

Abstract
Pituitary tumor transforming gene (Pttg) is induced in pituitary tumors and associated with increased tumor invasiveness. Pttg–null mice do not develop tumors, but exhibit pituitary hypoplasia, while mice heterozygous for the retinoblastoma (Rb) deletion develop pituitary tumors with high penetrance. Pttg-null mice were therefore cross-bred with Rb+/− mice to test the impact of pituitary hypoplasia on tumor development. Prior to tumor development, Rb+/−Pttg−/− mice have smaller pituitary glands with fewer cycling pituitary cells and exhibit induction of pituitary p21 levels. Pttg silencing in vitro with specific shRNAi in AtT20 mouse corticotrophs led to a marked induction of p21 mRNA and protein levels, decreased RB phosphorylation, and subsequent 24% decrease in S-phase cells. Eighty six percent of Rb+/−Pttg+/+ mice develop pituitary adenomas by 13 months, in contrast to 30% of double-crossed Rb+/−Pttg−/− animals (P<0.01). Pituitary hypoplasia, associated with suppressed cell proliferation, prevents the high penetrance of pituitary tumors in Rb+/− animals, and is therefore a protective determinant for pituitary tumorigenesis.
Keywords: Pituitary adenoma, corticotroph cell, Pttg
Introduction
Pituitary tumors are invariably benign and exhibit high prevalence and potential for significant morbidity (1). Like other differentiated neuroendocrine tissues, the pituitary gland displays trophic hormone cell plasticity in response to physiological and homeostatic demands (2, 3). Intrapituitary growth factors, hypothalamic hormones, inactivating tumor suppressor genes or activated oncogene mutations have been implicated in the spectrum of pathogenetic events leading to pituitary hyperplasia and adenoma development (1, 4, 5).
Mice bearing a single Rb mutant allele develop pituitary tumors with almost complete penetrance (6-8). Analysis of mutant mouse strains for the retinoblasoma gene (Rb1) has underscored the importance of retinoblastoma for tumor suppression. In mammalian cells, proliferation control is primarily achieved in the G1-phase of the cell cycle. RB is phosphorylated in a cell cycle dependent manner, and G1 cyclin/cyclin dependent kinase (Cdk) complexes phosphorylate RB and RB-related pocket binding proteins. RB hyperphosphorylation promotes subsequent release of E2F transcription factors resulting in S phase cell cycle progression (9). Cyclin-dependent kinases integrate extracellular signals into the cell-cycle machinery (10-12). Cyclin/Cdk complexes are regulated by multiple mechanisms, including cyclin-dependent kinase inhibitors (CdkI) (11). Cdk4(6) actions are regulated specifically by Ink4-type inhibitors (p16, p15, p18, p19) whereas Cdk2 is inhibited by Cip/Kip-type p21, p27 and p57 inhibitors (12, 13). By inhibiting cyclin/Cdk activity CdkIs govern the G1-to-S transition. Perturbed G1 control is a critical step for cellular transformation and tumorigenesis (14-16).
Pituitary tumor transforming gene (Pttg) behaves as a mammalian securin homolog facilitating sister chromatid separation during metaphase (17). Pttg exhibits oncogene properties as over-expression causes cell transformation, induces aneuploidy (18, 19), promotes tumor formation in nude mice, induces basic fibroblast growth factor (bFGF) and activates angiogenesis (20, 21). Pttg initially isolated from pituitary tumor cells, is over-expressed in pituitary tumors and correlates with tumor invasiveness (22). Mice lacking Pttg are viable and fertile, and exhibit testicular and splenic hypoplasia, thymic hyperplasia, and pancreatic ß cell hypoplasia (23, 24), while pituitary-directed transgenic Pttg over-expression results in focal pituitary hyperplasia and adenoma formation (25) To elucidate the PTTG role in tumorigenesis, we generated compound Rb x Pttg mutant mice to determine effects of deficient PTTG on tumor development in Rb+/− animals.
Results
Pttg deletion results in selective decreased organ weight
Compound Rb+/−Pttg−/− mutant mice have lower body weights as compared to Rb+/−Pttg+/+ animals (P<0.05), but similar to single Pttg−/−-deficient animals. At 4 months of age, before tumor development these animals weighed 32 g as compared to Rb+/−Pttg+/+ (41.3 ± 2.4 g, P<0.05) littermates (Table 1). Pttg deficiency resulted in organ-specific decreased Rb+/− weights. Spleen (P<0.01), pancreas (P<0.05), testis (P<0.05) and pituitary (P<0.05) dry weights were lower in compound Rb+/−Pttg−/− mutant mice than in Rb+/−Pttg+/+ and WT mice, and did not differ from single Pttg mutant animals. Similar organ-specific weight patterns were apparent when determined as a percentage of body mass (Table 1). Liver, brain and heart weights did not differ between genotypes (data not shown).
Table 1.
Body and organ dry weight in male mice at 4 months.
| Rb+/+Pttg+/+ | Rb+/+Pttg−/− | Rb+/−Pttg+/+ | Rb+/−Pttg−/− | |
|---|---|---|---|---|
| BW (g) | 36.5 ±1.7 | 32.1 ± 1.6§ | 41.3 ± 2.4 | 31.9 ± 2.0§ |
| Pituitary (mg) | 2.8 ± 0.1 | 1.5 ± 0.1§ | a5.0 ± 0.6* | 2.1 ± 0.3§ |
| Pituitary (%BWx10−4) | 7.5 ± 0.4 | 4.6 ± 0.4§* | 12 ± 1.5* | 6.5 ± 1.5§ |
| Spleen (g) | 0.18 ± 0.004 | 0.07 ± 0.05§** | 0.11 ± 0.01** | 0.05 ± 0.01§* |
| Spleen (% BW) | 0.50 ± 0.02 | 0.20 ± 0.03§* | 0.27 ± 0.03* | 0.14 ± 0.04§** |
| Testes (g) | 0.17 ± 0.03 | 0.10 ± 0.03§** | 0.19 ± 0.0013 | 0.12 ± 0.02§** |
| Testes (% BW) | 0.46 ± 0.01 | 0.34 ± 0.02§** | 0.45 ± 0.01 | 0.28 ± 0.01§** |
| Pancreas (g) | 0.27 ± 0.02 | 0.19 ± 0.02§** | 0.28 ± 0.02 | 0.20 ± 0.02§** |
| Pancreas (% BW) | 0.74 ± 0.01 | 0.62 ± 0.01* | 0.67 ± 0.01 | 0.62 ± 0.01* |
n=7-8 animals/group
P<0.05,
P<0.01 vs Rb+/+Pttg+/+
P<0.05,
P<0.01 vs Rb+/−Pttg+/+
, some Rb+/−Pttg+/+ pituitary glands may already exhibit pre-tumorous hyperplasia
Pttg deletion results in decreased cell proliferation in the pre -tumorous pituitary gland
Pituitary weight of Pttg−/− animals was low (1.5 ± 0.07 mg, P<0.01 vs WT), and at 4 months, before tumor development, pituitary dry weight was lower in Rb+/−Pttg−/− than in Rb+/−Pttg+/+ mice (2.1 ± 0.8 vs 5.0 ± 0.6 mg, p<0.05) and did not differ from WT (2.8 ± 0.08 mg) (Table 1). Pituitary cell proliferation rate evaluated by BrdU incorporation and Ki67 immunolabeling was attenuated in mice deficient in PTTG. In young 1- month single mutant Rb+/+Pttg−/− mice pituitary BrdU incorporation was lower (0.1 ± 0.04%) than in Rb+/−Pttg−/− and WT animals (0.76 ± 0.037% and 0.8 ±0.09% respectively), and lower than in Rb+/−Pttg+/+ pituitary glands (1.2 ± 0.22%, P<0.05) (Fig. 1a). These results were supported by finding low immunolabeled Ki67 expression in pituitary sections derived from Rb+/+Pttg−/− mice (2.7 ± 0.09% P<0.01). In double mutant mice the Ki67 count did not differ from WT animals (13.2 ± 3.2% and 7.8 ± 1.3% respectively) but was markedly lower than in Rb+/−Pttg+/+ controls (26.1 ± 5.3%, P<0.05) (Fig. 1b).
Fig 1. Pttg deletion results in decreased cell proliferation in pre-tumorous pituitary.
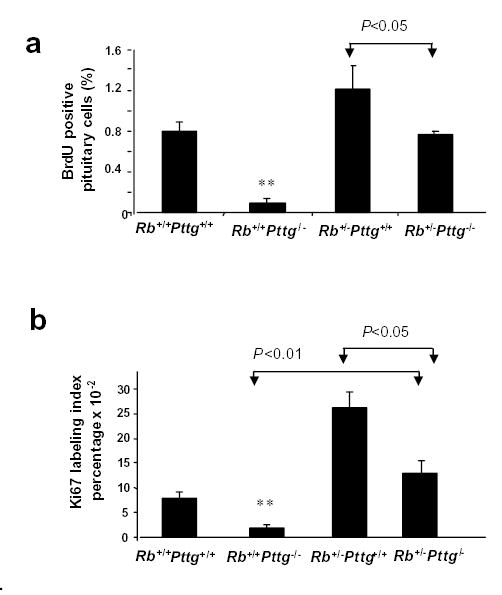
a) BrdU positive pituitary cells in 4 week old Rb+/+Pttg+/+, Rb+/−Pttg+/+, Rb+/+Pttg−/− and Rb+/−Pttg−/− mice sacrificed 24 hours after BrdU injection. Each value represents mean percentage of positive cells ± SE (5-7 fields/section, 3 sections/animal, n=3 animal/genotype analyzed); b) Ki67 labeling index in 4 week old Rb+/+Pttg+/+, Rb+/−Pttg+/+, Rb+/+Pttg−/− and Rb+/−Pttg−/− mice. Each value represents mean ± SE (10 fields/animal n=3 animal/genotype analyzed); In a and b: **, P<0.01 in Rb+/+Pttg−/− mice vs three other genotypes.
Pttg is up-regulated in pre-tumorous Rb+/−Pttg+/+ pituitary gland
Pituitary Pttg mRNA levels were 2-fold higher (p<0.05) and PTTG immunoreactivity was stronger in Rb+/−Pttg+/+ compared to WT pituitary gland (Fig.2 a,b) when tested at 2-4 months of age.
Fig 2. Pttg is overexpressed in pre-tumorous Rb+/− pituitary gland.
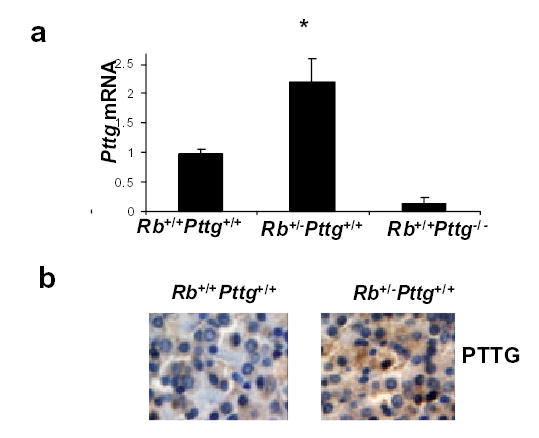
a)Real time PCR analysis of pituitary Pttg mRNA in 2-4 month old pre-tumorous mice. Values are expressed as mean ± SE of triplicate measurements for each experimental group (n=4-6 animals per group). *, P<0.05 vs WT; b) Pituitary immunohistochemistry for PTTG expression in WT and Rb+/−Pttg +/+mice. Representative sections are shown.
Pttg deletion increases p21 expression in pre -tumorous pituitary gland
p21 belongs to the Cip/Kip family of cyclin-dependent kinase (Cdk) inhibitors which regulate cell cycle progression (12, 26, 27). p21 restrains Cdk2 activity, and decreased phosphorylation of Cdks leads to decreased RB phosphorylation (11, 12, 26, 27), which consequently slows cell cycle progression. Young 2 month old Rb+/+Pttg−/− and compound Rb+/−Pttg−/− mice exhibit increased pituitary p21 mRNA (Fig.3a) and protein levels, while phosphorylated Cdk2 is reduced. Pituitary glands derived from Pttg-deficient animals demonstrate abundant p21 nuclear staining relative to WT and Rb+/−Pttg+/+ mice (Fig.3b). Conversely, in transgenic mice over-expressing pituitary PTTG, p21 protein levels are very low, and phoshorylated Cdk2 is increased (Fig.3c).
Fig 3. Pttg deletion increases pituitary p21expression in PTTG-deficient mice.
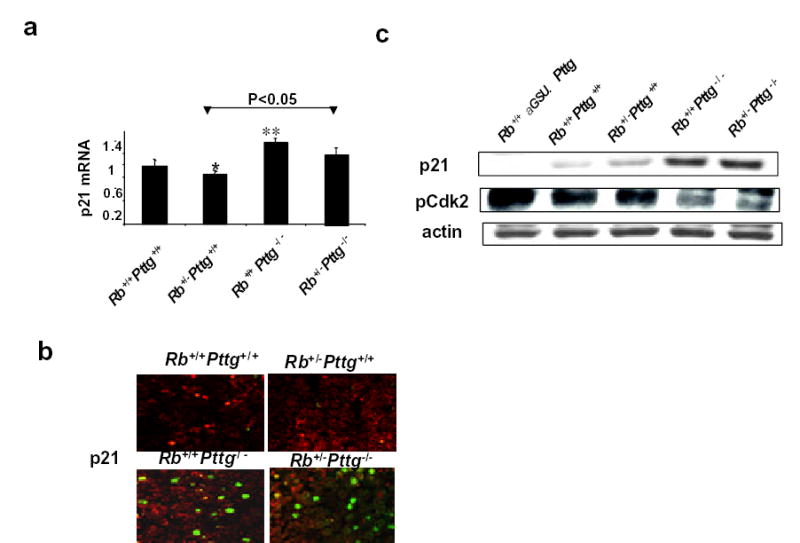
2-4 month old pre-tumorous Rb+/+Pttg+/+, Rb+/−Pttg+/+, Rb+/+Pttg−/− and Rb+/−Pttg−/− mice are analyzed. a) Real time PCR of p21 mRNA. 1- Rb+/+Pttg+/+, 2- Rb+/−Pttg+/+ , 3- Rb+/−Pttg−/−, 4- Rb+/−Pttg−/− . Values are expressed as mean ± SE of triplicate measurements for each experimental group (n=4-6 animals per group).*, P<0.05; **, P<0.01 vs Rb+/+Pttg+/+ ; b) immunohistochemistry for pituitary p21 expression. Green fluorescence indicate intra-nucleus localization of p21. Representative sections are shown; c) Western blot analysis of pituitary p21 and phosphoCdk2 protein levels.
PTTG regulates p21 expression in murine corticotroph cells
To confirm that the observed decreased cell proliferation in PTTG deficient mice is a result of Pttg deficiency, two short hairpin interfering RNAs were specifically designed from residues 497 to 521 (shRNAi I) and from residues 394-413 (shRNAi II) of the mouse Pttg mRNA coding region. AtT20 corticotroph cells were transfected with shRNAi I or shRNAi II or mismatched control shRNAi. Both specific shRNAi constructs suppressed Pttg expression by ~90% suggesting that most cells were successfully transfected. In cells where Pttg was silenced, p21 mRNA and protein levels were up-regulated, indicating an apparent inverse relationship between PTTG and p21 expression (Fig.4a). p21 induction was associated with decreased levels of phosphorylated RB. Silencing Pttg in AtT20 cells with shRNAi I resulted in ~24% reduction in the number of BrdU- incorporated cells (cells in S-phase) as assessed by FACS analysis (30.3 ± 2.1% cells transfected with mismatched shRNAi vs 23.3 ± 3.1% cells transfected with specific Pttg shRNAi, p<0.05). Transfection with shRNAi II resulted in ~20% reduction in the number of BrdU-incorporated cells (32.3±1.1% cells transfected with mismatched shRNAi vs 26.3±2.6% cell transfected with specific Pttg shRNAi, p<0.05) (Fig.4b). Thus, disrupted Pttg expression results in decreased murine pituitary cell proliferation rates.
Fig 4. PTTG regulates pituitary corticotroph p21 expression.
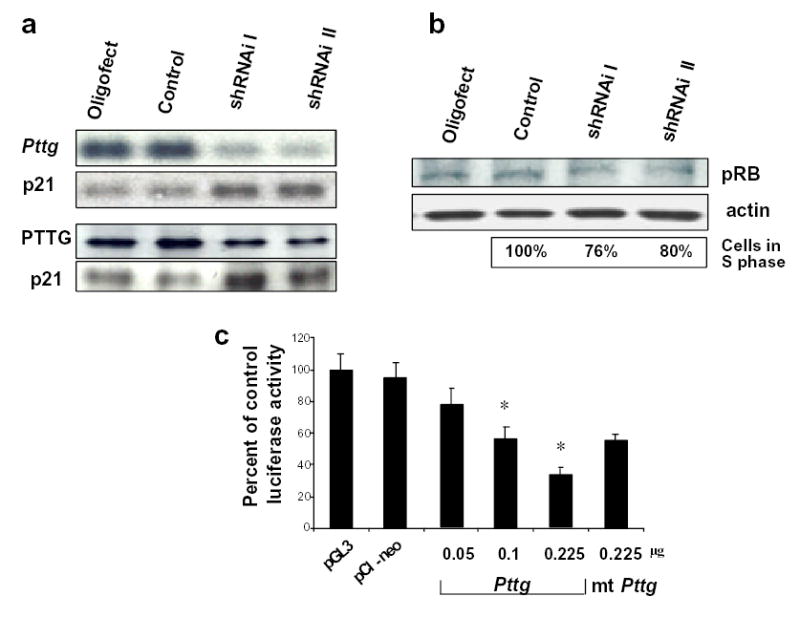
a) Northern (upper panel) and Western (lower panel) blot analysis of AtT20 cells transfected with anti-Pttg shRNAi. After hybridization with Pttg probe (Northern ) or anti-PTTG antibodies (Western) membranes were stripped and re-blotted with p21 probe (Northern) or anti-p21 antibodies (Western); b) Western blot analysis of AtT20 cells transfected with anti-Pttg shRNAi and hybridized with anti-phosphoRB antibodies. Lower panel: decreased number of cells in S phase (% of control) after transfection with shRNAi. c) Suppression of murine p21 promoter activity by increasing doses of human Pttg in CHO cells; mt- mutant Pttg *, P<0.05 vs basic vector.
The abundance of endogenous Pttg in experimental pituitary cell lines makes it difficult to accurately interpret tissue-specific effects of Pttg on p21 promoter activity. To explore the possibility that Pttg transcriptionally suppresses p21, CHO cells were therefore tested and transfected with murine a p21 promoter- luciferase reporter construct and co-transfected with increasing amounts of human wild type Pttg. p21 promoter activity was suppressed dose-dependently by the Pttg expression plasmid but not by either control plasmids. Mutated human Pttg (28) partially suppressed p21 promoter activity relative to wt Pttg (33% in wt Pttg vs 59% in mutant Pttg vs 100% in basic vector, P<0.05 between wt Pttg and basic vector ) (Fig.4c).
These results suggest that PTTG restrains p21 expression in pituitary corticotrophs, and Pttg deletion decreases pituitary cell proliferation in young Rb+/−Pttg−/− animals prior to visible tumor development by inducing pituitary p21.
Pttg deletion suppresses pituitary tumor development in Rb+/− mice
Rb heterozygous mice die mostly from pituitary tumors at 8-12 months of age depending on their genetic background (6, 29-31). Rb+/−Pttg+/+ mice developed pituitary tumors starting from 4 months of age, and by 13 months 25 of 29 (86%) Rb+/−Pttg+/+ mice had pituitary tumors. The appearance of pituitary tumors was delayed in Rb+/−Pttg−/− mice; of 57 doubly mutant mice, only 20 % harbored tumors at 13 months, and by 17 months 30% had tumors. These adenomas did not differ morphologically from Rb+/− tumors. In WT mice spontaneous pituitary tumors were observed in 4 of 28 animals (14%) starting at 9 months of age. Of 23 Rb+/+Pttg−/− mice, three animals (13%) harbored pituitary tumors at 16 months. While Rb+/− mice do not survive more than 13 months, compound Rb+/−Pttg−/− animals have now survived for more than 18 months (Fig. 5a). Kaplan-Meier survival analysis (log-rank test) of the time of death with evidence of pituitary tumor in the different genotypes showed significant differences between Rb+/−Pttg−/−and Rb+/−Pttg+/+ (P <0.01), between Rb+/−Pttg−/− and Rb+/+Pttg−/− (P< 0.05), and between Rb+/−Pttg+/+ and Rb+/+Pttg−/− mice (P < 0.01).
Fig 5. Pttg deletion suppresses pituitary tumor development in Rb+/−mice.
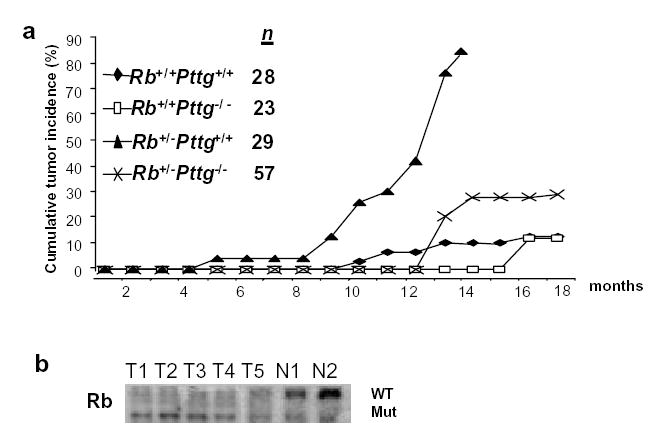
a) Development of pituitary tumors in Rb+/+Pttg+/+, Rb+/−Pttg+/+, Rb+/+Pttg−/− and Rb+/−Pttg−/− mice over time, n=total number of animal sacrificed; b) Southern-based Rb LOH analysis of DN A extracted from either tumor or tail tissue; mut-mutant, WT-wild type allele. T1, T3, T5-tissue derived from Rb+/−Pttg−/− pituitary tumors, N1, N2-tail tissue from WT mice.
Consistent with results previously shown for Rb+/− tumors (32), Rb LOH was observed in five of seven compound tumors analyzed, indicating that loss of the single Rb allele in Rb+/−Pttg−/− animals is present in these tumors (Fig.5b).
Discussion
The pathogenesis of pituitary neoplasms has been extensively studied to identify oncogene or growth factor mutations, or signaling defects (33, 34). Pttg originally isolated from experimental pituitary tumor cells, is expressed in several malignancies (21), is associated with increased tumor invasiveness of pituitary adenomas (22), epithelial neoplasias (35) and colorectal carcinomas (22), and in the pituitary is especially induced by estrogen (36). Pttg is also a component of a 17- gene expression signature marker of metastatic potential for human tumors (37). As mice heterozygous for Rb show enhanced predisposition to pituitary and thyroid tumors (8, 29, 31), Rb+/−Pttg−/− compound mutant mice were employed to determine the impact of PTTG loss on development and progression of pituitary tumors in Rb-deficient animals.
Pituitary Pttg mRNA and protein levels were induced in pre-tumorous Rb +/- mice. Pttg deletion leads to slowing of pituitary cell proliferation and induction of the Cdk inhibitor, p21, in young pre-tumorous pituitary glands, and in mouse AtT20 corticotroph cells. Conversely, mice with pituitary directed Pttg overexpression exhibit very low pituitary p21 levels. Compound mice with deleted Pttg develop pituitary tumors with markedly lower frequency than Rb heterozygous animals. High p21 levels likely restrain tumor initiation and progression in Pttg-deficient compound animals. The results suggest that pituitary cell proliferation capacity is required for early high penetrance of pituitary tumor formation in Rb heterozygous mice.
Pre-tumorous compound Rb+/−Pttg−/− animals had lower selective organ weights consistent with splenic, testicular and pancreatic ß cell hypoplasia observed in Pttg−/− mice (23, 24), indicating the requirement for PTTG in post-development growth control of selected cell types. Hypoplastic organs appeared developmentally normal with appropriate differentiated gene expression; although the testes are hypoplastic, males are fertile (23). Pttg-disrupted MEF or pancreatic ß cells do not exhibit higher rates of apoptosis (23, 24). The relation between PTTG and apoptosis is not clear. PTTG overexpression caused p53-dependent and p53 independent apoptosis (18), and p53 suppresses Pttg promoter activity in response to DNA damage (38). While pituitary weights were lower in PTTG-deficient mice, apoptosis rates were extremely low in young pre-tumorous pituitary glands and no differences in apoptotic rates were noted between genotypes as assessed by TUNEL assay (data not shown).
Low pituitary and other selected organ weights in animals lacking PTTG might result from a proliferation defect. Slow pituitary cell proliferation is evident by low pituitary BrdU incorporation as well as low immunolabeling with Ki67. Ki67 is expressed during both G1 and S phases of proliferation, but not in quiescent cells (39). Additional support for a proliferative defect was derived from experiments showing that Pttg suppression in AtT20 cells by shRNAi decreased the percentage of cells in S-phase. Thus, PTTG deletion slows pituitary cell proliferation, while up-regulated Pttg mRNA and protein levels observed in the pre-tumorous Rb+/−Pttg+/+ pituitary gland may promote cell cycle entry.
Mechanisms underlying organ-specific decreased Pttg−/− cell proliferation are not clear. In humans, two additional Pttg homologs have been identified (40): the index Pttg, and homologous Pttg2 and Pttg3. Although Pttg is most abundantly expressed in normal testes, Pttg2 is preferentially expressed in spleen, liver, heart and pituitary, and Pttg3 in the kidney and prostate. PTTG may be important for neuro-endocrine cell proliferation, while in other tissues PTTG requirement could be less essential, or Pttg function may be substituted by other Pttg family members. Similarly, acute RB loss in quiescent pituitary cells is compensated by Rb-related associated pocket binding protein p107 (41).
Negative regulation of cell cycle progression, particularly during development, could depend on cell-specific combinations of Cdk inhibitors (42). No differences in the expression of Cdk inhibitors p27 and p18 were found between genotypes (data not shown). Therefore, a mechanism for decreased pituitary cell proliferation in mice lacking PTTG could be induced specifically by pituitary p21 expression. Cell proliferation control is primarily achieved in G1, when RB and p21 are critical components (26). Sequential activation of cyclin/Cdk complexes regulates progression through the cell cycle. In vitro, p21 has a high affinity for cyclin E/Cdk2 complexes and 95% of active Cdk2 in normal fibroblasts is associated with p21(27, 43). A recent model describes G1 progression as occurring in two discrete stages controlled by Cdk4(6) under RB regulation and Cdk2 under p21 regulation. Inhibition of either stage attenuates cell progression (26). Rb+/−p21−/− mice exhibit alteration of both stages and have accelerated pituitary tumor development compared to Rb heterozygous animals (26). In our experiments, induced p21 leads to a decline in phosphorylated Cdk2 levels which likely affect pituitary cell proliferation. These results indicate that p21 function limits tumor cell growth and that the delay in tumor progression observed in compound Rb+/−Pttg−/− animals might arise as a consequence of pituitary p21 overexpression. Mutually exclusive patterns of Ki67 and p21 occur in gastrointestinal epithelium with p21 apparently restraining epithelial proliferation (39). Similarly, our data showing high p21 and low Ki67 expression suggest a restraining role for p21 in pituitary cell proliferation in the young PTTG-deficient pituitary gland.
Increased p21 expression in Rb+/+Pttg−/− and Rb+/−Pttg−/− animals is probably due to Pttg ablation. Our in vitro experiments demonstrate that silencing Pttg in AtT20 mouse corticotrophs by shRNAi leads to marked p21 gene and protein induction. High p21, in turn, is associated with decreased RB phosphorylation with subsequent diminished S phase cell number. PTTG might also directly affect the p21 promoter as PTTG over-expression dose-dependently decreased p21 promoter activity.
An alternative explanation of our results would be that PTTG-derived mitotic alteration could activate checkpoint signals, leading to p53 stimulation and consequent p21 induction (39, 44, 45). Indeed, PTTG has been shown to interact with p53 and inhibit its transcriptional ability after DNA damage (46). In this and previous (23, 24) studies however we did not observe p53-dependent increased pituitary apoptosis in PTTG-deficient mice. In undamaged cells, p21 may negatively control proliferation in a p53-independent manner(39). Thus, our results indicate that PTTG deficiency has significant consequences for cell proliferation, and imply that PTTG regulation of the pituitary cells involves p21-dependent mechanisms.
Striking similarities are apparent between Pttg−/− and Cdk4–deficient mice. Cdk4−/− animals have hypoplastic pituitary glands and develop diabetes mellitus associated with pancreatic islet degeneration (47). At least in part, Cdk4 controls S-phase transition via negative regulation of p27, another Cdk inhibitor (42, 48). PTTG negatively regulates p21, and similar to Cdk4 promotes cell cycle entry. Cooperation of p27 and p21 appears critical for tissue-specific withdrawal from the cell cycle (42).
High p21 levels likely restrain tumor formation and progression in compound double mutant mice. In this study we show that by 12 months pituitary tumors were evident in 86% of Rb+/−Pttg+/+ mice. Pttg absence suppresses and delays progression of Rb-related tumors resulting in extended murine life span. Thus, while Rb+/−Pttg+/+ mice invariably die by 13 months, only 30% of Rb+/−Pttg−/− develop tumors by 18 months.
Both humans and mice harboring a germ line Rb mutation develop tumors with almost complete penetrance, and tumor development is accompanied by tumor loss of the wild type allele (30, 32, 49). In the absence of PTTG, the proportion of individual cells that eliminate the remaining WT allele of Rb during tumor development could be lower. However, as 5 of 7 tumors derived from Rb+/−Pttg−/− compound mice do in fact exhibit Rb LOH, it is unlikely that PTTG regulates the frequency of loss of the remaining Rb allele in these tumors. However, we cannot exclude the effect of PTTG as a securin protein on chromatin exchange, leading to accelerated LOH and tumor formation. Aneuploidy is a ubiquitous feature of human solid tumors, causes genetic instability, and also promotes further aneuploidy. PTTG is a mammalian securin, localizes in the interphase nucleus and mitotic spindles and binds to and inhibits separin, which cleaves cohesin binding of sister chromatids (17). At the end of metaphase, PTTG is degraded, allowing equal separation of sister chromatids. PTTG overexpression induces aneuploidy by inhibiting equal chromatid segregation (19) and increasing the number of aneuploid cells leading to genomic instability. Paradoxically, abnormal nuclei, increased aneuploidy and premature centromere division are also observed in fibroblasts derived from Pttg−/− mice (23). Therefore, both Pttg excess as observed in tumors, and Pttg loss lead to cell cycle disruption and aneuploidy. These features point to Pttg as a caretaker gene ensuring genomic stability (50, 51). It is not yet apparent whether aneuploidy is a contributing cause or secondary consequence of cell transformation (51). Chromosamal instability can also arise from defects in cell cycle transformation (52). Despite increased aneuploidy, the incidence of pituitary tumors in Pttg-null mice are notably lower than in Rb heterozygous animals.
Our results are in contrast with an earlier in vitro study showing that PTTG overexpression induced growth arrest in human lung cancer cells by a p21-dependent mechanism (53). However, low pituitary weight, decreased cell proliferation, induction of pituitary p21 in PTTG-deficient mice, very low p21 protein levels in mice with pituitary-directed PTTG-overexpression, high levels of PTTG in pre-tumorous pituitary glands of Rb-heterozygous mice and marked decrease in tumor incidence in Rb+/− mice with Pttg deletion, all observed in our study indicate that in vivo PTTG promotes the pituitary cell cycle via p21 arrest and thus may induce or potentiate pituitary tumor formation. The contrasting results could be explained by strong tissue-specific properties of p21 (for example, RB stimulates p21 promoter in epithelial cells, but not in fibroblasts) (54). Thus the effect of PTTG-deficiency on p21 over-expression and cell cycle arrest may also be pituitary-specific. The extent to which such tissue specificity underlies the relations hip between PTTG and p21 requires further study.
In summary, the results show that placing Rb+/− mice into a Pttg- deficient background reduces and delays the progression of pituitary tumors. Absent PTTG allows expression of p21. The observed results, taken together with the in vivo finding that pituitary-directed transgenic Pttg overexpression causes focal hyperplasia (25) suggest that overexpressed pituitary PTTG in Rb+/− mice influences tumor initiation and progression by enhancing cell proliferation. We conclude that pituitary hypoplasia is an important determinant for protection against pituitary tumor formation.
Materials and Methods
Animals
Experiments were approved by the Institutional Animal Care and Use Committee. Pttg−/− mice were generated on a mixed C57BL/6x129/Sv genetic background and backcrossed to a C57BL/6 parental genotype. Rb+/− mice on a 129/Sv genetic background were purchased from Jackson Laboratory. Compound Rb+/−Pttg−/− mice were bred by crossing Rb+/−Pttg+/− females and Rb+/−Pttg+/− males. Four genotypes were obtained from the same breeding: Rb+/−Pttg−/−, Rb+/−Pttg+/+, Rb+/+ Pttg−/−, and Rb+/+Pttg+/+ (wild type, WT). Animals were genotyped by PCR for Pttg (23) and Rb loci as described (6). Transgenic mice with aGSU promoter driving PTTG expression (25) were cross-bred with Rb+/− animals.
Anatomic and histological analysis
Animals were sacrificed and subjected to necropsy at the first indication of morbidity (weight loss, dehydratation or ataxia). Others were sacrificed as age- matched controls. For histological analyses tissues were fixed, paraffin-embedded, and sections stained with hematoxylin-eosin and periodic acid-Schiff.
Immunohistochemistry
The streptavidin-biotin-peroxidase complex technique was used with polyclonal PTTG antibodies (rabbit anti-human, Zymed, san Francosco, California) (55), and for p21 detection goat anti- mouse p21 polyclonal antibodies conjugated with Alexa 488 fluorescent dye was used (Molecular Probe, Eugine, Oregon). Antigen retrieval was performed by heating; control reactions lacked primary antibodies or were stained with blocking antibodies.
BrdU and Ki67 labeling
One month old mice were injected with BrdU (50 μg/g BW, Sigma, St Louis, Missouri) and sacrificed 24 h later. Pituitary sections were stained for BrdU (mouse anti-BrdU antibody, Becton Dickinson, Franklin Lake, New Jersey), counterstained with hematoxylin, and positive cells detected with ABC peroxidase system (Vector, Burlingame, California). Five to seven randomly chosen visual fields/per section were counted, and three sections/per animal derived from three animals of each genotype were analyzed.
Ki67 labeling index (MIB-1 antibody, Immunotech, Westbrook, Minnesota) was determined based on the number of positively stained nuclei divided by the total number of nuclei counted. Ten fields containing approximately 100 cells were counted from each animal and three animals from each genotype were analyzed.
Loss of heterozygosity (LOH)
Rb loss was determined by Southern blotting of DNA prepared from tumor tissues derived from either Rb+/−Pttg+/+ or Rb+/−Pttg−/− animals. DNA was digested with Pst1/Kpn1, and hybridized with a probe spanning exon 3 of the Rb locus (generous gift of Dr. T. Jacks, MIT, Cambridge).
Quantitative PCR
Quantitative real time PCR was performed (56) to detect p21 and Pttg mRNA expression. The following specific primers were used: p21 forward 5’-CAGTACTTCCTCTGCCCTGC-3’, p21 reverse 5’-AATCTGTCAGGCTGGTCTGC-3’. Pttg forward 5′-CGTCCTCAATGCCAATATCC-3′, reverse 5′-TCAACCCATCCTTAGATGCC-3′; 18S forward 5′-AAACGGCTACCACATCCAAG-3′, reverse 5′-CCTCCAATGGATCCTGGTTA-3′. Relative quantification of each gene in experimental samples was determined from the corresponding standard curve, normalized to 18S, and expressed as arbitrary units.
Short hairpin interfering RNA (shRNAi)
For suppression of cellular Pttg expression, two shRNAis that specifically targeted Pttg mRNA were designed according to the manufacturer’s protocol (Epicentra, Madison, Wisconsin). The sense sequence of shRNAi I spanning residues 497 -521 of mouse Pttg coding region was 5’-GGACAGTCAACAGAGTTGCCGAAAC-3’. The sense sequence of shRNAi II spanning residues 394 -413 was 5’-CTAGTGTCAAGGCCTTAGATC-3’. AtT20 murine corticotroph cells (American Type Culture Collection, Rockville, Maryland) were transfected with 100 nM Pttg shRNAi or mismatched shRNA using Oligofectamine (Invitrogen, Gaithersburg, Maryland), and cellular expression analyzed 24 hours later.
Northern and Western blot analysis
Northern analysis of pituitary Pttg and p21 expression was performed as described (56). Membrane was hybridized with 32P-labeled fragment of murine Pttg (23), stripped and re-hybridized with a murine p21 fragment (obtained by PCR, gene bank accession number U24173).
For Western blot, pituitaries or cells were processed according to manufacturer’s instruction (Immunoprecipitation Kit, Roche Diagnostics, Germany). Proteins were separated by SDS-PAGE, electroblotted onto Millipore membranes (Millipore, Massachusetts ), and incubated with anti-PTTG (Zymed, San Francisco, California) or anti-p21, p18, p27 (Santa Cruz, California ) or anti-phosphoCdk2 (Thr160) and -phosphorRB (Ser807/811) (Cell Signaling Technology, ‘Massachusetts) antibodies overnight, and then with corresponding secondary antibodies. Immunoreactive bands were detected by ECL immunodetection system.
Cell proliferation assay
Asynchronized AtT20 cells were pulsed with 10μM BrdU (Sigma, St Louis, Missouri) in PBS for 10 min at 37°C. Cells were washed three times with 1% BSA in PBS, harvested, fixed in 75% ethanol and analyzed by FACScan (Becton Dickinson, Mountain View, California ). The results depict the mean of three independent experiments ± SE.
Transfection and luciferase assay
Hamster ovarian carcinoma cells (CHO, ATCC, Rockville, Maryland) were plated in 6-well plates 12 hours before transient transfection in triplicate with 0.225 μg murine p21 promoter- luciferase reporter construct in pGL 3 (kindly provided by Dr. J. Pelling, University of Kansas) and co-transfected with increasing amounts of wild type or mutated human Pttg in pCI-neo. As a control, cells were co-transfected with reporter and expression vectors and each sample was co-transfected with LacZ control plasmid (Promega, San Louis Obispo, California ). 0.5 μg cDNA (including 0.05 μg LacZ) was transfected using Effectin (Qiagen, Valencia, California). Total DNA was kept constant by adding the required amount of pGL 3. Cells were harvested 24 hours after transfection, assayed for luciferase activity, results were normalized to ß-galactosidase activity and represent the average of three independent transfections ± SE. Luciferase activity in cells co- transfected with p21 and pGL3 basic vector is represented as 100%.
Statistical analysis: Comparisons of pituitary tumor incidences in the respective genotypes were made by Kaplan-Meier survival analysis (log-rank test). Body and organ weights, quantitative PCR, BrdU-and Ki67 labeling indices were analyzed using ANOVA followed by non-parametric t-test (Mann-Whitney) or Student t-test with a probability of P<0.05 considered significant.
Acknowledgments
We thank Fabio Rotondo, Anastasia Kariagina, Ph.D and Dana Alon for technical assistance, and Dr. I. Donangelo for providing Rb+/+ aGSU.Pttg mice
Footnotes
Supported by NIH grant CA 75979 (S.M.) and The Doris Factor Molecular Endocrinology Laboratory
References
- 1.Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112:1603–18. doi: 10.1172/JCI20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanislaus D, Pinter JH, Janovick JA, Conn PM. Mechanisms mediating multiple physiological responses to gonadotropin-releasing hormone. Mol Cell Endocrinol. 1998;144:1–10. doi: 10.1016/s0303-7207(98)00126-9. [DOI] [PubMed] [Google Scholar]
- 3.Goffin V, Binart N, Touraine P, Kelly PA. Prolactin: the new biology of an old hormone. Annu Rev Physiol. 2002;64:47–67. doi: 10.1146/annurev.physiol.64.081501.131049. [DOI] [PubMed] [Google Scholar]
- 4.Cruz-Soto ME, Scheiber MD, Gregerson KA, Boivin GP, Horseman ND. Pituitary tumorigenesis in prolactin gene-disrupted mice. Endocrinology. 2002;143:4429–36. doi: 10.1210/en.2002-220173. [DOI] [PubMed] [Google Scholar]
- 5.Lania A, Mantovani G, Spada A. Genetics of pituitary tumors: Focus on G-protein mutations. Exp Biol Med (Maywood) 2003;228:1004–17. doi: 10.1177/153537020322800904. [DOI] [PubMed] [Google Scholar]
- 6.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 7.Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–7. [PubMed] [Google Scholar]
- 8.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910–7. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 9.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 10.Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–8. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 11.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–4. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 12.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 13.Kiyokawa H, Koff A. Roles of cyclin-dependent kinase inhibitors: lessons from knockout mice. Curr Top Microbiol Immunol. 1998;227:105–20. doi: 10.1007/978-3-642-71941-7_5. [DOI] [PubMed] [Google Scholar]
- 14.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–8. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 16.Sherr CJ. Cell cycle control and cancer. Harvey Lect. 2000;96:73–92. [PubMed] [Google Scholar]
- 17.Zou H, McGarry TJ, Bernal T, Kirschner MW. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science. 1999;285:418–22. doi: 10.1126/science.285.5426.418. [DOI] [PubMed] [Google Scholar]
- 18.Yu R, Heaney AP, Lu W, Chen J, Melmed S. Pituitary tumor transforming gene causes aneuploidy and p53-dependent and p53- independent apoptosis. J Biol Chem. 2000;275:36502–5. doi: 10.1074/jbc.C000546200. [DOI] [PubMed] [Google Scholar]
- 19.Yu R, Lu W, Chen J, McCabe CJ, Melmed S. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology. 2003;144:4991–8. doi: 10.1210/en.2003-0305. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa H, Heaney AP, Yu R, Horwitz GA, Melmed S. Human pituitary tumor-transforming gene induces angiogenesis. J Clin Endocrinol Metab. 2001;86:867–74. doi: 10.1210/jcem.86.2.7184. [DOI] [PubMed] [Google Scholar]
- 21.Heaney AP, Horwitz GA, Wang Z, Singson R, Melmed S. Early involvement of estrogen-induced pituitary tumor transforming gene and fibroblast growth factor expression in prolactinoma pathogenesis. Nat Med. 1999;5:1317–21. doi: 10.1038/15275. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Horwitz GA, Heaney AP, et al. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J Clin Endocrinol Metab. 1999;84:761–7. doi: 10.1210/jcem.84.2.5432. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Yu R, Melmed S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol. 2001;15:1870–9. doi: 10.1210/mend.15.11.0729. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Moro E, Kovacs K, Yu R, Melmed S. Pituitary tumor transforming gene- null male mice exhibit impaired pancreatic beta cell proliferation and diabetes. Proc Natl Acad Sci U S A. 2003;100:3428–32. doi: 10.1073/pnas.0638052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abbud R TI, Baker E.,Ren S-G., Chen D-Y, Wawrowsky K, Melmed S. 2005 Early Multipotential Pituitary Focal Hyperplasia in αGSU-Driven Pituitary Tumor Transforming Gene (PTTG) Transgenic Mice. Mol Endocrinol in press [DOI] [PubMed]
- 26.Brugarolas J, Bronson RT, Jacks T. p21 is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J Cell Biol. 1998;141:503–14. doi: 10.1083/jcb.141.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation- induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–7. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 28.Horwitz GA, Miklovsky I, Heaney AP, Ren SG, Melmed S. Human pituitary tumor-transforming gene (PTTG1) motif suppresses prolactin expression. Mol Endocrinol. 2003;17:600–9. doi: 10.1210/me.2001-0006. [DOI] [PubMed] [Google Scholar]
- 29.Jacks T. Tumor suppressor gene mutations in mice. Annu Rev Genet. 1996;30:603–36. doi: 10.1146/annurev.genet.30.1.603. [DOI] [PubMed] [Google Scholar]
- 30.Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 31.Vooijs M, Berns A. Developmental defects and tumor predisposition in Rb mutant mice. Oncogene. 1999;18:5293–303. doi: 10.1038/sj.onc.1202999. [DOI] [PubMed] [Google Scholar]
- 32.Nikitin A, Lee WH. Early loss of the retinoblastoma gene is associated with impaired growth inhibitory innervation during melanotroph carcinogenesis in Rb+/− mice. Genes Dev. 1996;10:1870–9. doi: 10.1101/gad.10.15.1870. [DOI] [PubMed] [Google Scholar]
- 33.Heaney A, Melmed S. Molecular targets in pituitary tumors. Nature Review Cancer. 2004;4:285–295. doi: 10.1038/nrc1320. [DOI] [PubMed] [Google Scholar]
- 34.Lloyd RV, Erickson LA, Jin L, et al. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am J Pathol. 1999;154:313–23. doi: 10.1016/S0002-9440(10)65277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saez C, Japon MA, Ramos-Morales F, et al. hpttg is over-expressed in pituitary adenomas and other primary epithelial neoplasias. Oncogene. 1999;18:5473–6. doi: 10.1038/sj.onc.1202914. [DOI] [PubMed] [Google Scholar]
- 36.Heaney AP, Fernando M, Melmed S. Functional role of estrogen in pituitary tumor pathogenesis. J Clin Invest. 2002;109:277–83. doi: 10.1172/JCI14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Y, Mehta KR, Choi AP, Scolavino S, Zhang X. DNA damage- induced inhibition of securin expression is mediated by p53. J Biol Chem. 2003;278:462–70. doi: 10.1074/jbc.M203793200. [DOI] [PubMed] [Google Scholar]
- 39.el-Deiry WS, Tokino T, Waldman T, et al. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–9. [PubMed] [Google Scholar]
- 40.Prezant TR, Kadioglu P, Melmed S. An intronless homolog of human proto-oncogene hPTTG is expressed in pituitary tumors: evidence for hPTTG family. J Clin Endocrinol Metab. 1999;84:1149–52. doi: 10.1210/jcem.84.3.5658. [DOI] [PubMed] [Google Scholar]
- 41.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 42.Jirawatnotai S, Moons DS, Stocco CO, et al. The cyclin-dependent kinase inhibitors p27Kip1 and p21Cip1 cooperate to restrict proliferative life span in differentiating ovarian cells. J Biol Chem. 2003;278:17021–7. doi: 10.1074/jbc.M301206200. [DOI] [PubMed] [Google Scholar]
- 43.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 44.Wu RC, Schonthal AH. Activation of p53-p21waf1 pathway in response to disruption of cell- matrix interactions. J Biol Chem. 1997;272:29091–8. doi: 10.1074/jbc.272.46.29091. [DOI] [PubMed] [Google Scholar]
- 45.Weiss RH. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell. 2003;4:425–9. doi: 10.1016/s1535-6108(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 46.Bernal JA, Luna R, Espina A, et al. Human securin interacts with p53 and modulates p53- mediated transcriptional activity and apoptosis. Nat Genet. 2002;32:306–11. doi: 10.1038/ng997. [DOI] [PubMed] [Google Scholar]
- 47.Moons DS, Jirawatnotai S, Parlow AF, Gibori G, Kineman RD, Kiyokawa H. Pituitary hypoplasia and lactotroph dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology. 2002;143:3001–8. doi: 10.1210/endo.143.8.8956. [DOI] [PubMed] [Google Scholar]
- 48.Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–9. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 1998;14:223–9. doi: 10.1016/s0168-9525(98)01470-x. [DOI] [PubMed] [Google Scholar]
- 50.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 51.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 52.Mishima M, Pavicic V, Gruneberg U, Nigg EA, Glotzer M. Cell cycle regulation of central spindle assembly. Nature. 2004;430:908–13. doi: 10.1038/nature02767. [DOI] [PubMed] [Google Scholar]
- 53.Mu YM, Oba K, Yanase T, et al. Human Pituitary Tumor Transforming Gene (hPTTG) Inhibits Human Lung Cancer A549 Cell Growth through Activation of p21(WAF1/CIP1 ) Endocr J. 2003;50:771–81. doi: 10.1507/endocrj.50.771. [DOI] [PubMed] [Google Scholar]
- 54.Decesse JT, Medjkane S, Datto MB, Cremisi CE. RB regulates transcription of the p21/WAF1/CIP1 gene. Oncogene. 2001;20:962–71. doi: 10.1038/sj.onc.1204169. [DOI] [PubMed] [Google Scholar]
- 55.Vidal S, Kovacs K, Bell D, Horvath E, Scheithauer BW, Lloyd RV. Cyclooxygenase-2 expression in human pituitary tumors. Cancer. 2003;97:2814–21. doi: 10.1002/cncr.11387. [DOI] [PubMed] [Google Scholar]
- 56.Kariagina A, Romanenko D, Ren SG, Chesnokova V. Hypothalamic-pituitary cytokine network. Endocrinology. 2004;145:104–12. doi: 10.1210/en.2003-0669. [DOI] [PubMed] [Google Scholar]