

Abstract
Anthracene-9-carboxylic acid (9-AC) has been reported to show both potentiation and inhibitory effects on guinea-pig cardiac cAMP-activated chloride channels via two different binding sites, and inhibition of Mg2+-sensitive protein phosphatases has been proposed for the mechanism of 9-AC potentiation effect. In this study, we examined the effects of 9-AC on wild-type and mutant human cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels expressed in NIH3T3 or CHO cells. 9-AC inhibits whole-cell CFTR current in a voltage-dependent manner, whereas the potentiation effect is not affected by membrane potentials. Anthracene-9-methanol, an electro-neutral 9-AC analog, fails to block CFTR, but shows a nearly identical potentiation effect, corroborating the idea that two chemically distinct sites are responsible, respectively, for potentiation and inhibitory actions of 9-AC. 9-AC also enhances the activity of ΔR-CFTR, a constitutively active CFTR mutant whose R-domain is removed. In excised inside-out patches, 9-AC increases Po by prolonging the mean burst durations and shortening the interburst durations. We therefore conclude that two different 9-AC binding sites for potentiation and inhibitory effects on CFTR channels are located outside of the R-domain. We also speculate that 9-AC potentiates CFTR activity by directly affecting CFTR gating.
Keywords: Chloride channel, Cystic fibrosis, Patch clamp, Single channel
Introduction
Anthracene-9-carboxylic acid (9-AC), an aromatic compound, has been widely used as an anion probe to study various chloride channels such as cardiac cAMP-activated chloride currents in guinea pig ventricular myocytes [33], swelling-activated chloride currents in canine ventricular myocytes [6], CLC-1 chloride channel expressed in Xenopus oocytes [11], and Ca2+-activated chloride currents in mouse kidney inner medullary collecting duct cells [25]. Most of these studies show that 9-AC blocks the anion permeation pathway. However, Zhou et al. [33] reported dual effects of 9-AC on the cardiac isoform of cystic fibrosis transmembrane conductance regulator (CFTR). In addition to a voltage-dependent block of whole-cell cardiac CFTR currents, 9-AC voltage-independently increases the cAMP-dependent chloride conductance. They concluded that 9-AC blocks cardiac CFTR Cl− conductance from the extracellular side but enhances the conductance from the intracellular side by inhibiting a Mg2+-sensitive protein phosphatase [33, 35].
CFTR, a member of the ABC (ATP binding cassettes) superfamily [27], is a chloride channel that is regulated by protein kinase A (PKA)-dependent phosphorylation of the regulatory (R)-domain, and gated by ATP binding/hydrolysis at its nucleotides binding domains [14]. Thus, inhibition of cellular protein phosphatases can increase the steady-state phosphorylation level of CFTR and thereby increases the activity of the channel [19, 31]. However, many well-characterized CFTR activators, including isoflavonoids [21], benzimidazolone compounds [2, 16, 23], and benzoquinolizinium compounds [3, 15] seem to potentiate CFTR activity by directly binding to the channel protein [4, 10, 13, 29, 30].
Although 9-AC inhibits Mg2+-sensitive protein phosphatase isolated from cardiac myocytes [35], whether this is the sole mechanism for the potentiation effect of 9-AC on CFTR remains unclear. Considering the fact that many structurally divergent compounds can affect CFTR gating independently of phosphorylation/dephosphorylation, we set out to test the hypothesis that 9-AC directly modulates CFTR gating. In this paper, we first confirmed the dual effects of 9-AC on CFTR using human epithelial CFTR exogenously expressed in NIH3T3 and CHO cells and then focused on the mechanisms of CFTR potentiation by 9-AC mainly using single-channel recording techniques.
Materials and methods
Cell culture
NIH3T3 cells stably expressing wild-type CFTR (NIH3T3/CFTR) or K1250A-CFTR mutant channels were grown as previously described [32] at 37°C and 5% CO2 in DMEM supplemented with 10% FBS. CHO cells were maintained as described in Powe et al. [24]. For whole-cell experiments, cell suspensions were prepared with trypsinization (0.25% trypsin and 1 mM EGTA in PBS). For excised inside-out experiments, cells were grown on small glass chips in a 35-mm Petri dish 1–2 days prior to use.
Construction of ΔR-CFTR mutant
The plasmids, pGEMHE-1–633 and pGEMHE-837–1480 [8], were gifts from Dr. David Gadsby’s laboratory (Rockefeller University, New York, N.Y., USA). We obtained the pBudCE4.1 CFTR1–633 by subcloning the 2-kb PstI-XhoI fragment from pGEMHE1–633 subcloned into PstI and SalI sites of pBudCE4.1 expression vector (Invitrogen, Carlsbad, Calif., USA). The 1.9-kb KpnI-XhoI fragment from pGEMHE837–1480 was then ligated to KpnI and XhoI sites of pBudCE4.1 CFTR1–633. This final construct, pBudCE4.1 ΔR-CFTR, allows, in a single plasmid, the expression of the N-terminal half of CFTR (amino acids 1–633) under CMV promoter and the expression of the C-terminal half of CFTR (amino acids 837–1480) under human elongation factor 1α (EF1α) promoter. All constructs were confirmed by automated sequencing (DNA Core, University of Missouri, Columbia, Mo., USA).
Transient expression of ΔR-CFTR
To transiently express ΔR-CFTR, CHO cells were grown in 35-mm tissue culture dishes with or without glass chips 1 day prior to transfection. The plasmid pBudCE4.1 split ΔR-CFTR was co-transfected with pEGFP-C3 (Clontech, Palo Alto, Calif., USA) encoding green fluorescent protein using SuperFect transfection reagent (Qiagen, Valencia, Calif., USA) according to the manufacturer’s protocols. The Po of the ΔR-CFTR expressed in CHO cells is consistently higher than that reported by Csanády et al. [8]. Perhaps different expression systems could account for this discrepancy.
Patch-clamp experiments
Pipette electrodes were made from Corning 7056 glass capillaries (Warner Instruments, Hammed, Conn., USA). The pipette resistance was ~3 MΩ in the bath solution. For whole-cell experiments, the membrane potential was held with an EPC9 or 10 amplifier (HEKA, Lambrecht/Pfalz, Germany) at 0 mV, following break-in with suction. Repetitive ramp voltages (±100 mV, 2 s in duration, every 6 or 10 s) were generated with Pulse software (HEKA) to create I-V relationships. Currents traces were filtered at 1 kHz with a built-in four-pole Bessel filter and then digitized at 2 kHz. The currents were recorded at room temperature (~25°C). The pipette solution contained (in mM): 10 EGTA, 121 TEA-Cl, 10 MgATP, 2 MgCl2, 5.5 glucose, and 10 HEPES (pH 7.4 with CsOH). The bath solution contained (in mM): 119 NaCl, 2 MgCl2,1 CaCl2, 5 glucose, and 5 HEPES (pH 7.4 with NaOH). Sucrose (20 mM) was added to the bath solution to prevent activation of swelling-induced currents.
Single-channel CFTR currents were recorded at room temperature (~25°C) with an EPC10 patch-clamp amplifier. Data were filtered at 100 Hz with an eight-pole Bessel filter (Warner Instruments) and captured onto a hard disk at 500 Hz. To obtain recordings with a high signal/noise ratio, we discarded patches with a seal resistance <20 GΩ. The pipette solution contained (in mM): 140 N-methyl-d-glucamine chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2, and 5 HEPES (pH 7.4 with NMDG). After the membrane patch was excised into an inside-out mode, the pipette was continuously perfused (~0.8 ml/minute) with a solution containing (in mM): 150 NMDG-Cl, 2 MgCl2, and 5 Trizma base (pH 7.4 with NMDG). PKA and/or 1 mM Mg-ATP were applied to activate wild-type CFTR or ΔR-CFTR activity (see Results).
Reagents
Forskolin, purchased from Alexis (Lausen, Switzerland), was stored as 20 mM stock in DMSO at 4°C. Anthracene-9-carboxylic acid was purchased from Aldrich (Milwaukee, Wis., USA) and stored as 1 M stock in DMSO at −20°C. 9-Anthracene-methanol (9-AM) was also purchased from Aldrich and stored as 100 mM stock in DMSO at −20°C. Mg-ATP was purchased from Sigma (St Louis, Mo., USA) and stored as 250 mM stock in H2O at −20°C.
Data analysis
Whole-cell and part of the single-channel analysis was done with Igor software (Wavemetrics, Lake Oswego, Ore., USA). For single-channel kinetic analysis, we used the program suite developed by Csanády [7]. Briefly, current traces were baseline corrected, idealized and fitted to a three-state model:
where O and C are open and closed states, respectively; B is a blocked state induced by an intrinsic blocker [34]; and rCO, rOC, rOB, and rBO are corresponding rate constants. Mean interburst, burst durations, and channel open probability (Po) were calculated as τib = 1/rCO, τb = (1/rOC) (1 + rOB/rBO), Po = 1/(1 + rOC/rCO + rOB/rBO), respectively. All values are presented as mean±SEM. Student’s t-test was performed with Sigmaplot (SPSS Science, Chicago, Ill., USA). P<0.05 was considered significant.
Results
Effects of 9-AC on whole-cell CFTR channel currents
The effects of 9-AC on wild-type CFTR were examined first with the whole-cell patch-clamp technique. Figure 1A shows a continuous whole-cell current trace from an NIH3T3 cell stably expressing wild-type CFTR. The basal conductance in the absence of forskolin is minimal (a) and the application of forskolin elicits a conductance with a linear I-V relationship (b–a). 9-AC, added in the presence of forskolin, increases the whole-cell CFTR currents at positive membrane potentials, but inhibits inward currents at a pipette potential of −100 mV (c–a). These dual effects of 9-AC are very similar to those reported previously on cardiac CFTR Cl− channels [33].
Fig. 1A–D.
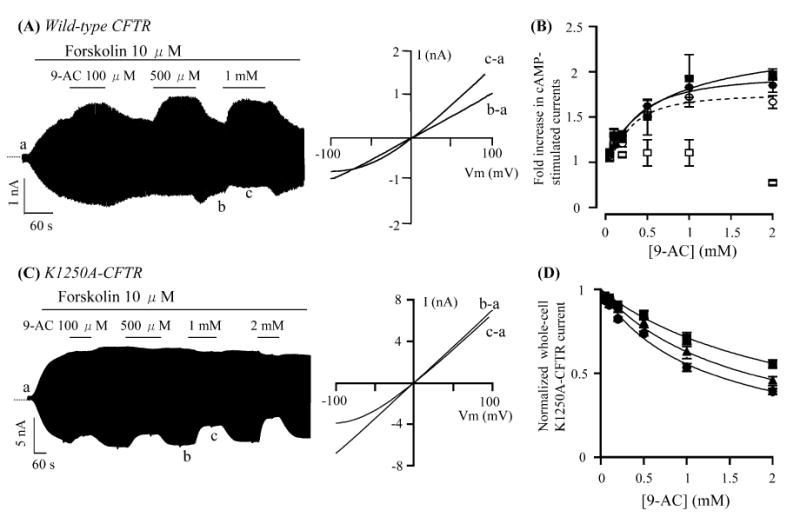
Effects of anthracene-9-carboxylic acid (9-AC) on whole-cell cystic fibrosis transmembrane conductance regulator (CFTR) currents. A A whole-cell wild-type CFTR current trace in the presence of 10 μM forskolin and various concentrations of 9-AC. I-V relationships shows net CFTR currents in different conditions as marked in the left panel. B Dose-response relationships of 9-AC potentiation effects. Net potentiation effects in wild-type CFTR were calculated by correcting for the inhibitory effects obtained from recordings of 9-AC on K1250A-CFTR currents. ycomp = yraw/(1 − x/100), where ycomp, is the compensated value; yraw, the measured fold increase; x, the percentage inhibition of whole-cell K1250A-CFTR currents with 1 mM 9-AC at a pipette potential (Vp) of +100 mV or −100 mV. Open circles: yraw for 1 mM 9-AC at +100 mV; filled circles: ycomp at +100 mV; open squares: yraw at −100 mV; filled squares: ycomp at −100 mV. C A whole-cell K1250A-CFTR current trace. I-V relationships show net K1250A-CFTR currents in the presence or absence of 9-AC. D Dose-response relationships of the inhibitory effect on K1250A-CFTR currents at three different voltages. Filled squares: −60 mV; filled triangles: −80 mV; filled circles: −100 mV. All values are represented by mean±SEM (n=3). Lines were fitted using the Hill equation: y=1+(ymax − 1)/[1+(Kd/x)n] or y=1/[1+(x/Ki)n], where y is normalized current; ymax the maximal response; x the concentration of 9-AC; Kd the half-maximal concentration of potentiation; Ki the half-maximal concentration of inhibition; and n is the Hill coefficient
The whole-cell CFTR currents in the presence of different concentrations of 9-AC at a positive and negative pipette potentials +100 mV and −100 mV were normalized to the current level with 10 μM forskolin and plotted against 9-AC concentrations (open symbols in Fig. 1B). The enhancement effects were underestimated because of the inhibitory effect. Since K1250A-CFTR channels exhibit a long-lived locked-open state (Po > 0.85 [17, 24, 34], cf. [5, 26]), we reason that 9-AC may show a negligible potentiation effect on this mutant and thus the inhibitory effect can be isolated and quantified. Figure 1C shows the whole-cell current trace from an NIH3T3 cell stably expressing K1250A-CFTR. Indeed, 9-AC only exerts a voltage-dependent inhibitory effect on this mutant. Figure 1D shows the dose-response relationships of this inhibitory effect of 9-AC on K1250A-CFTR at three different membrane potentials (−60, −80, and −100 mV). A fit to the Hill equation yielded Ki values of 2.55 mM, 1.70 mM, and 1.26 mM, and a Hill coefficient of 0.98, 0.98, and 0.95 (at −60, −80, and −100 mV, respectively).
Once the inhibitory effect of 9-AC was quantified, we used the resulting parameters to correct the underestimated enhancement effects. Compensated values for potentiation at +100 mV and −100 mV nearly overlap (closed symbols in Fig. 1B), indicating that, unlike the inhibitory effect, the potentiation effect is voltage-independent. It is also interesting to note that the Kd for the potentiation effect (compensated Kd: 291 μM at 100 mV) is lower than those for the inhibitory effect, suggesting the presence of two distinct binding sites. We now focus on exploring the mechanisms of potentiation.
Effects of 9-AM on wild-type CFTR
The voltage-dependent inhibition by 9-AC suggests that the negative charge moiety is important for binding of 9-AC to the inhibitory site. If so, elimination of the negative charge of 9-AC may abolish its inhibitory effects. Indeed, 9-AM, which has an electrically neutral side chain, no longer inhibits the CFTR currents (Fig. 2A). However, the potentiation effect is still observed (Fig. 2B). Maximally dissolvable concentration of 9-AM (100 μM) enhanced CFTR currents by 1.25±0.07 fold (n=4), a value very close to the potentiation by 100 μM 9-AC (see dose-response relationships in Fig. 1B). These results indicate that the negative charge is not required for the potentiation effect and the potentiation site is chemically distinct from the inhibitory site.
Fig. 2A–D.
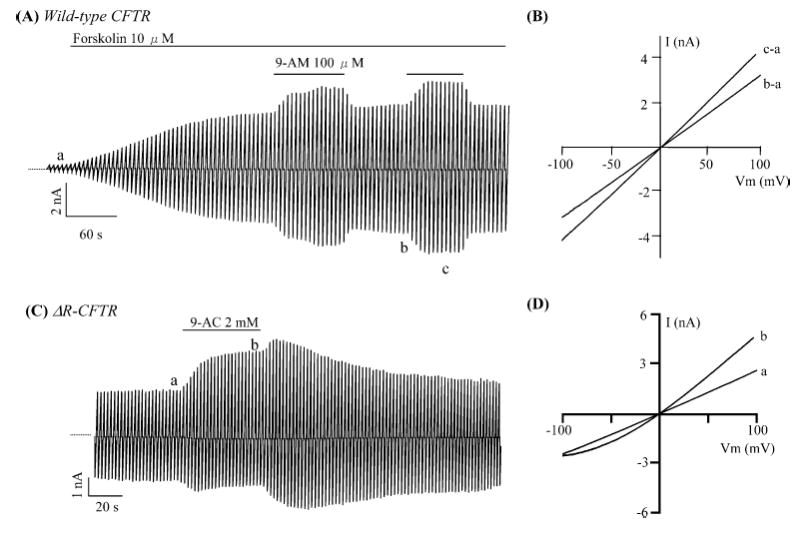
Effects of 9-anthracene compounds on CFTR activity. A Effects of 9-anthracene methanol (9-AM) on whole-cell cAMP-stimulated wild-type CFTR currents (n=4). B I-V relationships in different conditions as marked in A. C Effects of 9-AC on whole-cell ΔR-CFTR currents (n=5). Notice that basal currents were observed in the absence of cAMP agonists. D I-V relationships in different conditions as indicated in C
Effects of 9-AC on constitutively active CFTR mutant
It has been reported that several lipophilic CFTR activators such as genistein and MPB compounds potentiate CFTR currents by phosphorylation-independent mechanisms [10, 29]. The R-domain is the main regulatory domain for CFTR channel activity via phosphorylation/dephosphorylation process [14]. We next tested 9-AC on ΔR-CFTR, a CFTR construct whose regulatory domain (amino acids 634–836) was completely removed, and therefore is constitutively active [8]. Figure 2C shows significant basal Cl− conductance from a CHO cell transiently expressing ΔR-CFTR. This conductance is not affected by the addition of forskolin or CPT-cAMP (data not shown). However, 9-AC exerts a similar potentiation effect as that on wild-type CFTR as seen with the outwardly rectified I-V curve in the presence of 9-AC (Fig. 2D).
Effects of 9-AC on CFTR gating in excised inside-out patches
Next we examined effects of 9-AC on CFTR gating in excised inside-out patches. Once wild-type CFTR currents were activated by exogenous PKA (25 U/ml) and ATP (1 mM), the channel activity is well maintained in the presence of ATP alone, e.g., [32]. Subsequent application of 9-AC altered the CFTR gating both at positive (+50 mV, Fig. 3A) and negative membrane potential (−50 mV, Fig. 3B). In the presence of 9-AC, the open channel noise is higher and the single-channel current amplitudes are smaller at −50 mV. This inhibition, typical of a fast flickering channel block, is voltage dependent (Fig. 3C). Figure 3D shows that the fold increases of NPo at +50 mV and −50 mV are very similar, again demonstrating the voltage-independent nature of the potentiation site. Thus, these results firmly establish that 9-AC acts on CFTR by interacting with two different binding sites.
Fig. 3A–D.
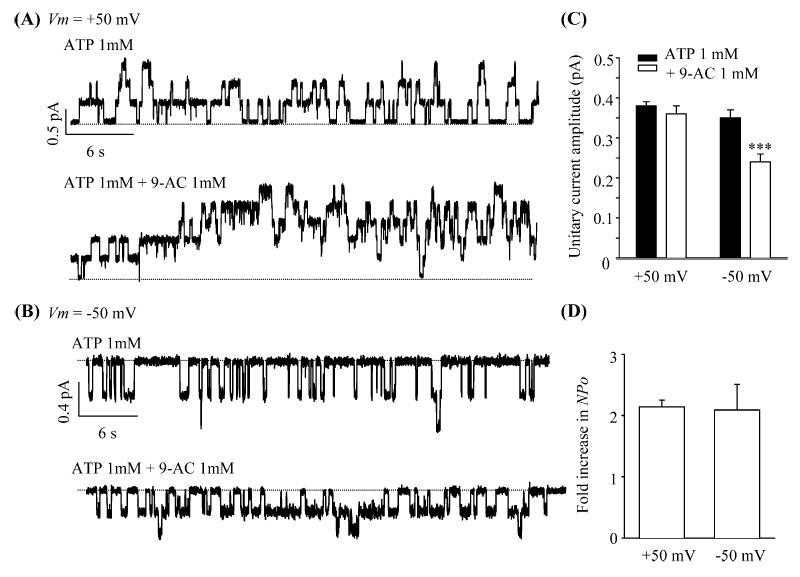
Direct effects of 9-AC on CFTR gating. Single-channel traces of wild-type CFTR in excised inside-out patches in the presence or absence of 9-AC at two different membrane potentials: +50 mV (A) and −50 mV (B). C Summary of unitary current amplitudes. D Fold increase in NPo at two different potentials. NPo was calculated by dividing the steady-state mean currents with unitary current amplitudes. All values are mean±SEM (n=5). ***P<0.001 versus ATP 1 mM
Effects of 9-AC on single-channel kinetics of ΔR-CFTR
To further quantify the effects of 9-AC on CFTR gating, we recorded single-channel current of ΔR-CFTR in excised inside-out patches. ΔR-CFTR was chosen because it is technically easier to obtained patches containing few channels (maximal current steps ≤ 2) and this channel is resistant to run-down by membrane-associated protein phosphatases. The current was recorded at +50 mV to ensure a high signal/noise ratio, an essential requirement for rigorous kinetic analysis. Figure 4A shows a continuous current trace of ΔR-CFTR in an excised inside-out patch. The addition of 1 mM 9-AC slightly decreases the single-channel amplitude (also see Fig. 4B) but significantly increases the Po of the CFTR channel. As summarized in Fig. 4C, 9-AC (1 mM) increased Po by prolonging the mean burst durations (τb) and shortening the mean interburst durations (τib) significantly.
Fig. 4A–C.
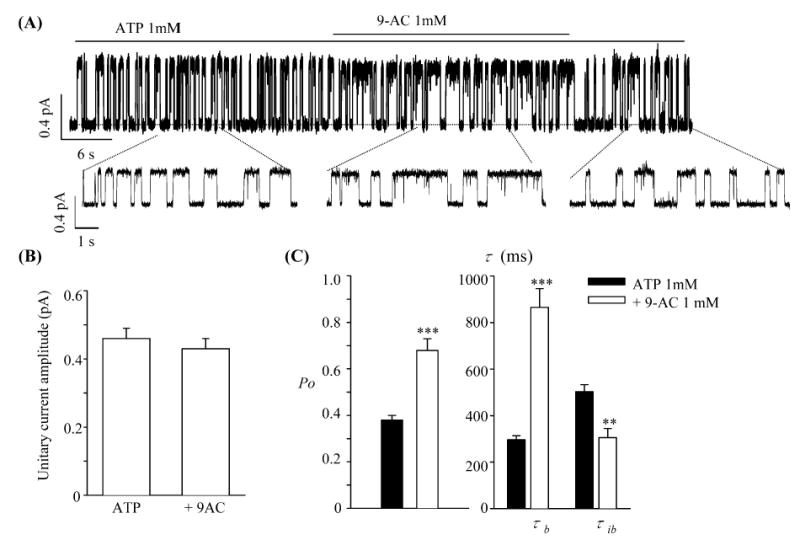
Effects of 9-AC on single-channel kinetics of ΔR-CFTR. A A single-channel ΔR-CFTR current trace in an excised inside-out patch (Vm=50 mV). B Summary of unitary current amplitudes in the presence or absence of 9-AC. C Summary of single-channel kinetics parameters. All values are mean±SEM (n=6). **P<0.01, ***P<0.001 versus ATP 1 mM
Discussion
A Cl− channel blocker, 9-AC, has been previously reported to have a voltage-independent potentiation effect as well as a voltage-dependent inhibitory effect on guinea pig cardiac CFTR channels using whole-cell recordings [33]. In this study, we focused on the mechanism of the potentiation effect of 9-AC on human CFTR channels exogenously expressed in NIH3T3 or CHO cells mainly using single-channel recordings. We largely confirmed the results previously reported by Zhou et al. [33], but also obtained several original and some different results from those in their reports.
Are protein phosphatases involved in 9-AC potentiation?
PKA-dependent phosphorylation of consensus serine/threonine residues in the R-domain is a prerequisite to activate CFTR [14]. Therefore, inhibiting protein phosphatases is a potential mechanism for enhancing CFTR activity. Several different types of phosphatases have been implicated in dephosphorylating CFTR. Luo et al. [22] showed that phosphatase 2C can deactivate CFTR in excised patches. Coimmunoprecipitation experiments suggest that phosphatase 2C and CFTR form a stable complex to facilitate regulation of the channel [36]. Travis et al. [28] indicated that phosphatase 2C reduced CFTR activity in epithelia by increasing inactivation rate. Our previous studies showed that okadaic acid and calyculin A, both inhibitors of phosphatase 1 and 2A, enhance cAMP-dependent whole-cell CFTR currents in guinea pig ventricular myocytes or cell-attached CFTR expressed in insect Hi-5 cells [19, 31]. Inhibition of protein phosphatase may also account for the pharmacological effects of other CFTR activators, such as deltamethrin [12] and bromotetramisole [23].
Zhou et al. [35] showed that the magnitude of cAMP-activated cardiac whole-cell CFTR conductance is inversely proportional to intracellular free [Mg2+]. Since the potentiation effect of low [Mg2+] mimics that of 9-AC, they propose that 9-AC enhances CFTR activity by inhibiting Mg2+-sensitive protein phosphatase(s). By measuring phosphatase activity in the presence of various [Mg2+], they also showed that 9-AC inhibits phosphatase activity in a Mg2+-dependent manner.
In this paper, we have demonstrated that 9-AC can affect ATP-dependent gating of wild-type CFTR in excised inside-out membrane patches. Similar potentiation of CFTR gating by 9-AC was also observed for ΔR-CFTR, a construct whose R-domain is completely removed. Although removal of the R-domain eliminates most of the consensus sites for PKA- and PKC-dependent phosphorylation, CFTR protein is known to possess potential phosphorylation sites outside the R-domain [10, 14]. Therefore, our ΔR-CFTR experiments can not completely rule out the possibility of involvement of phosphorylation/dephosphorylation in 9-AC action in whole-cell recordings. However, it is worth noting that ΔR-CFTR expressed in our systems using CHO cells does not respond to cAMP agonists in whole-cell and cell-attached modes, whereas it has been reported that ΔR-CFTR expressed in oocytes can still be enhanced by cAMP stimulation [8]. Exogenous PKA also fails to increase ATP-dependent ΔR-CFTR activity in excised inside-out patches (T. Ai et al., unpublished observations). Moreover the magnitude of Po increase by 9-AC for wild-type and ΔR-CFTR activity in excised inside-out patches is very similar to those obtained in the whole-cell configuration. Thus, while we cannot rule out the possibility of additional involvement of protein phosphatases in 9-AC potentiation, the simplest explanation for these results is that 9-AC, like numerous other CFTR activators characterized previously (e.g., isoflavonoids, benzimidazolone compounds), modulates CFTR gating independently of phosphorylation/dephosphorylation.
Our proposition that 9-AC affects CFTR seems to contradict the conclusion reached by Zhou et al. [33]. It should be noted, however, that the experiments were performed on CFTRs expressed in different systems. We obtained a Kd value of ~290 μM for the potentiation effect which was more than tenfold higher than that reported by Zhou et al. (~13 μM) [33]. This suggests that the mechanisms underlying 9-AC-induced potentiation in guinea pig cardiac myocytes might be different from what we report here.
How does 9-AC modify CFTR gating?
Zhou et al. [33] previously reported the existence of two different effects of 9-AC: one is an extracellular inhibitory effect with a low affinity and another is an intracellular potentiation effect with a high affinity. Our data also clearly demonstrated that 9-AC acts on two different binding sites for its potentiation and inhibitory effects. This conclusion is supported by several observations: (1) the apparent affinity for the potentiation effect is higher than that of the inhibitory effect; (2) while the inhibition is voltage-dependent, the potentiation is not; and (3) removing the negative charge in 9-AC completely abolishes the block but does not affect the potentiation effect.
It is also noteworthy that 9-AC blocks CFTR channels from the cytoplasmic side of the membrane in our experiments (Fig. 3), whereas Zhou et al. [33] did not observe an inhibitory effect upon cytoplasmic application of 9-AC and concluded that 9-AC blocks CFTR only from the extracellular side. The 5.3% and 31.4% decreases of the single-channel amplitude at +50 and −50 mV, respectively (Fig. 3C), are consistent with a cytoplasmic-accessible binding site ~50% across the field. This is perhaps the same binding site thoroughly characterized for numerous organic anion blockers (reviewed by Hwang and Sheppard [18]).
The exact molecular nature of the binding site for potentiation effect is unclear. It is, however, interesting to note that all the CFTR gating modifiers (including genistein and capsaicin ) we have characterized so far show very similar actions on the single-channel kinetics. They all increase the Po by shortening the closed time and prolonging the open time [1, 2, 20, 29]. These studies also indicate that the binding site is not located in the R-domain or at the interface between the R-domain and other domains. As proposed previously [4, 29], the binding site might be in the nucleotide binding domains for their obvious roles in CFTR gating. The structural simplicity of anthracene compounds makes them useful tools for future explanation of the molecular basis of gating modification.
Acknowledgments
The authors are grateful to Shenghui Hu for his technical assistance. This work was supported by the National Institutes of Health (TCH, DK55835, HL53445). Dr. Ai is a recipient of Postdoctoral Fellowship from the American Heart Association, Heartland Affiliate. Dr. Bompadre is the recipient of a NRSA from the National Institutes of Health (DK062565). Dr. Sohma is the recipient of a grant from the Japan Society for the Promotion of Science (15590196).
References
- 1.Ai T, Bompadre SG, Wang X, Hu S, Li M, Hwang TC. Capsaicin potentiates wild-type and mutant CFTR chloride channel currents. Mol Pharmacol. 2004;65:1415–1426. doi: 10.1124/mol.65.6.1415. [DOI] [PubMed] [Google Scholar]
- 2.Al-Nakkash L, Hu S, Li M, Hwang TC. A common mechanism for cystic fibrosis transmembrane conductance regulator protein activation by genistein and benzimidazolone analogs. J Pharmacol Exp Ther. 2001;296:464–472. [PubMed] [Google Scholar]
- 3.Becq F, Mettey Y, Gray MA, Galietta LJ, Dormer RL, Merten M, Metaye T, Chappe V, Marvingt-Mounir C, Zegarra-Moran O, Tarran R, Bulteau L, Derand R, Pereira MM, McPherson MA, Rogier C, Joffre M, Argent BE, Sarrouilhe D, Kammouni W, Figarella C, Verrier B, Gola M, Vierfond JM. Development of substituted benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J Biol Chem. 1999;274:27415–27425. doi: 10.1074/jbc.274.39.27415. [DOI] [PubMed] [Google Scholar]
- 4.Cai Z, Sheppard DN. Phloxine B interacts with the cystic fibrosis transmembrane conductance regulator at multiple sites to modulate channel activity. J Biol Chem. 2002;277:19546–19553. doi: 10.1074/jbc.M108023200. [DOI] [PubMed] [Google Scholar]
- 5.Carson MR, Travis SM, Welsh MJ. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J Biol Chem. 1995;270:1711–1717. doi: 10.1074/jbc.270.4.1711. [DOI] [PubMed] [Google Scholar]
- 6.Clemo HF, Stambler BS, Baumgarten CM. Persistent activation of a swelling-activated cation current in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res. 1998;83:147–157. doi: 10.1161/01.res.83.2.147. [DOI] [PubMed] [Google Scholar]
- 7.Csanády L. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys J. 2000;78:785–799. doi: 10.1016/S0006-3495(00)76636-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Csanády L, Chan KW, Seto-Young D, Kopsco DC, Nairn AC, Gadsby DC. Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J Gen Physiol. 2000;116:477–500. doi: 10.1085/jgp.116.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahan D, Evagelidis A, Hanrahan JW, Hinkson DA, Jia Y, Luo J, Zhu T. Regulation of the CFTR channel by phosphorylation. Pflugers Arch. 2001;443:S92–S96. doi: 10.1007/s004240100652. [DOI] [PubMed] [Google Scholar]
- 10.Derand R, Bulteau-Pignoux L, Mettey Y, Zegarra-Moran O, Howell LD, Randak C, Galietta LJ, Cohn JA, Norez C, Romio L, Vierfond JM, Joffre M, Becq F. Activation of G551D CFTR channel with MPB-91: regulation by ATPase activity and phosphorylation. Am J Physiol. 2001;281:C1657–C1666. doi: 10.1152/ajpcell.2001.281.5.C1657. [DOI] [PubMed] [Google Scholar]
- 11.Estevez R, Schroeder BC, Accardi A, Jentsch TJ, Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 2003;38:47–59. doi: 10.1016/s0896-6273(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 12.Fischer H, Illek B, Machen TE. Regulation of CFTR by protein phosphatase 2B and protein kinase C. Pflugers Arch. 1998;436:175–181. doi: 10.1007/s004240050620. [DOI] [PubMed] [Google Scholar]
- 13.French PJ, Bijman J, Bot AG, Boomaars WE, Scholte BJ, de Jonge HR. Genistein activates CFTR Cl− channels via a tyrosine kinase- and protein phosphatase-independent mechanism. Am J Physiol. 1997;273:C747–C753. doi: 10.1152/ajpcell.1997.273.2.C747. [DOI] [PubMed] [Google Scholar]
- 14.Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- 15.Galietta LJ, Springsteel MF, Eda M, Niedzinski EJ, By K, Haddadin MJ, Kurth MJ, Nantz MH, Verkman AS. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem. 2001;276:19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- 16.Gribkoff VK, Champigny G, Barbry P, Dworetzky SI, Meanwell NA, Lazdunski M. The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J Biol Chem. 1994;269:10983–10986. [PubMed] [Google Scholar]
- 17.Gunderson KL, Kopito RR. Conformational states of CFTR associated with channel gating: the role of ATP binding and hydrolysis. Cell. 1995;82:231–239. doi: 10.1016/0092-8674(95)90310-0. [DOI] [PubMed] [Google Scholar]
- 18.Hwang TC, Sheppard DN. Molecular pharmacology of the CFTR Cl− channel. Trends Pharmacol Sci. 1999;20:448–453. doi: 10.1016/s0165-6147(99)01386-3. [DOI] [PubMed] [Google Scholar]
- 19.Hwang TC, Horie M, Gadsby DC. Functionally distinct phospho-forms underlie incremental activation of protein kinase-regulated Cl− conductance in mammalian heart. J Gen Physiol. 1993;101:629–650. doi: 10.1085/jgp.101.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang TC, Wang F, Yang IC, Reenstra WW. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am J Physiol. 1997;273:C988–C998. doi: 10.1152/ajpcell.1997.273.3.C988. [DOI] [PubMed] [Google Scholar]
- 21.Illek B, Fischer H, Santos GF, Widdicombe JH, Machen TE, Reenstra WW. cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am J Physiol. 1995;268:C886–C893. doi: 10.1152/ajpcell.1995.268.4.C886. [DOI] [PubMed] [Google Scholar]
- 22.Luo J, Pato MD, Riordan JR, Hanrahan JW. Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am J Physiol. 1998;274:C1397–C1410. doi: 10.1152/ajpcell.1998.274.5.C1397. [DOI] [PubMed] [Google Scholar]
- 23.Luo J, Zhu T, Evagelidis A, Pato MD, Hanrahan JW. Role of protein phosphatases in the activation of CFTR (ABCC7) by genistein and bromotetramisole. Am J Physiol. 2000;279:C108–C119. doi: 10.1152/ajpcell.2000.279.1.C108. [DOI] [PubMed] [Google Scholar]
- 24.Powe A, AL-Nakkash A, Li M, Hwang T-C. Mutations of the Walker A lysine 464 in CFTR reveal functional interaction between its two nucleotide binding domains. J Physiol (Lond) 2002;539:333–346. doi: 10.1113/jphysiol.2001.013162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qu Z, Wei RW, Hartzell HC. Characterization of Ca2+-activated Cl− currents in mouse kidney inner medullary collecting duct cells. Am J Physiol. 2003;285:F326–F335. doi: 10.1152/ajprenal.00034.2003. [DOI] [PubMed] [Google Scholar]
- 26.Ramjeesingh M, Li C, Garemi E, Galley KA, Huan LJ, Wang Y, Bear CE. Walker mutations reveal loose relationship between catalytic and channel-gating activities of purified CFTR (cystic fibrosis transmembrane conductance regulator) Biochem. 1999;38:1463–1468. doi: 10.1021/bi982243y. [DOI] [PubMed] [Google Scholar]
- 27.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 28.Travis SM, Berger HA, Welsh MJ. Protein phosphatase 2C dephosphorylates and inactivates cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci U S A. 1997;94:11055–11060. doi: 10.1073/pnas.94.20.11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Zeltwanger S, Yang IC, Nairn AC, Hwang TC. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating. Evidence for two binding sites with opposite effects. J Gen Physiol. 1998;111:477–490. doi: 10.1085/jgp.111.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinreich F, Wood PG, Riordan JR, Nagel G. Direct action of genistein on CFTR. Pflugers Arch. 1997;434:484–491. doi: 10.1007/s004240050424. [DOI] [PubMed] [Google Scholar]
- 31.Yang IC, Cheng TH, Wang F, Price EM, Hwang TC. Modulation of CFTR chloride channels by calyculin A and genistein. Am J Physiol. 1997;272:C142–C155. doi: 10.1152/ajpcell.1997.272.1.C142. [DOI] [PubMed] [Google Scholar]
- 32.Zeltwanger S, Wang F, Wang GT, Gillis KD, Hwang TC. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J Gen Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou SS, Takai A, Tominaga M, Okada Y. Phosphatase-mediated enhancement of cardiac cAMP-activated Cl− conductance by a Cl− channel blocker, anthracene-9-carboxylate. Circ Res. 1997;81:219–228. doi: 10.1161/01.res.81.2.219. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Z, Hu S, Hwang TC. Voltage-dependent flickery block of an open cystic fibrosis transmembrane conductance regulator (CFTR) channel pore. J Physiol (Lond) 2001;532:435–448. doi: 10.1111/j.1469-7793.2001.0435f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou SS, Takai A, Okada Y. Regulation of cardiac CFTR Cl(−) channel activity by a Mg(2+)-dependent protein phosphatase. Pflugers Arch. 2002;444:327–334. doi: 10.1007/s00424-002-0822-0. [DOI] [PubMed] [Google Scholar]
- 36.Zhu T, Dahan D, Evagelidis A, Zheng S, Luo J, Hanrahan JW. Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. J Biol Chem. 1999;274:29102–29107. doi: 10.1074/jbc.274.41.29102. [DOI] [PubMed] [Google Scholar]