

Abstract
γ-Secretase, which is responsible for the intramembranous cleavage of Alzheimer β-amyloid precursor protein and the signaling receptor Notch, is a multiprotein complex consisting of at least four components: presenilin (PS); nicastrin (Nct); APH-1 (anterior pharynx-defective-1); and presenilin enhancer-2 (PEN-2). Presenilin 1 (PS1) is known to be essential for the stability, interaction, and trafficking of the other PS1/γ-secretase components. However, the precise functions of the other components remain elusive. Here, we investigated the functions of Nct within the PS1/γ-secretase complex. We demonstrated that the loss of Nct expression in the embryonic fibroblast cells (Nct KO cells) results in dramatically decreased levels of APH-1, PEN-2, and PS1 fragments accompanied by a significant accumulation of full-length PS1. In the absence of Nct, PEN-2 and full-length PS1 are subjected to proteasome-mediated degradation, whereas the degradation of APH-1 is mediated by both proteasomal and lysosomal pathways. Unlike the case of wild type cells in which the γ-secretase complex mainly locates in the trans-Golgi network, the majority of residual PEN-2, APH-1, and the uncleaved full-length PS1 in Nct KO cells reside in the endoplasmic reticulum, which remain associated with each other in the absence of Nct. Interestingly, significant amounts of full-length PS1 and PEN-2, but not APH-1, are detected on the plasma membrane in Nct KO cells, suggesting the Nct-independent cell surface delivery of the PEN-2·PS1. Finally, the diminished PEN-2 protein level in Nct-deficient cells can be partially restored by overexpression of exogenous PS1, APH-1, or PEN-2 individually or collectively, indicating a dispensable role for Nct in controlling PEN-2 level. Taken together, our study demonstrates a critical role of Nct in the stability and proper intracellular trafficking of other components of the PS1/γ-secretase complex but not in maintaining the association of PEN-2, APH-1, and full-length PS1.
An important pathological feature of Alzheimer disease is the formation of extracellular senile plaques in the brain whose major constituents are 40–42 amino acid β-amyloid peptides derived from sequential proteolyses of β-amyloid precursor protein (APP)1 by β-secretase and γ-secretase (1). Biochemical evidence demonstrates that γ-secretase activity resides in a high molecular weight multiprotein complex composed of at least four components: presenilin 1 or 2 (PS1 or PS2), nicastrin (Nct), APH-1a or b and PEN-2 (2, 3). These four components are necessary and probably are sufficient for the γ-secretase activity (2–6).
Full-length presenilins (PS1 and PS2) are short-lived multi-transmembrane proteins that undergo endoproteolytic cleavage by an unknown protease (named presenilinase) to generate stable amino-terminal fragments (NTF) and carboxyl-terminal fragments (CTF) (7). PS NTF/CTF heterodimer physically interacts with Nct, APH-1, and PEN-2 to assemble in an appropriate manner into a functional γ-secretase (2, 6). It is generally accepted that PS1 heterodimers contain the active site, namely the catalytic core of the γ-secretase complex (3, 7).
The PS1/γ-secretase complex is a delicately regulated system in which the four components counterregulate each other and orchestrate coordinately to fulfill the γ-secretase function. PS1 deficiency leads to a reduced level of PEN-2 (8, 9), which is due to ubiquitin-proteasome-mediated degradation (10, 11). Recently, using biochemical approaches such as RNAi and protein overexpression, we and others (6, 8) have demonstrated that PEN-2 is required for the endoproteolysis of PS1 and that APH-1 plays an important role in stabilizing PS1. In addition to its role in PS1 endoproteolysis, PEN-2 is also required for the stability of PS1 fragment (12). On the other hand, Nct and APH-1 are relatively stable and remain associated even in the absence of PSs or when PEN-2 level is reduced (13–15). Moreover, Nct appears to interact more closely with APH-1 because down-regulation of Nct by small interfering RNA (siRNA) results in a reduced APH-1 level (15, 16). Based on these observations, it has been proposed that Nct may first bind to APH-1 to form a Nct·APH-1 precursor subcomplex (17, 18) prior to further association with PS1 and PEN-2 or with PS1·PEN-2 subcomplex. This model is further supported by the existence of the Nct·APH-1 subcomplex detected by blue-native gel electrophoresis (17). Our recent finding that PEN-2 and PS1 traffic together, separately from Nct, from the trans-Golgi network (TGN) to the cell surface upon treatments with certain γ-secretase inhibitors further sustains this notion (19).
Endogenous PS1, Nct, PEN-2, and APH-1 have been described to mainly localize to the endoplasmic reticulum (ER) and Golgi (8, 13), and the properly assembled complex is transported through the secretory pathway to localize predominantly in the Golgi and, to a lesser extent, at the cell surface and in endocytic compartments (21–24). Consistent with the localization of the active γ-secretase complex, Aβ has been shown to be generated primarily in the Golgi/TGN (25–27), although other cellular compartments including ER (25, 27–29), endosomes (29, 30), and plasma membrane (22, 31) may also be involved. Recent studies from several groups including our own have demonstrated that, in the absence of PS1, Nct fails to be post-translationally glycosylated and the immature Nct and PEN-2 are sequestered in the ER and not transported to Golgi/TGN (10, 11, 19, 32). These results suggest that, in addition to its critical role in the γ-secretase activity, PS1 may regulate intracellular trafficking/maturation of other γ-secretase components as well as the substrate APP (33, 34). Whether Nct is also important for the proper localization of the γ-secretase complex is unclear. In fact, immature Nct remains stable when other components are disrupted (12, 32, 35), implicating a possible scaffold function of Nct in retaining other PS1/γ-secretase components in the proper subcellular locations.
Although it is becoming clear that a proper assembly of nascent PS1, Nct, APH-1, and PEN-2 within the ER is a prerequisite for post-translational maturation, stabilization, and trafficking of the complex to its final destination, the precise functions of these components, especially Nct, in these processes have only been scarcely elucidated. Given the fact that Nct forms a subcomplex with APH-1 independent of PS1 and PEN-2 (14, 17, 18), it is of interest to understand how Nct deficiency affects the stability, trafficking, and interaction of the other components of the PS1/γ-secretase complex. Since earlier published studies on the function of Nct within the γ-secretase complex utilized the RNAi approach, which allows the residual Nct molecules to still exert their functions, the data as obtained require further validation using cells completely lacking Nct expression. For example, fibroblast cells isolated from two independently generated Nct KO mouse strains (36, 37) both demonstrated undetectable PS1 fragments accompanied by a significant accumulation of full-length PS1 (36), whereas experiments using the RNAi approach to down-regulate Nct only revealed a reduction of PS1 fragments but not the accumulation of full-length PS1 (6, 38). In these Nct KO mice, it is reported that PS/γ-secretase complex was diminished, resulting in abolished APP processing (36, 37). It is necessary to further investigate the mechanisms underlying these processes upon Nct deficiency. Therefore, in this study, we utilized the embryonic fibroblasts isolated from an Nct KO mouse line (37) as a model to study the effect of the complete removal of Nct on the stability, trafficking, and interaction of each of the endogenous APH-1, PEN-2, and PS1.
MATERIALS AND METHODS
Cell Cultures and Transfection
Embryonic fibroblast cells isolated from Nct KO mouse (37) and its wild type counterpart (Nct WT) cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics. Mouse embryonic stem cells isolated from PS1/PS2 double knockout (PS 2KO) as well as wild type mouse (PS WT) (a kind gift from Dr. H. Zheng, Baylor College of Medicine) were maintained in Dulbecco’s modified Eagle’s medium with 20% fetal bovine serum, non-essential amino acid, ESGRO (Chemicon), and β-mercaptoethanol. Mouse neuroblastoma (N2a) cells were maintained in medium containing 50% Dulbecco’s modified Eagle’s medium and 50% Opti-MEM supplemented with 5% fetal bovine serum and antibiotics. The expression constructs encoding PS1, PEN-2, APH-1aL, and Nct have been described previously (7, 8, 35, 39). Transient transfection was performed using FuGENE 6 (Roche Applied Science) following the manufacturer’s protocol. Nct KO cells stably expressing human Nct were selected with 0.4 mg/ml hygromycin (39).
Antibodies
Rabbit anti-APH-1a antibody (from Zymed Laboratories Inc.) was raised against the COOH-terminal region of mammalian APH-1aL as described previously (35). Rabbit polyclonal antibodies Ab14 and PNT2, which recognize the NH2 terminus of PS1 (7) and the first 26 amino acids of PEN-2 (8), respectively, were generated in our laboratory. The antibody against α-tubulin (Sigma) was purchased, and anti-Nct antibody 716 and anti-APP COOH-terminal antibody 369 were described previously (27, 32).
Statistical Comparison of Protein Expression Levels
We used a simple method to statistically compare the expression levels of various proteins in different pairs of cell lines. An equal amount of total proteins from various cell lysates was loaded on the same gel for SDS-PAGE/Western blot. After exposure, the film was scanned as grayscale with a resolution of 300 pixels/inch. Protein bands of interest on the image were measured for the mean gray value after subtracting background using Scion Image software (Scion Corporation). Three independent samples were measured for each protein. After normalizing to the wild type control, protein expression levels in various cell systems were statistically compared using Student’s t test.
Pharmacological Treatment and RNA Interference
In the presence of 500 μm cycloheximide (CHX), Nct KO cells were treated with lactacystin (2.5, 10, and 20 μm) or imipramine (10, 40, and 80 μm) to inhibit the proteasomal or lysosomal activity, respectively. Alternatively, both Nct WT and Nct KO cells were treated with 100 μm CHX for different time periods (0, 1, 2, 4, 6, and 10 h). After incubation, cells were harvested and lysed in Nonidet P-40 lysis buffer (phosphate-buffered saline, pH 7.4, 0.5% Nonidet P-40, and 0.5% deoxycholate supplemented with protease inhibitors). Equal amounts of proteins then were analyzed by SDS-PAGE and Western blotting. Down-regulation of PEN-2 levels in both Nct WT and Nct KO cells using siRNA was described by Luo et al. (8), and the treatments with CHX for different time periods were mentioned above.
Subcellular Fractionation
Cells were homogenized using a ball-bearing cell cracker (40) and then centrifuged at 800 × g for 5 min, and the resulting supernatant was fractionated by sucrose density gradient as described previously (25, 27, 41).
Cell Surface Protein Biotinylation
To biotinylate cell surface proteins, cells were washed with ice-cold phosphate-buffered saline containing 1 mm each of CaCl2 and MgCl2 and incubated at 4 °C with 0.5 mg/ml Sulfo-NHS-LC-biotin (Pierce) for 20 min and the process was repeated once. Cell lysate was prepared in Nonidet P-40 lysis buffer. After affinity precipitation with streptavidin beads (Pierce), the biotinylated proteins were eluted with SDS-PAGE sample buffer (Invitrogen) and loaded directly on gels for electrophoresis followed by Western blot analysis with antibodies specific to APH-1, PEN-2, and PS1.
Deconvolution Immunofluorescence Microscopy
Cells were fixed in 2% paraformaldehyde and permeabilized as previously published (42). They then were incubated with polyclonal antibodies against PS1 (Ab14) at room temperature for 1 h. Subsequent secondary antibody incubation was carried out using Alexa Fluor 594-conjugated goat anti-rabbit IgG (Molecular Probe). Specimens were examined, and fluorescence images were collected with an Applied Precision DeltaVision imaging system (Issaquah, WA) coupled to an Olympus (I×70) fluorescence microscope. Cross-sectional images of cells were obtained with a 200-nm step width to optimize reconstruction of the center plane image. Deconvolution was done on a Silicon Graphics Octane® visual workstation (SGI, Mountain View, CA) equipped with Delta Vision reconstruction software.
Coimmunoprecipitation
Cells were lysed in 1% CHAPSO-containing buffer (25 mm HEPES, pH 7.4, 150 mm NaCl, and 2 mm EDTA supplemented with a mixture of protease inhibitors). After centrifugation, the supernatants were immunoprecipitated with rabbit preimmune serum, anti-PS1 (Ab14), anti-PEN-2 (PNT2), or anti-APH1 antibodies, and analyzed by Western blot using specific antibodies.
RESULTS
Nct Is Essential for the Stability of APH-1, PEN-2, and PS1
It has been reported that Nct KO results in undetectable proteolytic PS1 fragments (36, 37). Other studies using RNAi to down-regulate Nct confirmed this observation (6, 15, 16, 38) and showed a further reduction of PEN-2 and APH-1 (15, 16). Thus, it is necessary to examine the effect of Nct deficiency in the expression of the other three components of γ-secretase in a quantitative manner.
We established a quantitative Western blot-based assay system to analyze the protein expression level of the specific components and compare the levels in the Nct KO and PS 2KO cells. Consistent with previous notions that the Nct KO exhibits similar phenotypes to those of the PS double KO (36), we found that the levels of PS1 NTF, APH-1, and PEN-2 were dramatically decreased in Nct KO cells. On the other hand, the full-length PS1 accumulated to readily detectable levels in the absence of Nct (Fig. 1). Although PEN-2 levels were reduced in both Nct KO fibroblasts and PS 2KO cells, APH-1 level was dramatically reduced in Nct KO cells but remained stable in PS 2KO cells. The latter finding agrees with the previous results (13, 15) and supports the concept that APH-1 preferably associates with Nct rather than PEN-2 or PS1. As expected, the loss of Nct expression severely impaired the γ-secretase activity, resulting in the accumulation of the APP CTFs (Fig. 1a).
Fig. 1. Nicastrin deficiency destabilizes APH-1, PEN-2, and PS1 fragments.
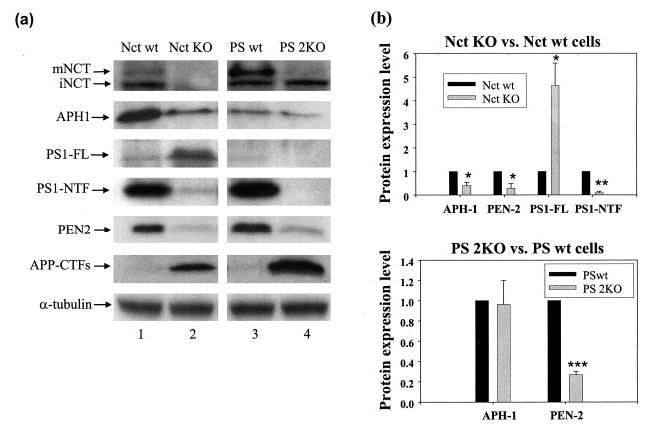
a, the expression levels of γ-secretase complex components and APP CTFs were analyzed in Nct KO, PS 2KO, and their respective wild-type counterparts (Nct wt and PS wt, respectively). Antibodies used here were rabbit anti-APH1a antibody, Ab14 (for PS1-full length (FL) and PS1-NTF), PNT2 (for PEN-2), 716 (for nicastrin), and 369 (for APP CTFs). α-Tubulin was used for loading control. mNCT, mature Nct; iNCT, immature Nct. b, statistical comparison of APH-1, PEN-2, and PS1 (FL and NTF) expression levels. Equal amounts of total proteins from different cell lysates were used. Quantitative data represent an average of three experiments. Student’s t test was performed for statistical comparison. Asterisks, different from the respective control (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Full-length PS1 and PEN-2 Are Subjected to Proteasome-mediated Degradation, Whereas APH-1 Is Degraded via Both Proteasomal and Lysosomal Pathways in Nct KO Cells
To explore the mechanisms underlying the diminished protein levels of the other three γ-secretase components, we investigated the protein degradation pathways involved in degrading APH-1, PEN-2, and PS1 in Nct KO cells using proteasome- and lysosome-specific inhibitors in the presence of an inhibitor of de novo protein synthesis (cycloheximide). Treating Nct KO cells for 4 h with 10 or 20 μm lactacystin, a specific proteasome inhibitor, considerably rescued PEN-2 (Fig. 2a, top panel) and full-length PS1 (Fig. 2a, middle panels), whereas imipramine, a specific lysosome inhibitor, had little effect on PEN-2 and PS1 degradation. Interestingly, both lactacystin and imipramine showed significant effects on rescuing APH-1 degradation (Fig. 2a, bottom panel). These data suggest that, in the absence of Nct, PEN-2, APH-1 and full-length PS1 are subjected to proteasome degradation presumably in the ER, whereas a portion of APH-1 may escape from the ER and be targeted from the Golgi to endosomal/lysosomal compartment for further degradation (see below for discussion). We also observed higher levels of PS1 NTF in cells treated with lactacystin (Fig. 2a, middle panels).
Fig. 2. PEN-2 and full-length PS1 degradation are subjected to the proteasomal pathway, whereas APH-1 degradation is subjected to both proteasomal and lysosomal pathways.
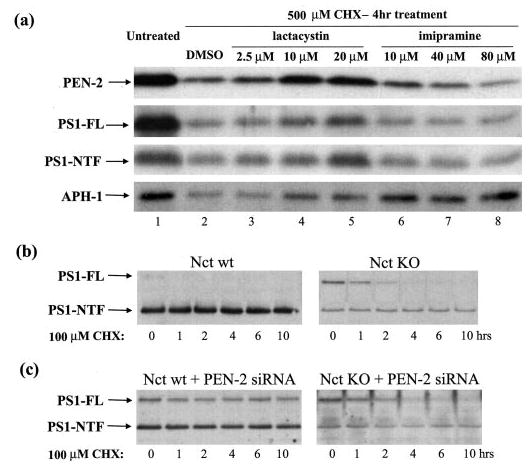
a, Nct KO cells were treated for 4 h with Me2SO (DMSO) (lane 2), the proteasomal specific inhibitor lactacystin (2.5, 10, and 20 μm; lanes 3–5), or with the lysosomal specific inhibitor imipramine (10, 40, and 80 μm; lanes 6–8) in the presence of 500 μm CHX. Nontreated Nct KO cells were used as control (lane 1). Antibodies PNT2, Ab14, and anti-APH-1a were used to detect PEN-2 (top panel), PS1 (full length (FL) and NTF, middle panels), and APH-1 (bottom panel), respectively. b, Nct WT and Nct KO cells were treated with 100 μm CHX for 0, 1, 2, 4, 6, and 10 h, respectively. PS1 (FL and NTF) levels then were analyzed using antibody Ab14. c, after using siRNA to down-regulate PEN-2 levels in both Nct WT and Nct KO cells, the cells were treated and analyzed as in b.
To further ascertain the kinetics of PS1 stability, cells were treated with protein synthesis inhibitor cycloheximide for different time periods (Fig. 2b). Despite a reduction in its protein level, PS1 NTF in Nct KO cells possesses a long half-life similar to that in Nct WT cells, suggesting that Nct may not be important for PS1 NTF stability. In contrast to WT cells, the full-length PS1 in Nct-deficient cells accumulates to readily detectable levels and is degraded within 2–4 h (Fig. 2b). We have previously shown that down-regulation of PEN-2 in N2a cells by siRNA increases the stability of full-length PS1 (8). Consistent with the study in N2a cells, full-length PS1 accumulated and remained stable for up to 10 h upon PEN-2 siRNA treatment in WT fibroblasts (Fig. 2c, left panel). However, attenuation of PEN-2 function by siRNA in Nct KO cells resulted in only an initial accumulation of full-length PS1, which then was quickly degraded (Fig. 2c, right panel). These data imply that Nct may be required for the stability of full-length PS1 polypeptide (6). On the other hand, our previous study has suggested a role for APH-1 in stabilizing full-length PS1 based on the observation that down-regulation of APH-1 diminishes the accumulation of full-length PS1 caused by PEN-2 siRNA (8). Together with the current observation that Nct deficiency destabilizes APH-1 (Fig. 1), we hypothesize that both APH-1 and Nct, probably in their subcomplex formation, are required for full-length PS1 stabilization.
The accumulation of full-length PS1 in Nct KO cells is probably a result of a dynamic balance between reduced PS1 endoproteolysis caused by diminished PEN-2 levels and proteasome-dependent degradation of full-length PS1. The partial instead of full recovery of full-length PS1 upon proteasome inhibitor treatment may be a net result of the two opposite effects. An increased PS1 endoproteolysis resulted from increased PEN-2 level and a reduced full-length PS1 degradation because of proteasome inhibition. The inhibition of lysosomal degradation by imipramine, which rescued APH-1 level on the other hand, exhibited little effect on full-length PS1, suggesting that, in the absence of Nct, APH-1 alone may not be sufficient for full-length PS1 stabilization.
Altered Subcellular Localization of APH-1, PEN-2, and PS1 in the absence of Nct
Previous studies have established a role for PS1 in regulating trafficking of select membrane proteins including APP, PEN-2, and Nct (10, 11, 19, 32, 33, 34). To investigate whether Nct may also be involved in subcellular localization of the other components, we examined the patterns of the ER and Golgi localization of PEN-2, APH-1, and PS1 in Nct KO and WT cells using a well established sucrose gradient fractionation method. Although a large amount of endogenous PS1 fragments, PEN-2, and APH-1 was localized in the Golgi-enriched fractions in WT cells (Fig. 3, left panels), PEN-2, APH-1, and full-length PS1 were largely accumulated in the fractions corresponding to the ER compartments in Nct KO cells (Fig. 3, right panels). The observed alteration in subcellular localization patterns suggests that Nct is required for the intracellular trafficking of APH-1, PEN-2, and PS1 from the ER to Golgi and further supports the notion that complete assembly and interaction among γ-secretase components are essential for the trafficking and maturation of the complex.
Fig. 3. Nct affects subcellular localization of APH-1, PEN-2, and PS1.
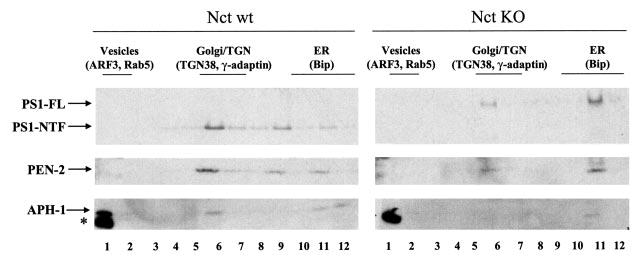
Nct KO and WT cells were homogenized and then fractionated by sucrose gradient sedimentation. Samples were analyzed by Western blotting using anti-APH-1a antibody (for APH-1), PNT2 (for PEN-2), and Ab14 (for PS1), respectively. The enrichment of protein markers for various organelles across the gradients is indicated. Asterisk indicates nonspecific cross-reactive bands.
Differential Cell Surface Localization of Full-length PS1, PEN-2, and APH-1 in Nct KO Cells
A bulk of available data indicate that, whereas all four γ-secretase components are synthesized and initially assembled in the ER, the mature enzymatically active complex seems to reside in late Golgi/TGN and, to a lesser extent, in the plasma membrane (21–24). Having demonstrated that Nct is important for ER exit and steady-state localization in post-ER compartments of a large fraction of PS1, PEN-2, and APH-1, we next examined whether Nct deficiency may exert impacts on plasma membrane delivery (from the Golgi) of those molecules that have escaped from the ER. Our results showed that, similar to PEN-2 and PS1 fragments (19), there is a significant amount of APH-1 that appears on the cell surface in Nct WT cells, whereas in Nct KO cells, only a negligible amount of APH-1 was detected on the cell surface by biotinylation (Fig. 4, top panel). Nct deficiency does not significantly affect the proportion of PEN-2 molecules that is on the plasma membrane (Fig. 4, bottom panel). Quantitative analysis of our data showing identical ratios of PEN-2 level in total lysates versus that of biotinylated PEN-2 between Nct WT cells and Nct KO cells also supported this conclusion. Interestingly, although PS1 NTFs on the cell surface of Nct KO cells were not detected, probably because of the very low levels of PS1 NTFs in these cells, we noticed a significant amount of uncleaved full-length PS1 accumulated on the plasma membrane (Fig. 4, middle panel).
Fig. 4. Cell surface distribution of APH-1 but not PS1 or PEN-2 is diminished in the absence of Nct.
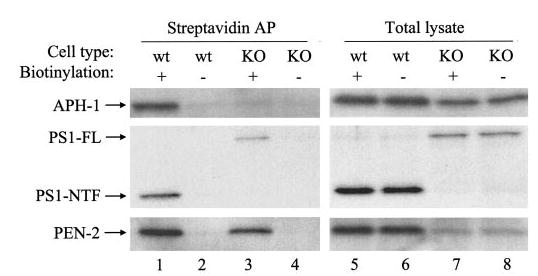
Nct KO and WT cells were incubated in the presence or absence (as a negative control) of biotin followed by affinity precipitation (AP) using streptavidin (lanes 1–4). APH-1, PEN-2, and PS1 then were analyzed using Western blot with anti-APH-1a antibody, PNT2 (for PEN-2), or Ab14 (for PS1-NTF and PS1-full length (FL)). Five percent of cell lysates was loaded as input (lanes 5–8).
The cell surface localization of PS1 was corroborated by immunofluorescence deconvolution microscopic study using PS1 antibody Ab14. Plasma membrane staining of PS1 was striking in Nct KO cells (Fig. 5, middle panel). On the contrary, in WT cells, cell surface PS1 staining is notable (indicated by white arrows in Fig. 5, left panel), but the majority of PS1 staining is observed on intracellular membranes. Stable expression of human Nct reversed the intensity of cell surface PS1 staining in Nct KO cells to a level similar to that in Nct WT cells, confirming the specificity of the immunostaining of PS1 antibody Ab14 (Fig. 5, right panel). The levels of cell surface biotinylated full-length PS1 were also diminished after stable transfection of human Nct into Nct KO cells (data not shown). In addition, cell surface staining of PEN-2 in Nct KO cells was also evident (data not shown); however, cell surface immunostaining of APH-1 has failed in our experiments, probably because the APH-1 antibody is not suitable for immunofluorescence labeling of endogenous mouse APH-1.
Fig. 5. Marked cell surface localization of PS1 in Nct KO cells.
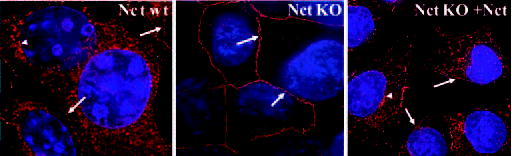
Nct WT cells, Nct KO cells, and Nct KO cells stably expressing human nicastrin (+Nct) were immunostained using anti-PS1 antibody Ab14. After subsequent secondary antibody incubation with Alexa Fluor 594-conjugated goat anti-rabbit IgG, cells were examined with deconvolution microscopy. Arrows show plasma membrane profile of PS1. Arrowheads indicate intracellular perinuclear-like staining of PS1.
Nct Is Not Essential for Association among APH-1, PEN-2, and PS1
Previous studies have shown that, in the functional γ-secretase complex, the four components are associated with each other to form a high molecular weight complex (for review, Ref. see 3). To investigate whether the absence of Nct may affect the association of the remaining components, we performed coimmunoprecipitation experiments using Nct WT and KO cells. As shown in Fig. 6a, PEN-2 antibody coimmunoprecipitated PS1-NTF in WT cells (lane 5) and full-length PS1 in Nct KO cells (lane 6), confirming the binding of PEN-2 to both full-length and fragments of PS1 (8) and further demonstrating association between PEN-2 and full-length PS1, even in the absence of Nct. This is further supported by the observation that PS1 antibody Ab14 was able to coimmunoprecipitate PEN-2 in both Nct WT and KO cells (Fig. 6b, lanes 5 and 6). Similarly, anti-APH-1a antibody coimmunoprecipated PS1-NTF and, to a much lesser extent, full-length PS1 in WT cells (Fig. 6a, lane 7). The APH-1 antibody was still able to coimmunoprecipitate full-length PS1, even in the absence of Nct (Fig. 6a, lane 8). Moreover, APH-1a was shown to remain associated with PEN-2 in both WT and Nct KO cells (Fig. 6b, lanes 7 and 8). Together, these results suggest that, in the absence of Nct, APH-1, PEN-2, and PS1 still associate with each other, although PS1 exists in its full-length form in such association.
Fig. 6. Association between APH-1, PEN-2, and PS1 in the absence of Nct.
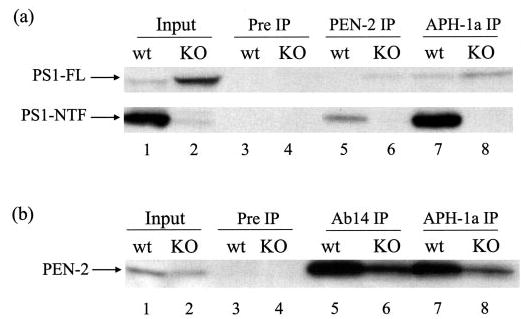
Nct WT and KO cells were lysed in 1% CHAPSO buffer. a, association of PS1 with PEN-2 and APH-1. Lysates were immunoprecipitated with rabbit preimmune serum (lanes 3 and 4), PEN-2 antibody PNT2 (lanes 5 and 6), or anti-APH-1a antibody (lanes 7 and 8) followed by Western blotting using PS1 antibody Ab14. b, association of PEN-2 with PS1 and APH-1. Lysates were immunoprecipitated (IP) with rabbit preimmune serum (lanes 3 and 4), PS1 antibody Ab14 (lanes 5 and 6), or anti-APH-1a antibody (lanes 7 and 8) followed by immunoblotting using PEN-2 antibody PNT2. 5% total lysates was used as input (lanes 1 and 2). FL, full length.
The Diminished PEN-2 Level in Nct KO Cells Can Be Partially Restored by Overexpressing APH-1, PEN-2, or Full-length PS1 Individually or Collectively
To study whether reductions of PS1 fragments, PEN-2, or APH-1 in the absence of Nct can be rescued, we transiently overexpressed PS1, APH-1, or PEN-2 (HA-tagged) individually or the three molecules collectively in Nct KO cells. As shown in Fig. 7, the level of endogenous PEN-2 in Nct KO cells was increased by overexpression of APH-1 (~1.8-fold, lane 3 versus lane 2), PEN-2 (~5-fold, lane 4 versus 2), or PS1 (~2-fold, lane 5 versus lane 2). The PEN-2 level was restored to a level comparable to that in WT cells by overexpression of APH-1, PEN-2, and PS1 collectively (lane 6 versus lane 1). Similar to the endogenous PEN-2, the levels of endogenous PS1 and APH-1 were also partially rescued but to various degrees by individually or collectively overexpressing exogenous APH-1, PEN-2, and PS1 (data not shown). Although the preceding data clearly demonstrated that Nct is crucial for maintaining the stability of the other γ-secretase components, this function of Nct may be bypassed by non-physiologically overexpressing the other components.
Fig. 7. Exogenous (Exo) APH-1, PEN-2 and/or PS1 expression partially restore endogenous (Endo) PEN-2 level in Nct KO cells.
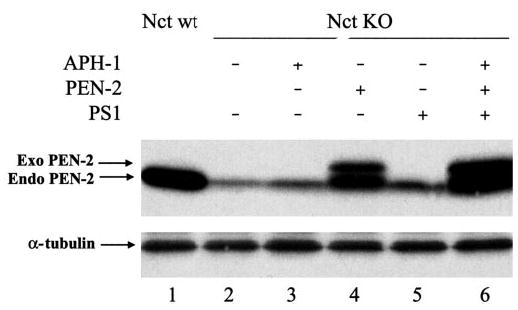
APH-1, PEN-2, or PS1 expression constructs was transiently transfected into Nct KO cells individually or collectively. Antibody PNT2 was used to detect both endogenous PEN-2 and exogenous hemagglutinin-PEN-2. α-Tubulin was used as control.
DISCUSSION
Among the four γ-secretase components, Nct has been suggested as a scaffold protein during complex assembly (3, 15) because it is relatively stable in PS KO cells or when PEN-2 or APH-1 expression is down-regulated (by RNAi) (12, 32, 35). Our present studies from Nct KO cells, as well as results from Nct RNAi knock-down studies (6, 15, 16, 38), demonstrate that the protein levels of the other three components are dramatically affected by the level of Nct, further supporting an essential role for Nct in maintaining the stability of the complex. Unlike the results from down-regulation of Nct by RNAi (6, 38), we observed significant accumulation of full-length PS1 in Nct KO cells, which is probably the result of reduced conversion of full-length PS1 to its fragments. We speculate that the reduced endoproteolysis of full-length PS1 to its fragments may be mediated by the effect of Nct on the stability of PEN-2, which has been demonstrated to play an essential role in the PS1 endoproteolysis (8) and to be required for the stability of PS1 fragments (12). However, the possibility that Nct may also directly mediate PS1 endoproteolysis cannot be ruled out and further experiments are needed to address this issue.
The potential role of Nct in maintaining the stability of the complex predicts an effect of Nct deficiency on intracellular trafficking/distribution of other γ-secretase components. In this study, we found that the patterns of intracellular localization of those molecules are significantly altered when Nct is absent. In contrast to their predominant Golgi localization in WT cells, the majority of the non-degraded PS1, PEN-2, and APH-1 are found in the ER fractions and, to a lesser extent, in the Golgi/TGN fractions. It is conceivable that a correct conformation of the complex that is required for exiting the ER and for further transport along the secretary pathway is not achieved in the absence of Nct, although the other three components still bind to each other (Fig. 6 and see below for discussion). The incomplete complex that accumulates in the ER may be targeted to the proteasome degradation machinery as shown by our study that the protein levels of PEN-2, APH-1, and PS1 can be rescued by the proteasome inhibitor lactacystin (Fig. 2). However, it is unclear whether the unassociated monomeric polypeptides or the incompletely assembled PS1·PEN-2·APH-1 complex is more prone to degradation by the proteasome.
The steady-state level of a protein in the Golgi/TGN is a combinatory result of the balance between incoming traffic from the ER and outgoing transport to the plasma membrane. Whereas our fractionation studies suggested reduced transport of the three proteins from the ER to the Golgi, we also found significant amounts of PEN-2 and full-length PS1 along with a negligible amount of APH-1 on the cell surface of Nct KO cells (Figs. 4 and 5). Thus, our results suggest a possible role for Nct in strengthening and retaining the γ-secretase complex in Golgi. In Nct KO cells, a fraction of the trimeric PS1·PEN-2·APH-1 assembly escapes from ER-associated proteasome degradation and exits the ER but remains susceptible for dissociation. However, following detachment from the trimeric assembly, APH-1 may be discriminated by the sorting machinery in the TGN and targeted to endosomal/lysosomal degradation, consistent with our demonstration that APH-1 may be subjected to both proteasomal and lysosomal degradation (Fig. 2). On the other hand, the PEN-2·PS1 subcomplex (43) proceeds to traffic to the plasma membrane without being retained in the Golgi. The differential trafficking of PS1·PEN-2 and APH-1 is consistent with our recent report of a coordinate surface delivery of PS1 and PEN-2, but not NCT, upon γ-secretase inhibitor treatments (19). Furthermore, the significant accumulation of full-length PS1 at the cell surface raises a possibility that NCT may also affect internalization of full-length PS1. Taken together, these findings reflect a tighter relationship between PS1 and PEN-2, further supporting the existence of NCT·APH-1 precursor and PS1·PEN-2 intermediate subcomplexes (13, 17, 43).
The above model is based on our observations that APH-1 and PEN-2·PS1 exhibit distinct patterns in subcellular localization, trafficking, and degradation. Accordingly, one would anticipate that APH-1 may not associate with PEN-2 and PS1 in the absence of its partner Nct. Surprisingly, our coimmunoprecipitation experiments showed a relatively minor yet clear binding of APH-1 with both PEN-2 and full-length PS1 in Nct KO cells. Although this observation is difficult to explain with our current knowledge, one may speculate that APH-1 binds PEN-2 and full-length PS1 early in the ER, which stabilizes both full-length and fragments of PS1 and makes the trimeric complex competent for ER exit. The presence of Nct further strengthens the complex and facilitates achieving the conformation conducive for endoproteolysis of PS1. In fact, our previous results showed that APH-1 plays a role in stabilizing both full-length and fragments of PS1 (8), providing support to this model. When APH-1 fails to associate with Nct, the majority of APH-1 dissociates from the trimeric assembly and gets targeted to ER-associated degradation as well as lysosomal degradation. Alternatively, in the absence of Nct, the majority of APH-1 may become unavailable to stabilize nascent PEN-2 and PS1 polypeptides in the ER (and hence affect PS1 endoproteolysis). However, a small amount of APH-1 may persist as a PS1·PEN-2·APH-1 complex in Nct KO cells and contribute to the existence of the residual PEN-2 and PS1 fragments.
Finally, when exogenous APH-1, PS1, or PEN-2 was expressed either individually or collectively into Nct KO cells, the diminished levels of PEN-2 (Fig. 7), APH-1, and PS1 (data not shown) were partially rescued. These results indicate an important but dispensable role of Nct in maintaining the stability of the other components of the γ-secretase complex and further support the notion that the γ-secretase components regulate each other in a coordinated way (8–16).
Taken together, our results strongly support an essential role for Nct in stabilizing and in intracellular trafficking of other γ-secretase components during biogenesis as well as following their assembly into a functional multimeric complex. Nevertheless, extensive future efforts are necessary to elucidate the more detailed mechanisms underlying the regulation and function of the γ-secretase.
Footnotes
This work was supported by National Institutes of Health Grants NS046673 (to H. X.), AG021173 (to H. X.), F32 AG024895-01A1 (to Y. Z.), F32 AG023432-01 (to W. L.), AG021495 (to G. T.), and by Alzheimer’s Association (to H. X. and G. T.).
The abbreviations used are: APP, amyloid precursor protein; Nct, nicastrin; PS, presenilin; NTF, NH2-terminal fragment; RNAi, RNA interference; CTF, COOH-terminal fragment; siRNA, small interfering RNA; ER, endoplasmic reticulum; TGN, trans-Golgi network; KO, knockout; PS 2KO, PS1/PS2 double knockout; WT, wild type; CHX, cycloheximide; CHAPSO, 3-[(3-holamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid; APH-1, anterior pharynx-defective-1; PEN-2, presenilin enhancer-2.
References
- 1.Steiner H, Haass C. Nat Rev Mol Cell Biol. 2000;1:217–224. doi: 10.1038/35043065. [DOI] [PubMed] [Google Scholar]
- 2.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Proc Natl Acad Sci U S A. 2003;100:6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Strooper B. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 4.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 5.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch RD, Ruble C, Nye JS, Curtis D. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 6.Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- 7.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 8.Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H. J Biol Chem. 2003;278:7850–7854. doi: 10.1074/jbc.C200648200. [DOI] [PubMed] [Google Scholar]
- 9.Steiner H, Winkler E, Edbauer D, Prokop S, Basset G, Yamasaki A, Kostka M, Haass C. J Biol Chem. 2002;277:39062–39065. doi: 10.1074/jbc.C200469200. [DOI] [PubMed] [Google Scholar]
- 10.Bergman A, Hansson E, Pursglove SE, Farmery MR, Lannfelt L, Lendahl U, Lundkvist J, Naslund J. J Biol Chem. 2004;279:16744–16753. doi: 10.1074/jbc.M313999200. [DOI] [PubMed] [Google Scholar]
- 11.Crystal AS, Morais VA, Fortna RR, Carlin D, Pierson TC, Wilson CA, Lee VMY, Doms RW. Biochemistry. 2004;43:3555–3563. doi: 10.1021/bi0361214. [DOI] [PubMed] [Google Scholar]
- 12.Prokop S, Shirotani K, Edbauer D, Haass C, Steiner H. J Biol Chem. 2004;279:23255–23261. doi: 10.1074/jbc.M401789200. [DOI] [PubMed] [Google Scholar]
- 13.Gu Y, Chen F, Sanjo N, Kawarai T, Hasegawa H, Duthie M, Li W, Ruan X, Luthra A, Mount HT, Tandon A, Fraser PE, St George-Hyslop P. J Biol Chem. 2003;278:7374–7380. doi: 10.1074/jbc.M209499200. [DOI] [PubMed] [Google Scholar]
- 14.Morais VA, Crystal AS, Pijak DS, Carlin D, Costa J, Lee VM, Doms RW. J Biol Chem. 2003;278:43284–43291. doi: 10.1074/jbc.M305685200. [DOI] [PubMed] [Google Scholar]
- 15.Shirotani K, Edbauer D, Kostka M, Steiner H, Haass C. J Neurochem. 2004;89:1520–1527. doi: 10.1111/j.1471-4159.2004.02447.x. [DOI] [PubMed] [Google Scholar]
- 16.Shirotani K, Edbauer D, Capell A, Schmitz J, Steiner H, Haass C. J Biol Chem. 2003;278:16474–16477. doi: 10.1074/jbc.C300095200. [DOI] [PubMed] [Google Scholar]
- 17.LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ. J Biol Chem. 2003;278:37213–37222. doi: 10.1074/jbc.M303941200. [DOI] [PubMed] [Google Scholar]
- 18.Hu Y, Fortini ME. J Cell Biol. 2003;161:685–690. doi: 10.1083/jcb.200304014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Luo WJ, Zhang YW, Li YM, Thinakaran G, Greengard P, Xu H. J Biol Chem. 2004;279:40560–40566. doi: 10.1074/jbc.M404345200. [DOI] [PubMed] [Google Scholar]
- 20.Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W. J Cell Biol. 2001;154:731–740. doi: 10.1083/jcb.200104045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaether C, Lammich S, Edbauer D, Ertl M, Rietdorf J, Capell A, Steiner H, Haass C. J Cell Biol. 2002;158:551–561. doi: 10.1083/jcb.200201123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, Selkoe DJ, Kopan R, Goate AM. J Biol Chem. 1999;274:36801–36807. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 24.Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlisny MB, Fraser P, Hyslop PS, Selkoe DJ, Haass C. Mol Med. 1996;2:673–691. [PMC free article] [PubMed] [Google Scholar]
- 25.Greenfield JP, Xu H, Greengard P, Gandy S, Seeger M. J Biol Chem. 1999;274:33843–33846. doi: 10.1074/jbc.274.48.33843. [DOI] [PubMed] [Google Scholar]
- 26.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S. Proc Natl Acad Sci U S A. 1997;94:3748–3752. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- 29.Soriano S, Chyung AS, Chen X, Stokin GB, Lee VM, Koo EH. J Biol Chem. 1999;274:32295–32300. doi: 10.1074/jbc.274.45.32295. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chyung JH, Selkoe DJ. J Biol Chem. 2003;278:51035–51043. doi: 10.1074/jbc.M304989200. [DOI] [PubMed] [Google Scholar]
- 32.Leem JY, Vijayan S, Han P, Cai D, Machura M, Lopes KO, Veselits ML, Xu H, Thinakaran G. J Biol Chem. 2002;277:19236–19240. doi: 10.1074/jbc.C200148200. [DOI] [PubMed] [Google Scholar]
- 33.Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, Sisodia SS, Thinakaran G, Xu H. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- 34.Kim SH, Leem JY, Lah JJ, Slunt HH, Levey AI, Thinakaran G, Sisodia SS. J Biol Chem. 2001;276:43343–43350. doi: 10.1074/jbc.M108245200. [DOI] [PubMed] [Google Scholar]
- 35.Lee SF, Shah S, Li H, Yu C, Han W, Yu G. J Biol Chem. 2002;277:45013–45019. doi: 10.1074/jbc.M208164200. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Fici GJ, Mao CA, Myers RL, Shuang R, Donoho GP, Pauley AM, Himes CS, Qin W, Kola I, Merchant KM, Nye JS. J Biol Chem. 2003;278:33445–33449. doi: 10.1074/jbc.M301288200. [DOI] [PubMed] [Google Scholar]
- 37.Li T, Ma G, Cai H, Price DL, Wong PC. J Neurosci. 2003;23:3272–3277. doi: 10.1523/JNEUROSCI.23-08-03272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Z, Romano DM, Kovacs DM, Tanzi RE. J Biol Chem. 2004;279:34130–34137. doi: 10.1074/jbc.M401094200. [DOI] [PubMed] [Google Scholar]
- 39.Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H, Greengard P, Gandy S. J Biol Chem. 1995;270:23243–23245. doi: 10.1074/jbc.270.40.23243. [DOI] [PubMed] [Google Scholar]
- 41.Gasparini L, Gouras GK, Gross RS, Beal MF, Wang R, Greengard P, Xu H. J Neurosci. 2001;21:2561–2570. doi: 10.1523/JNEUROSCI.21-08-02561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin P, Fischer T, Weiss T, Farquhar MG. Proc Natl Acad Sci U S A. 2000;97:674–679. doi: 10.1073/pnas.97.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, Wolfe MS. Biochemistry. 2004;43:323–333. doi: 10.1021/bi035748j. [DOI] [PubMed] [Google Scholar]