

Abstract
The fatty acid ω-hydroxylation regiospecificity of CYP4 enzymes may result from presentation of the terminal carbon to the oxidizing species via a narrow channel that restricts access to the other carbon atoms. To test this hypothesis, the oxidation of 12-iodo, 12-bromo-, and 12-chlorododecanoic acids by recombinant CYP4A1 has been examined. Although all three 12-halododecanoic acids bind to CYP4A1 with similar dissociation constants, the 12-chloro and 12-bromo fatty acids are oxidized to 12-hydroxydodecanoic acid and 12-oxododecanoic acid, whereas the 12-iodo analogue is very poorly oxidized. Incubations in H2 18O show that the 12-hydroxydodecanoic acid oxygen derives from water whereas that in the aldehyde derives from O2. The alcohol thus arises from oxidation of the halide to an oxohalonium species that is hydrolyzed by water, whereas the aldehyde arises by a conventional carbon hydroxylation-elimination mechanism. No irreversible inactivation of CYP4A1 is observed during 12-halododecanoic acid oxidation. Control experiments show that CYP2E1, which has an ω−1 regiospecificity, primarily oxidizes 12-halododecanoic acids to the ω-aldehyde rather than alcohol product. Incubation of CYP4A1 with 12, 12 - [2H]2-12-chlorododecanoic acid causes a two to three-fold increase in halogen versus carbon oxidation. The fact that the order of substrate oxidation (Br > Cl ≫ I) approximates the inverse of the intrinsic oxidizability of the halogen atoms is consistent with presentation of the halide terminus via a channel that accommodates the chloride and bromide but not iodide atoms, which implies an effective channel diameter greater than 3.90 Å but smaller than 4.30 Å.
Fatty acids are readily oxidized by cytochrome P450 enzymes in an isoform specific manner to hydroxy fatty acids and, in the case of unsaturated fatty acids, also to epoxides (1–3). The hydroxylation of fatty acids in general contributes to fatty acid homeostasis, but the oxidation of arachidonic acid gives rise to products that have specific functions in signaling pathways. For example, the ω-hydroxylated product, 20-hydroxyeicosatetraenoic acid (20-HETE), is a vasoconstrictor (4, 5) whereas the ω−1 hydroxylation product 19-HETE and some of the arachidonic acid epoxides are vasodilators (2, 5, 6). The regiochemistry of oxidation of fatty acids, particularly arachidonic acid, is therefore of critical physiological importance.
The oxidation of fatty acids at internal sites along the fatty acid hydrocarbon chain is mediated by a variety of cytochrome P450 enzymes. The ω−1 position on the carbon adjacent to the terminal carbon of the hydrocarbon chain is the usual primary default position for the oxidation of fatty acids by P450 enzymes not specifically evolved as fatty acid hydroxylases (7, 8). Hydroxylation of the terminal (ω) carbon does not occur, or is a very minor product, with such P450 enzymes. This specificity reflects the fact that the ω−1 position is intrinsically more reactive than the terminal (ω) position because a secondary C–H bond is several Kcal weaker than a primary C–H bond, and thus is more susceptible to oxidation by P450 enzymes (9, 10). Furthermore, in steric terms, the ω−1 carbon is only second to the terminal carbon in steric accessibility. The ω-hydroxylation of fatty acids is therefore a difficult reaction that must be actively promoted by the active site structure.
The CYP4 family of P450 enzymes is unique in that it preferentially catalyzes the ω-hydroxylation of fatty acids. These enzymes therefore reflect the evolution of a strategy that predominantly directs the reaction to the terminal carbon. We have previously postulated that this strategy involves a constricted access channel to the ferryl species that disfavors presentation of any atom other than the terminal methyl for oxidation (7). This hypothesis is supported by the fact that (a) different chain length fatty acids are preferentially oxidized at the ω- terminus, ruling out mechanisms that involve sensing the distance of the ω-carbon from the carboxyl group (11); (b) the oxidation of terminal triple bonds inactivates the CYP4 enzymes via a reaction that involves addition of the ferryl oxygen to the terminal carbon of the triple bond, yielding a ketene, rather than to the internal carbon, which would result in heme alkylation (7, 12); and (c) placement of bulky substituents, such as a phenyl group, at the fatty acid terminus suppresses oxidation of the substrate (13).
In addition to hydrocarbon hydroxylation, P450 enzymes catalyze a diversity of other reactions, including the oxidation of heteroatoms to zwitterionic products: e.g., of trialkyl amines to trialkyl amine oxides and dialkylsulfides to dialkylsulfoxides (14). However, P450 enzymes are powerful oxidative catalysts and evidence exists that they can oxidize not only nitrogen and sulfur functions but also the halogen atom in aryl- and alkyliodides, -bromides, and possibly even chlorides. Conclusive evidence for oxidation of a halide atom is only available for aryliodides. It has been demonstrated that the P450-catalyzed, iodosobenzene-dependent oxidation of labeled iodobenzene results in the formation of labeled iodosobenzene (15). Using a substrate that stabilizes the iodoso structure, Guengerich has furthermore directly demonstrated formation of the iodoso derivative in a normal P450-catalyzed reaction (16). The evidence for bromide and chloride oxidation is less direct. The oxidation of 1,2-dichloroethane and propyl halides by liver microsomes produced a range of products, including compounds whose formation is compatible with (but does not require) a mechanism involving oxidation of the halide atom (17, 18). The formation of a metabolite in the oxidation of CCl4 that reacts as an electrophilic chloride atom has also been postulated from the formation of a p-chlorophenol in the presence of a phenol trap (19).
We report here preparation of the 12-iodo-, 12-bromo-, and 12-chlorododecanoic acids and analysis of their oxidation by recombinant CYP4A1, a rat enzyme that highly favors ω-hydroxylation of dodecanoic (lauric) acid. The hypothesis behind this study is that the halogen atom becomes the “ω” position in these substrates, and therefore its oxidation, if possible, should be enforced by the same active site features that promote ω hydroxylation of dodecanoic acid. The results have provided important insights into the mechanism used by the CYP4 family of enzymes to favor ω- over (ω−1) hydroxylation of fatty acids.
EXPERIMENTAL SECTION
Materials
Ampicillin, δ-aminolevulinic acid, glycerol, lysozyme, DLPC, glutathione, catalase, NADPH, and dodecanoic acid were from Sigma (St. Louis, MO). 11-Dodecenoic acid was from Nu Chek Prep (Elysian, MN), and Tris from Fisher Scientific. [1-14C]-dodecanoic acid (55 mCi/mmol, 18 mM) was from American Radiolabeled Chemicals (St. Louis. MO). CYP4A1 was expressed and purified as previously reported (20). Human CYP2E1 (+ P450 reductase + cytochrome b5 Supersomes™) and control insect cell membranes were purchased from Gentest, BD Sciences. The human NADPH-cytochrome P450 reductase cDNA was provided by Stephen M. Black and the protein was expressed and purified as previously described (20). Purified rat liver cytochrome b5 was a gift from Lester Bornheim (University of California, San Francisco). Human liver cytochrome b5 was purchased from Promega (Fitchburg, WI).
Preparation of Probes
12-Iodododecanoic acid (3) was synthesized from 12-hydroxydodecanoic acid using the procedure of Apparu et al. (21) (trimethylsilylchloride/NaI) to afford 3 as a white solid (mp: 59.0–60.0 °C; Lit. mp: 59.0–61.0 °C (22)). 12-Bromododecanoic acid (2) was synthesized from 12-hydroxydodecanoic acid using the procedure of Jocelyn and Polar (23) (HBr/H2SO4) to afford 2 as a white solid (mp: 52.0–53.0 °C; Lit. mp: 52.0 °C (24); 53.0 °C (25)). 12-Chlorododecanoic acid (1) was synthesized from 2 using the procedure of Peyrat et al. (26) (trimethylsilylchloride/dimethylformamide) to afford 1 as a white solid (mp: 40.0–41.0 °C; Lit. mp: 39.5–40.5°C (27)).
12,12-[2H]2-12-Chlorododecanoic acid (7) was synthesized from 12-hydroxydodecanoic acid in four steps beginning with esterification of the free acid with diazomethane and protection of the alcohol as the t-butyldimethylsilyl ether (90% yield over two steps). Reduction of the ester with lithium aluminum deuteride (89% yield) was followed by chlorination of the alcohol with triphenylphosphine/carbon tetrachloride (79% yield). Acidic cleavage of the silyl ether was followed by oxidation to the carboxylic acid 7 using 8 N Jones’ Reagent. Purification by flash chromatography (silica gel, 10% ethyl acetate in hexanes, 0.5% acetic acid) afforded 7 (60% yield) as a white solid (mp 38.0–39.0 °C). 1H NMR (500 MHz, CDCl3) δ 1.20–1.45 (14H, m), 1.59 (2H, quint, J1 = 7.5 Hz), 1.71 (2H, quint, J1 = 7.5 Hz), 2.31 (2H, t, J1 = 7.5 Hz), 3.50 (0.04H, residual CH2Cl, t, J1 = 7.2 Hz), 9.80 (1H, brs). 13C NMR (125 MHz, CDCl3) δ 24.6, 26.8, 28.8, 29.0, 29.1, 29.30, 29.35, 29.38, 32.4, 34.0, 44.4 (quint, 1JC-D 22.8 Hz), 180.5. GC/MS: 238/236 (M+·, 0.2/0.4), 195/193 (1.3, 4.0), 129 (12.0), 85 (10.5), 73 (67.3), 71 (20.3), 60 (100), 55 (41.9).
12-Oxododecanoic acid (4) was prepared by the procedure of Corey and Suggs (28). A suspension of 12-hydroxydodecanoic acid (1.07 g, 5 mmol) in 12 ml of CH2Cl2 was combined with pyridinium chlorochromate (1.63 g, 7.5 mmol) in 10 ml of CH2Cl2 and the resulting mixture was stirred under nitrogen at ~25 °C for 2 h. Diethyl ether (100 ml) was then added and the black reaction mixture was filtered. The filtered ether fraction was passed through Florisil, the ether was removed under vacuum at a rotary evaporator, and the residue was purified by silica gel flash chromatography. The GC-MS of the trimethylsilyl (TMS) derivative of aldehyde 4 had a retention time of 10.40 min and a molecular ion at m/z 286.
Spectroscopic Methods
Reduced CO-difference spectra and spectroscopic substrate binding studies were performed on a Varian Cary 1E UV/visible dual-beam spectrophotometer. Absolute spectra were recorded on a Hewlett-Packard 8452 diode array spectrophotometer. Both instruments were equipped with temperature control accessories. The cytochrome P450 content was determined by the method of Omura and Sato (29). To obtain substrate binding difference spectra, the probes dissolved in methanol were titrated into the CYP4A (0.5 μM) solution using a 10 μl Hamilton syringe, resulting in a final sample volume change of less than 2% and introduction of less than 2% methanol into the solution. All spectra were obtained at 25 °C. The spectral binding constant Ks was determined from the hyperbolic plot of the respective differences between the absorbance at 417 and 390 nm versus the ligand concentration.
Measurement of CYP4A1 Hydroxylation Activity
CYP4A1 ω- and ω−1 hydroxylation was measured by mixing the stock phosphate buffer solutions of 10 μg of DLPC, 3.12 to 15 pmol of CYP4A1, 31.25 to 250 pmol of cytochrome P450 reductase, 3.13 to 25 pmol of cytochrome b5, and 5 μg of catalase. This mixture was incubated for 10 min at room temperature before 50 mM Tris (pH 7.4) containing 250 mM NaCl and 10% glycerol was added to a final volume of 490 μl. Finally, 5 μl of a 10 mM solution of [1-14C]-dodecanoic acid (final concentration 100 μM) was added. The reaction was started after an additional incubation time of 2 min at 37 °C by addition of 10 μl of 100 mM NADPH. The mixture was incubated at 37 °C. Aliquots (50 μl) were removed at time periods between 0–10 min and were quenched by adding 100 μl of ice-cold 94:6 acetonitrile:acetic acid (v/v). The precipitated proteins were pelleted out by centrifugation in a microcentrifuge for 2 min and the samples were analyzed by HPLC coupled to a flow scintillation analyzer.
HPLC Dodecanoic Acid Hydroxylation Assay
The hydroxydodecanoic acids were separated from dodecanoic acid on a C18 Alltech Econosil column (3.2 × 150 mm) operated isocratically with a 90:10 mixture of acetonitrile (0.1% trifluoroacetic acid): water (0.1% trifluoroacetic acid). The HPLC system was coupled to a Packard Radiomatic Flow-One model A500 radioisotope detector. Under these conditions, the hydroxydodecanoic acids eluted at 3.3 min, whereas dodecanoic acid eluted at 4.6 min.
Assay for Inactivation of CYP4A1
A two-stage procedure was used to evaluate time-dependent inactivation of CYP4A1 in the presence of 12-halododecanoic acids. In the first stage, 10-fold concentrated incubation solutions were incubated at 37 °C with 0–200 μM concentrations of each of the three 12-halododecanoic acids either in the presence or absence of 10 mM NADPH. At specified time intervals, an aliquot (5 μl) was diluted 10-fold by the addition of 50 μl of a second stage assay mixture consisting of the same buffer, 10 mM NADPH, and 100 μM [1-14C]-dodecanoic acid. The diluted mixture was incubated for a further 11 min at 37 °C and was then terminated by the addition of ice-cold 94:6 acetonitrile:acetic acid (v/v). The precipitated proteins were pelleted by centrifugation in a microcentrifuge for 2–5 min, and the samples were stored at −20 °C until analyzed by HPLC.
Measurement of CYP4A Oxidation Activity by GC-MS
CYP4A1 ω-hydroxylation was measured by mixing together 10 μg of DLPC, 12.5 pmol of CYP4A1, 125 pmol of cytochrome P450 reductase, 12.5 pmol of cytochrome b5, and 5 μg of catalase, followed by incubation of the mixture for 10 min at room temperature. The required amount of substrate (dodecanoic acid, 12-chlorododecanoic acid (1), 12-bromododecanoic acid (2), 12-iodododecanoic acid (3) or 12,12-[2H]2-12-chlorododecanoic acid (7): usually 100 μM final concentration) was then added, followed by sufficient 50 mM Tris (pH 7.4) buffer containing 250 mM NaCl, 0.02% sodium cholate, and 20% glycerol to bring the volume to 245 μl. After an additional incubation time of 2 min at 37 °C, the reaction was started by adding 5 μl of 100 mM NADPH. The mixture was then incubated at 37 °C for a period of 0–150 min. The reaction was terminated by adding 20 μl of ice-cold 6 M HCl, immediately mixing the contents of the vial and placing it on ice. The internal standard (phenylacetic acid, 100 μM final concentration) was then added and the samples were mixed again before they were transferred to solid-phase extraction.
Solid-Phase Extraction
In a modification of a published procedure (30), Bond-Elut C18 cartridges (100 mg, 3 ml, Varian) were sequentially washed with 2 ml of CHCl3, methanol, and water. The incubation samples were transferred to the cartridges and the solvent was slowly pushed through under nitrogen pressure. The cartridges were then washed with 2 ml of water and were thoroughly dried for 1 h by nitrogen at 20 psi. The analytes were eluted into 5 ml glass screw-top tubes with Teflon-lined caps with two 1-ml aliquots of CHCl3. The CHCl3 was evaporated to dryness under a stream of nitrogen. The samples were then converted to the trimethylsilyl derivatives by incubation with 50 μl of N, O-bis(trimethylsilyl)trifluoroacetamide) at 70 °C for 10–15 min. The derivatized samples were allowed to cool, vortexed, and transferred to sealed Teflon-capped glass vials for either manual or auto injection into the GC-MS.
GC - MS
Product analysis was preformed on an Agilent GC6850 Series II GC coupled to an Agilent 5973 Network Mass Spectrometry Detector. This GC-MS system was equipped with an Agilent 7683 Series autosampler interfaced with a PC computer running Agilent MSD Productivity ChemStation Revisions D.01.02 software. A 30-m DB-5 capillary column coupled with a 5-m guard column (Agilent, USA) (0.25 mm i.d., 0.1 μm film) was used with helium as the carrier gas at a head pressure of 50 psi. Injection (3 μl) was in the split mode (10:1) with the all-glass injector temperature at 250 °C. The GC oven temperature was programmed for 1 min at 70 °C followed by a rise to 170 °C at 25 °C/min, to 200 °C at 5 °C/min and to 280 °C at 20 °C/min. The oven was finally held at 280 °C for 5 min. The MS source and interface were maintained at 250 °C and 280 °C, respectively, and a solvent delay of 4 min was used. The MS was set for a full scan from 30–500 mass units. The analytes were identified from a total ion chromatogram.
The following are the retention times and molecular masses of the trimethylsilyl derivatives of the authentic standards of the substrates and products: dodecanoic acid-TMS, 8.62 min, MR 272; 1-TMS, 12.14 min, MR 306; 2-TMS, 12.98 min, MR 350, 352; 3-TMS, 13.79 min, MR 398; 4-TMS, 11.40 min, MR 286; 5-diTMS, 13.50 min, MR 374; 6-diTMS, 12.82 min, MR 360; 7-TMS, 12.14 min, MR 308; 8-diTMS, 12.15 min, MR 360; 9-TMS, 8.42 min, MR 270.
CYP2E1 Oxidation Activity by GC-MS
Standard incubations (250 μl total volume) consisted of 50 pmol CYP2E1 microsomes (human CYP2E1 + P450 reductase + cytochrome b5 Supersomes®), substrate (100 μM), DLPC, superoxidase dismutase, catalase, and NADPH (2 mM) in 0.1 M phosphate buffer (pH 7.4). The substrate (dodecanoic acid, 1 or 7) was coated on the walls of an Eppendorf tube with methanol and the methanol was removed by a stream of nitrogen before the other components were added. The NADPH was added to initiate the reaction after preincubation of the other components for 5 min at 37 °C. The incubation was continued for 2.5 h at 37 °C and was then quenched with 6 M HCl. The internal standard (phenylacetic acid, 100 μM final concentration) was then added and the samples were mixed again before they were transferred to solid-phase extraction for GCMS analysis.
Measurement of CYP4A1 Activity in 18O-Labeled Water
The procedure was nearly identical to that used for the measurement of CYP4A1 activity in normal buffer. To prepare the 18O-buffer (265 μl), 1.6 M Tris (pH 7.4 at 0 °C, 7.8 μl) was mixed with 3.5 mg of NaCl, 50 μl of glycerol, 1 μl of sodium cholate (5% in H2O) and finally 188.5 μl of H2 18O. In a separate tube, the CYP4A1 system was prepared by mixing CYP4A1 (12.5 pmol), P450 reductase (125 pmol), cytochrome b5 (12.5 pmol), DLPC (10 μl, 0.5 g/l) and catalase (2.5 μl, 10 g/l) in their storage phosphate buffer together to a total volume of 15.6 μl. This CYP4A1 system was mixed with the 18O-buffer and incubated for 10 min at 0 °C followed by 10 min at 25 °C. The substrate (1 or 2) was then added and the mixture was preincubated at 37 °C for 2 min before NADPH was added. The final 18O-content was approximately 87.5%. The incubation was continued for 2.5 hr at 37 °C and was then quenched with 6 M HCl. The internal standard (phenylacetic acid, 100 μM final concentration) was then added and the samples mixed before they were transferred to solid-phase extraction for GCMS analysis. In a different protocol, after the incubation was finished, the quenching step was omitted and during the solid phase extraction, the solid phase extraction column was not washed with water to minimize possible exchange of the 18O in metabolites with 16O water.
RESULTS
Binding of 12-Halododecanoic acids to CYP4A1
12-Chloro-, 12-bromo-, and 12-iodododecanoic acids (Fig. 1) were titrated into a cuvette containing a solution of ferric CYP4A1 and the spectroscopic changes were recorded. Each of these three 12-halododecanoic acids caused the appearance of a binding spectrum (Fig. 2). The difference between the absorbance at 390 nm and 417 nm as a function of the ligand concentration gave rise to a hyperbolic plot from which the spectroscopic dissociation constant Ks was calculated (Fig. 2 inset). These measurements gave the following values for the dissociation constants of the three halododecanoic acids: 12-chloro, Ks = 11.8 μM; 12-bromo, Ks = 7.9 μM; and 12-iodo, Ks = 6.0 μM. Comparison of these values with Ks = ~ 30 μM for dodecanoic acid itself indicates that all three 12-halododecanoic acids bind to CYP4A1 with similar affinities but 3 to 4-fold more tightly than the parent dodecanoic acid (20).
Figure 1.
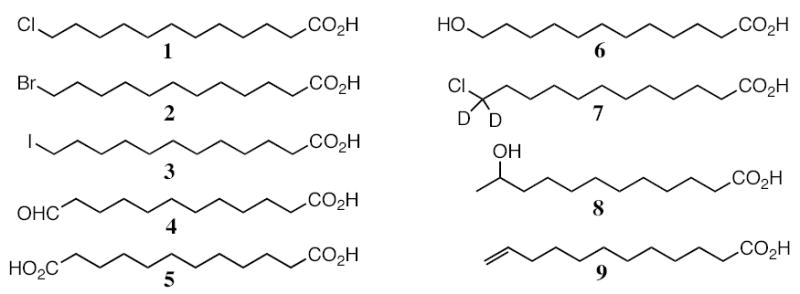
Structures of the substrates and products discussed in this study.
Figure 2.
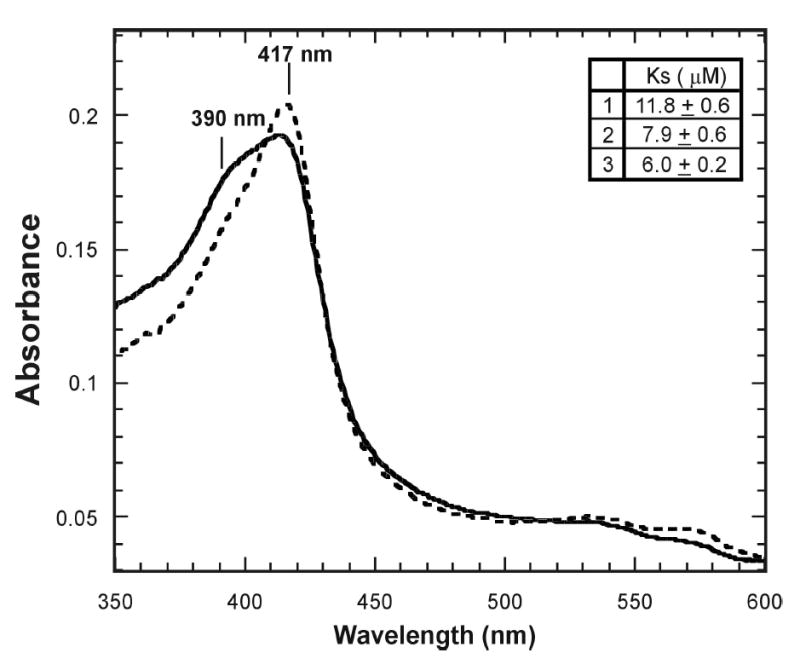
Spectrum of the binding of 12-iodododecanoic acid to CYP4A1. The spectrum of the protein before binding of a substrate (-----) and after binding of 12-iodododecanoic acid (_____). The inset shows the Ks values for the three 12-halododecanoic acids. The spectroscopic binding studies were carried out in the following buffer: 50 mM Tris (pH 7.4) buffer containing 250 mM NaCl, 0.02% sodium cholate, and 20% glycerol.
Products Formed in the CYP4A1-Catalyzed Oxidation of 12-Halododecanoic Acids
Analysis of the products formed in incubations of CYP4A1 with the 12-halododecanoic acids identified three catalytically generated products: 12-hydroxydodecanoic acid (6), 12-oxododecanoic acid (4), and 1,12-dodecanedioic acid (5) (Fig. 3a, Table 1). These peaks were the only ones present that were not also present in an incubation from which NADPH had been omitted. The identities of the products, after conversion to the trimethylsilyl derivatives, were confirmed by chromatographic and mass spectrometric comparison with similarly derivatized authentic samples. 11-Dodecenoic acid (9) was not detected as a metabolite. A small amount of dodecanoic acid was observed in all the incubations at time zero, but its concentration did not change with incubation time. This suggests that the dodecanoic acid is present as a contaminant in one of the incubation components. To confirm that the trace dodecanoic acid observed is actually a contaminant, four set of control experiments were separately performed: a) without NADPH, b) without the substrate, c) without the CYP4A1 system and d) under anaerobic conditions. In all of these control experiments, dodecanoic acid was detected and its ratio versus the substrate remained constant (data not shown). In view of the control experiments, the source of the contaminating dodecanoic acid is likely to be the DLPC.
Figure 3.
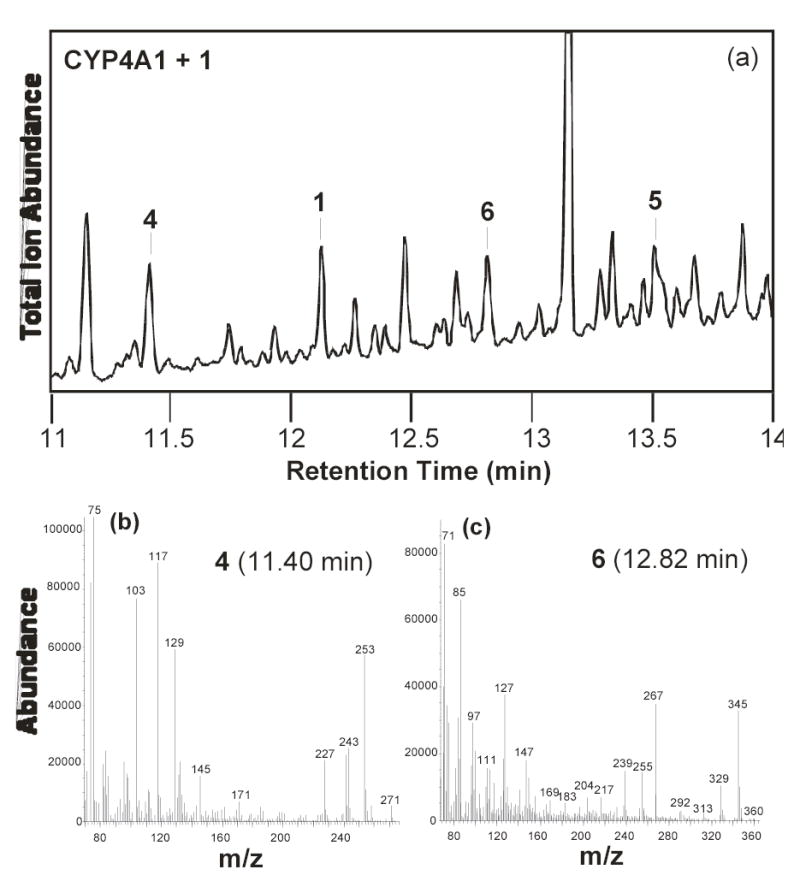
GC-MS of the products formed in the incubation of CYP4A1 with 12- chlorododecanoic acid: (a) GC-MS trace of the products, (b) mass spectrum of product 4 at 11.40 min, and (c) mass spectrum of product 6 at 12.82 min.
Table 1.
Incubations of 1, 2, 3 and 7 with recombinant human CYP4A1 and baculovirusmicrosomes containing recombinant human CYP2E1.a
| CYP4A1 | |||||
|---|---|---|---|---|---|
| 4(%) | 5 (%) | 6 (%) | Conversion (%) | ω/ω–1 | |
| 1 | 30 ± 1 | 11 ± 2 | 11 ± 1 | 52 ± 3 | 0.27 ± 0.02 |
| 2 | 14 ± 1 | 11 ± 1 | 13 ± 1 | 38 ± 1 | 0.51 ± 0.04 |
| 3 | 1.2 ± 0.2 | 1.5 ± 0.1 | 2.9 ± 0.1 | 5.6 ± 0.3 | 1.12 ± 0.16b |
| 7 | 10 ± 1 | 10 ± 1 | 12 ± 1 | 33 ± 1 | 0.61 ± 0.04 |
| CYP2E1 | |||||
| 1 | 10 ± 1 | 5.9±0.4 | 10 ± 0.3 | 26±1 | nac |
na: not available;
All the data were obtained after a 2.5 hr incubation.
This value is not reliable because the low turnover for 3 yielded data points with relatively large error limits;
6 appears to arise from the ω−1 hydroxylation product.
Rates of Halododecanoic Acid Metabolism by CYP4A1
The rates of formation of 12-hydroxydodecanoic acid, 12-oxododecanoic acid, and 1,12-dodecanedioic acid were followed as a function of time of incubation to estimate the relative rates of metabolism of the three 12-halododecanoic acids. As shown in Fig. 4a and summarized in Table 2, the formation of 12-oxododecanoic acid occurs substantially more rapidly with 12-chlorododecanoic acid than with 12-bromododecanoic acid. Formation of this product is hardly observed with 12-iodododecanoic acid (the amount of 12-oxododecanoic acid observed is on a par with that obtained in a control experiment without NADPH in the incubation). As shown in Fig. 4b, the formation of 1,12-dodecanedioic acid occurs with a similar trend as that seen for the formation of 12-oxododecanoic acid, as might be expected if the diacid is obtained by secondary oxidation of the 12-oxododecanoic acid metabolite. With 12-iodododecanoic acid formation of 1,12-dodecanedioic acid was not observed and is therefore not plotted on Fig. 4b.
Figure 4.
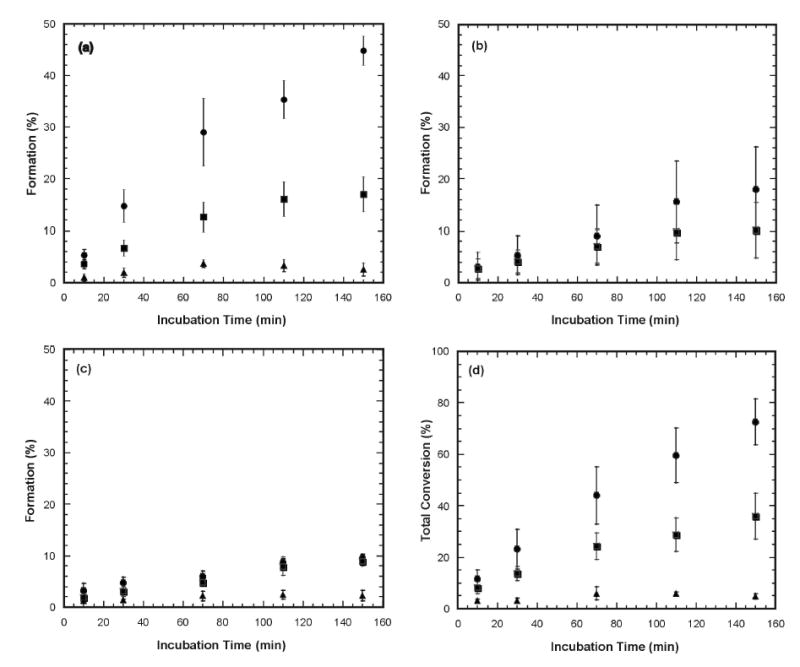
Product formation as a function of time in incubations of the complete CYP4A1 system with 12-chloro- (•), 12-bromo- (▪), and 12-iodododecanoic acid (▴): (a) Formation of 12-oxododecanoic, (b) Formation of 1,12-dodecanedioic acid acid, (c) Formation of 12- hydroxydodecanoic acid, and (d) Combined formation of 12-oxododecanoic acid, 1,12-dodecanedioic acid, and 12-hydroxydodecanoic acid. Due to differences in incubation conditions, the final conversion ratio was slightly different from that of the single-time-point (i.e. 2.5 hrs) incubation.
Table 2.
Turnover numbers for the metabolites (min−1)
| 4 | 5 | 6 | Total | |
|---|---|---|---|---|
| 1 | 6.0 | 2.4 | 1.3 | 9.7 |
| 2 | 2.3 | 1.3 | 1.2 | 4.8 |
| 3 | 0.3 | Nd | 0.3 | 0.6 |
The turnover numbers were estimated from the data in Fig. 4, the fact that the incubations contained 12.5 pmol of CYP4A1 and 25 nmol of 12-halododecanoic acid, and the incubation time of 150 min.
A related but less marked trend is observed for oxidation of the 12-halododecanoic acids to 12-hydroxydodecanoic acid. Thus, the 12-hydroxy product is formed at a similar rate with the 12-chloro- and 12-bromododecanoic acids, but only to a low extent with 12-iodododecanoic acid (Fig. 4c).
If one plots the oxidation of the 12-halododecanoic acids, as judged by the combined formation of the three metabolites, one obtains the plot in Fig. 4d. It is clear from these results, and most clearly shown by the final plot, that 12-chlorododecanoic acid is oxidized more rapidly than 12-bromododecanoic acid, whereas 12-iodododecanoic acid is hardly a substrate for CYP4A1.
Source of the Oxygen in the 12-Halododecanoic Acid Metabolites
To determine the origin of the hydroxyl group in the products formed from the oxidation of 12-chloro- and 12-bromododecanoic acid, these substrates were incubated with the enzyme system in a medium estimated to contain 87.5% of H2 18O. Mass spectrometric analysis of the products from these 12-halododecanoic acids indicated that the hydroxyl oxygen atom in the 12-hydroxydodecanoic acid from both 12-halododecanoic acids was approximately 75% 18O-labeled. This conclusion derives from a comparison of the relative intensities of the peaks at m/z 345 and 347, which correspond to M+-15 for the unlabeled and labeled 12-hydroxylauric acid diTMS derivatives, respectively (Fig. 5a). This value is consistent with the percent of 18O-label in the water used for the incubation. In contrast, in both cases the oxygen in the 12-oxododecanoic acid product was only slightly labeled or not labeled at all, as shown by the fact that the parent ion peak is at m/z 271, which corresponds to M+-15 for the 12-oxododecanoic acid TMS derivative (Fig. 5b). If the aldehyde oxygen were labeled with 18O, this peak should have been at m/z 273. In a control experiment in which we simply incubated 12-oxododecanoic acid in H2 18O, it was found that the aldehyde oxygen of 12-oxododecanoic acid readily exchanges with the oxygen of water. Therefore a different work-up protocol was tried in which the quenching and the washing steps in the solid phase extraction procedure were omitted to minimize solvent (H2 16O) exchange in the work-up stage. The results showed again that the hydroxyl oxygen atom in the 12-hydroxydodecanoic acid from both 12-halododecanoic acids was approximately 75–80% 18O-labeled and the oxygen in the 12-oxododecanoic acid product was ~11% labeled, presumably as a result of a small extent of exchange with the solvent during incubation in the H2 18O medium. Furthermore, about 10% 18O label was observed in the 1,12-dodecanedioic acid that results from the further oxidation of the aldehyde, consistent with the ratio seen with the aldehyde itself.
Figure 5.
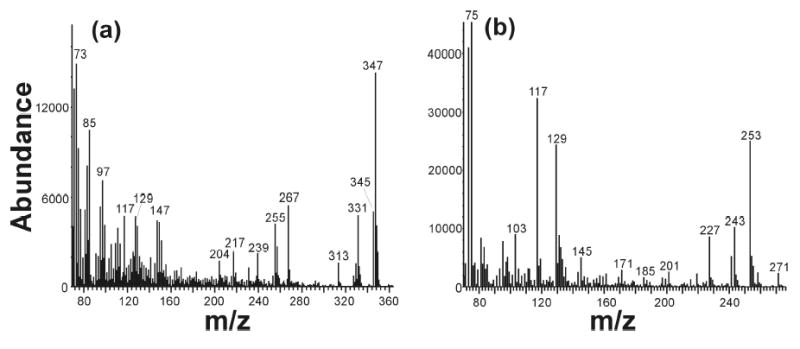
Mass spectra of a) the 12-hydroxydodecanoic acid and (b) 12-oxododecanoic acid obtained from an incubation of 12-chlorododecanoic acid with the complete CYP4A1 system in 18O-labeled medium.
Assay for Inactivation of CYP4A1
The possible formation of an electrophilic intermediate in the oxidation of the 12-halododecanoic acids suggested that CYP4A1 might be inactivated during turnover of these substrates due to alkylation of the heme or protein. Ten-fold concentrated solutions of the complete CYP4A1 system were therefore preincubated with each of the three 12-halododecanoic acids and the residual activity of the enzyme after a tenfold dilution was evaluated with the standard dodecanoic acid hydroxylation assay as a function of time. No inactivation in excess of that obtained in the absence of 12-chlorododecanoic acid was observed over a preincubation period of up to 25 min with up to 200 μM concentrations of 12-chlorododecanoic acid. 12-Bromo- and 12-iodododecanoic acids were also tested and no inactivation was apparent, although the data points were more scattered than in the case of 12-chlorododecanoic acid and a small amount of inactivation would not have been detected.
CYP2E1 Oxidation of 12-Chlorododecanoic
CYP2E1 is known to catalyze the ω−1 hydroxylation of fatty acids and was therefore examined as a control. This enzyme was first tested with dodecanoic acid and the ω/(ω−1) hydroxylation ratio was found to be ~0.16. Thus, 86% of the oxidation occurred at the ω−1 position and 14% at the ω-position. We then tested this enzyme with 12-chlorododecanoic acid, for which the overall conversion to products was 26% as compared to 52% for CYP4A1 (Table 1). The products observed in the reaction were again 12-hydroxydodecanoic, 12-oxododecanoic, and 1,12-dodecanedioc acids. Unexpectedly, given the ω−1 regiospecificity of the enzyme, 12-hydroxydodecanoic acid constituted a large proportion of the products. However, the CYP2E1 enzyme employed for these studies was the commercially available human CYP2E1 that is provided in baculovirus transfected insect cell microsomes. As the insect cell microsomes contain no other P450 enzyme but to contain other enzymes, we incubated the enzyme preparation with 12-oxododecanoic acid itself and found that it is readily reduced to 12-hydroxydodecanoic acid in the presence of NADPH. In a control experiment, insect cell microsomes not expressing CYP2E1 were found to have not catalytic activity towards lauric acid or 12-chlorododecanoic acid.
Product formation in incubations of this same enzyme preparation with 12,12-[2H]2-12-chlorododecanoic acid was greatly attenuated due to a deuterium isotope effect. Nevertheless, we observed a roughly 4–5% (≈1 nmol using 25 pmol protein) conversion of the deuterated starting material to products. The molecular mass of the 12-hydroxydodecanoic acid formed in the reaction was 361, as indicated by a peak at m/z 346 (M+-15, data not shown), in agreement with retention of only one of the two original deuterium atoms. This finding indicates that the alcohol product from 12,12-[2H]2-12-chlorododecanoic acid arose by reduction of 12[2H]-12-oxododecanoic acid, the aldehyde obtained by normal ω−1 hydroxylation of the substrate by CYP2E1. If the alcohol had arisen from direct oxidation of the halide, both deuterium atoms would have been preserved in the product, yielding 12,12-[2H]2-12-hydroxydodecanoic acid with a molecular mass of 362, as was found with CYP4A1.
DISCUSSION
The binding of substrates to the resting ferric state of P450 enzymes can usually be monitored by spectroscopic changes associated with substrate binding (31, 32). These changes normally involve changes in the coordination of the heme iron, such as dissociation of the distal water ligand or coordination of an atom of the substrate with the iron, that result in shifts between the pentacoordinated and hexacoordinated states of the enzyme (31, 32). As shown here, the three 12-halododecanoic acids, like dodecanoic acid itself, cause a readily monitored change in the spectrum of CYP4A1 involving a decrease in absorption at 417 nm and a corresponding increase at 390 nm (Fig. 2). This type of spectroscopic shift, known as a Type I binding spectrum (31), strongly suggests that the three substrates, like dodecanoic acid itself, promote a shift from the hexacoordinated to the pentacoordinated state of the heme iron, presumably by promoting dissociation of a distal water ligand. Titration of the spectroscopic change indicates that all three 12-halododecanoic acids bind more tightly than dodecanoic acid itself (Ks of 6–12 μM compared to ~30 μM for dodecanoic acid). Tighter binding is consistent with the fact that all the 12-halododecanoic acids involve extension of the dodecanoic acid chain by the addition of a hydrophobic atom: the nonaromatic lipophilicity constants for Cl, Br, and I substituents (water versus octanol) are, respectively, π = +0.39, +0.60, and +1.00. (33). Binding to P450 enzymes is generally enhanced by increased lipophilicity (34).
The principal surprise of this study was that the 12-chloro- and 12-bromo derivatives of dodecanoic acid were oxidized by CYP4A1, whereas the 12-iodo derivative was almost inert despite its demonstrated ability to bind to the enzyme and to promote dissociation of the distal water ligand. Two types of oxidative reactions were observed. The first is a conventional reaction in which the enzyme hydroxylates the halogen-substituted carbon to give the 12-halo-12-hydroxydodecanoic acid, which undergoes intramolecular elimination of HX (X = halide ion) to yield the aldehyde. The resulting aldehyde is then partially oxidized in a second step by CYP4A1 via a well-established reaction to the acid (35, 36). This sequence of events is confirmed by the observation that the aldehyde and the diacid formed from 12-chlorododecanoic acid in a medium in which the water is ~87.5% 18 O-labeled incorporate only a trace of the 18O-label.
CYP4A1 is an ω-hydroxylase that preferentially oxidizes fatty acids at the ω- (i.e., terminal) carbon atom. However, this specificity is not absolute and a small amount of the (ω−1)-hydroxylated fatty acid (8) is also produced (11). In the case of dodecanoic acid, the ratio of ω- to (ω−1)-hydroxylation is as high as 40:1, although this specificity decreases for myristic acid, which is two carbons longer, to a ratio of 3:1 (11). In both cases, the CYP4A enzymes favor the thermodynamically difficult ω-hydroxylation over oxidation of the intrinsically more reactive ω−1 position. To explain this selectivity, we have proposed that the active site involves presentation of the terminal atom to the oxidizing ferryl species via a narrow access channel that favors oxidation of the terminal carbon at the expense of the (ω−1) carbon (7). In this model, the small amount of (ω−1)-hydroxylation results from a limited degree of active site flexibility that occasionally allows the (ω−1)-carbon to become accessible to the ferryl species. The results with the 12-halododecanoic acids are readily understood within this proposed active site paradigm. The halogen in the 12-halododecanoic acids is effectively the terminal atom of the chain, so that aldehyde production from the 12-halododecanoic acids is the result of an (ω−1)-hydroxylation process. The observed decrease in formation of the aldehyde product from the 12-halododecanoic acids in the order Cl > Br ≫ I correlates with the increasing size of the ω-halogen atom; i.e., the larger the terminal atom, the less “play” there is in the active site and the less the enzyme is able to circumvent the halide atom to oxidize the (ω−1)-position. The alternative explanation that the C-H bond next to the chloride is broken more readily than that next to the bromide is contradicted by a body of literature which shows that the bond strength, and reactivity, of the C-H bond is higher if a bromide rather than a chloride is present on the carbon (37).
Even more impressive and definitive is the finding that 12-chlorododecanoic acid and 12-bromododecanoic acid are oxidized by CYP4A1 to the corresponding alcohols, whereas the 12-iodo is again very poorly oxidized by this pathway. The mechanism of this oxidation is unambiguously established by the demonstration, using H2 18O, that the hydroxyl group in the product derives essentially quantitatively from the water of the medium. The alcohol therefore must arise by a mechanism independent of formation of the aldehyde by (ω−1)-hydroxylation, as only minor (11%) 18O-incorporation is observed for the aldehyde as a result of exchange with the solvent). It is evident that the mechanism involves direct oxidation of the halide by the P450 ferryl intermediate, producing the corresponding oxohalonium species (Scheme 1) that is displaced by water to give the alcohol with incorporation of an oxygen from the medium. The hydrolysis of oxohalonium substituents is a well-established chemical reaction (38).
The view that oxidation of the terminal halide atom and hydroxylation of the carbon to which it is attached are competing reaction pathways for the ferryl intermediate is supported by studies with 1,12-[2H2]-12-chlorododecanoic acid. Oxidation of this deuterated substrate gives similar products to oxidation of the non-deuterated analogue, with the alcohol retaining both deuterium atoms, but the ratio of the alcohol and aldehyde plus acid products indicates that oxidation of the halide atom increased approximately two to three-fold. This metabolic switching phenomenon, which reflects the higher energy of the C–2H than C–H bond, is a characteristic of a P450 reaction in which the two sites of reaction compete for the oxidizing ferryl species (39). In a related experiment, we have shown that the 12-halododecanoic acids are oxidized by CYP2E1, a non-specific P450 enzyme that primarily catalyzes the ω−1 hydroxylation of dodecanoic acid, to the corresponding aldehyde product with only minimal direct formation of the alcohol product. Although alcohol product was formed, control experiments and experiments with the 12,12-dideuterated 12-chlorododecanoic acid clearly establish that with this enzyme the initial P450 product is the aldehyde that is subsequently reduced by an adventitious aldehyde dehydrogenase activity in the baculovirus microsomes. This emphasizes the critical role that is played by the CYP4A1 protein in channeling the catalytic turnover towards oxidation of the terminal atom in the fatty acid chain.
The most interesting finding is that the order of oxidation of the halogen in the 12-halododecanoic acid is Br > Cl ≫ I. This is particularly informative because the oxidation potentials of the halides follow a somewhat reverse order: i.e., iodide is more oxidizable than bromide, which in turn is more oxidizable than chloride (40). Thus, intrinsic reactivity would predict that oxidation of the 12-halododecanoic acids would occur in the order I > Br > Cl. This paradox is readily explained by the hypothesis that the ω-specificity of the enzyme is determined by presentation of the terminal atom of the fatty acid chain for oxidation through a sterically restricted channel or opening. The van der Waal’s radius of a chloro atom (1.8 Å) is slightly smaller than that of a methyl (2.0 Å), and the radius of a bromo substituent (1.95 Å) is approximately the same size, but that of an iodide (2.15 Å) is somewhat larger (41). Thus, if the terminal atom must be presented via a tightly constrained channel, it is not surprising that the chloride is readily presented and (despite its high oxidation potential) oxidized to the oxochloronium substituent. The bromide can also be presented, but with less clearance, so that although its oxidation to the ω-alcohol is not affected (it is actually increased a little compared to the chloride), oxidation to the aldehyde is attenuated due to the increased difficulty in accessing the carbon to which the halide is attached. Finally, the iodide is apparently sufficiently large that it no longer fits into the access channel and, if at all, is very poorly oxidized. As already noted, the van der Waal’s radii of the bromo and iodide atoms are 1.95 and 2.15 Å, respectively. This suggests that the orifice through which the terminal atom must be presented for oxidation by the ferryl species has an effective diameter larger than 3.90 Å but smaller than 4.30 Å. A very tight tolerance of the presentation channel is implied by the ω-specificity of the enzyme, as the looser the fit the more the enzyme would favor the thermodynamically easier ω−1 hydroxylation.
As already noted, all three 12-halododecanoic acids displace the distal water ligand, or at least cause the spectroscopic change characteristic of a Type II binding event, even though the iodide apparently cannot approach the heme iron atom close enough to physically displace the water ligand from the iron. This suggests that a conformational change or a significant change in active site polarity triggers loss of the water ligand or, alternatively, that access to the ferryl species only becomes restricted during catalysis, perhaps as a result of conformational changes associated with the active site reorganization that attends oxygen activation (42). We have recently reported that computational docking of imidazole ligands to cytochrome P450cam roughly predicts inhibitory activity but neither the docking nor catalytic results correlate with the spectroscopically measured binding of the ligand (43). These combined results suggest that a greater allowance may need to be made for the dynamic nature of the P450 active site in interpreting spectroscopically determined changes in the distal iron ligand.
Formation of the oxochloronium and oxobromonium substituents, both of which are excellent leaving groups, could result not only in their displacement by water to give the alcohol metabolite but also in reaction with an active site nucleophile, resulting in inactivation of the enzyme. Alkyl oxohalonium species such as these are known to chemically react with nucleophiles (38, 44). However, analysis of the protein activity after preincubation with the 12-halododecanoic acids indicates that there is no loss of activity over and above that observed in the absence of the halogenated probe molecules. Thus, either water rapidly traps the oxohalonium intermediate, either within the active site or after its dissociation into the medium, before it can react with a critical protein nucleophile, or the activated metabolite only reacts with non-essential amino acid residues in the protein.
In summary, oxidation of the 12-halododecanoic acids by CYP4A1 has provided (a) unambiguous evidence that halogens, including iodide, bromide, and chloride, can be oxidized by P450 enzymes to the corresponding 12-oxohalonium products that are readily hydrolyzed to the alcohols, and (b) direct evidence that the ω-regiospecificity of CYP4A1, and presumably all the other CYP4A enzymes, is enforced by requiring the terminal atom of the chain to be threaded for oxidation through a narrow channel that tolerates a methyl, chloro, or bromo but not iodo atom. This implicates an effective channel diameter larger than 3.90 Å but smaller than 4.30 Å.
Figure 6.
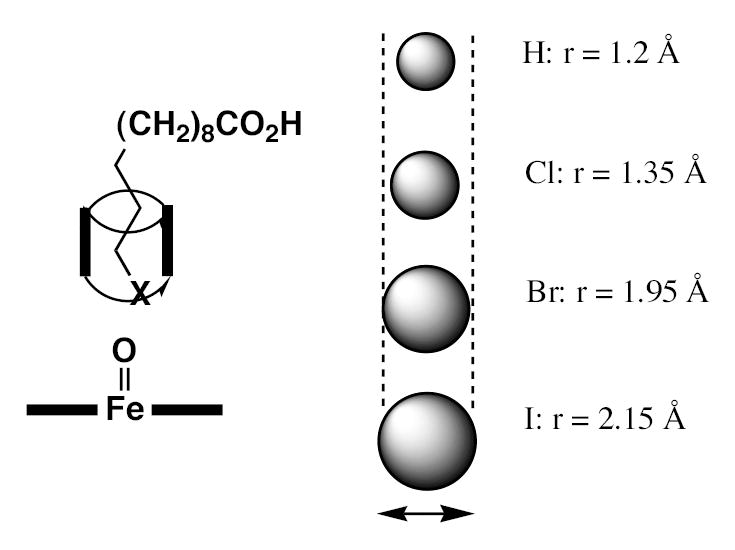
Proposed mechanism for discrimination between the chloro-, bromo-, and iodo terminal atom in the 12-halododecanoic acids. The CYP4A1 ferryl species is represented by the Fe=O in the figure. The increasing size of the halide atoms relative to the diameter of the channel is illustrated schematically and the Van der Waal’s radius of H, Cl, Br, and I is cited. This mechanism directly relates to the ω- versus (ω−1)-specificity of CYP4A1 and other CYP4 family enzymes.
Scheme 1.
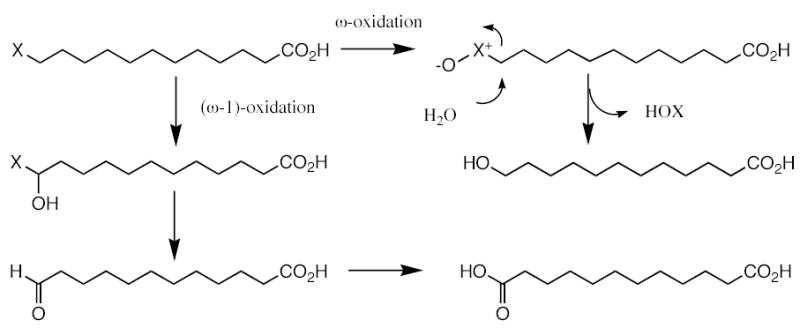
Mechanism proposed for oxidation of the 12-halododecanoic acids to 12-hydroxydodecanoic acid with incorporation of the oxygen form water into the hydroxyl group of the product (ω-oxidation), and to 12-oxododecanoic acid and eventually 1,12-dodecanedioic acid with incorporation of oxygen from molecular oxygen but not from water (ω−1 hydroxylation).
Acknowledgments
We would like to acknowledge early preliminary work on this problem using protein isolated from rat liver microsomes by Dr. Claire Cajacob.
Footnotes
This work was supported by grant GM25515 from the National Institutes of Health (PROM), a University of Queensland Francine Kroesen Travel Award (MJC) and Grant DP0210635 from the Australian Research Council (JJDV).
The abbreviations used are: heme, iron protoporphyrin IX regardless of oxidation and coordination state; CYP, cytochrome P450; DLPC, dilauroylphosphatidylcholine; TMS, trimethylsilyl.
References
- 1.Capdevila, J. H. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd Ed. (Ortiz de Montellano, P. R., Ed.) Plenum Klewer, New York, 531–551 (2005).
- 2.Zeldin DC. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 3.Simpson AECM. Gen Pharmac. 1997;28:351–359. doi: 10.1016/s0306-3623(96)00246-7. [DOI] [PubMed] [Google Scholar]
- 4.Imig JD, Zou AP, Stec DE, Harder DR, Falck JR, Roman RJ. Am J Physiol. 1996;270:R217–227. doi: 10.1152/ajpregu.1996.270.1.R217. [DOI] [PubMed] [Google Scholar]
- 5.Ma YH, Gebremedhin ML, Schwartzman ML, Falck JR, Clark JE, Masters BS, Harder DR, Roman RJ. Circ Res. 1993;72:126–136. doi: 10.1161/01.res.72.1.126. [DOI] [PubMed] [Google Scholar]
- 6.Escalante B, Falck JR, Yadagiri P, Sun LM, Laniado-Schwartzman M. Biochem Biophys Res Commun. 1988;152:1269–1274. doi: 10.1016/s0006-291x(88)80422-4. [DOI] [PubMed] [Google Scholar]
- 7.CaJacob CA, Chan W, Shephard E, Ortiz de Montellano PR. J Biol Chem. 1988;263:18640–18649. [PubMed] [Google Scholar]
- 8.Alterman MA, Hanzlik RP. FEBS Lett. 2002;512:319–322. doi: 10.1016/s0014-5793(02)02260-3. [DOI] [PubMed] [Google Scholar]
- 9.Korzekwa KR, Jones JP, Gillette JR. J Am Chem Soc. 1990;112:7042–7046. [Google Scholar]
- 10.De Visser SP, Kumar D, Cohen S, Shacham R, Shaik S. J Am Chem Soc. 2004;126:8362–8363. doi: 10.1021/ja048528h. [DOI] [PubMed] [Google Scholar]
- 11.Hoch U, Zhang Z, Kroetz DL, Ortiz de Montellano PR. Arch Biochem Biophys. 2000;373:63–71. doi: 10.1006/abbi.1999.1504. [DOI] [PubMed] [Google Scholar]
- 12.Chan WK, Ortiz de Montellano PR. Chem Res Toxicol. 1993;6:38–45. doi: 10.1021/tx00031a006. [DOI] [PubMed] [Google Scholar]
- 13.Bambal RB, Hanzlik RP. Arch Biochem Biophys. 1996;334:59–66. doi: 10.1006/abbi.1996.0429. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz de Montellano, P. R., and De Voss, J. J. (2005) In Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd Ed. (Ortiz de Montellano, P. R., Ed.) Kluwer Academic/Plenum Publishers, New York, 183–245.
- 15.Burka LT, Thorsen A, Guengerich FP. J Am Chem Soc. 1980;102:7615–7616. [Google Scholar]
- 16.Guengerich FP. J Biol Chem. 1989;264:17198–17205. [Google Scholar]
- 17.Guengerich FP, Crawford WM, Domoradzki JY, Macdonald TL, Watanabe PG. Toxicol Appl Pharmacol. 1980;55:303–317. doi: 10.1016/0041-008x(80)90092-7. [DOI] [PubMed] [Google Scholar]
- 18.Tachizawa H, MacDonald TL, Neal RA. Molec Pharmacol. 1982;22:745–751. [PubMed] [Google Scholar]
- 19.Mico BA, Branchflower RV, Pohl LR. Biochem Pharmacol. 1983;32:2357–2359. doi: 10.1016/0006-2952(83)90188-0. [DOI] [PubMed] [Google Scholar]
- 20.Dierks EA, Davis SC, Ortiz de Montellano PR. Biochemistry. 1998;37:1839–1847. doi: 10.1021/bi972458s. [DOI] [PubMed] [Google Scholar]
- 21.Apparu M, Comet M, Leo PM, Mathieu JP, Du Moulinet d’Hardemare A, Pasqualini R, Vidal M. Bull Soc Chim France. 1988;1:118–124. [Google Scholar]
- 22.Matsuyama H, Nakamura T, Kamigata N. J Org Chem. 1989;54:5218–5223. [Google Scholar]
- 23.Jocelyn, P. C., and Polgar, N. In Organic Synthesis, Wiley, New York, Volume 1, 29 (1941).
- 24.Zakharkin LI, Churilova IM, Anikina EV. Zhurnal Organicheskoi Khimii. 1988;24:77–80. [Google Scholar]
- 25.Mirviss SB. J Org Chem. 1989;54:1948–1951. [Google Scholar]
- 26.Peyrat JF, Figadère B, Cavé A. Synthetic Commun. 1996;26:4563–4567. [Google Scholar]
- 27.Logemann E, Rissler K, Schill G, Fritz H. Chem Berichte. 1981;114:2245–2260. [Google Scholar]
- 28.Corey EJ, Suggs W. Tet Letters. 1975;31:2647–2650. [Google Scholar]
- 29.Omura T, Sato R. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 30.Holmes VE, Bruce M, Shaw PN, Bell DR, Qi FM, Barrettt DA. Anal Biochem. 2004;325:354–363. doi: 10.1016/j.ab.2003.10.046. [DOI] [PubMed] [Google Scholar]
- 31.Orrenius, S., Wilson, B. J., von Bahr, C., and Schenkman, J. B. (1972) In Biological Hydroxylation Mechanisms, Biochem. Soc. Symp. 34 (Boyd, G. S., and Smellie, R. M. S., Eds) Acad. Press, NY, pp. 55–77. [PubMed]
- 32.Schenkman, J. B., and Jansson, I. (1998) In Cytochrome P450 Protocols (Phillips, I. R., and Shephard, E. A., Eds.) Humana Press, Totowa, NJ, pp. 25–33.
- 33.Hansch, C., Leo, A., and Hoekman, D. (1995) In Exploring QSAR. Hydrophobic, Electronic, and Steric Constants, American Chemical Society, Washington, DC, pp. 219–220.
- 34.Lewis DFV, Dickins M. Drug Metab Rev. 2002;35:1–18. doi: 10.1081/dmr-120018245. [DOI] [PubMed] [Google Scholar]
- 35.Soberman RJ, Sutyak JP, Okita RT, Wendelborn DF, Roberts LJ, Austen KF. J Biol Chem. 1988;263:7996–8002. [PubMed] [Google Scholar]
- 36.Cali JJ, Russell CW. J Biol Chem. 1991;266:7774–7778. [PubMed] [Google Scholar]
- 37.Kubic VL, Anders MW. Drug Metab Dispos. 1975;3:104–112. [PubMed] [Google Scholar]
- 38.Macdonald TL, Narasimhan N, Burka LT. J Am Chem Soc. 1980;102:7760–7765. [Google Scholar]
- 39.Harada N, Miwa G, Walsh J, Lu A. J Biol Chem. 1984;259:3005–3010. [PubMed] [Google Scholar]
- 40.Hunsberger, J. F. (1972) In CRC Handbook of Chemistry and Physics, 53rd Ed., CRC Press, Cleveland, OH, pp. D111–D112.
- 41.Pauling, L., (1960) The Nature of the Chemical Bond, 3rd Ed., Cornell University Press, Ithaca, NY, pp. 257–264.
- 42.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 43.Verras A, Kuntz ID, Ortiz de Montellano PR. J Med Chem. 2004;47:3572–3579. doi: 10.1021/jm030608t. [DOI] [PubMed] [Google Scholar]
- 44.Crambie RC, Chambers D, Lindsay BG, Rutledge PS, Woodgate PD. J. Chem. Soc. Perkins I. 1980:822–827. [Google Scholar]