

Abstract
Our previous studies have shown that cells conditionally deficient in Tsg101 arrested at the G1/S cell cycle checkpoint and died. We created a series of Tsg101 conditional knock-out cell lines that lack p53, p21Cip1, or p19Arf to determine the involvement of the Mdm2-p53 circuit as a regulator for G1/S progression and cell death. In this new report we show that the cell cycle arrest in Tsg101-deficient cells is p53-dependent, but a null mutation of the p53 gene is unable to maintain cell survival. The deletion of the Cdkn1a gene in Tsg101 conditional knock-out cells resulted in G1/S progression, suggesting that the p53-dependent G1 arrest in the Tsg101 knock-out is mediated by p21Cip1. The Cre-mediated excision of Tsg101 in immortalized fibroblasts that lack p19Arf seemed not to alter the ability of Mdm2 to sequester p53, and the p21-mediated G1 arrest was not restored. Based on these findings, we propose that the p21-dependent cell cycle arrest in Tsg101-deficient cells is an indirect consequence of cellular stress and not caused by a direct effect of Tsg101 on Mdm2 function as previously suggested. Finally, the deletion of Tsg101 from primary tumor cells that express mutant p53 and that lack p21Cip1 expression results in cell death, suggesting that additional transforming mutations during tumorigenesis do not affect the important role of Tsg101 for cell survival.
The tumor susceptibility gene 101 (Tsg101) encodes a multido-main protein that mediates a variety of biological functions. Some of these functions have been postulated from the predicted protein structure, its intracellular localization, the identification of Tsg101-binding proteins, and very recently, the generation of Tsg101-deficient mouse models. These functions include a role in ubiquitination (1, 2), transcriptional regulation (3, 4), endosomal trafficking (5–8), proliferation (9–12), and cell survival (12, 13). The C-terminal coiled-coil domain of Tsg101, formally known as CC2, was isolated first in a yeast two-hybrid screen with the cell growth regulating protein Stathmin (14). This region is a potential co-repressor that is able to modulate the transcriptional activity of steroid receptors (3, 4, 15). Furthermore, Tsg101 possesses a proline-rich sequence known to exist in activation domains of transcription factors (16). The N-terminal region of Tsg101 (UEV domain) is similar to the catalytic domain of ubiquitin-conjugating (E2) enzymes. Because Tsg101 lacks a key cysteine residue found in authentic E2 enzymes, it has been postulated that this protein might serve as a negative regulator for the ubiquitin-mediated degradation of other proteins (1, 2). Suggested targets of important factors for cell cycle regulation whose stability and function were affected by Tsg101 are Mdm2 and p21Cip1 (17, 18).
New insights about the biological role of Tsg101 in vivo were obtained from genetically engineered mice that lack Tsg101 completely (11, 13) or in selected cell types (12, 13). The deletion of the promoter and the first coding exon of Tsg101 resulted in embryonic lethality around implantation (13). Tsg101-deficient embryos lacking exons 8 and 9 die around day 6.5 of gestation due to a defect in cell proliferation and mesoderm formation (11). The WAP1-Cre-mediated deletion of Tsg101 from differentiating mammary epithelial cells (conditional knock-out) in adult females resulted in increased cell death, impaired mammary development, and a lactation-deficient phenotype (13). The excision of the Tsg101 gene in primary cultures of mouse embryonic fibroblasts (MEFs) or mammary epithelial cells (MECs) revealed that Tsg101-deficient cells arrested at the G1/S transitional phase of the cell cycle before they underwent cell death (12, 13). In contrast to previous reports (16, 19), neither haplo-insufficiency of Tsg101 nor the deletion of both Tsg101 alleles in the conditional knock-out models (in vitro and in vivo) resulted in neoplastic transformation, suggesting that a null mutation of Tsg101 is not an initiating event for tumorigenesis (11–13).
Cell cycle arrest at the G1/S checkpoint and cell death were two major phenomena that we observed in the Tsg101 conditional knock-out model (12, 13). The analysis of crucial regulators of the cell cycle revealed that Tsg101 deficiency resulted in growth arrest through the inactivation of cyclin-dependent kinase 2 (Cdk2). Consequently, DNA replication was not initiated in Tsg101-deficient cells before they died (12). A crucial regulatory mechanism, which is commonly associated with G1 arrest and cell death, is the p19Arf-Mdm2-p53 tumor-surveillance or stress-response pathway (20). The goal of this study was to further discriminate the p53-dependent and p53-independent mechanisms that lead to either cell cycle arrest or cell death as a consequence of Tsg101 deficiency. We now show that the G1 arrest in Tsg101-deficient cells is dependent upon the presence of functional p53 and its downstream mediator p21Cip1. Our findings suggest that, in contrast to keratinocytes (18), Tsg101 is not required for p21Cip1 protein stability and function in proliferating fibroblasts. The absence of either p53 (and, therefore, p21) or p21Cip1 alone is, however, unable to sustain cell survival. In addition to these findings in vitro, a null mutation of the p53 gene does not restore normal mammogenesis and lactation in Tsg101 mammary-specific knockout mice (WAP-Cre Tsg101fl/fl p53−/ −). To address the proposed role of Tsg101 as a crucial regulator for Mdm2 function (17), we have generated a conditional double-knock-out of Tsg101 and Cdkn2a. The deletion of Tsg101 in immortalized fibroblasts that lack expression of p19Arf does not alter Mdm2 function and does not restore the p21-mediated G1 arrest. In summary, our data do not support a biologically relevant function of Tsg101 as a stabilizer for Mdm2. Therefore, we propose that the p21-mediated induction of the G1 arrest might be an indirect consequence of Tsg101 deficiency due to cellular dysfunction and stress. Finally, we show that the deletion of Tsg101 from tumorigenic cells that express mutant p53 and that lack p21Cip1 expression results in cell death. This observation suggests that additional, transforming mutations during tumorigenesis do not affect the important role of Tsg101 for cell survival.
EXPERIMENTAL PROCEDURES
Mouse Models, Genotyping Protocols, and Whole Mount Analysis of Mammary Glands
The PCR protocol for genotyping the whey acidic protein (WAP)-Cre mice (TgN(Wap-cre)11738Mam) as well as the generation and phenotypic characterization of Tsg101 conditional knock-out mice (Tsg101tm1Kuw) have been described earlier (13, 21). Mutant mice with a targeted deletion of the p53 gene (Trp53tm1Brd) (22) were purchased from Taconic Farms, Inc. Cdkn1a (23) and Cdkn2a (24) knock-out mice (Cdkn1atm1Tyj and Cdkn2atm1Rdp, respectively) were obtained from Jackson Laboratories and the repository of the Mouse Model for Human Cancer Consortium (MMHCC). Athymic nude mice (NCr strain, NCI) were used for transplantation studies. The preparation and staining of mammary gland whole mounts were described previously (13, 25). All animals used in the studies were treated humanely and in accordance with federal guidelines and institutional policies.
Primary Cell Cultures and Retroviral Expression Vectors
MEFs from 13.5- or 14.5-day-old Tsg101fl/fl, Tsg101fl/fl p53−/−, Tsg101fl/fl Cdkn1a−/−, or Tsg101fl/fl Cdkn2a−/− embryos and their littermate controls were explanted and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mm glutamine, 0.1 mm nonessential amino acids, 10 μg/ml gentamycin, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cells at passages 2–4 were plated at a density of 4 × 105 cells/10-cm culture dish and infected with the pBabe and pBabe-Cre constructs. The generation of these retroviral vectors was published previously (12, 13). Forty-eight hours after infection cells were selected in complete medium containing 7 μg/ml puromycin (Sigma) or 200 μg/ml hygromycin B (Invitrogen).
Cell Cycle Analysis and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
Approximately 106 MEFs were harvested 4 days after puromycin selection to determine cell cycle progression using a flow cytometric analysis. Cells were washed in 1× phosphate-buffered saline and fixed in ice-cold 70% ethanol for 30 min. After an additional washing step, cells were stained overnight with propidium iodide as described previously (26). The stained MEFs were analyzed in a FACScalibur (BD Biosciences) flow cytometer. The software packages CELLquest (BD Biosciences) and Modfit LT (Verity) were used for data acquisition. The cell cycle analysis was repeated three or four times for each experimental setting, and a t test was performed to validate statistically significant differences between the various double knock-out cell lines and their controls. An MTT growth assay was performed as described earlier (27) to determine the growth properties of Tsg101/p53 double knock-out cells. The MTT was obtained from Sigma. 2 × 104 cells of each genotype were seeded in triplicates in a 96-well microtiter plate. Absorbance was measured at 570 nm with an Elx 808 (Bio-Tek Instruments) enzyme-linked immunosorbent assay reader.
Western Blot Analysis and Cdk2 Kinase Assay
MEFs were pelleted and lysed on wet ice for 30 min in 1× phosphate-buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride, 0.4 units/ml aprotinin, 1 mm NaF, 0.1 mm sodium orthovanadate. Protein was quantified using a Bradford assay (Pierce) according to the manufacture’s protocol. Approximately 50 –100 μg of protein per lane was resolved on a 4 –20% SDS-PAGE gradient gel and blotted onto polyvinylidene fluoride membranes (Invitrogen). The membranes were blocked for 1 h in 1× Tris-buffered saline, 0.1% Tween 20, and 5% dry milk. Subsequently, membranes were incubated with primary antibodies in blocking buffer at 4 °C overnight, washed 3 times for 15 min in washing buffer (1× Tris-buffered saline, 0.1% Tween 20), and incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies in blocking buffer. Membranes were washed again 3 times in washing buffer and once for 15 min in 1× Tris-buffered saline without Tween 20. Protein bands were detected using the ECL chemiluminescence kit for Western blot analysis (Amersham Biosciences) according to the manufacture’s protocol. Membranes were stripped using 0.2 m NaOH for consecutive detection of various proteins. The following antibodies were used in this study: α-Tsg101 (C-2), α-cyclin B1 (M-20), α-cyclin A2 (C-19), α-p16Ink4a (M-156), and α-ActB (I-19) from Santa Cruz Biotechnology as well as α-p21Cip1 (SX118) from Pharmingen and α-p19Arf (Ab-1) from Oncogene at a 1:1000 dilution. Horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology and used at a 1:1000 dilution. A Cdk2 kinase assay was performed using the α-Cdk2 (M-2) antibody from Santa Cruz Biotechnology and Histone H1 (1 μg/μl) from Sigma.
RESULTS
A Null Mutation of p53 Does Not Rescue the Deleterious Phenotype Caused by Tsg101 Deficiency in Vitro and in Vivo
The Cre-mediated excision of both Tsg101 floxed alleles from proliferating primary fibroblasts (MEFs) and MECs resulted in cell cycle arrest and cell death (12, 13). The activation of the p19Arf-Mdm2-p53 stress-response pathway is frequently associated with both phenomena. To further discriminate the p53-dependent and p53-independent mechanisms that lead to either cell cycle arrest or cell death as a consequence of Tsg101 deficiency, we derived MEFs from Tsg101 conditional knockout mice (Tsg101fl/fl) that carry a targeted null mutation of the p53 gene (22). Cells treated with a control virus (pBabe) or a retrovirus expressing Cre recombinase (pBabe-Cre) were grown for 7 days after infection to quantitatively examine the growth of Tsg101 knock-out cells in the presence or absence of p53. The multiplication of Tsg101-deficient MEFs and their controls were determined using an MTT assay between days four through seven post-infection when optimal puromycin selection, expression of Cre, and excision of Tsg101 had been achieved (Fig. 1A). The Cre-mediated deletion of Tsg101 resulted in a severe growth inhibition and reduction in the number of viable cells. More importantly, we did not observe a significant modification of growth properties or prolonged cell survival in the presence or absence of one or both copies of the p53 gene. Our data suggested that a p53 null mutation was neither able to rescue growth inhibition nor significantly modify the rate of cell death of Tsg101-deficient primary fibroblasts. We have shown in a control experiment that the expression of Cre recombinase can be excluded as a cause or possible modifier of this phenotype. Tsg101 wild type cells (Tsg101+/+) infected with the retroviral pBabe-Cre vector did not exhibit growth retardation in an MTT assay. In addition, we were able to rescue the deleterious phenotype of Tsg101−/− cells through the expression of exogenous, hemagglutinin-tagged Tsg101 from a retroviral vector (12).
Fig. 1. p53 deficiency does not rescue cell death in the Tsg101 conditional knock-out.
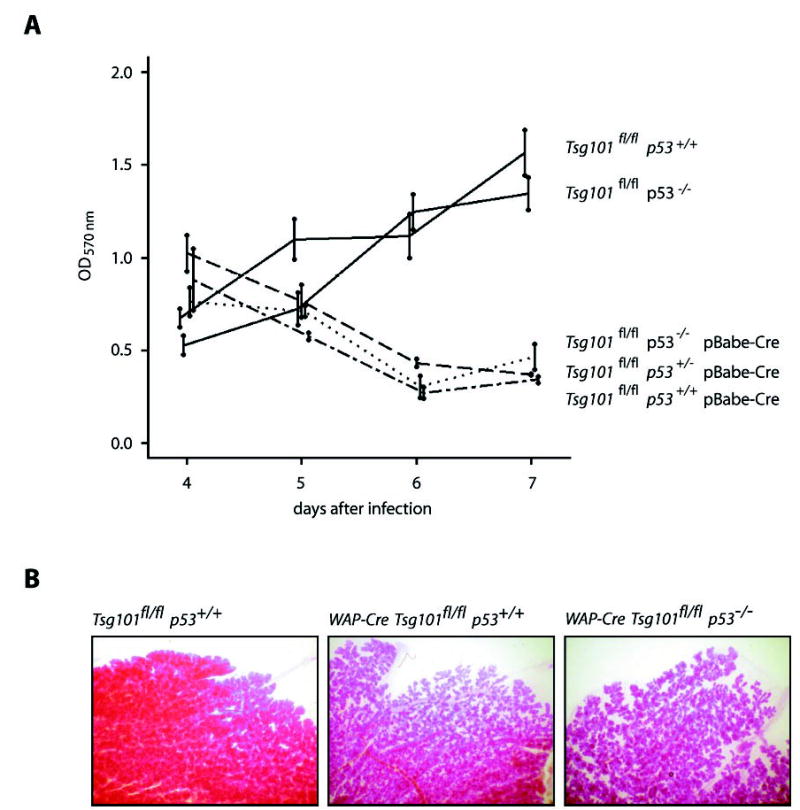
A, in vitro analysis. An MTT color assay was performed to determine the growth rates of pBabe and pBabe-Cre-infected Tsg101fl/fl MEFs, which lack one or two alleles of the p53 gene (p53+/− and p53−/ −) and their wild type controls (p53+/+). The A570 nm values that correspond to total cell numbers decrease in the Tsg101 knock-out (Tsg101fl/fl pBabe-Cre) between 4 and 7 days after retroviral Cre infection regardless of the p53 mutation status. In contrast, the number of cells in the pBabe-infected controls increase steadily. Error bars correspond to S.D. B, in vivo analysis. Whole mounts of carmine alum-stained mammary glands from tissue-specific Tsg101 knock-out mice (WAP-Cre Tsg101fl/fl p53+/+), females that are conditionally deficient in both, Tsg101 and p53 (WAP-Cre Tsg101fl/fl p53−/ −), and their controls (Tsg101fl/fl p53+/+). Mammary glands were taken, fixed, and stained for several hours post-partum (magnification 40×). Note that alveolargenesis is severely impaired in the Tsg101 conditional knock-outs regardless of whether p53 is expressed or not.
To verify these initial findings in a different cell type and in vivo, we bred two mutant p53 alleles into mammary-specific Tsg101 conditional knock-out mice (WAP-Cre Tsg101fl/fl p53−/−). The expression of Cre under the WAP promoter is largely confined to differentiating mammary epithelial cells in late-pregnant and lactating females (21, 25). In contrast to littermate controls (Tsg101fl/fl p53+/+), the WAP-Cre-mediated deletion of Tsg101 in a p53 wild type background (WAP-Cre Tsg101fl/fl p53+/+) resulted in impaired mammogenesis at the onset of lactation (Fig. 1B). The introduction of two mutant copies of the p53 gene into the Tsg101 conditional knock-out (WAP-Cre Tsg101fl/fl p53−/−) was unable to rescue impaired mammogenesis caused by the WAP-Cre-mediated excision of the two floxed Tsg101 alleles. Therefore, our observations in mutant female mice were consistent with the cell culture studies on Tsg101/p53 double mutant MEFs. Taken together, the outcome of both studies (in vitro and in vivo) suggests that the inability of a p53 null mutation to rescue a Tsg101-deficient phenotype is neither a cell culture phenomenon nor is it caused by a possible difference in p53 function between different cell types of mesodermal and ectodermal origin (i.e. fibroblasts and mammary epithelial cells).
Deletion of p53 in Tsg101-deficient Cells Restores the Activity of Cdk2 and Progression into the S Phase
Despite p53-independent mechanisms that trigger the dominant phenotype (i.e. lethality) of Tsg101−/− cells, we postulated that deficiency of p53 might modify or even eliminate the cell cycle arrest in Tsg101-deficient cells before they die. To address this hypothesis, we analyzed the cell cycle progression of Tsg101 null cells lacking one or two copies of the p53 gene. The Cre-mediated deletion of Tsg101 in MEFs with a heterozygous null mutation of p53 (Tsg101fl/fl p53+/− pBabe-Cre) leads to a significant reduction in the number of cells in S phase (Fig. 2A) (p < 0.01; t test), a decrease of the cyclin A2 protein level (Fig. 2B, third lane), and the inactivation of the Cdk2 (Fig. 2C). Consequently, the inhibition of only one p53 allele had no effect on impaired G1/S progression of Tsg101−/− cells, and the consequences of Tsg101 deficiency were identical to cells with two functional copies of p53 (12). In contrast, the deletion of both p53 alleles in Tsg101 conditional knock-out MEFs (Tsg101fl/fl p53−/− pBabe-Cre) re-established a normal G1/S progression (Fig. 2A) (p < 0.02; t test), increased the expression of cyclins A2 (Fig. 2B), and restored the activation of Cdk2 (Fig. 2C). In summary, these observations confirm our previously stated hypothesis that p53 deficiency is able to lift the G1 cell cycle arrest and to revert part of the complex phenotype of Tsg101 knock-out cells despite its inability to sustain cell survival.
Fig. 2. A p53 null mutation restores G1/S progression and the activity of Cdk2 in Tsg101-deficient cells.
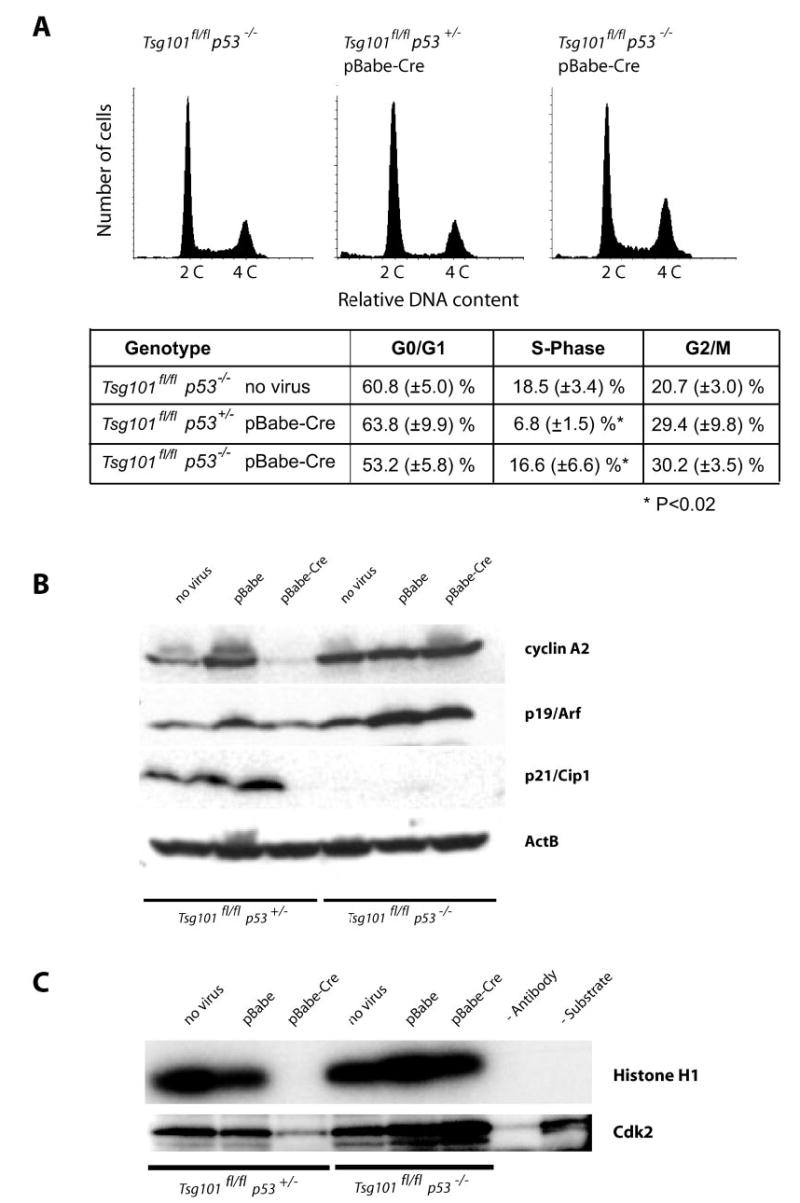
A, flow cytometric analysis of the DNA content of viable Tsg101-deficient MEFs (Tsg101fl/fl, pBabe-Cre) that lack one or two alleles of the p53 gene (p53+/− and p53−/ −) and their uninfected controls (Tsg101fl/fl p53−/ − ). The sub-G1 population of apoptotic cells was gated out in this assay. Note that the relative number of cells in S phase is reduced only in p53 heterozygous knock-out cells lacking Tsg101 but not in Tsg101-deficient MEFs carrying two mutant p53 alleles. B, Western blot analysis of cyclin A (S-phase cyclin) and regulators of G1/S progression (p19Arf and p21Cip) in Tsg101-deficient MEFs that lack one or two copies of p53 and their uninfected controls. Note that cyclin A2 was markedly down-regulated in Tsg101-deficient MEFs that carry at least one functional allele of p53. Immortal p53/p21Cip1-deficient MEFs express high levels of cyclin A2 and p19Arf regardless of the Tsg101 mutation status. C, Cdk2 activity assay using histone H1 as a substrate for phosphorylation. Only Tsg101 knock-out MEFs that carry at least one functional p53 allele exhibit a reduced activity of Cdk2. The complete deletion of p53 and, subsequently, the absence of the Cdk2 inhibitor p21Cip1 restore a normal activity of Cdk2 in Tsg101-deficient cells.
P21Cip1 Is a Mediator of the G1 Cell Cycle Arrest in Tsg101 Conditional Knock-out Cells
The activity of Cdk2 in complex with cyclin E is negatively regulated by the cell cycle inhibitor p21Cip1 whose expression level is dependent upon the transcriptional activation of p53 (28–30). Therefore, we hypothesized that p21Cip1 might be a component of the p53-dependent cell cycle arrest in response to Tsg101 deficiency. To address this issue, we bred Cdkn1a−/− mice (23) into the Tsg101-floxed background (Tsg101fl/fl p21−/−) to generate MEFs that lack expression of p21 and Tsg101 after infection with pBabe-Cre and selection with puromycin (Fig. 3A). The infection of p21Cip1-deficient cells with the control vector pBabe and selection with puromycin had little or no consequences on the viability and multiplication of primary cells. The Cre-mediated excision of only one floxed allele of Tsg101 (Tsg101fl/+ p21−/− pBabe-Cre) also did not affect the survival and G1/S progression of p21Cip1-deficient MEFs as determined by flow cytometry (Fig. 3B). The deletion of two floxed alleles of Tsg101 (Tsg101fl/fl p21−/− pBabe-Cre) progressively led to the death of Tsg101-deficient MEFs within 7 days after infection with the retroviral Cre vector (data not shown). Like the deletion or functional inhibition of p53, a null mutation of the Cdkn1a gene was, however, able to restore the G1/S progression in cells lacking Tsg101 (Fig. 3B). The average number of cells in S phase was not significantly different between the double knock-out cells and their controls (p > 0.05). The relative increase in a subset of cells at the G2/M phase is probably caused by the puromycin selection since we observed the same phenomenon when these cells where infected with the pBabe vector control. Collectively our findings confirmed the working hypothesis, which predicted that p21Cip1 is a mediator of the G1 cell cycle arrest in Tsg101-deficient cells.
Fig. 3. p21Cip1 is a mediator of the cell cycle block at the G1 checkpoint in Tsg101 conditional knock-out cells.
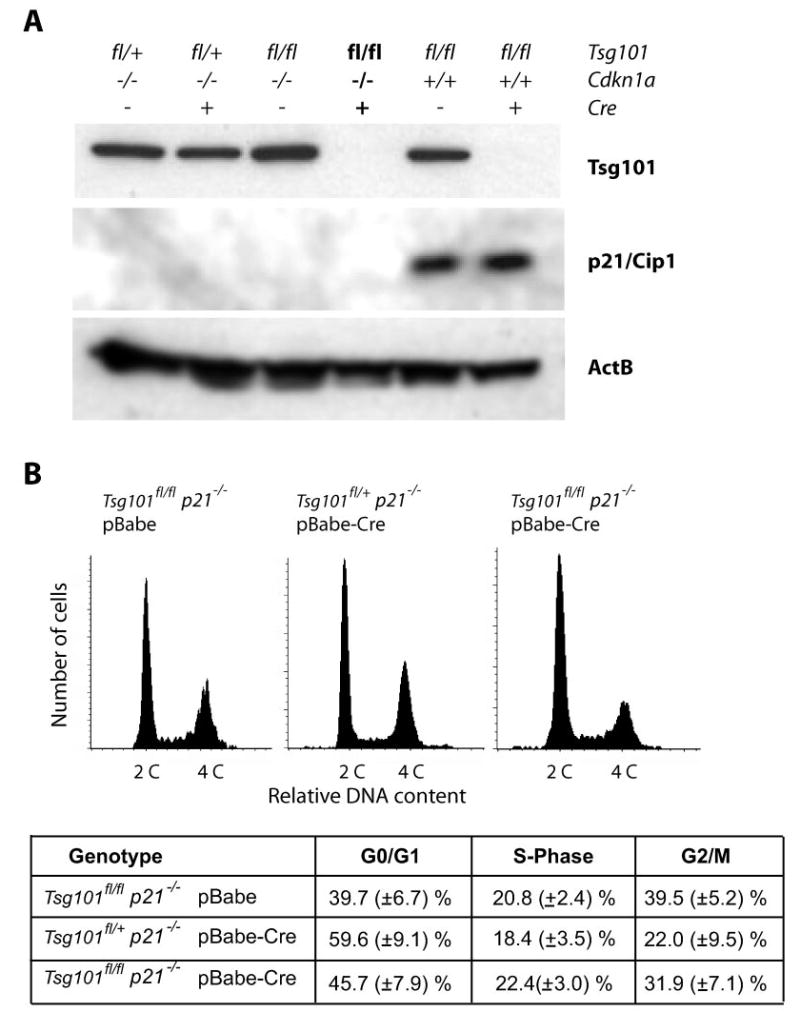
A, Western blot analysis of Tsg101 and p21Cip1 to monitor effective Cre-mediated excision and down-regulation of Tsg101 in MEFs lacking one or two copies of the Cdkn1a gene and their controls. B, flow cytometric analysis of the DNA content in cells doubly deficient in Tsg101 and p21Cip1 (Tsg101fl/fl p21−/− pBabe-Cre) and their controls. The sub-G1 population of apoptotic cells was gated out in this assay. Note that the relative number of cells in S phase is normal in Tsg101-deficient MEFs carrying two mutant Cdkn1a alleles compared with their controls (p > 0.05).
The Deletion of Tsg101 in Immortalized Fibroblasts That Lack p19Arf Does Not Alter the Ability of Mdm2 to Sequester p53
Several reports propose a potential role for Tsg101 as an important negative regulator for the ubiquitin-mediated turnover of Mdm2 and p21Cip1 in various cells lines and in differentiating primary keratinocyctes (11, 17, 18). In addition, Oh et al. (28) suggest that Tsg101 is critical for the p21-mediated inhibition of cyclin-Cdk complexes in proliferating keratinocyctes (18). The cell cycle analysis in double knock-out MEFs lacking Tsg101 and Cdkn1a revealed that p21Cip1 is important for the G1 arrest (see the previous paragraph). This observation suggests that a knock-out of Tsg101 does not impair the stability, ubiquitin-mediated proteolysis, or function of p21Cip1 in proliferating fibroblasts. To address the proposed function of Tsg101 as a stabilizer for Mdm2, we designed a new set of experiments illustrated in Fig. 4. Thus far, we determined the role of p53 and p21Cip1 as regulators for the G1/S progression in Tsg101-deficient cells. Both proteins act downstream of Mdm2 (20). In this new experimental design we planned to immortalize primary MEFs upstream of Mdm2 through deletion of the Cdkn2a gene (Fig. 4A, left). The Cdkn2a locus encodes p19Arf, which is known to negatively regulate Mdm2 (20). Therefore, p19Arf-deficient immortalized cells also lack expression of p53 and p21Cip1. The level of unrestrained Mdm2 protein for the sequestration of p53 is crucial in immortal p19Arf-null cells.2 This causal relationship is probably best illustrated by the fact that the deletion of p19Arf alone has no influence on the survival of Mdm2 mutant mice, whereas p53/Mdm2 double knockouts (31, 32) or p19Arf/Mdm2/p53 triple mutant mice are viable (33). According to this mechanism, the destabilization and down-regulation of Mdm2 as a consequence of Tsg101 deficiency (Fig. 4A, right) should result in an accumulation of p53 and p21Cip1. In turn, the up-regulation of p53 and p21Cip1 should inhibit cell cycle progression in Cdkn2a mutant cells. In conclusion, if Tsg101 is essential for the stability and function of Mdm2 as previously suggested (17), then its deletion in immortalized MEFs lacking p19Arf should impair G1/S progression in a very similar fashion as in Tsg101-deficient nonimmortalized cells with normal p53 and p21Cip1 levels.
Fig. 4. The p19Arf-p53 tumor surveillance and stress response pathway and its proposed interaction with Tsg101.
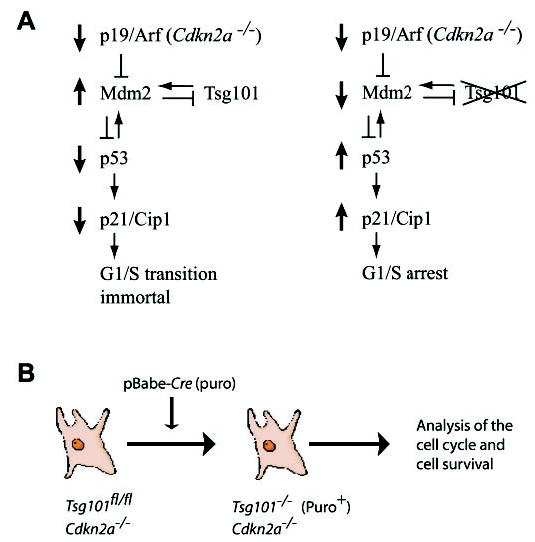
A, Mdm2 is negatively regulated by p19Arf. As a ubiquitin ligase, Mdm2 regulates the ubiquitin-mediated degradation of the tumor suppressor protein p53. Therefore, p19Arf-deficient cells are immortal and lack expression of p53 and p21Cip1. In normal cells, p21Cip1 is transcriptionally regulated by p53. It has been suggested recently that Tsg101 is a positive regulator for Mdm2 protein levels and function. Therefore, the deletion of the Tsg101 gene (right panel) should destabilize Mdm2 and induce a p53/p21Cip1-mediated G1 arrest in cells lacking p19Arf. B, experimental design to study the proposed interaction of Tsg101 with Mdm2/p53 in immortalized cells lacking p19Arf. The Cre-mediated deletion of Tsg101 in Cdkn2a−/− immortalized cells should have a biologically relevant effect on Mdm2 and p53 protein levels and restore the cell cycle arrest.
To generate a conditional double-knock-out of Tsg101 and Cdkn2a, we decided to cross Tsg101-floxed animals with Cdkn2a knock-out mice (24) that lack both tumor susceptibility proteins (p19Arf and p16Ink4a) encoded by this locus. Our decision was based on the fact that Tsg101 and its proposed biological functions were first described in murine 3T3 fibroblasts and their derived tumorigenic SL6 cells (16). These cell lines lack p16Ink4a in addition to p19Arf as determined in a preliminary study (Fig. 5A). To generate primary Tsg101/p19Arf double knock-out cells, we infected MEFs carrying two floxed alleles of Tsg101 in addition to two null alleles of Cdkn2a (Tsg101fl/fl Cdkn2a−/−) with the pBabe-Cre virus (Fig. 5B). Tsg101 knock-out cells in a Cdkn2a heterozygous background (Tsg101fl/fl Cdkn2a+/−) or Tsg101 heterozygous mutants carrying two Cdkn2a null alleles (Tsg101fl/+ Cdkn2a−/−) were used as controls. The infection of these control cell lines with the pBabe retroviral control vector and puromycin selection had also no influence on cell survival (data not shown). As expected, the expression of Cre also had no effect on cell proliferation and cell survival of MEFs with a Tsg101-floxed heterozygous mutation whether cells express p19Arf or not. However, the ablation of Tsg101 in p19Arf-deficient cells using the pBabe-Cre construct (Tsg101fl/fl Cdkn2a−/− pBabe-Cre) resulted in cell death shortly after excision of Tsg101, and the phenotype was comparable to those observed in Tsg101/p53 and Tsg101/p21 double mutants (data not shown). This observation suggested that none of the genes of the p19Arf-Mdm2-p53-p21Cip1 circuit alone or in combination with p16Ink4a seem to be required for the initiation of cell death of Tsg101-deficient MEFs.
Fig. 5. Cell cycle regulation in immortal cells that lack Cdkn2a and Tsg101.
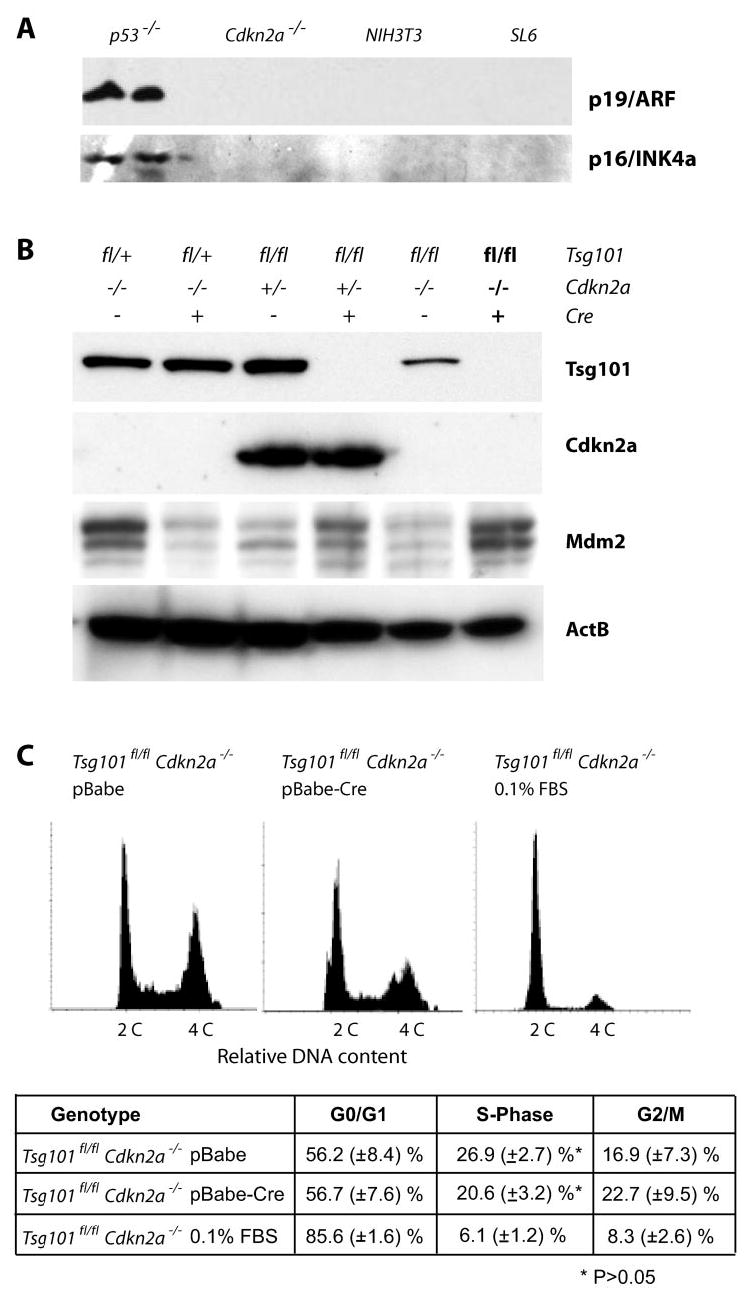
A, the tumorigenic, Tsg101-anti-sense-expressing SL6 cell line and its parental mouse 3T3 fibroblast cell line lack expression of p19Arf and p16Ink4a. Cdkn2a-deficient cells were used as negative controls, and immortal p53−/− MEFs served as positive controls for p19Arf and p16Ink4a protein expression. B, Western blot analysis of Tsg101 and Cdkn2a to monitor effective Cre-mediated down-regulation of Tsg101 in MEFs lacking one or two copies of the Cdkn2a gene and their controls. The steady-state levels of Mdm2 did not decrease and were slightly elevated in cells lacking Tsg101. C, flow cytometric analysis of the DNA content in cells doubly deficient in Tsg101 and p19Arf (Tsg101fl/fl Cdkn2a−/− pBabe-Cre) and their controls. The sub-G1 population of apoptotic cells was gated out in this assay. P19Arf-deficient cells deprived of essential growth factors (Tsg101fl/fl Cdkn2a−/− 0.1% fetal bovine serum (FBS)) served as an additional positive control to monitor the accumulation of cells at G1 and the relative decrease in the number of cells at the S-phase of the cell cycle. Note that the deletion of Tsg101 in a p19Arf-deficient background does not cause a cell cycle arrest, whereas growth factor withdrawal from these cells results in a sharp reduction of cells in S phase.
Our previous studies showed that the conditional deletion of the Tsg101 gene did not negatively affect the steady-state level of Mdm2 in primary fibroblasts (12). Similarly, the steady-state level of Mdm2 was not reduced but was slightly elevated in Cdkn2a+/− or Cdkn2a−/− MEFs lacking Tsg101 expression (Fig. 5B). We performed a flow cytometric analysis to study whether the loss of Tsg101 function was able to restore the G1 arrest in p19Arf-deficient cells before they died (Fig. 5C). In addition, we used p19Arf-deficient cells (Tsg101fl/fl Cdkn2a−/−) deprived of essential growth factors (0.1% fetal bovine serum) as a positive control to monitor the accumulation of cells at G1 and the relative decrease in the number of cells at the S phase of the cell cycle. Cells deficient in Tsg101 and p19Arf did not arrest at G1 and had approximately the same relative number of cells in S phase as MEFs lacking Tsg101 in addition to p53 and/or p21Cip1 (see Fig. 2A and 3B; p > 0.05).
To verify these findings in a different experimental setting, we immortalized Tsg101fl/fl MEFs and their wild type controls by inhibiting the expression of p19Arf through overexpression of the T-box protein 2 (data not shown). T-box protein 2 is a known transcriptional repressor for the mouse and human CDKN2A(ARF) promoters (34). MEFs expressing T-box protein 2 exhibited reduced levels of p19Arf and p21Cip1 at passages seven and eight. After Cre-mediated recombination of the floxed Tsg101 locus, these immortalized cells died within 7 days post-infection. The steady-state levels of Mdm2 did not decrease, and p21Cip1 protein expression levels remained low and unchanged in the Tsg101 knock-out cells lacking functional p19Arf (data not shown). In summary, these results are consistent with our findings in Tsg101 knock-out cells with a targeted deletion of the entire Cdkn2a locus. Therefore, Tsg101 deficiency seemed to not alter Mdm2 function in a biologically relevant manner that would influence the Mdm2/p53 negative feedback loop.
Transforming Mutations during Neoplastic Transformation Do Not Affect the Important Role of Tsg101 for Cell Survival
Li and Cohen (16) report that a functional knock-out of Tsg101 using a conventional antisense approach and inducible overexpression of Tsg101 resulted in reversible neoplastic transformation of mouse 3T3 fibroblasts. Our published observations using a site-directed, targeted knock-out in animal models and derived cell lines suggested that a loss-of-function of Tsg101 is insufficient to trigger neoplastic transformation in vitro and in vivo (12, 13). Because Tsg101 seems not to be a primary tumor suppressor, we hypothesized that this gene might function as a modifier for neoplastic transformation (12). Null or inactivating mutations in p19Arf, p53, or p21Cip1 are able to partially rescue the complex phenotype caused by Tsg101 deficiency (i.e. the G1/S progression), and it is, therefore, logical to test whether additional mutations during neoplastic transformation have an impact on the crucial role of Tsg101 as a survival factor in tumorigenic cells. Fig. 6A illustrates our experimental design to address this issue. We previously described the generation of immortalized Tsg101fl/fl MEFs using a standard 3T3 protocol (12). These cells carry an E255D mutation in the DNA binding domain of p53, and consequently, the p21Cip1 protein is not expressed. The deletion of Tsg101 in this immortalized cell line resulted in instant cell death (12). The G1/S transition is, however, restored due to the lack of p21Cip1. Hence, the phenotype of these cells relating to cell cycle regulation and cell death is equivalent to double-mutant MEFs with targeted deletions of p53 and Tsg101.3 Immortal Tsg101fl/fl 3T3 cells were passaged numerous times, infected with a pBabe-hygro retrovirus, and selected for hygromycin resistance. Next, 8 × 105 cells were injected subcutaneously into athymic nude mice (NCr strain) to select mutants that were able to grow in vivo and to form solid tumors. Tumorigenesis was observed in all animals after a medium latency of 3–5 weeks (Fig. 6B). Histopathologically, these lesions were well vascularized, and neoplastic cells invaded into adjacent normal tissues such as the fat pad of the thoracic mammary gland (Fig. 6C). Tumor cells were explanted and grown in culture in hygromycin-containing media to remove all nontumorigenic cell types such as endothelial cells, epithelial cells, and tumor-associated fibroblasts of the host. Next, neoplastic cells were infected with pBabe-Cre to excise both floxed copies of Tsg101. Infection of these cells with the control vector pBabe and selection with puromycin had little effect on their viability, whereas the deletion of Tsg101 resulted in growth arrest and cell death (Fig. 6D and E). These data suggested that additional transforming mutations during neoplastic transformation did not rescue the lethal phenotype caused by Tsg101 deficiency, and therefore, Tsg101 might be important for the survival of both normal and neoplastic cells.
Fig. 6. Tsg101 deficiency causes cell death of tumorigenic cells.
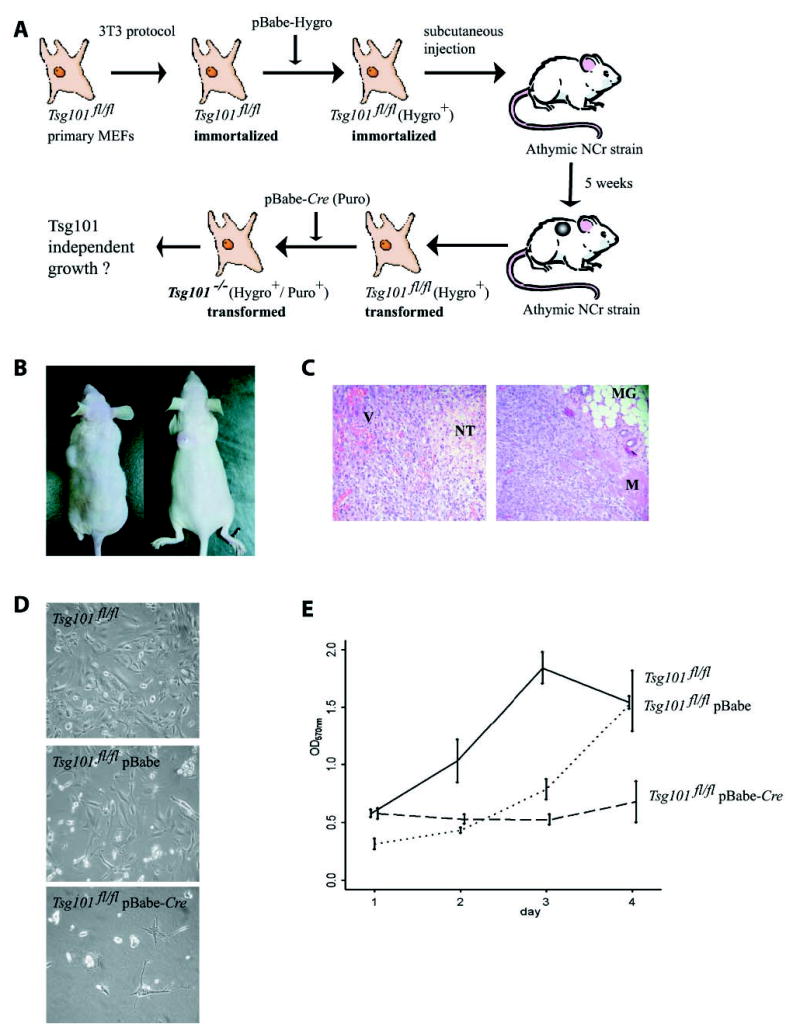
A, experimental design to address whether Tsg101 is dispensable or essential for the survival of neoplastic cells. B, the subcutaneous injection of immortal Tsg101fl/fl 3T3 fibroblasts expressing mutant p53 into whn−/− nude mice (NCr strain) form solid tumors. C, hematoxylin and eosin staining of histological sections from solid tumors (magnification 100×). Note that these lesions are highly vascularized, and tumor cells invade into adjacent normal tissues. V, vasculature; NT, necrotic tumor tissue; MG, mammary gland; M, muscle. D, explanted tumor cells carrying two floxed alleles of Tsg101 (Tsg101fl/fl) infected with pBabe (control) or the pBabe-Cre retroviral vector to excise both copies of Tsg101 (magnification 200×). Note that additional, transforming mutations during neoplastic transformation are incapable of rescuing lethality caused by Tsg101 deficiency. E, MTT color assay to quantify the lack of cell growth of tumor cells lacking Tsg101 compared with their controls. Error bars correspond to S.D.
DISCUSSION
Tsg101 Deficiency and Cellular Stress Response
We previously reported that Tsg101 is indispensable for the survival of cells in vitro and in vivo (12, 13). The collective data presented in this new report demonstrated that targeted mutations to abolish the function of p19Arf, p16Ink4a, p53, and p21Cip1 were insufficient to maintain the survival of Tsg101 knock-out cells. These observations suggest that the p19Arf-Mdm2-p53 tumor surveillance or stress response pathway does not control cellular mechanisms that lead to the death of Tsg101-deficient cells. These results are in full agreement with our previous studies, in which we have demonstrated that the functional inhibition of p53, through expression of E6 or a spontaneous mutation within the DNA binding domain of p53, had no effect on the viability of Tsg101 knock-out cells (12). By analyzing conventional knock-out mice that die very early in utero, Ruland et al. (11) established that Tsg101 is required for cell proliferation and that p53 is a crucial player for the replication defect of Tsg101-deficient cells. Using our conditional knock-out model, we established that the deletion of Tsg101 resulted in a sustained cell cycle arrest at the G1 checkpoint before these cells died (12). Based on the findings by Ruland et al. (11) and due to the fact that Tsg101 conditional knock-out cells lacked active Cdk2, we hypothesized that the p53 and its downstream mediator p21Cip1 may at least in part contribute to the complex phenotype. We proposed that cell cycle arrest and cell death were controlled by separate, p53-dependent and p53-independent, pathways (13). In this report we show that, in contrast to mechanisms leading to cell death, the G1 arrest in Tsg101-deficient cells is dependent upon expression of functional p53. We also determined that p21Cip1 is an integral part of this phenotype. Recent reports suggested a potential role for Tsg101 as a dominant-negative regulator for Mdm2 and p21Cip1 protein turnover (17, 18). Evidently, Tsg101 cannot serve simultaneously as a stabilizer for both regulators of the cell cycle. According to the model by Li et al. (17) and Ruland et al. (11), functional levels of p21Cip1 were required to mediate the cell cycle arrest. Tsg101 deficiency leads to destabilization of Mdm2, accumulation of active p53, and subsequently, the transcriptional activation of p21Cip1. If Tsg101 is required for p21 stability or the recruitment of p21 to the cyclin-Cdk2 complex as proposed by Oh et al. (18), then the combined effects of a Tsg101 knock-out on Mdm2 and p21Cip1 should nullify each other.
The overexpression of Tsg101 or its conditional deletion results in a G1 arrest and cell death, suggesting that the level of Tsg101 protein in a given cell is tightly regulated and that a high or low amount of Tsg101 outside a narrow range cannot be tolerated (9, 10, 12, 13, 35, 36). Interestingly, p21Cip1 seems to be a key player for the G1 arrests not only in the overexpression model (18) but also the conditional knock-out. As demonstrated in this report, a knock-out of the Cdkn1a gene (p21Cip1) eliminated the G1 arrest and restored the G1/S progression of Tsg101-deficient cells. Based on the fact that p21Cip1 levels did not change in the Tsg101 conditional knock-out (Ref. 12 and Figs. 2B, second and third lanes, and 3A, fifth and sixth lanes), we presume that Tsg101 deficiency leads directly to cellular conditions where p21 ability to bind to the Cdk2 is elevated. The pathways that link the function of Tsg101 and p21Cip1 need to be identified. There is increasing evidence in the recent literature that Tsg101 is essential for endosomal trafficking and other cellular processes in addition to the regulation of ubiquitination (5–8). It is, therefore, very likely that Tsg101 deficiency or its overexpression will cause a severe negative impact on a number of biological processes. Those might subsequently trigger the activation of cellular stress responses such as the recruitment of p21Cip1 to the Cdk2 complex to arrest cells in the G1 phase. Ruland et al. (11) suggest that p53 is the upstream regulator of p21Cip1, which mediates the cell cycle arrest in Tsg101-deficient cells. Based on data by Li et al. (17), the authors proposed a direct interaction of Tsg101 with members of the p53 stress response pathway, in particular Mdm2. In the conditional knock-out model we were, however, unable to detect higher levels of p53 or changes in the p53 transcriptional activation of p21Cip1 (12). Moreover, we questioned the suggested role of Tsg101 as a positive regulator for Mdm2 function based on the evidence that Tsg101/p53 double mutant mice still die early in utero (11), whereas p53 deficiency results in a complete rescue of embryonic lethality in Mdm2 knock-out mice (31, 32). To rigorously examine the proposed function of Tsg101 as a stabilizer for Mdm2, we generated cells lacking Tsg101 and p19Arf. The functional inhibition of p19Arf leads to increased Mdm2-mediated sequestration of p53 and immortalization (20). We utilized the complete deletion of the Cdkn2a locus and the T-box protein 2-mediated transcriptional repression of p19Arf as two alternative approaches to immortalize MEFs carrying two conditional Tsg101 knock-out alleles. In both model systems the deletion of Tsg101 had no effect on Mdm2 steady-state levels and function. In particular, the p53-mediated transcriptional activation of p21Cip1 and induction of a G1 arrest was not restored. In addition, we were unable to co-precipitate Mdm2 with endogenous Tsg101 or transgenic hemagglutinin-tagged Tsg101 expressed at near physiological levels,4 suggesting that under normal conditions these proteins show weak or no interaction. In summary, results obtained from three double knock-out models (Tsg101/p53, Tsg101/p21, and Tsg101/p19Arf) suggest that a direct interaction of Tsg101 with members of the Mdm2-p53 circuit is unlikely. We propose that the activation of a p21Cip1-mediated G1 arrest might be an indirect effect of Tsg101 deficiency on other cellular processes that subsequently trigger stress response pathways.
Normal Tsg101 Function Is Crucial for the Survival of Primary, Immortalized, and Neoplastic Cells
The role of the TSG101 gene as a tumor suppressor or oncogene in human malignancies is still controversial. After cloning the mouse Tsg101 locus and revising the human TSG101 gene structure, we found that many of the previously described aberrant splice variants in human malignancies represent alternative splice forms that originate solely from exon skipping (35, 37). Although the transforming capability of Tsg101 as an oncogene has not been examined in an animal model to date, we and others could show that neither haplo-insufficiency of Tsg101 (11, 13) nor the deletion of both Tsg101 alleles in selected cell types of conditional knock-out mice resulted in tumorigenesis (12, 13). In contrast to a previous report (16), we demonstrated that a null mutation of Tsg101 is not an initiating event for neoplastic transformation of primary cells (12, 13). Therefore, we hypothesized that this gene might function as a modifier for tumorigenesis (12). This earlier assumption was based on the fact that the role of Tsg101 in neoplastic transformation was initially established in immortalized 3T3 cells (16). In preliminary studies we could show that these cells lacked expression of other important tumor suppressor proteins, in particular p19Arf and p16Ink4a (see Fig. 5). As discussed above, Tsg101 was also suggested to be a crucial regulator of the Mdm2-p53 circuit and p21Cip1. In contrast to our earlier working hypothesis, we can demonstrate in this study that none of the immortalizing mutations (i.e. knock-out of p19Arf, p16Ink4a, p53, and/or p21Cip1) or additional sporadic mutations causing neoplastic transformation and tumorigenesis had an impact on the important role of Tsg101 for cell survival. In agreement with these findings in primary cell cultures, the deletion of the p53 gene in Tsg101 mammary-specific knock-out mice (WAP-Cre Tsg101fl/fl p53−/−) did not rescue the survival of Tsg101-deficient epithelial cells. More importantly, haplo-insufficiency of p53 in the Tsg101 somatic knock-out model did not cause mammary tumorigenesis in 12-month-old animals. Unlike in our Brca1 mammary-specific knock-out mice (38), the deletion of Tsg101 in p53 heterozygous mutants (WAP-Cre Tsg101fl/fl p53+/−) seemed not to cause genomic alterations that lead to the loss of heterozygosity of the wild type p53 allele and neoplastic transformation.4 Mammary tumors were also not observed in females carrying two mutant p53 alleles (WAP-Cre Tsg101fl/fl p53−/−). Like p53 single knockout mice, they succumbed to lymphoma after a latency period of approximately six months. Finally, we have confirmed the importance of Tsg101 as a survival factor for tumorigenic cells in a murine cancer model that carries a mouse mammary tumor virus (MMTV)-driven Her2/neu oncogene (39) in addition to the somatic knock-out of Tsg101 (MMTV-neu WAP-Cre Tsg101fl/fl). In this animal model the deletion of Tsg101 did not accelerate but delayed neoplastic transformation (40). In conclusion to the data presented in this study, we propose that Tsg101 is an important factor for cell survival in normal, immortalized, and neoplastic cells. Because Tsg101 is essential for cell survival, the ability to target tumor cells and inhibit Tsg101 function should result in the death of neoplastic cells regardless of the functional inactivation of the Mdm2-p53 circuit or sporadic mutations in the p53, p19Arf, or p16Ink4a tumor susceptibility loci.
Acknowledgments
We thank Malinda D. Henry for technical assistance and animal husbandry. We also thank Charles Kuszynski and Linda Wilkie of the University of Nebraska Medical Center Flow Cytometry Facility for the cell cycle analysis and Joerg Rahnenfuehrer (University of California, Berkeley) for help in the evaluation of the MTT assay. We are grateful to Dr. Alan Diehl (University of Pennsylvania) for reading this manuscript and valuable suggestions.
Footnotes
This work was supported by NCI, National Institutes of Health Public Health Service Grant CA93797 (to K.-U. W.).
The abbreviations used are: WAP, whey acidic protein; Cre, site-specific recombinase in bacteriophage P1 (catalyzes recombination between loxP sites (loxP, locus of X-ing over)); Mdm2, transformed mouse 3T3 double minute 2 protein; MEF, mouse embryonic fibroblast; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Cdk2, cyclin-dependent kinase 2; MEC, mammary epithelial cell.
C. Eischen (University of Nebraska Medical Center), J. Weber (Washington University), and G. Zambetti (St. Judes Children’s Research Hospital), personal communications.
A. Krempler and K.-U. Wagner, unpublished information.
A. Krempler, A. A. Triplett, and K.-U. Wagner, unpublished information.
References
- 1.Koonin EV, Abagyan RA. Nat Genet. 1997;16:330 –331. doi: 10.1038/ng0897-330. [DOI] [PubMed] [Google Scholar]
- 2.Ponting CP, Cai YD, Bork P. J Mol Med. 1997;75:467–469. [PubMed] [Google Scholar]
- 3.Watanabe M, Yanagi Y, Masuhiro Y, Yano T, Yoshikawa H, Yanagisawa J, Kato S. Biochem Biophys Res Commun. 1998;245:900 –905. doi: 10.1006/bbrc.1998.8547. [DOI] [PubMed] [Google Scholar]
- 4.Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ. EMBO J. 1999;18:5380 –5388. doi: 10.1093/emboj/18.19.5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babst M, Odorizzi G, Estepa EJ, Emr SD. Traffic. 2000;1:248 –258. doi: 10.1034/j.1600-0854.2000.010307.x. [DOI] [PubMed] [Google Scholar]
- 6.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 7.Lu Q, Hope LW, Brasch M, Reinhard C, Cohen SN. Proc Natl Acad Sci U S A. 2003;100:7626 –7631. doi: 10.1073/pnas.0932599100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bache KG, Brech A, Mehlum A, Stenmark H. J Cell Biol. 2003;162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong Q, Chen Y, Jones D, Lee WH. Cancer Res. 1998;58:2699 –2702. [PubMed] [Google Scholar]
- 10.Xie W, Li L, Cohen SN. Proc Natl Acad Sci U S A. 1998;95:1595–1600. doi: 10.1073/pnas.95.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruland J, Sirard C, Elia A, MacPherson D, Wakeham A, Li L, Luis DLP, Cohen SN, Mak TW. Proc Natl Acad Sci U S A. 2001;98:1859 –1864. doi: 10.1073/pnas.98.4.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krempler A, Henry MD, Triplett AA, Wagner KU. J Biol Chem. 2002;277:43216 –43223. doi: 10.1074/jbc.M207662200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner KU, Krempler A, Qi Y, Park K, Henry MD, Triplett AA, Riedlinger G, Rucker EB, Hennighausen L. Mol Cell Biol. 2003;23:150 –162. doi: 10.1128/MCB.23.1.150-162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maucuer A, Camonis JH, Sobel A. Proc Natl Acad Sci U S A. 1995;92:3100 –3104. doi: 10.1073/pnas.92.8.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Z, Pan J, Hope WX, Cohen SN, Balk SP. Cancer. 1999;86:689 –696. doi: 10.1002/(sici)1097-0142(19990815)86:4<689::aid-cncr19>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 16.Li L, Cohen SN. Cell. 1996;85:319 –329. doi: 10.1016/s0092-8674(00)81111-3. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Liao J, Ruland J, Mak TW, Cohen SN. Proc Natl Acad Sci U S A. 2001;98:1619 –1624. doi: 10.1073/pnas.98.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh H, Mammucari C, Nenci A, Cabodi S, Cohen SN, Dotto GP. Proc Natl Acad Sci U S A. 2002;99:5430 –5435. doi: 10.1073/pnas.082123999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Li X, Francke U, Cohen SN. Cell. 1997;88:143–154. doi: 10.1016/s0092-8674(00)81866-8. [DOI] [PubMed] [Google Scholar]
- 20.Sherr CJ. Genes Dev. 1998;12:2984 –2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- 21.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 23.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 24.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 25.Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. Development. 2002;129:1377–1386. doi: 10.1242/dev.129.6.1377. [DOI] [PubMed] [Google Scholar]
- 26.Telford WG, King LE, Fraker PJ. Cell Prolif. 1991;24:447–459. doi: 10.1111/j.1365-2184.1991.tb01173.x. [DOI] [PubMed] [Google Scholar]
- 27.van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM. J Immunol Methods. 1994;174:311–320. doi: 10.1016/0022-1759(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 28.el Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 29.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 30.Sherr CJ, Roberts JM. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 31.Jones SN, Roe AE, Donehower LA, Bradley A. Nature. 1995;378:206 –208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 32.Montes de Oca LR, Wagner DS, Lozano G. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 33.Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. Genes Dev. 2000;14:2358 –2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs JJ, Keblusek P, Robanus-Maandag E, Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ, Koh EY, Daley GQ, van Lohuizen M. Nat Genet. 2000;26:291–299. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- 35.Wagner KU, Dierisseau P, Rucker EB, Robinson GW, Hennighausen L. Oncogene. 1998;17:2761–2770. doi: 10.1038/sj.onc.1202529. [DOI] [PubMed] [Google Scholar]
- 36.Feng GH, Lih CJ, Cohen SN. Cancer Res. 2000;60:1736 –1741. [PubMed] [Google Scholar]
- 37.Wagner KU, Dierisseau P, Hennighausen L. Cytogenet Cell Genet. 1999;84:87–88. doi: 10.1159/000015221. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 39.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Proc Natl Acad Sci U S A. 1992;89:10578 –10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.M. D. Henry Henry, M. D., Triplett, A. A., Oh, K.B., Smith, G. H., and Wagner, K.-U. (2004) Oncogene, in press [DOI] [PubMed]