

Abstract
Purpose:
Membrane depolarization and subsequent synaptic release of L-glutamate have been implicated in ischemic retinal damage. However, the mechanisms that lead to retinal damage are poorly understood. In this study, we injected KCl, a classical membrane depolarizing agent, and have investigated the role of matrix metalloproteinase-9 in KCl-induced retinal damage.
Methods:
Normal adult CD-1 mice were treated with KCl via intravitreal injection. MMP activity in retinal protein extracts was determined by gelatin-zymography. Tissue localization of MMP-9 in the retina was determined by immunohistochemistry. MMP-9, MMP-2, TIMP-1, TIMP-2, Bax, and BCl-2 proteins in retinal extracts were determined by western blot analysis. Apoptotic cell death in the retina was determined by TUNEL assays. Retinal damage was assessed by immunolocalization studies using antibodies against neurofilament (NF-L) and calretinin.
Results:
Depolarizing concentrations of KCl induced a dose- and time-related up-regulation in MMP-9 activity and protein in the retina. KCl-mediated MMP-9 up-regulation was associated with an increase in pro-apoptotic protein Bax and apoptotic death of cells in the ganglion cell layer (GCL) and inner nuclear layer (INL), and subsequent loss of NF-L-positive ganglion cells and calretinin-positive amacrine cells. Intravitreal injection of KCl along with a NMDA-type glutamate receptor antagonist, MK-801, and a non-NMDA-type glutamate receptor antagonist, NBQX, resulted in a reduction in KCl-mediated MMP-9 up-regulation in the retina. Furthermore, a synthetic MMP inhibitor inhibited KCl-mediated MMP-9 up-regulation and this led to a significant attenuation against KCl-induced retinal damage.
Conclusion:
These results suggest that MMP-9 up-regulation, in part, plays a causative role in KCl-induced retinal damage.
Keywords: Retina, KCl, depolarization, matrix metalloproteinases, apoptosis, ganglion cells, and retinal degeneration
Abbreviations used: ECM, extracellular matrix; GCL, ganglion cell layer; INL, inner nuclear layer; MK-801, (5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate; MMPs, matrix metalloproteinases; NBQX, 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo [f]quinoxaline-7-sulfonamide; NMDA, N-methyl-D aspartate; TUNEL, TdT-mediated dUTP nick-end labeling; AMPA, Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; KCl, potassium chloride; TEA-Cl, tetraethyl ammonium chloride
Introduction
A number of previous studies have implicated that ischemia, in part, plays a degenerative role in human blinding retinal diseases such as glaucoma and diabetic retinopathy1–7. Although the mechanisms that underlie ischemia-induced retinal damage are less clear, a wealth of information accumulated so far suggest that ischemic damage starts with a reduction in cellular ATP levels that in turn results in rapid failure of the Na/K-ATPase pumps and subsequent depolarization of the cell membrane8–10. Membrane depolarization then leads to two additional responses in affected cells: a) a large increase in calcium influx through voltage-gated channels, and b) release of physiological neurotransmitter L-glutamate into the extracellular space10–12. Accumulation of extracellular glutamate may then over-stimulate (excitotoxicity) NMDA and kainate-type ionotropic glutamate receptors and lead to retinal damage13,14 due to increased calcium influx15. However, the precise mechanisms involved in ischemia-induced retinal degeneration are poorly understood.
Results from this laboratory have previously demonstrated that an extracellular matrix (ECM) modulating protease, matrix metalloproteinase-9 (MMP-9), mediates ischemia- and excitotoxicity-mediated retinal damage16–18. Matrix metalloproteinases (MMPs) are a family of proteases that play an important role in modulation or degradation of extracellular matrix (ECM). So far more than 28 members of MMP family have been identified19–21. MMPs play a role not only in ECM remodeling but also in cell migration, invasion22–24, angiogenesis25, and wound healing 26–29. MMP activity is controlled at multiple levels, such as transcriptional regulation, proenzyme activation and inhibition by TIMPS (tissue inhibitors of metalloproteinases)22,23,30–32. Although MMPs are essential for physiological remodeling, excessive levels of these proteases can modulate or degrade ECM22,33–36 and contribute to cell death. In this regard, mounting evidence indicates that MMPs play a degenerative role in the CNS37 and optic nerve38,39. In addition, recent studies have reported that MMP-9 alone causes apoptosis of neuronal cells in vitro40
In spite of previous studies from this laboratory that ischemia-mediated MMP-9 contributes to retinal damage, the mechanisms and the key events that lead to up-regulation of MMP-9 under ischemic conditions are poorly understood. Since, depolarization seems to be an earliest event during ischemic retinal damage, a hypothesis was put forward that depolarization might be one of the mechanisms that leads to an up-regulation in MMP-9 in the retina and MMP-9 in turn promotes retinal damage. Therefore, three specific questions were addressed in this study: 1) Does a membrane depolarizing agent, KCl (potassium chloride), cause retinal damage? 2) Is an up-regulation in MMP-9, in part, plays a role in KCl-induced retinal damage? 3) Is MMP-9 up-regulation mediated by glutamate receptors? These questions were addressed using an animal model in which a classical membrane depolarizing KCl was injected into the vitreous humor of normal adult CD-1 mice. This is the first time, to our knowledge, that KCl, a classical depolarizing agent, was injected into the vitreous humor to create experimental depolarization in the retina.
Methods
Intravitreal injections.
All the experiments on mice were performed under general anesthesia according to institutional protocol guidelines and the ARVO (Association for Research in Vision and Ophthalmology) statement for the use of Animals in Ophthalmology and Vision Research. Normal adult CD-1 mice (6–8 weeks old; Charles River Breeding Labs, Wilmington, MA) were anesthetized by an intraperitonial injection of 1.25% avertin (2,2,2-tribromoethanol in tert-amyl alcohol; 17 ul/g body weight). Two membrane depolarizing agents, KCl41–43 and TEA-Cl (Tetraethyl ammonium chloride)41, were injected into the vitreous humor in CD-1 mice according to previously described methods44,45. It is pointed out that these previously published procedures describe in general an intravitreal injection technique and they are not specific to induction of depolarization in the retina. In control experiments, eyes (n=6) were injected with 2.5 ul of 0.1M phosphate buffered saline alone (PBS, pH 7.4) and in treatment groups, eyes (n=6) were injected with 2.5 ul of 8, 80, and 160 mM KCl (corresponding to 20, 200 and 400 nmoles final concentration), 2.5 ul of 160 mM TEA (corresponding to 400 nmoles), 2.5 ul of 160 mM NaCl (corresponding to 400 nmoles) prepared in PBS. In separate sets of experiments, eyes (n=6) were injected with 2.5 ul of 160 mM KCl (corresponding to 400 nmoles) plus 100 and 200 mM NBQX (corresponding to 100 and 200 nmoles; Tocris, Ellisville, MO), or KCl plus 100 and 200 mM (+) - MK801 maleate (corresponding to 200 and 400 nmoles; Tocris, Ellisville, MO).
Protein extraction.
At 6 h, 12 h, 1 day and 2 days after intravitreal injection, eyes were enucleated from animals anesthetized with an overdose of avertin. Enucleated eyes were cut in half at the equator and the lenses were removed. Retinas were carefully peeled off using forceps and washed three times with phosphate buffered saline (pH 7.4) to remove vitreous that may have adhered to the retina. Three to four retinas each were placed in Eppendorff tubes containing 40 μl of extraction buffer (1% nonidet-P40, 20 mM Tris-HCl, 150 mM NaCl, 1 mM Na3VO4, pH 7.4) and the tissues were homogenized. Tissue homogenates were centrifuged at 10,000 rpm for 5 min at 4°C and the supernatants were collected. Protein concentration in supernatants was determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Gelatin Zymography.
MMP activity was determined by gelatin-zymography assays according to the methods described previously 16,17,45. Briefly, aliquots containing equal amounts of retinal protein extracts (25 ug) were mixed with SDS gel-loading buffer and loaded without heating onto 10% SDS polyacrylamide gels containing 0.2% gelatin as MMP substrate. After electrophoresis, the gels were washed three times with 2.5% Triton-X 100 (15 min each time), and placed in activation buffer containing 10 mM CaCl2 (pH 7.4) and incubated overnight at 37°C to allow proteolysis of the substrates in the gels. The gels were stained with 0.1% Coomassie Brilliant Blue-R250 and then de-stained with a solution containing 25% methanol and 10% acetic acid. Samples containing a mixture of murine MMP-9 and MMP-2 were co-electrophoresed for comparison (data not shown). A reduced molecular weight size standard was also included on all gels (data not shown; Life Technologies, Gaithersburg, MD).
Immunohistochemistry.
At indicated times after intravitreal injections, eyes were enucleated, fixed with 4% paraformaldehyde for 1 h at room temperature, and embedded in OCT compound (Sakura Finetek USA, Torrance, CA). Traverse, 10 micron-thick cryostat sections were cut and placed onto super-frost plus slides (Fisher Scientific, Pittsburgh, PA). Sections were subsequently processed for indirect immunofluorescent localization using antibodies against murine MMP-9 (1:1000 dilution in PBS, a kind gift from Dr. Robert Senior, St. Louis, MO), neurofilament-light (NF-L, 1:100 dilution in PBS, Chemicon, Temecula, CA), and calretinin (1:100 dilution in PBS, Chemicon, Temecula, CA). Sections were incubated with appropriate Alexa FluorR-568-conjugated secondary antibodies (1:200 dilution in PBS; Molecular Probes, Eugene, OR) for 1 h at room temperature and mounted with a coverslip. Sections were observed under a Nikon bright field microscope equipped with epifluorescence, and digitized images were obtained using a SPOT digital camera. Images were processed and compiled using Adobe Photoshop Software, versions 5.5 and 7.0 (Adobe system Incorporated, CA).
Apoptosis assay.
Apoptotic cell death in the retina was determined as previously described 16,45. Briefly, 10 micron-thick cryostat sections prepared as described above and apoptotic cell death was detected by TdT-mediated dUTP nick-end labeling (TUNEL) assay, using a kit (In situ cell death detection kit with fluorescein; Roche Biochemicals, Mannheim, Germany) and the protocol provided by the manufacturer. Tissue sections were examined using a Nikon microscope equipped with epifluorescence, and digital images were obtained with a SPOT digital camera and images were compiled using Adobe Photoshop Software, versions 5.5 and 7.0 (Adobe System Incorporated, CA). TUNEL-positive cells in retinal cross sections were counted in four microscope fields from two independent experiments. Statistical significance was analyzed by ANOVA, followed by a post hoc Tukey’s test (GB-Stat Software, Dynamic Microsystems, Silver Spring, MD) and expressed as the mean+/− SEM.
Western blot analysis.
Aliquots containing an equal amount of protein (25 ug) were mixed with gel loading buffer, and separated on 10% SDS-polyacrylamide gels. For Bcl-2, Bax and caspase-3, aliquots containing an equal amount of protein (25 ug) were separated on 4–20% gradient Tris-glycine gels (Bio-Rad Laboratories, Richmond, CA). After electrophoresis, the proteins were transferred onto nylon membranes and non-specific binding was blocked with 10% non-fat dry milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T). Membranes were then probed with antibodies against mouse MMP-9 (1:2500 dilution in TBS, Triple Point Biologics, Forest Grove OR), MMP-2 (1:000 dilution in TBS, NeoMarkers, CA, data not shown), TIMP-1 (1:500 dilution in TBS, Santa Cruz Biotechnology, Santa Cruz, CA), TIMP-2 (1:1000 dilution in TBS, Sigma, St. Louis, MO), Bcl-2 (1:1000 dilution in TBS, BD Pharmingen, San Diego, CA), and Bax (1:1000 dilution in TBS, Cell Signaling Technology, Beverly, MA). After incubation with the primary antibodies, membranes were washed with TBS-T, and incubated with appropriate horse-radish peroxidase (HRP)-conjugated secondary antibodies (1:2500 dilution in TBS) at room temperature for 1 h. Finally, the proteins on the membranes were detected using an ECL chemiluminescence kit (Amersham Pharmacia Biotech, Piscataway, NJ) and exposing the membranes to an X-ray film. Purified MMP-9 (Triple Point Biologics, Forest Grove, OR) and MMP-2 (Lab Vision, Fremont, CA) were co-electrophoresed as positive standards (data not shown). For Bax and BCl-2, protein bands on X-ray films were scanned by a densitometer and the data from two independent experiments was represented as mean arbitrary units +/− SEM. Statistical significance was analyzed by using a non-parametric Tukey/Kramer procedure (GB-Stat Software, Dynamic Microsystems, Silver Spring, MD).
Results
Effect of KCl on retinal degeneration.
Since depolarization seems to be an earliest event during ischemic damage, experiments were performed to determine whether depolarization, induced by intravitreal injection of a classical depolarizing agent, KCl, causes retinal damage. Three different assays were performed to assess KCl-induced retinal damage. In the first experiment phosphate buffered saline (PBS), 400 nmoles of KCl and TEA (a concentration sufficient to induce depolarization) or NaCl (400 nmoles) were injected into the vitreous humor of normal CD-1 mice. At one day after injection, total retinal proteins were extracted and changes in pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2 levels were determined by western blot analysis (Figure 1A). Low levels of Bax and BCl-2 proteins were observed in retinal protein extracts prepared after intravitreal injection of PBS. In contrast, Bax protein levels were increased in retinal protein extracts prepared after intravitreal injection of KCl and TEA but not after intravitreal injection of NaCl. No significant changes in Bcl-2 protein levels were observed (Figure 1A and B).
Figure 1. KCl- induces changes in pro-apoptotic regulators in the retina.
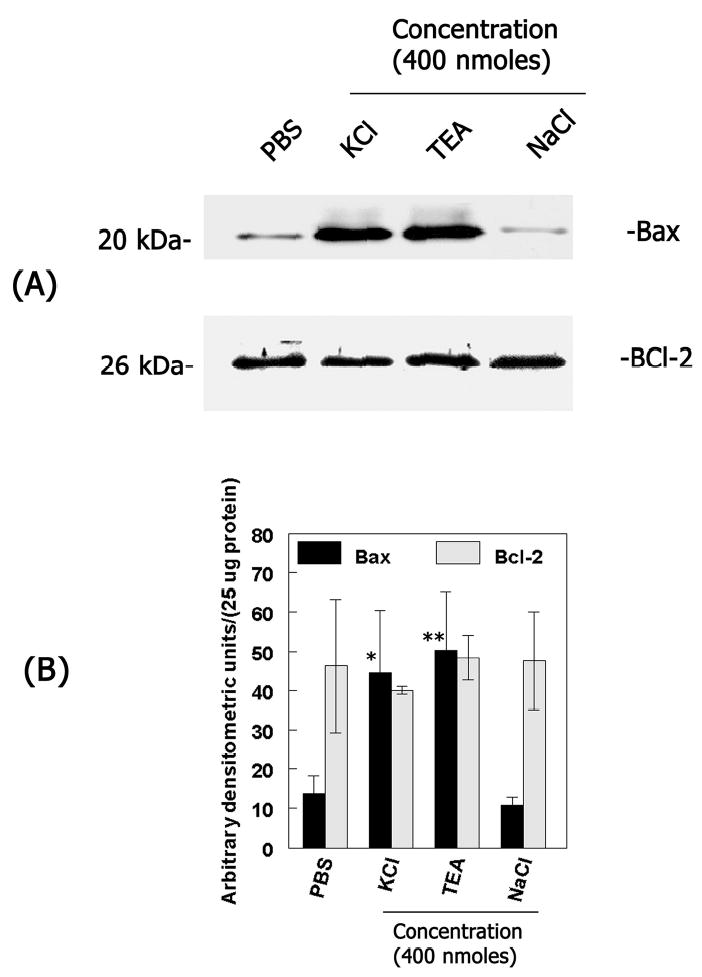
(A) At one day after intravitreal injection of PBS or KCl, TEA, NaCl (400 nmoles each) total retinal proteins were extracted and aliquots containing an equal amount of retinal proteins (25 ug) from each treatment were subjected to western blot analysis using antibodies against Bax and Bcl-2. (B) Bax and Bcl-2 protein bands were scanned by a densitometer and results from two independent experiments were represented as arbitrary densitometric units per 25 ug protein. Bax, *P<0.05 PBS vs KCl; Bax, **P<0.05 PBS vs TEA. Results indicate that intravitreal injection of KCl and TEA but not NaCl results in increased Bax protein levels (but not Bcl-2) in the retina.
In the second experiment, retinal cross sections were prepared at various times after intravitreal injection of KCl and apoptotic cell death was determined by TdT-mediated dUTP nick-end labeling (TUNEL) assays (Figure 2A). Examination of retinal cross sections indicated TUNEL-positive apoptotic cells in the ganglion cell layer as early as 6 h after intravitreal injection of KCl. At one day after KCl injection, TUNEL-positive cells were observed in the GCL and INL. Two days after intravitreal injection of KCl, increased numbers of TUNEL-positive cells were observed in the INL. TUNEL-positive cells were absent in retinal cross sections prepared from un-injected control or PBS or NaCl-injected eyes. In contrast, TEA-injected eyes showed TUNEL-positive cells both in the GCL and INL similar to that observed in retinal cross sections prepared from KCl injected eyes (Figure 2A). Quantitation of TUNEL-positive cells in retinal cross sections indicated a significant increase in the number of apoptotic cells both in the GCL and in the INL after intravitreal injection of KCl and TEA, but not after NaCl (Figure 2B).
Figure 2. KCl- induces retinal degeneration.
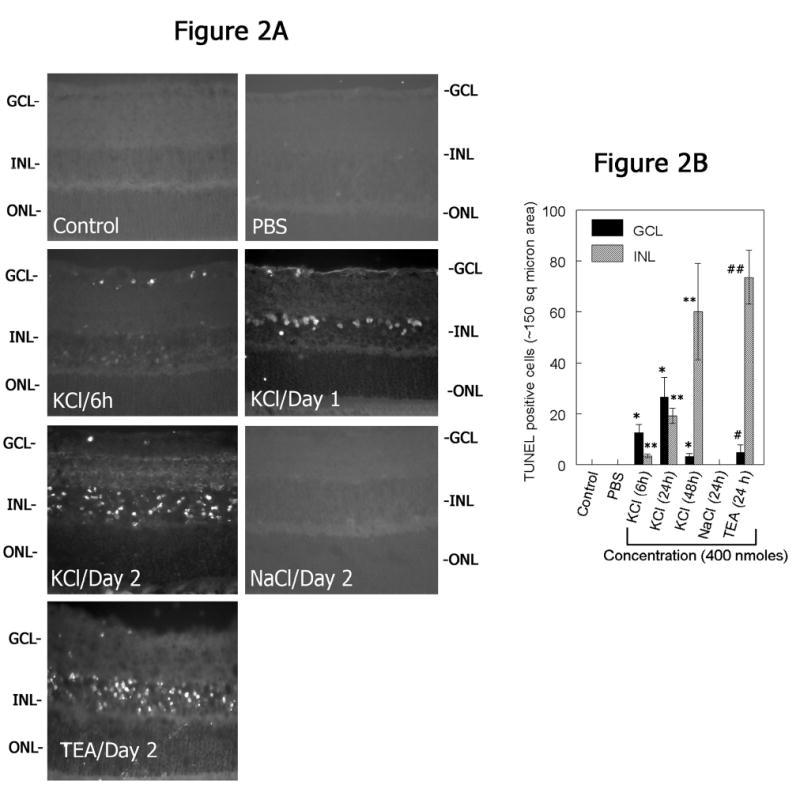
(A) Retinal cross sections were prepared at 6 h, day 1, and 2 days after intravitreal injection of KCl (400 nmoles) or PBS or NaCl (400 nmoles/day 2) or TEA (400 nmoles/day 2) and assayed for apoptotic cell death by TUNEL assays. (B) At various times after intravitreal injection of KCl, TEA, and NaCl (400 nmoles each), retinal cross sections were prepared and TUNEL assays were performed to determine apoptotic cell death in the retina. The numbers of TUNEL-positive cells in the ganglion cell layer (GCL) and in the inner nuclear layer (INL) were quantified and plotted as a bar graph. Results indicate that depolarizing agents both KCl and TEA induces apoptotic cell death in the retina. *P<0.05, PBS vs KCl/TEA in the GCL; **P<0.05, PBS vs KCl/TEA in the INL.
In the third experiment, retinal cross sections prepared after intravitreal injection of KCl or NaCl were immunostained with antibodies against ganglion cell marker, neurofilament-light (NF-L), and amacrine cell marker, calretinin (Figure 3). Examination of retinal cross sections indicated loss of NF-L-positive immunostaining one day and two days after KCl injection (400 nmoles) but not after intravitreal injection of similar concentrations of NaCl (Figure 3A). In addition, loss of calretinin-positive amacrine cells was also observed in retinal cross sections prepared at one day and two days after intravitreal injection of KCl (Figure 3B). Taken together, the above three experiments indicate that intravitreal injection of KCl leads to apoptotic death of both ganglion cells and amacrine cells in the retina.
Figure 3. KCl-induces loss of ganglion- and amacrine cells.
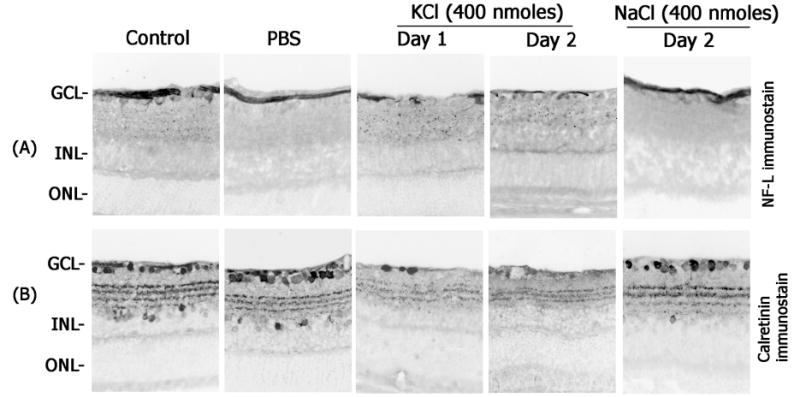
Retinal cross sections prepared after intravitreal injection of KCl or NaCl (400 nmoles each) were immunostained with antibodies against neurofilament-light (NF-L; top panels) and calretinin (lower panels). Note a decrease in NF-L- and calretinin-immunoreactivity after intravitreal injection of KCl but not after intravitreal injection of NaCl. All the photographs were taken at 40× magnification.
Effect of KCl on MMP activity in the retina.
To determine whether KCl-mediated depolarization modulates MMP levels in the retina, varying concentrations of KCl (20, 200, and 400 nmoles) were injected into the vitreous humor of CD-1 mice. Retinal proteins were extracted at 2 days after intravitreal injection and aliquots containing an equal amount of protein (25 ug) from each treatment were then loaded onto SDS-polyacrylamide gels containing gelatin as a substrate for MMPs. Gelatin-zymography assays indicated low and constitutive levels of MMP-9 in retinal proteins extracted from un-injected control or PBS-injected eyes (Figure 4A). Intravitreal injection of KCl resulted in an up-regulation in MMP-9 activity but only at concentrations higher than 20 nmoles. Intravitreal injection of 400 nmoles KCl-resulted in a dramatic increase in MMP-9 activity over un-injected control or PBS injected eyes. Up-regulation of MMP-9 was also observed in retinal extracts after intravitreal injection of TEA (400 nmoles). Western blot analysis confirmed the increase in MMP-9 protein levels in response to KCl and TEA injections (Figure 4A). Similar concentrations of NaCl failed to up-regulate MMP-9 activity in the retina (Figure 4A).
Figure 4. KCl-up-regulates MMP-9 activity.
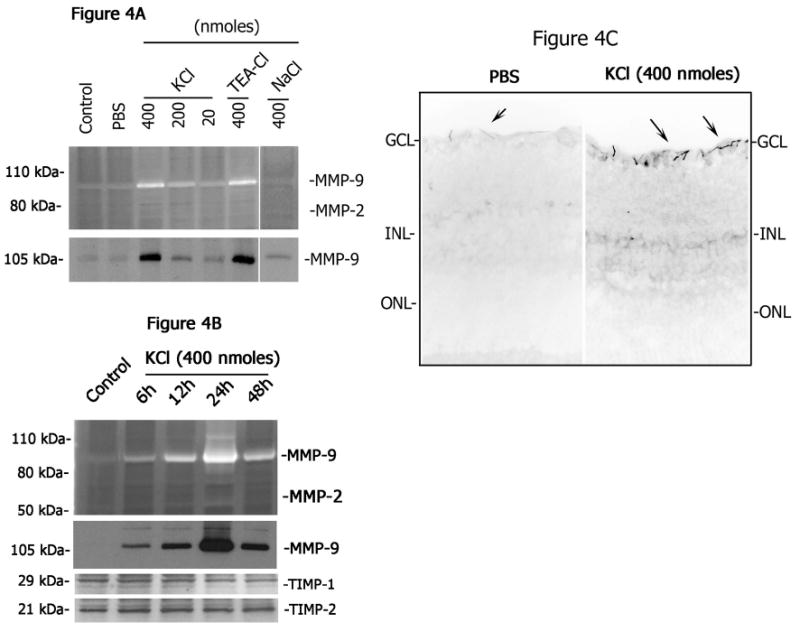
(A) Aliquots containing an equal amount of retinal proteins (25 ug) extracted at two days after intravitreal injection of KCl, TEA, NaCl or PBS or from un-injected control eyes were subjected to gelatin zymography (upper panel) and western blot analysis (lower panel). (B) At various times after intravitreal injection of KCl (400 nmoles) retinal protein extracts were extracted and aliquots containing an equal amount of retinal protein (25 ug) were subjected to gelatin zymography (upper panel) and western blot analysis (lower panel). (C) Retinal sections prepared from PBS-injected or KCl-injected eyes were immunostained using antibodies against MMP-9. Note a dose-and time-related increase in MMP-9 activity and protein levels in the retina after intravitreal injection of KCl and MMP-9 tissue localization in the ganglion cell layer. Also, note that TIMP-1 and TIMP-2 levels remained unchanged.
To determine whether KCl-mediates time-related up-regulation in MMP-9 activity in the retina, 400 nmoles of KCl were injected into the vitreous humor of CD-1 mice, retinal proteins were extracted at various times after injection and gelatin-zymography assays were performed to determine MMP activity and western blot analysis was performed to determine MMP-9 protein levels. Gelatin zymography and western blotting analysis indicated a time-related up-regulation in MMP-9 activity and protein levels in the retina after intravitreal injection of KCl (Figure 4B). MMP-9 activity and protein levels were increased as early as 6 h, reached peak levels around day 1, and then returned to lower levels by day 2 (Figure 4B). No other gelatinolytic MMPs were observed after KCl injection. Immunolocalization studies performed on retinal cross sections prepared 1 day and 2 days after intravitreal injection of KCl indicated that astrocytes in the ganglion cell/nerve fiber layer express MMP-9 protein (Figure 4C). Western blot analysis indicated no major changes in TIMP-1 and TIMP-2 protein levels in the retina after intravitreal injection of KCl (Figure 4B). In addition, no significant changes in MMP-2 protein levels were observed (data not shown). These results indicate that KCl induces MMP-9 up-regulation in the retina.
Effect of glutamate receptor antagonists on MMP-9 activity.
Previous reports have suggested that membrane depolarization causes the release of endogenous glutamate into the extracellular space, which then over-stimulates postsynaptic retinal neurons (known as “excitotoxicity”). Although extracellular glutamate levels in the retina were not determined in this study, it was reasoned that use of glutamate receptor antagonists might help to determine whether glutamate (that might be released after depolarization) plays a role in KCl-induced MMP-9 up-regulation in the retina. Therefore, using an indirect approach, the role of glutamate receptors in MMP-9 up-regulation was determined by injecting 400 nmoles KCl along with a NMDA-type glutamate receptor antagonist, MK-801 (200 and 400 nmoles), or a non-NMDA-type glutamate receptor antagonist, NBQX (100 and 200 nmoles), into the vitreous humor in CD-1 mice. Total retinal proteins were extracted at one day after injection and MMP activity was determined by gelatin zymography assays. An increase in MMP-9 activity was observed in retinal proteins extracted after KCl injection, as expected (Figure 5). In contrast, a decrease in MMP-9 activity in the retina was observed after intravitreal injection of KCl along with MK-801 or NBQX. Intravitreal injection of receptor antagonists alone (without KCl) failed to up-regulate MMP-9 activity in the retina. These results indicate that depolarization-induced MMP-9 activity is mediated, in part, by both NMDA and non-NMDA-type glutamate receptors.
Figure 5. Glutamate receptor antagonism inhibits KCl-induced MMP-9 activity.

Retinal proteins extracted at one day after intravitreal injection of KCl (400 nmoles) with or without indicated concentrations of NMDA receptor antagonist (MK-801) and non-NMDA receptor antagonists (NBQX) were subjected to gelatin zymography. Note that both receptor antagonists inhibited KCl-induced MMP-9 activity in the retina.
Effect of MMP inhibitor on KCl-induced retinal degeneration
Although the results described above implicated an association between KCl-mediated up-regulation of MMP-9 and subsequent retinal degeneration, it was not completely clear whether MMP-9 played a causative role in KCL-induced retinal degeneration. Therefore, a synthetic MMP inhibitor (N-hydroxy-1,3-di-[4-metoxybenzenesulfonyl)-5,5-dimethy;-[1,3]-piperazine-2-carboxamide; MMP inhibitor II; Calbiochem, San Diego, CA; IC50=2.7 nmoles for MMP-9) was used to investigate whether inhibition of MMP-9 activity attenuates KCl-induced retinal damage. MMP inhibitor (0.6 nmoles and 6 nmoles) along with KCl was injected into the vitreous humor and retinal proteins were extracted at 1 day after injection. Aliquots containing equal amount of proteins (25 ug) were then subjected to gelatin-zymography assays to determine MMP activity in the retina (Figure 6A). Zymography assays indicated an up-regulation in MMP-9 in the retina after intravitreal injection of KCl but not after intravitreal injection of DMSO (a vehicle used to dissolve the MMP inhibitor). In contrast, intravitreal injection of 6.0 nmoles of MMP inhibitor (MMP-I) along with KCl resulted in a reduction in MMP-9 activity levels whereas intravitreal injection of DMSO along with KCl failed to inhibit KCl-induced MMP-9 activity in the retina (data not shown). To determine whether inhibition of MMP-9 observed in retinal proteins extracted after intravitreal injection of MMP inhibitor also attenuates retinal degeneration, retinal cross sections were prepared at 1 day after intravitreal injection of KCl with or without MMP inhibitor, and apoptotic cell death was determined by TUNEL assays (Figure 6B). Examination of retinal cross sections indicated the appearance of TUNEL-positive apoptotic cells both in the GCL and in the INL after KCl injection as expected. In contrast, a significant decrease in the number of TUNEL-positive cells was observed after intravitreal injection of KCl along with MMP inhibitor (Figure 6B & C). TUNEL-positive cells were absent in retinal cross sections prepared from both un-injected control and DMSO injected eyes. In addition, intravitreal injection of MMP inhibitor along with KCl also attenuated loss of NF-L-positive ganglion cells and calretinin-positive cells in the retina (Figure 6D). These results indicate that MMP-9, in part, plays a causative role in KCl-induced retinal degeneration.
Figure 6. Inhibition of MMP activity attenuates KCl-induced retinal degeneration.
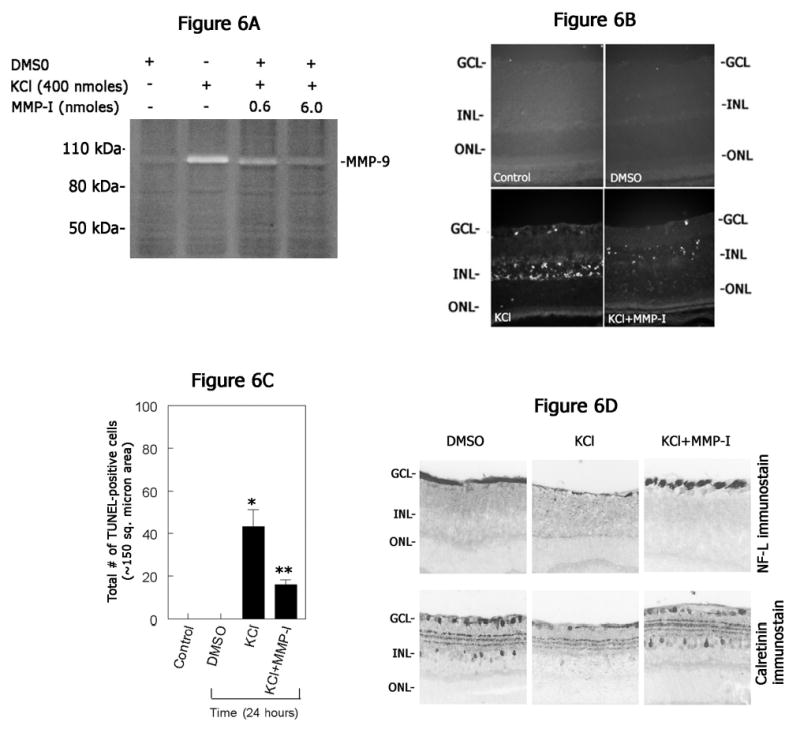
(A) Retinal proteins were extracted at one day after intravitreal injection of KCl (400 nmoles) along with indicated concentrations of MMP inhibitor (MMP-inhibitor-I). Aliquots containing an equal amount of proteins (25 ug) were subjected to gelatin zymography. Note a decrease in KCl-mediated MMP-9 activity after intravitreal injection of MMP inhibitor. (B) Retinal cross sections prepared at one day after intravitreal injection of KCl (400 nmoles) with or without MMP inhibitor (6 nmoles; MMP-inhibitor-I) were subjected to TUNEL assays and compared with un-injected control or vehicle (DMSO)-injected eyes. (C) TUNEL positive cells were counted and plotted as a bar graph. Results indicate a significant reduction in the number of TUNEL positive cells in retinal cross sections after intravitreal injection of KCl along with MMP inhibitor. *P<0.05, controls vs KCl; **P<0.05 KCl vs KCl plus MMP inhibitor; ANOVA, followed by a post hoc Tukey’s test. (D) Retinal cross sections prepared after intravitreal injection of KCl along with or without MMP inhibitor were immunostained with antibodies against neurofilament-light (NF-L) and calretinin. Compared KCl injected eyes, retinal cross sections prepared after intravitreal injection of KCl along with MMP inhibitor showed attenuation in ganglion and amacrine cell loss.
Discussion
Although membrane depolarization and subsequent synaptic release of L-glutamate have been implicated in ischemic retinal damage, the mechanisms involved in retinal damage are poorly understood. In this study, we report a novel finding that injection of a classical depolarizing agent, KCl, into the vitreous humor in CD-1 mice up-regulates MMP-9 activity and protein in the retina and that MMP-9 in turn promotes KCl-induced retinal damage. In support of this hypothesis, depolarizing concentrations of KCl (more than 20 nmoles) injected into the vitreous humor led to a transient up-regulation of MMP-9 activity in the retina. TEA-Cl, another depolarizing agent also up-regulated MMP-9 activity but similar concentrations of NaCl failed to induce this up-regulation. Although MMP-9 pro-form observed in the zymograms can be converted into an active form during retinal damage, our zymography results showed an increase in pro-form of MMP-9, consistent with a number of previous investigations. This is due, in part, to the fact that activated MMP-9 is unstable and undergoes rapid degradation. Nevertheless, KCl-induced an up-regulation of not only in MMP-9 activity but it was also associated with an increase in pro-apoptotic protein Bax, and subsequent apoptotic death of both ganglion cells and amacrine cells in the retina.
Since MMP-9 was up-regulated in response to KCl we reasoned that KCl-might act by releasing endogenous L-glutamate into the extracellular space via membrane depolarization of retinal neurons (mediated by KCl). Although, determination of glutamate levels might provide some information whether glutamate directly play a role in MMP-9 up-regulation, total glutamate levels determined in the retina does not provide enough information whether glutamate exists extracellularly to act on its receptors. In addition, local concentrations of glutamate seem to be more important in promoting retinal damage than the average glutamate levels present in the retina. Therefore, we used an indirect approach in this study based on the notion that endogenous glutamate released in to the extracellular space (by KCl-mediated depolarization) might act on both NMDA and non-NMDA type glutamate receptors to up-regulate MMP-9 activity in the retina. Therefore, we used two specific glutamate receptor antagonists to test this possibility: 1) MK-801 (a specific NMDA glutamate receptor antagonist) and 2) NBQX (a specific non-NMDA glutamate receptor antagonist). Intravitreal injection of MK-801 or NBQX along with KCl, in deed, resulted in reduced MMP-9 activity, compared to KCl-injection alone, indicating that both types of glutamate receptors play a role in depolarization-induced MMP-9 up-regulation.
Although the precise mechanisms that lead to MMP up-regulation after KCl-injection are not clear at this time, previous studies have suggested that KCl induces changes in the synthesis of mRNAs encoding c-Fos protein46,47. Interestingly, c-Fos controls the transcription of a number of immediate early genes, including MMP-948,49. Once MMP-9 is up-regulated, it can proteolytically modulate the extracellular matrix protein laminin in the retina, and can lead to apoptotic cell death. Interestingly, MMP-9 up-regulation was associated with degradation of laminin in the retina (data not shown) as previously reported16,45. Therefore, we reasoned that inhibition of MMP-9 may have protective effect on KCl-induced retinal damage. To test this possibility, a synthetic MMP inhibitor was injected into the vitreous humor to inhibit MMP activity in the retina. Although the inhibitor used in this study is not specific in inhibiting a particular MMP, we used this inhibitor because of KCl caused an up-regulation in MMP-9 alone. Intravitreal injection of MMP inhibitor along with KCl not only reduced MMP-9 activity in the retina but also attenuated KCl-induced retinal damage. This was evident by a) a reduction in the number of TUNEL-positive apoptotic cells and b) by number of remaining NF-L-and calretinin-positive cells in the retina after intravitreal injection of MMP inhibitor along with KCl. These observations support the hypothesis that MMP-9, in part, plays a causative role in promoting KCl-induced retinal damage. It is possible that cell loss could be due to just plain toxicity of KCl. However, the reduction in KCl-induced apoptosis and protection against KCl-induced retinal damage observed by a MMP inhibitor rules out this possibility. Although, additional mechanisms such as inflammation might play a role in KCl-induced retinal damage, our immunolocalization studies using antibodies against macrophage origin (data not shown) failed to detect inflammatory cells in the retina ruling out the possible role of inflammation in KCl-induced retinal degeneration. There is one caveat in this study. Although, we used KCl, a classical depolarizing agent, to induce retinal damage, we have not determined whether KCl directly induces membrane depolarization in the retina (by electrophysiological studies). The results presented in this study, however, shows that MMP-9, in part, plays a role in KCl- induced retinal damage that, in part, is mediated by ionotropic glutamate receptors indicating the classical depolarizing actions of KCl in the retina. The results presented in this study are also consistent with recent studies on the CNS in which depolarization and subsequent up-regulation in MMP-9 have been shown to play a causative role in cortical damage. In addition, a recent study indicated that cortical spreading depression (CSD) associated with depolarization also activates and up-regulates MMP-9 in the CNS51.
In conclusion, the data presented above provides, to our knowledge, the first evidence that intravitreous injection of a membrane depolarization drug, KCl, causes retinal degeneration via an up-regulation in MMP-9.
Acknowledgments
This work was supported by a National Eye Institute project grant, EY13643 (to SKC) and a Vision Research Infrastructure Development Grant EY014803.
References
- 1.Buchi ER. Cell death in rat retina after pressure-induced ischaemia-reperfusion insult: electron microscopic study. II. Outer nuclear layer. JpnJOphthalmol. 1992;36(1):62–68. [PubMed] [Google Scholar]
- 2.Levin LA, Louhab A. Apoptosis of retinal ganglion cells in anterior ischemic optic neuropathy. Arch Ophthalmol. 1996;114(4):488–91. doi: 10.1001/archopht.1996.01100130484027. [DOI] [PubMed] [Google Scholar]
- 3.Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci. 1995;36(5):774–786. [PubMed] [Google Scholar]
- 4.Quigley HA, Miller NR, Green WR. The pattern of optic nerve fiber loss in anterior ischemic optic neuropathy. Am J Ophthalmol. 1985;100(6):769–76. doi: 10.1016/s0002-9394(14)73365-3. [DOI] [PubMed] [Google Scholar]
- 5.Osborne NN, Ugarte M, Chao M, Chidlow G, Bae JH, Wood JP, Nash MS. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol. 1999;43 (Suppl 1):S102–28. doi: 10.1016/s0039-6257(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 6.Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23(1):91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(2):283–90. doi: 10.1016/S0278-5846(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 8.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 9.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330(9):613–22. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 10.Nishizawa Y. Glutamate release and neuronal damage in ischemia. Life Sci. 2001;69(4):369–81. doi: 10.1016/s0024-3205(01)01142-0. [DOI] [PubMed] [Google Scholar]
- 11.Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43(5):1369–74. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- 12.Wahl F, Obrenovitch TP, Hardy AM, Plotkine M, Boulu R, Symon L. Extracellular glutamate during focal cerebral ischaemia in rats: time course and calcium dependency. J Neurochem. 1994;63(3):1003–11. doi: 10.1046/j.1471-4159.1994.63031003.x. [DOI] [PubMed] [Google Scholar]
- 13.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11(10):465–9. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 14.Chidlow G, Osborne NN. Rat retinal ganglion cell loss caused by kainate, NMDA and ischemia correlates with a reduction in mRNA and protein of Thy-1 and neurofilament light. Brain Res. 2003;963(1–2):298–306. doi: 10.1016/s0006-8993(02)04052-0. [DOI] [PubMed] [Google Scholar]
- 15.Katayama Y, Kawamata T, Tamura T, Hovda DA, Becker DP, Tsubokawa T. Calcium-dependent glutamate release concomitant with massive potassium flux during cerebral ischemia in vivo. Brain Res. 1991;558(1):136–40. doi: 10.1016/0006-8993(91)90730-j. [DOI] [PubMed] [Google Scholar]
- 16.Chintala SK, Zhang X, Austin JS, Fini ME. Deficiency in matrix metalloproteinase gelatinase B (MMP-9) protects against retinal ganglion cell death after optic nerve ligation. J Biol Chem. 2002;277(49):47461–8. doi: 10.1074/jbc.M204824200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Chintala SK. Influence of interleukin-1 beta induction and mitogen-activated protein kinase phosphorylation on optic nerve ligation-induced matrix metalloproteinase-9 activation in the retina. Exp Eye Res. 2004;78(4):849–60. doi: 10.1016/j.exer.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Cheng M, Chintala SK. Optic nerve ligation leads to astrocyte-associated matrix metalloproteinase-9 induction in the mouse retina. Neurosci Lett. 2004;356(2):140–4. doi: 10.1016/j.neulet.2003.10.084. [DOI] [PubMed] [Google Scholar]
- 19.Woessner JF. In: Matrix Metalloproteinases, Parks, W C, Mecham,R.P, Editor. 1998, Academic Press: San Diego. p. 1–14.
- 20.Nagase H, Suzuki K, Itoh Y, Kan CC, Gehring MR, Huang W, Brew K. Involvement of tissue inhibitors of metalloproteinases (TIMPS) during matrix metalloproteinase activation. Adv Exp Med Biol. 1996;389:23–31. doi: 10.1007/978-1-4613-0335-0_3. [DOI] [PubMed] [Google Scholar]
- 21.Woessner JF., Jr Matrix metalloproteinase inhibition. From the Jurassic to the third millennium. Ann N Y Acad Sci. 1999;878:388–403. doi: 10.1111/j.1749-6632.1999.tb07697.x. [DOI] [PubMed] [Google Scholar]
- 22.Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 2000;14(17):2123–33. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 23.Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 1998;21(2):75–80. doi: 10.1016/s0166-2236(97)01169-7. [DOI] [PubMed] [Google Scholar]
- 24.Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. NatRevNeurosci. 2001;2(7):502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA. Matrix metalloproteinases: a review. Crit RevOral BiolMed. 1993;4(2):197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- 26.Fini ME, Girard MT, Matsubara M. Collagenolytic/gelatinolytic enzymes in corneal wound healing. Acta Ophthalmol Suppl, 1992(202): 26–33. [DOI] [PubMed]
- 27.Fitch J, Fini ME, Beebe DC, Linsenmayer TF. Collagen type IX and developmentally regulated swelling of the avian primary corneal stroma. Dev Dyn. 1998;212(1):27–37. doi: 10.1002/(SICI)1097-0177(199805)212:1<27::AID-AJA3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 28.Sivak JM, Fini ME. MMPs in the eye: emerging roles for matrix metalloproteinases in ocular physiology. Prog Retin Eye Res. 2002;21(1):1–14. doi: 10.1016/s1350-9462(01)00015-5. [DOI] [PubMed] [Google Scholar]
- 29.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crawford HC, Matrisian LM. Mechanisms controlling the transcription of matrix metalloproteinase genes in normal and neoplastic cells. Enzyme Protein. 1996;49(1–3):20–37. doi: 10.1159/000468614. [DOI] [PubMed] [Google Scholar]
- 31.Murphy G, Stanton H, Cowell S, Butler G, Knauper V, Atkinson S, Gavrilovic J. Mechanisms for pro matrix metalloproteinase activation. Apmis. 1999;107(1):38–44. doi: 10.1111/j.1699-0463.1999.tb01524.x. [DOI] [PubMed] [Google Scholar]
- 32.Kleiner DE, Stetler-Stevenson WG. Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol. 1999;43 (Suppl):S42–51. doi: 10.1007/s002800051097. [DOI] [PubMed] [Google Scholar]
- 33.Matrisian LM, Wright J, Newell K, Witty JP. Matrix-degrading metalloproteinases in tumor progression. Princess Takamatsu Symp. 1994;24:152–61. [PubMed] [Google Scholar]
- 34.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29(5):1020–30. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg GA, Dencoff JE, McGuire PG, Liotta LA, Stetler-Stevenson WG. Injury-induced 92-kilodalton gelatinase and urokinase expression in rat brain. Lab Invest. 1994;71(3):417–22. [PubMed] [Google Scholar]
- 36.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4(2):228–31. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 38.Agapova OA, Ricard CS, Salvador-Silva M, Hernandez MR. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human optic nerve head astrocytes. Glia. 2001;33(3):205–16. doi: 10.1002/1098-1136(200103)33:3<205::aid-glia1019>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 39.Agapova OA, Kaufman PL, Lucarelli MJ, Gabelt BT, Hernandez MR. Differential expression of matrix metalloproteinases in monkey eyes with experimental glaucoma or optic nerve transection. Brain Res. 2003;967(1–2):132–43. doi: 10.1016/s0006-8993(02)04234-8. [DOI] [PubMed] [Google Scholar]
- 40.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297(5584):1186–90. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 41.Gualandris A, Jones TE, Strickland S, Tsirka SE. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci. 1996;16(7):2220–5. doi: 10.1523/JNEUROSCI.16-07-02220.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12(6):1207–21. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 43.Levitan ES, Gealy R, Trimmer JS, Takimoto K. Membrane depolarization inhibits Kv1. 5 voltage-gated K+ channel gene transcription and protein expression in pituitary cells. J Biol Chem. 1995;270(11):6036–41. doi: 10.1074/jbc.270.11.6036. [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Schlamp CL, Nickells RW. Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 1999;40(5):1004–8. [PubMed] [Google Scholar]
- 45.Zhang X, Cheng M, Chintala SK. Kainic acid-mediated upregulation of matrix metalloproteinase-9 promotes retinal degeneration. Invest Ophthalmol Vis Sci. 2004;45(7):2374–83. doi: 10.1167/iovs.03-1239. [DOI] [PubMed] [Google Scholar]
- 46.Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322(6079):552–5. doi: 10.1038/322552a0. [DOI] [PubMed] [Google Scholar]
- 47.Black IB, Adler JE, Dreyfus CF, Friedman WF, LaGamma EF, Roach AH. Biochemistry of information storage in the nervous system. Science. 1987;236(4806):1263–8. doi: 10.1126/science.2884727. [DOI] [PubMed] [Google Scholar]
- 48.Crowe DL, Brown TN. Transcriptional inhibition of matrix metalloproteinase 9 (MMP-9) activity by a c-fos/estrogen receptor fusion protein is mediated by the proximal AP-1 site of the MMP-9 promoter and correlates with reduced tumor cell invasion. Neoplasia. 1999;1(4):368–72. doi: 10.1038/sj.neo.7900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaczmarek L, Lapinska-Dzwonek J, Szymczak S. Matrix metalloproteinases in the adult brain physiology: a link between c-Fos, AP-1 and remodeling of neuronal connections? Embo J. 2002;21(24):6643–8. doi: 10.1093/emboj/cdf676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Gertten C, Holmin S, Mathiesen T, Nordqvist AC. Increases in matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 mRNA after cerebral contusion and depolarisation. J Neurosci Res. 2003;73(6):803–10. doi: 10.1002/jnr.10729. [DOI] [PubMed] [Google Scholar]
- 51.Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, Wang X, Rosenberg GA, Lo EH, Moskowitz MA. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113(10):1447–55. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]