


Abstract
We have developed a method for transferring exogenous DNA molecules into isolated mammalian mitochondria using bacterial conjugation. In general, we accomplish this by (i) inserting an origin of DNA transfer (oriT) sequence into a DNA construct, (ii) transforming the construct into an appropriate Escherichia coli strain and then (iii) introducing the mobilizable DNA into mitochondria through conjugation. We tested this approach by transferring plasmid DNA containing a T7 promoter sequence into mitochondria that we had engineered to contain T7 RNA polymerase. After conjugation between E.coli and mitochondria, we detected robust levels of T7 transcription from the DNA constructs that had been transferred into the mitochondria. This approach for engineering DNA constructs in vitro and subsequent transfer into mitochondria by conjugation offers an attractive experimental system for studying many aspects of vertebrate mitochondrial gene expression and is a potential route for transforming mitochondrial networks within mammalian cells.
INTRODUCTION
The typical animal cell contains a complex mitochondrial network that produces the majority of the cell's ATP through oxidative phosphorylation (1). The 13 proteins, 2 rRNAs and 22 tRNAs encoded by the mitochondrial DNA (mtDNA) genome are critical for normal cellular energy metabolism, and mutations in this mtDNA molecule are known to cause a wide range of human diseases (2).
An elegant and effective method for the genetic transformation of mitochondria using a biolistic ‘gene gun’ apparatus has been described previously (3,4), but to date this approach has been limited to the transformation of mitochondria in yeast and closely related organisms. Despite the high level of interest in mammalian mitochondrial genomes, no practical methods have yet been developed for introducing biologically active exogenous DNA into the mitochondrial networks of mammalian cells. Relatively small linear DNA fragments have been transported into mitochondria by covalently linking either oligonucleotides (5) or double-stranded DNA (dsDNA) (6) to mitochondrial-targeting peptides, and recently cationic liposome- (7) and peptide nucleic acid-mediated DNA transfer (8,9) have been tried. Much larger plasmid DNA molecules have been introduced into isolated mammalian mitochondria by electroporation (10,11) but the electroporation conditions used in these procedures appear to cause irreparable damage to the mitochondria that prevents their reincorporation back into mammalian cells (Y.G. Yoon, unpublished data). A recent paper has reported that exogenous DNA can be introduced into the mitochondria of human cells using a process dubbed ‘protofection’ (12), but the data presented to support this claim are not conclusive and the process has not yet been duplicated by other investigators.
In this report, we describe and demonstrate the feasibility of a new mitochondrial transformation system involving bacteria-to-mitochondria conjugation. Because conjugation is a relatively gentle process, we reasoned that this type of procedure would enable us to transfer large DNA constructs into mitochondria without damaging the structural integrity of the mitochondria. Although conjugation typically involves bacterium-to-bacterium DNA transfer via pili, the conjugation process is driven entirely by molecular machinery in the donor cell (13) and so an amazingly broad range of cell types can serve as the DNA recipient, including yeast (14,15), plants (16) and mammalian cells (17,18).
When a conjugative-competent donor cell contacts a suitable recipient, the donor bacteria forms a mating bridge and a nick is made within the origin of DNA transfer (oriT) sequence in the mobilizing plasmid. Extension of the 3′ end of the oriT nick displaces a single strand that is then transferred into the recipient. The TraI enzyme that catalyzes both the first and the second nicking events binds covalently to the 5′ end of the single-stranded DNA (ssDNA) and guides the DNA from the donor cell to the recipient through the mating bridge (13). The second nicking event at the mating bridge liberates one unit length of the plasmid and supplies the 3′ terminal end for circularization (13). DNA molecules as large as the entire 4.7 Mb Escherichia coli chromosome can be transferred by conjugation (19), but smaller molecules are transferred more quickly and efficiently.
We chose to utilize the transfer genes and the oriT sequence from the conjugative plasmid RP4 (13,20,21) in this mitochondrial transformation procedure because the conjugative functions of this plasmid are both well characterized and efficient. We tested the ability of the RP4 conjugative system to transfer DNA into the matrix of mammalian mitochondria by using a mobilizable plasmid containing a T7 promoter sequence and purified mouse mitochondria that we had engineered to contain T7 RNA polymerase (T7RNAP) within their matrix. After conjugation between E.coli and these isolated mitochondria, we detected robust levels of T7 transcription from the DNA constructs that had been transferred into the mitochondrial matrix. To our knowledge, this is the first example of conjugative DNA transfer into mitochondria.
MATERIALS AND METHODS
E.coli strains, cell lines and culture media
E.coli strain DH5α, used as a host for all cloning experiments, was grown at 37°C in Luria–Bertani (LB) medium, supplemented with 12.5 µg tetracycline/ml or 50 µg ampicillin/ml for TcR plasmid- or AmpR plasmid-containing strains. E.coli S17-1 (ATCC 47055) (20) was used as the donor strain for conjugative crosses. The mouse cell line LL/2 (ATCC CRL-1642) was grown in DMEM (Life Technologies, Rockville, MD) in the presence of heat-inactivated 10% fetal bovine serum (FBS) at 37°C in a humidified 10% CO2 incubator. HeLa 229 (ATCC CCL-2.1) was cultured in MEM alpha (Life Technologies) with 10% heat-inactivated FBS at 37°C in a humidified 5% CO2 incubator.
DNA manipulation
Mini-scale preparations of plasmid DNA were prepared by the alkaline lysis method (22) and large quantities of plasmid DNA were prepared by the PEG precipitation method and other recombinant DNA techniques were performed essentially as described previously (22). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA) and used as recommended by the manufacturer. Pfu Turbo DNA polymerase was purchased from Stratagene (La Jolla, CA) and AmpliTaq DNA polymerase was purchased from Applied Biosystems (Branchburg, NJ). Deoxyoligonucleotides were synthesized from Life Technologies (Rockville, MD) or Integrated DNA Technologies (Coralville, IA).
Construction of plasmids
For the construction of mitochondria-targeting gene expression vector, the 24 N-terminal amino acids mitochondrial leader sequence of the mouse mitochondrial transcription factor A (TFAM) (23) was amplified by PCR using primers 5′-musmtTFA (5′-AG GAATTC GCTAGC GTC GGC CCG AGC GAT GGC-3′, EcoRI and NheI restriction sites underlined, respectively) and mtTFA3′NruI (5′-GG GGTACC TCGCGA GGA AAA ACA CTT CGG AAT-3′, KpnI and NruI restriction sites underlined, respectively) and cloned between the NheI and KpnI sites of pcDNA6/HisA after digestion of the PCR products with NheI and KpnI. The 2.7 kb T7 RNA polymerase (T7RNAP) gene obtained by PCR amplification was then cloned into NruI and BamHI sites of the vector, resulting in pcDNA6-musTFAML-T7RNAP (Figure 2A). The GFP gene was also cloned between NruI and XhoI sites of the vector to test the mitochondrial-targeting function of the leader sequence, resulting in pcDNA6-musTFAML-GFP (Figure 2A). The mobilizing plasmids that contain the T7 promoter sequences were constructed using pTANTS (24). The GFP gene was amplified by PCR, in which one PCR primer contains the T7 promoter (PT7) sequence (SmaT7ProGFP, 5′-GGG TAA TAC GAC TCA CTA TAG GGA TGG TGA GCA AGG GCG AGG A-3′, T7 promoter sequence underlined; GFP3′Sal, 5′-AGT GTC GAC TTA CTT GTA CAG CTC GTC CA-3′, SalI restriction sites underlined) and cloned in SmaI/SalI sites of pTANTS. T7 terminator sequence was also synthesized, annealed and cloned in SalI/SphI sites. Then, the complementary sequences of T7 promoter (T7hp) were synthesized, annealed and cloned in the EcoRI/SmaI sites (T7ds1, 5′-CCG GGA GTG AGT CGT ATT ACC CTC CCA TGT TGA TGG GA-3′; T7ds2, 5′-TCC CAT CAA CAT GGG AGG GTA ATA CGA CTC ACT CCC GGG TAC-3′), resulting pT7hpGFP (Figure 4A). The oriT sequence was amplified from plasmid pRK290 (21) by PCR (HIIIOriT, 5′-AGC CAT AAG CTT GCC CTC ATC TGT TAC-3′, HindIII restriction sites underlined; SphOriT, 5′-GTA GAG CAT GCA CAA AAC AGC AGG GAA-3′, SphI restriction sites underlined) and cloned in the SphI/HindIII sites, resulting in pT7hpGFP+oriT (Figure 4A).
Figure 2.
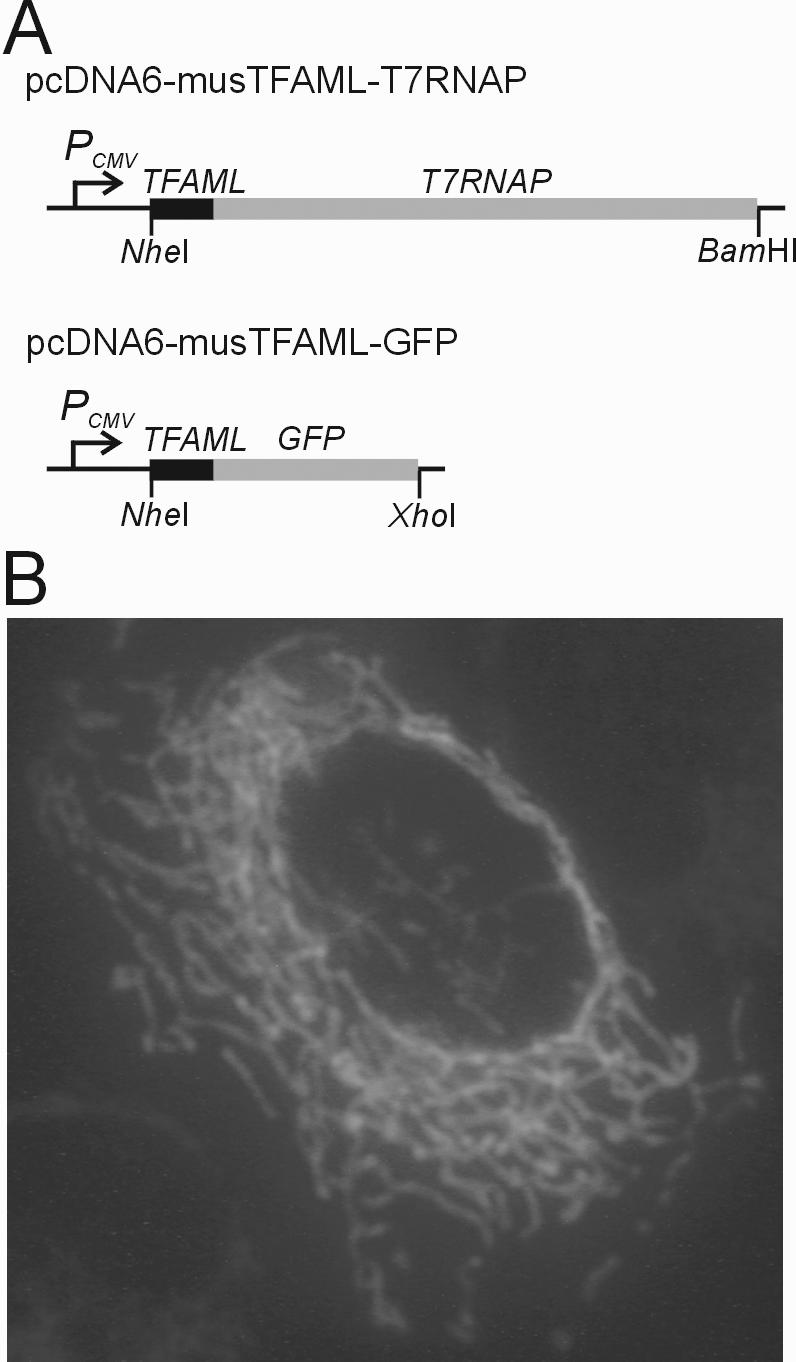
Construction of cell lines expressing fusion proteins targeted to the mitochondrial matrix. (A) Structure of two chimeric gene expression vectors. The mitochondrial leader sequence from the mouse mitochondrial transcription factor A (TFAML) was cloned at the 5′ end of the each structural gene in frame using the pcDNA6/HisA vector. (B) Fluorescence microscopy of HeLa 229 cells expressing GFP targeted to the mitochondrial matrix. The HeLa 229 cells were transfected with the TFAML-GFP chimeric gene expression vector and subjected to fluorescence microscopy after 24 h incubation at 37°C in a 5% humidified CO2 incubator.
Figure 4.

Plasmids for assaying conjugative DNA transfer between E.coli and T7RNAP-mitochondria. (A) Structures of control and mobilizing plasmids, pT7hpGFP and pT7hpGFP+oriT, respectively. The T7hp sequence was cloned in front of the T7 promoter (PT7) and was designed to form a double-stranded hairpin-loop T7 promoter after transferring the single-stranded plasmid DNA into the mitochondria by conjugation. Only the mobilizing plasmid contains the oriT sequence. (B) A schematic representation for the formation of a double-stranded hairpin-loop T7 promoter. Once a DNA strand is transferred to T7RNAP-mitochondria by conjugation, spontaneous annealing around the T7 promoter region in the ssDNA will form a fully duplexed T7 promoter inside the mitochondria. T7RNAP can recognize this duplexed sequence (27,28) and initiate transcription of an RNA molecule (red characters).
Generation of cell lines expressing mitochondria-targeted T7RNAP
To make a mouse cell line expressing T7RNAP targeted to mitochondria, 10 µg of the expression vector pcDNA6-musTFAML-T7RNAP (Figure 2A) was linearized with BglII and transfected into the mouse LL/2 cells. Transfection of the cell was performed using the calcium phosphate method (25), and stable cell lines expressing mitochondria-targeted T7RNAP were selected with blasticidin (5 µg/ml) for 2 weeks. A clonal LL/2-T7mt cell line was obtained by diluting these blasticidin-resistant cells to a single cell per well on 96-well plates. The cloned cells were then cultured in normal medium supplemented with 3 µg/ml of blasticidin. To verify the mitochondrial-targeting function of the mouse TFAM leader peptide, HeLa 229 was transiently transfected with pcDNA6-musTFAML-GFP (Figure 2A), and the green fluorescent protein (GFP) expression and localization were examined in these cells (Figure 2B).
Electroporation of mitochondria
These experiments were performed using the method described previously (11). Cells were harvested by centrifugation, washed twice with 1 mM Tris–HCl, pH 7.0, 0.13 M NaCl, 5 mM KCl and 7.5 mM MgCl2. The cell pellet was resuspended in half the cell volume with 1/10× IB (4 mM Tris–HCl, pH 7.4, 2.5 mM NaCl and 0.5 mM MgCl2) and the cells were broken using a Pellet Pestle® tissue grinder (Kontes, Vineland, NJ). The homogenate was mixed with one-ninth of the packed cell volume of 10× IB resulting in a buffer concentration of ∼1× IB. The unbroken cells and nuclei were removed by two consecutive low-speed centrifugations (5 min at 320 g). The supernatant was placed into new 1.5 ml tubes and centrifuged at full speed (10 min at 15 800 g) to obtain a crude mitochondrial pellet. Mitochondria were rinsed once with 500 µl of 1× IB, centrifuged again at full speed for 10 min and resuspened into 0.33 M sucrose/10% glycerol (100 mg of mitochondrial protein/ml) for electroporation. An aliquot of 1 µg of plasmid DNA was added to 50 µl of 100 mg/ml of mitochondrial suspension and the mixture was transferred into a cold electroporation cuvette (0.1 cm gap cuvette; Bio-Rad). Electroporation was then carried out using a Bio-Rad Gene Pulser™ at a capacitance of 25 µF, a resistance of 400 Ω and field strength of 12 kV/cm. After the electric pulse, 1 ml of incubation buffer (40 mM Tris–HCl, pH 7.4, 25 mM NaCl, 5 mM MgCl2 and 10% glycerol) was added to the cuvette, rapidly mixed by pipetting, transferred to a new tube and washed three times with the incubation buffer by centrifugation.
In organello RNA synthesis
The final mitochondrial pellets were resuspended in 50 µl of the incubation buffer supplemented with 1 mM pyruvate, 1 mM ATP and 1 mg/ml BSA and incubated at 37°C for 2 h (26). For labeling of mitochondrial RNA, 10 µCi of [α-32P]UTP was also added to the incubation buffer (26). After incubation, the mitochondrial suspension was pelleted by centrifugation and washed twice with incubation buffer. The pellets were suspended in 200 µl of DNase I buffer (10% glycerol, 10 mM Tris–HCl, pH 8.0, 1 mM MgCl2) and incubated with 200 Kunitz units of DNase I (Sigma) at 37°C for 30 min. The mitochondria were then pelleted and washed twice with washing buffer (10% glycerol, 10 mM Tris–HCl, pH 7.4, 1 mM EDTA) to inactivate and remove the nucleases. Total RNA was then prepared directly from the mitochondria using Trizol Reagent (Life Technologies).
Conjugation between E.coli and isolated T7RNAP-mitochondria
Conjugative crosses were performed by mixing mid-logarithmic phase E.coli (OD600 = 0.5∼0.6) and isolated mitochondria in LB medium. Briefly, 100 µl of 100 mg/ml mitochondrial suspension, which was isolated from the LL/2-T7mt cell line (T7RNAP-mitochondria), were mixed with 200 µl of rapidly growing E.coli. After discarding the supernatant by centrifugation, the mating mixture was resuspended with 25 µl of fresh LB. Then, 25 µl of 1% low-melting point agarose was added to the mating mixture and the mating mixture/agarose mixture was put on ice for 1 min to make an agarose block. Fresh LB (500 µl) was added to the mating mixture/agarose block and incubated at 37°C by gentle shaking for 5 h. After 5 h incubation, the mating mixture/agarose block was incubated for additional 2 h at 37°C with 200 µg/ml of ampicillin, 200 Kunitz units of DNase I and 50 µg/ml of RNase A to lyse E.coli and to remove any nucleic acids outside of the mitochondria. Supernatant from the mating mixture/agarose block was removed gently by pipetting, and the agarose block was washed twice with fresh LB. After removal of the LB media, 1 ml of Trizol was added directly to the mating mixture/agarose block and total RNA was prepared as recommended by the manufacturer (Trizol Reagent, Life Technologies).
RT–PCR analysis
Total RNAs from the T7RNAP-mitochondria mating mixture or from electroporated mitochondria were collected by isopropyl alcohol precipitation, and residual DNA contaminants were removed using RNase-free DNase I (Promega, Madison, WI) as directed by the manufacturer. RT–PCR analysis was then carried out using a SuperScript First-Strand Synthesis system (Life Technologies) and the following primers: EGFPinternal (5′-GAC GAC GGC AAC TAC AAG AC-3′) and GFP3′Sal (5′-AGT GTC GAC TTA CTT GTA CAG CTC GTC CA-3′) for the GFP gene transcription and CmR-F (5′-GTA CCT ATA ACC AGA CCG TTC AGC-3′) and CmR-R (5′-CAG CGG CAT CAG CAC CTT GTC-3′) for the CAT gene transcription. The following PCR cycle parameters were used for each of the RT–PCRs: 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 55°C or 60°C for 45 s and 72°C for 30 s, and finally 72°C for 10 min.
RESULTS
Experimental design
We have devised a method for transferring engineered DNA constructs into mitochondria using bacterial conjugation (Figure 1). We used E.coli S17-1, which carries the genes for DNA transfer functions from the broad host range plasmid RP4 (20), as the donor bacterial strain and cloned the oriT sequence from RP4 (21) into our DNA constructs in order to specifically target them for mobilization. To test this approach, we designed an assay system that used isolated mitochondria containing T7RNAP in their matrix as conjugative recipients and cloned a T7 transcription promoter into our DNA constructs. Because the DNA transferred by conjugation is single stranded (13), we constructed the mobilizing plasmid in such a manner that a double-stranded hairpin-loop containing a transcriptionally active T7 promoter (27,28) will form from the mobilized ssDNA (Figure 4B). When these DNA molecules are transferred from E.coli into the mitochondrial matrix, the T7RNAP initiates transcription from the T7 promoter in the hairpin-loop and generates RNA transcripts that we can readily assay by RT–PCR. We ensure that only RNA transcribed in structurally intact mitochondria are measured in this RT–PCR assay by selectively lysing the E.coli donor cells and then subjecting the mitochondrial solution to high levels of DNase and RNase prior to isolating the in organello RNA transcripts.
Figure 1.

Conjugation between E.coli and mitochondrion containing T7 RNA polymerase (T7RNAP). When a conjugative-competent E.coli contacts a mitochondrion, the donor bacteria forms a mating bridge (arrows) and a nick is made within the oriT sequence (black box) in the plasmid by the TraI enzyme (open circle), which binds covalently to the freed 5′ end (13). Extension of the 3′ end of the oriT nick displaces a single strand and TraI guides the DNA from E.coli to the mitochondrion through the mating bridge. The second nicking event at oriT liberates 1 U length of the plasmid and supplies the 3′ terminal end for circularization. T7RNAP then starts transcription from the T7 promoter in the transferred DNA and the newly synthesized RNA transcripts (dotted lines) are assayed by RT–PCR.
Targeting of T7RNAP into the mitochondrial matrix
We constructed expression vectors to selectively deliver polypeptides into the mitochondrial matrix by fusing the mitochondrial leader sequence from the mouse Tfam gene (23) in frame with the T7RNAP or GFP coding sequence. Maps of these two vectors are shown in Figure 2A. Because the mitochondrial leader peptide from TFAM has not yet been well characterized, the subcellular localization of recombinant polypeptides from this novel expression vector was confirmed by microscopic analysis of GFP targeted to mitochondria. HeLa 229 cells that were transiently transfected with the chimeric TFAML-GFP vector (Figure 2A) are shown in Figure 2B. We found that the GFP expressed in these cells was localized in the mitochondrial network, as expected, without detectable levels of cytoplasmic expression.
We then used the TFAML-T7RNAP vector to construct a stable cell line expressing T7RNAP targeted to mitochondria using mouse LL/2 cells with blasticidin selection and cloned a single cell that showed correct expression of T7RNAP by RT–PCR analysis. To test whether or not the T7RNAP targeted to these mitochondria can initiate transcription from exogenously transferred DNA, we electroporated a control plasmid (pUC-T7CAT, Figure 3A) into mitochondria isolated from LL/2-T7mt using previously described conditions (see Materials and Methods) (11). We then incubated the electroporated mitochondria in a buffer suitable for in organello RNA synthesis for 2 h at 37°C, washed three times and added 200 Kunitz units of DNase I to remove any DNA outside the electroporated mitochondria. Total RNA from these mitochondria was purified and RT–PCR analyses were performed using the CAT-specific primers CmR-R and CmR-F (see Materials and Methods) to identify the T7RNAP-generated RNA transcripts from the electroporated plasmid DNA. As shown in Figure 3B (lane 4), RT–PCR CAT products of the expected size (470 bp) were clearly detected in the electroporated T7RNAP-mitochondria. No signal was detected in the control reactions without reverse transcriptase (−RT) (Figure 3B, lanes 1 and 3) or in the ‘no electroporation’ control, in which plasmid pUC-T7CAT was mixed with isolated T7RNAP-mitochondria without electroporation (Figure 3B, lanes 1 and 2).
Figure 3.
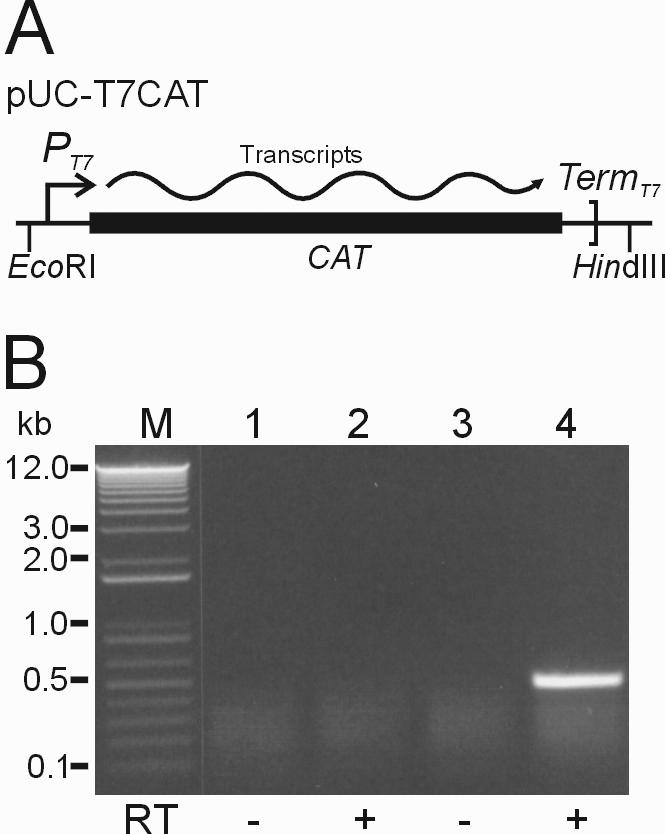
T7RNAP-driven transcription assay by in organello RNA synthesis after electroporation into isolated mouse T7RNAP-mitochondria. (A) Structure of pUC-T7CAT, a control plasmid containing the prokaryotic CAT gene and T7 promoter (PT7). The CAT gene is transcribed from the T7 promoter when this plasmid is introduced into transcriptionally active T7RNAP-mitochondria. (B) RT–PCR analysis of the CAT transcripts in electroporated mouse T7RNAP-mitochondria. Plasmid pUC-T7CAT was electroporated into the isolated mouse T7RNAP-mitochondria and in organello RNA syntheses of these electroporated mitochondria were performed in incubation buffer for 2 h at 37°C. After isolating total RNA from the mitochondria, RT–PCR was carried out using CAT-specific primers CmR-F and CmR-R (see Materials and Methods). Expected band size of the RT–PCR CAT products was 470 bp. Lane M, 1 kb plus DNA ladder (Life Technologies); lanes 1 and 2, no electroporation control with plasmid; lanes 3 and 4, 12 kV/cm electroporation with plasmid. Control lanes in which reverse transcriptase (RT) was omitted are indicated below by minus signs.
Construction of mobilizing plasmids and donor E.coli strains
Maps of the mobilizing plasmid with the oriT sequence (pT7hpGFP+oriT) and a control plasmid without the oriT sequence (pT7hpGFP) are shown in Figure 4A. We used the oriT sequence amplified from pRK290 (21) to construct the mobilizing plasmid. In order to allow a double-stranded hairpin-loop containing an active T7 promoter sequence to form in mobilized ssDNA, we also added appropriately designed inverted repeat sequences that include the T7 promoter sequence (i.e. T7hp and PT7, Figure 4B) (27). Finally, to minimize transcription from E.coli promoter sequences in these plasmids, we flanked the T7hp promoter and GFP assay sequence with the strong E.coli transcriptional terminator sequences λtL3, λtI and rrnBt1t2 (24). The mobilizing and control plasmids were transformed into the conjugative E.coli strain S17-1 (20), and transformants were selected on LB-Tet plates (12.5 µg/ml tetracycline).
In organello RNA synthesis in various mating buffers
In order for us to conduct these conjugation experiments, we needed to find a suitable incubation buffer system that would support both E.coli growth and in organello RNA synthesis inside the mitochondria. We tested 11 buffers that combined components of standard conjugation and in organello RNA synthesis buffer systems (Table 1) for their ability to support growth of an overnight E.coli culture and for their ability to promote in organello RNA synthesis in isolated mitochondria (Figure 5). In incubation buffer 11, E.coli did not grow but good in organello RNA synthesis was observed (Figure 5, lane 11). In incubation buffers 1, 2, 4, 5, 7 and 8, poor levels of both E.coli growth and in organello RNA syntheses were observed (Figure 5, lanes 1, 2, 4, 5, 7 and 8). Addition of yeast extracts to these buffers (incubation buffers 3, 6 and 9) significantly improved E.coli growth and improved in organello RNA syntheses in two of them (Figure 5, lanes 3 and 9). Finally, we tested standard LB medium (buffer 10) as the mating and in organello RNA synthesis buffer and found both high level of in organello RNA synthesis and the best E.coli growth of the buffers tested (Figure 5, lane 10). Based on these results, we decided to use LB as the mating buffer for our conjugation experiment.
Table 1.
Various mating and transcription buffer conditions for E.coli growth and in organello RNA synthesis
| Incubation buffers | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TB | + | + | + | + | |||||||
| M9 Salts | + | + | + | + | + | ||||||
| PBS | + | + | + | ||||||||
| 0.5% YE | + | + | + | ||||||||
| 1% Glucose | + | + | + | + | + | + | + | + | + | ||
| 1 mg/ml Thiamin proline | + | + | + | + | + | + | |||||
| 20 Amino acids mixture | + | + | + | + | + | + | |||||
| 1 mM ATP | + | + | + | + | + | + | + | ||||
| 1 mM ADP | + | + | + | ||||||||
| 1 mM Pyruvate | + | + | + | + | + | + | + | ||||
| 1 mM Succinate | + | + | + | ||||||||
| LB | + | ||||||||||
| E.coli growth (OD600) | 0.6 | 0.7 | 1.2 | 0.2 | 0.3 | 0.9 | 0.6 | 0.7 | 1.3 | 1.8 | 0 |
TB, transcription buffer (10% glycerol, 40 mM Tris–HCl, pH 7.4, 25 mM NaCl, 5 mM MgCl2 and 1 mg/ml BSA); M9 salts, 12.8 g of Na2HPO4·7H2O, 3.0 g of KH2PO4, 0.5 g of NaCl and 1.0 g of NH4Cl in 1 liter of deionized H2O (22); PBS, phosphate-buffered saline; YE, yeast extracts; LB, Luria–Bertani (10 g of bacto-trypton, 5 g of bacto-yeast extracts, 5 g of NaCl in 1 liter of deionized H2O, pH 7.4).
Figure 5.
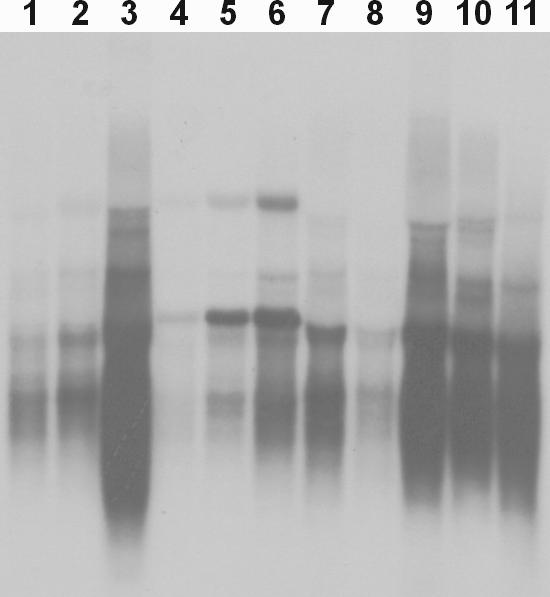
In organello RNA syntheses in various incubation buffers using isolated mouse mitochondria. The mitochondria were isolated from the mouse LL/2 cell line and incubated in 11 modified incubation buffers (Table 1) with 10 µCi of [α-32P]UTP at 37°C for 2 h by shaking. Total RNA was isolated from the mitochondria and fractionated on a 1% of NorthernMax-Gly (Ambion, Austin, TX) agarose gel at 5 V/cm. The gel was then dried and exposed directly on an X-ray film.
Transfer of exogenous DNA into mitochondria by conjugation
Donor E.coli S17-1 cells harboring the mobilizing and the control plasmids, respectively, were mixed with the T7RNAP-containing mitochondria isolated from the LL/2-T7mt cell line. To improve the mating efficiency and ensure good contact between E.coli and mitochondria, we added 1% low-melting point agarose to the mating mixture of E.coli and T7RNAP-mitochondria to form a small agarose block, which was agitated by shaking at 37°C. After incubating for 5 h, we added ampicillin (200 µg/ml) to the mating mixture to lyse the E.coli cells and DNase I and RNase A (50 µg/ml) for 2 h at 37°C to remove any DNA that had not been transferred into the mitochondria and any RNA that may have been synthesized outside of the mitochondria.
For control experiments, we used E.coli S17-1 harboring a control plasmid that has no oriT sequence (pT7hpGFP, Figure 4A) and performed mating with T7RNAP-mitochondria with concurrent in organello RNA synthesis (Figure 6, lanes 1 and 2). No signal was detected by RT–PCR in these samples. We clearly see the expected RT–PCR product (Figure 6, lane 4), however, in the conjugation reactions between T7RNAP-mitochondria and E.coli S17-1 harboring a mobilizable plasmid that has the oriT sequence (pT7hpGFPT+oriT, Figure 4A). No signal was detected in the control reactions without reverse transcriptase (−RT) (Figure 6, lanes 1 and 3). We performed multiple repetitions of this mating experiment to confirm that the procedure described is reproducible and robust.
Figure 6.
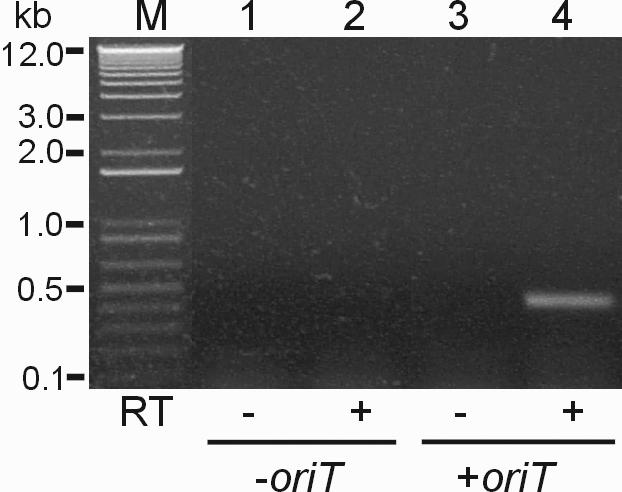
T7RNAP-driven transcription assay after conjugation between E.coli and T7RNAP-mitochondria. E.coli S17-1 harboring pT7hpGFP (lanes 1 and 2) and pT7hpGFP+oriT (lanes 3 and 4), respectively, were used as the donors. All mating samples were treated with 200 µg/ml of ampicillin, 200 U of DNase I and 50 µg/ml of RNase A for 2 h at 37°C after 5 h mating in an agarose block. After isolating total RNA from the mating mixture, RT–PCR was carried out using GFP-specific primers (see Materials and Methods). Expected band size of the RT–PCR GFP products was 423 bp. Lane M, 1 kb plus DNA ladder (Life Technologies). Control lanes (lanes 1 and 3) in which reverse transcriptase (RT) was omitted are indicated by minus signs.
DISCUSSION
We have found that exogenous plasmid DNA can be transferred into the mitochondrial matrix by means of conjugation between bacteria and mitochondria. Mitochondria arose from an intracellular bacterial symbiont of the first ancestral eukaryotic cells (29) and have both an inner and outer membrane that are generally similar to many bacteria. Bacterial conjugative transfer evolved to transfer DNA through this type of double membrane, and so it is perhaps not surprising that this system can transfer DNA constructs into mitochondria as well.
In order to determine if bacterial conjugation could serve as an effective means of transforming mitochondria, we first designed an assay to specifically detect DNA that was transferred into the matrix of structurally intact mitochondria (Figure 1). In this assay, the transferred DNA is transcribed by an RNA polymerase that is present only in the mitochondrial matrix and these transcripts are specifically detected by RT–PCR. Any DNA or RNA outside of the mitochondria is eliminated by specifically lysing the bacterial DNA donor and adding high levels of DNase and RNase prior to in organello RNA isolation. Although this assay could perhaps have been performed by placing a mitochondrial promoter on the mobilizable DNA construct and by relying on the endogenous mitochondrial RNA polymerase to generate transcripts from transferred DNA, we instead chose to use the robust and well-characterized phage T7 transcription system for our experiments.
T7RNAP is a single 100 kDa polypeptide that recognizes a simple 18 nt promoter and has been widely used for both in vivo and in vitro expression systems (27,28) and has been shown to be transcriptionally active when introduced into the mitochondrial matrix in yeast (30). We used the mitochondrial leader peptide of the mouse mitochondrial transcription factor A (TFAM) to target T7RNAP into the mitochondrial network in mouse cells (Figure 2A). TFAM is encoded by a nuclear gene and is imported into the mitochondrial matrix, where it functions as a key regulator of mammalian mtDNA transcription (31). We confirmed that the TFAM leader sequence (TFAML) that we used for mitochondrial targeting did in fact efficiently target exogenous proteins as expected by fusing the GFP reading frame with that of TFAML and confirming that the GFP was localized to the mitochondrial network (Figure 2B). We also confirmed that the T7RNAP was active inside the mitochondria by electroporating control plasmids containing a T7 promoter into isolated T7RNAP-containing mitochondrial fractions (Figure 3A and B). We detected specific transcripts from electroporated DNA using RT–PCR (Figure 3B, lane 4), but no transcripts were detected when the DNA and mitochondria were mixed without electroporation (Figure 3B, lane 2).
DNA that is transferred to the recipient cell by conjugation is single stranded and is typically replicated back into dsDNA using the replication machinery of the recipient cells (13). However, because the replication of foreign DNA introduced into isolated mitochondria has not yet been documented, we did not want to make our assay system dependent on this additional complex process. Instead, we designed and constructed a plasmid that will spontaneously form a double-stranded hairpin structure containing a transcriptionally active dsDNA T7 promoter sequence (27,28) even when the plasmid is transferred as ssDNA into the mitochondria (Figure 4B). The transcription assay using this construct, therefore, is a direct indicator of transfer of the DNA construct into the mitochondrial matrix containing the T7RNAP and is not dependent on uncharacterized secondary events, such as DNA replication.
To remove the E.coli after conjugative transfer of plasmid DNA into mitochondria, we added ampicillin (200 µg/ml) to the mating mixture to lyse the E.coli selectively. Beta-lactam antibiotics, such as ampicillin, exert their bactericidal effects by inhibiting the cross-linking step (transpeptidation) of bacterial cell wall biosynthesis, resulting in cell lysis (32). In contrast, the mitochondria are not sensitive to ampicillin and remain intact after the addition of this antibiotic. By also adding RNase A and DNase I to the mating mixture, we could eliminate any false background RT–PCR products that might arise from RNA transcripts generated either in the E.coli or outside of the mitochondria in the mating mixture. The RNAs transcribed by the T7RNAP from the DNA transferred into the mitochondrial matrix, on the other hand, are protected from RNase A attack by intact mitochondrial membranes.
We performed mating reactions between mitochondria and E.coli with plasmid constructs that contain oriT transfer sequences or, as a control, E.coli with plasmids that do not have an oriT sequence (Figure 4). These latter reactions were identical to the experimental samples in every way except for the absence of the plasmid DNA sequences necessary to target the plasmid for conjugative DNA transfer. These -oriT reactions, therefore, serve as a control for all of the sources of background RT–PCR signal that do not involve conjugative transformation. As expected, these control reactions did not generate a signal in this assay (Figure 6, lanes 1 and 2), indicating that no DNA was transferred into the mitochondria in the absence of conjugation and that the assay did not have a detectable level of background. When identical conjugative experiments were performed with plasmids that did contain the oriT sequence, however, a robust signal was obtained (Figure 6, lane 4).
Electroporation is the only practical method that has previously been shown to be capable of introducing DNA constructs into isolated mitochondria. The harsh experimental conditions required for these transformations, however, appear to irreversible damage the structural integrity of the mitochondria and kill mammalian cells when applied to whole cells rather than mitochondrial preparations (Y.G. Yoon, personal observation). Bacterial conjugation, on the other hand, is a relatively gentle procedure that does not affect the structural integrity of the recipient mitochondria. This should allow some in organello experiments to be performed that have not been possible with electroporation. More importantly, adapting conjugative DNA transfer to the transformation of mitochondrial networks within living cells should also be possible. E.coli strains capable of invading and replicating within the cytoplasm of mammalian cells are becoming increasingly well characterized (33), and a conjugative, invasive strain of E.coli would make an excellent tool for transferring DNA into the mitochondrial networks of mammalian tissue culture cells.
We believe that conjugative transfer of DNA constructs into mitochondria has important practical applications for both applied and basic research. The ssDNA that is transferred in the conjugation reaction would be an excellent substrate for recombination into endogenous mitochondrial genomes, both for the purposes of characterizing mitochondrial recombination systems and for potentially modifying those genomes. Alternatively, complete mammalian mitochondrial genomes, which we have previously shown can be cloned and modified in E.coli (11), could be transferred into mitochondria via conjugation reactions. Although these genomes would initially be introduced as ssDNA, the endogenous mtDNA replication system may synthesize the second DNA strand from the origins of DNA replication present in the genome, much in the same manner as typically occurs in bacterial conjugation. If neither of the two-replication origins proves to be functional as purely ssDNA, it should be possible to engineer appropriate hairpin sequences (e.g. see Figure 4B) to provide the required dsDNA to initiate DNA synthesis.
Acknowledgments
The authors would like to thank Michael Sadowsky for the gifts of plasmids and E.coli strains, and the generous financial support from the Minnesota Medical Foundation, the Academic Health Center and the Institute of Human Genetics of the University of Minnesota. Funding to pay the Open Access publication charges for this article was provided by a grant from NIH (NINDS grant number NS052612).
Conflict of interest statement. None declared.
REFERENCES
- 1.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 2.Wallace D.C. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 3.Fox T.D., Sanford J.C., McMullin T.W. Plasmids can stably transform yeast mitochondria lacking endogenous mtDNA. Proc. Natl Acad. Sci. USA. 1988;85:7288–7292. doi: 10.1073/pnas.85.19.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston S.A., Anziano P.Q., Shark K., Sanford J.C., Butow R.A. Mitochondrial transformation in yeast by bombardment with microprojectiles. Science. 1988;240:1538–1541. doi: 10.1126/science.2836954. [DOI] [PubMed] [Google Scholar]
- 5.Vestweber D., Schatz G. DNA–protein conjugates can enter mitochondria via the protein import pathway. Nature. 1989;338:170–172. doi: 10.1038/338170a0. [DOI] [PubMed] [Google Scholar]
- 6.Seibel P., Trappe J., Villani G., Klopstock T., Papa S., Reichmann H. Transfection of mitochondria: strategy towards a gene therapy of mitochondrial DNA diseases. Nucleic Acids Res. 1995;23:10–17. doi: 10.1093/nar/23.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geromel V., Cao A., Briane D., Vassy J., Rotig A., Rustin P., Coudert R., Rigaut J.P., Munnich A., Taillandier E. Mitochondria transfection by oligonucleotides containing a signal peptide and vectorized by cationic liposomes. Antisense Nucleic Acid Drug Dev. 2001;11:175–180. doi: 10.1089/108729001300338708. [DOI] [PubMed] [Google Scholar]
- 8.Chinnery P.F., Taylor R.W., Diekert K., Lill R., Turnbull D.M., Lightowlers R.N. Peptide nucleic acid delivery to human mitochondria. Gene Ther. 1999;6:1919–1928. doi: 10.1038/sj.gt.3301061. [DOI] [PubMed] [Google Scholar]
- 9.Flierl A., Jackson C., Cottrell B., Murdock D., Seibel P., Wallace D.C. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol. Ther. 2003;7:550–557. doi: 10.1016/s1525-0016(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 10.Collombet J.M., Wheeler V.C., Vogel F., Coutelle C. Introduction of plasmid DNA into isolated mitochondria by electroporation: a novel approach toward gene correction for mitochondrial disorders. J. Biol. Chem. 1997;272:5342–5347. doi: 10.1074/jbc.272.8.5342. [DOI] [PubMed] [Google Scholar]
- 11.Yoon Y.G., Koob M.D. Efficient cloning and engineering of entire mitochondrial genomes in Escherichia coli and transfer into transcriptionally active mitochondria. Nucleic Acids Res. 2003;31:1407–1415. doi: 10.1093/nar/gkg228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan S.M., Bennett J.P., Jr Development of mitochondrial gene replacement therapy. J. Bioenerg. Biomembr. 2004;36:387–393. doi: 10.1023/B:JOBB.0000041773.20072.9e. [DOI] [PubMed] [Google Scholar]
- 13.Waters V.L. Conjugative transfer in the dissemination of beta-lactam and aminoglycoside resistance. Front. Biosci. 1999;4:D433–456. doi: 10.2741/waters. [DOI] [PubMed] [Google Scholar]
- 14.Heinemann J.A., Sprague G.F., Jr Bacterial conjugative plasmids mobilize DNA transfer between bacteria and yeast. Nature. 1989;340:205–209. doi: 10.1038/340205a0. [DOI] [PubMed] [Google Scholar]
- 15.Nishikawa M., Yoshida K. Trans-kingdom conjugation offers a powerful gene targeting tool in yeast. Genet. Anal. Biomol. Eng. 1998;14:65–73. doi: 10.1016/s1050-3862(97)10003-1. [DOI] [PubMed] [Google Scholar]
- 16.Piers K.L., Heath J.D., Liang X., Stephens K.M., Nester E.W. Agrobacterium tumefaciens-mediated transformation of yeast. Proc. Natl Acad. Sci. USA. 1996;93:1613–1618. doi: 10.1073/pnas.93.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunik T., Tzfira T., Kapulnik Y., Gafni Y., Dingwall C., Citovsky V. Genetic transformation of HeLa cells by Agrobacterium. Proc. Natl Acad. Sci. USA. 2001;98:1871–1876. doi: 10.1073/pnas.041327598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waters V.L. Conjugation between bacterial and mammalian cells. Nature Genet. 2001;29:375–376. doi: 10.1038/ng779. [DOI] [PubMed] [Google Scholar]
- 19.Simon R. High frequency mobilization of gram-negative bacterial replicons by the in vitro constructed Tn5-Mob transposon. Mol. Gen. Genet. 1984;196:413–420. doi: 10.1007/BF00436188. [DOI] [PubMed] [Google Scholar]
- 20.Simon R., Priefer U., Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. BioTechnology. 1983;1:784–791. [Google Scholar]
- 21.Ditta G., Schmidhauser T., Yakobson E., Lu P., Liang X.W., Finlay D.R., Guiney D., Helinski D.R. Plasmids related to the broad host range vector, pRK290, useful for gene cloning and for monitoring gene expression. Plasmid. 1985;13:149–153. doi: 10.1016/0147-619x(85)90068-x. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 23.Larsson N.G., Garman J.D., Oldfors A., Barsh G.S., Clayton D.A. A single mouse gene encodes the mitochondrial transcription factor A and a testis-specific nuclear HMG-box protein. Nature Genet. 1996;13:296–302. doi: 10.1038/ng0796-296. [DOI] [PubMed] [Google Scholar]
- 24.Koob M., Shaw A., Cameron D.C. Minimizing the genome of Escherichia coli. Ann. N. Y. Acad. Sci. 1994;745:1–3. doi: 10.1111/j.1749-6632.1994.tb44359.x. [DOI] [PubMed] [Google Scholar]
- 25.Kingston R.E., Chen C.A., Okayama H. Calcium phosphate transfection. In: Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K., editors. Short Protocols in Molecular Biology. 3rd ed. New York: John Wiley and Sons, Inc.; 1987. pp. 9-5–9-8. [Google Scholar]
- 26.Gaines G.L., III In organello RNA synthesis system from HeLa cells. Methods Enzymol. 1996;264:43–49. doi: 10.1016/s0076-6879(96)64007-5. [DOI] [PubMed] [Google Scholar]
- 27.Diaz G.A., Rong M., McAllister W.T., Durbin R.K. The stability of abortively cycling T7 RNA polymerase complexes depends upon template conformation. Biochemistry. 1996;35:10837–10843. doi: 10.1021/bi960488+. [DOI] [PubMed] [Google Scholar]
- 28.Maslak M., Martin C.T. Kinetic analysis of T7 RNA polymerase transcription initiation from promoters containing single-stranded regions. Biochemistry. 1993;32:4281–4285. doi: 10.1021/bi00067a017. [DOI] [PubMed] [Google Scholar]
- 29.Gray M.W., Burger G., Lang B.F. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 30.Pinkham J.L., Dudley A.M., Mason T.L. T7 RNA polymerase-dependent expression of COXII in yeast mitochondria. Mol. Cell. Biol. 1994;14:4643–4652. doi: 10.1128/mcb.14.7.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clayton D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell. Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- 32.Waxman D.J., Strominger J.L. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu. Rev. Biochem. 1983;52:825–869. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 33.Sinai A.P., Bavoil P.M. Hyper-invasive mutants define a novel Pho-regulated invasion pathway in Escherichia coli. Mol. Microbiol. 1993;10:1125–1137. doi: 10.1111/j.1365-2958.1993.tb00982.x. [DOI] [PubMed] [Google Scholar]