

Abstract
γ-Secretase facilitates the regulated intramembrane proteolysis of select type I membrane proteins that play diverse physiological roles in multiple cell types and tissue. In this study, we used biochemical approaches to examine the distribution of amyloid precursor protein (APP) and several additional γ-secretase substrates in membrane microdomains. We report that APP C-terminal fragments (CTFs) and γ-secretase reside in Lubrol WX detergent-insoluble membranes (DIM) of cultured cells and adult mouse brain. APP CTFs that accumulate in cells lacking γ-secretase activity preferentially associate with DIM. Cholesterol depletion and magnetic im-munoisolation studies indicate recruitment of APP CTFs into cholesterol- and sphingolipid-rich lipid rafts, and co-residence of APP CTFs, PS1, and syntaxin 6 in DIM patches derived from the trans-Golgi network. Photoaffinity cross-linking studies provided evidence for the preponderance of active γ-secretase in lipid rafts of cultured cells and adult brain. Remarkably, unlike the case of APP, CTFs derived from Notch1, Jagged2, deleted in colorectal cancer (DCC), and N-cadherin remain largely detergent-soluble, indicative of their spatial segregation in non-raft domains. In embryonic brain, the majority of PS1 and nicastrin is present in Lubrol WX-soluble membranes, wherein the CTFs derived from APP, Notch1, DCC, and N-cadherin also reside. We suggest that γ-secretase residence in non-raft membranes facilitates proteolysis of diverse substrates during embryonic development but that the translocation of γ-secretase to lipid rafts in adults ensures processing of certain substrates, including APP CTFs, while limiting processing of other potential substrates.
Sequential processing of amyloid precursor protein (APP)1 by β - and γ-secretases releases the 39–42-amino acid-long β-amy-loid (Aβ) peptides, which accumulate in the brains of aged individuals and patients with Alzheimer disease (AD) (1). The major β-secretase in neurons is an aspartyl protease termed BACE-1, which cleaves APP within the luminal domain, generating the N terminus of Aβ (2). The C terminus of Aβ is generated by intramembranous cleavage of APP C-terminal fragments (CTFs) by γ-secretase, a multiprotein complex made of four essential components, presenilin (PS) 1 (or PS2), nicastrin, PEN-2, and APH-1 (3). Mutations in PSEN1 and PSEN2, encoding multipass membrane proteins PS1 and PS2, respectively, co-segregate with the majority of cases of autosomal dominant familial early-onset AD (4). Familial AD-linked PS1 and PS2 variants elevate the production of highly fibrillogenic Aβ42 peptides (1). A role for PS1 in Notch function was first discovered in Caenorhabditis elegans screens and involves intramembranous cleavage of the Notch receptor, analogous to APP processing (5, 6). Nicastrin is a type I membrane protein independently identified as a novel component of the GLP-1/Notch signaling pathway in C. elegans early embryos and in the biochemical characterization of proteins that interacted with PS1 (7, 8). Multitransmembrane protein APH-1 and the two-transmembrane protein PEN-2 were also identified in C. el-egans screens as genes essential for Notch signaling (9, 10). The four components of the γ-secretase interact early during biogenesis and cooperatively exit the ER. Nicastrin then undergoes complex glycosylation, and PS1 undergoes endoproteolysis to generate N- and C-terminal derivatives to generate the functional γ-secretase (3).
Compelling evidence from the transition state analog inhibitors show that γ-secretase is a transmembrane aspartyl protease, and two Asp residues essential for γ-secretase activity have been localized to the transmembrane domains 6 and 7 of PS1 (11). In contrast to the complex nature of the functional γ-secretase assembly, intramembranous cleavage by γ-secretase shows poor sequence specificity and does not appear to involve strict rules for substrate recognition. However, efficient cleavage by γ-secretase requires the removal of the extracellular domain by juxtamembrane proteolysis, leaving membrane-bound CTFs (12). Consequently, several type I membrane proteins that undergo ectodomain shedding, mediated by a set of metalloproteases termed “α-secretases,” are also substrates for intramembranous cleavage by the γ-secretase. To date, known γ-secretase substrates include APP homologs, Notch1 and homologs, Notch ligands Jagged2 and Delta, ErbB4, CD44, low density lipoprotein receptor-related protein, N- and E-cadherins, nectin-1α, deleted in colorectal cancer (DCC), p75 neurotrophin receptor, syndecan 3, etc. Exactly how such diverse sets of molecules residing in multiple subcellular sites are recognized by the γ-secretase is not well understood. Similarly, with the exception of Notch signaling, there is only limited information available on the functional consequence downstream of intramembranous cleavage of several γ-secretase substrates.
We reported recently on the cholesterol-dependent association of each of the γ-secretase components with lipid rafts in post-Golgi and endosome membranes enriched in syntaxin 6, syntaxin 13, and VAMP4 (13). Lipid rafts are cellular membrane microdomains rich in cholesterol and sphingolipids (14). These specialized detergent-insoluble microdomains (DIM) contribute to trafficking of proteins and lipids in the secretory and endocytic pathways by regulating vesicle sorting and formation (15). In addition, lipid rafts serve as versatile platforms wherein certain plasma membrane receptors and kinases regulate signal transduction (14). There is growing evidence to suggest that amyloidogenic processing by secretases may also be compartmentalized in lipid rafts by targeting BACE-1, γ-secretase, and APP to DIMs (13, 16–20). Most interestingly, only a small fraction of endogenous APP is found in DIMs, raising concerns whether γ-secretase is active exclusively within ordered lipid raft domains or not. Furthermore, the role of lipid rafts in intramembranous cleavage of other γ-secretase substrates has not been investigated.
In this study we investigated the distribution of select γ-secretase substrates in cholesterol-rich raft domains isolated by sucrose density gradient fractionation of detergent-insoluble membranes. We report that the majority of APP CTFs and active γ-secretase resides in lipid rafts in cultured cells and adult mouse brain. On the other hand, CTFs of Notch1, Jagged2, and DCC are predominantly found in non-raft membrane domains. While addressing this apparent discrepancy, we found that PS1, APP, and other γ-secretase substrates, including DCC and N-cadherin, localize to non-raft membranes in developing embryonic brain. We propose that spatial segregation of γ-secretase within cholesterol- and sphingolipid-rich ordered lipid domains limits access to certain γ-secretase substrates in the adult.
EXPERIMENTAL PROCEDURES
Cell Culture
Mouse N2a neuroblastoma stable cell lines were cultured in 45% Dulbecco’s modified Eagle’s medium and 50% Opti-MEM (Invitrogen) supplemented with 5% fetal bovine serum. The stably transfected N2a cell line Swe.10 expressing c-Myc-tagged human APP695 harboring “Swedish” double mutation (APPSwe), N2a cell line Wt.11 co-expressing APPSwe and human wild-type (wt) PS1, and pooled stable N2a transfectants co-expressing APPSwe and wt PS1 or PS1 D385A have been described previously (21–23). Wild-type and PS1−/−/PS2−/− embryonic fibroblasts, NIH 3T3 cells stably expressing human Jagged2, and PS1+/− mouse embryonic fibroblasts stably expressing Notch1–6mycGFP were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (24–26). For γ-secretase inhibitor treatments, cells grown to 70% confluency were treated either with Me2SO, 10 nM compound E (CompE), or 1 μM L-685,458 for 18 h. For depleting cholesterol, confluent cultures were washed once with warm phosphate-buffered saline and incubated with 5 mM methyl-β-cyclodextrin (MβCD) in serum-free medium for 2 h at 37 °C (13).
Antibodies
The following antibodies were used in this study: PS1NT was raised against residues 1–65 of PS1 (27); antibody Ab14 was raised against residues 1–25 of PS1 (28) (provided by Dr. Sam Gandy, Thomas Jefferson University; Dr. Huaxi Xu, The Burnham Institute); αPS1Loop was raised against residues 263–407 of PS1 (28); SP716 was raised against residues 62–93 of nicastrin (29); 3925 recognizes C-terminal 19 amino acids of nicastrin (30) (provided by Dr. Philip Wong, The Johns Hopkins University School of Medicine); PNT2 was raised against residues 1–26 of PEN-2 (13); O2C2 recognizes C-terminal 20 amino acids of the long isoform of human APH-1a (provided by Drs. Paul Fraser and Peter St George-Hyslop, the University of Toronto) (31); anti-BACE-1 was raised against N-terminal 46–163 amino acids (32) (provided by Dr. Philip Wong); 369 was raised against the cytosolic tail of APP (33) (provided by Dr. Sam Gandy, Thomas Jefferson University); and 26D6 is a monoclonal antibody raised against Aβ residues 1–12 (Merck) (34). The following antibodies were purchased from commercial sources: flotillin-2, syntaxin 6, γ-adaptin, Notch1, N-cadherin, and DCC were from BD Transduction Laboratories; OKT8 and 9E10 were from the ATCC; caveolin-1 was from Santa Cruz Biotechnology; and calnexin was from Stressgen.
Lipid Raft Isolation
Lipid rafts were isolated from Lubrol WX ly-sates of cultured cells by discontinuous flotation density gradients as described previously (13). PS1+/− mouse embryonic fibroblasts stably expressing Notch1–6mycGFP were washed with Hanks’ buffered salt solution and treated with 10 mM EDTA for 30 min to induce Notch cleavage (26) prior to lipid raft isolation. Lipid rafts were isolated from mouse forebrain as described previously (35) with some modifications. Briefly, 200–300 mg of tissue from brains of 12-month-old wt mice or E14–16 embryos generated by intercrossing C57Bl/6J PS1+/− mice (36) were homogenized with a glass-Teflon homogenizer in 2 ml of buffer A containing 20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 250 mM sucrose, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor mixture (Sigma). After passage through a 25-gauge needle three times, homogenates were spun at 960 × g for 10 min at 4 °C. The supernatant was collected, and the pellet was resuspended in 1 ml of buffer A, which was passed through a 25-gauge needle five times and spun as described above. The two supernatants were pooled and adjusted to 0.5% Lubrol WX, 25 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 5 mM EDTA and mixed at 4 °C for 20 min. The lysate was then adjusted to 45% final concentration of sucrose and centrifuged as described previously (13).
Immunoisolation of Raft Patches Using Magnetic Beads
Mono-clonal syntaxin 6 antibody was bound to Dynabeads M-280 beads pre-coated with anti-mouse IgG (Dynal) according to the manufacturer’s instructions. Antibody OKT8 (recognizes CD8α) was used as negative control to establish the specificity of the immunoisolation procedure. Syntaxin 6 containing raft patches were immunoisolated from pooled lipid raft fractions as described previously (13).
Photoaffinity Cross-linking
Flotation density gradient fractions 4 and 5 (raft) or 8–12 (non-raft) were pooled and incubated with 1 μM L-852,505, a photoactive, biotinylated derivative of the aspartyl protease transition state analog of L-685,458 (37), at 4 °C for 1 h followed by irradiation with 365 nm UV light, using a Stratalinker (Stratagene), on ice for 90 min. Biotinylated proteins were captured with streptavidin-agarose beads (Pierce) at 4 °C and eluted by incubation in Laemmli sample buffer. Eluted proteins as well as 1/40th of the input were fractionated by SDS-PAGE and analyzed by Western blot analysis with αPS1Loop antibody.
RESULTS
Association of γ-Secretase Components and APP CTFs in Lubrol-resistant Lipid Rafts in the Adult Brain
Previous studies have suggested that processing of APP to the amyloid-β peptide occurs predominantly in lipid rafts and that BACE-1 is the rate-limiting enzyme in this process. In order to gain insights into membrane subdomains involved in γ-secretase processing of APP CTFs, we isolated detergent-insoluble membrane microdomains (DIMs) by discontinuous flotation density gradient centrifugation of mouse brain lysates prepared in Lubrol WX. Lubrol WX-resistant buoyant light density membranes, enriched in the classical lipid raft-associated protein flotillin-2 (38), were found in fractions 4 and 5, at the interface between 5 and 35% sucrose. Consistent with previous reports, we found that ~20% of mature BACE-1 and the vast majority of PS1, mature nicastrin, and PEN-2 were present in these raft fractions (Fig. 1). In agreement with previous reports, less than 5% of full-length mature APP was recovered in Lubrol WX-resistant fractions (17, 20, 39), whereas the majority of full-length mature and immature APP was found in the denser, non-raft fractions 9–12 (Fig. 1). Most interestingly, about 70% of APP C-terminal fragments (CTFs) were recovered in the DIM fractions (Fig. 1). The above results suggest that a small fraction of full-length APP partitions into lipid rafts, whereas APP CTFs preferentially associate with lipid raft microdomains in the adult mouse brain.
Fig. 1. DIM association of BACE-1, γ-secretase, and APP CTFs in mouse brain.

12-Month-old adult mouse brain was homogenized and solubilized in a buffer containing 0.5% Lubrol WX at 4 °C for 30 min. The lysates were then subject to flotation sucrose density gradient centrifugation as described under “Experimental Procedures.” The gradients were harvested from the top, and equal volume of each fraction was analyzed by Western blotting with antibodies against APP C terminus, BACE-1, PS1, nicastrin, and PEN-2. Fractions 4 and 5 represent the interface between 5 and 35% sucrose in the gradient and are enriched in lipid raft marker flotillin-2. Fractions 8–12 contain detergent-soluble proteins, marked by the presence of non-raft proteins γ-adaptin and calnexin.
APP CTFs Accumulate in Lipid Rafts in Cells Lacking PS1/PS2 Expression
Although the presence of γ-secretase components and APP CTFs in DIMs indicates that lipid raft microdomains are likely the sites of γ-secretase processing of APP CTFs, it was important to determine whether the lack of γ-secretase processing promoted APP CTFs to accumulate in Lubrol WX-resistant microdomains, thus validating our prediction. To this end, we examined raft association of endogenous APP CTFs in mouse fibroblasts with targeted deletions in PSEN1 and PSEN2 (PS1−/−/PS2−/−). Components of the γ -secretase complex cooperatively mature and exit the ER (3). When one of the core components of the γ-secretase complex is not expressed in a given cell, the others remain highly unstable and/or fail to exit the ER. In PS1−/−/PS2−/− cells, nicastrin exists only as core glycosylated immature polypeptide, whereas the steady-state levels of PEN-2 are markedly diminished. More importantly, the subunits that still remain in PS1−/−/PS2−/− cells reside exclusively in detergent-soluble non-raft membranes (13). Raft fractionation of wt fibroblasts showed the presence of endogenous APP CTFs both in the raft and non-raft fractions (Fig. 2). Examination of PS1−/−/PS2−/− cells revealed striking accumulation of endogenous APP CTFs in lipid raft fractions as compared with non-raft fractions (Fig. 2). Similarly, in PS1−/− and NCT−/− cells stably transfected with APPswe, APP CTFs markedly accumulate in lipid raft fractions (data not shown). These findings suggest that APP α- and β-CTFs preferentially accumulate in lipid raft microdomains of membranes in cells lacking functional γ-secretase activity.
Fig. 2. DIM accumulation of APP CTFs in cells lacking γ-secretase activity.

wt and PS1−/−/PS2−/− fibroblasts were solubilized in 0.5% Lubrol WX and analyzed by sucrose gradient fractionation and Western blotting with antibodies against APP, flotillin-2, and cavelolin-1. In the absence of PS1 and PS2 expression, endogenous wt APP CTFs become Lubrol WX-resistant and were recovered mainly in raft fractions 4 and 5, identified by the presence of raft markers.
APP CTFs Accumulate in Lubrol-resistant Lipid Rafts in PS1 D385A Cells
To verify independently the above findings, we examined membrane microdomain localization of APP CTFs in stable N2a cell lines co-expressing human APPswe and human wt PS1 or the dominant-negative PS1 D385A mutant. Unlike wt PS1, D385A variant fails to be endoproteolytically processed but forms stable complexes with nicastrin, APH-1, and PEN-2 (40–42). Flotation gradient analysis showed that similar to wt PS1-derived N-terminal fragment, a fraction of PS1 D385A variant was recovered in Lubrol WX-resistant DIM fractions 4 and 5 (Fig. 3). Further analyses revealed the presence of endogenous mature nicastrin, APH-1, and PEN-2 in lipid raft fractions in PS1 D385A cells, indicating that the mutation of PS1 Asp-385 residue did not impair localization of the catalytically inactive PS1 molecule or other γ-secretase components in lipid raft microdomains (Fig. 3). As described previously, APP CTFs accumulate in D385A cells due to the absence of γ-secretase activity (40). As predicted from the PS1−/−/PS2−/− findings described above, the majority of APP CTFs in D385A cells partitioned into lipid raft fractions 4 and 5 (Fig. 4). Similarly, the majority of CTFs derived from endogenous wt APP in stable HEK293 cells expressing PS1 D385A also accumulated in lipid raft fractions, whereas less than 10% of full-length mature glycosylated APP partitioned into lipid raft microdomains (data not shown). Together, these results demonstrate that APP CTFs accumulate to a greater extent in Lubrol-resistant lipid raft microdomains in cells expressing the catalytically inactive PS1 D385A mutant. Most interestingly, we also observed the raft accumulation of a small fraction of mature full-length APP in PS1 D385A cells, indicating the potential effects of dominant-negative PS1 expression on selective turnover or processing of raft-associated full-length APP.
Fig. 3. DIM localization of γ-secretase components and APP CTFs in stable PS1 D385A cells.

Stably transfected N2a APPSwe cells co-expressing wt PS1 or D385A PS1 were subject to flotation sucrose density gradient centrifugation and analyzed by Western blotting with antibodies against PS1, nicastrin, APH-1, PEN-2, and APP. Note that APP CTF in PS1 wt cells is detectable in non-raft fractions upon longer exposure of the immunoblots to the film. mat, mature; imm, immature.
Fig. 4. APP CTFs associate with DIM in cholesterol-sensitive manner and co-reside with syntaxin 6 and PS1.

A, N2a Swe.10 cells were treated with Me2SO or 1 μM L685,458 for 16 h. Lipid raft association of full-length APP and APP CTFs was assessed by flotation sucrose density gradient centrifugation. Note the marked accumulation of APP CTFs in raft fractions in cells treated with the inhibitor. B, Swe.10 cells were exposed to 10 nM CompE for 16 h and then treated with Me2SO (vehicle control) or 5 mM MβCD for 2 h at 37 °C prior to fractionation. Note that DIM accumulation of APP CTFs is sensitive to cholesterol depletion. C, pooled fractions 4 and 5 from the indicated cell lines were fractionated on 16.5% Tris-Tricine gels and sequentially probed with APP C-terminal polyclonal antibody 369 and mAb 26D6 (raised against epitopes 1–12). Antibody 369 reacts with β-, β′ (+11)-, and α-CTFs, whereas mAb 26D6 only reacts with 3-CTF. D, DIM fractions from Swe.10 cells treated with CompE were incubated with magnetic beads coated with syntaxin 6 or OKT8 antibody. Bound DIMs were analyzed by Western blotting using 369, mAb 26D6, and PS1NT. An aliquot of the input (1/30th volume) was also fractionated in the same gel for comparison. Note that APP CTFs, but not full-length APP, co-reside with syntaxin 6 in DIMs.
Cholesterol-dependent Accumulation of APP CTFs in Lipid Rafts of Cells Treated with γ-Secretase Inhibitors
It has been reported previously that APP CTFs accumulate on the cell surface and other intracellular organelles in cells lacking PS1 expression or γ-secretase activity (43–45). In order to determine whether accumulation of APP CTFs in lipid rafts in cells lacking γ-secretase components is directly related to the lack of γ-secretase activity, we turned our attention to pharmacological γ-secretase inhibitors. L685,458 and CompE represent two classes of inhibitors that have been characterized; L685,458 is a transition state analog, and CompE is a small molecule inhibitor (46, 47). Most interestingly, both inhibitors have been successfully cross-linked to PS1, demonstrating that PS1 is the direct target of either of these highly potent γ-secretase inhibitors. Treatment of stable N2a cell line expressing human APP-swe (Swe.10) with L685,458 or CompE results in marked accumulation of APP CTFs without discernible change in the levels of full-length APP. Flotation gradient centrifugation revealed the preponderance of APP CTFs in Lubrol-resistant lipid raft fractions, whereas the vast majority of immature and mature full-length APP was recovered in non-raft fractions (Fig. 4A and B). Together, these results indicate that upon pharmacological inhibition of the γ-secretase activity, the substrate APP CTFs accumulate in Lubrol-resistant microdomains.
We considered the possibility that the topographical relationships between APP CTFs and distinct lipid domains may not be adequately preserved when native membranes are disturbed by the biochemical detergent extraction and fractionation methods. To address this issue, we incubated Swe.10 cells with MβCD, a drug known to selectively deplete biological membranes of cholesterol. Cholesterol plays an important role in stabilizing lipid raft microdomains, and depletion of cholesterol disrupts lipid raft integrity and imparts detergent solubility on certain raft-associated proteins, including γ-secretase components and PrP (13, 48, 49). Incubation of CompE-treated Swe.10 cells with MβCD prior to raft isolation caused the displacement of APP CTFs from the low density Lubrol WX-resistant membrane domains (Fig. 4B) to the denser Lubrol WX-soluble membrane fractions, providing evidence that APP CTF-rich buoyant membranes indeed represent bona fide cholesterol-rich lipid rafts. In addition to APP CTFs, we also observed the accumulation of a small fraction (5–10%) of mature full-length APP in lipid raft fractions, which is also sensitive to MβCD treatment. Thus we conclude that in the absence of γ-secretase processing, a significant fraction of APP CTFs associates with lipid rafts in a cholesterol-dependent manner.
APPγ-CTFs Co-reside with Syntaxin 6 in DIMs
The findings described above imply that DIMs are the likely platforms where 3-secretase processing of APP occurs. Recently, we characterized the co-residence of components of the γ-secretase complex with syntaxin 6 in Golgi/trans-Golgi network/endosome lipid raft membrane microdomains (13). Hence we predicted that in the absence of γ-secretase processing, APP CTFs likely accumulate in DIMs of intracellular organelles where γ-secretase resides. To test this idea directly, we carried out antibody-mediated magnetic immunoisolation of syntaxin 6 containing DIM patches from a pool of Lubrol WX-resistant DIMs from Swe.10 cells treated with CompE. Western blot analysis of the bound fraction revealed the presence of APP β-CTFs and PS1 CTF (Fig. 4D). More importantly, we failed to detect full-length APP molecules in syntaxin 6-positive DIMs. These results are consistent with the co-residence of APP CTFs and γ-secretase components in DIMs of trans-Golgi network/late endosomes rich in syntaxin 6.
Detection of Active γ-Secretase in Raft and Non-raft Fractions
Although the above data implicate the presence of active γ-secretase in DIMs, we performed the following set of experiments to formally establish this notion by direct labeling of PS1 using an active site-directed photoaffinity probe. As described previously (37), L-852,505 is a photoreactive, biotinylated derivative of the highly specific and potent transition state analog inhibitor of γ-secretase L-685,458 (46); photoactivated L-852,505 covalently labels PS1 CTF. We prepared DIMs (pooled fractions 4 and 5) and non-raft fractions (pooled fractions 8–12) from wt mouse fibroblasts, and we incubated these samples at 4 °C for 60 min with 1 μM L-852,505. Reaction mixtures were subject to photoactivation at 350 nm, and bioti-nylated polypeptides were recovered with immobilized streptavidin and subjected to Western blot analysis with αPS1Loop antibody. The photoprobe readily labeled PS1 CTF in raft fractions, but labeling was undetectable in non-raft fractions (Fig. 5A, right panel). However, as noted by the paucity of PS1 CTF in non-raft input relative to its abundance in the raft input, we considered the possibility that photoprobe labeling of PS1 CTF in non-raft fractions was beyond the limit of detection. To address this issue, we repeated this experiment by using raft and non-raft preparations from N2a cells overexpressing PS1, and we increased the amount of non-raft input. Following photoactivation, readily discernable levels of PS1 CTF were labeled by L-852,505 in both raft and non-raft preparations (Fig. 5B). Nevertheless, a comparison of photoprobe-labeled CTF to input indicated preferential labeling of CTF in DIMs. Finally, to confirm the presence of active γ-secretase in brain DIMs, we incubated raft and non-raft preparations from adult mouse brain with the L-852,505 photoprobe. As shown in Fig. 5C, the large majority of labeled PS1 CTF was found in the raft fraction. The above findings suggest that the preponderance of active γ-secretase complex is present in DIMs of untransfected fibroblasts and adult mouse brain.
Fig. 5. Covalent labeling of PS1 by active site directed 3-secretase inhibitor L-852,505.
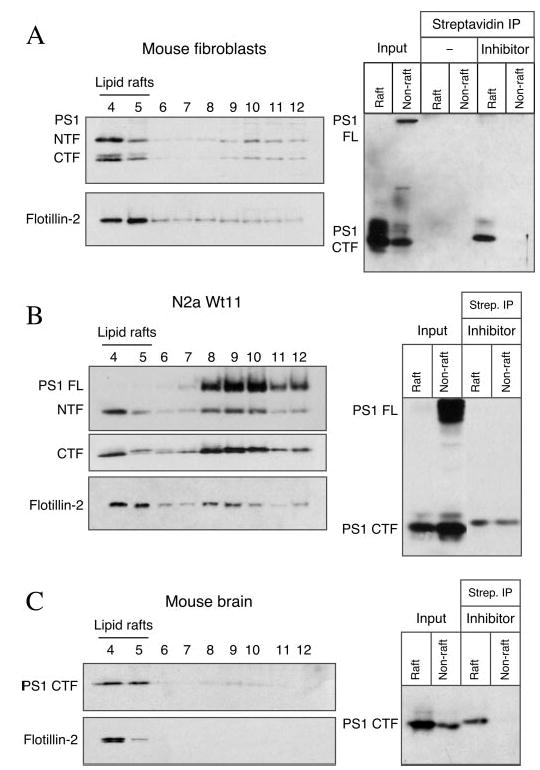
Pooled raft and non-raft fractions from wt mouse embryonic fibroblasts (A), N2a wt.11 cells (B), or adult mouse brain (C) were incubated with Me2SO (− inhibitor) or L-852,505 (+ inhibitor), followed by irradiation with 365 nm UV light on ice for 90 min to allow photoaffinity cross-linking. Biotinylated proteins were captured with streptavidin-agarose beads and eluted by incubation at 95 °C for 5 min in Laemmli/SDS sample buffer. The eluates (lanes 5–9) as well as 1/40th of the solubilized membranes used for the streptavidin (Strep.) precipitation (input) were analyzed by immunoblotting using αPS1Loop antibody. The left panels show the distribution of PS1 and flotillin-2 in the flotation density gradient fractions used for the in vitro cross-linking experiment. IP, immunoprecipitation.
Non-raft Localization of Notch1 and Jagged2 CTFs
Although the studies described above are consistent with the presence of active γ-secretase and APP CTFs in Lubrol-resistant DIMs, these results are insufficient to conclude that γ-secretase processing of all substrates occurs exclusively within DIMs. Therefore, we set out to determine directly whether other γ-secretase substrates also associate with DIMs. Similar to our characterization of APP CTFs, we asked whether the inhibition of γ-secretase activity leads to DIM accumulation of Notch C-terminal derivative termed S2/NEXT (Notch extra-cellular truncation), which is the immediate precursor for γ-secretase processing (50). For these studies, we employed a mouse embryonic fibroblast line stably expressing a chimeric full-length Notch1–6mycGFP molecule (26). In this fibroblast line, immunoblotting with mAb 9E10 detects ~350-kDa full-length Notch and ~80-kDa furin cleaved Notch CTF termed S1/TMIC (transmembrane-bound intracellular domain). Treatment with EDTA stimulates sequential cleavage of S1/TMIC by an ADAM protease and γ-secretase, resulting in robust production of membrane-tethered C-terminal derivatives S2/NEXT and S3/NICD (Notch intracellular domain), respectively (26).
We decided to employ the ligand-independent, Ca2+-depletion paradigm to facilitate S2 and S3 cleavage of Notch (51). We treated stably transfected Notch-6mycGFP fibroblasts with EDTA for 20 min and analyzed lysates by flotation gradient centrifugation. As shown in Fig. 6A, the majority of full-length Notch and a greater fraction of TMIC and NEXT polypeptides (which migrate as a close doublet in our gel system) were associated with detergent-soluble heavier fractions (fractions 8–12), and about 15–20% of TMIC/NEXT polypeptides were present in DIM fractions (fractions 4 and 5). As expected, preincubation of Notch1–6mycGFP cells with CompE resulted in complete loss of NICD and a proportional increase in the levels of TMIC/NEXT polypeptides. Most interestingly, we observe a consistent increase in the TMIC/NEXT polypeptide intensity in the non-raft fractions, whereas the signal intensity of TMIC/NEXT in DIM fractions remains largely unaffected (Fig. 6A, right panel). Furthermore, in contrast to TMIC/NEXT polypeptides, endogenous APP CTFs preferentially accumulated in DIM fractions of Notch1–6mycGFP fibroblasts pre-treated with CompE. It should be noted that although the majority of PS1 and mature nicastrin was found in the raft fractions, it is still plausible that the low levels of PS1/γ-secretase in the non-raft fractions are catalytically active in promoting intramembranous proteolysis of Notch 1 CTFs. In any event, our results clearly demonstrate differential accumulation of APP and Notch1 CTFs in raft versus non-raft fractions of fibroblasts.
Fig. 6. Flotation density gradient analysis of Notch1 and Jagged2 derivatives.

A, mouse embryonic fibroblasts stably expressing Notch-6mycGFP were treated or not with 10 nM CompE for 18 h and incubated with 10 mM EDTA for 30 min to induce ligand-independent activation of Notch processing (51) prior to lysis in 0.5% Lubrol WX and fractionation. Full-length Notch- and C-terminal derivatives were visualized by immunoblotting with mAb 9E10. Note that the Notch cleavage products S1/TMIC (indicated by filled arrowhead) and S2/NEXT (open arrowhead) migrate as a close doublet, and S3/NICD (open circle) is absent in cells treated with CompE. Endogenous APP CTF was analyzed by combined immunoprecipitation and immunoblotting with APP antibody 369. Lower panels represent immunoblots probed with PS1 and nicastrin antibodies. mat, mature; imm, immature. B, 3T3 fibro-blasts stably transfected with Myc-tagged Jagged2 were incubated with CompE, and distribution of Jagged2 and derivatives was analyzed by fractionation and immunoblotting with mAb 9E10. CompE-treated cells lack JICD but have increased levels of Jagged2 CTF in non-raft fractions.
We further extended our analysis to Jagged2, one of the Notch1 ligands that has been shown to undergo γ-secretase processing (25, 52). Flotation density gradient analysis of stably transfected NIH 3T3 fibroblasts revealed the presence of Jagged2-CTF and JICD (Jagged intracellular domain) in lipid raft fractions 4 and 5 (Fig. 6B). When the analysis was performed in cells incubated with CompE to inhibit γ-secretase processing of Jagged2, the band corresponding to JICD was not detected. Concomitantly, we observe an obvious increase in the Jagged2-CTF signal intensity in non-raft fractions (fractions 8–12), whereas the signal intensity in DIM fractions (fractions 4 and 5) remained unchanged. Thus, we conclude that upon inhibition of γ-secretase processing APP CTFs accumulate in DIM fractions, whereas CTFs derived from Notch1 and Jagged2 preferentially accumulate in detergent-soluble, non-raft fractions.
Non-raft Localization of γ-Secretase and Substrates in Embryonic Brain
Since γ-secretase plays an essential role in mammalian embryonic development, we performed a series of studies using brain tissue from PS1+/− and PS1−/− embryos harvested at E15.5. First, we examined the distribution of Notch1 using a C-terminal-specific antibody. Immunoblot analysis of fractions from embryonic brain homogenates that were subjected to flotation gradient centrifugation showed predominant non-raft distribution of full-length Notch1 as well as Notch1 C-terminal derivatives TMIC and NEXT polypeptides both in PS1+/− and PS1−/− embryonic brain (Fig. 7). As expected from previous reports, the polypeptide corresponding to NICD was not detectable in PS1−/− embryonic brain lysates. These results are consistent with our findings from stably transfected fibroblasts described above. Next, we turned our attention to other γ-secretase substrates that have been reported to play important roles in the nervous system. We recently reported that DCC, the netrin-1 receptor, is subject to proteolysis within the ectodomain segment and that the residual membrane-tethered DCC “stub” is subsequently processed by γ-secretase to generate DCC-intracellular domain (53, 54). In PS1+/− embryonic brain, full-length DCC and the vast majority of DCC CTF were recovered in non-raft fractions with only a minor fraction of each species present in DIM raft fractions (Fig. 7). In PS1−/− embryonic brain, wherein γ-secretase activity is greatly reduced, we readily observed an increase in the levels of DCC CTF. Furthermore, the relative abundance of DCC CTF in the non-raft fractions of PS1−/− embryonic brain was higher when compared with that of PS1+/− embryonic brain. We also examined the distribution of N-cadherin, another γ-secretase substrate (55), and found that the majority of full-length N-cadherin and its CTFs was present in non-raft fractions of embryonic brain. Consistent with the lack of γ-secretase cleavage, we observed increased levels of CTFs in non-raft fractions of PS1−/− embryonic brain compared with that of the PS1+/− brain. The above results suggest that γ-secretase processing of DCC and N-cadherin CTFs occurs in non-raft fractions in embryonic brain.
Fig. 7. Non-raft association of Notch1, DCC, and N-cadherin CTFs.
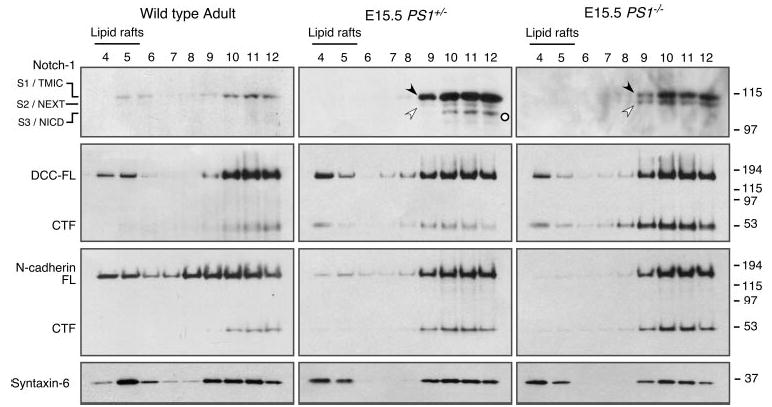
Brain tissue from 12-month-old adult mouse or E15.5 embryos were homogenized in 0.5% Lubrol WX lysis buffer and subject to flotation sucrose density gradient centrifugation. Aliquots of the gradients were analyzed by Western blotting with antibodies against C terminus of Notch1, DCC, N-cadherin, and syntaxin 6. Notch cleavage products S1/TMIC (indicated by filled arrowhead), S2/NEXT (open arrowhead), and S3/NICD (open circle) are indicated. Note that in PS1−/− embryos S3/NICD is absent, and the levels of S2/NEXT as well as DCC and N-cadherin CTFs are higher relative to wt embryonic brain.
As we had observed in the embryonic brain, the majority of TMIC peptides was present in the non-raft fractions of adult mouse brain (Fig. 7). However, and in contrast to the findings in embryonic brain where we failed to detect any TMIC in raft fractions, we observed low levels of TMIC in the raft fractions from adult brain (Fig. 7). These findings raise the possibility that lipid raft association of proteins could markedly differ between embryonic and adult brain tissue. Hence, we examined the raft/non-raft distribution of full-length APP and APP CTFs in embryonic brain lysates, and we found that the vast majority of full-length APP molecules was predominantly associated with non-raft membranes, consistent with the results from analysis of the adult brain. Most interestingly, and in contrast to the adult brain in which the vast majority of APP CTFs was present in raft fractions (see Fig. 1), we found that significant amounts of APP CTFs were present in non-raft fractions in embryonic brain (Fig. 8).
Fig. 8. Non-raft association of PS1 and APP CTFs in embryonic mouse brain.
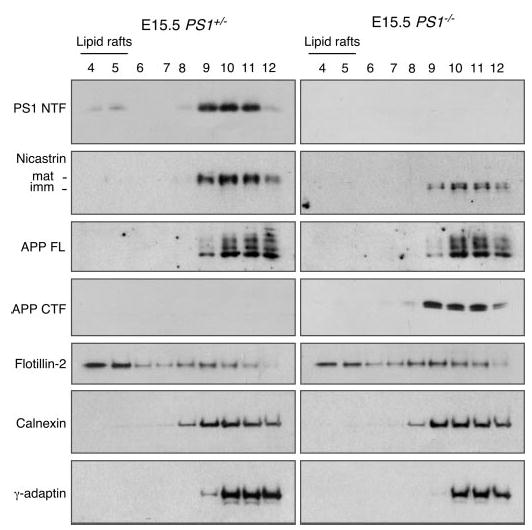
Raft and non-raft distribution of PS1, nicastrin, APP, flotillin-2, calnexin, and γ-adaptin in flotation density gradient fractions from E15.5 embryonic brains were analyzed by Western blotting. Note the predominant non-raft localization of PS1, nicastrin, and APP CTFs. mat, mature; imm, immature.
Further evaluation of embryonic brain samples revealed the surprising finding that the large majority of PS1 and nicastrin also resided in non-raft domains amenable to solubilization in Lubrol WX. Immunoblotting with raft marker flotillin-2, and raft-associated t-SNARE syntaxin-6, documented that reproducible recovery of PS1 and APP in non-raft fractions of embryonic brain did not result from the absence of Lubrol WX-resistant DIM in embryonic brain tissue or inadvertent disruption of DIM domain integrity during the biochemical isolation procedure (Fig. 8). These results are in accord with γ-secretase processing of substrates in both raft and non-raft subdomains of cellular membranes during embryonic development.
DISCUSSION
Several lines of evidence suggest a role for lipid rafts in amyloidogenic processing of APP. In this study we characterized the raft versus non-raft distribution of CTFs derived from APP and other select γ-secretase substrates (Notch1, Jagged2, DCC, and N-cadherin) and report several novel insights regarding γ-secretase processing. First, loss of γ-secretase processing leads to preferential association of APP CTFs with DIMs. Association of APP CTFs is sensitive to depletion of cholesterol by using methyl-β-cyclodextrin, strongly suggesting their recruitment into cholesterol- and sphingolipid-rich lipid rafts. By using a photo-cross-linking approach, we also demonstrate the preponderance of active γ-secretase in lipid raft fractions of cultured cells and adult mouse brain. These findings implicate lipid rafts as the principal sites in cellular membranes where Aβ is generated. Second, we show that the large majority of CTFs generated by ectodomain cleavage of Notch1, Jagged2, DCC, and N-cadherin remain Lubrol WX-soluble when γ-secretase cleavage is inhibited (in cultured cells) or absent (in PS1−/− embryonic brain), indicating that these substrates are likely cleaved by γ-secretase in non-raft membrane domains. Third, we show that significant amount of PS1, nicastrin, and APP CTFs remain Lubrol WX-soluble in the embryonic brain. Together, these results suggest that in cultured cells and in adult brain the majority of active γ-secretase and APP CTFs is sequestered in lipid rafts away from several other γ-secretase substrates, and that catalytic levels of γ-secretase are sufficient to facilitate processing of substrates such as Notch1 and Jagged2 in non-raft membranes.
The functional significance of lipid rafts in regulating cellular signaling is well established (reviewed in Ref. 14). For example, tumor necrosis factor (TNF) receptor 1 is recruited to lipid rafts within minutes after TNFα binding, and this translocation is essential for NF-κB activation (56). Disruption of the lipid raft sensitizes cells to TNF receptor 1-induced cell death by blocking ubiquitination of adaptor proteins in the TNF receptor 1 signaling complex and preventing NF-κB activation. Similarly, immunoglobulin E signaling and T-cell antigen receptor signaling in immune cells involves redistribution into lipid rafts and clustering of raft components (14). The examples above also address serious concerns regarding the relationship between lipid ordered domains in living cells and light buoyant density membranes resulting from extraction of cells in cold non-ionic detergents. First, whereas detergent insolubility can be an inherent property of certain proteins, differential behavior of surface receptors following ligand binding argues in favor of their dynamic association with detergent-resistant membrane microdomains in live cells during signal transduction. Second, experimental manipulations that are used to deplete cholesterol in cellular membranes modulate the functional outcome of ligand-induced signaling, strongly suggesting the involvement of cholesterol-rich lipid raft microdomains in spatial control of signaling in live cells. Thus, cholesterol-dependent association with light buoyant density membranes is an acceptable criterion for raft localization of proteins.
There has been considerable debate regarding the levels of full-length APP that partitions into lipid rafts (20, 57–59). We find that only a small fraction of endogenous full-length APP becomes associated with Lubrol WX-resistant raft fractions in mouse brain and in cultured cell lines. In marked contrast, loss of γ-secretase activity, resulting from a lack of PS1/PS2 expression, expression of a dominant-negative PS1 mutant, or exposure to highly potent γ-secretase inhibitors, leads to preferential accumulation of APP α- and β-CTFs in Lubrol WX-resistant fractions. Data from wt adult mouse brain demonstrate that raft association of α-and β-CTFs does not depend on compromised γ-secretase activity. One plausible explanation for the accumulation of β-CTF in rafts is that BACE cleaves full-length APP mainly within lipid rafts, and once generated, β-CTFs continue to remain associated with lipid raft microdomains until they are processed by γ-secretase. There is sufficient evidence to indicate the presence of mature BACE and amyloidogenic processing of APP by BACE in lipid rafts (17, 18, 39, 60). For example, antibody-induced co-patching of APP and BACE-1 at the cell surface promotes Aβ production in a cholesterol-dependent manner (17). Direct evidence demonstrating the presence of α-secretase(s) or α-secretase processing of APP in lipid rafts is still lacking. However, based on zinc metalloprotease-mediated α-secretase-type shedding of PrP, a glycosylphosphatidylinositol-anchored plasma membrane protein localized to lipid rafts (61), we cannot formally rule out the possibility of α-secretase cleavage of APP within lipid rafts. Alternatively, accumulation of α-CTF within lipid rafts upon inhibition of γ-secretase activity might suggest that APP CTFs have the intrinsic property to segregate into cholesterol- and sphingolipid-rich microdomains regardless of the ectodomain cleavage of APP in raft or non-raft membranes.
By extending our previous studies on raft association of γ-secretase components (13), we performed photoaffinity cross-linking studies using a biotinylated, benzophenone-derivatized γ-secretase inhibitor and report the predominant localization of active γ-secretase in lipid raft microdomains in cultured cell lines and in adult mouse brain. Nevertheless, this direct labeling approach also reveals detectable levels of active γ-secretase in non-raft fractions of neuroblastoma cells overexpressing human PS1. Most interestingly, the dominant-negative PS1 variant lacking the critical aspartate residue Asp-385 (40) also localizes to DIMs, suggesting that PS1 complex is recruited into lipid raft microdomains independent of the catalytic potential of the γ-secretase complex. Thus, although the large majority of each of the four components of the γ-secretase is associated with lipid rafts in cultured cells, the residence in raft is neither required for γ-secretase activity nor indicative of functional γ-secretase.
Our results show that CTFs derived from Notch1, Jagged2, DCC, and N-cadherin accumulate mainly in Lubrol WX-soluble membrane fractions in the absence of γ-secretase activity, indicating that these substrates are likely processed by γ-secretase in non-raft regions of cellular membranes. It is somewhat surprising that in embryonic brain, the majority of PS1 and nicastrin, as well as APP CTFs, remains Lubrol WX-soluble. These findings support the notion that γ-secretase cleavage is not restricted to proteins that are localized within lipid rafts. Furthermore, predominant non-raft localization of PS1 and nicastrin in embryonic brain may be indicative of efficient γ-secretase processing of diverse substrates such as Notch1 and DCC that are known to play essential roles during embryonic development (62). These results also suggest that in the adult tissue, signaling resulting from regulated intramembrane proteolysis of certain substrates is regulated by efficient partitioning of active γ-secretase in lipid raft domains. How this developmental regulation is achieved is a focus of our future investigations. One possibility that might explain our findings is that protein and lipid components of raft domains are not sufficiently stabilized in embryonic brain, rendering them susceptible to solubilization in Lubrol WX. Changes in lipid composition and organization have been documented during in vitro differentiation of cultured neurons (63). However, raft association of full-length DCC, syntaxin 6, and flotillin-2 seems to be largely unaffected in embryonic brain, arguing against this notion of “immature” status of lipid raft microdomains in embryonic brain tissue. Nevertheless, it is clear that γ-secretase complex and APP CTFs have some yet unidentified properties that allow them to get recruited and remain associated with lipid raft microdomains of cultured cells and adult brain tissue. Finally, our results on the spatial resolution of APP and Notch processing also have potential implication in therapeutics aimed at reducing the Aβ burden. By selectively targeting γ-secretase inhibitors to cholesterol- and sphingo-lipid-rich membrane domains, it may be possible to inhibit Aβ production without adverse effects on non-raft resident substrates.
Acknowledgments
We thank Drs. Wim Annaert and Bart De Strooper for generously providing wild-type and PS1−/−/PS2−/− fibroblasts and Drs. Sam Gandy, Huaxi Xu, Paul Fraser, Peter St George-Hyslop, and Philip C. Wong for providing antibodies. We are grateful to Dr. Mark Shearman (Merck) for the L-685,458 and L-852,505 compounds and Dr. Todd E. Golde for compound E.
Footnotes
This work was supported in part by National Institutes of Health Grants AG021495, AG019070 (to G. T.), and AG021494 (to S. S. S.), the Alzheimer’s Association (to G. T. and A. T. P.), and the American Health Assistance Foundation (to G. T.).
The abbreviations used are: APP, amyloid precursor protein; Aβ, β-amyloid; BACE1, β-site APP-cleaving enzyme; CompE, compound E; CTF, C-terminal fragment; DCC, deleted in colorectal cancer; DIM, detergent-insoluble membrane; ER, endoplasmic reticulum; JICD, Jagged intracellular domain; PS, presenilin(s); NICD, Notch intracellular domain, NEXT, Notch extracellular truncation; TMIC, transmembrane-bound intracellular domain; TNF, tumor necrosis factor; wt, wild type; AD, Alzheimer disease; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; mAb, monoclonal antibody; PS, presenilin; MβCD, methyl-β-cyclodextrin.
References
- 1.Sisodia SS, St George-Hyslop PH. Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- 2.Vassar R. J Mol Neurosci. 2004;23:105–114. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- 3.Iwatsubo T. Curr Opin Neurobiol. 2004;14:379–383. doi: 10.1016/j.conb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Tanzi RE, Bertram L. Neuron. 2001;32:181–184. doi: 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- 5.Levitan D, Greenwald I. Nature. 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 6.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 7.Goutte C, Hepler W, Mickey KM, Priess JR. Development (Camb) 2000;127:2481–2492. doi: 10.1242/dev.127.11.2481. [DOI] [PubMed] [Google Scholar]
- 8.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- 9.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 10.Goutte C, Tsunozaki M, Hale VA, Priess JR. Proc Natl Acad Sci U S A. 2002;99:775–779. doi: 10.1073/pnas.022523499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfe MS, Kopan R. Science. 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- 12.Struhl G, Adachi A. Mol Cell. 2000;6:625–636. doi: 10.1016/s1097-2765(00)00061-7. [DOI] [PubMed] [Google Scholar]
- 13.Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simons K, Toomre D. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 15.Ikonen E. Curr Opin Cell Biol. 2001;13:470–477. doi: 10.1016/s0955-0674(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 16.Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 17.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Proc Natl Acad Sci U S A. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wada S, Morishima-Kawashima M, Qi Y, Misono H, Shimada Y, Ohno-Iwashita Y, Ihara Y. Biochemistry. 2003;42:13977–13986. doi: 10.1021/bi034904j. [DOI] [PubMed] [Google Scholar]
- 20.Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG. J Cell Biol. 2004;167:953–960. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. J Biol Chem. 1996;271:9390–9397. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- 22.Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- 23.Sato N, Urano F, Leem JY, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, Sisodia SS, Thinakaran G. Nat Cell Biol. 2000;2:863–870. doi: 10.1038/35046500. [DOI] [PubMed] [Google Scholar]
- 24.Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- 25.Ikeuchi T, Sisodia SS. J Biol Chem. 2003;278:7751–7754. doi: 10.1074/jbc.C200711200. [DOI] [PubMed] [Google Scholar]
- 26.Martys-Zage J, Kim SH, Berechid B, Bingham S, Chu S, Nye J, Sisodia SS. J Mol Neurosci. 2001;15:189–204. doi: 10.1385/jmn:15:3:189. [DOI] [PubMed] [Google Scholar]
- 27.Thinakaran G, Regard JB, Bouton CML, Harris CL, Price DL, Borchelt DR, Sisodia SS. Neurobiol Dis. 1998;4:438–453. doi: 10.1006/nbdi.1998.0171. [DOI] [PubMed] [Google Scholar]
- 28.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 29.Leem JY, Vijayan S, Han P, Cai D, Machura M, Lopes KO, Veselits ML, Xu H, Thinakaran G. J Biol Chem. 2002;277:19236–19240. doi: 10.1074/jbc.C200148200. [DOI] [PubMed] [Google Scholar]
- 30.Li T, Ma G, Cai H, Price DL, Wong PC. J Neurosci. 2003;23:3272–3277. doi: 10.1523/JNEUROSCI.23-08-03272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu Y, Chen F, Sanjo N, Kawarai T, Hasegawa H, Duthie M, Li W, Ruan X, Luthra A, Mount HT, Tandon A, Fraser PE, St George-Hyslop P. J Biol Chem. 2003;278:7374–7380. doi: 10.1074/jbc.M209499200. [DOI] [PubMed] [Google Scholar]
- 32.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 33.Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, Liao F, Checler F, Buxbaum JD, Chait BT, Thinakaran G, Sisodia SS, Wang R, Greengard P, Gandy S. Nat Med. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- 34.Lamb BA, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, Sisodia SS, Hoeger EJ. Nat Neurosci. 1999;2:695–697. doi: 10.1038/11154. [DOI] [PubMed] [Google Scholar]
- 35.Ma L, Huang YZ, Pitcher GM, Valtschanoff JG, Ma YH, Feng LY, Lu B, Xiong WC, Salter MW, Weinberg RJ, Mei L. J Neurosci. 2003;23:3164–3175. doi: 10.1523/JNEUROSCI.23-08-03164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 37.Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Proc Natl Acad Sci U S A. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lang DM, Lommel S, Jung M, Ankerhold R, Petrausch B, Laessing U, Wiechers MF, Plattner H, Stuermer CA. J Neurobiol. 1998;37:502–523. doi: 10.1002/(sici)1097-4695(199812)37:4<502::aid-neu2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 39.Riddell DR, Christie G, Hussain I, Dingwall C. Curr Biol. 2001;11:1288–1293. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- 40.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 41.Yu G, Chen F, Nishimura M, Steiner H, Tandon A, Kawarai T, Arawaka S, Supala A, Song YQ, Rogaeva E, Holmes E, Zhang DM, Milman P, Fraser PE, Haass C, George-Hyslop PS. J Biol Chem. 2000;275:27348–27353. doi: 10.1074/jbc.M002982200. [DOI] [PubMed] [Google Scholar]
- 42.Nyabi O, Bentahir M, Horre K, Herreman A, Gottardi-Littell N, Van Broeckhoven C, Merchiers P, Spittaels K, Annaert W, De Strooper B. J Biol Chem. 2003;278:43430–43436. doi: 10.1074/jbc.M306957200. [DOI] [PubMed] [Google Scholar]
- 43.Chen F, Yang DS, Petanceska S, Yang A, Tandon A, Yu G, Rozmahel R, Ghiso J, Nishimura M, Zhang DM, Kawarai T, Levesque G, Mills J, Levesque L, Song YQ, Rogaeva E, Westaway D, Mount H, Gandy S, St George-Hyslop P, Fraser PE. J Biol Chem. 2000;275:36794–36802. doi: 10.1074/jbc.M006986200. [DOI] [PubMed] [Google Scholar]
- 44.Leem JY, Saura CA, Pietrzik C, Christianson J, Wanamaker C, King LT, Veselits ML, Tomita T, Gasparini L, Iwatsubo T, Xu H, Green WN, Koo EH, Thinakaran G. Neurobiol Dis. 2002;11:64–82. doi: 10.1006/nbdi.2002.0546. [DOI] [PubMed] [Google Scholar]
- 45.Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, Sisodia SS, Thinakaran G, Xu H. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- 46.Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL. Biochemistry. 2000;39:8698–8704. doi: 10.1021/bi0005456. [DOI] [PubMed] [Google Scholar]
- 47.Seiffert D, Bradley JD, Rominger CM, Rominger DH, Yang F, Meredith JE, Jr, Wang Q, Roach AH, Thompson LA, Spitz SM, Higaki JN, Prakash SR, Combs AP, Copeland RA, Arneric SP, Hartig PR, Robertson DW, Cordell B, Stern AM, Olson RE, Zaczek R. J Biol Chem. 2000;275:34086–34091. doi: 10.1074/jbc.M005430200. [DOI] [PubMed] [Google Scholar]
- 48.Klein U, Gimpl G, Fahrenholz F. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- 49.Harder T, Simons K. Curr Opin Cell Biol. 1997;9:534–542. doi: 10.1016/s0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- 50.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. Mol Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 51.Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster JC. Mol Cell Biol. 2000;20:1825–1835. doi: 10.1128/mcb.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LaVoie MJ, Selkoe DJ. J Biol Chem. 2003;278:34427–34437. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- 53.Taniguchi Y, Kim SH, Sisodia SS. J Biol Chem. 2003;278:30425–30428. doi: 10.1074/jbc.C300239200. [DOI] [PubMed] [Google Scholar]
- 54.Parent AT, Barnes NY, Taniguchi Y, Thinakaran G, Sisodia SS. J Neurosci. 2005;25:1540–1549. doi: 10.1523/JNEUROSCI.3850-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 56.Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Immunity. 2003;18:655–664. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 57.Bouillot C, Prochiantz A, Rougon G, Allinquant B. J Biol Chem. 1996;271:7640–7644. doi: 10.1074/jbc.271.13.7640. [DOI] [PubMed] [Google Scholar]
- 58.Parkin ET, Hussain I, Turner AJ, Hooper NM. J Neurochem. 1997;69:2179–2188. doi: 10.1046/j.1471-4159.1997.69052179.x. [DOI] [PubMed] [Google Scholar]
- 59.Parkin ET, Turner AJ, Hooper NM. Biochem J. 1999;344:23–30. [PMC free article] [PubMed] [Google Scholar]
- 60.Kametaka S, Shibata M, Moroe K, Kanamori S, Ohsawa Y, Waguri S, Sims PJ, Emoto K, Umeda M, Uchiyama Y. J Biol Chem. 2003;278:15239–15245. doi: 10.1074/jbc.M208611200. [DOI] [PubMed] [Google Scholar]
- 61.Parkin ET, Watt NT, Turner AJ, Hooper NM. J Biol Chem. 2004;279:11170–11178. doi: 10.1074/jbc.M312105200. [DOI] [PubMed] [Google Scholar]
- 62.Lai EC. Development (Camb) 2004;131:965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 63.Prinetti A, Chigorno V, Prioni S, Loberto N, Marano N, Tettamanti G, Sonnino S. J Biol Chem. 2001;276:21136–21145. doi: 10.1074/jbc.M010666200. [DOI] [PubMed] [Google Scholar]