

Abstract
Osteoblast-derived IL-6 functions in coupled bone turnover by supporting osteoclastogenesis favoring bone resorption instead of bone deposition. Gene regulation of IL-6 is complex occurring both at transcription and post-transcription levels. The focus of this paper is at the level of mRNA stability, which is important in IL-6 gene regulation. Using the MC3T3-E1 as an osteoblastic model, IL-6 secretion was dose dependently decreased by SB203580, a p38 MAPK inhibitor. Steady state IL-6 mRNA was decreased with SB203580 (2μM) ca. 85% when stimulated by IL-1β (1 5ng/ml). These effects require de novo protein synthesis as they were inhibited by cycloheximide. p38 MAPK had minor effects on proximal IL-6 promoter activity in reporter gene assays. A more significant effect on IL-6 mRNA stability was observed in the presence of SB203580. Western blot analysis confirmed that SB203580 inhibited p38 MAP kinase, in response to IL-1β in a dose dependent manner in MC3T3-E1 cells. Stably transfected MC3T3-E1 reporter cell lines (MC6) containing green fluorescent protein (GFP) with the 3′ untranslated region of IL-6 was constructed. Results indicated that IL-1β, TNFα, LPS but not parathyroid hormone (PTH) could increase GFP expression of these reporter cell lines. Endogenous IL-6 and reporter gene eGFP-IL-6 3′ UTR mRNA was regulated by p38 in MC6 cells. In addition, transient transfection of IL-6 3′ UTR reporter cells with immediate upstream MAP kinase kinase-3 and -6 increased GFP expression compared to mock transfected controls. These results indicate that p38 MAPK regulates IL-1β-stimulated IL-6 at a post transcriptional mechanism and one of the primary targets of IL-6 gene regulation is the 3′ UTR of IL-6.
Keywords: IL-6, IL-1β, osteoblasts, gene expression, p38 MAPK, mRNA Stability, GFP, ARE
INTRODUCTION
Bone is a dynamic tissue that constantly undergoes a remodeling process where bone resorption and bone deposition are balanced. When chronic inflammation occurs in bone, and other mineralized tissues, this balance is disrupted, thus favoring net bone loss. Diseases involving an intimate combination of bone loss and inflammation include periodontal disease and rheumatoid arthritis or metabolic bone diseases such as osteoporosis, Paget’s disease, and multiple myeloma where cytokine disregulation is observed. Current therapeutic targets aimed at treatment and management of inflammatory bone diseases have focused on cytokines, many of which play central and critical roles in inflammatory bone disease pathophysiology.
Interleukin-6 (IL-6) is the first identified centrally important cytokine for osteoclastogenesis and thus bone resorption (33). IL-6 is a member of a family of structurally related cytokines that use the gp130 signal transducer in their receptor complex. Besides IL-6, the family includes IL-11, oncostatin M (OSM), leukemia inhibitory factor (LIF), and cardiotropin 1. Several investigators have demonstrated that both estrogens and androgens suppress the production of IL-6 as well as the expression of the two subunits of the IL-6 receptor, IL-6R and gp130, in cells of the bone marrow stromal/osteoblastic lineage (1, 14, 34). Similar results were obtained subsequently by others in rats as well as in humans, in the bone marrow and in the peripheral blood (35, 51). Importantly, it was shown that neutralization of IL-6 with antibodies or knockout of the IL-6 gene in mice prevents the upregulation of granulocyte macrophage-colony stimulating factor (GM-CSF) in the marrow, and the expected increase of osteoclast numbers in trabecular bone sections; and also protects the loss of bone following loss of sex steroids (1, 21).
This cytokine is produced by osteoblasts and is inducible by the pro-inflammatory cytokines IL-1, TNF, or bacterial-derived lipopolysacchride (LPS) as well as other osteotrophic hormones including parathyroid hormone (PTH) (6, 12, 19, 26). IL-6 as well as IL-β, TNF-α, and macrophage-colony-stimulating factor (M-CSF) appear to be the major target genes for osteoclastogenesis (53). More recently, it has been recognized that all of these pro-inflammatory cytokines converge to stimulate receptor activator of NF-kß ligand (RANKL), which in the presence of low concentrations of M-CSF is necessary and sufficient for the complete osteoclast differentiation. In addition, most of these same cytokines decrease the production of osteoprotegerin (OPG), a potent anti-osteoclastogenic factor. OPG acts as a decoy, blocking the binding of RANKL to its receptor, RANK, which is expressed in committed pre-osteoblastic cells (18). IL-6 and IL-11 may influence osteoclastogenesis by stimulating the self-renewal and inhibiting the apoptosis of osteoclast progenitors (22). Because of the interdependent nature of the production of IL-1, IL-6, and TNF, a significant increase in one of them may amplify, in a cascade fashion, the effect of the others (22).
However, production and secretion of IL-6 and RANKL, e.g., are the final result of different stimuli acting on the cell surface. These stimuli generate a message or signal, which has to be taken to the cell nucleus where it will initiate the appropriate response. Mitogen-activated protein kinases (MAPK) are key enzymes in the signal transduction cascade from the extracellular environment to the nucleus of essentially every eukaryotic cell type (36). Three groups of MAPKs have been identified in mammalian cells (3, 9). These are the extracellular signal regulated kinases (ERKs), the c-Jun N-terminal kinases (JNKs) and the p38 MAPKs.
Recent studies of both osteoblasts and chrondocytes have shown that the IL-1- or TNF-induced IL-6 production can be blocked with p38 MAPK inhibitors (4, 5, 39). The functional consequence of blocking IL-6 was demonstrated further when p38 MAPK inhibitors were shown to prevent IL-1- or TNF-mediated bone resorption in an in vitro model (28).
The more obvious inference of such information is that p38 MAPK inhibitors prevent the signal generated by IL-1 or TNF to reach the nucleus, thereby acting on gene transcription. However, regulation of mRNA turnover is now recognized to be one of the centrally important mechanisms of controlling the level of cytoplasmic mRNA, and consequently, gene expression. The stability of different mRNAs varies considerably and in many cases is regulated in response to extracellular stimuli. Expression of cytokines is usually transient because their mRNAs are inherently unstable. Rapid mRNA turnover is mediated by cis-acting elements that are distributed throughout the mRNA molecule (49). For example, a common destabilizing element found in the 3′ untranslated region (UTR) of short-lived mRNAs is the AU-rich element (ARE), consisting of multiple copies of the pentanucleotide, AUUUA, and a high content of U residues (7, 52). Although an ARE or an UUAUUUAUU nonamer can direct rapid mRNA turnover by promoting deadenylation followed by decay of the mRNA body, the mechanism underlying this activity has not been elucidated and has been the focus of several investigators.
Extracellular stimuli may also positively or negatively regulate ARE-directed mRNA decay (20, 55). Specific cis elements present in some cytokine mRNAs are recognized by signal responsive trans-acting factors that modulate or determine ARE-directed decay. The signaling pathways that regulate ARE-directed mRNA decay have not been widely explored, and the relevant cis elements that mediate signal-induced mRNA stabilization have only started to be examined. In addition, mRNA stabilization may require cooperation between multiple RNA elements, suggesting that either proper mRNA folding is necessary for signal-induced stabilization or that interactions between different transacting factors can modulate ARE-directed mRNA decay (2). Herein, we show that p38 MAPK decreases IL-1β-induced IL-6 gene expression by influencing mRNA stability, and also that a main target of this regulation is the 3′UTR of the IL-6 gene.
METHODS AND MATERIALS
Cell culture of MC3T3-E1 cells
MC3T3-E1 osteoblastic cells were obtained from RIKEN (Japan). Cells were cultured in DMEM (Invitrogen Life Technologies, Grand Island, NY), supplemented with 10% fetal bovine serum (Sigma), 100 U/ml penicillin, and 100μg/ml streptomycin, in a humidified atmosphere of 5% CO2 in air at 37°C. Osteoblast phenotypic mRNAs, including bone sialoprotein and osteocalcin, were routinely assayed by reverse transcription polymerase chain reaction (RT-PCR) to verify osteoblastic phenotype expression in these cells.
Gene Expression assessed by Real Time Polymerase Chain Reaction
To assess IL-6 gene expression after stimulation with IL-1β in the presence and absence of p38 MAPK inhibitor (SB203580), we employed real time PCR. Briefly, total RNA was isolated using Trizol reagent (Invitrogen). Total RNA was visualized for intactness by ethidium bromide staining following gel electrophoresis. RNA was quantitated by spectroscopy (SmartSpec 3000, BioRad). PCR primers were commercially synthesized (Sigma-Genosys, Woodland, TX) for IL-6 (Genbank accession # NM-031168), eGFP (Clontech), and the house-keeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Genbank #AF106860). Primer sequences that were employed for IL-6 were 5′- GACAACTTTGGCATTGTGG -3′ for forward sequence and 5′- ATGCAGGGATGATGTTCTG -3′ for reverse sequence. The primer sequences for eGFP were 5′-CAACTACAACAGCCACAACG-3′ for forward sequence and 5′-GGTCACGAACTCCAGCAG -3′ for reverse sequence. The sequences for GAPDH were 5′- CAAAGCCAGAGTCCTTCAGA-3′ for forward primer sequence and 5′- GATGGTCTTGGTCCTTAGCC -3′ for reverse primer sequence. Products sizes for IL-6 were 160 base pairs (bp) and 132 bp for GAPDH. Real-time PCR was performed using the SYBR-Green dye system on an iCycler thermocycler (BioRad). Total RNA is isolated using TRIZOL (Invitrogen) from MC3T3-E1 cells following treatments. RT is performed using Superscript II with oligo dT. Real Time PCR is performed on the RT product using Taq Polymerase (Invitrogen) and primers specific for mouse IL-6 and GAPDH cDNAs. IL-6 is amplified for 50 cycles and GAPDH was amplified for 50 cycles. Threshold values are assigned and used with Amplification Efficiencies to calculate IL-6 expression levels. Q-gene (http://www.biotechniques.com/softlib/qgene.html) quantitative software was used to analyze gene expression based on cycle threshold values normalized to GAPDH expression. Real time PCR products were also verified through electrophoresis on 2% agarose gel and visualized using ethidium bromide staining to ensure that they were of the correct size.
For analysis the influence of mRNA stability on IL-6 gene expression in the presence or absence of SB203580, we used the DNA polymerase inhibitor, actinomycin D. In these experiments, cells were pre-treated with SB2303580 (2μM) for 1h and then treated with IL-1β for 4h. Subsequently, actinomycin D was added to prevent further transcription. Total RNA was harvested and analyzed by real time PCR as outlined above after 0, 30, 60 minutes of treatment with actinomycin D (2μg/ml).
To determine whether protein synthesis was required for the SB203580 mediated effects on IL-6 mRNA levels, cells were incubated with cycloheximide (CHX; 2μg/ml) prior to treatment with SB203580 and IL-1β. RT-PCR analysis for IL-6 and GAPDH expression was conducted as described above.
Transient transfection and reporter gene assays
IL-6 pCAT promoter constructs were originally obtained from Dr. L. Vales (UMDNJ) (23, 45). IL-6 promoter regions were subcloned into pGL-3 Basic (Promega, Madison, WI, USA), a reporter vector containing the firefly luciferase gene. MC3T3-E1 cells were cultured to ca. 50% confluency in 60 mm2 dishes, washed twice with Opti-MEM media (Life Technologies) mixture according to manufacturer’s instructions. The cells were transiently transfected with L-luciferase reporter constructs containing -225 and -138 bps of the IL-6 proximal promoter using Lipofectamine Plus (Invitrogen). Six hours post transfection, cells were treated with SB203580 (5μM; Calbiochem) and stimulated with IL-1β (1ng/ml) for 24 hours. Cell extracts were normalized by total protein content as determined by Bradford method. Luminescence was measured using a Berthold Orion Microplate luminometer using the Luminescent kit (Promega) following manufacturers’ instructions.
Western Blot Analysis
MC3T3-E1 cells were exposed to SB203580 (1–20μM) for ~30 minutes, then stimulated with IL-1β (1–5 ng/ml) for 20–30 minutes. Cells were rinsed with ice-cold PBS and whole cell lysates harvested directly in SDS-PAGE buffer (BioRad). In some cases, protein concentrations were measured by Bradford method (BioRad). Ten micrograms of each sample were electrophoresed on 10% denatured SDS-PAGE gels and electrotransferred to nitrocellulose membranes (BioRad). Antibodies against phosphorylated and non-phosphorylated forms of p38 MAPK kinase (Cell Signaling Technologies) were used as primary antibodies in these studies. Primary antibodies were detected using HRP-conjugated secondary antibodies and LumniGlo (Cell Signaling) chemiluminescence detection and quantitated using a computerized imaging system.
Stable Reporter System for IL-6 3′UTR
To evaluate the relative contribution of the IL-6 3′UTR on mRNA stability, we established MC3T3-E1 cells which expressed enhanced green fluorescent protein (eGFP) as a reporter system under the control of the IL-6 3′UTR. Briefly, a 411 bp Xho1-Mlu1fragment containing the IL-6 3′UTR was cloned into peGFP-C1 (Clontech) vector to replace the poly-adenylation site from the vector. Neomycin-resistant stable transfectants of MC3T3-E1 cells were obtained by clonal selection. Clones that generated eGFP activity upon LPS or cytokine stimulation and low background in unstimulated cultures were used in these studies. GFP expression in these cells was determined using flow cytometry (BD Biosciences FACSCalibur Cytometer) and also by fluorescent microscopy (Zeiss Axiophot Fluorescence Microscopy System). In all flow cytometry experiments, 10,000 cells were examined for eGFP expression following stimulation with IL-1β (1ng/ml), TNFα (5ng/ml), E.coli LPS (10μg/ml), and parathyroid hormone (PTH; 10nM). In some cases, cells with constitutively active expressing upstream activators of the p38 MAPK, MKK-3b and -6b (obtained from Dr. Matsumo, Japan (37)) were used in transient transfection experiments as described above. Expression of eGFP in these cells was measured by flow cytometry as previously described.
RESULTS
SB203580 dose dependently inhibits IL-1β-induced IL-6 secretion in MC3T3-E1 cells
To determine the relative role of p38 MAPK on IL-6 production in MC3T3-E1 osteoblastic cells, quantitative ELISA specific for mouse IL-6 was utilized. As shown in Figure 1, IL-1β-induced IL-6 secretion was dose dependently inhibited by SB203580. In all experiments, SB203580 was added to culture media ca. 30 minutes prior to stimulation with IL-1β. In other preliminary experiments SB203580 was added at the same time as IL-1β resulting in diminished IL-6 inhibition (data not shown). The experimentally determined IC50 for SB203580 inhibition of IL-1β-induced IL-6 secretion was approximately ~2.5μM. In subsequent studies we used SB203580 at a final concentration of 2μM to evaluate p38 effects on IL-6 mRNA expression. These data are consistent with previous findings where IL-6 and cyclooxygenase mRNAs were inhibited by SB203580 at concentrations of ~1μM, which selectively inhibit p38 MAP kinase (39, 46).
Figure 1. IL-6 secretion is dose dependently inhibited by SB203580 in MC3T3-E1 cells.
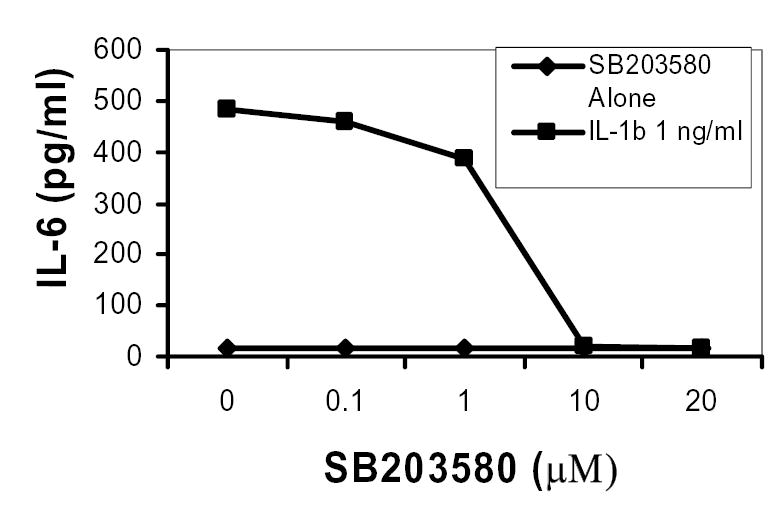
MC3T3-E1 cells were treated with SB203580 at indicated concentrations for 30 minutes and then stimulated with IL-1β for 24 hours. Cell cultured supernatants were harvested and analyzed for IL-6 by ELISA. Data are presented as means of duplicate experiments measured in triplicate.
p38 is required for IL-1β-induced IL-6 mRNA Induction
IL-6 mRNA steady state gene expression was determined by real time RT-PCR. Using this technique, we have shown that p38 is necessary for IL-1β-induced IL-6 mRNA synthesis. When MC3T3-E1 cells were pretreated with SB203580 (2μM) and then stimulated with IL-1β (0.5ng/ml) we observed an 8-fold reduction in IL-1β-induced IL-6 mRNA levels compared to IL-1β treated cells only (see Figure 3A). Minimal effects were seen with SB203580-only treated cells. Less inhibition was observed when cells were treated with SB203580 and stimulated with IL-1β at the same time (data not shown).
Figure 3A. p38 MAP kinase is required for IL-1β-induced IL-6 gene expression.
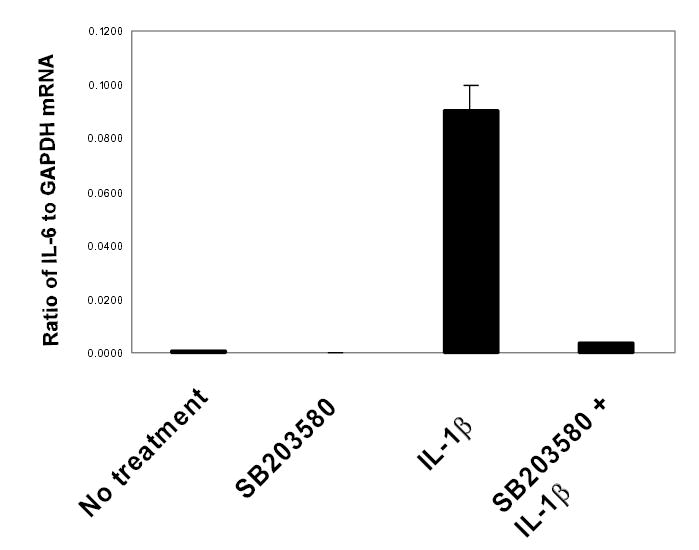
MC3T3-E1 cells were treated with SB203580 (2μM), then stimulated with IL-1βfor 18 hours. Total RNA was isolated and real time RT-PCR performed for IL-6 and GAPDH using SybrGreen dye (ABI) on an iCycler PCR machine (BioRad). Threshold cycle values were used to quantitate normalized expression through QGene Software and expressed as ratio of target gene to reference gene (IL-6: GAPDH). Mean normalized expression ratios from 4 indepednent experiments are presented.
De novo protein synthesis is necessary for IL-1β-induced IL-6 mRNA expression
To explore further the mechanism of p38 MAPK in IL-1β-induced IL-6 expression, we performed experiments in the presence of the protein synthesis inhibitor cycloheximide (CHX) using SB203580 to inhibit IL-1β-induced IL-6 mRNA synthesis. As shown in Figure 3B, CHX blocked the ability of SB203580 to inhibit IL-1β-induced IL-6 mRNA production. In addition, CHX was able to dramatically affect IL-6 mRNA levels in the presence or absence of IL-1β. We have observed a similar effect in IL-6 mRNA expression in primary rat calvarial-derived osteoblasts (25). This effect of CHX has been attributed to ‘superinduction’ phenomenon observed with a variety of cytokine genes, including IL-6, where treatment of cells with CHX causes the ribosomes to ‘freeze’ on the mRNA, potentially shielding it from degradation by cytoplasmic RNases (43).
Figure 3B. De novo protein synthesis is required for p38 MAPK-mediated IL-6 mRNA synthesis in MC3T3-E1 Cells.
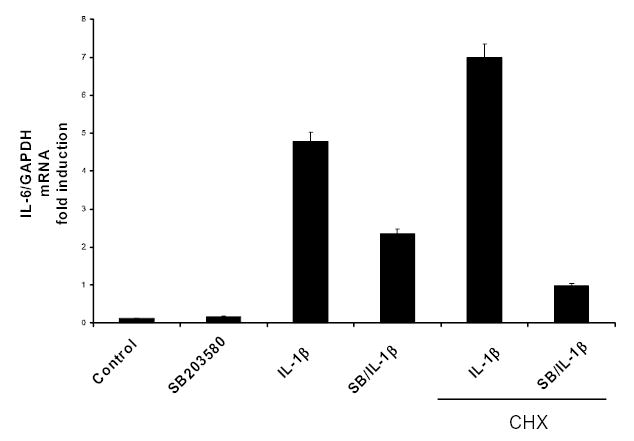
MC3T3-E1 cells with treated with IL-1 (1.0ng/ml) and SB203580 (2μM) and in the presence or absence of cycloheximide (CHX; 2μg/ml). Semi-quantitative RT-PCR was performed to evaluate IL-6 mRNA expression. See text for details on CHX effects. Means from two indepednent experiments are presented.
SB203580 has minimal effect of proximal IL-6 promoter activity
To determine if p38 MAPK induces IL-6 promoter activity after IL-1β stimulation, transient transfection with mouse IL-6 promoter constructs in MC3T3-E1 cells were performed. Following transient transfection the −225 or with the −138 bp IL-6 promoter constructs driving the reporter L-luciferase (Luc) gene, cells were incubated in the SB203580 (5μM) in the presence or absence of IL-1β for 24 hours and then assayed for Luc activity. Figure 4 shows that cells transfected with either the −225 or the −138 constructs expressed relatively low constitutive Luc activity. Both IL-6 promoter constructs were stimulated in the presence of IL-1β (1ng/ml) by ca. 6-fold or <2-fold, for the −225 and −138 constructs, respectively (n=2). However, with the proximal constructs used in these studies, we observed only slight to modest repression of IL-6 promoter activity in the presence of SB203580 (see Figure 4).
Figure 4. p38 MAP kinase inhibition has minimal effect on proximal IL-6 promoter function.
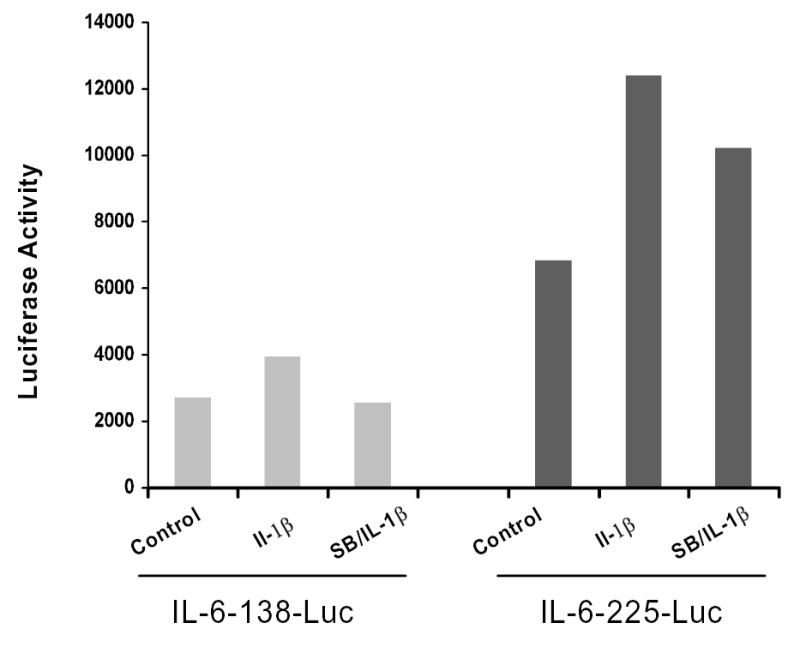
MC3T3-E1 cells were transiently transfected with L-luciferase reporter constructs containing -225 and -138 bps of the IL-6 proximal promoter using Lipofectamide Plus (Invitrogen). Six hrs following transfection, cells were treated with SB203580 (5μM; Calbiochem) and stimulated with IL-1β (1ng/ml) for 24 hours. Cell extracts were normalized by cotransfection with an R-luciferase vector. Luminesence was measured using a Berthold Orion luminometer using the Dual luminescent kit (Promega) following manufacturers’ instructions. Representative data is presented. These experiments were performed twice independently with similar results.
p38 is needed to mediate IL-1β-induced IL-6 mRNA stability
To determine if p38 MAPK was involved in IL-1β induced IL-6 mRNA stability, we used actinomycin D, which blocks further transcription and allows the rates of mRNA decay to be determined. MC3T3-E1 cells were pretreated with SB203580 (2μM) and then stimulated with IL-1β (1.0 ng/ml). During the last period of incubation, actinomycin D (2μg/ml) was added for 0.5 and 1 hr. IL-6 mRNA levels were then analyzed by RT-PCR. The results indicate that IL-1β increases the half-life of IL-6 (Figure 5), with calculated half-lives of >90 minutes, consistent with previous reports where IL-1β prolonged the half-life of IL-6 mRNA (25, 42). However, in the presence of SB20380, the half-life of IL-6 was decreased to 24 minutes following normalization to GAPDH, which has a relatively long half-life (~8 hours) (11).
Figure 5. p38 MAP kinase mediates IL-1β-induced IL-6 mRNA stability.
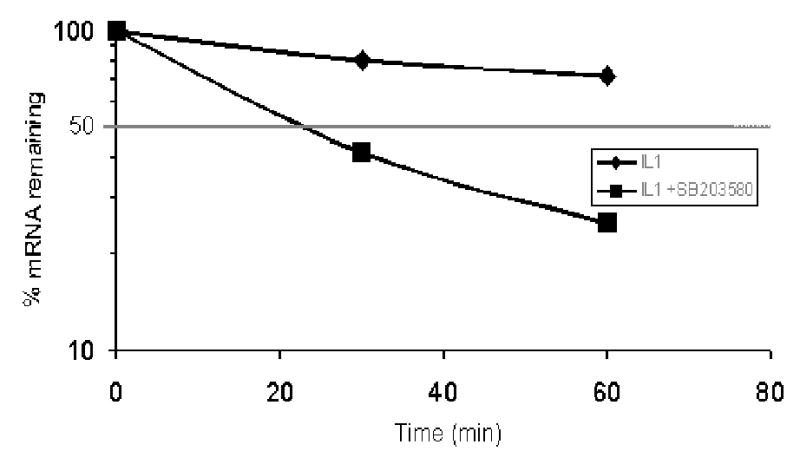
MC3T3-E1 cells were treated as in Figure 4, and then actinomycin D (2μg/ml) was added for 30 and 60 minutes. Total RNA was harvested and analyzed by real time RT-PCR as above. Results are expressed as percentage of IL-6 mRNA remaining after normalized to GAPDH. IL-6 mRNA t½ was calculated to be >120 minutes with IL-1β treatment and reduced to ~24min with SB203580 (2μM). Means from two indepedent experiments are presnented.
IL-1β increases GFP-IL-6 3′UTR reporter activity
To understand the molecular nature of p38 signaling intermediates and how this pathway regulates IL-6 mRNA stability, it was important to establish reporter systems in MC3T3-E1 cells which could be used as readouts to determine cis-acting elements and signaling intermediates of the MAPK pathway which could regulate particular cis elements of the IL-6 gene. From the literature to date and our own preliminary data, the 3′UTR of the IL-6 gene is an obvious starting point for these studies (41, 60). Thus, in order to monitor p38-sensitive turnover regulation in cells, we have generated reporter constructs in which the CMV promoter is driving the expression of a reporter gene containing the coding sequence for the green fluorescent protein (GFP) fused with the 3′UTR of IL-6 (Figure 6A). In brief, the 411bp Mlu1-NotI fragment was cloned into the peGFP vector which had the SV40 polyadenylation sequence removed by Mlu1-NotI. Using this eGFP-3′UTR IL-6 expression vector, we stably transfected MC3T3-E1 osteoblasts and selected clones which were neomycin resistant. Several cell lines were screened for increased GFP expression following IL-1β stimulation. One of these clones, referred to as MC6, was used in subsequent experiments to determine if p38 MAPK affects the IL-6 3′UTR in osteoblasts. Following treatment of the MC6 cell line with IL-1β, we observed a marked increase in GFP expression by flow cytometry (Figure 6B). Other agents, TNFα, E.coli LPS, but not PTH, also enhanced GFP-IL-6 3′UTR reporter expression. These preliminary experiments provide evidence for p38 regulation of GFP expression in MC3T3-E1 cells under control of defined IL-6 3′UTR cis elements. Importantly, we demonstrated that IL-6 and eGFP mRNA expression were regulated in MC6 reporter cells as IL-6 was regulated in the parent cell line MC3T3-E1 cells (see Figures 6C and 6D). In both cases, SB203580 at either 2μM or 5 μM was able to inhibit IL-1β induced IL-6 or eGFP-IL-6 3′UTR expression.
Figure 6A. Diagram of the IL-6 gene structure and GFP-IL-6 3′UTR Reporter Construction.
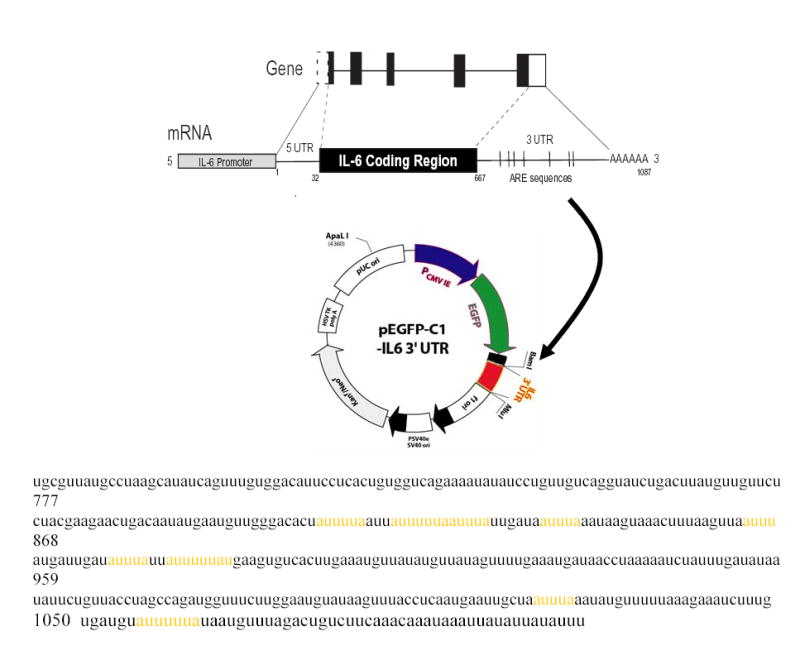
Relevant cis elements (5′UTR, coding region and 3′UTR) discussed in proposal are indicated. As part of the preliminary data presented in this section, the murine IL-6 3′UTR (~390 bps) has been cloned into pEGFP-C1 vector where the SV40 poly adenylation site has been removed permitting expression of the EGFP gene under control of the CMV promoter. Stable clones of expressing this construct have been obtained in MC3T3-E1, NIH 3T3, and MC3T3-E1cells immortalized with SV40 large T antigen (designated MCT).
Figure 6B. Various cytokines increase GFP expression of CMV-eGFP-IL-6 3′UTR construct in MC3T3-E1 cells.
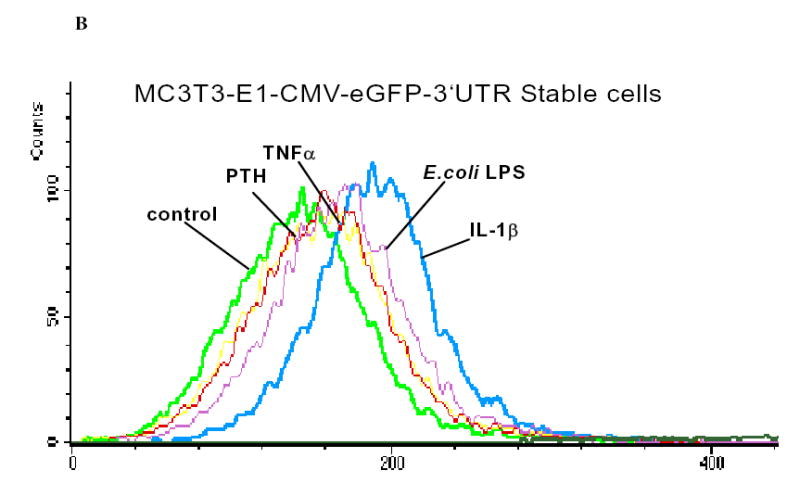
MC3T3-E1CMV-eGFP-IL-63′UTR cells were treated with DMSO (control), PTH (10 nM), TNF-α (5ng/ml), E.Coli LPS (10μg/ml), or IL-1β (1 ng/ml) for 48 hours. Cells were harvested and visualized by flow cytometry on a FACScan. Histogram plots showing the entire GFP-expressing population are overlapped to indicate an increase in the GFP expressing cells when cells are stimulated with IL-1β or LPS. Lesser changes were observed with PTH or TNFα in this experiment. 10,000 cells were gated in this experiment for each treatment group.
Figure 6C. p38 MAP kinase is required for IL-1β-induced IL-6 gene expression in MC6 cells.
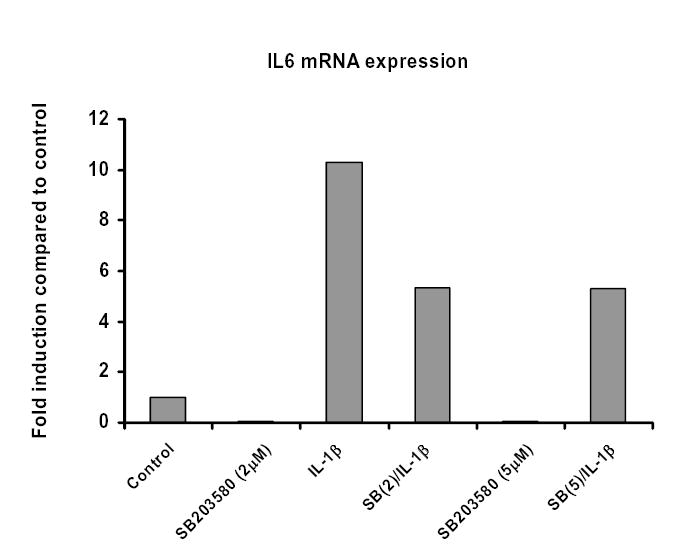
MC6 cells were treated with SB203580 (2 or 5μM), then stimulated with IL-1β (1ng/ml) for 18 hours. Total RNA was isolated and real time RT-PCR performed for IL-6 and GAPDH using SybrGreen dye (ABI) on an iCycler PCR machine (BioRad). Threshold cycle values were used to quantitate normalized expression through QGene Software and expressed as ratio of target gene to reference gene (IL-6: GAPDH) compared to control untreated cells.
Figure 6d. p38 MAP kinase is required for IL-1β-induced eGFP gene expression in MC6 cells.
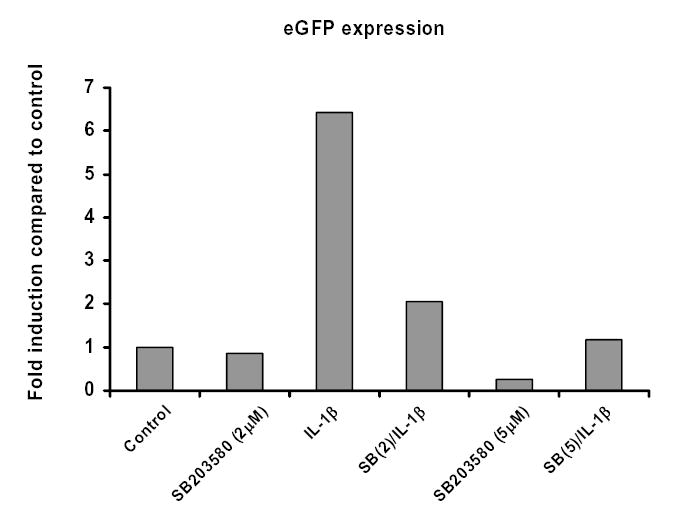
MC6 cells were treated with SB203580 (2 or 5μM), then stimulated with IL-1β (1ng/ml) for 18 hours. Total RNA was isolated and real time RT-PCR performed for eGFP and GAPDH using SybrGreen dye (ABI) on an iCycler PCR machine (BioRad). Threshold cycle values were used to quantitate normalized expression through Q-Gene Software and expressed as ratio of target gene to reference gene (eGFP: GAPDH) compared to control untreated cells.
To confirm that the p38 MAPK pathway mediates enhanced GFP-IL-6 3′ UTR activity, we transiently transfected MC6 reporter cells with upstream p38 pathway intermediates (Figure 6E). Both constitutively active MKK-3 and-6 constructs were able to enhance GFP compared to empty vector (mock transfected) controls. These results indicate that upstream kinases of p38 could specifically enhance GFP-IL-6 3′ UTR reporter expression.
Figure 6E. Upstream signaling intermediates of p38 MAPK increase GFP expression in MCT-GFP-IL-6 3′UTR cells.
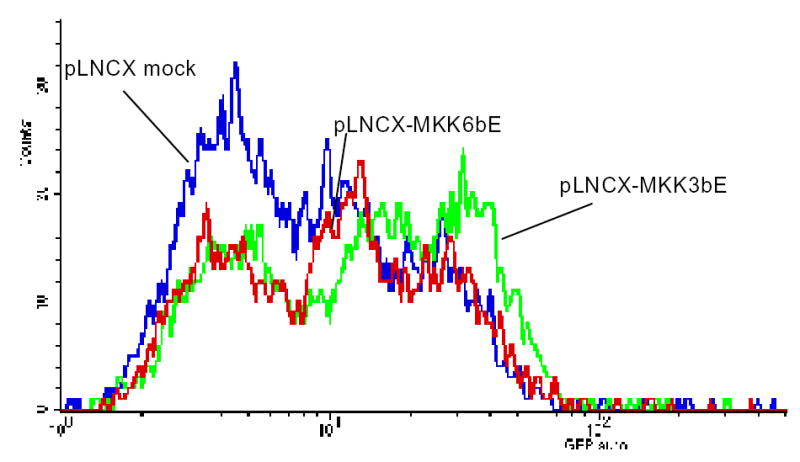
Cells were transduced with viral particles containing constitutively active MKK-6, MKK-3, or empty vector viral particles in the presence of polybream for 48 hours. GFP expression was measured by FACscan (10,000 cells were sorted in each group). Data indicates that both MKK-6 and MKK-3 can increase GFP expression in MCT-GFP-IL-6 3′UTR cells. These data suggest that both immediate upstream signaling intermediates of p38 MAPK are able to increase IL-6 mRNA reporter activity.
DISCUSSION
IL-6 regulation in bone is extremely important for tissue homeostasis. Inflammatory mediators, including IL-1β, TNFα, and gram negative LPS promote shifts in this balance towards bone resorption. One of the pathways for this is represented by increased expression of IL-6 in osteoblasts. Although many studies have interpreted this as a function of enhanced transcriptional initiation and promoter function of the IL-6 gene, relatively few studies have addressed post-transcriptional regulation of IL-6 mRNA. Rapid turnover of short-lived mRNA such as those coding cytokines is mediated by cis-acting elements that are distributed throughout the mRNA molecule (49). Stabilization of short-lived ARE-containing mRNAs, such as IL-6, is part of a pathological mechanism to amplify gene expression in chronic inflammatory conditions. Thus, the understanding of the pathogenesis of such conditions may be greatly improved by the study of the mechanisms involved in signaling pathways utilized to prolong mRNA stability. By using pharmacological inhibitors of p38 MAPK, we have shown that the IL-6 mRNA half-life is prolonged through a p38-dependent mechanism. Through reporter gene assays we showed that the 3′UTR region of IL-6 gene is, at least, one of the major targets of the p38 pathway to increase mRNA stabilization.
Herein, we provide evidence that a specific p38 MAPK inhibitor can dose dependently inhibit IL-1β-induced IL-6 secretion in osteoblastic cells. Similar findings have been observed with other agonists of IL-6 in osteoblasts and other cell types (4, 17, 26). The calculated IC50 is ~2.5μM of SB203580. This concentration of SB203580 has been shown to be specific for p38 and not inhibit other MAP kinases such as JNK or ERK (8). SB203580 binds with high affinity to p38 near the ATP binding site, thus rendering p38 inactive (10). In addition, additional studies showed that inhibition of p38 by the pharmacological inhibitor SB203580 or by a dominant-negative mutant of MAP kinase kinase-6 (MKK6), an upstream activator of the p38 pathway, interfered with NF-κB-dependent gene expression but not with its DNA-binding activity (59).
In Figure 2, we show that SB203580 can inhibit the ability of IL-1β to induce p38 phosphorylation in our model system by western blot analysis. Note that these changes in phosphorylated p38 were specific for phosphorylated p38 and not non-phosphorylated levels of p38. These effects are consistent with numerous other studies. Although we do not observe a significant decrease in phosphorylated p38 levels at 2.5μM, we do see significant amount of IL-6 secretion suppression at that concentration. This disparity reflects to time course difference in experiments. The IL-6 secretion inhibition occurs over 18–24 hour time frame versus the < 1 hour for the western blot data.
Figure 2. p38 MAPK phosphorylation is dose-dependently inhibited by SB203580.
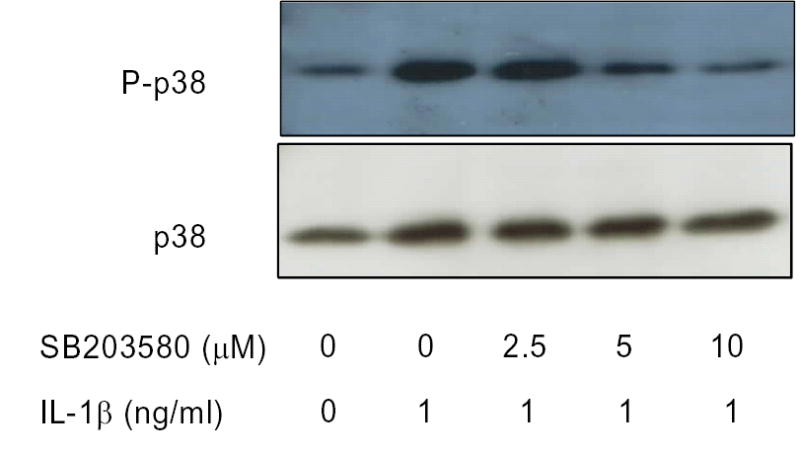
MC3T3-E1 cells were serum starved, then treated with SB203580 at indicated concentrations for 30 minutes, and then stimulated for 20 minutes with IL-1β (1ng/ml). Whole cell extracts were analyzed by Western blot with antibodies against phosphorylated and non-phosphorylated p38 MAPK. Data is representative experiment preformed independently three times with similar results.
We have additionally shown that inhibition of the p38 pathway has a significant effect on steady state IL-6 mRNA levels in MC3T3-E1 cells. In Figure 3 SB203580 significantly inhibited IL-1β-induced IL-6 mRNA synthesis by nearly 8-fold. These results were observed rather consistently in MC3T3-E1 cells. However, we have not been able to observe the same results in all cell types utilized in our laboratory (unpublished data). In addition, we have observed SB203580 inhibition of IL-1β-induced IL-6 mRNA expression when both SB203580 and IL-1β are added at the same time, but the effects of the inhibitor are greatly diminished as opposed to what is observed by pretreatment with SB203580 (data not shown). Moreover, we show that de novo protein synthesis is required for p38-induced IL-6 gene expression (Figure 3B) consistent with results obtained in human synoviocytes (39). SB203580 (2 μm) inhibits approximately 50% of IL-1β-induced IL-6 expression in MC3T3-E1 osteoblastic cells, and that this effect depends on de novo protein synthesis as these effects were inhibited with cycloheximide (CHX). Increased IL-6 mRNA levels of IL-6 in the presence of CHX have been attributed to CHX causing the ribosomes to shield mRNA, protecting mRNA from degradation by cytoplasmic RNases (43).
Importantly, p38 MAP kinase appears to mediate its effects on enhancing IL-6 gene expression mainly through the stabilization IL-6 mRNA. As shown in Figure 4, SB203580 reduces the half-life of the IL-6 message from >120 minutes to less that 25 minutes in IL-1-β-stimulated cells in the presence of actinomycin D, an inhibitor of DNA-primed RNA polymerase, to arrest transcription. Thus, these data strongly implicate the p38 MAP kinase pathway as a central signaling pathway which controls IL-6 mRNA stability. Other investigators have also shown that p38 or the immediate downstream kinase MAPKAPK2 (MK2) plays a role in IL-6 mRNA stability (39, 56, 60). In addition, these data are consistent with other findings where defects of IL-6 mRNA stability were observed in animals lacking the immediate downstream substrate of MK2 (41). In these MK2−/− animals, absence of MK2 leads to a 10-fold increased degradation rate of IL-6 mRNA in LPS-stimulated macrophages. Furthermore, MK2 can stabilize an mRNA reporter construct carrying the IL-6 3′-UTR, which contains ARE-like motifs (41). However, it is also possible that at least part of the increase in IL-6 mRNA steady state expression results from enhanced IL-6 promoter activity and concomitant transcription initiation.
To help determine the relative contribution of p38 on IL-6 gene expression, we performed IL-6 gene promoter assays with the proximal IL-6 promoter region. We investigated the level at which p38 MAPK regulates expression of IL-6 in osteoblastic cells by transiently transfecting MC3T3-E1 cells with two different (−225/+11 and a −138/+11) IL-6 promoter constructs coupled to a Luciferase reporter vector. The -225 construct contains the NFκB site and a pivotal C/EBP site necessary for robust IL-6 transcription (38). Using these constructs, we have found that p38 had relatively minimal effects on IL-6 proximal promoter activity. These results contrast with those reported with TNF-induced human IL-6 promoter function in a human osteoblastic cell model (58). These studies showed strong inhibition of TNF-induced IL-6 secretion in the presence of SB203580, but only ca. 50% inhibition of IL-6 promoter activity. Moreover, the authors did not address IL-6 mRNA levels or mRNA stability in these studies. In our studies, we have observed strong inhibition of IL-1β-induced IL-6 mRNA levels and marked decrease in IL-6 mRNA stability in the presence of p38 inhibitors without significant inhibition of IL-6 promoter activity. These data argue that p38 regulation of IL-1β-induced IL-6 occurs more at the post-transcriptional rather than transcriptional level.
We have started to extend our studies on p38 regulation of IL-6 mRNA stability through the use of reporter gene systems. One of the reporter cell lines selected, MC6, which we report here has utilized the GFP reporter gene fused to the 3′UTR of IL-6 shown schematically in Figure 6A. The 3′UTR of IL-6 has several AU-rich elements in the 3′UTR common to many inflammatory genes and early immediate genes, which are critical for post-transcriptional regulation (16). These sequences are formed by the presence of one or more copies of the pentamer AUUUA and bind to proteins involved in stabilizing or destabilizing these mRNAs. In the case of IL-6, these elements are necessary for mRNA stability (47, 60), but the 3′UTR for TNFα mediates translational efficiency (27). In the present study, GFP-IL-6 3′UTR stable MC3T3-E1 osteoblastic cells (MC6) were generated to gain experimental insight of p38 effects on the 3′UTR of IL-6. Using these reporter cells, we have shown that IL-1β and to a lesser extent LPS and TNFα can increase GFP reporter expression through flow cytometry analysis (see Figure 6B). Interestingly, PTH did not increase GFP-IL-6 3′UTR reporter expression. Previous studies have shown that PTH stimulates IL-6 production in osteoblasts through signaling intermediates including protein kinase (PK) A and PKC (24, 50), but not p38 MAPK. IL-1β, TNFα, and LPS all have been shown to increase p38 signaling in response to stimulation in osteoblasts (4, 25, 26, 28). Addition data shows that IL-6 and eGFP mRNA expression are regulated in MC6 cells through p38 (Figure 6C and 6D). We believe that eGFP regulation through p38 pathway in MC6 cells reflects regulation of the IL-6 3′UTR in mRNA stability which will increase translational efficiency. However, we cannot rule out p38 effects on the AP-1 sites within the CMV promoter will drives eGFP expression in this construct. These data provide supportive data for the IL-6 3′UTR as being one of the main targets of the p38 signaling pathway
We have further expanded on the relevant role of the p38 pathway in IL-6 mRNA stability by transfecting upstream signaling intermediates of p38 in MC6 cells (Figure 6C). The activities of p38 kinases are controlled by upstream kinases, namely MKK-3, -4, and -6. Among these, MKK-3 and -6 are specific for p38 whereas MKK-4 also activates cJun-N-terminal kinases (JNKs) (54). As shown here both constitutively active forms of MKK-3 and MKK-6 can increase GFP expression in MC6 cells whereas the empty vector mock control had no effect on GFP expression. These data provide additional evidence that the p38 MAPK pathway impacts on IL-6 regulation, at least in part, through the 3′UTR of IL-6. However, the exact mechanism of this regulation remains to be elucidated.
It is assumed that regulation of mRNA stability is mediated through trans-acting RNA-binding factors, which interact with cis-elements, including AREs. A number of proteins that selectively bind AREs have been identified and characterized. ARE-binding regulators of mRNA stability include: AUF1 (heterogeneous nuclear ribonucleoprotein (hnRNP-D), four members of the ELAV family [HuR (human)/HuA (murine), HuB/Hel-N1, HuC, HUD], glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), hnRNPA1, hnRNPC, and tristeraprolin (TTP) (15, 16). We have shown that of those RNA binding proteins, AUF1, HuR (HuA-like), and TTP are expressed in MC3T3-E1 cells (unpublished data). At present, we do not know the target of p38 regulation of IL-6 mRNA stability in osteoblasts. Recently, affinity ‘pull-down’ experiments using the TNFα ARE has identified ca. twenty different proteins which selectively bind to TNFα ARE mRNA in LPS-stimulated macrophages (48). Many of the proteins identified contain RNA recognition motifs (RRMs) and some of these were from the hnRNP A/B family which possess arginine/glycine rich domains known to be involved in protein-protein interactions as well as nuclear-cytoplasmic shuttling and RNA processing (2). One of the identified proteins, hnRNP A0 was identified as a downstream substrate of MAPKAPK2. In vitro kinase assays indicated that hnRNP A0 was phosphorylated by MAPKAPK2 and sensitive to SB203580. Most notably, other mRNAs, including COX-2 and macrophage inflammatory protein (MIP)-2, but not IL-6 were bound to immunoprecipitated hnRNP A0. Another recent paper has shown that poly(A)-binding protein (PABP)-1 was the downstream substrate of MAPKAPK2 which could bind to the GM-CSF ARE. The IL-6 ARE has been shown to bind to HuR in glioma tissues (40), however, HuR is does not appear to be target of p38 signaling (57).
In summary, we have demonstrated that p38 MAPK is a critical signaling intermediate in IL-6 mRNA synthesis in osteoblasts. A major point of p38-dependent IL-6 regulation occurs at the level of mRNA stability. One of the main targets of IL-1β-induced IL-6 mRNA stability regulation is the IL-6 ARE. The immediate downstream substrate in the p38MAPK/ARE axis of regulation of IL-6 mRNA stability is MAPKAPK2 (41). However, the downstream substrates including RNA binding proteins which regulate IL-6 mRNA stability in a p38-dependent manner remain to be elucidated. Ongoing studies are currently addressing these vital issues of IL-6 post-transcriptional gene regulation. Once identified, these trans-acting factors of IL-6 mRNA stability regulation may provide novel therapeutic targets in the management of inflammatory bone diseases such as rheumatoid arthritis and periodontitis which are mediated, in part, through sustained IL-6 production.
Acknowledgments
Support for this research was provided by the grant #1-R03-DE-1460-1A1 from the National Institutes of Dental and Craniofacial Research, P.I. Dr. Keith Kirkwood, State University of New York at Buffalo (presently University of Michigan).
References
- Bellido T, Jilka RL, Boyce BF, Girasole G, Broxmeyer H, Dalrymple SA, Murray R, Manolagas SC. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. The role of the androgen receptor. J Clin Invest. 1995;95:2886–2895. doi: 10.1172/JCI117995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J Cell Physiol. 2003;195:356–372. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci U S A. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae HJ, Chae SW, Chin HY, Bang BG, Cho SB, Han KS, Kim SC, Tae KC, Lee KH, Kim DE, Im MK, Lee SJ, Chang JY, Lee YM, Kim HM, Kim HH, Lee ZH, Kim HR. The p38 mitogen-activated protein kinase pathway regulates interleukin-6 synthesis in response to tumor necrosis factor in osteoblasts. Bone. 2001a;28:45–53. doi: 10.1016/s8756-3282(00)00413-0. [DOI] [PubMed] [Google Scholar]
- Chae HJ, Kim SC, Chae SW, An NH, Kim HH, Lee ZH, Kim HR. Blockade of the p38 mitogen-activated protein kinase pathway inhibits inducible nitric oxide synthase and interleukin-6 expression in MC3T3E-1 osteoblasts. Pharmacol Res. 2001b;43:275–283. doi: 10.1006/phrs.2000.0778. [DOI] [PubMed] [Google Scholar]
- Chaudhary LR, Spelsberg TC, Riggs BL. Production of various cytokines by normal human osteoblast-like cells in response to interleukin-1 beta and tumor necrosis factor-alpha: lack of regulation by 17 beta-estradiol. Endocrinology. 1992;130:2528–2534. doi: 10.1210/endo.130.5.1572280. [DOI] [PubMed] [Google Scholar]
- Chen CY, Xu N, Shyu AB. mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Mol Cell Biol. 1995;15:5777–5788. doi: 10.1128/mcb.15.10.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerk A, Harrison JG, Long CS, Sugden PH. Pro-inflammatory cytokines stimulate mitogen-activated protein kinase subfamilies, increase phosphorylation of c-Jun and ATF2 and upregulate c-Jun protein in neonatal rat ventricular myocytes. J Mol Cell Cardiol. 1999;31:2087–2099. doi: 10.1006/jmcc.1999.1040. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Dani C, Piechaczyk M, Audigier Y, El Sabouty S, Cathala G, Marty L, Fort P, Blanchard JM, Jeanteur P. Characterization of the transcription products of glyceraldehyde 3-phosphate-dehydrogenase gene in HeLa cells. Eur J Biochem. 1984;145:299–304. doi: 10.1111/j.1432-1033.1984.tb08552.x. [DOI] [PubMed] [Google Scholar]
- Feyen JH, Elford P, Di Padova FE, Trechsel U. Interleukin-6 is produced by bone and modulated by parathyroid hormone. J Bone Miner Res. 1989;4:633–638. doi: 10.1002/jbmr.5650040422. [DOI] [PubMed] [Google Scholar]
- Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S, Kawai S, Faucheu C, Huet L, Baron R, Roman-Roman S. Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone. 2001;28:491–498. doi: 10.1016/s8756-3282(01)00415-x. [DOI] [PubMed] [Google Scholar]
- Girasole G, Jilka RL, Passeri G, Boswell S, Boder G, Williams DC, Manolagas SC. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest. 1992;89:883–891. doi: 10.1172/JCI115668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouble A, Morello D. Synchronous and regulated expression of two AU-binding proteins, AUF1 and HuR, throughout murine development. Oncogene. 2000;19:5377–5384. doi: 10.1038/sj.onc.1203910. [DOI] [PubMed] [Google Scholar]
- Grzybowska EA, Wilczynska A, Siedlecki JA. Regulatory functions of 3′UTRs. Biochem Biophys Res Commun. 2001;288:291–295. doi: 10.1006/bbrc.2001.5738. [DOI] [PubMed] [Google Scholar]
- Hedges JC, Singer CA, Gerthoffer WT. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am J Respir Cell Mol Biol. 2000;23:86–94. doi: 10.1165/ajrcmb.23.1.4014. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000;15:2–12. doi: 10.1359/jbmr.2000.15.1.2. [DOI] [PubMed] [Google Scholar]
- Ishimi Y, Miyaura C, Jin CH, Akatsu T, Abe E, Nakamura Y, Yamaguchi A, Yoshiki S, Matsuda T, Hirano T, et al. IL-6 is produced by osteoblasts and induces bone resorption. J Immunol. 1990;145:3297–3303. [PubMed] [Google Scholar]
- Iwai Y, Bickel M, Pluznik DH, Cohen RB. Identification of sequences within the murine granulocyte-macrophage colony-stimulating factor mRNA 3′-untranslated region that mediate mRNA stabilization induced by mitogen treatment of EL-4 thymoma cells. J Biol Chem. 1991;266:17959–17965. [PubMed] [Google Scholar]
- Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boyce B, Broxmeyer H, Manolagas SC. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC. Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- Kannabiran C, Zeng X, Vales LD. The mammalian transcriptional repressor RBP (CBF1) regulates interleukin-6 gene expression. Mol Cell Biol. 1997;17:1–9. doi: 10.1128/mcb.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GS, Kim CH, Choi CS, Park JY, Lee KU. Involvement of different second messengers in parathyroid hormone- and interleukin-1-induced interleukin-6 and interleukin-11 production in human bone marrow stromal cells. J Bone Miner Res. 1997;12:896–902. doi: 10.1359/jbmr.1997.12.6.896. [DOI] [PubMed] [Google Scholar]
- Kirkwood K, Martin T, Andreadis ST, Kim YJ. Chemically modified tetracyclines selectively inhibit IL-6 expression in osteoblasts by decreasing mRNA stability. Biochem Pharmacol. 2003;66:1809–1819. doi: 10.1016/s0006-2952(03)00450-7. [DOI] [PubMed] [Google Scholar]
- Kondo A, Koshihara Y, Togari A. Signal transduction system for interleukin-6 synthesis stimulated by lipopolysaccharide in human osteoblasts. J Interferon Cytokine Res. 2001;21:943–950. doi: 10.1089/107999001753289550. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Kumar S, Votta BJ, Rieman DJ, Badger AM, Gowen M, Lee JC. IL-1- and TNF-induced bone resorption is mediated by p38 mitogen activated protein kinase. J Cell Physiol. 2001;187:294–303. doi: 10.1002/jcp.1082. [DOI] [PubMed] [Google Scholar]
- Lee JC, Badger AM, Griswold DE, Dunnington D, Truneh A, Votta B, White JR, Young PR, Bender PE. Bicyclic imidazoles as a novel class of cytokine biosynthesis inhibitors. Ann N Y Acad Sci. 1993;696:149–170. doi: 10.1111/j.1749-6632.1993.tb17149.x. [DOI] [PubMed] [Google Scholar]
- Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors--mechanisms and therapeutic potentials. Pharmacol Ther. 1999;82:389–397. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]
- Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47:185–201. doi: 10.1016/s0162-3109(00)00206-x. [DOI] [PubMed] [Google Scholar]
- Li X, Udagawa N, Itoh K, Suda K, Murase Y, Nishihara T, Suda T, Takahashi N. p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology. 2002;143:3105–3113. doi: 10.1210/endo.143.8.8954. [DOI] [PubMed] [Google Scholar]
- Manolagas SC. The role of IL-6 type cytokines and their receptors in bone. Ann N Y Acad Sci. 1998;840:194–204. doi: 10.1111/j.1749-6632.1998.tb09563.x. [DOI] [PubMed] [Google Scholar]
- Manolagas SC, Jilka RL, Girasole G, Passeri G, Bellido T. Estrogen, cytokines, and the control of osteoclast formation and bone resorption in vitro and in vivo. Osteoporos Int. 1993;3(Suppl 1):114–116. doi: 10.1007/BF01621882. [DOI] [PubMed] [Google Scholar]
- Manolagas SC, Kousteni S, Jilka RL. Sex steroids and bone. Recent Prog Horm Res. 2002;57:385–409. doi: 10.1210/rp.57.1.385. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-kappa B ligand (RANKL) J Biol Chem. 2000;275:31155–31161. doi: 10.1074/jbc.M001229200. [DOI] [PubMed] [Google Scholar]
- Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, Akira S. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci U S A. 1993;90:10193–10197. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin-1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J Biol Chem. 1998;273:24832–24838. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002;277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- Ng SB, Tan YH, Guy GR. Differential induction of the interleukin-6 gene by tumor necrosis factor and interleukin-1. J Biol Chem. 1994;269:19021–19027. [PubMed] [Google Scholar]
- Ragheb JA, Deen M, Schwartz RH. The destabilization of IL-2 mRNA by a premature stop codon and its differential stabilization by trans-acting inhibitors of protein synthesis do not support a role for active translation in mRNA stability. J Immunol. 1999;163:3321–3330. [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Ray A, Sassone-Corsi P, Sehgal PB. A multiple cytokine- and second messenger-responsive element in the enhancer of the human interleukin-6 gene: similarities with c-fos gene regulation. Mol Cell Biol. 1989;9:5537–5547. doi: 10.1128/mcb.9.12.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley SH, Dean JL, Sarsfield SJ, Brook M, Clark AR, Saklatvala J. A p38 MAP kinase inhibitor regulates stability of interleukin-1-induced cyclooxygenase-2 mRNA. FEBS Lett. 1998;439:75–80. doi: 10.1016/s0014-5793(98)01342-8. [DOI] [PubMed] [Google Scholar]
- Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. Embo J. 2002;21:6505–6514. doi: 10.1093/emboj/cdf639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB. Messenger RNA degradation in eukaryotes. Cell. 1993;74:413–421. doi: 10.1016/0092-8674(93)80043-e. [DOI] [PubMed] [Google Scholar]
- Sanders JL, Stern PH. Protein kinase C involvement in interleukin-6 production by parathyroid hormone and tumor necrosis factor-alpha in UMR-106 osteoblastic cells. J Bone Miner Res. 2000;15:885–893. doi: 10.1359/jbmr.2000.15.5.885. [DOI] [PubMed] [Google Scholar]
- Scheidt-Nave C, Bismar H, Leidig-Bruckner G, Woitge H, Seibel MJ, Ziegler R, Pfeilschifter J. Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J Clin Endocrinol Metab. 2001;86:2032–2042. doi: 10.1210/jcem.86.5.7445. [DOI] [PubMed] [Google Scholar]
- Shaw G, Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- Srivastava BI, Srivastava A, Srivastava MD. Phenotype, genotype and cytokine production in acute leukemia involving progenitors of dendritic Langerhans’ cells. Leuk Res. 1994;18:499–511. doi: 10.1016/0145-2126(94)90088-4. [DOI] [PubMed] [Google Scholar]
- Stanton, L.A., Sabari, S., Sampaio, A.V., Underhill, T.M. and Beier, F. (2003) p38 MAP kinase signaling is required for hypertrophic chondrocyte differentiation. Biochem J, Pt [DOI] [PMC free article] [PubMed]
- Stoecklin G, Hahn S, Moroni C. Functional hierarchy of AUUUA motifs in mediating rapid interleukin-3 mRNA decay. J Biol Chem. 1994;269:28591–28597. [PubMed] [Google Scholar]
- Stoecklin G, Stoeckle P, Lu M, Muehlemann O, Moroni C. Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. Rna. 2001;7:1578–1588. [PMC free article] [PubMed] [Google Scholar]
- Sully, G., Dean, J.L., Wait, R., Rawlinson, L., Santalucia, T., Saklatvala, J. and Clark, A.R. (2003) Structural and functional dissection of a conserved destabilizing element of cyclooxygenase 2 mRNA: evidence against involvement of AUF-1, AUF-2, TTP, HuR or FBP1. Biochem J, Pt [DOI] [PMC free article] [PubMed]
- Webb SJ, McPherson JR, Pahan K, Koka S. Regulation of TNF-alpha-induced IL-6 production in MG-63 human osteoblast-like cells. J Dent Res. 2002;81:17–22. doi: 10.1177/002203450208100105. [DOI] [PubMed] [Google Scholar]
- Wesselborg S, Bauer MK, Vogt M, Schmitz ML, Schulze-Osthoff K. Activation of transcription factor NF-kappaB and p38 mitogen-activated protein kinase is mediated by distinct and separate stress effector pathways. J Biol Chem. 1997;272:12422–12429. doi: 10.1074/jbc.272.19.12422. [DOI] [PubMed] [Google Scholar]
- Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. Embo J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]