

Abstract
Human arginase I is a potential target for therapeutic intervention in diseases linked to compromised l-arginine homeostasis. Here, we report high-affinity binding of the reaction coordinate analogue inhibitors 2(S)-amino-6-boronohexanoic acid (ABH, Kd = 5 nM) and S-(2-boronoethyl)-l-cysteine (BEC, Kd = 270 nM) to human arginase I, and we report x-ray crystal structures of the respective enzyme–inhibitor complexes at 1.29- and 1.94-Å resolution determined from crystals twinned by hemihedry. The ultrahigh-resolution structure of the human arginase I–ABH complex yields an unprecedented view of the binuclear manganese cluster and illuminates the structural basis for nanomolar affinity: bidentate inner-sphere boronate–manganese coordination interactions and fully saturated hydrogen bond networks with inhibitor α-amino and α-carboxylate groups. These interactions are therefore implicated in the stabilization of the transition state for l-arginine hydrolysis. Electron density maps also reveal that active-site residue H141 is protonated as the imidazolium cation. The location of H141 is such that it could function as a general acid to protonate the leaving amino group of l-ornithine during catalysis, and this is a revised mechanistic proposal for arginase. This work serves as a foundation for studying the structural and chemical biology of arginase I in the immune response, and we demonstrate the inhibition of arginase activity by ABH in human and murine myeloid cells.
Keywords: boronic acid, metalloenzyme, protein crystallography
Arginase is a trimeric binuclear manganese metalloenzyme that catalyzes the hydrolysis of l-arginine to form l-ornithine and urea (1–3). Two isozymes have been identified in mammals: arginase I catalyzes the final cytosolic step of the urea cycle in liver, and arginase II is a mitochondrial enzyme that functions in l-arginine homeostasis in nonhepatic tissues. Notably, arginase I is also expressed in certain nonhepatic tissues where it, too, can function in l-arginine homeostasis. For example, arginase I may regulate substrate l-arginine bioavailability to NO synthase in the immune response. Macrophage arginase I and NO synthase are reciprocally regulated at the level of transcription: NO synthase is induced by T-helper type 1 (TH1) cytokines, and arginase I is induced by T-helper type 2 (TH2) cytokines (4–7). As a modulator of NO-dependent macrophage cytotoxicity, arginase I is implicated in the regulation of macrophage activity in wound healing (8) and the suppression of the tumoricidal activity of macrophages (9) and T cells (10). Notably, arginase I is very highly up-regulated in the murine spinal cord during experimental autoimmune encephalomyelitis, an animal model for human multiple sclerosis (11), and it is up-regulated in the inflammatory regions of the asthmatic lung (12–14).
Arginase I in the immune response is also implicated in cancer biology: arginase I is significantly up-regulated and promotes tumor cell growth in breast cancer (15, 16) and colorectal cancer (17). Rodriguez et al. (10, 18, 19) recently demonstrated that macrophages stimulated by interleukins 4 and 13 produce high levels of arginase I and cause T-cell anergy by decreasing the expression of the T-cell receptor CD3ζ chain; furthermore, arginase inhibition blocks the growth of lung carcinoma in a murine model. Thus, human arginase I is a potential target for therapeutic intervention in the modulation of the immune response.
To date, the crystal structures of rat arginase I, human arginase II, and the arginase from Bacillus caldovelox have been solved (20–22). The structure of rat arginase I has been exceptionally useful for understanding inhibitor structure–affinity relationships (23–25). However, given the potential pharmaceutical importance of the human enzyme, an experimentally determined structure is critical for guiding the structure-based design of potent and isozyme-selective inhibitors. Human arginase I is a 322-residue protein with an amino acid sequence 87% identical to that of rat arginase I and 55% identical to that of human arginase II (26, 27). We now report the x-ray crystal structures of human arginase I complexed with the inhibitors 2(S)-amino-6-boronohexanoic acid (ABH) at 1.29-Å resolution and S-(2-boronoethyl)-l-cysteine (BEC) at 1.94-Å resolution. This work serves as a foundation for studying the structural and chemical biology of arginase I in the immune response, and we also demonstrate the inhibition of arginase activity by ABH in human and murine myeloid cells.
Materials and Methods
Crystal Structure Determination. The full length cDNA of human arginase I was inserted into the pBS(KS) vector (Stratagene) as a BspHI/BamHI, expressed in Escherichia coli BL21(DE3), and purified as described (27). Soluble protein extracts were further purified on a carboxymethyl-cellulose column equilibrated and washed with 10 mM Tris·HCl, pH 7.5/1 mM 2-mercaptoethanol/2 mM MnCl2 and then eluted with 0–300 mM NaCl.
The human arginase I–ABH and –BEC complexes were crystallized by the sitting drop vapor diffusion method at 21°C and 4°C, respectively. Drops containing 4.0 μl of enzyme solution (5.0 mg/ml arginase I/5 mM MnCl2/2 mM ABH or BEC/50.0 mM bicine, pH 8.5) and 4.0 μl of precipitant buffer [0.1 M Bis-Tris, pH 6.5/14–28% (wt/vol) polyethylene glycol–monomethyl ether 5000] were equilibrated against a 1-ml reservoir of precipitant buffer. Rod-like crystals appeared in ≈2–3 days with typical dimensions of 0.3 × 0.3 × 0.5 mm. Crystals of the ABH complex yielded diffraction data to 1.29-Å resolution at the Cornell High Energy Synchrotron Source, and crystals of the BEC complex yielded diffraction data to 1.94-Å resolution at the Brookhaven National Laboratory (beamline X12B). Diffraction intensities measured from these crystals exhibited symmetry consistent with the apparent space group P6 (unit cell parameters a = b = 91.4 Å and c = 69.6 Å). These crystals were isomorphous with those obtained of the rat arginase I–ABH and –BEC complexes (23, 24). Data reduction was achieved with denzo and scalepack (28). Statistical analyses of intensity data revealed that the true crystallographic P3 symmetry was convoluted by hemihedral twinning.
The structure of the human arginase I–ABH complex was solved by the difference Fourier method using the structure of the rat arginase I–ABH complex [PDB accession code 1D3V (24); less inhibitor atoms, water molecules, and the side chains of mutated residues] as an initial model for rigid body refinement using the program cns (29). To calculate electron density maps, structure factor amplitudes derived from twinned data were deconvoluted by using the structure-based algorithm of Redinbo and Yeates (30, 31). The initial omit electron density map revealed clear peaks corresponding to the side chains of mutated residues as well as the inhibitor ABH bound as the tetrahedral boronate anion. Iterative cycles of refinement using torsion angle dynamics with a starting temperature of 5,000 K in the initial stages of refinement and model building using cns and o (32) improved the protein structure as monitored by Rtwin and Rfree/twin. In the final stages of refinement, when Rtwin = 0.182 and Rtwin/free = 0.224, full-matrix least-squares refinement was performed against twinned data by using the program shelx (33). Several peaks in difference electron density maps were observed for hydrogen positions on various amide and imidazole NH groups, as well as ordered CH2 groups. Accordingly, hydrogen atoms were placed in calculated positions. The Rfree value dropped 0.003 after refinement with hydrogen atoms. Isotropic B factors were used in refinement with the exception of the manganese ions and sulfur atoms, which were refined anisotropically. The twin fraction refined to 0.46, consistent with nearly perfect hemihedral twinning. Segments M1–K4 and P320–K322 of monomer A and M1–K4 and P314–K322 of monomer B were absent in the experimental electron density, presumed disordered, and omitted from the final model. Data reduction and refinement statistics are recorded in Table 1.
Table 1. Data collection and refinement statistics.
| Structure | Human arginase I—ABH complex | Human arginase I—BEC complex |
|---|---|---|
| Resolution range, Å | 50.0-1.29 | 15.4-1.94 |
| Unique reflections | 150499 | 46041 |
| Rmerge* | 0.063 (0.56)† | 0.075 (0.34)† |
| I/σ(I) | 9.6 (1.9)† | 8.7 (2.0)† |
| Completeness, % | 91.8 | 99.2 |
| Reflections used in refinement, test set | 150499/7526 | 45844/4119 |
| R/Rfree‡ | 0.137/0.150§ | 0.155/0.213¶ |
| Water molecules∥ | 383 | 201 |
| Inhibitor atoms∥ | 26 | 26 |
| Manganese ions∥ | 4 | 4 |
| Bond lengths (rmsd), Å | 0.012 | 0.007 |
| Bond angles (rmsd), ° | 2.79 | 1.42 |
| Dihedral angles (rmsd), ° | 25.7 | 22.9 |
| Improper dihedral angles (rmsd), ° | 1.78 | 0.91 |
rmsd, rms deviation.
Rmerge = Σ|I — 〈I〉|/ΣI, where I is the observed intensity, and 〈I〉 is the average intensity calculated for replicate data.
Number in parentheses refers to the outer 0.1-Å shell of data.
R = Σ∥Fobs| — |Fcalc∥/Σ|Fobs|, where |Fobs| and |Fcalc| are the observed and calculated structure factors amplitudes, respectively, for the twinned structure. The same expression describes Rfree as calculated for test-set reflections excluded from refinement.
Values calculated with SHELX for reflections with I > 2σ(I); using all reflections, R = 0.165 and Rfree = 0.179.
R = Rtwin = Σ∥Fobs| — (|Fcalc/A|2+|Fcalc/B|2)1/2|/Σ|Fobs| for reflections contained in the working set. Fcalc/A and Fcalc/B are the structure factor amplitudes calculated for the separate twin domains A and B, respectively. Rtwin underestimates the residual error in the model over the two twin-related reflections by a factor of ≈0.7. The same expression describes Rtwin/free as calculated for test-set reflections excluded from refinement with CNS.
Per asymmetric unit.
The structure of the human arginase I–BEC complex was solved in similar manner and refined with cns by using group B factors. Data reduction and refinement statistics are recorded in Table 1. Disordered segments at the N and C termini (M1–K4 and P314–K322) were absent in the experimental electron density and are omitted from the final model.
Enzyme Activity and Inhibitor-Binding Assays. Human arginase I activity was measured by using the radioactive assay of Rüegg and Russell (34), as described, for the study of rat arginase I activity (23). Inhibitor affinity was measured by using isothermal titration calorimetry with an MCS isothermal titration calorimeter (Microcal Software, Northampton, MA), and a procedure identical to that described for the assay of rat arginase I–inhibitor affinity (23). For the measurement of cellular arginase activity, cell lysates from murine peritoneal macrophages activated by interleukins 4 and 13 (19), murine tumor-associated myeloid cells (10), and human myeloid suppressor cells isolated from peripheral blood of a patient with renal cell carcinoma (35) were assayed for l-ornithine production. Briefly, cell lysates in the presence or absence of 50 μM ABH were added to 50 μl of 50 mM Tris·HCl (pH 7.5)/10 mM MnCl2 and heated at 55–60°C for 10 min. The hydrolysis of l-arginine to form l-ornithine was identified by a colorimetric assay after the addition of ninhydrin solution and incubation at 37°C for 1 h (35).
Results and Discussion
Enzyme Kinetics and Inhibitor-Binding Affinities. Wild-type human arginase I exhibits KM values for l-arginine hydrolysis of 0.08 mM at pH 8.5 and 0.02 mM at pH 9.5. In comparison, rat arginase I exhibits a KM value of 1.4 mM at pH 9.0 (36). Calorimetric measurement of the dissociation constant for the human arginase I–ABH complex yields Kd = 5.0 nM at pH 8.5 (Fig. 6, which is published as supporting information on the PNAS web site), which is ≈20-fold smaller than that measured for the rat arginase I–ABH complex (37) (Table 2). For the human arginase I–BEC complex, Kd = 270 nM at pH 8.5 (Fig. 7, which is published as supporting information on the PNAS web site), which is nearly 10-fold smaller than that measured for the corresponding complex with rat arginase I (23) (Table 2). It is notable that substrate and inhibitors bind more tightly to human arginase I than to rat arginase I.
Table 2. Arginase I affinities at pH 8.5.
| l-arginine | ABH | BEC | |
|---|---|---|---|
 |
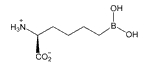 |
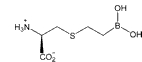 |
|
| Arginase I | KM (mM) | Kd (nM) | Kd (nM) |
| Rat* | 1.4 | 110 | 2,220 |
| Human | 0.08 | 5.0 | 270 |
Human Arginase I Structure and Mechanism. The asymmetric unit of the unit cell in the human arginase I–ABH complex contains two monomers from two different trimers. The rms deviation is 0.55 Å for 308 Cα atoms between the human and rat enzymes. The greatest structural difference of 0.71 Å is rather modest and is observed for the polypeptide segments flanking E42 and N69, which contain several mutations.
The binuclear manganese cluster in the human arginase I–ABH complex (Fig. 1a) is nearly identical to that in the rat arginase I–ABH complex (24), and ABH boronate hydroxyl group O1 nearly symmetrically bridges 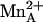 and
and 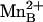 (average metal–oxygen separation = 2.17 Å). However, ABH boronate hydroxyl group O2 makes a true inner-sphere coordination interaction (
(average metal–oxygen separation = 2.17 Å). However, ABH boronate hydroxyl group O2 makes a true inner-sphere coordination interaction (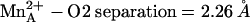 ), which is not quite the case in the rat enzyme complex (
), which is not quite the case in the rat enzyme complex (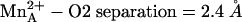 ) or in the human and rat enzyme complexes with BEC (
) or in the human and rat enzyme complexes with BEC (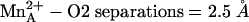 ). This significantly impacts the understanding of
). This significantly impacts the understanding of 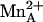 function in catalysis. Because ABH undergoes the same chemistry as the substrate l-arginine in the arginase active site (nucleophilic attack by the metal-bridging hydroxide ion), and because the boronate anion of ABH mimics the tetrahedral intermediate and its flanking transition states in catalysis, ABH boronate hydroxyl group O2 corresponds to the η2-NH2 group of substrate l-arginine. Accordingly, we conclude that
function in catalysis. Because ABH undergoes the same chemistry as the substrate l-arginine in the arginase active site (nucleophilic attack by the metal-bridging hydroxide ion), and because the boronate anion of ABH mimics the tetrahedral intermediate and its flanking transition states in catalysis, ABH boronate hydroxyl group O2 corresponds to the η2-NH2 group of substrate l-arginine. Accordingly, we conclude that 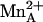 engages the developing sp3 lone electron pair of the η2-NH2 group through a strong inner-sphere bidentate coordination interaction as the transition state is approached (Fig. 2).
engages the developing sp3 lone electron pair of the η2-NH2 group through a strong inner-sphere bidentate coordination interaction as the transition state is approached (Fig. 2).
Fig. 1.

Arginase I-inhibitor complexes. (A) Omit electron density map of the wild-type human arginase I–ABH complex (contoured at 2.5σ). Atoms are color-coded as follows: carbon (yellow), oxygen (red), nitrogen (blue), manganese (pink), and boron (light green). Water molecules appear as red spheres, and manganese ions A and B appear as larger pink spheres. (B) Omit electron density map of the wild-type human arginase I–BEC complex (contoured at 2.5σ). Atoms are color-coded as in a, with sulfur being dark green.
Fig. 2.

Mechanism of human arginase I. New features suggested by the 1.29-Å resolution structure of the complex with ABH are the inner-sphere coordination of η2-NH2 to 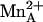 in the tetrahedral intermediate, and the protonation of the amino leaving group of l-ornithine by the conformationally flexible imidazolium group of general acid H141. D128 may also donate a proton to l-ornithine before product release. In the final step of catalysis, the H141 imidazole may serve as a general base to abstract a proton from the metal-bridging water molecule (possibly through an intervening solvent molecule). For clarity, only the side chain guanidinium group of substrate l-arginine and the side chain amino group of product l-ornithine are indicated.
in the tetrahedral intermediate, and the protonation of the amino leaving group of l-ornithine by the conformationally flexible imidazolium group of general acid H141. D128 may also donate a proton to l-ornithine before product release. In the final step of catalysis, the H141 imidazole may serve as a general base to abstract a proton from the metal-bridging water molecule (possibly through an intervening solvent molecule). For clarity, only the side chain guanidinium group of substrate l-arginine and the side chain amino group of product l-ornithine are indicated.
In the structure of inactivated, 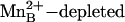 Bacillus caldovelox arginase complexed with l-arginine, Bewley et al. (22) measure an average
Bacillus caldovelox arginase complexed with l-arginine, Bewley et al. (22) measure an average 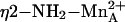 separation of 2.5 Å and conclude that η2-NH2 sp2→sp3 rehybridization facilitates
separation of 2.5 Å and conclude that η2-NH2 sp2→sp3 rehybridization facilitates 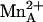 coordination. However, 2.5 Å is somewhat long to be considered inner-sphere metal coordination, so it is unlikely that
coordination. However, 2.5 Å is somewhat long to be considered inner-sphere metal coordination, so it is unlikely that 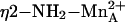 coordination is substantial in the precatalytic enzyme–substrate complex. Moreover, such an interaction is difficult to reconcile with the insensitivity of substrate KM values to perturbations in the binuclear manganese cluster (38).
coordination is substantial in the precatalytic enzyme–substrate complex. Moreover, such an interaction is difficult to reconcile with the insensitivity of substrate KM values to perturbations in the binuclear manganese cluster (38).
Importantly, the structure of the human arginase I–ABH complex provides striking mechanistic inferences on the catalytic function of H141. The imidazole group of H141 is proposed to serve as a proton shuttle that facilitates the ionization of a metal-bridging water molecule to regenerate the nucleophilic metal-bridging hydroxide ion (20). The structure of the human arginase I–ABH complex reveals that H141 donates a hydrogen bond to Oε2 of E277, with an average Nδ–Oε2 separation of 2.83 Å. The carboxylate side chain of E277 also accepts a weak hydrogen bond from boronate hydroxyl O2, but this is longer (average Oε2–O2 separation = 3.22 Å) and more poorly oriented. Significantly, difference electron density maps indicate the presence of a proton on the Nε atom of H141 (Fig. 3). Thus, we conclude that H141 is stabilized with a positively charged imidazolium group by its hydrogen bond with E277. Given the proximity of H141 to the ε-CH2 group of ABH (≈3.5 Å), which corresponds to the leaving amino group of l-ornithine in the tetrahedral intermediate, H141 could serve as a general acid catalyst to donate the proton required for leaving group departure. This function would require an ≈90° conformational change about side chain torsion angle χ2 to orient H141 for proton transfer, which would also acidify the imidazolium proton by disrupting the E277 hydrogen bond (Fig. 2). The H141 imidazole could then serve as a general base/proton shuttle to regenerate the nucleophilic metal-bridging hydroxide ion in the final step of catalysis. The mechanistic function of H141 as a general acid/general base would be consistent with the 5,000-fold loss of catalytic activity measured for the H141L variant of human arginase I identified in a patient suffering from hyperargininemia, a hereditary disease of liver arginase deficiency (39).
Fig. 3.

H141-E277 interaction. Electron density map calculated with Fourier coefficients 2|Fobs|–|Fcalc| (blue, contoured at 1.0σ) indicates the presence of hydrogen atoms on H141 and E277; the Nε-H atom of H141 is confirmed in an omit map contoured at 3.9σ (red). Atoms are colored as in Fig. 2 with hydrogen atoms colored in magenta. Given that Nδ–H donates a hydrogen bond to E277, this provides direct structural evidence for the stabilization of H141 as the positively charged imidazolium cation.
Carboxylate–histidine imidazolium pairs serve as general acids to protonate amino leaving groups in other hydrolytic metalloenzymes. For example, consider the D246–H265 pair in the zinc deacetylase LpxC. The side chain of H265 is protonated at physiological pH and protonates the amino leaving group in the collapse of the tetrahedral intermediate to form UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine and acetate (40, 41). Similarly, a role for H141 of human arginase I is proposed in view of crystallographic evidence for its stabilization in a carboxylate–histidine imidazolium pair. Analysis of all arginase structures determined to date yields H141–E277 interactions ranging 2.8–5 Å, largely due to the conformational flexibility of H141; such flexibility would be consistent with the general acid function contemplated for H141 (K. Jude and D.W.C., unpublished results).
As previously noted, the substrate l-arginine and the inhibitors ABH and BEC bind more tightly to human arginase I than to rat arginase I (Table 2). Given the potential pharmaceutical importance of the human enzyme, it is important to understand the structural basis of its higher affinity for ligands. Using the 1.29-Å resolution structure of the human arginase I–ABH complex and the 1.70-Å resolution structure of the rat arginase I–ABH complex (24) as the basis for a detailed molecular comparison, it is clear that many enzyme–inhibitor hydrogen bond interactions are slightly shorter in the active site of the human enzyme (Fig. 4). Additionally, the metal coordination interactions of the ABH boronate anion are also slightly different in the human and rat arginase I complexes, in that boronate hydroxyl group O2 is more strongly coordinated to 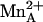 (as previously discussed). We conclude that multiple shorter hydrogen bond interactions with the α-amino and α-carboxylate groups of substrate and inhibitors are consistent with higher-affinity binding to the human enzyme compared with the rat enzyme.
(as previously discussed). We conclude that multiple shorter hydrogen bond interactions with the α-amino and α-carboxylate groups of substrate and inhibitors are consistent with higher-affinity binding to the human enzyme compared with the rat enzyme.
Fig. 4.

Arginase I–ABH complexes. Summary of average intermolecular interactions in the human arginase I–ABH complex (gray numbers) and the rat arginase I–ABH complex (orange numbers). Note that the higher resolution of the human arginase I–ABH structure allows for more accurate hydrogen bond distance measurements. Manganese coordination interactions are indicated by green dashed lines, and hydrogen bonds are indicated by black dashed lines. Several slightly shorter and stronger enzyme-inhibitor hydrogen bond interactions are observed in the human enzyme, consistent with its enhanced affinity for ABH (Table 2).
The structure of the human arginase I–BEC complex is similar to that of the arginase I–ABH complex. An omit electron-density map of this complex is shown in Fig. 1b. Some differences are evident in the binding of BEC to the active sites of human arginases I and II. The α-carboxylate and α-amino groups of BEC are anchored to the active site of human arginase I by three direct and four water-mediated hydrogen bonds, whereas these interactions consist of two direct and five water-mediated hydrogen bonds in the human arginase II complex (21). By inference, then, water-mediated hydrogen bonds make the majority of interactions with the α-carboxylate and α-amino group of the substrate l-arginine. Differences among these interactions in human arginases I and II may provide clues to the design of isozyme-specific inhibitors.
Arginase I in the Immune Response. Inhibition of arginase activity was tested in cytoplasmic extracts of murine and human cells known to have high levels of arginase activity. These include murine peritoneal macrophages stimulated with interleukins 4 and 13 (19), myeloid suppressor cells infiltrating murine 3LL lung carcinoma (10), and myeloid suppressor cells from the peripheral blood of a cancer patient (35). The results shown in Fig. 5 demonstrate that 50 μM ABH significantly inhibited arginase activity of the murine cells and completely inhibited human arginase function. That human arginase I activity is so sensitive to the presence of ABH may reflect the higher affinity of ABH for the human enzyme.
Fig. 5.

Arginase activity in the immune response. Measurements of arginase activity as a function of l-ornithine production in cytoplasmic extracts of murine peritoneal macrophages (PM) activated with interleukins 4 and 13 (IL4/IL13), murine tumor-associated myeloid cells (TAMC), and human myeloid suppressor cells (MSC). Notably, murine arginase activity is significantly attenuated, and human arginase activity is obliterated in the presence of 50μM ABH.
Conclusion
It is clear that arginase I plays a significant role in modulating the immune response, and previous work demonstrates that arginase inhibition blocks the growth of lung carcinoma in a murine model (10, 18, 19). Here, we demonstrate that the boronic acid substrate analogue ABH is a highly potent inhibitor of human arginase I. Therefore, ABH may be a potentially useful agent for blocking tumor cell growth, and preliminary experiments in our laboratory demonstrate that ABH attenuates proliferation in estrogen receptor-negative human breast cancer cell line MDA-MB-468 (H. A. Gennadios and D.W.C., unpublished results). Analysis of the 1.29-Å resolution structure of the human arginase I–ABH complex reveals that nanomolar affinity is a consequence of bidentate inner-sphere boronate–manganese coordination interactions and strong enzyme–inhibitor hydrogen bond networks. Such structure-affinity relationships are important to consider as arginase I is explored as a potential target for new therapies directed toward inflammatory and immunological disorders and especially so as boronic acid-based inhibitors establish a niche in the greater family of enzyme-targeted drugs (42).
Supplementary Material
Acknowledgments
We thank the Cornell High Energy Synchrotron Source and National Synchrotron Light Source at the Brookhaven National Laboratory (beamline X12B) for beamline access. This work was supported by National Institutes of Health Research Grants GM49758 (to D.W.C.) and CA107974 (to A.C.O.).
Author contributions: L.D.C., G.S., A.M., P.C.R., A.C.O., F.C., and D.W.C. designed research, performed research, contributed new reagents/analytic tools, analyzed data, and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TH1/2, T-helper 1/2; ABH, 2(S)-amino-6-boronohexanoic acid; BEC, S-(2-boronoethyl)-l-cysteine.
Data deposition: The atomic coordinates and structure factors for human arginase I complexed with ABH and BEC have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2aeb and 1wva, respectively).
References
- 1.Christianson, D. W. (2005) Acc. Chem. Res. 101, 191–201. [DOI] [PubMed] [Google Scholar]
- 2.Ash, D. E., Cox, J. D. & Christianson, D. W. (2000) Met. Ions Biol. Syst. 37, 407–428. [PubMed] [Google Scholar]
- 3.Morris, S. M., Jr. (2002) Ann. Rev. Nutr. 22, 87–105. [DOI] [PubMed] [Google Scholar]
- 4.Corraliza, I. M., Soler, G., Eichmann, K. & Modolell, M. (1995) Biochem. Biophys. Res. Commun. 206, 667–673. [DOI] [PubMed] [Google Scholar]
- 5.Wang, W. W., Jenkinson, C. P., Griscavage, J. M., Kern, R. M., Arabolos, N. S., Byrns, R. E., Cederbaum, S. D. & Ignarro, L. J. (1995) Biochem. Biophys. Res. Commun. 210, 1009–1016. [DOI] [PubMed] [Google Scholar]
- 6.Boutard, V., Havouis, R., Fouqueray, B., Phillipe, C., Moulinoux, J.-P. & Baud, L. (1995) J. Immunol. 155, 2077–2084. [PubMed] [Google Scholar]
- 7.Modolell, M., Corraliza, I. M., Link, F., Soler, G. & Eichmann, K. (1995) Eur. J. Immunol. 25, 1101–1104. [DOI] [PubMed] [Google Scholar]
- 8.Albina, J. E., Abate, J. A. & Mastrofrancesco, B. (1993) J. Surg. Res. 55, 97–102. [DOI] [PubMed] [Google Scholar]
- 9.Keller, R., Gehri, R., Keist, R., Huf, E. & Kayser, F. H. (1991) Cell Immunol. 134, 249–256. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez, P. C., Quiceno, D. G., Zabaleta, J., Ortiz, B., Zea, A. H., Piazuelo, M. B., Delgado, A., Correa, P., Brayer, J., Sotomayor, E. M., et al. (2004) Cancer Res. 6 4, 5839–5849. [DOI] [PubMed] [Google Scholar]
- 11.Xu, L., Hilliard, B., Carmody, R. J., Tsabary, G., Shin, H., Christianson, D. W. & Chen, Y. H. (2003) Immunology 110, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann, N., King, N. E., Laporte, J., Yang, M., Mishra, A., Pope, S. M., Muntel, E. E., Witte, D. P., Pegg, A. A., Foster, P. S., et al. (2003) J. Clin. Invest. 111, 1863–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Endo, M., Oyadomari, S., Terasaki, Y., Takeya, M., Suga, M., Mori, M. & Gotoh, T. (2003) Am. J. Physiol. Lung Cell Mol. Physiol. 285, 313–321. [DOI] [PubMed] [Google Scholar]
- 14.Morris, C. R., Poljakovic, M., Lavrisha, L., Machado, L., Kuypers, F. A. & Morris, S. M., Jr. (2004) Am. J. Respir. Crit. Care Med. 170, 148–153. [DOI] [PubMed] [Google Scholar]
- 15.Singh, R., Pervin, S., Karimi, A., Cederbaum, S. & Chaudhuri, G. (2000) Cancer Res. 60, 3305–3312. [PubMed] [Google Scholar]
- 16.Chang, C.-I., Liao, J. C. & Kuo, L. (2001) Cancer Res. 61, 1100–1106. [PubMed] [Google Scholar]
- 17.Leu, S.-Y. & Wang, S.-R. (1992) Cancer 70, 733–736. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez, P. C., Zea, A. H., Culotta, K. S., Zabaleta, J., Ochoa, J. B. & Ochoa, A. C. (2002) J. Biol. Chem. 277, 21123–21129. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez, P. C., Zea, A. H., DeSalvo, J., Culotta, K. S., Zabaleta, J., Quiceno, D. G., Ochoa, J. B. & Ochoa, A. C. (2003) J. Immunol. 171, 1232–1239. [DOI] [PubMed] [Google Scholar]
- 20.Kanyo, Z. F., Scolnick, L. R., Ash, D. E. & Christianson, D. W. (1996) Nature 383, 554–557. [DOI] [PubMed] [Google Scholar]
- 21.Cama, E., Colleluori, D. M., Emig, F. A., Shin, H., Kim, S. W., Kim, N. N., Traish, A. M., Ash, D. E. & Christianson. D. W. (2003) Biochemistry 42, 8445–8451. [DOI] [PubMed] [Google Scholar]
- 22.Bewley, M. C., Jeffrey, P. D., Patchett, M. L., Kanyo, Z. F. & Baker, E. N. (1999) Structure (London) 7, 435–448. [DOI] [PubMed] [Google Scholar]
- 23.Kim, N. N., Cox, J. D., Baggio, R. F., Emig, F. A., Mistry, S. K., Harper, S. L., Speicher, D. W., Morris, S. M., Jr., Ash, D. E. & Traish, A., et al. (2001) Biochemistry 40, 2678–2688. [DOI] [PubMed] [Google Scholar]
- 24.Cox, J. D., Kim, N. N., Traish, A. M. & Christianson, D. W. (1999) Nat. Struct. Biol. 6, 1043–1047. [DOI] [PubMed] [Google Scholar]
- 25.Colleluori, D. M. & Ash, D. E. (2001) Biochemistry 40, 9356–9362. [DOI] [PubMed] [Google Scholar]
- 26.Haraguchi, Y., Takiguchi, M., Amaya, Y., Kawamoto, S., Matsuda, I. & Mori., M. (1987) Proc. Natl. Acad. Sci. USA 84, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mora, A., del Ara Rangel, M., Fuentes, J. M., Soler, G. & Centeno, F. (2000) Biochim. Biophys. Acta 1476, 181–190. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 306–326. [DOI] [PubMed] [Google Scholar]
- 29.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 30.Redinbo, M. R. & Yeates, T. O. (1993) Acta Crystallogr. D 49, 375–380. [DOI] [PubMed] [Google Scholar]
- 31.Yeates, T. O. (1997) Methods Enzymol. 276, 344–358. [PubMed] [Google Scholar]
- 32.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 33.Sheldrick, G. M. & Schneider, T. R. (1997) Methods Enzymol. 277, 319–343. [PubMed] [Google Scholar]
- 34.Rüegg, U. T. & Russell, A. S. (1980) Anal. Biochem. 102, 206–212. [DOI] [PubMed] [Google Scholar]
- 35.Zea, A. H., Rodriguez, P. C., Atkins, M. B., Hernandez, C., Signoretti, S., Zabaleta, J., McDermott, D., Quiceno, D., Youmans, A., O'Neill, A., et al. (2005) Cancer Res. 65, 3044–3048. [DOI] [PubMed] [Google Scholar]
- 36.Cavalli, R. C., Burke, C. J., Kawamoto, S., Soprano, D. R. & Ash, D.E. (1994) Biochemistry 33, 10652–10657. [DOI] [PubMed] [Google Scholar]
- 37.Baggio, R., Cox, J. D., Harper, S. L., Speicher, D. W. & Christianson, D. W. (1999) Anal. Biochem. 276, 251–253. [DOI] [PubMed] [Google Scholar]
- 38.Cama, E., Emig, F. A., Ash, D. E. & Christianson, D. W. (2003) Biochemistry 42, 7748–7758. [DOI] [PubMed] [Google Scholar]
- 39.Vockley, J. G., Goodman B. K., Tabor, D. E., Kern, R. M., Jenkinson, C. P., Grody, W. W. & Cederbaum, S. D. (1996) Biochem. Mol. Med. 59, 44–51. [DOI] [PubMed] [Google Scholar]
- 40.Coggins, B. E., McClerren, A. L., Jiang, L., Li, X., Rudolph, J., Hindsgaul, O., Raetz, C. R. & Zhou, P. (2005) Biochemistry 44, 1114–1126. [DOI] [PubMed] [Google Scholar]
- 41.Hernick, M., Gennadios, H. A., Whittington, D. A., Rusche, K. M., Christianson, D. W. & Fierke, C. A. (2005) J. Biol. Chem. 280, 16969–16978. [DOI] [PubMed] [Google Scholar]
- 42.Robertson, J. G. (2005) Biochemistry 44, 5561–5571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.