

Abstract
Research on fluorescent semiconductor nanocrystals (also known as quantum dots or qdots) has evolved over the past two decades from electronic materials science to biological applications. We review current approaches to the synthesis, solubilization, and functionalization of qdots and their applications to cell and animal biology. Recent examples of their experimental use include the observation of diffusion of individual glycine receptors in living neurons and the identification of lymph nodes in live animals by near-infrared emission during surgery. The new generations of qdots have far-reaching potential for the study of intracellular processes at the single-molecule level, high-resolution cellular imaging, long-term in vivo observation of cell trafficking, tumor targeting, and diagnostics.
The improved resolution, sensitivity, and versatility of fluorescence microscopy (1) and the development of fluorescent sensors and labeling of proteins in live cells have yielded a clearer understanding of the dynamics of intracellular networks, signal transduction, and cell-cell interaction (2). However, the monumental task of elucidating the role of all proteins and their mutual interactions in a given organism—that is, completing its proteome and its interactome—will require new approaches and tools. Single-molecule methods (3) will play a major role in this endeavor, but the complex milieu encountered inside live cells requires substantial adaptations of current in vitro techniques. Two problems associated with fluorescence microscopy—cell autofluorescence in the visible spectrum (which can mask signals from labeled molecules) and the requirement of long observation times—have created a need for new probes that emit in the near-infrared (NIR) region (wavelength >700 nm) and are more photo-stable than current organic fluorophores.
Properties of Qdots
Several characteristics distinguish qdots from the commonly used fluorophores. [A more detailed description of qdot properties can be found in (4) and references listed in the online supplement.] Colloidal semiconductor quantum dots are single crystals a few nanometers in diameter whose size and shape can be precisely controlled by the duration, temperature, and ligand molecules used in the synthesis (4). This process yields qdots that have composition- and size-dependent absorption and emission (Fig. 1A). Absorption of a photon with energy above the semiconductor band gap energy results in the creation of an electron-hole pair (or exciton). The absorption has an increased probability at higher energies (i.e., shorter wavelengths) and results in a broadband absorption spectrum, in marked contrast to standard fluorophores (Fig. 1B). For nanocrystals smaller than the so-called Bohr exciton radius (a few nanometers), energy levels are quantized, with values directly related to the qdot size (an effect called quantum confinement, hence the name ‘‘quantum dots’’) (4). The radiative recombination of an exciton [characterized by a long lifetime, >10 ns (5)] leads to the emission of a photon in a narrow, symmetric energy band (Fig. 1), another difference from the red-tailed emission spectra and short lifetimes of most fluorophores. The long fluorescence lifetime of qdots enables the use of time-gated detection (6) to separate their signal from that of shorter lived species (such as background autofluorescence encountered in cells).
Fig. 1.

(A) Emission maxima and sizes of quantum dots of different composition. Quantum dots can be synthesized from various types of semiconductor materials (II-VI: CdS, CdSe, CdTe. . .; III-V: InP, InAs . . .; IV-VI: PbSe. . .) characterized by different bulk band gap energies. The curves represent experimental data from the literature on the dependence of peak emission wavelength on qdot diameter. The range of emission wavelength is 400 to 1350 nm, with size varying from 2 to 9.5 nm (organic passivation/solubilization layer not included). All spectra are typically around 30 to 50 nm (full width at half maximum). Inset: Representative emission spectra for some materials. Data are from (12, 18, 27, 76–82). Data for CdHgTe/ZnS have been extrapolated to the maximum emission wavelength obtained in our group. (B) Absorption (upper curves) and emission (lower curves) spectra of four CdSe/ZnS qdot samples. The blue vertical line indicates the 488-nm line of an argon-ion laser, which can be used to efficiently excite all four types of qdots simultaneously. [Adapted from (28)] (C) Size comparison of qdots and comparable objects. FITC, fluorescein isothiocyanate; GFP, green fluorescent protein; qdot, green (4 nm, top) and red (6.5 nm, bottom) CdSe/ZnS qdot; qrod, rod-shaped qdot (size from Quantum Dot Corp.’s Web site). Three proteins—streptavidin (SAV), maltose binding protein (MBP), and immunoglobulin G (IgG)—have been used for further functionalization of qdots (see text) and add to the final size of the qdot, in conjunction with the solubilization chemistry (Fig. 2).
Surface defects in the crystal structure act as temporary ‘‘traps’’ for the electron or hole, preventing their radiative recombination. The alternation of trapping and untrapping events results in intermittent fluorescence (blinking) visible at the single-molecule level (7) and reduces the overall quantum yield, which is the ratio of emitted to absorbed photons. One way to overcome these problems, and to protect surface atoms from oxidation and other chemical reactions, is to grow a shell of a few atomic layers of a material with a larger band gap on top of the nanocrystal core. This shell can be designed carefully to obtain quantum yields close to 90% (8); this step also enhances qdots’ photostability by several orders of magnitude relative to conventional dyes (9).
Single qdots can be observed and tracked over an extended period of time (up to a few hours) with confocal microscopy (10), total internal reflection microscopy (11, 12), or basic wide-field epifluorescence microscopy (12–14). Single-molecule studies of qdots have revealed phenomena hidden in ensemble measurements, such as blinking (7), spectral jumps (15), or the existence of multiple long fluorescence lifetimes (16). Single-molecule microscopy is possibly one of the most exciting new capabilities offered to the biologist, as discussed below. A related technique, fluorescence correlation spectroscopy, has allowed determination of the brightness per particle and also provides a measurement of the average qdot size (17, 18) (see Fig. 1C for a comparison scale).
Qdots are also excellent probes for two-photon confocal microscopy because they are characterized by a very large absorption cross section (17). Naturally, they can be used simultaneously with standard dyes. In particular, qdots have a largely untapped potential as customizable donors of a fluorescence resonance energy transfer (FRET) pair (19).
Qdot Solubilization and Functionalization
Qdots are mostly synthesized in nonpolar organic solvents. If they are to be solubilized in aqueous buffers, their hydrophobic surface ligands must be replaced by amphiphilic ones. Different qdot solubilization strategies have been devised over the past few years (Fig. 2), including (i) ligand exchange with simple thiol-containing molecules (20, 21) or more sophisticated ones such as oligomeric phosphines (22), dendrons (23), and peptides (24); (ii) encapsulation by a layer of amphiphilic diblock (25) or triblock copolymers (26) or in silica shells (27, 28), phospholipid micelles (29), polymer beads (30), polymer shells (31), or amphiphilic polysaccharides (32); and (iii) combinations of layers of different molecules conferring the required colloidal stability to qdots (9, 33). Recent developments include a promising water-based synthesis method (34, 35) that yields particles that emit from the visible to the NIR spectrum and are intrinsically water-soluble, but the particles have yet to be tested in biological environments.
Fig. 2.

Qdot solubilization and functionalization. (A) Surface chemistries. TOPO (trioctylphosphine oxide)–passivated qdots can be solubilized in aqueous buffer by addition of a layer of amphiphilic molecules containing hydrophilic (w+) and hydrophobic (w–) moieties, or by exchange of TOPO with molecules that have a Zn-coordinating end (usually a thiol group, SH) and a hydrophilic end. Examples of addition include (a) formation of a cross-linked polymer shell (31), (b) coating with a layer of amphiphilic triblock copolymer (26), and (c) encapsulation in phospholipid micelles (29). Examples of exchange include (d) mercaptoacetic acid (MAA) (20), (e) dithiothreitol (DTT) (21), (f) dihydrolipoic acid (DHLA) (33), (g) oligomeric phosphines (22), (h) cross-linked dendrons (23), and (i) peptides (24). The curved arrow indicates sites available for further functionalization. (B) Peptide toolkit. The light blue segment contains cysteines and hydrophobic amino acids ensuring binding to the qdot (24) and is common to all peptides. S, solubilization sequence; P, PEG; B, biotin; R, peptide recognition sequence; Q, quencher; D, DOTA; X, any unspecified peptide-encoded function. Qdot solubilization is obtained by a mixture of S and P. Qdots can be targeted with B, R, or other chemical moieties. Qdot fluorescence can be turned on or off by attaching a Q via a cleavable peptide link. In the presence of the appropriate enzyme, the quencher is separated from the qdot, restoring the photoluminescence and reporting on the enzyme activity. For simultaneous PET and fluorescence imaging, qdots can be rendered radioactive by D chelation of radionuclides; for simultaneous MRI and fluorescence imaging, qdots can be rendered radioactive by D chelation of nuclear spin labels.
Unless used as a nonspecific fluorescent stain, qdots require some sort of biological ‘‘interfacing.’’ For simple applications such as qdot tagging of a target molecule, a single recognition moiety can be grafted to the qdot (e.g., DNA oligonucleotide or aptamer, antibody, etc.) or, simpler yet, used as the qdot solubilization ligand (36). Qdot ligands containing either an amine or a carboxyl group, for instance, offer the possibility of cross-linking molecules containing a thiol group (29, 37, 38) or an N-hydroxysuccinimyl ester moiety (24, 27) by means of standard bioconjugation reactions. Another approach uses electrostatic interactions between qdots and charged adapter molecules, or between qdots and proteins modified to incorporate charged domains (39). These functionalization steps can be repeated to add or change functionality. For instance, streptavidin-coated qdots can be used in combination with biotinylated proteins or antibodies (13, 24, 25, 40, 41). By extension, a generic three-layer approach [using (i) an antibody against a specific target, (ii) a biotinylated secondary antibody against the first, and (iii) a streptavidin-coated qdot] allows qdot labeling of most types of target (13, 25).
The large number (10 to 100) of potential surface attachment groups can be used to ‘‘graft’’ different functionalities to individual qdots, resulting in multipotent probes. For instance, in addition to a recognition moiety, qdots can be equipped with a membrane-crossing or cell-internalization capability, and/or an enzymatic function. The production of biologically synthesized qdots consisting of CdS cores coated by natural peptides (42) led us to investigate the peptide-coating approach for the surface modification of qdots (Fig. 2B). Peptides have the advantage of being easily customized, and with the correct choice of sequence, a single-step surfactant exchange can yield all necessary functions: (i) Protect the core/shell structure and maintain the original qdot photophysics, (ii) solubilize qdots, (iii) provide a biological interface, and (iv) allow the incorporation of multiple functions. The resulting particles have excellent colloidal properties, photophysics, and biocompatibility, and this ‘‘peptide toolkit’’ can easily be tailored to provide additional functionalities. Such functionalities can be improved by molecular evolution, a strategy that has proven extremely powerful for the recognition, synthesis, and self-assembly of nanocrystals (43).
Qdots in Cell Biology
Over the past few years, qdots have been tested in most biotechnological applications that use fluorescence, including DNA array technology, immunofluorescence assays [reviewed in (44)], and cell and animal biology. Some of the early and most successful uses of qdots have been in immunofluorescence labeling of fixed cells and tissues; immuno-staining of membrane proteins (9, 25, 37, 45, 46), microtubules (25), actin (25, 27), and nuclear antigens (25); and fluorescence in situ hybridization on chromosomes (21, 47, 48) or combed DNA (11). Qdots tend to be brighter than dyes because of the compounded effects of extinction coefficients that are an order of magnitude larger than those of most dyes (29, 49), comparable quantum yield, and similar emission saturation levels (18). But their main advantage resides in their resistance to bleaching over long periods of time (minutes to hours), allowing the acquisition of images that are crisp and well contrasted. This increased photostability is especially useful for three-dimensional (3D) optical sectioning, where a major issue is bleaching of fluorophores during acquisition of successive z-sections, which compromises the correct reconstruction of 3D structures. The gain obtained with qdots was illustrated by the high-resolution 3D confocal imaging of an unforeseen domain distribution of band3, a membrane protein involved in anion transport in erythrocytes (45) and that of clusters of overexpressed multidrug P-glycoproteins (Pgp’s) in breast adenocarcinoma cell membranes (9).
The most interesting property of qdots for immunofluorescence is the very small number of qdots necessary to produce a signal. Indeed, several studies have reported flickering of some specimens, a phenomenon due to the blinking of a small number of qdots (20, 47). This demonstrates that single qdots can still be observed in immunocytological conditions, with an ultimate sensitivity limit of one qdot per target molecule. Additionally, qdots are available in a virtually unlimited number of well-separated colors, all excitable by a single wavelength (Fig. 1). Aside from simplifying image acquisition, this property could be used in confocal microscopy to perform nanometer-resolution colocalization of multiple-color individual qdots (10).
Live-cell experiments introduce a few extra levels of complexity, depending on the application: whole-cell labeling, labeling of membrane-bound proteins, and cytoplasmic or nuclear target labeling. Whole-cell labeling is of particular interest for cell or pathogen detection, cell tracking, and cell lineage studies. This can be achieved without any functionalization through microinjection (29), electroporation, or even phagocytosis of qdots (40, 50). Such internalization was observed in phagokinetic human cancer cells (51) plated on a qdot-coated coverslip. Peptide translocation domains or cationic lipids have also been shown to be efficient facilitators of endocytosis, allowing rapid labeling of whole cell populations with specific colors (50). A better specificity and efficiency can be obtained with functionalized qdots. Transferrin was used to facilitate endocytosis by mammalian cells (20), a strategy also used successfully to label pathogenic bacteria and yeast cells (52), whereas Gram-positive bacteria were specifically detected by lectin-coated qdots (52). These and other studies have shown that qdots have a considerable advantage over standard dyes: the possibility of long-term observation with negligible photo-bleaching. In the experiment of Jaiswal et al. (40), Dictyostelium discoideum cells colored according to their starvation time could be tracked individually over several hours, allowing the determination that only starved cells contribute to the formation of aggregation centers characteristic of this species. Dubertret et al. (29) showed that in the Xenopus embryo, cells of the progeny of an injected cell still contain fluorescent qdots after several days of development. In all eukaryotic cell studies, however, qdots were observed to eventually end up in intracellular vesicles that could be identified in some cases as endosomes (40, 53, 54) or lysosomes (55) by organelle-specific labeling, or in a perinuclear region compatible with endosomal/lysosomal localization (29). Real-time and long-term observation of translocation of these vesicles was possible in some cases (40, 53).
Different types of functionalization have also been explored as a way to target qdots to cell surface proteins. Some examples include streptavidin (13, 25), secondary (56) or primary antibodies (40), receptor ligands such as epidermal growth factor (EGF) (46, 57) or serotonin (58), recognition peptides (56), and affinity pairs such as biotin-avidin after engineering of the target protein (24). Streptavidin-qdots were used to detect Her2 cancer markers on the surface of SK-BR-3 human breast cancer cells via a biotinylated secondary antibody to human and a humanized antibody to Her2. A similar strategy was also used in cultured spinal neurons to detect single glycine receptors, which could be successfully tracked with high resolution as they diffused in synaptic and extra-synaptic domains (13). The versatility of streptavidin-qdots makes them attractive, but the cumulative volume of qdot, streptavidin, and the two layers of antibodies may hinder their usefulness in studying the multimerization of receptors. Direct qdot functionalization with a secondary antibody reduces the number of detection layers to two; this method was used to detect the integrin αv subunit in SK-N-SH human neuroblastoma cells (56). Partial clustering of qdots was reported when compared to a direct integrin-recognition peptide qdot functionalization.
Another strategy consists of cross-linking primary antibodies to qdots. This was performed in two different ways in a study targeting Pgp in HeLa cells transiently transfected with a Pgp-encoding plasmid (40). The first approach involved the biotinylation of the Pgp primary antibody, which was subsequently attached to avidin-coated qdots. The second approach involved engineering an adaptor protein with both binding affinity to the Fc region of antibodies and electrostatic interactions with charged qdots (39, 40). Both experiments demonstrated specific surface labeling of the transfected cells and colocalization of the signal with that of an enhanced green fluorescent protein–fused Pgp.
In a further step to reduce the size of qdot probes, researchers have used ligands of surface receptors bound to qdots via a biotin-streptavidin link (46) or by direct cross-linking (57, 58). For instance, EGF-labeled qdots were used to study receptor-mediated signal transduction in different cancer cell lines (46). EGF-qdots bound to cell filopodia erbB1 receptors were observed to undergo a previously undetected retrograde transport (presumably along actin filaments) toward the cell body. Endocytosis of EGF-qdot–targeted erbB1 receptors in vesicles was also observed, followed by a rich dynamic pattern of diffusion, directed linear motion, and vesicular fusion.
Some proteins can be recognized by peptides, so it is attractive to use peptides for qdot functionalization. This strategy has been shown to work in human neuroblastoma with a short engineered peptide directed against integrin (56). This phenomenon was also proven to be relatively common by Akerman et al., who used three different peptides to target qdots to lung endothelial cells, brain endothelial cells, and human breast carcinoma cells in vitro and in vivo (37). In the absence of such peptide sequences or of an identified ligand, the target molecule can be engineered to include a recognizable polypeptide. We recently illustrated this approach with the fusion of an avidin polypeptide chain to the glycosylphosphatidylinositol (GPI)–anchoring sequence of human CD14 receptors (24). Biotinylated peptide–coated qdots were used to label the avidin receptors expressed in the cytoplasmic membrane of HeLa cells. The receptors were observed with single-molecule sensitivity and were tracked for several minutes as they diffused in the membrane of live cells and trafficked in the cytosol (Fig. 3) (12). This approach allowed us to study in detail the relationship between GPI-anchored receptors and lipid rafts in the membrane (12).
Fig. 3.

Single-particle tracking in a live cell. The brightness and photostability of qdots permit single qdot observation over a long period of time. Live HeLa cells stably transfected with a plasmid expressing a chimeric avidin-CD14 receptor (24) were grown on fibronectin-coated glass coverslips, incubated with biotin-qdots (emission 630 nm), and washed with the observation medium. The cells were observed by differential interference contrast (DIC) (A) and epifluorescence (B) on an inverted microscope (Axiovert 100, Zeiss) with a simple Hg lamp and imaged with a cooled monochrome charge-coupled device camera (CoolSnap HQ, Roper Scientific). Single qdots were observed to diffuse at characteristically different rates in different regions of the membrane or inside the cytosol (data not shown). (C) The 1000-step (100 ms/step) trajectory, R(t), of the qdot localized in the region marked in (A) and (B). (D) The corresponding qdot intensity, I(t). The blinking pattern (succession of on and off emission) demonstrates that a single qdot was observed.
In principle, the same concepts could be used to target and label cytoplasmic or nuclear targets. However, qdots need to (i) enter the cell cytoplasm and (ii) reach their target without being trapped in the endocytic pathway (40). A recent study showed that microinjection allows the delivery of qdots functionalized with the appropriate targeting peptide sequence to mitochondria or the cell nucleus (54). All other approaches seem to result in aggregation of qdots in the endosomes, but this is an area of very active research. A second level of difficulty is introduced by the impossibility of washing away probes that are in excess in the cytoplasm, as was possible with outer-membrane labeling experiments.
Qdots in Animal Biology
The long-term stability and brightness of qdots make them ideal candidates for live animal targeting and imaging. Two-photon excitation confocal microscopy was used to image blood vessels in live mice that had received qdots by intravenous injection, showing that higher contrast and imaging depth can be obtained at a lower excitation power than with organic dyes (17). A similar study that focused on long-term imaging of live mice revealed the importance of coating qdots with high molecular weight polyethylene glycol (PEG) molecules to reduce accumulation in the liver and bone marrow (49). After several months, qdots were still visible in the bone marrow and lymph nodes of animals, illustrating the outstanding stability of these probes.
Peptide-qdots were first used to target tissue-specific vascular markers (lung blood vessels and cancer cells) by intravenous injection in live mice (37). Histological sections of different organs after 5 or 20 min of circulation showed that peptide-qdots reach their targets and are internalized by endocytosis in target cells but not in surrounding tissues, probably because of their larger size relative to dye molecules (which would stain surrounding tissues). Live animal imaging of targeted qdot delivery was recently achieved in mice by Gao et al., who intravenously injected PEG-coated qdots functionalized with antibodies to prostate-specific membrane antigen (26). Because the qdots were emitting in the visible spectrum, a spectral demixing algorithm was used to separate tissue autofluorescence (59) from qdot signal in grafted tumors (26). This problem was eliminated altogether by Kim et al., who injected NIR-emitting qdots (850 nm) intradermally into live mice and pigs (60). Qdots rapidly migrated to nearby lymph nodes and could be imaged virtually background-free (Fig. 4C), allowing image-guided resection of a lymph node in a pig. This latter demonstration suggests that IR imaging of qdots could possibly aid surgical procedures in humans.
Fig. 4.

Animal use of qdots. (A and B) microPET and fluorescence imaging of qdots. Qdots having DOTA (a chelator used for radiolabeling) and 600-dalton PEG on their surface were radiolabeled with 64Cu (positron-emitting isotope with half-life of 12.7 hours). These qdots were then injected via the tail vein into nude mice (~80 μCi per animal) and imaged in a small animal scanner. (A) Rapid and marked accumulation of qdots in the liver quickly follows their intravenous injection in normal adult nude mice. This could be avoided by functionalizing qdots with higher molecular weight PEG chains, as other studies have shown (49). (B) Overlay of DIC and fluorescence images of hepatocytes from a mouse shows the accumulation of qdots within liver cells. Scale bar, 20 μm. A further step could involve TEM imaging of the precise localization of qdots in cells, illustrating the potential of qdots as probes at the macro-, micro-, and nanoscales. (C) Surgical use of NIR qdots. A mouse was injected intradermally with 10 pmol of NIR qdots in the left paw, 5 min after reinjection with 1% isosulfan blue and exposure of the actual sentinel lymph node. Left, color video; right, NIR fluorescence image. Isosulfan blue and NIR qdots were localized in the same lymph node (arrows). Copyright 2004 Nature Publishing Group. Reproduced with permission from (60).
Cytotoxicity
Cytotoxicity and the potential interference of qdot labeling with cellular processes are primary issues in any live-cell or animal experiment. These questions are complicated by the variety of published synthesis, solubilization, and functionalization protocols. Most of the reports discussed above did not find adverse effects on cell viability, morphology, function, or development over the duration of the experiments (from several hours to several days) at qdot concentrations optimized for labeling efficiency. At higher concentrations, however, effects on embryo development, for instance, were noticeable (29). The less protected the core or core/shell material is, the faster the appearance of signs of interference with cell viability or function (57), with release of Cd2+ or Se2– ion reported in both core and core-shell qdots (52, 57). Qdots are therefore not completely innocuous, but a safe range likely exists in which they can accomplish their task without major interference with the processes under study. Extensive scrutiny will naturally be needed before qdots can be used in medical procedures.
Perspectives
The current picture of qdot technology is bound to evolve rapidly on many levels. In synthesis, new compositions could entail qdots with properties such as (i) sensitivity to electric or magnetic fields (61, 62); (ii) narrower fluorescence emission and longer lifetimes (using lanthanide-doped qdots); (iii) smaller sizes and extension to the NIR spectrum, as demonstrated by ternary alloys (63); (iv) end-specific functionalizations of nanorod qdots (64); (v) suppression of blinking (14) and quantum yield enhancement (65); and (vi) built-in on-off switches or photoelectric biotransducers. An on-off switch was demonstrated for a maltose sensor (19) and can be obtained by associating the qdot to a fluorescence-quenching molecule that can be separated or cleaved upon binding to the target, or in the presence of a chemical species or enzyme. This would be of particular interest for intracellular applications, in which there is no way to get rid of unbound qdots (unlike outer cell membrane–labeling experiments). As an example of a biotransducer, light-excited qdots could transfer their charge to bound enzymes functioning as electron or hole acceptors, enabling their control by light activation. Reciprocally, qdots could be lit up by electron or hole donor enzymes through chemiluminescence (66). Peptide coating of nano-materials is in this respect a powerful tool for imparting novel functions to the organic-inorganic interface. The simultaneous engineering of the semiconductor’s band gap (by rational design) with the peptide’s redox potential (by molecular evolution) could be used to optimize qdot compositions and peptide sequences for binding and desired optical, electronic, magnetic, and chemical properties. In summary, different shapes, end specificities, and compositions will lead to more complex bioinorganic architectures that could be exploited as an optoelectronic interface to the cellular machinery.
Qdot functionalization is another area where new developments will be needed. Ligands of cellular proteins should be readily cross-linked to qdots by means of standard bioconjugation schemes. However, most of the potential extracellular or intracellular targets do not have any known ligands, or if they do, qdots may interfere with their interaction. The use of antibodies, which has proven successful for surface-bound proteins, may not be practical for intracellular targets because of the size of the qdot-antibody construct and delivery issues (see below). In vivo cross-linking strategies developed for dyes—such as the use of biarsenical ligands targeted against tetracysteine motifs (67), Ni2+–nitrilotriacetic acid moieties targeted against hexahistidine motifs (68), and other similar approaches (69, 70)—may be adapted for qdot functionalization. The development of affinity pairs orthogonal to the biotin-avidin pair, in which one component could be easily attached or fused to the target protein, is a promising approach. Several examples of such pairs developed by molecular evolution have been reported, with fusion peptides ranging from ~200 amino acids [single chain fragment antibody targeted against fluorescein, dissociation constant Kd ~ 50 fM (71)] down to ~30 amino acids [peptide hairpin against Texas Red, Kd ~ 25 pM (72)].
Intracellular delivery remains a challenge, and strategies to escape the endocytosis pathways will be needed. Two recent publications indicate that peptide-coated qdots could have many untapped virtues: (i) 20-nm gold nanoparticles coated with protein transduction domains could enter live cells, and further enter the nucleus with additional adenovirus nuclear localization and integrin binding domains (73); and (ii) after micro-injection into the cytoplasm, PEG-coated qdots with a nuclear or mitochondrial localization sequence were successfully targeted to the nucleus or mitochondria (54). Another interesting model system, based on adenosine triphosphate–triggered release of CdS qdots encapsulated in chaperonin molecules (74), suggests using a ‘‘Trojan horse’’ approach to avoid the cellular defense and recycling mechanisms. Cell-permeable virus capsids, vaults, or other biocapsules could be used to translocate encapsulated qdots in cells, provided that a simple release-on-demand mechanism could be engineered in the vehicle, such as a photolyzable or pH-sensitive lock.
Qdots’ photophysical properties (spectral range, brightness, long lifetimes) and their potential as single-molecule probes may justify the development of new detectors that would have the temporal resolution and sensitivity of avalanche photodiodes and the 2D spatial resolution of cameras for wide-field in vivo studies of protein dynamics and trafficking. Their large size and electronic density may be used for combined fluorescence and static high-resolution imaging techniques such as atomic force microscopy and electron and x-ray microscopy and tomography. Additional contrast mechanisms could be obtained by functionalization of qdots. Figure 4A illustrates this idea with 64Cu radioactive labeling of qdots coated with DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid) functionalized peptides. Micro–positron emission tomography (microPET) shows that qdots injected into the tail vein in mice are rapidly accumulated in the liver. Fluorescence imaging of qdots for tracking combined with the quantitative capability of microPET should therefore permit the elucidation of targeting mechanisms, biodistribution, and dynamics in living animals with high sensitivity. Nuclear spin labels for magnetic resonance imaging (MRI) could also be incorporated into qdot coatings with the use of similar chelating groups. It should therefore be possible to image targeted qdots at all scales, from whole-body down to nanometer resolution, using a single probe.
Beyond biotechnological and cell-imaging applications (Fig. 5A), and provided that cytotoxicity issues can be resolved, one can envision the use of intravenous injection of qdots to home in on and target cellular markers of diseased tissues and organs in the human body. Qdots could then be used as contrast reagents for functional imaging with a combination of MRI, PET, computed tomography, and IR fluorescence imaging (the latter by direct imaging through the epidermis or by a catheter-based confocal fiber microscope). In vivo optical biopsy could confirm the pathology, and therapy could then be performed selectively, locally, and temporally by depositing energy (monochromatic x-rays for k-shell absorption or laser IR radiation) into the targeted qdots, in a manner similar to the recently reported photothermal tumor ablation with gold nano-shells (75). Alternatively, it might be possible to graft therapeutic enzymes to the qdot surface and activate them by light, or produce free radicals (such as singlet oxygen) by optically cycling the qdots.
Fig. 5.
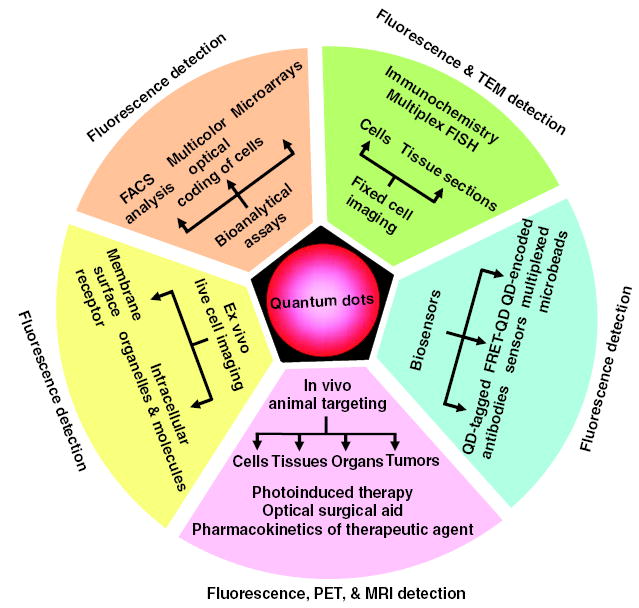
Applications of quantum dots as multimodal contrast agents in bioimaging.
Conclusion
Qdots as biological probes have lived up to the hopes of their initial promoters (20, 27). They will not replace the well-established fluorophores or fluorescent protein-fusion technologies, but will complement them for applications needing better photostability, NIR emission, or single-molecule sensitivity over long time scales. Undoubtedly, biologists will catch on to these exciting developments and will find as yet unforeseen applications for this new toolkit, thus enhancing and complementing their existing arsenal of bioimaging tools.
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/307/5709/538/DC1, References
References
- 1.Michalet X, et al. Annu Rev Biophys Biomol Struct. 2003;32:161. doi: 10.1146/annurev.biophys.32.110601.142525. [DOI] [PubMed] [Google Scholar]
- 2.Special issue on Biological Imaging, Science300, 75 (2003).
- 3.Weiss S. Science. 1999;283:1676. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 4.Alivisatos AP. Science. 1996;271:933. [Google Scholar]
- 5.Efros AL, Rosen M. Annu Rev Mater Sci. 2000;30:475. [Google Scholar]
- 6.Dahan M, et al. Opt Lett. 2001;26:825. doi: 10.1364/ol.26.000825. [DOI] [PubMed] [Google Scholar]
- 7.Nirmal M, et al. Nature. 1996;383:802. [Google Scholar]
- 8.Reiss P, Bleuse J, Pron A. Nano Lett. 2002;2:781. [Google Scholar]
- 9.Sukhanova A, et al. Anal Biochem. 2004;324:60. doi: 10.1016/j.ab.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 10.Lacoste TD. Proc Natl Acad Sci USA. 2000;97:9461. doi: 10.1073/pnas.170286097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalet X, et al. Single Mol. 2001;2:261. [Google Scholar]
- 12.F. Pinaud, X. Michalet, E. Margeat, H. P. Moore, S. Weiss, unpublished data.
- 13.Dahan M, et al. Science. 2003;302:442. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 14.Hohng S, Ha T. J Am Chem Soc. 2004;126:1324. doi: 10.1021/ja039686w. [DOI] [PubMed] [Google Scholar]
- 15.Empedocles SA, Norris DJ, Bawendi MG. Phys Rev Lett. 1996;77:3873. doi: 10.1103/PhysRevLett.77.3873. [DOI] [PubMed] [Google Scholar]
- 16.Kapitonov AM. J Phys Chem B. 1999;103:10109. [Google Scholar]
- 17.Larson D, et al. Science. 2003;300:1434. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 18.S. Doose, J. M. Tsay, F. Pinaud, S. Weiss, unpublished data.
- 19.Medintz IL. Nature Mater. 2003;2:630. doi: 10.1038/nmat961. [DOI] [PubMed] [Google Scholar]
- 20.Chan WCW, Nie SM. Science. 1998;281:2016. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 21.Pathak S, Choi SK, Arnheim N, Thompson ME. J Am Chem Soc. 2001;123:4103. doi: 10.1021/ja0058334. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Bawendi MG. J Am Chem Soc. 2003;125:14652. doi: 10.1021/ja0368094. [DOI] [PubMed] [Google Scholar]
- 23.Guo W, Li JJ, Wang YA, Peng XG. Chem Mater. 2003;15:3125. [Google Scholar]
- 24.Pinaud F, King D, Moore HP, Weiss S. J Am Chem Soc. 2004;126:6115. doi: 10.1021/ja031691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu XY. Nature Biotechnol. 2003;21:41. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 26.Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. Nature Biotechnol. 2004;22:969. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 27.Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998;281:2013. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 28.Gerion D, et al. J Phys Chem B. 2001;105:8861. [Google Scholar]
- 29.Dubertret B, et al. Science. 2002;298:1759. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Chan WCW, Nie S. J Biomed Opt. 2002;7:532. doi: 10.1117/1.1506706. [DOI] [PubMed] [Google Scholar]
- 31.Pellegrino T, et al. Nano Lett. 2004;4:703. [Google Scholar]
- 32.Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. J Am Chem Soc. 2004;126:6520. doi: 10.1021/ja048792a. [DOI] [PubMed] [Google Scholar]
- 33.Mattoussi H, et al. J Am Chem Soc. 2000;122:12142. [Google Scholar]
- 34.Rogach AL. Phys Status Solidi B. 2001;224:153. [Google Scholar]
- 35.Gaponik N, et al. J Phys Chem B. 2002;106:7177. [Google Scholar]
- 36.Patolsky F, et al. J Am Chem Soc. 2003;125:13918. doi: 10.1021/ja035848c. [DOI] [PubMed] [Google Scholar]
- 37.Akerman ME, Chan WCW, Laakkonen P, Bhatia SN, Ruoslahti E. Proc Natl Acad Sci USA. 2002;99:12617. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell GP, Mirkin CA, Letsinger RL. J Am Chem Soc. 1999;121:8122. [Google Scholar]
- 39.Goldman ER. Anal Chem. 2002;74:841. doi: 10.1021/ac010662m. [DOI] [PubMed] [Google Scholar]
- 40.Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nature Biotechnol. 2003;21:47. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]
- 41.Mansson A, et al. Biochem Biophys Res Commun. 2004;314:529. doi: 10.1016/j.bbrc.2003.12.133. [DOI] [PubMed] [Google Scholar]
- 42.Dameron CT. Nature. 1989;338:596. [Google Scholar]
- 43.Whaley SR, English DS, Hu EL, Barbara PF, Belcher AM. Nature. 2000;405:665. doi: 10.1038/35015043. [DOI] [PubMed] [Google Scholar]
- 44.Alivisatos AP. Nature Biotechnol. 2004;22:47. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]
- 45.Tokumasu F, Dvorak J. J Microsc. 2003;211:256. doi: 10.1046/j.1365-2818.2003.01219.x. [DOI] [PubMed] [Google Scholar]
- 46.Lidke DS. Nature Biotechnol. 2004;22:198. doi: 10.1038/nbt929. [DOI] [PubMed] [Google Scholar]
- 47.Xiao Y, Barker PE. Nucleic Acids Res. 2004;32:28E. doi: 10.1093/nar/gnh024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.L. A. Bentolila, S. Weiss, unpublished data.
- 49.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Bioconjug Chem. 2004;15:79. doi: 10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 50.Mattheakis LC. Anal Biochem. 2004;327:200. doi: 10.1016/j.ab.2004.01.031. [DOI] [PubMed] [Google Scholar]
- 51.Pellegrino T, et al. Differentiation. 2003;71:542. doi: 10.1111/j.1432-0436.2003.07109006.x. [DOI] [PubMed] [Google Scholar]
- 52.Kloepfer JA. Appl Environ Microbiol. 2003;69:4205. doi: 10.1128/AEM.69.7.4205-4213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parak WJ. Adv Mater. 2002;14:882. [Google Scholar]
- 54.Derfus AM, Chan WCW, Bhatia SN. Adv Mater. 2004;16:961. [Google Scholar]
- 55.Hanaki K, et al. Biochem Biophys Res Commun. 2003;302:496. doi: 10.1016/s0006-291x(03)00211-0. [DOI] [PubMed] [Google Scholar]
- 56.Winter JO, Liu TY, Korgel BA, Schmidt CE. Adv Mater. 2001;13:1673. [Google Scholar]
- 57.Derfus AM, Chan WCW, Bhatia SN. Nano Lett. 2004;4:11. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosenthal SJ. J Am Chem Soc. 2002;124:4586. doi: 10.1021/ja003486s. [DOI] [PubMed] [Google Scholar]
- 59.Lim YT. Mol Imaging. 2003;2:50. doi: 10.1162/15353500200302163. [DOI] [PubMed] [Google Scholar]
- 60.Kim S, et al. Nature Biotechnol. 2004;22:93. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shim M, Guyot-Sionnest P. Nature. 2000;407:981. doi: 10.1038/35039577. [DOI] [PubMed] [Google Scholar]
- 62.Mikulec FV. J Am Chem Soc. 2000;122:2532. [Google Scholar]
- 63.Bailey RE, Nie SM. J Am Chem Soc. 2003;125:7100. doi: 10.1021/ja035000o. [DOI] [PubMed] [Google Scholar]
- 64.Mokari T, Rothenberg E, Popov I, Costi R, Banin U. Science. 2004;304:1787. doi: 10.1126/science.1097830. [DOI] [PubMed] [Google Scholar]
- 65.J.M. Tsay, S. Doose , F. Pinaud, J.J. Li, S. Weiss, J. Phys. Chem. B, in press.
- 66.Poznyak SK, Talapin DV, Shevchenko EV, Weller H. Nano Lett. 2004;4:693. [Google Scholar]
- 67.Adams SR. J Am Chem Soc. 2002;124:6063. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 68.Kapanidis AN, Ebright YW, Ebright RH. J Am Chem Soc. 2001;123:12123. doi: 10.1021/ja017074a. [DOI] [PubMed] [Google Scholar]
- 69.Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Proc Natl Acad Sci USA. 2004;101:9955. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin J, Liu F, Li X, Walsh CT. J Am Chem Soc. 2004;126:7754. doi: 10.1021/ja047749k. [DOI] [PubMed] [Google Scholar]
- 71.Boder ET, Midelfort KS, Wittrup KD. Proc Natl Acad Sci USA. 2000;97:10701. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marks KM, Rosinov M, Nolan GP. Chem Biol. 2004;11:347. doi: 10.1016/j.chembiol.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 73.Tkachenko AG. Bioconjug Chem. 2004;15:482. doi: 10.1021/bc034189q. [DOI] [PubMed] [Google Scholar]
- 74.Ishii D, et al. Nature. 2003;423:628. doi: 10.1038/nature01663. [DOI] [PubMed] [Google Scholar]
- 75.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Cancer Lett. 2004;209:171. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 76.Yu WW, Falkner JC, Shih BS, Colvin VL. Chem Mater. 2004;16:3318. [Google Scholar]
- 77.Guzelian AA, Banin U, Kadavanich AV, Peng X, Alivisatos AP. Appl Phys Lett. 1996;69:1432. [Google Scholar]
- 78.Guzelian AA. J Phys Chem. 1996;100:7212. [Google Scholar]
- 79.Yu WW, Peng X. Angew Chem Int Ed. 2002;41:2368. doi: 10.1002/1521-3773(20020703)41:13<2368::AID-ANIE2368>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 80.Yu WW, Wang YA, Peng X. Chem Mater. 2003;15:4300. [Google Scholar]
- 81.Yu WW, Qu L, Guo W, Peng X. Chem Mater. 2004;16:560. [Google Scholar]
- 82.Tsay JM, Pflughoefft M, Bentolia LA, Weiss S. J Am Chem Soc. 2004;126:1926. doi: 10.1021/ja039227v. [DOI] [PubMed] [Google Scholar]
- 83.Supported by NIH grant 5-R01-EB000312, Keck Foundation grant 04074070, and Defense Advanced Research Projects Agency–Air Force Office of Scientific Research grant FA955004-10048. We are grateful for exchanges with our collaborators D. King, H.-P. Moore, A. P. Alivisatos, C. A. Larabell, D. Gerion, O. N. Witte, L. H. Rome, and G. Payne. Fluorescence microscopy images in Fig. 3 were obtained at the California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Shared Facility (ALMSSF), Department of Chemistry and Biochemistry, at UCLA. We thank K. Hamadani, G. Iyer, and the referees for helpful comments on the manuscript; E. Margeat for help with the experiments described in Fig. 3; and T. Olafsen for help with the experiments described in Fig. 4, A and B. Requests for materials should be sent to qdot-info@chem.ucla.edu.