

Abstract
The kinase Mirk/dyrk1B is essential for the differentiation of C2C12 myoblasts. Mirk reinforces the G0/G1 arrest state in which differentiation occurs by directly phosphorylating and stabilizing p27kip1 and destabilizing cyclin D1. We now demonstrate that Mirk is anti-apoptotic in myoblasts. Knockdown of endogenous Mirk by RNA interference activated caspase 3 and decreased myoblast survival by 75%, while transient overexpression of Mirk increased cell survival. Mirk exerts its anti-apoptotic effects during muscle differentiation at least in part through effects on the cell cycle inhibitor and pro-survival molecule p21cip1. Overexpression and RNA interference experiments demonstrated that Mirk phosphorylates p21 within its nuclear localization domain at Ser153 causing a portion of the typically nuclear p21 to localize in the cytoplasm. Phosphomimetic GFP-p21-S153D was pancellular in both cycling C2C12 myoblasts and NIH3T3 cells. Endogenous Mirk in myotubes, and overexpressed Mirk in NIH3T3 cells were able to cause the pancellular localization of wild-type GFP-p21, but not the non-phosphorylatable mutant GFP-p21-S153A. Translocation to the cytoplasm enables p21 to block apoptosis through inhibitory interaction with pro-apoptotic molecules. Phosphomimetic p21-S153D was more effective than wild-type p21 in blocking the activation of caspase 3. Transient expression of p21-S153D also increased myoblast viability in colony forming assays, while the p21-S153A mutant had no effect. This Mirk-dependent change in p21 intracellular localization is a natural part of myoblast differentiation. Endogenous p21 localized exclusively to the nuclei of proliferating myoblasts, but was also found in the cytoplasm of post-mitotic multinucleated myotubes and adult human skeletal myofibers.
Abbreviation: Mirk, minibrain-related kinase; GFP, green fluorescent protein; CDK, cyclin-directed kinase; PCNA, proliferating cell nuclear antigen; MAPK, mitogen-activated protein kinase; DM, differentiation medium; GM, growth medium
While there is a large body of work describing the functions of myogenic regulatory factors in skeletal muscle commitment and differentiation, much less is known regarding the signaling pathways that control the specific molecular events involved in terminal differentiation and maintenance of skeletal muscle. We recently demonstrated that a novel gene which we cloned in 1997, the kinase Mirk, plays a critical role in controlling C2C12 myoblast differentiation following the commitment stage of myogenesis (1). Depletion of endogenous Mirk by RNA interference blocked the transcription of myogenin and the subsequent myoblast differentiation program (1). Mirk controls the activation of the myogenin transcription factor MEF2 by regulating nuclear accumulation of the MEF2 inhibitors, class II histone deacetylases (2). However, it has been reported that embryonic knockout of Mirk did not block skeletal muscle development in the early embryo (3), suggesting that Mirk may have a more significant function in muscle repair than in initial myogenesis.
Mirk (Minibrain-related kinase; also known as Dyrk1B) is a member of an evolutionarily conserved family of proteins, the Dyrk/Minibrain family of arginine-directed serine/threonine protein kinases (4), (5) , (6), (7), that play roles in controlling the switch from proliferation to differentiation in a wide variety of organisms. The roles of Dyrk/Minibrain/Mirk homologues in yeast (Yak1) and slime mold (YakA) suggest that this group of kinases helps to regulate the transition from growth to differentiation in response to environmental stresses (8), (9).
Mirk is a unique, multifunctional kinase with primary activity in G0 and early differentiation. Specifically, Mirk elongates the G0 phase of the cell cycle in C2C12 myoblasts, Mv1Lu epithelial cells and NIH3T3 fibroblasts by stabilizing the CDK inhibitor p27kip1 (10) and by destabilizing the G1 cyclin, cyclin D1 (11). Mirk has also been shown to act as a transcriptional activator (12), (13), (2) and to inhibit cell motility (14). Because of these actions of Mirk on cell cycle regulators active in G0/G1 during myoblast differentiation, we extended our study of Mirk substrates to p21cip1. The CDK inhibitor p21 is highly expressed in differentiating muscle in vivo (15) and has been implicated as playing a central role in mediating the pre-differentiation growth arrest induced by the major muscle regulatory factor MyoD (16). It is believed that this effect of p21 during myogenesis is due to inhibition of G1 cyclin/CDK complexes and subsequent blockade of pRb phosphorylation (16).
Studies using mice deficient in members of the Cip/Kip CDK inhibitor family have demonstrated that in the absence of p21, mice can develop into normal adults (17). This appears to be due to redundant functions of p57, since mice deficient in both p21 and p57 demonstrate severely arrested muscle development (18). Specifically, the double mutant mice displayed increased proliferation and apoptosis of myoblasts and failed to form myotubes. Significantly, although p21-deficient mice appear to develop normally, p21 is essential for normal regeneration through the muscle progenitor (or satellite) cells that are responsible for repairing muscle following injury (19). It has been suggested that muscle differentiation during embryogenesis and during regeneration are fundamentally different, with p57 playing a dominant role in embryogenesis and p21 being the critical CDK inhibitor during regeneration (18).
In the current study, we demonstrate that in the C2C12 myoblast model of muscle regeneration, Mirk functions to promote cell survival during the initial stages of differentiation, similar to Mirk’s known function in mediating survival of colon carcinoma cells in serum-free conditions (20). Undifferentiated or partly differentiated cells are removed from cultures of fusing myoblasts by apoptosis, causing a loss of 20–30% of C2C12 myoblasts during the first 48 hours in differentiation medium (15). Mirk diminishes the extent of myoblast apoptosis during the differentiation process, at least in part by direct modulation of p21cip1 localization. Mirk phosphorylates p21 within its nuclear localization domain. This phosphorylation maintains a portion of p21 protein in the cytoplasm where p21 has been reported to be unable to mediate cell cycle arrest (21), and where p21 blocks caspase 3 activation (22).
EXPERIMENTAL PROCEDURES
Materials
Rabbit polyclonal antibodies were raised to unique sequences at the N-terminus (amino acids 1–19) (20) and C-terminus of Mirk (amino acids 595–624) and affinity purified. The C-terminal antibody was raised to a slightly longer peptide than the C-terminal antibody used in all our previous studies on Mirk (20), but was only homologous to Mirk/dyrk1B in BLAST analysis. This antibody detected all three Mirk splice variants found in normal muscle: 69, 70 and 75 kD, the 69 and 70 kDa variants found in C2C12 cells (1) and the inducible 70 kD Mirk form stably expressed in Mv1Lu cells (14), but no other proteins at the dilutions used in the current study (western blotting data not shown). The C-terminal antibody was labeled with Alexa Fluor 594 using the Xenon Rabbit IgG Labeling Kit (Molecular Probes) for use in direct immunofluorescence experiments. Alexa Fluor 488 and 594 (highly cross-adsorbed) secondary antibody conjugates and Alexa Fluor 594 phalloidin were purchased from Molecular Probes. Antibodies to p21 (C-19 and F-5), GFP, myogenin, and tubulin were from Santa Cruz Biotechnology. Rabbit monoclonal antibody to cleaved caspase 3 was from Cell Signaling. Antibody to the Flag epitope was from Sigma. Polyvinylidene difluoride transfer paper (Immobilon-P)was purchased from Millipore. LipofectAMINE, LipofectAMINE 2000 and PLUS reagent were from Invitrogen. All radioactive materials were purchased from PerkinElmer Life Sciences, and ECL reagents were from Amersham Biosciences. Tissue culture reagents were obtained from Mediatech. SB203580 was purchased from Calbiochem. All other reagents were from Sigma.
Plasmids
pCDNA3.1-HisA-Mirk and the kinase inactive forms pCDNA3.1-HisA-Mirk (KR) and pCDNA3.1-HisA-Mirk (YF) had been previously generated. pMT-5/p21Cip1 was the kind gift of Dr. M.-C. Hung. Wild-type p21Cip1 was subcloned into pGEX-4T1 (Amersham) and site-directed mutagenesis was performed using the GeneEditor in vitro site-directed mutagenesis system (Promega). Mutants were subsequently subcloned into pCMV-tag2B (Stratagene) to yield Flag-p21 and pEGFP-C2 (Clontech) to yield GFP-p21 for mammalian expression. All mutant p21 constructs were sequenced to confirm the mutated sequence.
RNA Interference
A sequence within the murine Mirk coding domain, si1: GACCTACAAGCACATCATT was used to knockdown endogenous Mirk, while another sequence mutant at 2 positions to murine Mirk, si2:GCTCTCTGTGGACCTCA; served as the mutant control (1), (10). Each was inserted into the pSilencer plasmid (Ambion).
Cell Culture
C2C12 mouse myoblasts and NIH3T3 cells were obtained from the ATCC and cultured according to their recommendations. C2C12 cells were maintained in growth medium (GM; Dulbecco's modified Eagle's medium, 4 mM L-glutamine, 4.5 g/L glucose, containing 20% fetal bovine serum) and induced to undergo differentiation by switching to differentiation medium(DM; Dulbecco's modified Eagle's medium containing 2% horseserum). NIH3T3 cells were maintained in Dulbecco’s modified Eagle’s medium, 4 mM L-glutamine, 4.5g/L glucose, containing 10% bovine calf serum. Cell cultures were typically maintained without the use of antibiotics. Cells were used for experiments only from passages 3-10 from our frozen stocks.
Transient Transfections
Two strategies were employed for transient transfections. In order to achieve high-level short term expression (for GFP localization and in vivo labeling) we used a modification of the manufacturer’s standard protocol for LipofectAMINE 2000 (Invitrogen). Specifically, we transfected cells with a 2:1 ratio (ul:ug) of LipofectAMINE 2000 to plasmid DNA immediately after plating freshly trypsinized cells. This increased transfection efficiency in the C2C12 cell line from an average of 20–30% to nearly 70–80%. LipofectAMINE 2000 (4–32 ul, generally 16 ul) and DNA (2–16 ug, generally 8 ug) were diluted in separate 500 ul portions of OptiMem I (Invitrogen) or serum free DMEM and combined after 5 minutes incubation at room temperature. The DNA/LipofectAMINE 2000 complexes were incubated for an additional 20 minutes at room temperature, then were added directly to 106 freshly plated cells in 3 ml of DMEM supplemented with 10% FBS in 60 mm culture dishes. Media was typically replaced with DMEM containing 10% FBS 24 hours after transfection. When lower levels of transfection were required, we transiently transfected cells using a 1:2:3 ratio of plasmid DNA: PLUS reagent: LipofectAMINE in serum free media. To lower the level of expression further for stability experiments, the p21 plasmids were diluted 1:5–1:10 with carrier plasmid DNA. The media was replaced with 10% FBS/DMEM after 4 hours of transfection in serum free media. Antibiotics were not used at any time with cells used for transfection.
GST Fusion Proteins
p21 constructs were subcloned into the pGEX-4T1 vector (Amersham Biosciences) and expressed and purified as we have previously described in detail (23). Mirk and kinase-inactive Mirk (YF) were subcloned into the pGEX-6P1 vector (Amersham) and expressed in the E. coli strain BL-21 (Amersham). GST-Mirk proved to be a difficult protein to purify using standard techniques as it was rapidly incorporated into inclusion bodies and tended to copurify with high quantities of the bacterial hsp70 homologue, the dnaK gene product. This protein has been reported to play a role in degradation of abnormally folded proteins in E. coli (24). The following protocol allowed the majority of the heat shock protein (hsp) to be eliminated from the final preparation. In addition, it appears that the conditions also promoted hsp-dependent refolding of GST-Mirk (25), (26), resulting in a highly purified and active enzyme preparation. 300 ml of LB medium was seeded with 10% of an overnight culture of BL-21 carrying the desired plasmidand incubated at 37 °C for 1–2 h. Protein expression was induced with 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 6 h at 24°C, and bacteria were collected by centrifugation and frozen at −70 °C. Pellets (averaging 2 g each) were resuspended in 5 ml of ice-cold sonication buffer (140 mM NaCl, 2.7 mM KCl, 10 mM NaH2PO4, 1.8 mM KH2PO4, 10 mM MgCl2, 5 mM ATP, 5 mM DTT, 20% glycerol, 2% Triton X-100, 1 mM PMSF, 100 ug/ml lysozyme, Roche EDTA-free Complete Protease Inhibitor Set, pH 7.3). The ATP and Mg2+ are required for the activity of the heat shock protein (25), (26). The resuspended bacteria were incubated on ice for 15 minutes, then the bacteria were lysed by sonication using a microtip (Misonix, setting 2.5) for 3 min with 5 s bursts separated by 5 s of rest. Lysates were clarified by centrifugation for 15 min at 15,000 x g to remove insoluble material. 13-ml portions of the cleared lysate were added to 1 ml (bed volume) of glutathione-sepharose4B (Amersham) in a 15 ml conical tube, then an additional 750 ul of 100 mM ATP, 150 ul of 1M MgCl2 and a Roche Complete Protease Inhibitor Mini-Tab was added to each sample. The beads were then incubated at 4 °C overnight with end-on rotation. Beads were washed 4 x 10 minutes with 10 ml of standard wash buffer (290 mM NaCl, 2.7 mM KCl, 10 mM NaH2PO4, 1.8 mM KH2PO4, 10% glycerol, 0.1% Triton X-100, pH 7.3) to remove the majority of contaminants from the beads. The beads were then washed 5 x 1 hour with 10 ml portions of Mg/ATP wash buffer (290 mM NaCl, 2.7 mM KCl, 10 mM NaH2PO4, 1.8 mM KH2PO4, 5 mM MgCl2, 2 mM ATP, 2 mM DTT, 10% glycerol, 0.1% Triton X-100, pH 7.3) to remove the majority of the heat shock protein contaminant. The beads were then washed 3 x 5 minutes with standard wash buffer. All washes and incubations were performed at 4°C with end-on rotation. The PreScission enzyme, which is active at 4°C was used to cleave Mirk from GST. The efficiency of PreScission protease cleavage was enhanced significantly by first eluting the GST-fusion proteins from the beads by the addition of 1 ml of glutathione elution buffer (75 mM HEPES, pH 7.4, 150 mM NaCl, 20 mM reduced glutathione, 5 mM DTT, 0.1 % Triton X-100). Elution was typically carried out overnight at 4°C with end-on rotation. The eluate was concentrated, the glutathione was dialyzed out and the elution buffer was exchanged for PreScission cleavage buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.01% Triton X-100, pH 7.0) by three concentrations with an Amicon Ultra-4 30,000 MWCO centrifugal filter device (Millipore). Protein concentration was measured using the Bradford protein assay and 20 units of PreScission enzyme were added per mg of protein. Proteolytic cleavage was carried out overnight at 4°C with end-on rotation. The cleaved protein solution was then incubated 2 x 4 hours at 4°C with equal volumes (bed volume) of glutathione-sepharose 4B in order to clear the sample of uncleaved protein, GST and the PreScission enzyme (also a GST-fusion protein). The resulting sample was concentrated and the cleavage buffer was replaced with PBS with 10% glycerol and protease inhibitors using centrifugal filtration. The product retained high activity for up to 1 year when stored at −20°C.
In Vitro Kinase Assays
2–5 ug of GST-p21 fusion proteins bound to glutathione-sepharose beads were washed twice with kinase assay buffer (10 mM Tris-HCl (pH 7.4),150 mM NaCl, 10 mM MgCl2, 0.5 mM dithiothreitol) and then incubated with 100 ng of recombinant Mirk for 20 min at 30 °C in 30 3l of kinase assay buffer containing 103M unlabeled ATP plus 5 μCi of [γ-32P]ATP. Reaction products were analyzed by SDS-PAGE and autoradiography.
In Vivo Labeling and Immunoprecipitations
2 x 106 C2C12 cells were plated in 60 mm dishes and transfected immediately with 12 ug of Flag-p21 constructs and 4 ug of HisA-Mirk using 32 ul of LipofectAMINE 2000 in DMEM containing 10% FBS. Following 24 hours of expression, cells were switched to differentiation media for 19 hours, then to phosphate free media for 1 hour, and then incubated with 400 uCi of 32P-orthophosphate for 4 hours in 2 ml of reduced phosphate medium (75% phosphate-free DMEM, 25% DMEM). Cells were washed twice and then lysed in 0.5 ml of buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10% glycerol, Roche Complete Protease Inhibitor Cocktail, and Sigma Phosphatase Inhibitor Cocktail I and II (lysis buffer). Lysates were precleared by rocking incubation at 4°C with 50 ul of protein A agarose (Santa Cruz) for 1 hour and then pelletedin a microcentrifuge at 10,000 x g for 20 min to remove insoluble material. Flag constructs were isolated using 15ul of pre-washed EZview Anti-Flag M2 Affinity Gel (Sigma), which was incubated with the cell lysate at 4 °C for 2 h and then washed five times with lysis buffer before analysis. Because both endogenous and Flag-p21 ran extremely close to the IgG light chain on SDS-PAGE, we used the TrueBlot anti-mouse IgG secondary detection system (eBioscience; 1:1000) for Western analysis of immunoprecipitated p21.
Two-dimensional Phosphopeptide Analysis
Chymotrypsin or trypsin cleavage and two-dimensional analysis of p21 phosphopeptides were carried out as described (23). Following an in vitro kinase assay or immunoprecipitation of phospho-labeled proteins, the substrate was resolved by SDS-PAGE and transferred to a nitrocellulose membrane, and the protein band was identified by autoradiography and excised from the membrane. Proteins were digested with sequencing grade chymotrypsin or trypsin (20 μg in 200 μl of freshly prepared 50 mM NH4HCO3 overnight at 37 °C), and the polypeptides were spotted onto 20 x 20-cm thin-layer cellulose plates and separated in the first dimension by electrophoresis for 45 min at 1000 Von an HTLE-7002 electrophoresis system (C.B.S. Scientific) using buffer containing 2.2% formic acid and 7.8% acetic acid at pH 1.9. Peptides were separated in the second dimension by chromatography for 9 h in phosphochromatography buffer (38%1-butanol, 25% pyridine, and 7.5% acetic acid). Dried plates were exposed to Eastman Kodak Biomax MS film for 2–8 days.
Immunodetection
Following treatment as indicated and washing with phosphate-buffered saline, cells were lysed as previously described (23). Depending upon the experiment, 20–50 μg of the cell lysate was resolved by SDS-PAGE and blotted onto polyvinylidene difluoride membranes. The blots were blocked in Tris-buffered saline containing 0.1% Tween 20 (TBST) and 5% nonfat dry milk for1 h and incubated for 1–2 h at room temperature with the primary antibody (1:1000 dilution of antibodies to p21Cip1, tubulin, and MyoD). Proteins were subsequently detected by enhanced chemiluminescence. Immunoblots were scanned using a Lacie Silverscanner DTP and densitometry was performed using the IP Lab Gel program (Scanalytics).
Localization of GFP-p21 Mutant Constructs
C2C12 myoblasts were plated in Lab-Tek 2-well chamber slides (3.5–5.0 x 105 cells per well) and then immediately transfected with GFP-p21: WT, nonphosphorylatable S153A, or the phosphomimetic S153D (3 ug plasmid DNA and 6 ul LipofectAMINE 2000 per well). NIH3T3 cells were plated at 1.5 X 105 cells per well, incubated overnight before transfection, then transfected with a mixture of 3 ug plasmid DNA, 5 ul PLUS reagent and 9 ul of LipofectAMINE per well. After 24 hours of expression, cells were washed 3X with PBS, fixed in 4% paraformaldehyde for 5 min, washed 2 x 5 minutes with PBS, permeabilized with 0.1% Triton in PBS (PBST) for 5 minutes, and blocked with 3%BSA/PBS for 30 minutes. Slides were incubated for 20 minutes with 2 units of Alexa Fluor 594 phalloidin (Molecular Probes) diluted in 3%BSA/PBS, washed twice, then incubated for 5 minutes with a 2 ng/ml solution of 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI) in PBST. Following 3 x 5 minute washes with PBST, slides were rinsed with distilled water, blotted dry and mounted with Biomedia Gel/Mount. Preparations were analyzed and photographed as described below.
Statistical Analysis of Localization
Cell counts were obtained at 400X (oil immersion) using a Nikon Eclipse E50i fluorescent microscope. For C2C12 cells, at least 300 cells were observed in 8–10 random fields in each of 4 separate preparations and the number of cells with localization of green fluorescence in the nucleus only and localization of green fluorescence in the nucleus and cytoplasm was determined. Two separate preparations of NIH3T3 fibroblasts were examined in the same way. Combined counts were analyzed by the Chi square test (Minitab) to determine the significance of differences between the WT and mutant constructs.
Immunofluorescence of Cultured Cells
C2C12 myoblasts were plated in Lab-Tek 2-well chamber slides (2 x 105 cells per well), cultured for one day in growth media, and then switched to differentiation media. After 24 hours of expression in GM or 48 hours in DM, cells were washed 2X with PBS, fixed in 4% paraformaldehyde for 5 min, washed 2 x 5 minutes with PBS, permeabilized with 0.1% Triton X-100/PBS (wash buffer – used in all subsequent washes), and blocked with 10% normal goat serum/PBS for 30 minutes. Dual staining for endogenous p21 and Mirk was accomplished by simultaneously incubating slides for 30 minutes with 1:500 dilutions of anti-p21 mouse monoclonal antibody (F-5, Santa Cruz) and anti-C-terminal Mirk antibody conjugated to Alexa Fluor 594. As a control for nonspecific antibody interactions, we also performed identical experiments with the C-19 polyclonal antibody (Santa Cruz) and our anti-N-terminal Mirk antibody. Additional controls included use of direct immunofluorescence (using the Zenon labeling system) and substitution of the primary antibody with non-specific mouse or rabbit IgG as appropriate. All labeling incubations were diluted in 10% normal goat serum/PBS. After 3 x 5 minute washes, cells were incubated with a 1:1000 dilution of goat anti-mouse antibody conjugated to Alexa Fluor 488 (Molecular Probes). Slides were washed 2 x 5 minutes. During single-labeling experiments for detection of p21, cells were incubated for 20 minutes with 2 units of Alexa Fluor 594 phalloidin (Molecular Probes) diluted in 3%BSA/PBS, and washed twice. When using direct labeling, cells were fixed a second time (4% paraformaldehyde for 5 min) to stabilize the Zenon label after the secondary antibody washes. Nuclei were counterstained with a 5 minute incubation by a 2 ng/ml solution of 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI). Following 3 x 5 minute washes, slides were rinsed with distilled water, blotted dry and mounted with Biomedia Gel/Mount. Images were obtained as described below.
Immunofluorescence of Human Muscle Frozen Sections
Anonymous samples of flash-frozen human muscle tissue on slides were obtained from the Department of Pathology, SUNY Upstate Medical University in accordance with institutional review procedures for clinical specimen use. Slides were thawed at room temperature for 15 minutes, hydrated with PBS for 15 minutes, and then fixed in 4% paraformaldehyde for 1 minute at room temperature, or ice-cold acetone for 10 minutes at 0°C. Slides were rinsed 3 x 5 minute with PBS. The sections were then demarcated with a PAP pen (Zymed) and permeabilized for 20 min with 0.2% Triton X-100 in PBS (wash buffer; used in all subsequent steps), and blocked with 10% normal goat serum/0.2% Triton/PBS for 30 minutes. Mirk was visualized with polyclonal antibody to either the N- or C-terminus of Mirk (both at 1:500 dilution for 1 hour), while p21 was visualized with either the polyclonal C-19 antibody (1:250 for 1 hour) or the monoclonal F-5 antibody (1:50 for 2 hours). Non-specific IgG isolated from the same species as the primary antibody diluted to an equivalent mass/concentration was used as a negative control. All labeling incubations were diluted in 10% normal goat serum/0.2% Triton/PBS. After 3 x 5 minute washes, cells were incubated for 30 minutes with a 1:1000 dilution of goat anti-IgG antibody Alexa Fluor conjugate (Molecular Probes). Sections were washed 2 x 5 minutes. In some experiments, sections were incubated for 20 minutes with 2 units of Alexa Fluor 594 phalloidin (Molecular Probes) diluted in 3%BSA/PBS, and washed twice. Nuclei were counterstained by a 5 minute incubation with a 2 ng/ml solution of 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI). Following 3 x 5 minute washes, slides were rinsed with distilled water, blotted dry and mounted with Biomedia Gel/Mount. Images were obtained as described below.
Imaging
Monochrome fluorescence images were obtained at 400X or 1000X (as indicated) using a Diagnostic Instruments SPOT RT camera mounted on a Nikon Eclipse E50i fluorescent microscope. SPOT RT Software v4.0.9 was used to pseudocolor the images, adjust the RGB histogram and merge the images. Image manipulation consisted of resetting the zero point of the RGB histogram of the green and red fluorescent channels to stretch the darker areas of the image yielding a uniform black background consistent with the image viewed through the microscope. Final figures were arranged using Adobe Photoshop v.7.0.
BrdU Incorporation Assay
C2C12 myoblasts were plated overnight in Labtek 2-well chamber slides (105 cells per well) and then transfected (2 ug plasmid DNA, 4 ul PLUS, and 4 ul LipofectAMINE per well) with GFP-vector, GFP-p21(WT), or the phosphomimetic GFP-p21(S153D). Cells were transfected in serum free media for 4 hours, then an equal volume of 20% FBS/DMEM was added. BrdU incorporation was measured using the Amersham Cell Proliferation Assay Kit. After 24 hours of expression, cells were incubated for 1 hour with BrdU labeling reagent diluted 1:500 in 10% FBS/DMEM. Cells were rinsed 2X with PBS, fixed for 30 minutes at room temperature with acid alcohol (90% absolute ethanol, 5% glacial acetic acid, 5% water). Cells were then rinsed 3X with PBS, blocked for 30 minutes in 10% NGS/PBST, then incubated for 1 hour at room temperature with a DNase I solution containing a 1:100 dilution of anti-BrdU antibody (Amersham). The denatured GFP protein was labeled by a 30 minute incubation with anti-GFP antibody (Santa Cruz 8334; 1:500). BrdU was visualized using anti-mouse Alexa Fluor 594; GFP was visualized with anti-rabbit Alexa Fluor 488. At least 300 GFP expressing cells were observed in 8-10 random fields in each of 3 separate preparations and the number of cells labeled for both GFP and BrdU was determined using a Green/Orange V2 filter set (Chroma) that allowed simultaneous visualization of both fluorophores. As an additional control, the proportion of cells incorporating BrdU was also assessed in a set of non-transfected slides stained with DAPI. Combined counts were analyzed by the Chi square test (Minitab) to determine the significance of differences between the GFP vector and GFP-p21 constructs.
RESULTS
Mirk promotes myoblast survival
Undifferentiated or partly differentiated cells are removed from cultures of fusing myoblasts by apoptosis. Programmed cell death of 20–30% of C2C12 myoblasts is seen during the first 48 hours after myoblasts are transferred from growth medium to differentiation medium (15). We had observed in earlier studies that the serine/threonine kinase Mirk afforded colon carcinoma cells increased survival capabilites, while mutant forms of kinase-inactive Mirk did not (20). Mirk is highly expressed in skeletal muscle cells and in myoblasts, so we speculated that one function of Mirk was to enhance the survival of differentiating myoblasts. Mirk is enriched in NIH3T3 cells in G0/G1, and downregulated when cells are treated with mitogens (10). Likewise, Mirk was found to be enriched in C2C12 myoblasts undergoing mitosis and in early G1 (Fig.1A). Levels of Mirk rapidly increase in C2C12 cells arresting in G0/G1 when placed in DM (1), (11). We hypothesized that if Mirk mediated cell survival, C2C12 cells expressing ectopic wild-type Mirk would survive in greater numbers during myoblast differentiation than cells expressing kinase-inactive YF-Mirk. C2C12 cells were transfected with either wild-type Mirk, YF-mutant Mirk, or vector alone. The constructs were allowed to express for 18 hours, and the cells were then transferred to differentiation medium for 0 to 48 hours. Expression of Mirk or YF-Mirk was determined by immunofluorescence analysis. Each construct was expressed in roughly the same percentage of C2C12 cells, 30–34%, after culture for 18 hours in growth medium. When cells were cultured in DM for 48 hours, 4 times as many cells in the differentiating cultures were found expressing ectopic wild-type Mirk as expressing kinase-inactive YF-Mirk. These data supported the hypothesis that Mirk provided some survival advantage to differentiating myoblasts. In parallel transfections, cell lysates were analyzed for expression of ectopic Mirk and YF-Mirk normalized to ß-tubulin by western blotting (Fig.1B, top). The amount of wild-type Mirk declined about 2-fold by 48 hours, during which time cells in the cultures were undergoing apoptosis. In contrast, the amount of YF-Mirk decreased far more, about l0-fold. To determine what percentage of Mirk left after 48 hours was endogenous, another set of transfections was performed. Cell lysates were analyzed for expression of ectopic YF-Mirk, and for endogenous Mirk from the cultures transfected with the vector alone (Fig.1B, bottom). These blots were exposed for a much longer time to visualize endogenous Mirk, which accounted for 2/3 of the Mirk detected in cultures transfected with YF-Mirk at the 48 hour point. Only about 8% of the YF-Mirk was left after 48 hours, while about 50% of wild-type Mirk was retained. Half-life measurements showed that this effect was not due to differences in protein stability (data not shown). Thus both biochemical analysis and immunofluoresence studies demonstrated that about 4-5 times as many C2C12 myoblasts survived apoptosis in cultures undergoing differentiation if they expressed ectopic wild-type Mirk than if they expressed ectopic kinase-inactive YF-Mirk.
Fig. 1.
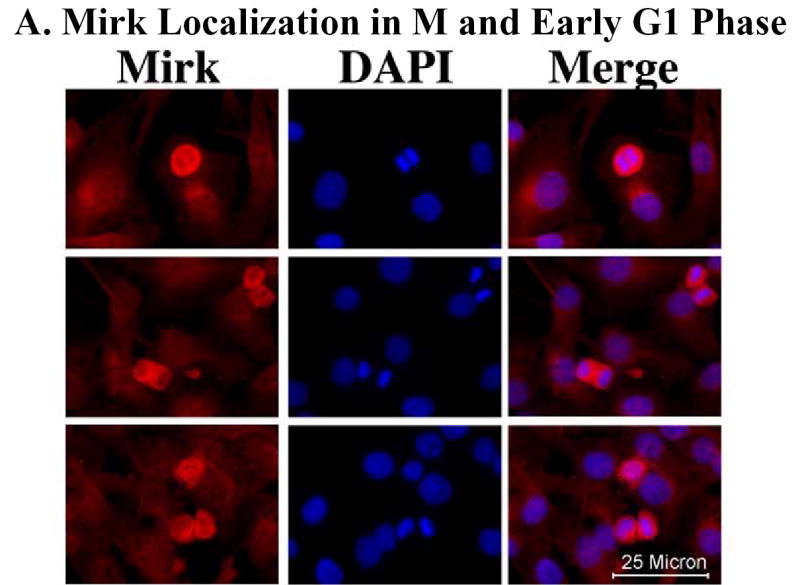
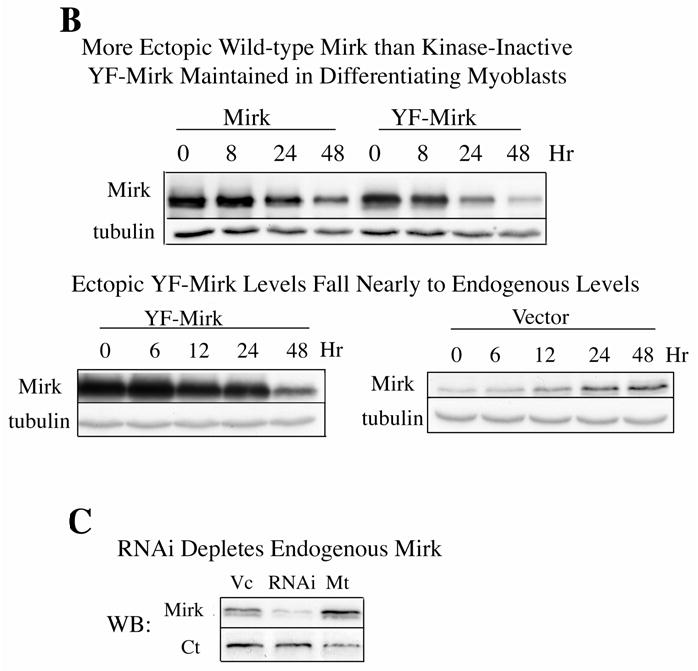
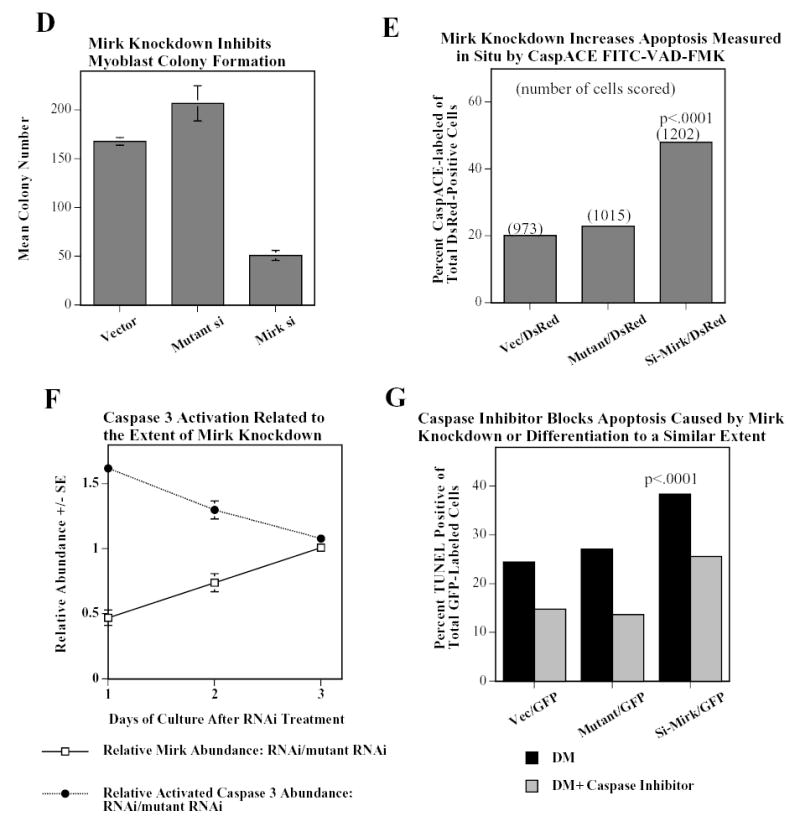
Mirk mediates myoblast survival. A. Mirk is expressed in cycling C2C12 myoblasts. Fluorescence microscopy demonstrates that Mirk expression is greatest in C2C12 cells in mitosis and in cells in early G1. Affinity-purified anti-peptide antibody to the Mirk C-terminus was conjugated to Alexa Fluor 594 and used to detect endogenous Mirk in C2C12 cells in growth medium. Nuclei were stained with DAPI, and identical fields were merged with SPOT RT software. Similar results were obtained with antibody directed to Mirk’s unique N-terminus. B. (top) Ectopic Mirk maintains the survival of differentiating C2C12 cells. Cells were transiently transfected with wild-type Mirk or kinase-inactive mutant YF-Mirk, allowed to express for 18 hours, then transferred to differentiation medium for up to 48 hours. Lysates were analyzed for expression of Mirk and ß-tubulin by western blotting. (bottom) To determine whether the amount of Mirk remaining in cells expressing ectopic YF-Mirk after 48 hours was either ectopic or endogenous Mirk, cells were transiently transfected with kinase-inactive mutant YF-Mirk, or vector (Vc), allowed to express for 18 hours, then transferred to differentiation medium for up to 48 hours. Lysates were analyzed for expression of Mirk and ß-tubulin by western blotting. The YF-Mirk and vector blots were exposed for the same amount of time to allow direct comparison. One of duplicate experiments with similar results is shown. C. Depletion of Mirk in cycling myoblasts by RNA interference reduced C2C12 cell viability in colony formation assays. Cells were plated at 5x105 per 60 mm dish, 3 dishes per point for 24 hours, then were transfected for 4 hours in serum-free medium with 5 ug pSilencer DNA encoding either RNAi to Mirk (RNAi), a mutant RNAi (Mt) or vector (Vc) together with 0.5 ug of neomycin resistance plasmid, using 10 ul each of Lipofectamine and Plus reagent. Fetal bovine serum was then added to 10% to maintain cell viability during expression. Cells were incubated for 20 hours before trypsinization, then lysed for western blotting. Ct, control, cross-reacting band showing similar loading. D. Same experiment as in panel C, but transfected cells were reseeded at the same cell density and selected in 400 ug/ml G418 for 3 weeks. Mean colony number +/SE is shown. E. Mirk knockdown increases apoptosis measured by fluorescence microscopy using an activated caspase 3 inhibitor. C2C12 myoblasts were plated overnight in LabTek 2-well chamber slides (2 x 105 cells per well) and then co-transfected with 0.5 ug pDsRed and 1.5 ug of pSilencer vector (Vec), Mutant si or RNAi to Mirk (4 ul PLUS, and 4 ul LipofectAMINE per well). Cells were transfected in serum free media for 4 hours, then an equal volume of 20% FBS/DMEM was added. Following 24 hours of expression, cells were incubated with differentiation media for 24 hours. Activated caspase was then labeled by a 30 minute incubation with 10 uM of CaspACE FITC-VAD-FMK in situ marker (Promega) diluted in DM. This is a fluorescent conjugate of the permeable caspase inhibitor VAD-FMK that was used as an in situ marker for apoptosis. Slides were washed and mounted with Biomedia GelMount. At least 200 DsRed expressing cells were observed in each of 4 separate preparations and the number of cells labeled for both DsRed and the FITC-VAD-FMK marker was determined using a Green/Orange V2 filter set (Chroma) that allowed simultaneous visualization of both fluorophores. Combined counts were analyzed by the Chi square test to determine the significance of differences between the RNAi constructs. Efficiency of cotransfection was determined to be greater than 85% in parallel experiments using a combination of GFP and DsRed. The number of cells scored per assay conditions is shown. F. Knockdown of endogenous Mirk by RNA interference inhibits the activation of caspase 3 in differentiating C2C12 cells. C2C12 cells (5 x105 per 60 mm dish) were transfected with pSilencer plasmid encoding RNAi to Mirk (RNAi) or mutant RNAi (Mt) for 24 hours, then switched to differentiation medium for 1, 2 and 3 days, as noted. The relative abundance of Mirk/tubulin and of activated caspase 3/tubulin in lysates were determined by western blotting on the upper and lower sections of same blot which was cut in half and then reunited for the exposure, with tubulin used as the loading control. The relative abundance of either protein in cells treated with RNAi to Mirk were then normalized to their relative abundance in cells treated with mutant RNAi. Mean +/− SE (if greater than 5%) is shown from data from 3 experiments. G. Apoptosis induced by differentiation medium or Mirk knockdown blocked to a similar extent by a caspase inhibitor. C2C12 myoblasts were plated, transfected and cultured as in panel E, except they were co-transfected with 0.5 ug pEGFP. A parallel set of cultures was incubated with DM containing 20 uM of the cell-permeable caspase inhibitor z-VAD.fmk (Promega). After 24 hours in DM, DNA breaks in apoptotic cells were labeled with tetramethyl-rhodamine-dUTP by TdT-mediated in situ end labeling (TUNEL) using the Roche In Situ Cell Death Detection Kit. At least 300 GFP expressing cells were observed in each of 4 separate preparations, so that an average of 1250 cells were scored per point. The number of GFP expressing cells labeled with the TUNEL marker was determined using a Green/Orange V2 filter set (Chroma) that allowed simultaneous visualization of both fluorophores. Combined counts were analyzed by the Chi square test. Efficiency of cotransfection was determined to be greater than 85% in parallel experiments using a combination of GFP and DsRed.
In a recent report, we showed that depletion of endogenous Mirk by RNA interference in C2C12 myoblasts undergoing differentiation blocked the induction of myogenin and contractile proteins, and subsequent myoblast fusion (1). In these experiments, we also observed a loss of those cells in which Mirk had been depleted, visualized by expression of cotransfected DsRed by fluorescence microscopy. To directly test whether Mirk played some role in myoblast survival, we depleted Mirk in C2C12 cells by RNA interference using RNAi to Mirk and a mutant RNAi as the control and measured cell viability by colony formation after Mirk depletion (Fig.1C,D). C2C12 cells were co-transfected with the neomycin-resistance gene and with the pSilencer vector encoding either RNAi to Mirk, a mutant RNAi or vector alone, and the extent of Mirk depletion was analyzed by western blotting (Fig.1C). The Mirk-depleted cells and controls were plated at single cell density, and selected in growth medium containing G418 for 3 weeks to determine the number of cells capable of proliferation to form a colony. Colony formation was reduced 75% in the C2C12 cells experiencing Mirk knockdown at the time of plating compared to cells transfected with the mutant RNAi or the vector control (Fig.1D). Parallel studies with NIH3T3 cells showed that the cells in colonies which did arise after Mirk knockdown expressed Mirk (data not shown). Thus these colonies presumably had arisen either from cells which had not been transfected or from cells which had lost the Mirk RNAi plasmid. These data indicate that knockdown of Mirk levels at the time C2C12 myoblasts were seeded as single cells had reduced their viability and thus their ability to give rise to colonies, and indicate that Mirk functions as a survival factor.
We next assayed whether depletion of Mirk would render cells more susceptible to apoptosis in short-term experiments without chemical selection of transfected cells (Fig.1E). We depleted Mirk in C2C12 cells by RNA interference, with a mutant RNAi and the empty vector as controls, and used co-transfected DsRed to mark the transfectants. After 24 hours to induce Mirk knockdown, we transferred the cells to differentiation medium for 1 day, and identified the apoptotic cells within the DsRed+ population by incubation with a fluorescent conjugate of the permeable caspase inhibitor VAD-FMK. 48% of cells with Mirk knockdown were undergoing apoptosis compared with 22.9% and 20.1% , respectively, of cells treated with mutant RNAi or vector, a highly significant difference (Fig.1E). A similar knockdown experiment was performed in a time-course format by measuring the abundance of Mirk and the apoptosis effector activated caspase 3 by western blotting (Fig. 1F). After 1 day, Mirk levels were depleted to about 50% of control values and caspase 3 was activated to 160% of control values. After 2 days, there was less Mirk knockdown and less activation of caspase 3. After 3 days, there was no Mirk knockdown and no activation of caspase 3 (Fig. 1F). Thus, depletion of Mirk protein correlated with increased abundance of activated caspase 3 in multiple experiments, and led to loss of cells by apoptosis. In another experiment, we measured the effect of the caspase inhibitor z-VAD-fmk on differentiation-induced apoptosis, as assayed by the TUNEL reaction (Fig. 1G). The caspase inhibitor blocked apoptosis to a similar extent, 40% in control cultures, 50% in mutant RNAi-treated cultures and 33% in cultures where Mirk knockdown occurred. These studies employing either Mirk knockdown or Mirk overexpression demonstrate that Mirk has anti-apoptotic survival functions in C2C12 myoblasts.
Mirk phosphorylates p21 in vivo
The CDK inhibitor p21cip1 has been implicated in the control of apoptosis during myoblast fusion (15), possibly through stabilization by ERKs (27). Mirk phosphorylates another CDK inhibitor p27kip1 in vivo (10), so we hypothesized that Mirk might also phosphorylate the closely related p21cip1. A wild-type p21 construct with an N-terminal Flag-epitope tag was cotransfected into NIH3T3 fibroblasts with either wild-type or kinase inactive Mirk, and allowed to co-express for 24 hours. After 4 hours of labeling with 32P-orthophosphate, Flag-p21 was immunoprecipitated with anti-Flag M2 antibody, and the immunoprecipitates were resolved by SDS-PAGE and analyzed by autoradiography (Fig.2). Flag-p21 migrated at the expected molecular weight, but was phosphorylated only in the co-transfections with wild-type Mirk. Flag-p21 was not labeled when co-expressed with kinase-inactive KR-Mirk or vector (Fig.2, upper panel). Western blotting for the Flag epitope (lower panel) demonstrated that equal amounts of Flag-p21 were immunoprecipitated. The lower band of the doublet was Flag-p21, while the upper band was light chain. The Flag-p21 protein exhibited retarded mobility only when co-expressed with wild-type Mirk (center lane), consistent with its phosphorylation. Similar data was obtained when Mirk was co-expressed with p21 in C2C12 myoblasts (data not shown). The many functions of p21 rely on its association with a variety of proteins, including cyclins, CDKs, PCNA, and others, which associate with p21 within specific domains. It was essential to determine Mirk’s phosphorylation site in p21 in order to be able to postulate the biological effect of this phosphorylation. For initial studies, we measured the capacity of Mirk to phosphorylate in vitro a series of GST-p21 constructs that were mutated to the nonphosphorylatable alanine residue at known phosphorylation sites: S130 (the p38MAPK site (23)), T57 (the GSK3ß site (28)), S146 (a PKCζ site (29) and one of two Akt sites (30)). Mirk phosphorylated each of these constructs to a similar degree (data not shown), indicating that none of these sites was the Mirk phosphorylation site.
Fig. 2.

Mirk phosphorylates transfected Flag-p21 in vivo. (Top panel) Autoradiogram after SDS-PAGE of in vivo phosphorylation of wild-type Flag-p21 cotransfected into NIH3T3 cells with either wild-type or kinase inactive Mirk, and allowed to co-express for 24 hours. After 4 hours of labeling, Flag-p21 was immunoprecipitated with anti-Flag M2 antibody, and the immunoprecipitates were resolved on SDS-PAGE with Flag-p21 migrating at the expected position. Flag-p21 was phosphorylated only in the co-transfections with wild-type Mirk. The nonspecific doublet seen at the top of each lane serves as the loading control. (Bottom panel) Anti-Flag western blot showed that equal amounts of Flag-p21 were immunoprecipitated. The upper band represents light chain, and the lower Flag-p21. There appears to be a slight decrease in mobility in the p21 phosphorylated by Mirk/dyrk1B.
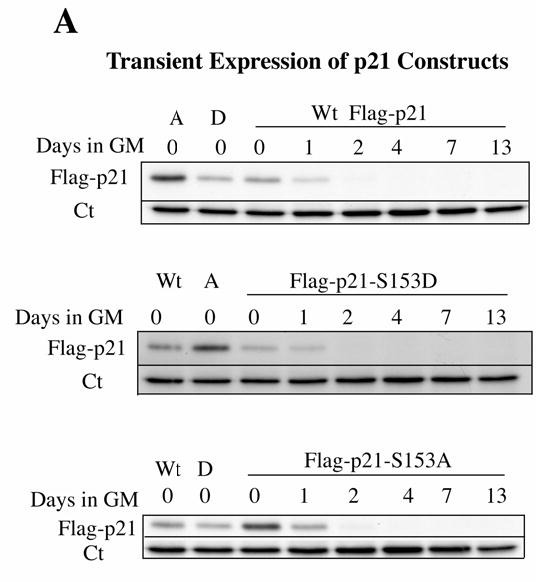
We then compared human and mouse p21 sequences for conserved threonine or serine residues within three or four residues of an arginine, the usual site of phosphorylation by Mirk, and mutated each of these six amino acids to the nonphosphorylatable alanine residue: S2A, S15A, S27A, T97A, T145A, S153A. Mutation of only one site, serine 153 to alanine, caused a reduction in in vitro phosphorylation by Mirk (data not shown). We confirmed that Mirk phosphorylated p21 at S153 in vitro by performing 2 dimensional phosphopeptide mapping following digestion of the phosphorylated p21 protein by trypsin. Three phosphopeptides were derived from wild-type p21 after in vitro phosphorylation by Mirk, but only one phosphopeptide was seen after phosphorylation of the mutant p21-S153A construct (Fig.3A). The slower migrating peptide on chromatography (long arrow) was probably a partial digestion product. The tryptic peptide containing serine-153 consists of amino acids 140–154 and is flanked by two KRR sequences, so trypsin could cleave at multiple points within both of these regions. Alternative cleavage may also have been promoted by steric hindrance resulting from phosphorylation of S153. Supporting this interpretation was the observation that the relative abundance of the two peptides varied in different experiments. However, the same general pattern was seen in three separate experiments, demonstrating that Mirk phosphorylates p21 at serine 153.
Fig. 3.
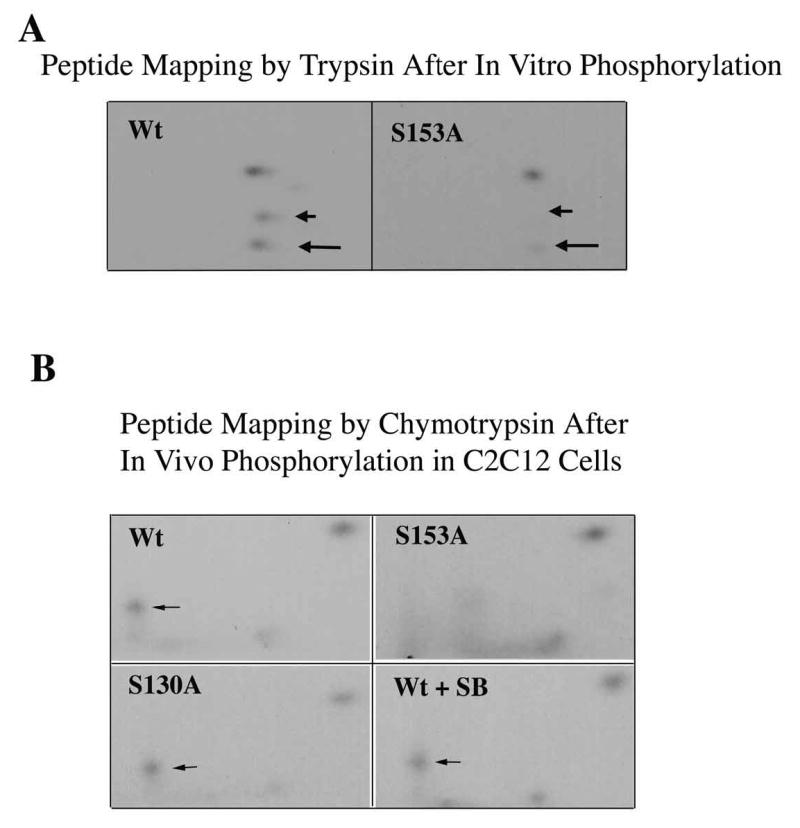
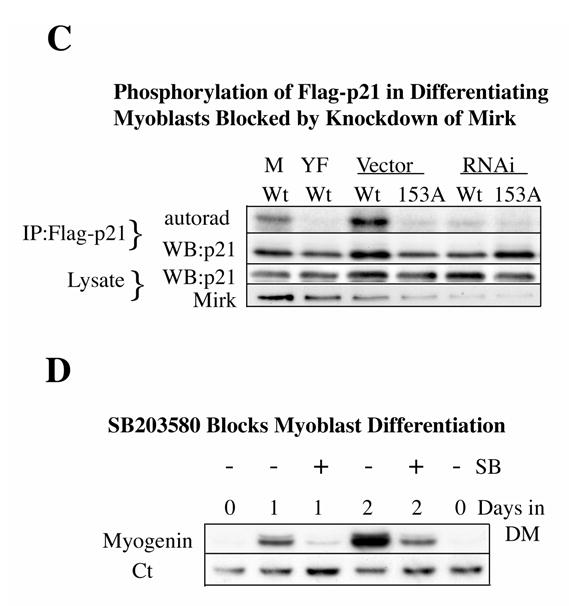
Mirk phosphorylates p21 at S153 in vitro and in differentiating myoblasts. A. Mirk phosphorylates p21 at S153 in vitro as shown by peptide mapping with trypsin. Wild-type (Wt) and mutant p21-S153A constructs were phosphorylated in vitro by purified recombinant Mirk, digested with trypsin and then subjected to two-dimensional peptide mapping. The peptide containing S153 and consisting of amino acids 143–154 is shown by a short arrow in both panels. The peptide identified by the longer arrow may be a partial digestion product containing S153, due to steric interference of the enzyme by the phosphate group. Similar data was seen in two additional peptide maps. B. Mirk phosphorylates p21 at S153 in vivo as shown by peptide mapping with chymotrypsin. Wild-type Flag-p21, mutant p21-S153A, and mutant p21-S130A were each cotransfected with wild-type Mirk into C2C12 cells. Constructs were allowed to express overnight, then cells were placed in myoblast differentiation medium for 24 hours, the last 4 hours of which cells were labeled with [32P]-orthophosphate. In one dish transfected with wild-type p21, 10 uM SB203580 was added at the same time as the label. Flag-p21 was immunoprecipitated with anti-Flag M2 antibody, the immunoprecipitates were digested with chymotrypsin, and then subjected to two-dimensional peptide mapping. The p21-S153A mutant incorporated about 40% as much label as wild-type p21 when normalized to the total amount of p21 immunoprecipitated. Chymotrypsin was used because it yields a simpler map with a broader distribution of peptides. The peptide indicated by the arrow is lost in the p21-S153A mutant, and is the presumed 152-159 amino acid peptide. C. Depletion of endogenous Mirk by RNA interference blocks the phosphorylation of transfected wild-type p21, but not p21-S153A mutant at the Mirk phosphorylation site. Cells were plated at 5x105 per 60 mm dish, cultured for 16 hours, then were transfected for 4 hours in serum-free medium with 2.5 ug of either Flag-p21 or Flag-p21-S153A, and 2.5 ug of pSilencer DNA encoding either RNAi to Mirk (RNAi) or vector. As controls, Flag-p21 was co-transfected with either wild-type Mirk (M) or kinase-inactive YF-Mirk (YF). Fetal bovine serum was then added to 10% to maintain cell viability during overnight expression, and cells were switched to OptiMEM differentiation medium for 24 hours, with 325 uCi 32P-orthophosphate added to each dish for the last 4 hours following 1 hour in phosphate-free medium. Anti-Flag epitope immunoprecipitates were separated by SDS-PAGE and the Flag-p21 bands were detected by autoradiography (top lanes), then by western blotting for Flag. The amount of p21 and Mirk in the lysates were determined by western blotting (lower 2 bands). D. SB203580 inhibits myoblast differentiation as measured by the level of expression of the myogenic regulatory factor myogenin which controls the differentiation program. C2C12 cells were placed in differentiation medium +/− 10 uM SB203580 for 1 and 2 days and the abundance of myogenin determined by western blotting. Ct, cross-reacting protein used as a loading control. E. Relative stability of mutant p21 constructs. C2C12 cells were transfected with either mutant Flag-p21-S153D, Flag-p21-S153A or wild-type Flag-p21, and constructs were allowed to express for 24 hours. Cycloheximide (CH) at 20 ug/ml was added to each culture and Flag-p21 and tubulin abundance were determined at the indicated times by western blotting, and normalized to the 0 time values. Mean +/− SE shown of 2 separate experiments.
In vivo phosphorylation of Flag-p21 by co-transfected Mirk occurs at S153 in differentiating myoblasts
In vivo phosphorylation is known to be more specific than in vitro phosphorylation, especially on a substrate such as p21 which exists in vivo in several different complexes, and is only found as a free molecule immediately after synthesis (31). In fact, in our previous study, p38MAPK phosphorylated p21 strongly at T57 and weakly at S130 in vitro, but in colon carcinoma cells only phosphorylated p21 at S130 (23). Mirk was co-transfected into proliferating myoblasts with either wild-type Flag-p21, the Mirk site mutant Flag-p21-S153A or the p38 site mutant Flag-p21-S130A. The cells were then switched to differentiation media for 24 hours, and labeled with [32P]-orthophosphate for the last 4 hours. The immunoprecipitated p21 constructs were digested with chymotrypsin before phosphopeptide mapping. Trypsin digestion yields 17 fragments from p21, while chymotrypsin yields 11, and only 6 of these peptide fragments contain either serine or threonine residues, resulting in a much simpler pattern. Additionally, the position of the peptide containing the p38MAPK phosphorylation site on p21 is readily identifiable in the chymotrypsin map (23). Mirk was found to phosphorylate p21 at S153 in differentiating myoblasts, in accordance with Mirk’s in vitro phosphorylation of p21. Mutation of p21 to S153A at the Mirk phosphorylation site blocked phosphorylation in vivo. Flag-p21 was phosphorylated on three phosphopeptides in vivo, while Flag-p21-S153A was phosphorylated on only two phosphopeptides (Fig.3B). The peptide containing S153 is indicated by an arrow in each panel. This peptide consists of amino acids 152–159 and contains only one serine and no threonines. Mutation of this peptide at serine 153 to alanine blocked its in vivo phosphorylation. Therefore, p21 is phosphorylated at S153 in differentiating myoblasts by Mirk.
Knockdown of endogenous Mirk by RNAi blocked phosphorylation of wild-type Flag-p21, but not Flag-p21-S153A, mutant at the Mirk phosphorylation site (Fig.3C), confirming that Mirk phosphorylates p21 at S153 in differentiating myoblasts in vivo. C2C12 cells were transfected with either wild-type or mutant p21 together with the pSilencer plasmid for Mirk RNAi or the vector alone. Parallel cultures were transfected with wild-type p21 and either wild-type Mirk or kinase-inactive YF-Mirk. Cells in DM were metabolically labeled with 32P-orthophosphate. The p21 constructs were immunoprecipitated by their Flag epitopes, and the extent of incorporation of the label determined by autoradiography after SDS-PAGE. Ectopic Mirk phosphorylated Flag-p21 in vivo while the kinase-inactive YF-Mirk did not (Fig.3C, first 2 lanes), and served as the positive and negative controls, respectively. Endogenous Mirk in the differentiating C2C12 cells phosphorylated wild-type p21, but not mutant p21-S153A (lanes 3 and 4), although similar amounts of p21 were immunoprecipitated. In the cultures in which Mirk knockdown was accomplished (last 2 lanes), neither wild-type Mirk nor the p21-S153A mutant was phosphorylated much above background levels. Therefore, endogenous Mirk phosphorylates p21 at S153 in vivo in differentiating myoblasts.
Mirk and p38MAPK interact in vivo. Mirk is activated by the p38MAPK kinase MKK3 (12) and p38MAPK can sequester Mirk in vivo (13). Thus, we questioned whether phosphorylation of p21 by p38MAPK occurred in differentiating myoblasts, as it does in colon carcinoma cells (23), and if so, whether it was an essential prerequisite for phosphorylation of p21 by Mirk. In one set of cultures in the peptide mapping studies (Fig.3B), 10 uM SB203580 was added to inhibit p38MAPK. During myoblast differentiation p38MAPK is activated, and in turn activates the myogenic regulatory factors MEF2A, MEF2C and MyoD (32), (33), (34). However, the in vivo peptide mapping studies (Fig.3B) show that p38MAPK does not appear to phosphorylate p21 in differentiating myoblasts, at least within the time frame of these experiments. Mutation of p21 at the p38MAPK site of S130 and treatment of cells with the p38 inhibitor SB203580 did not eliminate any of the three phosphorylated peptides. In addition, the S153-containing peptide was phosphorylated in the Flag-p21-S130A mutant construct to a similar extent as in wild-type p21, demonstrating that phosphorylation of p21 at S130 is not an essential prerequisite for phosphorylation by Mirk (Fig.3B). The SB203580 preparation used in these experiments was active because exposure of C2C12 cells in DM for 1 or 2 days to this drug prevented induction of myogenin, an essential myogenic regulatory factor, whose transcription is mediated by the p38MAPK substrate MEF2 (Fig.3D). Treatment of differentiating C2 myoblasts with SB203580 at 5 uM for 48 hours has been reported to block the kinase activity of p38 on the exogenous substrate MBP, but not to block the phosphorylation of p38 (35). Other investigators, as well as ourselves (data not shown) have also noted this discrepency. However, our studies do not entirely depend on the use of the inhibitor. There was no loss in phosphorylation of the p38 site mutant, p21-S130A, during myoblast differentiation since the peptide containing this mutant sequence was as equally phosphorylated as the wild-type peptide (Fig.3B). Therefore, we could detect no phosphorylation of p21 by p38MAPK during this initial period of myogenic differentiation.
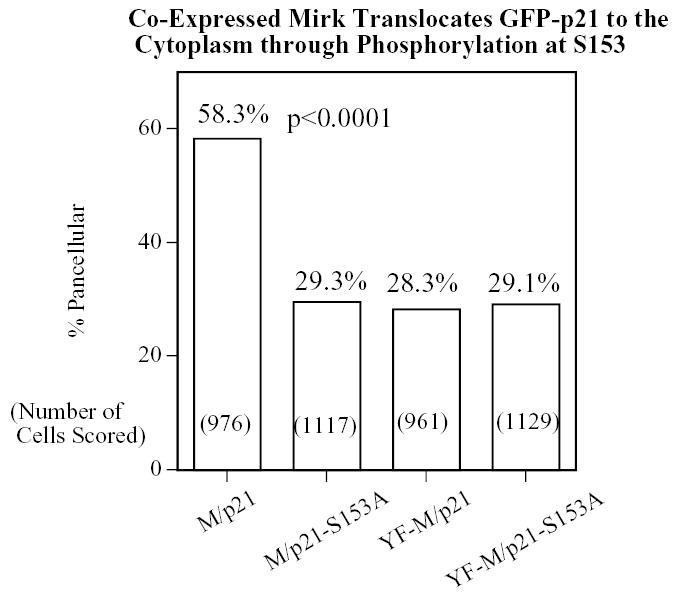
The phosphorylation of p21 by p38MAPK at S130 increased the stability of p21 in colon carcinoma cells (23). However, phosphorylation of p21 by Mirk did not stabilize p21 in C2C12 cells. In fact, p21-S153D was slightly less stable than wild-type p21, as demonstrated by cycloheximide arrest experiments (Fig.3E). Therefore, neither Mirk nor p38MAPK stabilizes p21 during the initial stages of myoblast differentiation. Furthermore, at this stage in differentiation when Mirk acts as a survival factor for myoblasts, only Mirk, not p38MAPK, phosphorylates p21.
Phosphomimetic p21-S153D is as effective as wild-type p21 in blocking cell cycle progression
Ectopic expression of p21 has been shown by others to decrease the percentage of myoblasts in S phase as assayed by BrdU incorporation using fluorescence microscopy (16). Similarly, we found that transient expression of GFP-p21 decreased the fraction of C2C12 myoblasts expressing GFP that showed BrdU incorporation from 30% in cells transfected with vector alone to 2.5% in cells expressing GFP-p21 (Table I). Transient expression of the phosphomimetic GFP-p21-S153D caused a similar arrest of cell growth, with only 1.9% of cells expressing this construct also positive for BrdU. Thus, p21 phosphorylated by Mirk remained capable of mediating growth arrest. These results were consistent with the known control of DNA replication by p21 and the retinoblastoma protein in terminally differentiated muscle cells (36).
Table 1.
BrdU Incorporation in C2C12 Cells Expressing GFP-p21 Constructs
| None | Vector | p21(WT) | p21(S153D) | |
|---|---|---|---|---|
| GFP | 910 | 973 | 922 | 935 |
| GFP+BrdU | 272 | 292 | 23 | 18 |
| % S-phase | 29.9 | 30.0 | 2.5 | 1.9 |
| (p<0.0001) | (p<0.0001) |
Phosphomimetic p21-S153D translocates to the cytoplasm
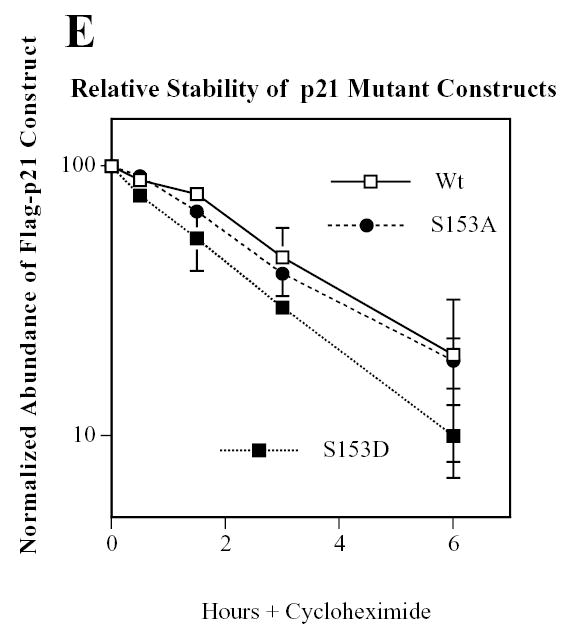
Serine 153 is within p21’s nuclear localization signal and within its PCNA binding domain (reviewed in (37)), so we predicted that phosphorylation at this residue would alter both the localization of p21 and its capacity to block DNA synthesis. In the current study we have concentrated on the localization of p21. We transiently transfected GFP-p21-wild-type, the phosphomimetic GFP-p21-S153D and the nonphosphorylatable GFP-p21-S153A into cycling C2C12 myoblasts (Fig.4A) and cycling NIH3T3 fibroblasts (Fig.4B). NIH3T3 cells were used to determine the generality of these observations since Mirk is widely distributed. Wild-type p21 and the nonphosphorylatable GFP-p21-S153A construct were predominately localized in the nucleus of both cell types. Nuclear localization was confirmed by counterstaining the nuclei with DAPI. The green fluorescent GFP and the blue fluorescent DAPI were merged in the last lane and showed overlap of each fluorochrome. In marked contrast to wild-type p21, the phosphomimetic GFP-p21-S153D construct was found in the cytoplasm as well as the nucleus in both C2C12 myoblasts and NIH3T3 cells (Fig.4). The GFP-p21-S153D was found distributed throughout the entire cell, allowing visualization of cytoplasmic extensions.
Fig. 4.
Fluorescence microscopy demonstrated that the Mirk-phosphorylation site phosphomimetic mutant p21-S153D is found in both the cytoplasm and the nucleus, while wild-type p21 and the non-phosphorylatable p21-S153A are localized exclusively in the nucleus in the majority of cells. GFP-p21 wild-type, GFP-p21-S153A which is not phosphorylated by Mirk, and GFP-p21-S153D, the Mirk site-phosphomimetic construct, were transiently expressed in C2C12 cells (panel A, 400X magnification) and in NIH3T3 cells (panel B, 1000X magnification). Nuclei were stained with DAPI and identical fields were photographed for GFP and DAPI. Images were merged with SPOT RT software. Scale bars = 50 um.
These observations were quantitated by counting 3824 transfected myoblasts and 1875 transfected fibroblasts (Table 2) and subjecting the data to the Chi square test. Since preliminary trials had demonstrated that GFP-p21 is always localized in the nucleus in this system, we used a 1-tailed analysis to compare the proportion of transfected cells expressing the GFP-constructs in both the cytoplasm and the nucleus to the proportion of cells expressing GFP-p21 exclusively in the nucleus. GFP-p21-wild-type was found in the cytoplasm in only 19.4% of myoblasts. Four times as many cells, 78.3%, expressed the Mirk-phosphomimetic GFP-p21-S153D construct in the cytoplasm. This gave a highly significant (p<0.0001) difference in localization. In contrast, the nonphosphorylatable GFP-p21-S153A construct remained localized in the nucleus in myoblasts, with only 15.6% of the cells expressing this construct in the cytoplasm, giving a statistically significant p value of p<0.02 compared to wild-type p21. Similar results were obtained with NIH3T3 cells (Table 2). Wild-type p21 was found in the cytoplasm of only 26.6% of transfected fibroblasts, while over twice as many transfected fibroblasts expressed the Mirk-phosphomimetic GFP-p21-S153D construct in the cytoplasm. This was also a highly significant difference in localization (p<0.0001). The nonphosphorylatable GFP-p21-S153A construct remained localized in the nucleus in fibroblasts, with only 22.4% of the cells containing this construct in the cytoplasm, a value which was not significantly different from cells expressing wild-type p21. These data clearly demonstrate that mutation of p21 to mimic phosphorylation by Mirk caused a much larger proportion of the p21 to localize in the cytoplasm in both myoblasts and fibroblasts. Mutation of an additional serine in the nuclear localization signal (S160 to S160A or S160D) did not affect the cellular distribution of GFP-p21 (data not shown).
Table 2.
Localization of GFP-p21 Constructs in C2C12 Cells and NIH3T3 Cells
| Cycling C2C12 Myoblasts | |||
|---|---|---|---|
| WT | S153A | S153D | |
| Nucleus+Cytoplasm | 246 | 200 | 998 |
| Nucleus Only | 1022 | 1082 | 278 |
| % Nucleus+Cytoplasm | 19.4 | 15.6 (p<0.02) | 78.3 (p<0.0001) |
| NIH3T3 Fibroblasts | |||
| WT | S153A | S153D | |
| Nucleus+Cytoplasm | 164 | 141 | 420 |
| Nucleus Only | 453 | 488 | 209 |
| % Nucleus+Cytoplasm | 26.6 | 22.4 | 66.8 (p<0.0001) |
Ectopic Mirk translocates GFP-p21 to the cytoplasm through phosphorylation at S153
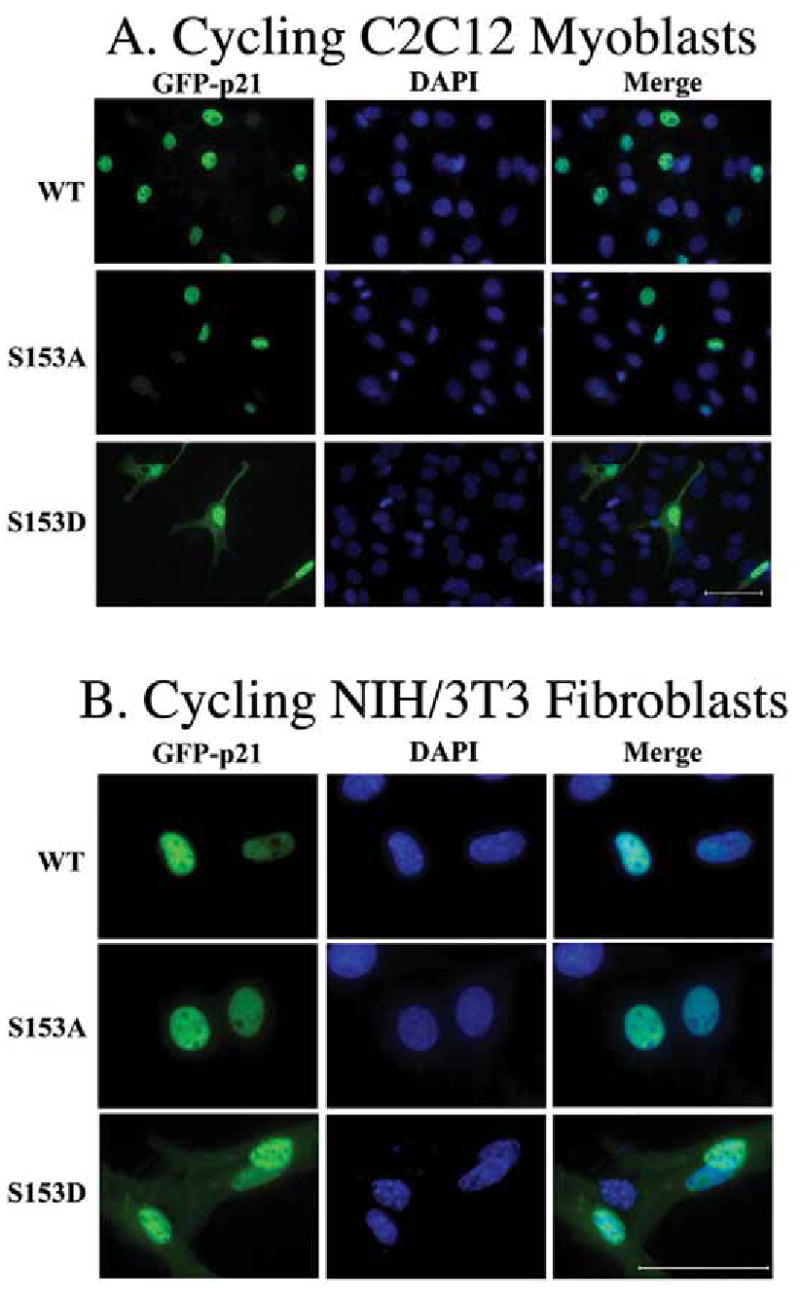
NIH3T3 fibroblasts were transfected with ectopic wild-type Mirk or kinase-inactive YF-Mirk and either wild-type GFP-p21 or GFP-p21-S153A. The cells expressing exogenous p21 and exogenous Mirk constructs were visualized by immunofluorescence. Cells expressing both constructs were visualized using a green/orange V2 filter set, and the percentage of such cells in which p21 was pancellular (versus restricted solely to the nucleus) was twice as large as the controls (Fig.5). Chi square analysis showed this to be a highly significant difference (p<0.0001). Expression of elevated levels of ectopic Mirk was sufficient to make the majority of wild-type p21 pancellular, while having no effect on the p21 mutant at the Mirk site (S153A). Moreover, the p21 translocation was dependent on Mirk’s kinase activity.
Fig. 5.
Overexpression of Mirk causes a portion of the wild-type p21 population to translocate to the cytoplasm in a Mirk kinase-dependent manner, but cannot translocate p21 mutated at the Mirk phosphorylation site. NIH3T3 fibroblasts were plated overnight in Labtek 2-well chamber slides (1.5 x 105 cells per well) and then transfected (2 ug plasmid DNA and 4 ul LipofectAMINE 2000 per well) with GFP-p21(p21) or the non-phosphorylatable GFP-p21(p21-S153A), together with either wild-type (M) or kinase inactive (YF-M) Mirk. Cells were transfected in DMEM containing 10% bovine calf serum. After 24 hours of expression, cells were fixed with 4% paraformaldehyde and the GFP-p21 protein was labeled by a 30 minute incubation with mouse anti-GFP monoclonal antibody (Santa Cruz 8334; 1:500), followed by anti-mouse Alexa Fluor 488. Mirk was visualized using rabbit polyclonal antibody to the Mirk C-terminus at 1:500, followed by anti-rabbit Alexa Fluor 594. An average of 1050 GFP-p21 expressing cells were observed for each transfectant. Cells were scored in 8–10 random fields in each of 3 separate preparations and the number of cells labeled for both GFP-p21 and transfected Mirk was determined using a Green/Orange V2 filter set (Chroma) that allowed simultaneous visualization of both fluorophores. Combined counts were analyzed by the Chi square test (Minitab) to determine the significance of differences between conditions. The total number of cells scored per condition is labeled on each bar.
More wild-type p21 is found in the cytoplasm when Mirk is induced during myoblast differentiation
The localization studies described above (Figs. 4 & 5) were performed in cycling myoblasts. We next transfected the same three GFP-p21 constructs into C2C12 cells, and then stimulated them to differentiate. Only the fused myoblasts were examined and cells were stained for actin so the myotubes would be unambiguous. As predicted, in each of four experiments wild-type GFP-p21 was also found in the cytoplasm of myotubes, while the nonphosphorylatable GFP-p21-S153A remained restricted to the nuclei of the multinucleated myotubes (Fig.6). The phosphomimetic GFP-p21-S153D served as the positive control and partitioned into both cytoplasm and nucleus as expected. Thus p21 was shifted from the nucleus into the cytoplasm by the presence of elevated levels of endogenous Mirk in differentiating myotubes.
Fig. 6.
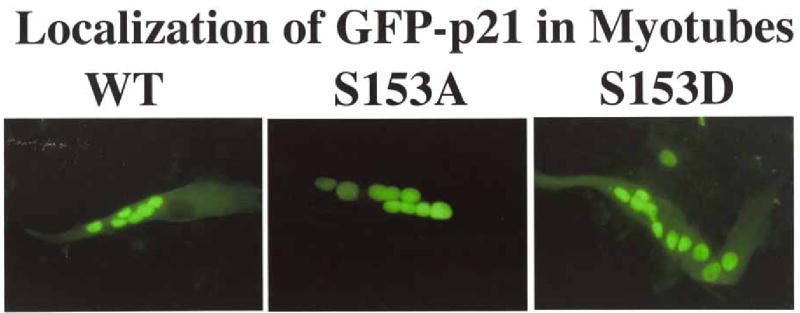
When Mirk is upregulated as a part of the myoblast differentiation program, both wild-type p21, as well as the Mirk-phosphorylation site phosphomimetic mutant p21-S153D, are translocated to the cytoplasm as shown by fluorescence microscopy. In contrast, the non-phosphorylatable p21-S153A is localized exclusively in the nuclei of the multinucleated myotubes. GFP-p21 wild-type, GFP-p21-S153A, and GFP-p21-S153D were transfected into C2C12 cells, which were then allowed to differentiate for 3 days. Similar data was seen in each of 4 experiments.
Changes in the localization of endogenous p21 during differentiation of C2C12 myoblasts
The data thus far confirmed that a significant proportion of exogenous p21 is localized in the cytoplasm of differentiating myoblasts following phosphorylation by Mirk. We next investigated the localization of endogenous p21 during differentiation of myoblasts. C2C12 myoblasts were cultured for one day in growth medium (GM), and then switched to differentiation medium (DM). After 24 hours in GM or 48 hours in DM, cells were fixed in paraformaldehyde, then endogenous p21 was visualized with the monoclonal antibody F-5. This antibody was raised against full length p21, so antibody binding should not be affected by phosphorylation at S153 in the C-terminus (Fig.7). As an additional control, p21 was labeled in separate experiments using the polyclonal antibody C-19 (data not shown). The anti-p21 antibody was detected with goat anti-mouse secondary antibody conjugated to green fluorescent Alexa Fluor 488. Cells were also incubated with Alexa Fluor 594 coupled to phalloidin to detect actin and DAPI for nuclear staining. All of the fields were merged to form the composite shown in the right-most column of the figure.
Fig. 7.
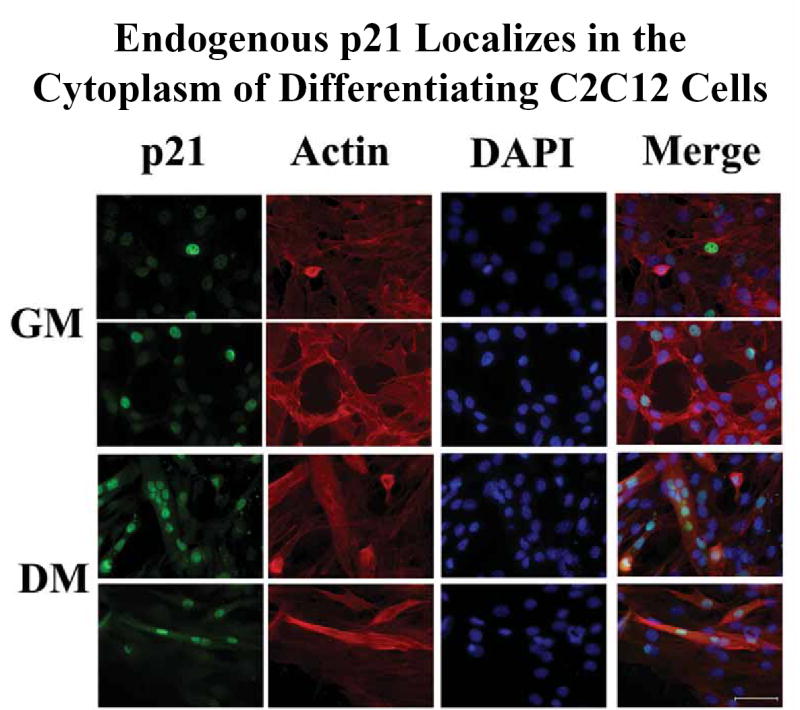
Endogenous p21 is exclusively nuclear in cycling C2C12 cells in growth medium (GM), but a portion of the endogenous p21 protein is translocated to the cytoplasm when C2C12 cells are treated for 48 hours with differentiation medium (DM). Endogenous p21 was visualized with the F-5 monoclonal antibody and goat anti-mouse antibody conjugated to Alexa Fluor 488, the nuclei were stained with DAPI, and actin within the entire cell body was visualized with phalloidin conjugated to Alexa Fluor 594. Identical fields were photographed for each fluorochrome (first three rows), then merged with SPOT RT software. Identical results were obtained with the C-19 polyclonal antibody. Scale bar = 50 um.
Endogenous p21 was exclusively localized in the nucleus in proliferating myoblasts in growth medium. The signals for p21 and for DAPI completely coincided (Fig.7, two fields of C2C12 cells are shown in the top 2 rows). When myoblasts were cultured for 2 days in differentiation medium, however, p21 was found in the cytoplasm as well as the nucleus in fused myoblasts (Fig.7, bottom two rows). Note in particular in the merged photo column, the large myotubes which are stained with actin to delineate the entire cell body and with DAPI to delineate nuclei. 11 nuclei were found in the myotube in the third row while 4 nuclei were found in the myotube in the fourth row. Both of these myotubes show extensive localization of endogenous p21 in their cytoplasm (Fig.7, p21 column, rows 3 and 4). These data clearly demonstrate that localization of endogenous p21 in the cytoplasm occurs as a natural part of myoblast differentiation. Thus the increase in concentration of the exogenous Flag-p21 within the cytoplasm of differentiating myotubes (Fig.6) is reflected in a similar cytoplasmic enrichment of the endogenous p21.
Endogenous p21 is found within the cytoplasm of adult human muscle
p21 is known to play critical roles in early muscle differentiation (18) and in the maintenance of adult muscle tissue through the muscle progenitor (satellite) cell population (19). However, there do not appear to be any extensive reports describing the presence or function of p21 in normal mature muscle. Therefore, we performed SAGE analysis using the NCBI/GEO database and found that p21 is expressed to a higher extent in muscle biopsies from young individuals, aged 21–31 years old, than in biopsies from most older patients, aged 62–77 (GDS156), but that all skeletal muscle biopsies contained p21 mRNA. We were unable to detect p21 in formalin-fixed paraffin sections of human muscle even after antigen retrieval with citrate, EDTA, or proteolytic digestion. This result is not unexpected given that p21 is normally found in complexes, where the epitope may not be accessible to the antibody, particularly after formalin fixation. We therefore performed immunofluorescence on frozen sections of adult human muscle. We were able to detect p21 in adult muscle using the C-19 antibody at a dilution of 1:200, followed by goat anti-rabbit Alexa Fluor 594 at 1:500 (Fig. 8). The distribution of p21 in the adult muscle fibers paralleled our observations of p21 localization in myotubes. p21 remained concentrated in the nucleus, but was distributed throughout the myofibrils. We were also able to see this pattern using the F-5 anti-p21 antibody, thus demonstrating the specificity of our immunolabeling results (data not shown). Therefore, examination of human skeletal muscle tissue confirmed that endogenous p21 partitioned into the cytoplasm as a natural part of muscle differentiation.
Fig. 8.
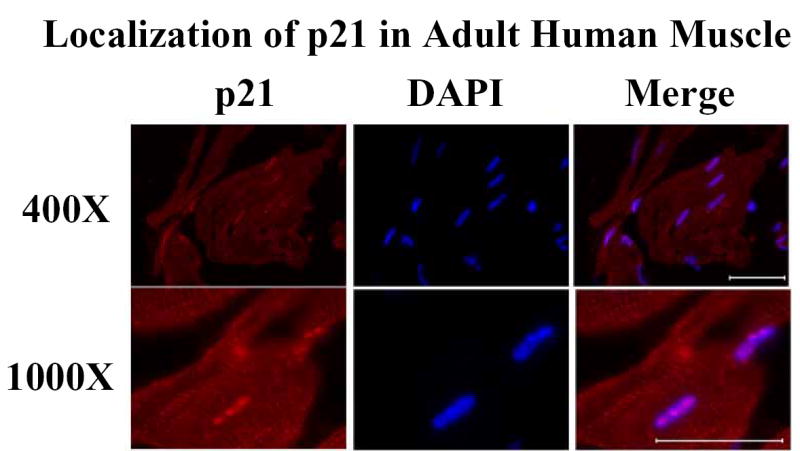
p21 localizes in both the cytoplasm and nucleus of adult skeletal muscle. Frozen sections of human muscle were analyzed for the localization of p21 by immunofluorescence using the polyclonal C-19 antibody (1:200) and goat anti-rabbit IgG Alexa Fluor 594 (1:500); nuclei were counterstained with DAPI. Scale bars = 50 um.
Phosphomimetic p21-S153D is more effective than wild-type p21 in blocking caspase 3 activation
Caspase 3 is the primary effector caspase in apoptosis. Caspase 3 is activated by cleavage of its precursor, procaspase 3. C2C12 myoblasts were transiently transfected with either wild-type Flag-p21, the phosphomimetic Flag-p21-S153D, the nonphosphorylatable Flag-p21-S153A, or vector alone. The constructs were allowed to express overnight, the cells were then cultured in differentiation medium for 16 hours, and the amount of active caspase 3, released by cleavage, was determined by western blotting. Wild-type p21 caused a small, 10% inhibition in the amount of activated caspase 3, while the Mirk phosphomimetic p21-S153D construct blocked caspase 3 activation 5 times as much (Fig.9). In contrast, little inhibition by the nonphosphorylatable construct was observed. In addition, the phosphomimetic p21 construct co-immunoprecipitated with ASK1 (data not shown), one upstream activator of this caspase cascade. ASK1 has been shown to be sequestered by wild-type p21 in other studies (22). Therefore, one mechanism by which Mirk blocks apoptosis in myoblasts is through phosphorylation of p21. A subpopulation of the p21 molecules is then retained in the cytoplasm where it blocks apoptosis through inhibition of the activation of caspase 3.
Fig. 9.
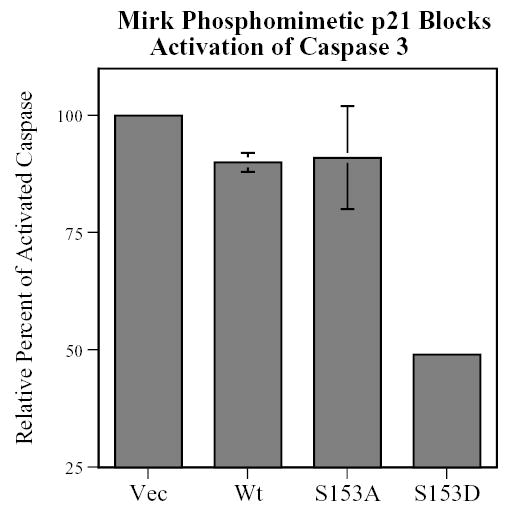
A p21 construct, phosphomimetic at the Mirk phosphorylation site, is twice as effective as wild-type p21 in blocking the activation of caspase 3. C2C12 myoblasts were transfected with Flag-p21 wild-type, Flag-p21-S153A, Flag-p21-S153D, or vector for 24 hours, cultured in differentiation medium for 16 hours, and then analyzed by SDS-PAGE and western blotting for activated caspase 3, normalized by western blotting for tubulin and the expression of exogenous Flag-p21 by western blotting for Flag. Mean +/− SD shown of 2 experiments.
Transient expression of phosphomimetic p21 aids myoblast survival
C2C12 cells were transiently transfected with either wild-type Flag-p21, the phosphomimetic Flag-p21-S153D or the nonphosphorylatable Flag-p21-S153A, and the length of time each construct was expressed was determined (Fig.10A). After 24 hours of transfection and expression, the cells were placed in growth medium for 0–13 days. All of the constructs expressed for only about 1 day in growth medium (Fig.10A), and then were lost. We used this transient expression to determine whether the phosphomimetic p21 construct could act as a survival factor in myoblasts at the time of plating at very low cell density for colony formation assays (Fig.10B). The same transfection protocol was used, except that a vector control was included. Transient expression of the phosphomimetic p21-S153D construct, and to a lesser extent the wild-type p21, increased myoblast colony formation (Fig.10B), a reflection of increased survival during the initial day on which cells were plated. Transient expression of p21-S153D for only one to two days (Fig.10A) thus inhibits caspase 3 activation during the initial plating period when cells are at very low density and susceptible to apoptosis. Loss of expression of the p21 construct after this period allows the cells to reenter the cell cycle and grow to form a discernible colony. In contrast, the phosphorylation site mutant p21-S153A had no survival effect compared to the vector (Fig.10B). Thus phosphorylation of p21 by Mirk enables p21 to oppose apoptosis and to enhance myoblast survival.
Fig. 10.
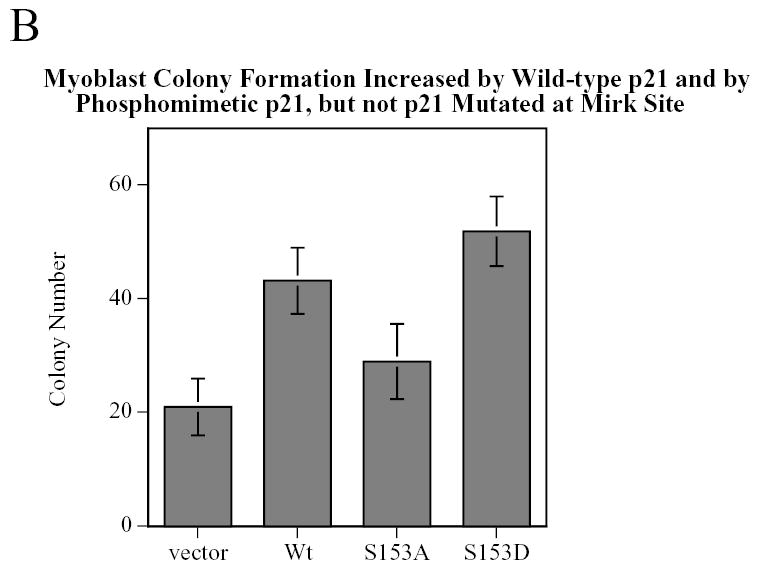
Transient expression of a p21 construct, phosphomimetic at the Mirk phosphorylation site, increases the viability of C2C12 cells. C2C12 myoblasts were transfected with Flag-p21 wild-type, Flag-p21-S153A, Flag-p21-S153D, or vector for 4 hours, allowed to express for 24 hours (0 point) then swtiched to serum-containing growth medium for 0–13 days. A. Each construct was expressed for only about 1 day in growth medium following the transfection and initial 24 hour expression period, as determined by western blotting for the Flag epitope. The zero time points for each construct were examined in parallel to the zero time points for the other two constructs on each blot as internal controls. Ct, cross-reacting protein used as a loading control. B. After 24 hours, the cells were trypsinized and plated at either 500 or 1000 cells in 100 mm tissue culture dishes, each in triplicate. Data shown is the mean of two such experiments, normalized to the number of colonies per 1000 cells plated.
DISCUSSION
In the current study we have demonstrated that the kinase Mirk mediates cell survival of myoblasts, during both growth and differentiation. These observations complement the known survival function of Mirk in colon carcinoma cells (20). We also show that at least some of the pro-survival functions of Mirk can be ascribed to its phosphorylation of the CDK inhibitor p21, and p21’s subsequent increased localization in the cytoplasm. Cytoplasmic p21 has a role in pro-survival signaling in differentiating myoblasts (Fig.9) and is maintained in the cytoplasm of mature myofibrils. The anti-apoptotic function of p21 in myogenesis is well-documented. In C2 myoblasts, ectopic expression of p21 has been shown to block apoptosis, while depletion of p21 by antisense oligonucleotides induced apoptosis (15). A role for cytosolic p21 has also been demonstrated in neuronal differentiation. After functioning in the nucleus to block cell proliferation, cytoplasmic p21 promotes neuronal differentiation where it regulates Rho-induced actin remodeling leading to neurite outgrowth (38). Cytoplasmic p21 has also recently been shown to enhance axonal regeneration and functional recovery after spinal cord injury (39).
Cell cycle progression is mediated by the sequential activation of members of the CDK protein kinase family. Cell cycling is blocked when cyclin/CDK complexes cannot form or when the catalytic activity of these complexes is blocked through binding of a CDK inhibitor molecule. One of these inhibitors is p21cip1/waf1 (40), which was originally identified as a PCNA binding protein (41). p21 inhibits DNA replication by preventing PCNA from contributing to DNA polymerase delta and epsilon function (42). The p21 gene is induced by the tumor suppressor p53 (43) and by cellular senescence (44). p21 is a major mediator of the G1 growth arrest induced by activation of the tumor suppressor p53 in response to DNA damage (reviewed in (45)). Other major functions for p21 are to promote assembly of cyclin D/CDK4 complexes and to increase cyclin D1 accumulation by direct inhibition of GSK3ß-triggered nuclear export (46), (47).
The cell cycle modulatory activity of p21 is tightly correlated with its nuclear localization. Movement of p21 from the nucleus to the cytoplasm blocks its cell cycle inhibitory activity and contributes to the cell growth induced by the oncogene Her-2/neu (21). Akt activated by Her-2/neu phosphorylates T145 and S146 within the nuclear-localization signal of p21 (21), (48), (30). Phosphorylation of p21 by Akt has been shown to promote association with 14-3-3 proteins, which function as nuclear export proteins (49). In the current study, we have documented that the kinase Mirk, like Akt, also promotes translocation of p21 to the cytoplasm by phosphorylation within the nuclear-localization signal of p21. In the case of Mirk, the phosphorylation site is serine 153, close to the Akt sites of threonine 145 and serine 146. A phosphomimetic mutant, p21-S153D, was found in the cytoplasm in 78% of C2C12 myoblasts and 67% of NIH3T3 fibroblasts, while wild-type p21 and the nonphosphorylatable mutant, p21-S153A were found in the cytoplasm in only 20–25% of transfected myoblasts and fibroblasts. The phosphomimetic mutant, p21-S153D, was still as capable of blocking myoblast cell cycling as ectopic wild-type p21 (Table 1). Because the p21-S153D construct was overexpressed, we speculate that enough p21-S153D molecules remained in the nucleus to maintain CDK2/cyclin complexes in the inactive form in these experiments. Differentiated myoblasts do remain in G0 arrest, moreover, suggesting that the elevated levels of endogenous p21 seen in myotubes are sufficient to enable adequate nuclear levels of p21 to be maintained to keep CDK2 activity in check. These elevated p21 levels would allow a fraction of the p21 population to be diverted to the cytoplasm to block apoptosis.
Endogenous p21 in differentiating myotubes displayed a similar localization pattern as the phosphomimetic construct. In growth medium where Mirk levels are low because most cells are cycling (1), endogenous p21 was restricted to the nucleus. When myoblasts arrest in G0/G1 and begin to differentiate, Mirk levels rise (1) and p21 begins to accumulate in the cytoplasm, though the majority of it is still found in the nucleus. This dual localization of p21 persists in adult human skeletal muscle. A number of recent reports suggest possible roles that Mirk and p21 might play in the cytoplasm of differentiating myotubes. In vascular smooth muscle cells, expression of a nuclear localization signal deficient p21 construct resulted in increased cytosolic levels of p21 and increased cell cycle transit in response to serum mitogens and to the mitogen PDGF-BB (50). There is also a growing body of evidence suggesting that breast cancer cell growth induced by the oncogene Her-2/neu is supported in part by a diminished capacity of p21 to act as a cell cycle inhibitor when it is removed from the nucleus as well as through the anti-apoptotic functions of p21. When p21 translocates to the cytoplasm it can function in the protection of cells against apoptosis through forming a physical complex with the apoptosis signal-regulating kinase 1 (ASK1) and by inhibiting activation of stress-activated ASK1 (22) and SAPK/JNK (51), (reviewed in (52)).
There may be a broader function for cytoplasmic translocation of p21 by Mirk in oncogenesis. Cytoplasmic p21 has been suggested to function as an oncoprotein (52), and Mirk is expressed in several solid tumors (53), (20). Cytoplasmic localization of p21 following phosphorylation by Akt has been identified in Her-2/neu overexpressing breast cancers where it correlates with poor prognosis (54). The effects of cytoplasmic p21 in tumors are likely to be wide-ranging and highly complex. In addition to the known effects of p21 in modulation of various apoptotic molecules, there is emerging evidence that cytoplasmic p21 may act upon several other signaling pathways. For example, accumulation of p21 in the cytoplasm was recently shown to mediate the cytoprotective effect of p21 against the chemotherapeutic drug paclitaxel (49). In addition, oncogenic H-RasG12V has been shown to induce cytoplasmic localization of p21 in ras-transformed NIH3T3 cells (55). This cytoplasmic p21 complexed with Rho kinase (ROCK) and blocked phosphorylation of cofilin, thus inhibiting the formation of actin stress fibers. As these examples demonstrate, localization of p21 in the cytoplasm of transformed cells might contribute not only to increased survival under environmental and/or pharmacological stress, but may also contribute to cell motility, and thus enhance tumor cell invasion.
Acknowledgments
We would like to thank Dr. Anna-Luise Katzenstein for providing us with specimens of normal muscle tissue; Dr. Howard Chang for assistance with frozen sections and Julie Lippa for assistance with paraffin section immunohistochemistry.
Footnotes
Supported by Public Health Service Award RO1 CA67405 to E.F.
References
- 1.Deng X, Ewton D, Pawlikowski B, Maimone M, Friedman E. J Biol Chem. 2003;278:41347–41354. doi: 10.1074/jbc.M306780200. [DOI] [PubMed] [Google Scholar]
- 2.Deng X, Ewton D, Mercer S, Friedman E. J Biol Chem. 2005;280:4894–4905. doi: 10.1074/jbc.M411894200. [DOI] [PubMed] [Google Scholar]
- 3.Leder S, Czajkowska H, Maenz B, de Graaf K, Barthel A, Joost H-G, Becker W. Biochem J. 2003;372:881–888. doi: 10.1042/BJ20030182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kentrup H, Becker W, Heukelbach J, Wilmes A, Schurman A, Huppertz C, Kainulainen H, Joost H-G. J Biol Chem. 1996;271:3488–3495. doi: 10.1074/jbc.271.7.3488. [DOI] [PubMed] [Google Scholar]
- 5.Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor F, Joost H-G. J Biol Chem. 1998;273:25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- 6.Himpel S, Tegge W, Frank R, Leder S, Joost H, Becker W. J Biol Chem. 2000;275:2431–2438. doi: 10.1074/jbc.275.4.2431. [DOI] [PubMed] [Google Scholar]
- 7.Leder S, Weber Y, Altafaj X, Estivill X, Joost H-G, Becker W. Biochem Biophys Res Com. 1999;254:474–479. doi: 10.1006/bbrc.1998.9967. [DOI] [PubMed] [Google Scholar]
- 8.Moriya H, Shimizu-Yoshida Y, Omori A, washita S, Katoh M, Sakai A. Genes Dev. 2001;150:1217–1228. doi: 10.1101/gad.884001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taminato A, Bagattini R, Gorjao R, Chen G, Kuspa A, Souza GM. Mol Biol Cell. 2002;13:2266–2275. doi: 10.1091/mbc.01-11-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng X, Mercer S, Shah S, Ewton D, Friedman E. J Biol Chem. 2004;279:22498–22504. doi: 10.1074/jbc.M400479200. [DOI] [PubMed] [Google Scholar]
- 11.Zou Y, Ewton D, Deng D, Mercer S, Friedman E. J Biol Chem. 2004;279:27790–27798. doi: 10.1074/jbc.M403042200. [DOI] [PubMed] [Google Scholar]
- 12.Lim S, Jin K, Friedman E. J Biol Chem. 2002;277:25040–25046. doi: 10.1074/jbc.M203257200. [DOI] [PubMed] [Google Scholar]
- 13.Lim S, Zou Y, Friedman E. J Biol Chem. 2002;277:49438–49445. doi: 10.1074/jbc.M206840200. [DOI] [PubMed] [Google Scholar]
- 14.Zou Y, Lim S, Lee K, Deng X, Friedman E. J Biol Chem. 2003;278:49573 – 49581. doi: 10.1074/jbc.M307556200. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Walsh K. Science. 1996;273:369–361. doi: 10.1126/science.273.5273.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo K, Wang J, Andres V, Smith R, Walsh K. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng C, Zhang P, Harper J, Elledge S, Leder P. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ. Genes Dev. 1999;13:213–224. doi: 10.1101/gad.13.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawke T, Meeson A, Jiang N, Graham S, Hutcheson K, DiMaio J, Garry D. Am J Physiol Cell Physiol. 2003;285:C1019–1027. doi: 10.1152/ajpcell.00055.2003. [DOI] [PubMed] [Google Scholar]
- 20.Lee K, Deng X, Friedman E. Cancer Research. 2000;60:3631–3637. [PubMed] [Google Scholar]
- 21.Zhou B, Liao Y, Xia W, Lee M-H, Hung M-C. Nature Cell Biology. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 22.Asada M, Yamada T, chijo H, Delia D, Miyazono K, Fukumuro K, Mitzutani S. EMBO J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim G-Y, Mercer S, Ewton D, Yan Z, Jin K, Friedman E. J Biol Chem. 2002;277:29792–29802. doi: 10.1074/jbc.M201299200. [DOI] [PubMed] [Google Scholar]
- 24.Sherman M, Goldberg A. PNAS. 1993;90:8648–8652. doi: 10.1073/pnas.90.18.8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mogk A, Tomoyasu T, Goloubinoff P, Rüdiger S, Röder D, Langen H, Bukau B. EMBO J. 1999;18:6934–6949. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamant S, Ben-Zvi AP, Bukau B, Goloubinoff P. J Biol Chem. 2000;275:21107–21113. doi: 10.1074/jbc.M001293200. [DOI] [PubMed] [Google Scholar]
- 27.Ostrovsky O, Bengal E. J Biol Chem. 2003;278:21221–21231. doi: 10.1074/jbc.M211357200. [DOI] [PubMed] [Google Scholar]
- 28.Rossig L, Badorff C, Holzmann Y, Zeiher AM, Dimmeler S. J Biol Chem. 2002;277:9684–9689. doi: 10.1074/jbc.M106157200. [DOI] [PubMed] [Google Scholar]
- 29.Scott M, ngram A, Ball K. EMBO J. 2002;21:6771–6780. doi: 10.1093/emboj/cdf684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Dowbenko D, Lasky L. J Biol Chem. 2002;277:11352–11361. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 31.Prall OWJ, Carroll JS, Sutherland RL. J Biol Chem. 2001;276:45433–45442. doi: 10.1074/jbc.M104752200. [DOI] [PubMed] [Google Scholar]
- 32.Yang S-H, Galanis A, Sharrocks AD. Mol Cell Biol. 1999;19:4028–4038. doi: 10.1128/mcb.19.6.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J. Mol Cell Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Z, Woodring P, Bhakta K, Tamura K, Wen F, Feramisco J, Karin M, Wang J, Puri P. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez I, Tripathi G, Carter EJ, Cobb LJ, Salih DAM, Lovett FA, Holding C, Pell JM. Mol Cell Biol. 2004;24:3607–3622. doi: 10.1128/MCB.24.9.3607-3622.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mal A, Chattopadhyay D, Ghosh MK, Poon RYC, Hunter T, Harter ML. J Cell Biol. 2000;149:281–292. doi: 10.1083/jcb.149.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dotto G. Biochim Biophys Acta. 2000;31:M43–56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka H, Yamashita T, Asada M, Mizutani S, Yoshikawa H, Tohyama M. J Cell Biol. 2002;158:321–329. doi: 10.1083/jcb.200202071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka H, Yamashita T, Yachi K, Fujiwara T, Yoshikawa H, Tohyama M. Neuroscience. 2004;127:155–164. doi: 10.1016/j.neuroscience.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Harper J, Adami G, Wei N, Keyomarsi K, Elledge S. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 41.Xiong Y, Zhang H, Beach D. Cell. 1992;71:505–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- 42.Waga S, Hannon G, Beach D, Stillman B. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 43.El-Deiry W, Tokino T, Velculescu V, Levy D, Parsons R, Trent J, Lin D, Mercer W, Kinzler K, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 44.Noda A, Ning Y, Tadich P, Venable S, Pereira-Smith O, Smith J. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 45.Scherr C, Roberts J. Genes & Devel. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 46.Alt J, Gladden A, Diehl J. J Biol Chem. 2002;277:8517–8523. doi: 10.1074/jbc.M108867200. [DOI] [PubMed] [Google Scholar]
- 47.LaBaer J, Garrett M, Stevenson L, Slingerland J, Sandhu C, Chou H, Fattaey A, Harlow E. Genes & Development. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 48.Rossig L, Jadidi A, Urbich C, Badorff C, Zeiher A, Dimmeler S. Mol Cell Biol. 2001;21:5644–5657. doi: 10.1128/MCB.21.16.5644-5657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heliez C, Baricault L, Barboule N, Valette A. Oncogene. 2003;22:3260–3268. doi: 10.1038/sj.onc.1206409. [DOI] [PubMed] [Google Scholar]
- 50.Dong Y, Chi S, Borowsky A, Fan Y, Weiss R. Cell Signalling. 2004;16:263–269. doi: 10.1016/s0898-6568(03)00136-0. [DOI] [PubMed] [Google Scholar]
- 51.Shim J, Lee H, Park J, Kim H, Choi E. Nature. 1996;381:804–806. doi: 10.1038/381804a0. [DOI] [PubMed] [Google Scholar]
- 52.Blagosklonny M. Cell Cycle. 2002;1:391–393. doi: 10.4161/cc.1.6.262. [DOI] [PubMed] [Google Scholar]
- 53.Lee K-M, Friedman E. Proc AACR. 1998;39:273. [Google Scholar]
- 54.Xia W, Chen J-S, Zhou X, Sun P-R, Lee D-F, Liao Y, Zhou BP, Hung M-C. Clin Cancer Res. 2004;10:3815–3824. doi: 10.1158/1078-0432.CCR-03-0527. [DOI] [PubMed] [Google Scholar]
- 55.Lee S, Helfman D. J Biol Chem. 2004;279:1885–1891. doi: 10.1074/jbc.M306968200. [DOI] [PubMed] [Google Scholar]