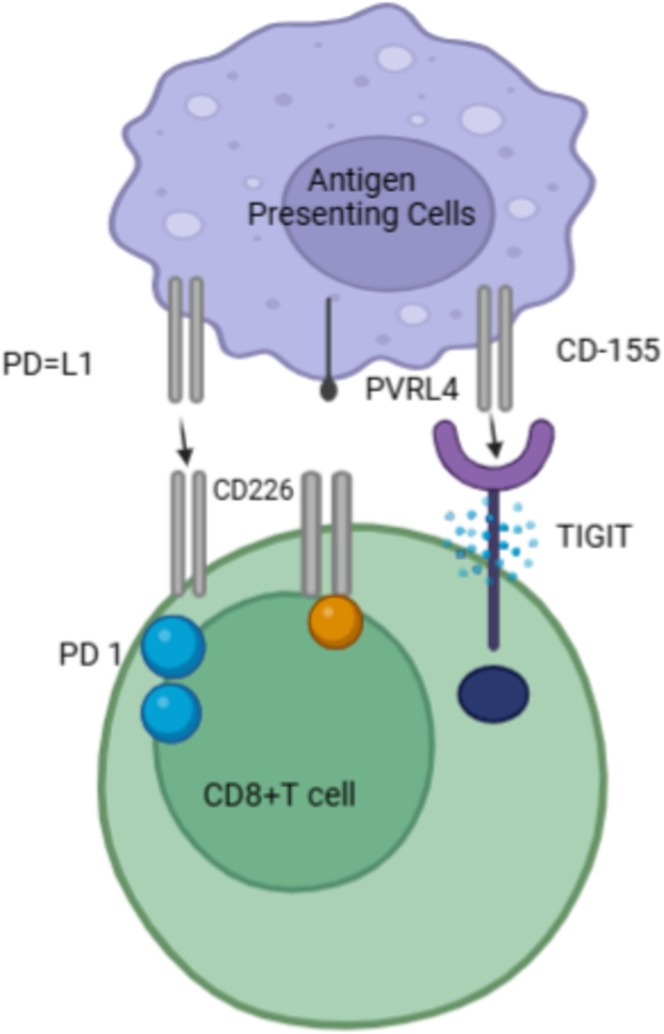
TIGIT receptors and ligands interaction. Modified from [15].
However, TIGIT can exogenously strengthen Treg cells' immunosuppressive properties. Treg cells are more prosperous in TIGIT and are linked to the ability of effector T cells to inhibit. On the other hand, CD226 stimulates the release of IFNγ and other effector cytokines while preventing the growth of Treg cells [18, 19]. By producing more interleukin‐10 (IL‐10) and fibrinogen‐like protein 2 (Fgl2), TIGIT expression on Treg cells also inhibits the growth of effector T cells and the response of pro‐inflammatory T helper 1 (Th1) and Th17 cells, but not Th2 cells [20]. Additionally, TIGIT can inhibit T cell activation by disrupting DCs and macrophage‐mediated cytokine production. In response to TIGIT's interaction with DC‐expressed CD155, extracellular signal‐regulated kinase (ERK) signaling phosphorylates CD155. This decreases pro‐inflammatory cytokine IL‐12 and increases the production of anti‐inflammatory cytokine IL‐10, impairing T cell function [21]. Through the nuclear translocation of c‐Maf, TIGIT also increases the secretion of IL‐10 and decreases the production of IFNγ and tumor necrosis factor α (TNFα), transforming macrophages from an M1 to an anti‐inflammatory M2 phenotype [22]. Furthermore, TIGIT either directly causes tumor‐infiltrating NK cells to become exhausted, resulting in a decrease in IFNγ and TNF production, or it indirectly aids in the exhaustion of CD8+ T cells, thereby compromising the anti‐tumor immune response (Figure 2).
FIGURE 2.
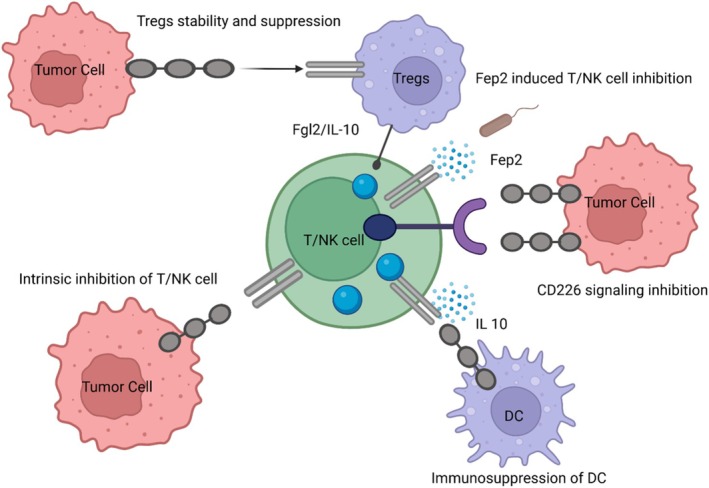
Mechanisms of TIGIT inhibition of T cells. TIGIT displays multiple inhibitory mechanisms in T cells. (a) TIGIT signaling in Tregs increases their immunosuppressive properties. (b) Fap2 protein from the Fusobacterium nucleatum binds TIGIT to trigger inhibitory signals. (c) TIGIT binds CD155 with greater affinity than CD226 or disrupts homodimerization of CD226 to impede CD226‐mediated T cell activation. (d) TIGIT adheres CD155 on APCs to spark IL‐10 production and decrease IL‐12 production, indirectly inhibiting T cells. (e) TIGIT binds CD155 and produces direct inhibitory signals in T cells. Modified from [15]. Re‐use permitted under CC BY‐NC.
Recent research has unveiled that the expression of TIGIT could differ, swayed by many factors. Numerous things can have an impact on TIGIT expression (Figure 3). Notably, 1α,25‐Dihydroxyvitamin D3 (1α,25(OH)(2)D(3)), GITR antibodies, interferon‐I (IFN‐I), and chemotherapy drugs have been shown to downregulate its expression [23]. On the other hand, it has been discovered that TIGIT expression is increased by microwave ablation, glucose deprivation, hypoxic circumstances, inhaled IL‐15, CCL23, IL‐10, and anti‐PD‐1/PD‐L1 treatments, all of which contribute to an immunosuppressive TME [24].
FIGURE 3.
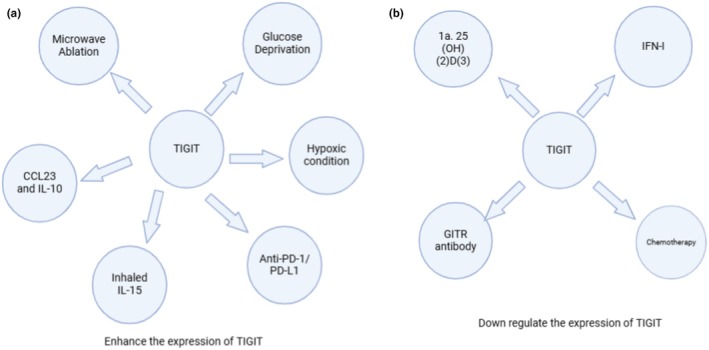
Factors affecting the expression of TIGIT. Preferably, (A) glucose deprivation, microwave ablation, hypoxic conditions, as well as the presence of CCL23, IL‐10, inhaled IL‐15, and anti‐PD‐1/PD‐L1 therapies would enhance the expression of TIGIT, In contrast, (B) chemotherapy agents, Interferon‐I (IFN‐I), GITR antibodies, and 1α,25‐Dihydroxyvitamin D3 would downregulate its expression, thereby paving towards an immunosuppressive TME. Modified from [15].
1.2. Second‐Generation Immunotherapeutic Targets: TIGIT (TIGIT)
The exploration of novel immune checkpoint receptors has been spurred by the immunotherapeutic successes in targeting the CTLA‐4 and PD‐1 pathways to expand the therapeutic repertory [25]. T cell immunoreceptors with Ig and ITIM domains (TIGIT) and T cell immunoglobulin and mucin‐domain containing‐3 (TIM‐3) are two of the expanding group of second‐generation immune checkpoint targets if CTLA‐4 and PD‐1 are to be regarded as first‐generation immune checkpoint targets [26].
Combination therapies have been spurred by promising pre‐clinical research involving anti‐TIGIT agents [27]. Anti‐TIGIT agents are currently being tested in 16 active clinical trials for a variety of conditions, such as solid and metastatic tumors, multiple myeloma, esophageal squamous cell carcinoma, cervical and ovarian cancer, and non‐small cell lung cancer (NSCLC) [28]. In one experiment, patients with non‐small cell lung cancer (NSCLC) were treated with tiragolumab (Roche), an anti‐TIGIT treatment, in combination with anti‐PD‐L1 atezolizumab. The combination produced twice as high objective response rates (31.3%) as atezolizumab monotherapy (16.2%). Additionally, adverse events were reported in 80.6% of patients receiving combination treatment, with high‐grade adverse events occurring in 14.9% of patients [29]. The FDA deemed this safety profile acceptable, and tiragolumab was awarded a Breakthrough Therapy Designation. This designation expedites developing and evaluating treatments for life‐threatening illnesses when preliminary data show a therapy's superiority over currently available treatments. Shortly, tiragolumab is likely the first anti‐TIGIT medication approved by the FDA [30].
2. Emerging Anti‐TIGIT Agents
Efforts to target TIGIT have led to the development of novel therapeutics aimed at disrupting its inhibitory signals. These agents exhibit several promising features [14].
2.1. Dual Mechanism of Action
Anti‐TIGIT agents have the ability to activate immunological effector functions in addition to blocking the inhibitory connection between TIGIT and its ligands. They improve the identification and elimination of tumor cells by enabling immune cells to fully realize their anti‐tumor potential.
2.2. Elective Targeting and Synergistic Potential
Latent warheads are engineered explicitly for selective tumor microenvironment (TME) activation, substantially reducing off‐target effects and systemic toxicity. This precise targeting augments the therapeutic index and enhances patient tolerance. Latent warheads exhibit synergistic potential to enhance therapeutic outcomes by integrating selective activation with diminished systemic toxicity.
Latent warheads facilitate targeted activation within the tumor microenvironment (TME), hence reducing systemic toxicity. Nectin‐4, a ligand for TIGIT, is crucial in tumor advancement and immune evasion. The overexpression of this component is utilized in antibody‐drug conjugates, improving precision targeting, therapeutic efficacy, and patient tolerance through selective activation and minimized off‐target effects.
2.3. Resilience Against Immune Evasion
By blocking TIGIT, malignancies may be able to thwart immune evasion tactics, such as the overexpression of alternative checkpoints like indoleamine 2,3‐dioxygenase (IDO) or programmed death‐ligand 1 (PD‐L1), which would strengthen the response's persistence.
Examples of TIGIT inhibitors exhibiting properties include Tiragolumab: which is a monoclonal antibody that targets TIGIT to augment T‐cell activation and increase anti‐tumor immunity.
Ociperlimab (BGB‐A1217): Inhibits TIGIT signaling to reinstate immune surveillance against neoplasms.
Etigilimab (OMP‐313 M32): Integrates TIGIT inhibition with checkpoint blockage to provide synergistic anti‐cancer action.
MK‐7684A: A TIGIT inhibitor combined with pembrolizumab for advanced‐stage malignancies.
Alternative immune checkpoint overexpression: Overexpression of alternative immune checkpoints, such as TIGIT or LAG‐3, facilitates immune evasion by malignancies, inhibiting T‐cell activation and allowing cancer growth despite suppressing the PD‐1/PD‐L1 pathway.
3. Anti‐TIGIT (TIGIT) Small Molecules
According to Zhou et al. [31], liothyronine administration in vivo might dramatically reduce tumor growth by boosting CD8+ T cell infiltration and immunological responses in tumor‐bearing mice. The immune cell depletion model demonstrated that CD4+ T cells, CD8+ T cells, and NK cells are necessary for liothyronine's antitumor actions. Azelnidipine has been reported by Zhou et al. [32] to co‐target SIRPα and PVR, hence dual blocking the TIGIT/PVR and CD47/SIRPα pathways. When co‐cultured with tumor cells, Azelnidipine may improve the phagocytosis of macrophages in vitro. Azelnidipine, by itself or in conjunction with radiation therapy, has the potential to slow the growth of MC38 tumors in vivo dramatically. By improving CD8+ T cell infiltration and function in the cancer as well as the systemic immune response in the tumor‐draining lymph node and spleen in a CD8+ T cell‐dependent manner, azelnidipine was also found to suppress the growth of CT26 tumors [32]. Using the T cell immunoglobulin and ITIM domain (TIGIT) target, Xiong et al. [33] utilized the DEL technology and produced unique hit compound 1 (IC50 = 20.7 μM). A machine learning (ML) modeling technique was developed to solve the problem of low sample distribution uniformity, which is also commonly encountered in DEL screening on new targets based on the screening data from DEL and hit derivatives. The data was reported by Shaw et al. [5], who also outlined the potential of elraglusib as an immune‐modulatory agent and illustrated the advantages of a sequential approach in melanoma, starting with immune checkpoint inhibition and ending with GSK‐3β inhibition. This approach also justified elraglusib's combination with immune checkpoint inhibitory molecules, such as those that target PD‐1, TIGIT, and LAG‐3, to be investigated clinically [5]. A sequential strategy for elraglusib in melanoma entails its initial application to target particular pathways that promote tumor growth, succeeded by combination medicines that augment immune response or mitigate resistance, so optimizing therapeutic results and improving patient prognosis [34].
Elraglusib is a powerful inhibitor of glycogen synthase kinase‐3 (GSK‐3), a crucial regulator of cellular functions including proliferation, differentiation, and death. Elraglusib alters tumor development and survival pathways by inhibiting GSK‐3, presenting therapeutic potential, especially in melanoma and other malignancies reliant on GSK‐3 activity [35].
Zhou et al. [36] conducted studies on the small molecule Hemin, which was screened using virtual molecular docking and a cell‐based blocking assay to disrupt the TIGIT/PVR interaction. Hemin was able to increase Jurkat‐hTIGIT cells' production of IL‐2 successfully. Hemin stimulated CD8+ T cells to rerelease IFN‐γ, and the increased IFN‐γ could work in concert with Hemin to cause tumor cells to undergo ferroptosis. By increasing the immunological response of CD8+ T cells and causing ferroptosis in the CT26 tumor model, Hemin suppressed the growth of the tumor. More significantly, in an anti‐PD‐1 resistant B16 tumor model, Hemin combined with PD‐1/PD‐L1 inhibition demonstrated increased antitumor activity (Table 1) [37].
TABLE 1.
Literature cited potential new TIGIT small molecule inhibitors.





3.1. Pharmacodynamics (PD)
Pharmacodynamics (PD) examines a drug's effects on the body, specifically how TIGIT (TIGIT) small molecule inhibitors engage with their target to elicit a therapeutic response. These inhibitors are essential in regulating immune responses to malignancies by targeting TIGIT and its corresponding ligands [38].
3.1.1. Mechanism of Action
TIGIT small molecule inhibitors function by interrupting the interaction between TIGIT on immune cells and its ligands, including CD155 (PVR), found on tumor and antigen‐presenting cells. This blockage obstructs the inhibitory signaling cascade that inhibits immune cell activation. By inhibiting TIGIT, these compounds reinstate and augment the functionality of effector T cells and natural killer (NK) cells.
Augmented Cytotoxicity: The inhibition yields heightened cytotoxic responses, resulting in more efficient tumor cell destruction. Increased synthesis of cytokines such as IFN‐γ and TNF‐α enhances the anti‐tumor immune response. TIGIT inhibition can enhance the efficacy of other immune checkpoint inhibitors, including anti‐PD‐1 and anti‐PD‐L1 treatments, by mitigating redundant immunosuppressive pathways.
3.1.2. Therapeutic Effects
TIGIT inhibitors enhance the immune system's capacity to identify and eliminate cancerous cells. Principal therapeutic effects encompass:
Tumor Regression: Activating T lymphocytes and NK cells results in decelerated tumor growth and possible tumor reduction.
Enhanced Treatment Outcomes: TIGIT inhibitors and other checkpoint inhibitors increase overall response rates (ORR) and disease control rates (DCR) across multiple malignancies.
Preclinical and Clinical Efficacy: Research has shown encouraging outcomes in preclinical models and current clinical studies for malignancies such as non‐small cell lung cancer and melanoma.
3.1.3. Biomarkers and Response Monitoring
The efficacy of TIGIT inhibitors can be monitored using distinct biomarkers and immunological signatures: Increased infiltration of activated T cells into the tumor microenvironment is a characteristic of successful TIGIT inhibition. Increased concentrations of IFN‐γ and IL‐2 signify heightened immunological activation. Molecular markers, including PD‐1/PD‐L1 co‐expression and TIGIT downregulation, facilitate the evaluation of the immune landscape and predict patient responses. Modification of Treatment: Continuous biomarker surveillance can inform dosage alterations or treatment combinations, assuring optimal results.
3.2. Pharmacokinetics (PK)
Pharmacokinetics (PK) examines pharmaceuticals' absorption, distribution, metabolism, and excretion (ADME), elucidating their interactions within the organism. Comprehending these features is essential for enhancing the therapeutic efficacy and safety of TIGIT small molecule inhibitors [39].
3.2.1. Absorption
A notable benefit of TIGIT small molecule inhibitors is their capacity for oral delivery.
Oral bioavailability: Elevated oral bioavailability guarantees effective absorption into systemic circulation. Strategies include the creation of prodrugs, which convert into active molecules post‐administration, or solubility improvement methods (e.g., nanoparticle formulations), that can enhance bioavailability.
Gastrointestinal Stability: Maintaining stability in the gastrointestinal environment is essential to avert early breakdown and optimize absorption.
3.2.2. Distribution
Effective tissue penetration is a characteristic of small molecule inhibitors, enabling them to target the tumor microenvironment precisely.
Volume of Distribution (V d): Optimizing V d is essential to guarantee that the medicine effectively targets primary and metastatic tumor locations while reducing off‐target effects.
Tumor Targeting: The enhanced permeability and retention (EPR) action in tumors can promote accumulation, enhancing the therapeutic index. Lipid‐based carriers, as advanced drug delivery vehicles, can enhance tissue‐specific distribution.
3.2.3. Metabolism
TIGIT inhibitors are predominantly processed by hepatic enzymes, specifically the cytochrome P450 (CYP450) family.
Metabolic Stability: Enhancing resistance to fast hepatic metabolism is crucial for sustaining therapeutic medication concentrations. This can be accomplished via structural changes that diminish CYP450‐mediated breakdown.
Metabolite Profiling: Identifying active or harmful metabolites aids in optimizing drug design and forecasting probable adverse effects. This profiling also helps reduce drug–drug interactions in combo therapy.
Initial Metabolic Processing: Reducing first‐pass metabolism can enhance systemic exposure and effectiveness.
3.2.4. Excretion
Excretion paths predominantly encompass renal (urine) or hepatic (bile) routes, which affect the drug's half‐life and dose schedule.
Elimination Half‐Life (t1/2): Optimizing the t1/2 establishes an optimal equilibrium between dosage frequency and therapeutic efficacy, enhancing patient compliance.
Clearing Mechanisms: Comprehending renal and hepatic clearance pathways is essential, especially in patients with impaired kidney or liver function. Modifying dosage or frequency in response to renal or hepatic impairment can reduce toxicity concerns.
Combination Strategies: Excretion studies guide the formulation of combination regimens that reduce cumulative toxicities and enhance synergy with other therapeutic medicines.
3.3. Optimizing PK and PD Properties
Enhancing pharmacokinetic (PK) and pharmacodynamic (PD) characteristics is essential for enhancing the therapeutic efficacy of TIGIT small molecule inhibitors and reducing side effects. Efficient optimization connects drug design with clinical effectiveness.
3.3.1. Balancing PK and PD
Optimizing the pharmacokinetic and pharmacodynamic characteristics guarantees therapeutic efficacy while minimizing adverse effects.
The therapeutic window: Sustaining medication concentrations within the therapeutic window is crucial for maximizing efficacy and reducing toxicity. This entails comprehending the lowest effective concentration (MEC) and the maximum tolerable concentration (MTC).
Dosing Regimen: Optimized regimens, such as once‐daily oral administration, can sustain stable medication concentrations, improve patient adherence, and streamline treatment. Sustained‐release formulations or depot injections can assist in maintaining consistent plasma concentrations.
Personalized Therapy: Dosing regimens can be customized according to patient‐specific variables such as metabolism, renal function, and concomitant drugs to attain an equilibrium between pharmacokinetics and pharmacodynamics.
3.3.2. Drug Design Considerations
Structural alterations in pharmacological design can substantially improve pharmacokinetic and pharmacodynamic properties.
Enhancing lipophilic areas of the molecule augments tissue penetration and facilitates the traversal of biological obstacles, including the tumor microenvironment.
Incorporating polar functional groups improves aqueous solubility, facilitating enhanced absorption and bioavailability.
Prodrug Strategies: Prodrugs are a novel methodology wherein inert precursors are digested in vivo to yield active pharmaceuticals. This enhances solubility, stability, and bioavailability while diminishing off‐target effects.
Molecular Stability: Improving resistance to enzymatic degradation (e.g., by CYP450) and minimizing the generation of reactive metabolites guarantees extended efficacy and mitigates toxicity risks.
3.3.3. Preclinical and Clinical Studies
Thorough preclinical and clinical assessments are essential for enhancing PK/PD profiles.
Preclinical Studies: Animal models offer critical insights into drug absorption, distribution, metabolism, excretion (ADME), and the therapeutic window. Pharmacological profiling at this juncture aids in forecasting human pharmacokinetic/pharmacodynamic interactions and toxicity.
Phase I Clinical Trials: Concentrate on safety, tolerability, and first pharmacokinetic/pharmacodynamic profile in healthy volunteers or a limited patient cohort. Determining the maximum tolerated dosage (MTD) and recognizing first pharmacokinetic patterns are primary aims.
Phase II and III Clinical Trials: These trials evaluate long‐term safety, appropriate dose regimens, and treatment effectiveness in larger, more heterogeneous patient groups. PK/PD connections are further elucidated to ascertain therapy response and resistance determinants.
Pharmacokinetic/Pharmacodynamic Modeling: Advanced computational modeling amalgamates data from preclinical and early‐phase trials to forecast clinical outcomes, refine dosing regimens, and discern biomarkers for response assessment.
3.4. Recent Advancements in the Development of TIGIT (TIGIT) Small Molecule Inhibitors
Recent advancements in the development of small molecule inhibitors targeting TIGIT focus on enhancing anti‐tumor immunity by blocking this inhibitory signal. Here are some key developments [40, 41].
3.4.1. Understanding TIGIT Structure and Function
Thorough structural analyses of TIGIT and its interactions with ligands, such as CD155 (PVR), have made the logical design of inhibitors easier. Essential insights into the binding surfaces and conformational dynamics required for efficient inhibition have been gained from these investigations. According to studies on its function in immune modulation, TIGIT is a promising therapeutic target in several cancers, including melanoma, lung cancer, and colorectal cancer.
3.4.2. Preclinical Studies
High‐throughput screening (HTS) methods have been used to find tiny compounds that can cause interactions between TIGIT and CD155 to break down. The capacity of libraries of various chemical compounds to block this pathway has been investigated. In preclinical research, chemical inhibitors have been shown to employ cellular assays and animal models to augment T cell and NK cell activity against malignancies, resulting in notable anti‐tumor effects.
3.4.3. Optimization of Lead Compounds
Improved potency, selectivity, and pharmacokinetic characteristics have been achieved by optimizing the first screening hits. The goal of medicinal chemistry is to make these molecules more drug‐like. Extensive SAR research has been carried out to pinpoint the essential functional groups and molecular characteristics necessary for these compounds' inhibitory action.
3.4.4. Clinical Development
Some small molecule TIGIT inhibitors have entered early‐phase clinical trials. These trials aim to assess the safety, pharmacokinetics, tolerability, and preliminary efficacy of these compounds in humans. There is a growing interest in combining TIGIT inhibitors with other immune checkpoint inhibitors (e.g., anti‐PD‐1/PD‐L1 antibodies) to enhance anti‐tumor responses. Clinical trials are underway to assess the synergistic effects of these combination therapies [42].
3.5. Advantages
3.5.1. Oral Bioavailability
Small compounds, as opposed to monoclonal antibodies, often requiring intravenous infusion, can be given orally. This enhances convenience and patient compliance. Because oral delivery eliminates the requirement for infusion clinic visits, it may result in cheaper healthcare expenses.
3.5.2. Tumor Penetration
Compared to bigger biologics, small molecules usually penetrate tissues and tumors more effectively, making it possible for them to reach and inhibit their targets within the tumor microenvironment more successfully.
3.5.3. Manufacturing and Scalability
Compared to biologics like monoclonal antibodies, which need intricate production and purification procedures, small molecules are typically more accessible and less expensive to produce on a large scale. Since small molecules are frequently made with high consistency and purity, batch‐to‐batch variability is decreased.
3.5.4. Modularity and Optimization
It is possible to chemically optimize small compounds to enhance their potency, selectivity, and half‐life, among other pharmacokinetic and pharmacodynamic qualities. Small compounds are more accessible for researchers to tweak to increase their specificity and binding affinity, which could result in more potent inhibitors.
3.5.5. Cost‐Effectiveness
Generally, the development and production costs for small molecule drugs are lower compared to biologics, which may translate to more affordable treatment options for patients.
3.6. Disadvantages
3.6.1. Selectivity and off‐Target Effects
Sometimes, interactions between small compounds and many targets might result in off‐target effects and related side effects. It is imperative to ensure high specificity for TIGIT to reduce these dangers. Throughout the medication development process, off‐target interactions must be closely monitored and managed, since they can have a harmful impact.
3.6.2. Shorter Half‐Life
Compared to monoclonal antibodies, small compounds frequently have shorter half‐lives, which means that additional doses may be needed to maintain therapeutic levels, which could affect patient adherence. The body rapidly metabolizes small compounds. Thus, formulations that maintain their presence at therapeutic levels for a sufficient time are required.
3.6.3. Drug Resistance
Small molecule inhibitors can cause cancer cells to become resistant to them through various processes, including target protein mutations or the activation of alternate signaling pathways. The continuous development of combination medicines or second‐generation inhibitors is necessary for this.
3.6.4. Immunogenicity
Small molecules can provoke immunological responses in certain people, while usually being less than biologics; this could result in diminished efficacy or worse side effects.
3.6.5. Complexity in Targeting
Small molecules have a more remarkable ability to enter cells than antibodies, but it can be challenging to ensure they reach the intracellular targets and block them successfully. Designing compounds that can pass through cell membranes and stay stable inside cells is difficult.
3.7. Structure–Activity Relationship (SAR) Studies
3.7.1. Core Structure Analysis
Pharmacophores–key functional groups–are identified as being in charge of binding and action. To learn more about how these pharmacophore modifications affect activity, they are investigated. Enhancing binding affinity and selectivity involves methodically altering the lead compound's core scaffold and retaining and optimizing structural motifs that interact well with the TIGIT binding site.
3.7.2. Functional Group Modification
The proportion of hydrophobic and hydrophilic groups must be balanced for the best binding and solubility. The modifications aim to preserve sufficient solubility while improving interactions with the hydrophobic pockets of TIGIT. Improved binding affinity and specificity are achieved by altering the electronic characteristics of substituents, such as electron‐donating or withdrawing groups, to maximize interactions with the TIGIT binding site.
4. Anti TIGIT Antibodies
Numerous anti‐TIGIT antibodies are in clinical trials currently, and others are in preclinical development. Therefore, more data are expected in the next few years regarding the efficacy of this new checkpoint inhibitor in multiple solid and hematologic malignancies. However, preliminary data are promising, and anti‐TIGIT treatment seems to confer more favorable responses when combined with anti‐PD‐1/anti‐PD‐L1 compared to either agent alone.
4.1. Clinical Trials
4.1.1. Advanced Solid Tumors
Tiraglobumab was administered in a dose‐escalation manner to 73 individuals with solid tumors in a phase Ia/Ib study [38]. Merely 4% of patients encountered treatment‐related adverse events of grade 3 or higher. The most frequently reported adverse events were anemia (31%) and fatigue (38%). There were no signs of dose‐limiting effects. ORR was 50%, and DCR was 79% in the metastatic NSCLC expansion cohort 9 (n = 14). The recommended dosage of tiragolumab was established at 600 mg every 3 weeks based on the maximal receptor occupancy and clinical activity seen in the phase 1 data [43, 44].
In PD‐L1 positive advanced/metastatic NSCLC without EGFR or ALK mutations, tiragolumab in conjunction with atezolizumab was investigated in the phase II CITYSCAPE (NCT03563716) trial [45, 46]. One hundred thirty‐five individuals were randomly assigned to receive atezolizumab + placebo or tiragolumab 600 mg plus 1200 mg IV every 3 weeks. The trial demonstrated a significant improvement in ORR (37% vs. 21%) and PFS (5.6 vs. 3.9 months; hazard ratio (HR) 0.59) at a median follow‐up of 10.5 months. Further stratified by high PD‐L1 status (PD‐L1 expression at least 50%), the combination's ORR rose to 66%, while atezolizumab alone was just 24% [47]. Serious adverse events attributable to treatment were experienced by 21% of participants in the combination group and 18% of individuals in the atezolizumab alone cohort. The most frequent adverse event, accounting for 3% of cases with atezolizumab and 9% of combinations, was a lipase rise. In the combination cohort, two treatment‐related fatalities (from infection and pyrexia) occurred. These results led to the start of the phase III SKYSCRAPER‐01 investigation.
In newly diagnosed metastatic NSCLC with PD‐L1 expression of at least 50%, the combination of tiragolumab and atezolizumab was examined in the phase III SKYSCRAPER‐01 trial (NCT04294810). Five hundred thirty‐four patients in all were randomly assigned to this investigation. The study's co‐primary endpoint of a PFS improvement was not met at the time of intermediate analysis. The overall survival (OS) data were still in their infancy during the analysis [48]. The study will continue until the following planned data analysis.
Phase III SKYSCRAPER‐02 (NCT04256421) is now underway in the SCLC space. In this trial, 490 patients with newly diagnosed extensive‐stage small cell lung cancer are randomized to receive 600 mg IV of tiragolumab every 3 weeks along with carboplatin and etoposide, either in combination (n = 247) or alone (n = 247). An interim analysis was conducted on 397 individuals with a median follow‐up of 14.3 months. PFS improvement was not the co‐primary outcome of this trial. With tiragolumab, the median PFS was 5.4 months (95% CI 4.7–5.5) as opposed to 5.6 months without this medication. By the anticipated final analysis, the other co‐primary endpoint of OS was not expected to attain statistical significance. For both groups, the median OS was 13.6 months (95% CI 10.8–14.9). Stratifying for brain metastases, lactate dehydrogenase levels, and Eastern Cooperative Oncology Group (ECOG) performance status (PS) did not significantly alter OS. Furthermore, there was no significant difference in ORRs between the cohorts (70.8% (95% CI 64.6–76.3) for tiragolumab and 65.6 (95% CI 59.3–71.4) for placebo). With tiragolumab, the duration of response (DOR) was 4.2 months (95% CI 4.1–4.4), but it was 5.1 months (95% CI 4.4–5.8) in the absence of the medication. The investigation will go on until the scheduled final OS analysis. In SKYSCRAPER‐02, adverse events were frequent; in both cohorts, 99.6% of patients reported side effects. 69.4% of patients receiving tiragolumab experienced grade 3 or more significant adverse events, compared to 70.3% of patients getting a placebo. 5.0% and 5.3% of patients receiving tiragolumab and placebo, respectively, withdrew due to toxicity. The most frequent side effects were alopecia, neutropenia, and anemia; however, the tiragolumab group experienced pruritus more frequently. Molecules ongoing clinical trials targeting TIGIT in advanced solid tumors are given in Table 2.
TABLE 2.
Ongoing clinical trials targeting TIGIT in advanced solid tumors and hematological malignancies.
| S. No | Target | Drug/intervention | Tumor type | Current status in clinical trial |
|---|---|---|---|---|
| 1. |
TIGIT + PD‐L1 |
Tiragolumab + atezolizumab |
Cervical cancer Gastric cancer and esophageal adenocarcinoma Urothelial carcinoma Pancreatic adenocarcinoma NSCLC SCLC HNSCC |
Ongoing (NCT04300647) Ongoing (NCT03281369) Ongoing (NCT03869190) Ongoing (NCT03193190) Ongoing (NCT03563716, NCT04619797) Ongoing (NCT04308785) |
| 2. |
Domvanalimab + zimberelimab |
NSCLC Advanced solid tumors |
Ongoing (NCT04262856) Ongoing (NCT03628677) |
|
| 3. | BMS‐986207 + nivolumab |
Advanced solid tumors |
Ongoing (NCT02913313, NCT04570839) | |
| Completed and ongoing phases III trials | ||||
|
4. |
TIGIT + PD‐1 |
Ociperlimab + tislelizumab vs. pembrolizumab | Locally advanced unresectable, or metastatic NSCLC, PD‐L1 ≥ 50%, first line |
Ongoing NCT04746924 |
|
5. |
TIGIT + PD‐1 |
Ociperlimab + tislelizumab + concurrent chemoradiotherapy (cCRT) followed by ociperlimab + tislelizumab vs. tislelizumab + cCRT followed by tislelizumab vs. cCRT followed by durvalumab |
Stage III NSCLC |
Ongoing NCT04866017 |
|
6. |
TIGIT + PD‐1 |
Atezolizumab + tiragolumab vs. atezolizumab |
Esophageal squamous cell Carcinoma after Chemo‐radiotherapy |
Ongoing SKYSCRAPER‐07 |
|
7. |
TIGIT + PD‐1 |
Atezolizumab + tiragolumab vs. atezolizumab |
Locally advanced unresectable or metastatic NSCLC |
No improvement in PFS SKYSCRAPER‐01 |
| 8. | TIGIT + PD‐1 |
Tiragolumab + atezolizumab + platinum‐pemetrexed vs. pembrolizumab + platinum |
Unresectable or metastatic NSCLC |
Ongoing SKYSCRAPER‐06 (NCT04619797) |
|
9. |
TIGIT + PD‐1 |
Tiragolumab + atezolizumab + carboplatin + Etoposide vs. placebo + atezolizumab + carboplatin + etoposide |
Extensive‐stage small cell lung cancer, first line |
HR for OS 1.04 [95% CI 0.79 to 1.36] HR for PFS 1.11 [95% CI 0.89 to 1.38] SKYSCRAPER‐02 |
|
10. |
TIGIT + PD‐1 |
Tiragolumab + atezolizumab + carboplatin + etoposide vs. placebo + atezolizumab + carboplatin + etoposide |
Extensive‐stage small cell lung cancer, first line |
Ongoing SKYSCRAPER‐02C (China) |
|
11. |
TIGIT + PD‐1 |
Atezolizumab + tiragolumab + paclitaxel + cisplatin vs. paclitaxel + cisplatin |
Recurrent, or metastatic esophageal Carcinoma, first line |
Ongoing SKYSCRAPER‐08 (NCT04540211) |
|
12. |
TIGIT + PD‐1 |
Tiragolumab + atezolizumab vs. durvalumab |
Unresectable stage III NSCLC |
Ongoing SKYSCRAPER‐03 (NCT04513925) |
| 13. | Pembrolizumab + vibostolimab vs. pembrolizumab |
Resected, high‐Risk Stage II‐IV melanoma, adjuvant |
Ongoing MK‐7684A‐010/ KEYVIBE‐010 |
|
| 14. |
Domvanalimab + zimberelimab + chemotherapy vs. Nivolumab + chemotherapy |
Locally advanced unresectable or metastatic gastric, gastroesophageal junction, and esophageal adenocarcinoma, first line |
Ongoing STAR‐221 |
|
| 15. |
Domvanalimab + zimberelimab + platinum‐based chemotherapy vs. pembrolizumab platinum‐based chemotherapy |
Metastatic NSCLC, first line |
Ongoing STAR‐121 |
|
| 16. |
Domvanalimab + durvalumab vs. durvalumab |
Stage III, unresectable stage III NSCLC |
Ongoing PACIFIC‐8 |
|
| Ongoing clinical trials targeting TIGIT in hematological malignancies | ||||
| 17. | TIGIT + PD‐1 | Tiragolumab combined with mosunetuzumab ± atezolizumab (anti‐PD‐L1 mAb) | Relapsed or refractory diffuse large B cell lymphoma, or follicular lymphoma | Ongoing, phase 1/2, (NCT05315713) |
| 18. | TIGIT + PD‐1 |
Tiragolumab Monotherapy/combined with rituximab/combined with daratumumab ± atezolizumab |
Relapsed or refractory B cell non‐Hodgkin's lymphoma/multiple myeloma | Ongoing, phase 1, (NCT04045028) |
| 19. | TIGIT + PD‐1 |
Ociperlimab (BGB A1217) Combined with rituximab, Combined with tislelizumab (anti‐PD‐1 mAb) |
Relapsed or refractory diffuse large B cell lymphom | Ongoing, phase 1/2, (NCT05267054) |
| 20. | TIGIT + PD‐1 | BMS‐986207 combined with pomalidomide and dexamethasone | Relapsed or refractory multiple myeloma | Ongoing, phase 1/2, (NCT04150965) |
| 21. | TIGIT + PD‐1 | Vibostolimab (MK7684A) combined with pembrolizumab (anti‐PD‐1 mAb) | Relapsed or refractory Hodgkin's lymphoma/B cell non‐Hodgkin's lymphoma/multiple myeloma | Ongoing, phase 2, (NCT05005442) |
| 22. | TIGIT + PD‐1 | COM902 monotherapy/combined with COM701 (anti‐PVRIG mAb) | Multiple myeloma | Ongoing, phase 1, (NCT04354246) |
| 23. | TIGIT + PD‐1 | SEA‐TGT monotherapy/combined with sasanlimab (anti‐PD‐1 mAb) |
Classical Hodgkin's lymphoma, relapsed or refractory diffuse large B cell lymphoma, peripheral T‐cell lymphoma |
Ongoing, phase 1, (NCT04254107) |
| 24. | TIGIT + PD‐1 | AB308, combined with zimberelimab (anti‐PD‐1 mAb) | Multiple myeloma, relapsed or refractory diffuse large B cell lymphoma | Ongoing, phase 1, (NCT04772989) |
| 25. | TIGIT + PD‐1 | EOS‐448 Monotherapy/Combined with iberdomide ± dexamethasone | Relapsed or refractory multiple myeloma; | Ongoing, phase 1/2, (NCT05289492) |
Abbreviations: IL, interleukin; LAG‐3, lymphocyte‐activation gene 3; N, number; NSCLC, non‐small cell lung cancer; NSCLC, non‐small‐cell lung cancer; ORR, objective response rate; OS, overall survival; pCR, pathologic complete response; PD‐1, programmed cell death protein 1; PD‐1, programmed cell death protein 1; PD‐L1, programmed death‐ligand 1; PD‐L1, programmed death‐ligand 1; PVRIG, poliovirus receptor‐related immunoglobulin domain‐containing; SCLC, small‐cell lung cancer; TIGIT, T‐cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine‐based inhibitory motif domains; TIGIT, T‐cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine‐based inhibitory motif domains.
4.1.2. Hematological Malignancies
Thus far, numerous studies have demonstrated the noteworthy anti‐tumor effects of TIGIT‐targeting immunotherapy. According to Catakovic, recombinant TIGIT‐Fc blocking of TIGIT would lower CLL viability in vitro by reducing the production of pro‐survival cytokines such as IL‐10 46. TIGIT expression caused CD8+ T cell death and suppressed cytokine production in AML [49, 50]. T cell function might be restored by TIGIT knockdown by siRNA by reducing apoptotic vulnerability and concurrently boosting TNFa and IFNg production. Moreover, TIGIT blockade greatly enhanced IFNg synthesis and NK cell degranulation, supporting NK cells' anti‐leukemia activities [51, 52, 53]. Likewise, elevated TIGIT expression encouraged T cell fatigue, which aided in advancing myeloma. On the other hand, the anti‐TIGIT treatment extended the lifetime of MM mice, inhibited tumor cell development, and avoided T cell exhaustion [54]. Guillerey also noted that anti‐MM CD8+ T cell survival in vivo was enhanced, and immunological function was restored by either TIGIT deficiency or mAb blockade [12, 13]. Furthermore, dual TIGIT and PD‐1 inhibition demonstrated possible synergistic immune‐killing effects. On the one hand, Wang found that AML patients had greater frequencies of TIGIT and PD‐1 dual expression, which was connected to worse remission rates and a higher frequency of FLT3‐ITD mutation. Research on Hodgkin lymphoma (HL) revealed that TIGIT and PD‐1 were co‐expressed in 68%–84% of T cells [55, 56]. FL and CLL also had a high frequency of TIGIT and PD‐1 dual expression [57, 58]. Zhang, however, found that TIGIT blockade alone only increased TNFa in TIGIT+ CD4+ T cells and IFNg, TNFa in TIGIT+ CD8+ T cells. On the other hand, in both CD4+ and CD8+ T cells, simultaneous inhibition of TIGIT, PD‐1, and Tim‐3 markedly increased IL‐2, IFNg, and TNFa, which may improve immune responses against leukemia [59]. For treating malignant lymphoma and multiple myeloma (Table 2), human anti‐TIGIT mAbs are being tested in phase 1/2 clinical trials either as a monotherapy or, in most studies, in combination with anti‐PD1/PD‐L1 antibodies or chemotherapies. This is due to the anti‐TIGIT mAbs' remarkable efficacy in solid tumors and potential immune‐killing effects mentioned above in preclinical research.
5. Toxicities Associated With TIGIT (TIGIT) Blockade
Despite the hopeful success of therapeutic strategies targeting TIGIT in hematological malignancies, over‐activated T cells can cause immune‐related adverse events (irAEs) that can lead to multiple organ dysfunction and a poor prognosis [60]. Two patients experienced adverse events (irAEs) in the first phase of the anti‐TIGIT antibody vibostolimab research, one of which was a severe skin response and the other an adrenal insufficiency [61]. An additional phase 1 trial with the combination of anti‐PD‐1 antibody tislelizumab and anti‐TIGIT antibody ociperlimab in advanced solid tumors revealed that 15 out of 26 patients experienced adverse events (irAEs), with three of those cases being severe (grade ≥ 3) [62]. The CITYSCAPE trial also found that skin rash was the most common adverse event (irAE) in 69% of patients treated with anti‐TIGIT tiragolumab and anti‐PD‐L1 antibody atezolizumab. Pancreatitis, hypothyroidism, colitis, and diabetes mellitus were the next most prevalent irAEs.
6. Critical Views
6.1. Promise in Cancer Therapy
The ability of TIGIT inhibitors to boost anti‐tumor immunity by reviving NK and T cell responses against malignancies is significant. Because small molecule inhibitors can enter tumors more effectively than larger biologics, they can be incredibly effective. Moreover, TIGIT inhibitors may work with other immune checkpoint inhibitors, such as PD‐1/PD‐L1 inhibitors, to produce synergistic effects. The combination can surmount resistance mechanisms that constrain the effectiveness of monotherapy.
6.2. Challenges and Concerns
Achieving high selectivity and specificity to prevent off‐target effects and related toxicity is a significant difficulty that has been identified. Ensuring the safety and efficacy of these inhibitors is crucial. Another issue is the possibility that cancer cells may become resistant to TIGIT inhibitors. This emphasizes the necessity of further study to comprehend and block these processes, potentially by creating next‐generation inhibitors or combination medicines. Although preclinical findings are encouraging, clinical solid trials are necessary to confirm TIGIT small molecule inhibitors' safety and effectiveness in various patient types.
6.3. Pharmacokinetics and Dosing
This research aims to maximize the pharmacokinetic characteristics of TIGIT inhibitors to guarantee adequate bioavailability, suitable half‐life, and low dosage frequency. The success of the patient's treatment and adherence depends on this.
7. Future Outlook
7.1. Refinement and Optimization
Creating TIGIT inhibitors of the next generation with enhanced pharmacokinetic, selective, and potent characteristics is probably the main focus of future research. In this procedure, sophisticated medicinal chemistry techniques will be crucial. Finding trustworthy biomarkers to forecast a patient's reaction to TIGIT inhibitors is essential for matching and tailored treatment. Ongoing research aims to identify and validate these biomarkers [15, 63].
7.2. Expanded Applications
TIGIT inhibitors may treat autoimmune disorders and persistent infections, among other illnesses marked by immunological dysfunction. This broader use of these inhibitors may lead to new treatment opportunities and increase their overall effect.
7.3. Combination Strategies
A significant area of interest is the combination of TIGIT inhibitors with other treatments, such as chemotherapy, targeted therapies, and other checkpoint inhibitors. These tactics seek to defeat resistance mechanisms and improve overall efficacy. Future medicines such as cytokines or vaccinations may combine TIGIT inhibitors with substances that further alter the larger immune milieu to enhance anti‐tumor responses.
7.4. Regulatory and Commercial Pathways
TIGIT minor molecule inhibitor regulatory approval is contingent upon completing rigorous clinical trials. The authors anticipate thoroughly assessing the safety and efficacy findings, which could soon result in commercial availability and possible approvals. TIGIT inhibitors may have a substantial effect on the market for cancer immunotherapies if they prove to be effective, giving patients additional alternatives and maybe enhancing prognoses across a range of cancer types.
8. Challenges in Clinical Development of Anti‐TIGIT
A few potential explanations for the early clinical trial failures in patients with SCLC and NSCLC were compiled based on the immune checkpoint blockades linked to TIGIT's mechanism of action. Tumor invasion, metastasis, and recurrence in different patient populations can be influenced by regulating the expression of PD‐L1, TIGIT, and other biomarkers, as well as the quantity and quality of TILs, circulating tumor cells, and TAMs. To recruit immune T cells to tumors for immunotherapy, patients must have an adequate number of these cells [57, 58]. Tumors with a deficiency of T cells are referred to as “cold” tumors, and cancers with an abundance of T cells are referred to as “hot” tumors, which may respond differently to TIGIT targeting therapy. Second, phase II results may not fully capture the genuine effects of the TIGIT‐blocking medication. Particularly in patients with advanced or high‐burden malignancies, TIGIT—the immune checkpoint regulation “braker”—in conjunction with another “braker,” PD‐1/PD‐L1, may not be enough to activate the immune system and kill tumor cells [64]. When combined with other “braker” medications, such as anti‐CTLA‐4, anti‐vascular endothelial growth factor (VEGF), TIGIT+PD‐1/PD‐L1 + VEGF, or a triple combination, chemotherapy may increase the efficacy of TIGIT cancer immunotherapy. Currently undergoing clinical development, several bispecific and trispecific antibodies exhibit encouraging initial therapeutic potential.
9. Conclusion
Preclinical and early clinical findings of the anti‐TIGIT agent are promising. However, there are still many obstacles to overcome. Successful clinical translation depends on identifying predictive biomarkers for patient classification, optimizing dosage regimens, and comprehending resistance mechanisms. Combination methods must also be carefully designed to minimize toxicity and maximize efficacy.
Following the period of anti‐PD‐(L)1 immunotherapy, anti‐TIGIT medications may represent new players in the development of cancer treatments. However, the encouraging preclinical findings have not yet fully translated into clinical trials. When used in isolation, anti‐TIGIT antibodies had minimal effect on advanced solid tumors. Nevertheless, their correlation with anti‐PD‐1 medications improved the effectiveness of the results. When tiragolumab and atezolizumab were combined in CITYSCAPE phase II, the ORR and PFS in the first line of advanced non‐small cell lung cancers were enhanced compared to when atezolizumab was used alone. The development of such medicines may be aided by identifying potentially intriguing subgroups outside of NSCLCs and using biomarkers to predict efficacies. In light of the emergence of adjuvant and neoadjuvant immune treatments, questions regarding the timing of administration are also available.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
J.P. conveys cordial thanks to G.S., S.N., D.P.B., A.R.D., N.K.D., GITAM (deemed to be university)‐Hyderabad, Datta Meghe Institute of Higher Education‐Wardha, NIPER‐Hyderabad, and Innatura Scientific Pvt. Ltd‐Hyderabad for carrying out this review article.
Funding: This work was supported by the SEED grant (2024/0306) sanctioned by GITAM (deemed to be University), Vishakhapatnam, Andhra Pradesh.
References
- 1. Rousseau A., Parisi C., and Barlési F., “Anti‐TIGIT Therapies for Solid Tumors: A Systematic Review,” ESMO Open 8, no. 2 (2023): 101184, 10.1016/j.esmoop.2023.101184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanmamed M. F. and Chen L., “A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization,” Cell 175, no. 2 (2018): 313–326, 10.1016/j.cell.2018.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen D. S. and Mellman I., “Elements of Cancer Immunity and the Cancer–Immune Set Point,” Nature 541, no. 7637 (2017): 321–330, 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 4. Cai L., Li Y., Tan J., Xu L., and Li Y., “Targeting LAG‐3, TIM‐3, and TIGIT for Cancer Immunotherapy,” Journal of Hematology & Oncology 16, no. 1 (2023): 101, 10.1186/s13045-023-01499-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaw G., Cavalcante L., Giles F. J., and Taylor A., “Elraglusib (9‐ING‐41), a Selective Small‐Molecule Inhibitor of Glycogen Synthase Kinase‐3 Beta, Reduces Expression of Immune Checkpoint Molecules PD‐1, TIGIT and LAG‐3 and Enhances CD8+ T Cell Cytolytic Killing of Melanoma Cells,” Journal of Hematology & Oncology 15, no. 1 (2022): 134, 10.1186/s13045-022-01352-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manieri N. A., Chiang E. Y., and Grogan J. L., “TIGIT: A Key Inhibitor of the Cancer Immunity Cycle,” Trends in Immunology 38, no. 1 (2017): 20–28, 10.1016/j.it.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 7. Xiao K., Xiao K., Li K., Xue P., and Zhu S., “Prognostic Role of TIGIT Expression in Patients With Solid Tumors: A Meta‐Analysis,” Journal of Immunology Research 2021 (2021): 1–8, 10.1155/2021/5440572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dougall W. C., Kurtuluş S., Smyth M. J., and Anderson A. C., “TIGIT and CD96: New Checkpoint Receptor Targets for Cancer Immunotherapy,” Immunological Reviews 276, no. 1 (2017): 112–120, 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 9. Gorvel L. and Olive D., “Targeting the ‘PVR‐TIGIT Axis’ With Immune Checkpoint Therapies,” F1000Research 9 (2020): 354, 10.12688/f1000research.22877.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. n M. M., Nair S. S., O'Leary J. G., et al., “Implication of TIGIT+ Human Memory B Cells in Immune Regulation,” Nature Communications 12, no. 1 (2021): 1534, 10.1038/s41467-021-21413-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rotte A., Sahasranaman S., and Budha N., “Targeting TIGIT for Immunotherapy of Cancer: Update on Clinical Development,” Biomedicine 9, no. 9 (2021): 1277, 10.3390/biomedicines9091277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckle I. and Guillerey C., “Inhibitory Receptors and Immune Checkpoints Regulating Natural Killer Cell Responses to Cancer,” Cancers 13, no. 17 (2021): 4263, 10.3390/cancers13174263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Josefsson S. E., Huse K., Kolstad A., et al., “T Cells Expressing Checkpoint Receptor TIGIT Are Enriched in Follicular Lymphoma Tumors and Characterized by Reversible Suppression of T‐Cell Receptor Signaling,” Clinical Cancer Research 24, no. 4 (2018): 870–881, 10.1158/1078–0432.ccr-17-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu X., “Pathway Analysis of Tumor Immunity by TIGIT and Recent Scientific Research Results,” Transactions on Materials Biotechnology and Life Sciences 3 (2024): 515–521, 10.62051/v490zv42. [DOI] [Google Scholar]
- 15. Chauvin J. M. and Zarour H. M., “TIGIT in Cancer Immunotherapy,” Journal for Immunotherapy of Cancer 8, no. 2 (2020): e000957, 10.1136/jitc-2020-000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodrigues R. R., Freitas V. S., Alves P. M., De Almeida Freitas R., De Souza L. B., and De Andrade Santos P. P., “Evaluation of the Presence of Th1 Response Through T‐Bet and IFN‐Gamma Immunohistochemical Expression in Lower Lip and Oral Tongue Squamous Cell Carcinomas,” Pathology, Research and Practice 253 (2024): 155010, 10.1016/j.prp.2023.155010. [DOI] [PubMed] [Google Scholar]
- 17. Liu Y., Zhang C., Jiang A., et al., “Potential Anti‐Tumor Effects of Regulatory T Cells in the Tumor Microenvironment: A Review,” Journal of Translational Medicine 22, no. 1 (2024): 293, 10.1186/s12967-024-05104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nair V. S. and Elkord E., “Immune Checkpoint Inhibitors in Cancer Therapy: A Focus on T‐Regulatory Cells,” Immunology and Cell Biology 96, no. 1 (2017): 21–33, 10.1111/imcb.1003. [DOI] [PubMed] [Google Scholar]
- 19. Ge Q., Zhao Z., Li X., et al., “Deciphering the Suppressive Immune Microenvironment of Prostate Cancer Based on CD4+ Regulatory T Cells: Implications for Prognosis and Therapy Prediction,” Clinical and Translational Medicine 14, no. 1 (2024): e1552, 10.1002/ctm2.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dittel L. J., Dittel B. N., and Brod S. A., “Ingested (Oral) Adrenocorticotropic Hormone (ACTH) Inhibits Interleukin‐17 in the Central Nervous System After Adoptive Transfer of T Helper (Th)1/Th17 T Cells in the Mouse Model of Multiple Sclerosis, Experimental Autoimmune Encephalomyelitis,” Journal of the Neurological Sciences 456 (2024): 122779, 10.1016/j.jns.2023.122779. [DOI] [PubMed] [Google Scholar]
- 21. Yuan S., “Analysis of the Role of Serum IL‐2R, IL‐6, IL‐8, IL‐10, and TNF‐α in the Treatment of Immune Escape Mechanisms in Patients With Minimal Ma Tumors,” International Journal of Frontiers in Medicine 6, no. 3 (2024): e060305, 10.25236/ijfm.2024.060305. [DOI] [Google Scholar]
- 22. Guo J., Tang X., Deng P., et al., “Interleukin‐4 From Curcumin‐Activated OECs Emerges as a Central Modulator for Increasing M2 Polarization of Microglia/Macrophage in OEC Anti‐Inflammatory Activity for Functional Repair of Spinal Cord Injury,” Cell Communication and Signaling 22, no. 1 (2024): 162, 10.1186/s12964-024-01539-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dunn G. P., Bruce A. T., Sheehan K. C. F., et al., “A Critical Function for Type I Interferons in Cancer Immunoediting,” Nature Immunology 6, no. 7 (2005): 722–729, 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 24. Cai L., Chen A., and Tang D., “A New Strategy for Immunotherapy of Microsatellite‐Stable (MSS)‐Type Advanced Colorectal Cancer: Multi‐Pathway Combination Therapy With PD‐1/PD‐L1 Inhibitors,” Immunology 10 (2024): e13785, 10.1111/imm.13785. [DOI] [PubMed] [Google Scholar]
- 25. Yin Z., Yu M., Ma T., et al., “Mechanisms Underlying Low‐Clinical Responses to PD‐1/PD‐L1 Blocking Antibodies in Immunotherapy of Cancer: A Key Role of Exosomal PD‐L1,” Journal for Immunotherapy of Cancer 9, no. 1 (2021): e001698, 10.1136/jitc-2020-001698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang C., Liu J., Wu Q., Wang Z., Hu B., and Bo L., “The Role of TIM‐3 in Sepsis: A Promising Target for Immunotherapy?,” Frontiers in Immunology 15 (2024): 1328667, 10.3389/fimmu.2024.1328667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Der Vlist M., Kuball J., Radstake T. R. D. J., and Meyaard L., “Immune Checkpoints and Rheumatic Diseases: What Can Cancer Immunotherapy Teach Us?,” Nature Reviews Rheumatology 12, no. 10 (2016): 593–604, 10.1038/nrrheum.2016.131. [DOI] [PubMed] [Google Scholar]
- 28. Reinoso G. P. G., Moscoso N. P. C., Goyo P. D. A., Rodríguez B. C., and Munoz J., “Advances in Cancer Immunotherapy: A Comprehensive Review of Current and Future Approaches in the Treatment of Solid and Hematologic Tumors,” International Journal of Medical Science and Dental Health 10, no. 2 (2024): 116–129, 10.55640/ijmsdh-10-02-16. [DOI] [Google Scholar]
- 29. Suzuki K., Yasui Y., Tsuchiya K., et al., “Impact of Immune‐Related Adverse Events in Patients With Hepatocellular Carcinoma Treated With Atezolizumab Plus Bevacizumab,” Journal of Gastroenterology and Hepatology 39, no. 6 (2024): 1183–1189, 10.1111/jgh.16532. [DOI] [PubMed] [Google Scholar]
- 30. Patwekar M., Sehar N., Patwekar F., et al., “Novel Immune Checkpoint Targets: A Promising Therapy for Cancer Treatments,” International Immunopharmacology 126 (2024): 111186, 10.1016/j.intimp.2023.111186. [DOI] [PubMed] [Google Scholar]
- 31. Zhou X., Du J., Wang H., et al., “Repositioning Liothyronine for Cancer Immunotherapy by Blocking the Interaction of Immune Checkpoint TIGIT/PVR,” Cell Communication and Signaling 18, no. 1 (2020): 1–14, 10.1186/s12964-020-00638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou X., Jiao L., Qian Y., et al., “Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPΑ and TIGIT/PVR Pathways for Cancer Immuno‐Therapy,” Biomolecules 11, no. 5 (2021): 706, 10.3390/biom11050706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiong F., Yu M., Xu H., et al., “Discovery of TIGIT Inhibitors Based on DEL and Machine Learning,” Frontiers in Chemistry 10 (2022): 982539, 10.3389/fchem.2022.982539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou X., Yang L., Zhang X., et al., “Hemin Blocks TIGIT/PVR Interaction and Induces Ferroptosis to Elicit Synergistic Effects of Cancer Immunotherapy,” Science China Life Sciences 67, no. 5 (2024): 996–1009, 10.1007/s11427-023-2472-4. [DOI] [PubMed] [Google Scholar]
- 35. Peng C., Xu Y., Wu J., Wu D., Zhou L., and Xia X., “TME‐Related Biomimetic Strategies Against Cancer,” International Journal of Nanomedicine 19 (2024): 109–135, 10.2147/ijn.s441135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dhanak D., Edwards J. P., Nguyen A., and Tummino P. J., “Small‐Molecule Targets in Immuno‐Oncology,” Cell Chemical Biology 24, no. 9 (2017): 1148–1160, 10.1016/j.chembiol.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 37. Abdel‐Rahman S. A. and Gabr M., “Small Molecule Immunomodulators as Next‐Generation Therapeutics for Glioblastoma,” Cancers 16, no. 2 (2024): 435, 10.3390/cancers16020435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang P., Liu X., Gu Z., et al., “Targeting TIGIT for Cancer Immunotherapy: Recent Advances and Future Directions,” Biomarker Research 12, no. 1 (2024): 7, 10.1186/s40364-023-00543-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brazel D., Ou S. I., and Nagasaka M., “Tiragolumab (Anti‐TIGIT) in SCLC: Skyscraper‐02, a Towering Inferno,” Lung Cancer 14 (2023): 1–9, 10.2147/lctt.s379389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cho B. C., Abreu D. R., Hussein M., et al., “Tiragolumab Plus Atezolizumab Versus Placebo Plus Atezolizumab as a First‐Line Treatment for PD‐L1‐Selected Non‐Small‐Cell Lung Cancer (CITYSCAPE): Primary and Follow‐Up Analyses of a Randomised, Double‐Blind, Phase 2 Study,” Lancet Oncology 23, no. 6 (2022): 781–792, 10.1016/S1470-2045(22)00226-1. [DOI] [PubMed] [Google Scholar]
- 41. Yu X., Harden K., Gonzalez L. C., et al., “The Structure of TIGIT–CD155 Interactions and Implications for Immunotherapy,” Nature Immunology 10, no. 11 (2009): 1073–1079.19701189 [Google Scholar]
- 42. Johnston R. J., Comps‐Agrar L., Yu X., et al., “The Immunological Role of TIGIT and Preclinical Evidence for Its Therapeutic Potential in Cancer,” Immunity 41, no. 5 (2014): 811–822. [Google Scholar]
- 43. American Association for Cancer Research , “Tiragolumab Impresses in Multiple Trials,” Cancer Discovery 10 (2020): 1086–1087. [DOI] [PubMed] [Google Scholar]
- 44. Bendell J. C., Bedard P., Bang Y.‐J., et al., “Abstract CT302: Phase Ia/Ib Dose‐Escalation Study of the Anti‐TIGIT Antibody Tiragolumab as a Single Agent and in Combination With Atezolizumab in Patients With Advanced Solid Tumors,” Cancer Research 80, no. 16_Supplement (2020): CT302, 10.1158/1538–7445.AM2020-CT302. [DOI] [Google Scholar]
- 45. Genentech , “Genentech Reports Interim Results for Phase III SKYSCRAPER‐01 Study in PD‐L1 High Metastatic Non‐Small Cell Lung Cancer,” (2022), accessed December 22, 2022, https://www.gene.com/media/press‐releases/14951/2022‐05‐10/genentech‐reports‐interim‐results‐for‐ph.
- 46. Rudin C. M., Liu S. V., and Lu S., “SKYSCRAPER‐02: Primary Results of a Phase III, Randomized, Double‐Blind, Placebo‐Controlled Study of Atezolizumab (Atezo) + Carboplatin + Etoposide (CE) With or Without Tiragolumab (Tira) in Patients (Pts) With Untreated Extensive‐Stage Small Cell Lung Cancer (ES‐SCLC),” Journal of Clinical Oncology 40, no. 17_suppl (2022): LBA8507, 10.1200/JCO.2022.40.17_suppl.LBA8507. [DOI] [Google Scholar]
- 47. Genentech , “Genentech Provides Update on Phase III SKYSCRAPER‐02 Study in Extensive‐Stage Small Cell Lung Cancer,” (2022), accessed December 22, 2022, https://www.gene.com/media/press‐releases/14947/2022‐03‐29/genentech‐provides‐update‐on‐phase‐iii‐s.
- 48. Catakovic K., Gassner F. J., Ratswohl C., et al., “TIGIT Expressing CD4+T Cells Represent a Tumor‐Supportive T Cell Subset in Chronic Lymphocytic Leukemia,” Oncoimmunology 7, no. 1 (2017): e1371399, 10.1080/2162402X.2017.1371399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu G., Zhang Q., Yang J., et al., “Increased TIGIT Expressing NK Cells With Dysfunctional Phenotype in AML Patients Correlated With Poor Prognosis,” Cancer Immunology, Immunotherapy 71, no. 2 (2021): 277–287, 10.1007/s00262-021-02978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brauneck F., Seubert E., Wellbrock J., Schulze Zur Wiesch J., Duan Y., and Magnus T., “Combined Blockade of TIGIT and CD39 or A2AR Enhances NK‐92 Cell‐Mediated Cytotoxicity in AML,” International Journal of Molecular Sciences 22, no. 23 (2021): 12919, 10.3390/ijms222312919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Minnie S. A., Kuns R. D., Gartlan K. H., et al., “Myeloma Escape After Stem Cell Transplantation Is a Consequence of T‐Cell Exhaustion and Is Prevented by TIGIT Blockade,” Blood 132, no. 16 (2018): 1675–1688, 10.1182/blood-2018-01-825240. [DOI] [PubMed] [Google Scholar]
- 52. Guillerey C., Harjunpää H., Carrie N., et al., “TIGIT Immune Checkpoint Blockade Restores CD8+ T‐Cell Immunity Against Multiple Myeloma,” Blood 132, no. 16 (2018): 1689–1694, 10.1182/blood-2018-01-825265. [DOI] [PubMed] [Google Scholar]
- 53. Annibali O., Bianchi A., Grifoni A., et al., “A Novel Scoring System for TIGIT Expression in Classic Hodgkin Lymphoma,” Scientific Reports 11, no. 1 (2021): 7059, 10.1038/s41598-021-86655-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li W., Blessin N. C., Simon R., et al., “Expression of the Immune Checkpoint Receptor TIGIT in Hodgkin's Lymphoma,” BMC Cancer 18, no. 1 (2018): 1209, 10.1186/s12885-018-5111-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hajiasghar‐Sharbaf R., Asgarian‐Omran H., Valadan R., et al., “CD8+ T‐Cells Co‐Expressing PD‐1 and TIGIT Are Highly Frequent in Chronic Lymphocytic Leukemia,” Iranian Journal of Allergy, Asthma, and Immunology 20, no. 6 (2021): 751–763, 10.18502/ijaai.v20i6.8027. [DOI] [PubMed] [Google Scholar]
- 56. Zhang X., Zhang H., Chen L., Feng Z., Gao L., and Li Q., “TIGIT Expression is Upregulated in T Cells and Causes T Cell Dysfunction Independent of PD‐1 and Tim‐3 in Adult B Lineage Acute Lymphoblastic Leukemia,” Cellular Immunology 344 (2019): 103958, 10.1016/j.cellimm.2019.103958. [DOI] [PubMed] [Google Scholar]
- 57. Niu J., Maurice‐Dror C., Lee D. H., et al., “First‐In‐Human Phase 1 Study of the Anti‐TIGIT Antibody Vibostolimab as Monotherapy or With Pembrolizumab for Advanced Solid Tumors, Including Non‐Small‐Cell Lung Cancer,” Annals of Oncology 33, no. 2 (2022): 169–180, 10.1016/j.annonc.2021.11.002. [DOI] [PubMed] [Google Scholar]
- 58. Frentzas S., Meniawy T., Kao S., et al., “ADVANTIG‐105: Phase 1 Dose‐Escalation Study of Anti‐TIGIT Monoclonal Antibody Ociperlimab (BGB‐A1217) in Combination With Tislelizumab in Patients With Advanced Solid Tumors,” Journal of Clinical Oncology 39 (2021): 15, 10.1200/JCO.2021.39.15_suppl.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rodriguez‐Abreu D., Johnson M. L., Hussein M. A., et al., “Primary Analysis of a Randomized, Double‐Blind, Phase II Study of the Anti‐TIGIT Antibody Tiragolumab (Tira) Plus Atezolizumab (Atezo) Versus Placebo Plus Atezo as First‐Line (1L) Treatment in Patients With PD‐L1‐Selected NSCLC (CITYSCAPE),” Journal of Clinical Oncology 38, no. 15_suppl (2020): 9503, 10.1200/JCO.2020.38.15_suppl.9503. [DOI] [Google Scholar]
- 60. Peterson C., Denlinger N., and Yang Y., “Recent Advances and Challenges in Cancer Immunotherapy,” Cancers 14, no. 16 (2022): 3972, 10.3390/cancers14163972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dhar R., Seethy A., Singh S. K., et al., “Cancer Immunotherapy: Recent Advances and Challenges,” Journal of Cancer Research and Therapeutics 17, no. 4 (2021): 834–844, 10.4103/jcrt.jcrt_1241_20. [DOI] [PubMed] [Google Scholar]
- 62. Florou V. and Garrido‐Laguna I., “Clinical Development of Anti‐TIGIT Antibodies for Immunotherapy of Cancer,” Current Oncology Reports 24, no. 9 (2022): 1107–1112, 10.1007/s11912-022-01281-5. [DOI] [PubMed] [Google Scholar]
- 63. Steeland S., Vandenbroucke R. E., and Libert C., “Nanobodies as Therapeutics: Big Opportunities for Small Antibodies,” Drug Discovery Today 21, no. 7 (2016): 1076–1113, 10.1016/j.drudis.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 64. Topalian S. L., Drake C. G., and Pardoll D. M., “Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy,” Cancer Cell 27, no. 4 (2015): 450–461, 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]