

Abstract
Protein aggregates are associated with numerous diseases. Here we report a platform for the rapid phenotypic selection of protein aggregation inhibitors from genetically encoded cyclic peptide libraries in E. coli based on phage-assisted continuous evolution (PACE). We developed a new PACE-compatible selection for protein aggregation inhibition and employed it to identify cyclic peptides that suppress amyloid-β42 (Aβ42) and human islet amyloid polypeptide (hIAPP) aggregation. Additionally, we integrated a negative selection that removes false positives and off-target hits, significantly improving cyclic peptide selectivity. We show that selected inhibitors are active when chemically re-synthesized in in vitro assays. Our platform provides a powerful approach for the rapid discovery of cyclic peptide inhibitors of protein aggregation and may serve as the basis for the future evolution of cyclic peptides with a broad spectrum of inhibitory activities.
Graphical Abstract

Introduction
The aggregation of amyloidogenic proteins has been associated with numerous human diseases.1 These proteins proceed through a series of misfolded intermediates to ultimately form the ordered, fibrillar structures known as amyloids, motivating the search for compounds that can disrupt their formation. However, structure-guided design of protein misfolding and aggregation inhibitors is complicated by the intrinsically disordered nature of many amyloidogenic species and the dearth of structural information on amyloid precursors.2,3
Inhibitor discovery by high-throughput screening or selection often requires minimal structural information, providing a powerful alternative to rational design.4 A recent and exciting example is the screening of small molecule libraries for their effects on the kinetics of amyloid formation in vitro using purified protein.5 Such screens have identified promising leads,6 but nevertheless can be limited by throughput and the technical challenges of producing high-quality target protein to ensure reproducibility.5 Additionally, small molecules can be poorly suited for binding tightly to disordered misfolded species7 or strongly inhibiting the protein-protein interactions involved in misfolding and aggregation.8
Macrocyclic peptides have increasingly been recognized as a promising source of chemical probes and potential therapeutics.9–11 These compounds can bind historically challenging targets, such as large or shallow protein surfaces, and enjoy improved stability and binding potency due to backbone rigidification.12,13 Platforms that select genetically encoded cyclic peptide libraries can search through an enormous number of sequences for ones that possess desired bioactivity. For example, mRNA and phage display methods have proven highly successful in identifying macrocyclic inhibitors for numerous targets.13,14 However, display techniques are typically constrained to identifying binders of immobilized proteins.13 A complementary approach, enabled by the rapidly emerging field of synthetic biology, involves the genetic or phenotypic selection of ribosomally synthesized cyclic peptide libraries in cells.15–18 Notably, this strategy can identify cyclic peptides with activities beyond single target binding,13 including inhibiting protein aggregation.19–21 In a groundbreaking example, the Lindquist group identified inhibitors of α-synuclein aggregation-induced toxicity by selecting cyclic peptides biosynthesized using the split-intein circular ligation of peptides and proteins (SICLOPPS) method15 in a yeast synucleinopathy model.19 More recently, the Skretas group used fluorescence-activated cell sorting to select a SICLOPPS library for rescue of a GFP folding reporter in E. coli to identify cyclic peptide inhibitors of amyloid-β (Aβ) and mutant superoxide dismutase aggregation.21,22
While powerful, these strategies for identifying cyclic peptide protein aggregation inhibitors are coupled to conventional selections such as cellular survival or fluorescence-based sorting, which are time- and labor-intensive and, in the case of cell sorting, require specialized equipment.23 Additionally, selections can suffer from false positives or uncover hits with undesirable properties,14 such as promiscuity or off-target activity. For example, the majority of hits selected in the Lindquist study were deemed to be false positives either arising from spontaneous mutations in the yeast strain or from off-target activity of the cyclic peptides themselves.19 Weeding out these false positive and off-target sequences then requires additional experiments, adding to time and labor.
To address these limitations, we have developed a platform for selecting cyclic peptide protein aggregation inhibitors based on phage-assisted continuous evolution (PACE).24 By linking cyclic peptide activity to phage reproduction, we can perform 1–2 rounds of selection per day, leading to rapid discovery of active sequences using only standard molecular biology techniques and equipment. We demonstrate the utility of our system by using it to identify cyclic peptide sequences that inhibit the aggregation of disease-associated proteins amyloid-β42 (Aβ42) and human islet amyloid polypeptide (hIAPP). We also report the implementation of a negative selection that purges hits with off-target activity, resulting in selective cyclic peptide sequences. Finally, we show that our identified hIAPP inhibitors are active in in vitro amyloid formation assays. We envision that the reported system will improve the speed and convenience of cellular cyclic peptide selections and complement existing strategies for identifying inhibitors of disease-associated protein aggregation.
Results
PACE and PACE-related systems24–29 use engineered M13 bacteriophage (selection phage, or SP) that carry the gene of interest (GOI) under selection in place of phage gene III (gIII). gIII encodes for protein III (pIII), which is essential for both infection of and viral escape from the E. coli host cell. A genetic selection carried on an “accessory plasmid” in the host cells provides pIII in response to some researcher-defined activity, linking phage reproduction to selection of the GOI for a desired trait. Finally, optional inclusion of a mutagenesis plasmid enables continuous diversification of the GOI. PACE and its related techniques differ from conventional directed evolution methods by their ability to explore large library sizes and greatly accelerate selection timelines. Non-continuous variants, such as PANCE (phage-assisted non-continuous evolution) and PANCS (phage-assisted non-continuous selection),25,27 deliver rapid selection without the need for specialized continuous flow equipment and are accessible to any laboratory with basic molecular biology infrastructure.
PACE and related techniques have been used to evolve a wide range of proteins,28 but to our knowledge have not been applied to cyclic peptides. Here, we envisioned a general strategy for phage-assisted cyclic peptide discovery, shown in Figure 1. We start with an SP-encoded cyclic peptide library generated by standard cloning techniques. This pool of SP is used to infect E. coli host cells with an accessory plasmid that links desired cyclic peptide activity with pIII expression. Upon infection, the E. coli biosynthesizes all SP-encoded genes, including the cyclic peptide. If the cyclic peptide possesses the activity under selection, it triggers pIII expression from the host cell, enabling phage propagation. Thus, only SP that carry active cyclic peptides enrich in the population.
Figure 1. PACE-based strategy for cyclic peptide discovery.

Selection phage (SP) carrying a library of genetically encoded cyclic peptides are used to infect host E. coli cells transformed with an “accessory plasmid.” The accessory plasmid encodes a selection that links a user-defined inhibitory activity to expression of phage protein pIII, which is required for phage reproduction. Upon infection, the cyclic peptide library member is expressed from the transduced SP genome and, if active, triggers production of pIII from the accessory plasmid. As a result, only SP that carry active library members propagate, allowing rapid enrichment of desired cyclic peptide sequences.
PACE-compatible selection for protein aggregation inhibitors
Previous strategies for selecting protein misfolding inhibitors in cells have utilized cell viability4,19,30,31 or signal from fluorescent protein fusions.22,32 These approaches cannot translate to PACE, which requires the selected activity to be coupled to gIII expression. Therefore, we turned to a split-T7 RNA polymerase (T7 RNAP) reporter used to evolve proteins with improved soluble expression.33 In our selection, host E. coli express an aggregation-prone protein (APP) fused to the N-terminal half of split-T7 RNAP (T7n). When the APP-T7n fusion is translated, it misfolds due to the APP, preventing T7n from associating with the C-terminal half of split-T7 RNAP (T7c; Figure 2a). If a SP-encoded inhibitor binds to and prevents APP aggregation, the fused T7n will remain folded and join with T7c to produce full-length T7 RNAP. The reconstituted T7 RNAP then transcribes gIII, which we place under control of the T7 promoter (PT7), producing pIII and allowing the SP to reproduce (Figure 2a–b).
Figure 2. Design of a selection for cyclic peptide protein aggregation inhibitors.
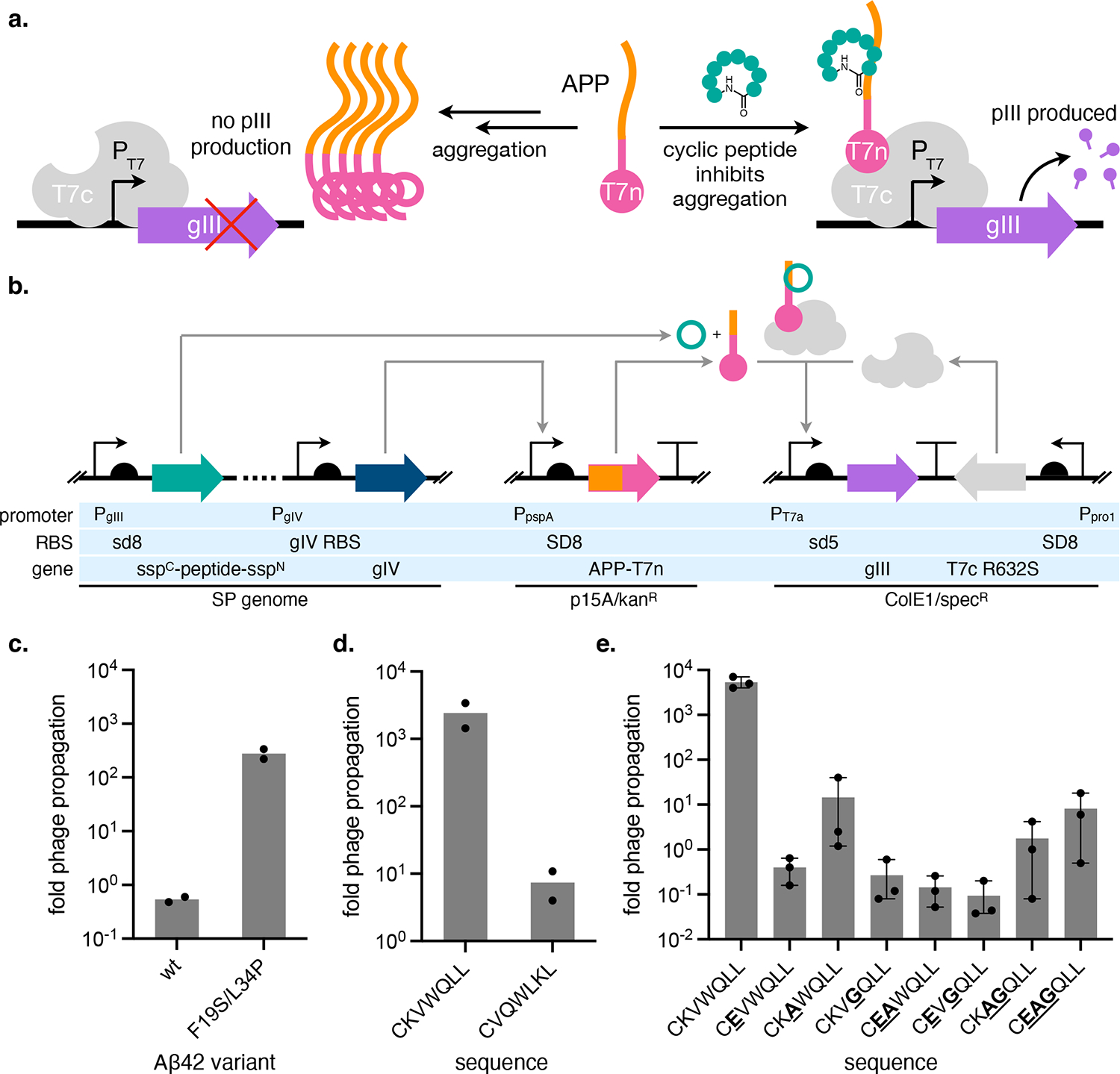
(a) Cartoon overview of a split T7 RNAP folding reporter strategy for identifying protein aggregation inhibitors. APP = aggregation-prone protein, T7n and T7c = N- and C-terminal halves of split-T7 RNAP, respectively. (b) Diagram of the genetic circuits that constitute the protein aggregation inhibitor selection. (c) Propagation activity of kanR-encoding phage on host cells encoding the APP-T7n selection, where the APP is either Aβ42 or its non-aggregating F19S/L34P mutant. (d) Comparison of phage propagation activities of SP carrying Aβ42 aggregation inhibitor cyclo-CKVWQLL or scrambled negative control cyclo-CVQWLKL on host cells encoding the Aβ42-T7n selection. (c-d) Data reflect two biological replicates plotted as individual values. (e) Effect of the mutation of key residues in the cyclo-CKVWQLL sequence on SP propagation on host cells encoding the Aβ42-T7n selection. Data reflects mean and standard error (s.e.) of three (e) biological replicates. Fold phage propagation is calculated as the number of phage generated from an infected culture divided by the number of phage (105) used to infect the culture.
We initially targeted amyloid-β42 (Aβ42) peptide, which is implicated in Alzheimer’s Disease, as the APP. Our choice was motivated by the recent identification of a cyclic peptide inhibitor of Aβ42 aggregation, cyclo-CKVWQLL,22,32 that would provide a valuable control for optimizing and benchmarking our system. When fused to T7n, Aβ42 should rapidly aggregate and prevent T7 RNAP reconstitution. Accordingly, this fusion produces low gIII transcription from PT7, and SP carrying a control gene (kanR) reproduce poorly on host cells encoding the selection circuit (Figure 2c). For comparison, we tested a fusion of a non-aggregating Aβ42 mutant (F19S/L34P),34 which gave rise to a ~500-fold increase in propagation of kanR SP (Figure 2c). These results suggest that the split-T7 RNAP folding reporter can translate the degree of Aβ42 aggregation to changes in gIII transcription.
Selection of SP-encoded SICLOPPS cyclic peptides
Next, we determined if our selection could discriminate between SP encoding cyclic peptides that inhibit Aβ42 aggregation and SP carrying inactive cyclic peptides. We used the SICLOPPS method to biosynthesize cyclic peptides that can be selected in E. coli.15 In SICLOPPS, a precursor fusion consisting of the C-terminal half of the Ssp DnaE split intein, the peptide sequence to be cyclized, and the N-terminal half of the same split intein undergoes intein splicing after translation to produce head-to-tail cyclized peptides (Supplementary Figure 1). To benchmark our system, we used positive control sequence cyclo-CKVWQLL, identified from a SICLOPPS cyclic peptide library to inhibit intracellular Aβ42-GFP aggregation and shown to be active when chemically re-synthesized in both in vitro and C. elegans Aβ42 aggregation models.22
We measured the reproduction of SP carrying SICLOPPS precursors for cyclo-CKVWQLL or a scrambled control cyclo-CVQWLKL on E. coli encoding the Aβ42-T7n selection (Figure 2b). CKVWQLL SP exhibited significantly increased (~500-fold) proliferation compared to SP carrying the scrambled control (Supplementary Figure 2a). However, we were concerned that the low overall phage amplifications observed (0.01- to 10-fold) could preclude selection of active cyclic peptides from a library. We thought that cyclic peptide maturation might not be fast enough to trigger pIII production during the SP life cycle (~15 min). Alternatively, SP backbone optimization could be required to coordinate the phage life cycle with our selection, which has been observed previously and can impose a prerequisite for implementing new PACE selections.28,35,36 To improve the overall fitness of cyclic peptide-encoding phage, we evolved the parent CKVWQLL SP on Aβ42-T7n selection cells in PACE (Supplementary Figure 2b). We chose SP clone 2 (Supplementary Figure 2c) for downstream experiments. This evolved SP exhibited significantly increased overall proliferation compared to the parent (Supplementary Figure 2d) yet maintained excellent (~300-fold) discrimination between CKVWQLL and scrambled control (Figure 2d). Installing point mutations at residues important for CKVWQLL activity22 produced decreases in SP propagation, further supporting dependence of phage fitness on cyclic peptide sequence (Figure 2e).
Cyclic peptide formation by SICLOPPS occurs through intein splicing. We tested whether phage propagation was splicing dependent using CKVWQLL SP where the first cysteine in the N-terminal intein was mutated to alanine, preventing formation of the first splicing intermediate (Supplementary Figure 1). Unexpectedly, intein inactivation did not impact CKVWQLL SP propagation (Supplementary Figure 2d), suggesting that both intein-bound and mature cyclic peptide forms of this sequence are active. The lack of splicing dependence could not be explained by our evolved SP because the unevolved SP also exhibited splicing-independent activity (Supplementary Figure 2d). Additionally, it was not an artifact of our phage-based selection, as we also observed it in experiments testing plasmid-expressed SICLOPPS precursors on an Aβ42-GFP folding reporter analogous to the one used to identify the CKVWQLL sequence (Supplementary Figure 2e–f).22 This suggests that, when selecting SICLOPPS libraries, the species undergoing selection may exist as a mixture of pre-splice, splicing intermediate, and spliced products. However, SICLOPPS libraries have repeatedly produced cyclic peptides that function when chemically re-synthesized and tested in orthogonal assays,20,21,37,38 suggesting that this ambiguity does not prevent the discovery of active hits.
Next, we tested whether we could select active cyclic peptides from a library of randomized sequences by subjecting a cyclo-CX6 SP library (X = NNK encoded amino acid) to PANCS27 using host cells encoding the Aβ42-T7n selection circuit (Figure 3a). A total of ~3 × 106 transformants was selected over seven rounds, over which we observed an increase in SP pool fitness (measured by fold phage propagation on the selection cells), suggesting enrichment of active cyclic peptide sequences (Figure 3b). Deep sequencing revealed that the final SP pool had strongly converged, with the top sequence (CRVWCAR) accounting for ~65% of total sequencing reads (Figure 3c). Alignment of the top 50 sequences (Supplementary Figure 3a) revealed high similarity to cyclo-CKVWQLL, while the top sequence (CRVWCAR) bears strong resemblance to sequences highly enriched by the selection that identified cyclo-CKVWQLL in a prior study (ex. CRVWCAL and CRVWQAL).20
Figure 3. Selection of cyclic peptide Aβ42 aggregation inhibitors.

(a) Graphical overview of selection procedure. (b) Phage titers (solid lines, plotted on left y-axis) and propagation activity (dashed lines, plotted on right y-axis) of successive rounds of selection of an SP-encoded cyclo-CX6 library on selection cells encoding Aβ42 as the target APP. “In” refers to the input SP pool used to infect selection cells and “out” refers to the output SP pool obtained after selection, for a given round of selection. pfu = plaque-forming units. (c) Top 10 enriched cyclic peptide sequences after 7 rounds of selection on Aβ42 as determined by high-throughput sequencing analysis. (d) Propagation activity of clonal phage encoding enriched sequences CRVWCAR and CRVYQVL on host cells encoding the Aβ42-T7n selection. Phage encoding active sequence CKVWQLL is shown for comparison. Data reflect two biological replicates plotted as individual values. (e) Propagation activity of clonal phage encoding single alanine substitutions of the CRVWCAR sequence on host cells encoding the Aβ42-T7n selection. (f) Evaluation of cyclic peptide sequences enriched by selection on Aβ42-T7n expressed from plasmid using the Aβ42-GFP folding reporter. CKVWQLL is shown for comparison. Cyclic peptides are induced at 0.04 mM IPTG. (e-f) Data reflects mean and s.e. of three biological replicates. (b, d-e) Fold phage propagation is calculated as the number of phage generated from an infected culture divided by the number of phage used to infect the culture. For (d-e), 105 input phage were used to infect.
We measured the fitness of clonal phage encoding the top two sequences, CRVWCAR and CRVYQVL, on the Aβ42-T7n selection (Figure 3d). SP carrying CRVWCAR exhibited robust propagation, comparable to that of CKVWQLL SP, while SP bearing CRVYQVL displayed more modest fitness. We also probed the sequence dependence of CRVWCAR through alanine substitutions (Figure 3e). Mutation of residues 2–4 to alanine greatly diminished activity, consistent with the strong enrichment of R, V, and W at these positions by our selection (Supplementary Figure 3a). However, substitution of the first cysteine with alanine, which prevents SICLOPPS splicing, had no discernable effect on Aβ42-T7n inhibition. This is consistent with our observations that CKVWQLL activity is also not splicing-dependent (Supplementary Figure 2d–f). Finally, we compared the activity of our sequences with CKVWQLL in the Aβ42-GFP folding reporter assay (Figure 3f). CRVWCAR had comparable activity to CKVWQLL, while CRVYQVL appeared inactive. However, induction of the CRVYQVL construct greatly decreased cell growth (Supplementary Figure 3b), which may convolute rescue of Aβ42-GFP fluorescence.
Together, these results suggest that our system can select for active cyclic peptide sequences from SP-encoded libraries.
Effect of continuous mutagenesis
In PACE, mutagenesis plasmids (ex. MP6) can diversify libraries beyond what is initially obtained through cloning.39 We tested the effect of continuous mutagenesis by selecting an SP-encoded cyclo-CZ6 SICLOPPS library (Z = NDT codon encoded amino acid: R, N, D, C, G, H, I, L, F, S, Y, or V) (~3 × 106 total sequences) for Aβ42-T7n aggregation inhibitors with MP6 (Supplementary Figure 4a). NDT codons exclude tryptophan, which is strongly represented in 7-mer cyclic peptides targeting Aβ42 (Supplementary Figure 3a and Ref20); thus, we wondered if MP6 could enable discovery of tryptophan-containing sequences not present in the starting library. Sequencing after four rounds of selection revealed convergence to sequences resembling CKVWQLL (Supplementary Figure 4b–d). However, the sequences from the +MP6 pool were highly similar to those from the -MP6 pool (Supplementary Figure 4b–c), and we did not observe enrichment of sequences outside of the NDT degenerate codon set used to clone the starting library. Therefore, MP6 had minimal effect on the outcome of this selection, potentially because the starting library already possessed active sequences.
Selection of cyclic peptide hIAPP aggregation inhibitors
Next, we examined if our system could discover inhibitors of other disease-associated APPs. We targeted human islet amyloid polypeptide (hIAPP), which forms amyloid deposits in patients with type-2 diabetes that are hypothesized to contribute to β-cell dysfunction.40 To create a selection for hIAPP aggregation inhibitors, we simply cloned hIAPP in place of Aβ42 in our selection circuit (Figure 2b). We used an SP-encoded CZ6 SICLOPPS library (Z = NDT codon encoded amino acid: R, N, D, C, G, H, I, L, F, S, Y, or V) (~3 × 106 total sequences). This codon set was chosen to reduce the theoretical library size and facilitate library cloning. The library (total of ~5 × 106 transformants) was cloned on a SP obtained from an additional 250 hours of PACE using a more stringent selection (Supplementary Figure 5 and Supplementary Note 1). This switch to a higher stringency selection and, correspondingly, a further evolved SP capable of performing at higher stringencies, was necessitated by the increased background produced by hIAPP-T7n relative to Aβ42-T7n (Supplementary Figure 6a).
We performed five rounds of selection, diluting the SP pool by ten-fold each round (Figure 4a). There was a modest increase in SP pool fitness over the course of the selection (~35-fold). To verify that we selected phage with higher activity, we measured the propagation of the SP from each round on hIAPP selection cells, normalizing input phage titers to 105 pfu/mL to enable head-to-head comparison between phage pools. We observed a ~50-fold increase in SP pool fitness (Figure 4b), suggesting enrichment of active cyclic peptides by the selection. Deep sequencing of the SP pool from round 5 revealed that a single sequence (CHVVGVI) accounted for ~95% of sequencing reads (Figure 4c–d), with the second most enriched sequence, CHVHSYL, exhibiting some similarity to the top hit. Aside from these two sequences, we did not observe any clear trends in the remaining enriched cyclic peptides.
Figure 4: Selection of cyclic peptide inhibitors of hIAPP aggregation.
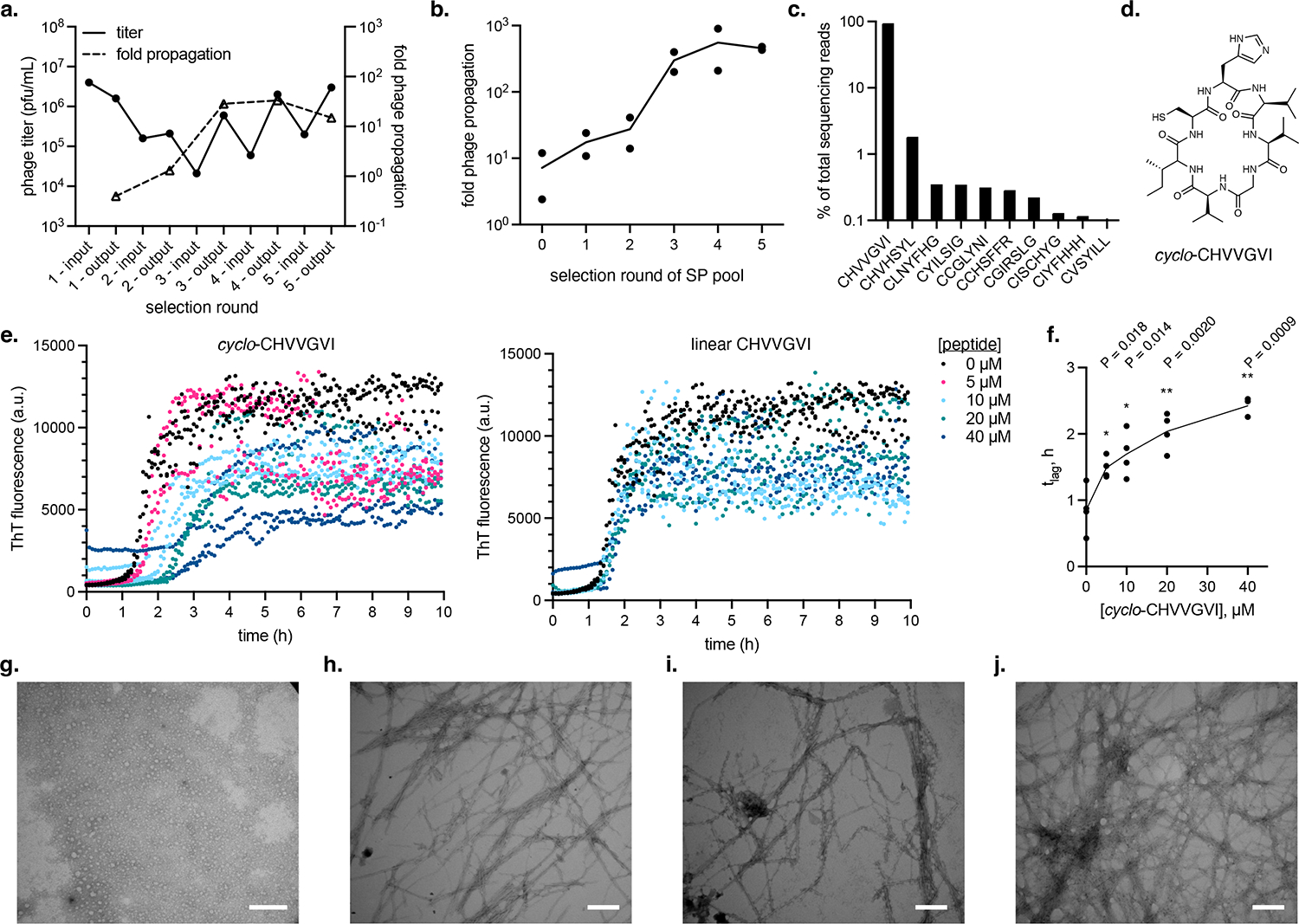
(a) Phage titer (solid line, plotted on left y-axis) and propagation activity (dashed line, plotted on right y-axis) of successive rounds of selection of an SP-encoded SICLOPPS library on selection cells targeting hIAPP aggregation. “In” refers to the input SP pool used to infect selection cells and “out” refers to the output SP pool obtained after selection, for a given round of selection. pfu = plaque-forming units. Fold phage propagation is calculated as the number of phage generated from an infected culture divided by the number of phage used to infect the culture. (b) Propagation activity of SP pools from each round of the selection shown in (a) where input phage titers are normalized to 105 pfu/mL. Data reflect two biological replicates plotted as individual values. (c) Top 10 enriched cyclic peptide sequences after 5 rounds of selection on hIAPP as determined by high-throughput sequencing analysis. (d) Structure of cyclo-CHVVGVI. (e) Effect of varying concentrations of chemically synthesized cyclo-CHVVGVI (left) or linear CHVVGVI (right) on aggregation of 10 μM hIAPP measured by ThT fluorescence. Assay was run in phosphate-buffered saline (PBS) pH 6.9 at 32 °C under quiescent conditions. Data reflect plots of individual values of three technical replicates. The hIAPP alone (0 μM peptide) data is the same in both plots. (f) Effect of cyclo-CHVVGVI on lag time (tlag) of hIAPP aggregation. Data reflects tlag fits from four biological replicates (4 separate ThT assays; see Supplementary Figure 7) plotted as individual values. P-values (two-tailed Student’s t-test between 0 μM peptide and each peptide concentration): *, P ≤ 0.05; **, P ≤ 0.01. (g-h) TEM analysis of 10 μM hIAPP incubated in the presence (g) of 40 μM cyclo-CHVVGVI or alone (h) for for 2 h. (i-j) TEM analysis of 10 μM hIAPP in the presence (i) of 40 μM cyclo-CHVVGVI or alone (j) for 14 h. (g-j) For compatibility with TEM, hIAPP incubations were run in 25 mM sodium phosphate buffer, pH 7.4; see Supplementary Figure 13 for hIAPP aggregation kinetics under TEM assay conditions. Scale bars = 200 nm. This experiment was not repeated.
To further investigate the outcome of this selection, we compared the activity of SP encoding six sequences from the post-selection pool and three sequences from the starting library but not detected in the post-selection pool (Supplementary Figure 6b). All six SP encoding post-selection sequences exhibited similarly high fitness on the hIAPP-T7n selection. In contrast, SP encoding sequences from the starting library were significantly less active. Alanine scanning of the dominant CHVVGVI sequence revealed that no single position produced large decreases in SP fitness when mutated (Supplementary Figure 6c). Together, these results indicate that we enriched for sequences with increased fitness on the hIAPP-T7n selection, but also suggest that this selection exhibits a lower stringency compared to the Aβ42-T7n selection.
We synthesized both cyclo- and linear CHVVGVI (Figure 4d) and measured their effects on hIAPP aggregation in vitro. cyclo-CHVVGVI inhibited hIAPP aggregation in thioflavin T (ThT) fluorescence assays (Figure 4e and Supplementary Figure 7), delaying the aggregation lag time (tlag) in a dose-dependent fashion (Figure 4f). In contrast, linear CHVVGVI showed greatly reduced ability to inhibit hIAPP (Figure 4e), suggesting that cyclization is important for activity. For comparison, we tested a 17-mer cyclic peptide previously shown to inhibit hIAPP aggregation and reduce hIAPP toxicity to cultured cells41 (“MCIP-2a,” Supplementary Figure 8), as well three cyclic peptides from our starting library not enriched by the hIAPP-T7n selection (cyclo-CLDCVFG, cyclo-CHLDIGC, and cyclo-CLSRRCG) (Supplementary Figure 9). MCIP-2a inhibited hIAPP tlag (Supplementary Figure 8b–d) with comparable activity to cyclo-CHVVGVI at lower concentrations but higher activity at the highest stoichiometry tested (Supplementary Figure 8e). cyclo-CLDCVFG and cyclo-CHLDIGC unexpectedly decreased overall ThT fluorescence but did not have a clear effect on tlag (Supplementary Figure 9). This decrease in ThT signal did not appear to reflect a reduction in fibril formation (Supplementary Figure 10), suggesting it is not due to an inhibitory effect of these peptides on hIAPP aggregation. cyclo-CLSRRCG exhibited suppression of hIAPP aggregation tlag (Supplementary Figure 9) despite not being enriched by our selection, thus representing a false negative.
We further characterized the interaction of cyclo-CHVVGVI with hIAPP. TEM analysis corroborated the ability of cyclo-CHVVGVI to delay hIAPP aggregation onset, showing that no fibrils are observed when hIAPP is incubated with cyclo-CHVVGVI for 2 h (Figure 4g and Supplementary Figure 12). In contrast, hIAPP produces numerous fibrils when incubated alone under the same conditions (Figure 4h and Supplementary Figure 12). No gross differences in morphology were observed between fibrils formed by hIAPP alone or with cyclic peptide after incubation at longer times when all hIAPP should be aggregated (Figure 4i–j), suggesting that cyclo-CHVVGVI is not being incorporated into hIAPP fibrils or sequestering hIAPP in a different form of aggregate. Finally, analysis of the interaction of cyclo-CHVVGVI with hIAPP by native mass spectrometry primarily detected a 1:1 complex between cyclo-CHVVGVI and hIAPP (Supplementary Figure 14), suggesting that cyclo-CHVVGVI inhibits hIAPP aggregation through binding to monomeric hIAPP, consistent with the APP-T7n selection mechanism (Figure 2a).
These results demonstrate that our system can be used to rapidly identify cyclic peptide inhibitors of a different aggregation-prone protein.
Negative selection identifies selective inhibitors
The top sequences enriched by the hIAPP selection (CHVVGVI and CHVHSYL) resembled previously identified Aβ42 aggregation inhibitors20 and sequences from our own selections on Aβ42-T7n (Supplementary Figure 4b). Because the amyloidogenic regions of Aβ42 and hIAPP share sequence similarity,42 we wondered if our selection for hIAPP inhibitors discovered peptides that are also active on Aβ42. Indeed, the hIAPP round 5 SP pool exhibited activity on both hIAPP-T7n and Aβ42-T7n (Figure 5a), suggesting that we had identified promiscuous sequences. However, the SP pools were inactive against a non-amyloidogenic target (an scFv that aggregates in E. coli cytosol). Non-specific hits and false positives are a concern for selection-based cyclic peptide discovery methods.14,43 Negative selection can increase selectivity and weed out false positives, but to our knowledge has not been explored in cellular cyclic peptide selections. These results motivated us to develop a negative selection that could quickly exclude cyclic peptides with non-selective activity or act outside of the APP in our selection circuit (for example, binding to and stabilizing T7n).
Figure 5. Negative selection to remove promiscuous cyclic peptide sequences.

(a) Propagation activity of the SP pool from round 5 of positive selection for hIAPP inhibition on selection cells encoding hIAPP, Aβ42, or an insoluble antibody fragment (scFv) fused to T7n. (b) Phage titer (solid line, plotted on the left y-axis) and propagation activity (dashed line, plotted on the right y-axis) of successive rounds of selection of the hIAPP round 5 SP pool on Aβ42-T7n negative selection cells. “In” refers to the input SP pool used to infect selection cells and “out” refers to the output SP pool obtained after selection, for a given round of selection. pfu = plaque-forming units. Fold phage propagation is calculated as the number of phage generated from an infected culture divided by the number of phage used to infect the culture. (c) Propagation activity of pre- and post-negative selection SP pools on selection cells encoding hIAPP or Aβ42 as the target APP. (d) Top 10 enriched cyclic peptide sequences after 4 rounds of negative selection as determined by high-throughput sequencing analysis. (e) Propagation of clonal phage encoding cyclic peptide sequences enriched by either positive selection on hIAPP only (CHVVGVI) or by hIAPP positive selection followed by negative selection against Aβ42 inhibitors (CDLGVFR and CRCVFSG) on selection cells encoding hIAPP or Aβ42 as the target APP. (a, c, e) Data reflect two biological replicates plotted as individual values. Propagations used an input of 105 phage.
To create a negative selection, we placed gIII-neg, a dominant-negative form of gIII,44 under PT7 in our selection circuit. Inhibition of a non-target APP (ex. Aβ42) triggers pIII-neg expression and poisons phage proliferation. A second plasmid produces small amounts of pIII from the phage shock promoter (Supplementary Figure 16a). All SPs infecting cells encoding this negative selection receive sufficient pIII to propagate. However, SP that carry a promiscuous or false positive cyclic peptide will also produce pIII-neg and generate non-infectious progeny (Supplementary Figure 16b–c). We subjected the hIAPP round 5 phage population to four rounds of negative selection, diluting the phage by 100-fold each round (Figure 5b). The starting fitness of the hIAPP SP pool on the negative selection was low, confirming presence of library members active on Aβ42. However, by the fourth round of negative selection, the SP pool fitness had increased by ~1000-fold, suggesting removal of promiscuous sequences (Figure 5b). Comparison of the activity of the pre- and post-negative selection SP pools revealed a ~1000-fold decrease in Aβ42-T7n aggregation inhibition, while the activity on hIAPP-T7n decreased modestly (< 10-fold; Figure 5c).
Deep sequencing of the post-negative selection pool (Figure 5d and Supplementary Table 1) revealed that CHVVGVI was absent from the top 10 sequences. Instead, > 50% of sequencing reads mapped to CDLGVFR and CRCVSFG (Figure 5d). These sequences were not highly enriched by positive selection (Figure 4c), illustrating the ability of negative selection to produce large shifts in the cyclic peptide sequence distribution. We compared the activity of clonal SP encoding CHVVGVI, CDLGVFR, or CRCVSFG on selection cells encoding either hIAPP-T7n or Aβ42-T7n (Figure 5e). While CHVVGVI SP propagated robustly on both targets, CRCVSFG SP were more selective, with ~10-fold higher activity on hIAPP vs. Aβ42 selection cells. SP encoding CDLGVFR were strikingly selective, exhibiting ~1000-fold greater propagation on hIAPP vs. Aβ42. We observed similar trends with the Aβ42-GFP folding reporter when CHVVGVI, CDLGVFR, and CRCVSFG were expressed from plasmid (Supplementary Figure 17), suggesting that selectivity is not an artifact of our phage system.
We examined the ability of chemically synthesized cyclo-CDLGVFR and cyclo-CRCVSFG (Figure 6a–b) to inhibit in vitro hIAPP aggregation. Both cyclic peptides inhibited hIAPP fibrillation and extended tlag (Figure 6c–f and Supplementary Figures 18–19). In contrast, linear CDLGVFR was completely inactive (Figure 6c), while linear CRCVSFG moderately inhibited hIAPP aggregation, but with reduced activity compared to cyclo-CRCVFSG (Figure 6e). Like cyclo-CHVVGVI, cyclo-CDLGVFR and cyclo-CRCVSFG exhibited similar activity to MCIP-2a at lower concentrations, but were less active than MCIP-2a at higher peptide:hIAPP stoichiometries (Supplementary Figure 8e). TEM analysis confirmed that both cyclic peptides delay hIAPP aggregation (Figure 6g–h and Supplementary Figure 12) and do not change the overall appearance of hIAPP fibrils (Figure 6i–j). Additionally, we observed 1:1 complexes of both cyclo-CDLGVFR and cyclo-CRCVSFG with hIAPP by native mass spectrometry (Supplementary Figure 20), suggesting that, like cyclo-CHVVGVI, these cyclic peptides are primarily hIAPP monomer binders.
Figure 6. Cyclic peptides identified by negative selection inhibit hIAPP aggregation.

(a-b) Structures of cyclo-CDLGVFR and cyclo-CRCVFSG. (c) Effect of varying concentrations of chemically synthesized cyclo-CDLGVFR (left) or linear CDLGVFR (right) on aggregation of 10 μM hIAPP measured by ThT fluorescence. Assay was run in phosphate-buffered saline (PBS) pH 6.9 at 32 °C under quiescent conditions. Data reflect plots of individual values of three technical replicates. The hIAPP alone (0 μM peptide) data is the same in both plots. (d) Effect of cyclo-CDLGVFR on lag time (tlag) of hIAPP aggregation. Data reflects tlag fits from four biological replicates (4 separate ThT assays; see Supplementary Figure 18) plotted as individual values. P-values (two-tailed Student’s t-test between 0 μM peptide and each peptide concentration): **, P ≤ 0.01. (e) Effect of varying concentrations of chemically synthesized cyclo- CRCVFSG (left) or linear CRCVFSG (right) on aggregation of 10 μM hIAPP measured by ThT fluorescence. Assay was run in phosphate-buffered saline (PBS) pH 6.9 at 32 °C under quiescent conditions. Data reflect plots of individual values of three technical replicates. The hIAPP alone (0 μM peptide) data is the same as in panel (c). One replicate of 40 μM linear CRCVFSG excluded due to high ThT signal at time = 0. (f) Effect of cyclo-CRCVFSG on lag time (tlag) of hIAPP aggregation. Data reflects tlag fits from four biological replicates (4 separate ThT assays; see Supplementary Figure 19) plotted as individual values. P-values (two-tailed Student’s t-test between 0 μM peptide and each peptide concentration): **, P ≤ 0.01. (g-h) TEM analysis of 10 μM hIAPP incubated in the presence of 40 μM cyclo-CDLGVFR (g) or cyclo-CRCVFSG (h) for 2 h. (i-j) TEM analysis of 10 μM hIAPP incubated in the presence of 40 μM cyclo-CDLGVFR (g) or cyclo-CRCVFSG (h) for 14 h. (g-j) For compatibility with TEM, hIAPP incubations were run in 25 mM sodium phosphate buffer, pH 7.4; see Supplementary Figure 13 for hIAPP aggregation kinetics under TEM assay conditions. Scale bars = 200 nm. This experiment was not repeated.
Together, these results confirmed that our negative selection had purged promiscuous sequences to successfully uncover active and selective cyclic peptide inhibitors of hIAPP aggregation.
Discussion
We have developed a platform for identifying cyclic peptide protein aggregation inhibitors that leverages elements of PACE to increase the speed and convenience of selection. This method does not require specialized equipment (such as a cell sorter) and, aside from the initial generation of the SP library, eliminates the need for additional cloning or transformation steps. As a demonstration of the platform’s utility, we applied it to identify new cyclic peptide inhibitors of hIAPP aggregation in less than a week. In addition, the observation that enriched inhibitors also exhibited activity on a different aggregation-prone protein, Aβ42, prompted us to create a negative selection strategy to remove promiscuous sequences. We used this negative selection to identify cyclic peptides that selectively inhibited hIAPP. Off-target activity or false positives can plague selections and high-throughput screens of chemical libraries for active compounds, resulting in extra time and labor expended on isolating desired hits. Thus, negative selections such as the one used here can improve hit quality by purging undesired library members and streamline the isolation of desirable cyclic peptide sequences.
To our knowledge, the cyclic peptide hIAPP inhibitors we report are the first to be identified through a selection-based method. Previously, macrocyclic peptides have been generated by rational design to inhibit hIAPP aggregation through displayed hIAPP sequence mimics41,45 or aromatic moieties.46,47 In a head-to-head comparison, our hits exhibit similar activity to one such rationally designed inhibitor, MCIP-2a,41 at low peptide:hIAPP stoichiometries, but have lower potency at the highest concentration we tested. Our cyclic peptides bear little sequence resemblance to rationally designed inhibitors, suggesting that unbiased selection can uncover new starting points for inhibitor development. They are also notably smaller in size (7 residues) compared to MCIP-2a (17 residues) and other rationally designed inhibitors (13–18 residues41,45–47), which is an advantage for bioavailability.48 Future efforts may improve the potency of hIAPP inhibitors identified by our platform through developing more stringent selections that require more active cyclic peptides to pass.
An unexpected observation made here is that the SICLOPPS cyclic peptide sequences from this work and other studies20 may not require intein splicing to inhibit target protein aggregation. Because splicing efficiency varies by extein sequence,49 it is difficult to predict the ratio of unspliced to spliced product for each cyclic peptide library member. Thus, the species under selection may be an intein-bound or spliced form of the peptide, or both. Our hits are active as chemically synthesized cyclic peptides and exhibit diminished activity when employed in linear form, suggesting that a cyclic conformation is important for activity in our selection for hIAPP inhibitors. However, other targets could behave differently, and moving forward, it may be preferable to switch the Ssp intein in SICLOPPS with with faster splicing homologs to minimize the time cyclic peptide sequences undergoing selection spend in the intein-bound form.49,50
One limitation of our platform is the size of the cyclic peptide libraries employed, which is restricted by cloning efficiencies. This constraint is not unique to our system and applies to other cellular selections.13,16 A potential route to increasing library size is to further mutate the SP using host cell-encoded mutagenesis plasmids.39 We examined the effects of selecting cyclic peptide libraries with MP6, but failed to observe enriched sequences that unambiguously arose from MP6 mutagenesis. We speculate that this is because our starting library already contained sequences with activity on the chosen target. Phage encoding such sequences will constitute a higher proportion of the total SP pool compared to phage randomly discovering an active sequence through MP6 mutagenesis; thus, the latter would likely be outcompeted by the former during the selection. This may not hold true for other targets, and future work may warrant revisiting the inclusion of mutagenesis plasmids.
Although here we exclusively select for protein aggregation inhibitors, in principle our platform could be used to select for any cyclic peptide activity that can be linked to a genetic selection. PACE selections have been developed for a wide range of biologically significant activities, such as protein-protein interaction,29,51 protein-DNA interaction,35,52,53 and ternary complex formation27 to name a few, and these selections could be adapted for use in cyclic peptide discovery. Our system may also be able to accommodate other types of genetically encoded peptide libraries.16,18 Thus, we anticipate that our work will provide a versatile and accessible new option for cyclic peptide discovery.
Materials and Methods
General methods
Antibiotics (Gold Biotechnology) were used at the following working concentrations: ampicillin, 50 μg/mL; spectinomycin, 100 μg/mL; chloramphenicol, 25 μg/mL; kanamycin, 50 μg/mL; tetracycline, 10 μg/mL; streptomycin, 50 μg/mL. HyClone water (GE Healthcare Life Sciences) was used for PCR reactions and cloning. For all other experiments, water was purified using a MilliQ purification system (Millipore). Phusion U Hot Start DNA polymerase (Thermo Fisher Scientific) or Q5 polymerase (New England Biolabs) were used for PCRs. A full list of plasmids used in this work is given in Supplementary Table 2. Key primers are provided in Supplementary Table 3. A full list of reagents and equipment used in this work is given in Supplementary Table 4. Plasmid maps are provided in Supplementary Data 1.
Strain S206035 was used for plasmid cloning, amplification, and phage assays. Strain S220835 was used for SP cloning and plaque assays. Plasmids and SPs were cloned by USER assembly or blunt-end ligation. Competent cells were prepared and transformed using the TSS method. Phage propagation and plaque assays were performed as previously described.54 Unless otherwise noted, phage propagation assays used an input of 105 phage to infect a 2 mL culture of host cells.
Phage-assisted continuous evolution
In general, PACE was set up and run as previously described.28
PACE 1.
S2060s transformed with pLY009, pTW6ap1a, and MP6 were maintained in a 40 mL chemostat. Lagoons (15 mL each) were infected with evolved spLY001 from PANCE at an initial titer of 4×104 pfu/mL and maintained at a flow rate of 0.5 V/h. Lagoon flow rates were increased to 1 V/h at 19 h, decreased to 0.8 V/h at 66 h, increased back to 1 V/h at 159.5 h, further increased to 1.5 V/h at 257 h, and finally to 2 V/h at 305 h. The experiment ended at 351 h.
PACE 2.
S2060s transformed with pTW357d5, pTW358b, and MP6 were maintained in a 40 mL chemostat. Lagoons (15 mL each) were infected with final phage populations collected from PACE 1 at an initial titer of 1×105 pfu/mL and maintained at a flow rate of 0.5 V/h. Lagoon flow rates were increased to 1 V/h at 42.5 h, 1.5 V/h at 117 h, followed by 2 V/h at 160.5 h then 2.5 h at 189 h, and finally 3 V/h at 208 h. The experiment ended at 254.5 h.
Aβ42-GFP fluorescence assay
Single colonies of S2060s transformed with pBL066c and SICLOPPS plasmid were used to make overnight cultures in 2xYT with maintenance antibiotics. The overnight culture was diluted (1:100) into 1 mL DRM on a 96 deep-well plate (VWR, 75870–796) with maintenance antibiotics and IPTG (0, 0.04, 0.2, or 1 mM). The diluted culture was incubated at 37 °C with shaking for 2.5 h, after which aTc (200 ng/mL) was added. 100 μL of the culture was transferred onto a black clear-bottom 96-well plate (VWR, 89131–680) to measure fluorescence signal 2, 3, 5 h after addition of aTc. GFP fluorescence signal (excitation wavelength = 485 nm, emission wavelength = 535 nm) and OD600 were then measured using a Tecan M Plex microplate reader.
Cyclic peptide library cloning
The reverse primer LY0003 and the degenerate forward primers LY0106 (CX6) or LY0107 (CZ6) (Supplementary Table 3) were used to clone the phage-encoded cyclic peptide libraries with spLY006b as the template. The resulting PCR product was purified by PCR clean-up and concentrated to 100 ng/μL. The purified PCR product was assembled by USER assembly. The USER assembled product was purified again by PCR clean-up and diluted to 50 ng/μL. For each aliquot, 250 ng of assembled DNA was transformed into chemicompetent S2208 cells. After heat shock, the aliquots were immediately combined into pre-warmed (37 °C) outgrowth media (2xYT containing 3 mM glucose, 500 μL for each aliquot) and incubated at 37 °C with shaking for 1 h. After outgrowth, the culture was centrifuged at 8,000 rcf for 2 min and the supernatant containing the phage library was collected. This process resulted in 3 × 106 and 5 × 106 independent clonal phage for the CX6 and CZ6 libraries, respectively, as measured by plaque assay.
Cyclic peptide library selection
The phage libraries obtained immediately after cloning were first amplified to introduce degeneracy: mid-log S2208 cells grown in Davis Rich Media (DRM) with maintenance antibiotics at 37 °C with shaking were infected with the phage library and phage propagation allowed to occur for 6 h before the culture was centrifuged at 8,000 rcf for 2 min and the supernatant containing the expanded phage library collected. The expanded phage libraries contained ~1011 total phage as measured by plaque assay.
Single colonies of host cells transformed with the appropriate selection plasmids were used to make overnight cultures in 2xYT with maintenance antibiotics. The overnight culture was diluted (1:100) into 4–5 mL DRM with maintenance antibiotics and incubated at 37 °C with shaking until the OD600 of the culture reached ~0.5, upon which the expanded phage library was added at a titer of 1 × 106 pfu/mL. The culture was incubated at 37 °C with shaking overnight (13–18 h). The next day, the culture was centrifuged at 8,000 rcf for 2 min to obtain the supernatant containing phage. The collected phage was diluted into fresh host cell cultures (10 to 1,000-fold) for the next round of selection, while the rest was stored at 4 °C for downstream analysis. This process was repeated until a significant increase in phage propagation was observed compared with the parent library. Both positive and negative selection rounds were performed using this procedure.
High-throughput sequencing and data analysis
Sample preparation.
Phage pools to be sequenced were first amplified using LY0145 and LY0146. The PCR product was purified, and adapters added using LY0147 and LY0148. The resulting PCR product was purified and barcoded through a final PCR using index primers to add unique indices for shared Illumina NovaSeq sequencing. The final PCR product was purified, and the concentration measured by Nanodrop. Further sample quality control was performed by the University of Wisconsin-Madison Biotechnology Center (UWBC).
High-throughput sequencing and data analysis.
High-throughput sequencing was performed by UWBC using a shared sequencing service on the NovaSeq 6000 platform. Sequencing reads were demultiplexed by UWBC and analyzed using custom-written Python scripts (Supplementary Note 5).
Solid-phase peptide synthesis.
General procedure for solid-phase peptide synthesis (SPPS).
(Refer to Supplementary Note 2 for abbreviations.) Dawson Dbz AM resin (Sigma-Aldrich) (for head-to-tail cyclized peptides) or Wang resin (CEM) (for linear peptides) was added to an empty SPE cartridge with pre-inserted frit (Agilent, 6 mL capacity). The resin was suspended in 2 mL DCM with shaking and allowed to swell for 40 min at RT. DCM was drained and the resin washed 3 times with 2 mL DCM, followed by 3 times with 2 mL DMF. The resin was deprotected by adding freshly prepared 20% piperidine in DMF and shaking for 30 min, followed by three washes with 2 mL DMF. Couplings were performed with 4 eq. Fmoc-protected L-amino acid, 3.9 eq. HATU, and 8 eq. DIPEA in 2 mL DMF for 1 h with agitation. After coupling, the resin was washed with 2 mL DMF three times. The deprotection and coupling steps were repeated until the peptide was completed. Couplings for Fmoc-L-Arg(Pbf)-OH was repeated once. For head-to-tail cyclic peptides, Boc-L-Cys(Trt)-OH was used as the final amino acid. Upon completion, the resin was washed 3 times with 2 mL DMF and 3 times with 2 mL DCM. Note: the resin was dried by N2 flow after every washing step in this procedure.
Dbz linker activation.
For head-to-tail cyclized peptides, 4-nitrophenyl chloroformate (4 eq) was dissolved in 2 mL DCM and added to the resin. After shaking for 30 min, the solution was drained, and the resin washed 3x with 2 mL DCM. The 4-nitrophenyl chloroformate addition and washing steps were repeated once. The resin was then washed 3x with 2 mL DMF. A solution of 110 μL DIPEA in 2 mL DMF was added and the resin agitated for 10 min, followed by draining the solution. This step was repeated twice. The resin was then washed 3x with 2 mL DMF, 3x with 2 mL DCM, and 3x with 2 mL diethyl ether, then allowed to dry under a flow of N2. Note: the resin was dried by N2 flow after every washing step in this procedure.
Deprotection and cleavage.
The resin was transferred to a 15 mL conical tube, then treated with cleavage solution (2 mL 90% TFA, 5% DCM, 2.5% H2O, and 2.5% TIPS) for 2 h with agitation. The solution was filtered into a 50 mL conical tube through an empty syringe plugged with cotton. The remaining resin was washed with another 2 mL cleavage solution and filtered combined into the 50 mL conical in the same manner. 40 mL diethyl ether was added into the conical, which was then incubated at −20 °C for 1 h to facilitate precipitation of peptide product. After precipitation, the solution was centrifuged at 1943 rcf for 10 min at 4 °C. The supernatant was decanted, and the precipitate was dried under N2 flow to evaporate remaining volatiles. The dried precipitate was then dissolved in 5 mL 20% aq. MeCN, frozen on dry ice for 1 h, and lyophilized.
Linear peptide purification.
Lyophilized linear peptide was dissolved in 2 mL 20% aq. MeCN. The solution was passed through a 0.22 μm syringe filter into a fresh 15 mL conical tube. Another 2 mL 20% aq. MeCN was used to wash the old conical and filtered into the new conical tube. All 4 mL of peptide-containing solution was purified by HPLC (Shimadzu, CBM-20A, LC-20AP, SPD-20AV, FRC-10A) in a single injection onto a preparative C18 column (Shimadzu, Premier Elite Polar 10 μ 150×30 mm). HPLC conditions: Flow rate, 25 mL/min. Mobile phase A: H2O containing 0.1% TFA. Mobile phase B: MeCN. A linear gradient of 10% to 50% mobile phase B over 32 min was used to purify the Nbz-containing linear peptide. Fractions were analyzed by MALDI-TOF to confirm mass of the Nbz-containing linear peptide, then combined and lyophilized.
Cyclization of Nbz-containing linear peptides.
Lyophilized linear peptide was dissolved in 4 mL cyclization buffer (0.1 M Na2HPO4, 6 M guanidinium chloride, and 20% v/v MeCN in H2O; pH 6.8–7.2) and incubated at 50 °C with rotation for 2–4 h.
Cyclic peptide purification.
After cyclization, the solution was directly injected onto a semi-preparative C18 column (Kromasil, Eternity-5-C18 10×250 mm) HPLC conditions: flow rate, 5 mL/min. Mobile phase A: H2O containing 0.1% TFA. Mobile phase B: MeCN containing 0.1% TFA. A linear gradient of 20% to 45% mobile phase B over 32 min was used to purify the cyclic peptide. Fractions were analyzed with MALDI-TOF to confirm desired mass of the cyclic peptide, then combined and lyophilized. After lyophilization, H2O was added to dissolve the cyclic peptide to a stock concentration of 1 mM. The prepared 1 mM cyclic peptide stock solution was quickly aliquoted into low-protein-binding Eppendorf tubes, frozen on dry ice for 1 h and lyophilized to dried peptide solid. Peptides were freshly dissolved before use. Additional freeze-thaw cycles were kept to a minimum to prevent degradation of cyclic peptide stocks.
Cyclic peptide characterization.
See Supplementary Note 3 for peptide characterization data. Analytical HPLC was performed using a HPLC system (Shimadzu, DGU-20A5R, LC-20AT, SIL-10AF, SPD-M20A, CTO-20A) equipped with an analytical C18 column (Kromasil, Eternity-5-C18 4.6×250 mm). HPLC conditions: at a flow rate of 1 mL/min. A binary solvent system with 90% mobile phase A (M.Q. H2O, 0.1% TFA) and 10% mobile phase B (ACN, 0.1% TFA) was used. A linear gradient of mobile phase B from 10% to 95% within 27 min was used to resolve purity of the sample. Mass of the cyclic peptide was confirmed by ESI-EMM conducted by mass spectrometry facilities in the Paul Bender Chemistry Instrumentation Center (UW-Madison Department of Chemistry).
Synthesis of MCIP-2a.
MCIP-2a was synthesized following reported procedures41 with the following modifications. Briefly, the linear peptide was synthesized on Rink amide resin (CEM) using a Liberty Blue HT24 System and cleaved from the resin as described above. Intramolecular disulfide bridge formation was performed by dissolving crude peptide (after cleavage and lyophilization) at 1 mg/mL in aqueous 0.1 M NH4HCO3 solution containing 40% DMSO. The reaction was allowed to proceed for 2 hours at room temperature with agitation. The peptide was then purified by RP-HPLC and characterized by ESI-MS as described above. Purified peptide was prepared in small aliquots, lyophilized, and stored at −80 °C until use.
ThT fluorescence assays
Preparation of ThT stock solution.
To prepare a stock solution of ThT (2–3 mM), 2–4 mg of ThT was added to 3 mL PBS (pH 6.9) in a 15 mL conical tube. The solution was sonicated for 5 min and the supernatant was filtered through a 0.22 μm syringe filter into a 1.5 mL Eppendorf tube. This was the ThT stock solution to be used in following experiments. 2 mL of a 1:200 diluted ThT solution was prepared by adding 10 μL ThT stock solution into 1990 μL PBS. The absorbance of the diluted ThT solution measured at 412 nm was used to calculate the ThT stock concentration using an extinction coefficient (ε) of 31600 M−1 cm−1.
Preparation of hIAPP stocks.
A 1 mg portion of amylin trifluoroacetate salt (Bachem) was purchased as lyophilized solid in a vial. The solid was dissolved in pre-chilled 2562 μL 35 mM sodium acetate (pH 5.3) to a stock concentration of 100 μM on ice. Aliquots of hIAPP were prepared by transferring 200 μL of the 100 μM hIAPP stock solution to low-protein-binding Eppendorf tubes on ice. The aliquots were then immediately snap frozen using liquid N2, lyophilized for 24 h, and stored at −80 °C until use.
Preparation of samples for ThT assays.
Using cyclic peptide stock solution (100 μM) and PBS, 400 μL aqueous solution of cyclic peptide was prepared in a low-protein-binding Eppendorf tube to achieve the desired cyclic peptide:hIAPP molar ratio (80 μM for 4:1, 40 μM for 2:1, 20 μM for 1:1, 10 μM for 0.5:1, 0 μM for 0:1). 30 μL of the solution was then loaded to wells on a black clear-bottom low-binding 96-well plate (Corning 3881) to make a triplicate for each condition. This plate loaded with aqueous solutions of cyclic peptides was then ready for adding hIAPP.
Preparation and addition of hIAPP for ThT assays.
Using ThT stock solution and PBS, 1 mL ThT solution (10 μM) was prepared in a low-protein-binding Eppendorf tube and chilled on ice. 0.5 mL of the pre-chilled ThT solution was added to one aliquot of lyophilized hIAPP, which was equilibrated to room temperature before opening the tube. Pipetting up and down four times was performed to facilitate complete dissolving of hIAPP. The 0.5 mL hIAPP-containing solution was immediately transferred to the remaining 0.5 mL ThT solution on ice. Pipetting up and down four times was performed to facilitate complete mixing. The resulting 1 mL hIAPP solution, where [hIAPP] = 20 μM, was immediately transferred to a 25 mL liquid reservoir. Using a multi-channel pipette, 30 μL of the hIAPP solution was quickly loaded into wells containing aqueous solutions of cyclic peptides on the 96-well plate. Final [hIAPP] = 10 μM, [ThT] = 5 μM.
ThT assay conditions.
The plate was then quickly covered and incubated in a Tecan M Plex microplate reader under quiescent condition at 32 °C for at least 12 h. ThT fluorescence at 480 nm was measured every 5 min through the bottom of the plate using an excitation wavelength of 440 nm.
Fitting of aggregation kinetics.
ThT aggregation kinetics data was fit to a sigmoidal function using GraphPad Prism and the aggregation lag time (tlag) and the apparent fibril elongation rate (kapp) calculated as described.55
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed at the University of Wisconsin School of Medicine and Public Health Electron Microscopy facility. hIAPP and cyclic peptide were dissolved in 25 mM sodium phosphate buffer (pH 6.8 with 0.4% DMSO) to achieve a molar ratio of 1:4 at concentrations of hIAPP = 10 μM and cyclic peptide = 40 μM. The sample was incubated quiescently at 32 °C before TEM analysis. 2 μL of the sample was placed onto Formvar-coated copper grids and the excess liquid was blotted away. 2 μL of diluted Nano-W solution was then added to stain the grids and the excess liquid was blotted away. The TEM analysis was performed on Philips CM120 at 80 kV and digital images were obtained with an AMT BioSprint12 camera.
nESI-MS analysis
hIAPP was dissolved in 200 mM ammonium acetate buffer (pH 7.4) to prepare 100 μM hIAPP stock solution. Cyclic peptide was first dissolved in DMSO to 10 mM, then diluted into the buffer to make 1 mM cyclic peptide stock solution. These stock solutions were diluted into the buffer to achieve final concentrations of [hIAPP] = 16 μM and [cyclic peptide] = 64 μM. The percentage of DMSO in the final solution was 0.64% for all the samples. Prepared samples were incubated quiescently for 20 min at 32 °C. Native ESI-MS analysis was then performed on a SELECT SERIES Cyclic IMS Q-TOF (Waters Corp., Wilmslow, U.K.) equipped with nano-ESI interface.
All the samples were analyzed using positive ionization ESI with a capillary voltage of 1.1 kV. The following instrumental parameters were used: source temperature 100 °C; desolvation temperature 250 °C; sampling cone 30 V; backing pressure 2.5 mbar; trap collision energy 6 V; trap DC −4 V; transfer collision energy 4 V. The system was calibrated with NaI cluster ions from a 2 μg/μL 50:50 2-propanol:water solution. Data were acquired over the m/z range of 50–8000 and processed using MassLynxV4.2 (Waters Corp., Wilmslow, U.K.).
Supplementary Material
Acknowledgements
This work was supported by the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison with funding from the Wisconsin Alumni Research foundation. L.L. acknowledges funding support of NIH shared instrument grants (NIH-NCRR S10RR029531, S10OD028473 and S10OD025084), NIH grants R01 DK071801, R01 AG052324, and R01AG078794, a Vilas Distinguished Achievement Professorship, and Charles Melbourne Johnson Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Footnotes
Competing interests
A patent application (18/906,707, status pending) has been filed by the University of Wisconsin on the PACE selection for protein aggregation inhibitors and new cyclic peptide sequences identified in this work, with L.Y. and T.W. as co-inventors. The remaining authors declare no competing interests.
Code availability
Python code for high-throughput sequencing analysis is provided in the Supporting Information (Supplementary Note 5).
Data availability
Data reporting the findings of this study are included in the Article, Supplementary Data, Source Data, and Supplementary Information. High-throughput sequencing information is available at the NCBI GEO database under accession code GSE279094. Plasmids used in this study are available through Addgene.
References
- 1.Chiti F & Dobson CM Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem 86, 27–68 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET & Kelly JW Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov 14, 759–780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cawood EE, Karamanos TK, Wilson AJ & Radford SE Visualizing and trapping transient oligomers in amyloid assembly pathways. Biophys. Chem 268, 106505 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saunders JC et al. An in vivo platform for identifying inhibitors of protein aggregation. Nat. Chem. Biol 12, 94–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sárkány Z, Rocha F, Damas AM, Macedo-Ribeiro S & Martins PM Chemical kinetic strategies for high-throughput screening of protein aggregation modulators. Chem. Asian J 14, 500–508 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Xu Y, Maya-Martinez R, Guthertz N, Heath GR, Manfield IW, Breeze AL, Sobott F, Foster R & Radford SE Tuning the rate of aggregation of hIAPP into amyloid using small-molecule modulators of assembly. Nat. Commun 13, 1040 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doig AJ et al. Why is research on amyloid-β failing to give new drugs for Alzheimer’s disease? ACS chemical neuroscience vol. 8 1435–1437 at (2017). [DOI] [PubMed] [Google Scholar]
- 8.Nie Q, Du X & Geng M Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin 32, 545–551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorzi A, Deyle K & Heinis C Cyclic peptide therapeutics: past, present and future. Curr. Opin. Chem. Biol 38, 24–29 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zhang H & Chen S Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol 3, 18–31 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naylor MR, Bockus AT, Blanco M-J & Lokey RS Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets. Curr. Opin. Chem. Biol 38, 141–147 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Li X, Craven TW & Levine PM Cyclic Peptide Screening Methods for Preclinical Drug Discovery: Miniperspective. J. Med. Chem 65, 11913–11926 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Sohrabi C, Foster A & Tavassoli A Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem 4, 90–101 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Wiedmann MM & Suga H RNA display methods for the discovery of bioactive macrocycles. Chem. Rev 119, 10360–10391 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Scott CP, Abel-Santos E, Wall M, Wahnon DC & Benkovic SJ Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci 96, 13638–13643 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang X, Lennard KR, He C, Walker MC, Ball AT, Doigneaux C, Tavassoli A & Van Der Donk WA A lanthipeptide library used to identify a protein--protein interaction inhibitor. Nat. Chem. Biol 14, 375–380 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King AM et al. Selection for constrained peptides that bind to a single target protein. Nat. Commun 12, 6343 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iannuzzelli JA & Fasan R Expanded toolbox for directing the biosynthesis of macrocyclic peptides in bacterial cells. Chem. Sci 11, 6202–6208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kritzer JA, Hamamichi S, McCaffery JM, Santagata S, Naumann TA, Caldwell KA, Caldwell GA & Lindquist S Rapid selection of cyclic peptides that reduce α-synuclein toxicity in yeast and animal models. Nat. Chem. Biol 5, 655–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delivoria DC et al. Bacterial Production and Direct Functional Screening of Expanded Molecular Libraries for Discovering Inhibitors of Protein Aggregation. Sci. Adv vol. 5 http://advances.sciencemag.org/ (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matis I et al. An integrated bacterial system for the discovery of chemical rescuers of disease-associated protein misfolding. Nat. Biomed. Eng 1, 838–852 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Delivoria DC et al. Bacterial production and direct functional screening of expanded molecular libraries for discovering inhibitors of protein aggregation. Sci. Adv 5, eaax5108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Packer MS & Liu DR Methods for the directed evolution of proteins. Nat. Rev. Genet 16, 379–394 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Esvelt KM, Carlson JC & Liu DR A system for the continuous directed evolution of biomolecules. Nature 472, 499–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth TB, Woolston BM, Stephanopoulos G & Liu DR Phage-assisted evolution of Bacillus methanolicus methanol dehydrogenase 2. ACS Synth. Biol 8, 796–806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brödel AK, Jaramillo A & Isalan M Engineering orthogonal dual transcription factors for multi-input synthetic promoters. Nat. Commun 7, 13858 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dewey JA, Azizi S-A, Lu V & Dickinson BC A System for the Evolution of Protein-Protein Interaction Inducers. ACS Synth. Biol 10, 2096–2110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller SM, Wang T & Liu DR Phage-assisted continuous and non-continuous evolution. Nat. Protoc 15, 4101–4127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zinkus-Boltz J, DeValk C & Dickinson BC A phage-assisted continuous selection approach for deep mutational scanning of protein-protein interactions. ACS Chem. Biol 14, 2757–2767 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Cheruvara H, Allen-Baume VL, Kad NM & Mason JM Intracellular screening of a peptide library to derive a potent peptide inhibitor of α-synuclein aggregation. J. Biol. Chem 290, 7426–7435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee LL, Ha H, Chang Y-T & DeLisa MP Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding and solubility. Protein Sci. 18, 277–286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matis I et al. An integrated bacterial system for the discovery of chemical rescuers of disease-associated protein misfolding. Nat. Biomed. Eng 1, 838–852 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Wang T, Badran AH, Huang TP & Liu DR Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol 14, 972–980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wurth C, Guimard NK & Hecht MH Mutations that reduce aggregation of the Alzheimer’s Aβ42 peptide: an unbiased search for the sequence determinants of Aβ amyloidogenesis. J. Mol. Biol 319, 1279–1290 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Hubbard BP et al. Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat. Methods 12, 939–942 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thuronyi BW et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol 37, 1070–1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ & Cohen SN Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag- TSG101 interaction. ACS Chem. Biol 3, 757–764 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Miranda E et al. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J. Am. Chem. Soc 135, 10418–10425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Badran AH & Liu DR Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun 6, 8425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milardi D et al. Proteostasis of islet amyloid polypeptide: a molecular perspective of risk factors and protective strategies for type II diabetes. Chem. Rev 121, 1845–1893 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spanopoulou A et al. Designed macrocyclic peptides as nanomolar amyloid inhibitors based on minimal recognition elements. Angew. Chemie 130, 14711–14716 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Krotee P et al. Common fibrillar spines of amyloid-β and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors. J. Biol. Chem 293, 2888–2902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horswill AR, Savinov SN & Benkovic SJ A systematic method for identifying small-molecule modulators of protein-protein interactions. Proc. Natl. Acad. Sci 101, 15591–15596 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlson JC, Badran AH, Guggiana-Nilo DA & Liu DR Negative selection and stringency modulation in phage-assisted continuous evolution. Nat. Chem. Biol 10, 216–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buchanan LE et al. Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci 110, 19285–19290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sivanesam K, Shu I, Huggins KNL, Tatarek-Nossol M, Kapurniotu A & Andersen NH Peptide Inhibitors of the amyloidogenesis of IAPP: verification of the hairpin-binding geometry hypothesis. FEBS Lett. 590, 2575–2583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao Y, Yu L, Yang R, Ma C, Qu L & de Harrington PB New peptide inhibitors modulate the self-assembly of islet amyloid polypeptide residues 11–20 in vitro. Eur. J. Pharmacol 804, 102–110 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Wang CK & Craik DJ Cyclic peptide oral bioavailability: lessons from the past. Pept. Sci 106, 901–909 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Shah NH, Dann GP, Vila-Perelló M, Liu Z & Muir TW Ultrafast protein splicing is common among cyanobacterial split inteins: Implications for protein engineering. J. Am. Chem. Soc 134, 11338–11341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Townend JE & Tavassoli A Traceless Production of Cyclic Peptide Libraries in E. coli. ACS Chem. Biol 11, 1624–1630 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Pu J, Zinkus-Boltz J & Dickinson BC Evolution of a split RNA polymerase as a versatile biosensor platform. Nat. Chem. Biol 13, 432–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller SM et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol 38, 471–481 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller SM, Wang T & Liu DR Phage-assisted continuous and non-continuous evolution. Nat. Protoc 15, 4101–4127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gade Malmos K, Blancas-Mejia LM, Weber B, Buchner J, Ramirez-Alvarado M, Naiki H & Otzen D ThT 101: a primer on the use of thioflavin T to investigate amyloid formation. Amyloid 24, 1–16 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reporting the findings of this study are included in the Article, Supplementary Data, Source Data, and Supplementary Information. High-throughput sequencing information is available at the NCBI GEO database under accession code GSE279094. Plasmids used in this study are available through Addgene.