

Abstract
Marine invertebrates are a rich source of structurally diverse secondary metabolites with broad biological activities, making them valuable for drug discovery. The genus Zoanthus is particularly noteworthy, producing numerous bioactive alkaloids, including the zoanthamines, which show promise in treating osteoporosis. Osteoporosis, a debilitating bone disease characterized by reduced bone mineral density and increased fracture risk, is linked to Wnt signaling pathway dysregulation. This highly conserved pathway maintains tissue homeostasis and is crucial for neurogenesis, synapse formation, and bone development. Dickkopf-1 (DKK1) and glycogen synthase kinase-3β (GSK-3β), key Wnt pathway regulators, are established therapeutic targets for osteoporosis. This study employed an integrated computational approach—combining molecular docking, extensive molecular dynamics (MD) simulations, and density functional theory (DFT) calculations—to assess the inhibitory potential of 69 zoanthamine-type alkaloids against DKK1 and GSK-3β. MD simulations, analyzing root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration, and free energy landscape, provided insights into protein-ligand complex stability and key interactions. Binding free energies were calculated using the MM-PBSA method combined with interaction entropy. DFT calculations further elucidated the electronic structure and reactivity of the most promising inhibitors (3α-hydroxyzoanthenamine, epioxyzoanthamine, 7α-hydroxykuroshine E, and norzoanthamine), which exhibited favorable binding interactions with key residues in target proteins. This integrative approach demonstrates the power of computational methods in drug discovery, highlighting the potential of zoanthamine alkaloids as lead compounds for innovative osteoporosis therapies.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-97537-8.
Keywords: Osteoporosis, DKK1, GSK-3β, Docking, Molecular dynamics, DFT
Subject terms: Drug discovery, Drug screening, Target identification
Introduction
Osteoporosis is a multifactorial bone metabolic disorder, distinguished by a reduction in bone mineral density, increased bone brittleness, impaired bone microarchitecture, and an elevated susceptibility to fracture1. The increasing prevalence of osteoporosis, particularly within the aging population, presents a significant public health concern, substantially impacting the quality of life for older adults. A considerable proportion of the population is at risk; studies indicate that approximately half of women over 50 and one-fifth of men worldwide are at risk of developing osteoporosis or experiencing fragility fractures2. Osteoporosis pathogenesis is characterized by an imbalance in bone remodeling, resulting from heightened osteoclast activity and reduced osteoblast function. This disruption of bone metabolism leads to progressive bone loss and structural deterioration3. Therapeutic strategies for osteoporosis predominantly focus on inhibiting bone resorption or promoting bone formation4, with bisphosphonates being a commonly employed class of drugs. However, vigilance in monitoring for gastrointestinal irritation and other adverse effects is crucial, as these treatments can lead to severe complications, including osteonecrosis of the jaw and profound suppression of bone turnover5.
Three Wnt signaling pathways—canonical Wnt/β-catenin, non-canonical Wnt/Ca²⁺, and planar cell polarity—are implicated in mesenchymal stem cell development, differentiation, and apoptosis, thereby influencing bone remodeling and contributing to the pathogenesis of bone loss6,7. In this study, we focused on the initial pathway involving the linkage of the Wnt protein to the F-class G protein-coupled transmembrane seven-helical Frizzled receptor and the co-receptor lipoprotein receptor-related protein 6 (LRP6). This interaction induces the abnormal phosphorylation of Disheveled, facilitating to the disassembly of the destruction complex, which consists of Axin, Adenomatous Polyposis Coli (APC), cyclin-dependent kinase inhibitor (CKI), and GSK-3β, ultimately resulting in the inactivation of GSK-3β. Consequently, the phosphorylation of β-catenin is inhibited, preventing its degradation via the ubiquitin-proteasome pathway. This leads to an increase in cytoplasmic β-catenin accumulation and its subsequent translocation into the nucleus, where it activates T-cell factor/lymphoid enhancer-binding factor (Tcf/Lef) transcriptional factors, which are essential for proper osteogenesis8–10.
LRP6 serves as a co-receptor for Wnt ligands, which, in conjunction with the Frizzled receptor, initiates the activation of the canonical Wnt/β-catenin signaling pathway. However, Wnt signaling pathway activity is initially inhibited by Dickkopf-1 (DKK1) protein, which binds to LRP6 in the extracellular matrix, thereby preventing Wnt interaction11. Assessing nucleotide sequence has demonstrated that LRP6 proteins consist of four intercellular YWTD domain repeats (P1–P4) interspersed with epidermal growth factor (EGF) repeats (E1-E4) spanning amino acids 21 to 1246, followed by three LDLR-type domains12. The four YWTD-EGF repeats form a functional recognition ectodomain responsible for binding to Wnt ligands and suppressors. Based on structural studies, these parallel repeats can be categorized into LRP6(1–2) and LRP6(3–4) units, with LRP6(1–2) identified as a active region for Wnt9b and Wnt1, while LRP6(3–4) preferentially binds Wnt3a. Furthermore, Dickkopf-1 (DKK1), a Wnt antagonist, belongs to the Dkk protein family, characterized by cysteine-rich Dkk_N and Dkk_C domains connected by a connector of approximately 50 residues. DKK1 binds to both sites: DKK1_N to LRP6(1–2) and DKK1_C to LRP6(3–4)13. In this study, the crystal structure of the DKK1-LRP6 complex (PDB ID: 3S2K), which confirms the upper surface of LRP6 as the target site for DKK114, was utilized to explore how inhibition of DKK1 could represent a potential therapeutic strategy for osteoporosis by modulating Wnt signaling.
As previously discussed, β-catenin is a key signaling molecule in the transduction process and serves as a central component linking the Wnt signaling pathway to bone-related cells. Specifically, an immediate influence is exerted by β-catenin on osteoblastic precursor cells and osteoblasts15. However, GSK-3β is the primary enzyme responsible for the phosphorylation of β-catenin, which subsequently leads to its ubiquitination and degradation. Therefore, inhibiting GSK-3β holds significant therapeutic potential for the treatment of osteoporotic patients.
Zoanthamines represent a unique family of non-aromatic alkaloids predominantly isolated from species within the genus Zoanthus16. Zoanthamines have been reported to exhibit a variety of pharmacological activities, including antiplatelet aggregation17, anti-inflammatory18, antibacterial19, and antiosteoporotic effects20,21. While zoanthamines exhibit diverse biological activities, the underlying mechanisms remain largely undefined except for norzoanthamine. Norzoanthamine shows promise as an anti-osteoporotic agent, demonstrably reducing bone loss by inhibiting IL-6 secretion20. This study employed in silico screening to evaluate the binding affinities of previously characterized alkaloids to key proteins within the Wnt signaling pathway—specifically DKK1 and GSK-3β—known to regulate bone remodeling. To further validate these interactions, molecular dynamics (MD) simulations, free energy landscape, and binding free energy calculations were performed using the molecular mechanics/Poisson–Boltzmann surface area (MM-PBSA) method to evaluate the stability of the docked complexes. Additionally, DFT analyses were employed to examine the chemical reactivity of the identified alkaloids. The identified alkaloid compounds, based on integrated computational analyses, warrant further investigation through in vitro and in vivo studies to confirm their therapeutic potential.
Materials and methods
Protein and ligand Preparation
The crystal structures involved in Wnt signaling inhibition through Dickkopf binding to LRP5/6 (PDB code: 3S2K)22, and glycogen synthase kinase-3β (PDB code: 1Q5K)23, complexed with the inhibitor n-(4-methoxybenzyl)-n′-(5-nitro-1,3-thiazol-2-yl)urea (TMU), were retrieved from the Protein Data Bank. These structures were imported and pre-processed using AutoDock Tools 1.5.6 by removing water molecules and adding hydrogen atoms. The proteins were then subjected to ionization and protonation calculations, followed by energy minimization, optimized for molecular docking. In the case of GSK-3β, the docking grid box was centered at the position of the original ligand (TMU) within the crystal structure.
Conversely, for DKK1, the grid was constructed based on the identification and selection of key residues. The identification of hotspot regions in the DKK1 protein revealed critical binding site residues, including THR221, CYS217, ARG237, CYS233, CYS253, LYS222, ARG224, ARG236, GLN184, HIS204, TRP206, ILE209, LYS211, VAL219, CYS220, ARG259, and LEU260, which are involved in interactions with the LRP6 protein. These interactions play a significant role in inhibiting the Wnt signaling pathway24,25. Thus, molecular docking simulations, employing ligand-specific grid dimensions (45 × 45 × 45, 0.375 Å spacing) scaled proportionally to each ligand’s radius of gyration, were centered on key residues within the DKK1 binding region to ensure comprehensive sampling of the interaction site26.
69 structures of all zoanthamine-type alkaloids16,27–29, alongside the control ligand (TMU) for the GSK-3β protein, were initially generated in two-dimensional (SDF) format using ChemBioDraw Ultra 13.3. These 2D structures were then converted into three-dimensional configurations and underwent energy minimization using Open Babel30. The energy-optimized structures were subsequently employed in molecular docking experiments.
Molecular Docking
Molecular docking is a computational technique used to predict the interactions between a protein and small molecules based on their geometric compatibility and binding scores. In this study, molecular docking was performed using the AutoDock 4.2 software31 to calculate the binding affinity between the receptor and the ligand. The docking results were visualized using Discovery Studio 2021, providing a detailed representation of the binding interactions, including hydrogen bonding, hydrophobic interactions, and involvement of specific amino acid residues in the binding process.
Molecular dynamics simulations
The consistency and dynamic characteristics of the protein-ligand interaction were assessed using GROMACS 2024.1 software32, with the aim of determining precise binding modes33. Simulations were conducted on proteins, utilizing the initial conformations derived from molecular docking studies. Protein structures were optimized using the CHARMM-GUI platform34, while ligands were fully hydrogenated using Discovery Studio Visualizer before generating topology files via the SwissParam web server (http://www.swissparam.ch)35. The CHARMM36 force field was employed to generate protein topologies using the GROMACS pdb2gmx module. This force field was utilized for all subsequent stages of the simulation36. Apo and protein-ligand complex systems were solvated in a cubic box using the TIP3P three-site water model37,38, maintaining a 10 Å buffer from the box edges. Charge neutralization was achieved through the addition of appropriate counterions39, and a physiological NaCl concentration of 0.15 M40 was employed to mimic the cellular environment. Following solvation and neutralization, the system’s geometry was optimized via energy minimization using the steepest descent algorithm for 50,000 steps to eliminate steric clashes and ensure structural integrity prior to molecular dynamics simulation41. All simulations employed a constant Number of particles, Volume, and Temperature (NVT) ensemble to maintain a reference temperature of 300 K. Subsequent equilibration at a reference pressure of 1 atm was achieved using a constant Number of particles, Pressure, and Temperature (NPT) ensemble; both NVT and NPT equilibration phases lasted for 100 ps42. The primary simulation was executed for 100 ns, with trajectories recorded at intervals of 0.01 ns. The outcomes of MD were subsequently analyzed to calculate the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA) as well as to assess connection between the ligands and main residues through the distribution of hydrogen bonds formation, visualized using VMD software43. In this study, hydrogen bonds were defined based on the criterion that the bonding angle between the hydrogen donor (D) and acceptor (A) (D-H⋯A) is greater than 120°, and the distance between the donor and acceptor to be no greater than the specified threshold of 3.5 Å44. In this study, the percentage occupancy of a type of contact between a ligand with residue was calculated by the formula:
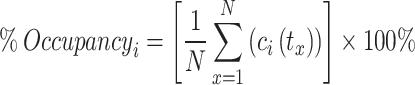 |
1 |
where N is the number of frames of the trajectory, ci is the total number of bonds of residue i with the ligand in frame x. The frequency values of residues can reach over 100% as they have formed multiple contacts with the ligand45.
Principal component and free energy landscape analysis
Principal component analysis (PCA)46 was applied to identify dominant collective motions within the protein upon ligand binding, transforming correlated atomic displacements into uncorrelated principal components (PCs) to facilitate analysis of essential conformational fluctuations. Analysis of the leading PCs (PC1 and PC2), visualized as a three-dimensional free energy landscape (FEL)47, provided insights into the relationship between protein dynamics, free energy, and structural stability, enabling the identification of energetically favorable binding modes and estimation of inhibitor efficiency.
MM-PBSA binding energy
The binding-free energy calculations were performed using the gmx_MMPBSA package, relying on the sole trajectory from GROMACS with the CHARMM36 force field48. This tool facilitates free energy calculations using the MM-PBSA method over the last 50 ns, with each nanosecond corresponding to a distinct frame, as described by the following equation49:
 |
2 |
And each G term is given by:
 |
3 |
Therefore, ΔGbind can be calculated as:
 |
4 |
In which:
 |
5 |
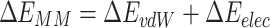 |
6 |
 |
7 |
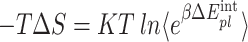 |
8 |
In the above equations, ΔEMM, ΔGsolvent, and TΔS are the changes in the gas phase molecular mechanics energy, solvation free energy, and conformational entropy upon ligand binding. ΔEvdW and ΔEelec are the van der Waals and the electrostatic interactions energy, respectively.
ΔGsolvent are calculated from polar ΔGPB (electrostatic solvation energy) and nonpolar ΔGSA between the solute and the continuum solvent. The entropy term (− TΔS) was calculated by the interaction entropy (IE) method previously described in48,50. Although MM-GBSA offers greater computational efficiency, MM-PBSA, with its more rigorous theoretical foundation, was deemed more suitable for determining the precise binding free energies. Consequently, MM-PBSA calculations were performed using MD trajectories for each protein-ligand complex49,51.
Reagents and chemicals
The reagents and chemicals employed for cell culture comprised Minimum Essential Medium α (MEM α, Thermo Scientific, Waltham, USA), fetal bovine serum (FBS, Merck, Darmstadt, Germany), penicillin-streptomycin (Thermo Scientific, Waltham, USA), sodium pyruvate (Thermo Scientific, Waltham, USA), non-essential amino acids (NEAA, Thermo Scientific, Waltham, USA), sodium bicarbonate (Sigma-Aldrich, St. Louis, USA), trypan blue (Sigma-Aldrich, St. Louis, USA), Dulbecco’s phosphate-buffered saline (PBS, Gibco, Waltham, USA), and trypsin (Thermo Scientific, Waltham, USA). Additionally, rutin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO) were sourced from Sigma-Aldrich (St. Louis, USA) for assessing cell viability. For the evaluation of alkaline phosphatase (ALP) activity, the following reagents were utilized: 0.9% isotonic sodium chloride solution (Taiwan Biotech, Taipei, Taiwan), Triton X-100 (J-T Baker, Phillipsburg, USA), p-nitrophenyl phosphate (Mallinckrodt, St. Louis, USA), BCA Protein Assay Kit (Pierce™, Thermo Scientific, Waltham, USA), and sodium hydroxide (NaOH, Honeywell, Muskegon, USA). Furthermore, the following reagents were utilized for mineralization: three of which are ascorbic acid, β-glycerophosphate, and alizarin red S obtained from Sigma-Aldrich (St. Louis, USA), along with cetylpyridinium chloride (Mallinckrodt, St. Louis, USA).
Cell viability assay
Cell viability was assessed using the MTT assay, as previously described in the literature52. Briefly, MG-63 cells were cultured in 96-well plates at a density of (4 × 103) cells per well. After 24 h, the culture medium was replaced with freshly formulated medium containing the test samples. Following a 72-h incubation period, the medium was removed, and MTT was added for 4 h. Subsequently, the wells were treated with 100 µL of DMSO, and the resulting dark-blue precipitate was dissolved by gentle mixing. Optical density (OD) measurements were obtained at 600 nm using a microplate reader (Bio Tek Instruments, Winooski, VT, USA), in accordance with the manufacturer’s instructions.
ALP activity assay
ALP activity in MG-63 cells was assessed using a colorimetric assay with p-nitrophenyl phosphate as the substrate53. Initially, the protocols for culturing MG-63 cells were consistent with those employed for the cell viability assessment. After a 72-h incubation, the medium was removed, and the cells were rinsed with 200 µL of normal saline before being treated with a lysis buffer containing 0.1% Triton X-100. Protein levels in the supernatant were quantified using a BCA assay, where the supernatants were mixed with BCA reagent for 30 min at room temperature, and absorbance was measured at 560 nm. Subsequently, ALP activity was evaluated by incubating the supernatants for 1 h at room temperature in a buffer solution composed of 0.2 M Tris-HCl (pH 9.5), supplemented with 0.1% Triton X-100, 1 mM MgCl2, and 6 mM p-nitrophenyl phosphate. The reaction was terminated by adding 0.5 M NaOH, and the absorbance was measured at 405 nm, allowing for the adjustment of ALP activity according to the protein levels of each sample.
Conceptual DFT
Density Functional Theory (DFT) serves as an indispensable tool for elucidating molecular and atomic structures by analyzing the energies of molecular orbitals, thereby offering critical insights into the structure-activity relationships of molecules54. This theoretical framework is fundamentally based on the Hohenberg–Kohn theorem55. In the present study, a subfield of DFT, termed Conceptual DFT, was employed to explore the chemical behavior of molecules through concepts derived from electron density56. Geometry optimizations and frequency calculations for the molecular structures of the compounds were conducted using DFT at the B3LYP/6–31G(d, p) level in the gas phase, utilizing the Gaussian 16 software suite (Gaussian Inc.; Wallingford, CT, USA). The absence of imaginary frequencies confirmed that all compounds analyzed in this study represented stable points on the potential energy surface (PES). Furthermore, various molecular characteristics, including molecular electrostatic potential (MEP) surfaces, the energy levels of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), as well as reactivity parameters from quantum chemistry, were calculated using DFT at the B3LYP/6–311++G(d, p) level, followed by below five discrete equations57:
 |
9 |
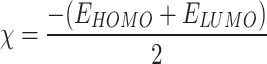 |
10 |
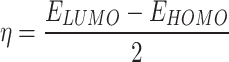 |
11 |
 |
12 |
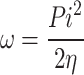 |
13 |
Results and discussion
Docking protocol validation
The generated grid was validated through a redocking procedure, wherein the co-crystallized ligand was re-docked using the same grid parameters. Following this, the resulting poses were superimposed, and the root mean square deviation (RMSD) was calculated for comparison. The redocking process was successfully performed, with the alignment of the docked poses shown in Fig. S1. Specifically, redocking of the co-crystallized ligand (TMU) from GSK-3β yielded a docking score of − 7.036 kcal/mol. The superimposition of the docked and co-crystallized ligands resulted in an RMSD of 1.406 Å, which falls within the acceptable threshold of < 2.0 Å. This confirms that the grid generation effectively targeted the inhibitor binding pocket and validates the accuracy of the docking approach used.
Molecular Docking
In this study, all compounds, illustrated in Table S1, were subjected to molecular docking with two distinct proteins: DKK1 and GSK-3β. Among these, the top ten ligands with the lowest docking scores for the DKK1 were selected for deeper scrutiny of their interactions and subsequent evaluation via MD. Similarly, for the GSK-3β protein, compounds exhibiting docking scores lower than that of the control ligand, TMU, were prioritized for further investigation. The detailed 3D and 2D interactions are illustrated in Figs. 1 and 2.
Fig. 1.

3D and 2D interactions between ligands and protein DKK1. (a) Kuroshine F. (b) Kuroshine C. (c) Epioxyzoanthamine. (d) Kuroshine B. (e) Zoanthenamine. (f) Norzoanthaminone. (g) 5α-methoxykuroshine E. (h) 18β-hydroxykuroshine J. (i) Zoanthamine. (j) Norzoanthamine.
Fig. 2.

3D and 2D interactions between ligands and protein GSK-3β. (a) Kuroshine E. (b) 7α-hydroxykuroshine E. (c) 11β-chloro-11-deoxykuroshine E. (d) 22-epi-28-deoxyzoanthenamine. (e) 18β-hydroxykuroshine J. (f) Epioxyzoanthenamine. (g) 7β-hydroxykuroshine (A) (h) 2-hydroxy-11-ketonorzoanthamide (B) (i) 3α-hydroxyzoanthenamine. (j) 27-hydroxykuroshine A. (k) TMU.
For the DKK1 protein, kuroshine F exhibited the lowest binding energy among the evaluated compounds (− 8.952 kcal/mol), followed closely by kuroshine C (− 8.950 kcal/mol) and epioxyzoanthamine (− 8.921 kcal/mol), as presented in Table 1. Among the top ten ligands, norzoanthamine achieved the highest docking score (− 8.718 kcal/mol) despite forming only a single hydrogen bond with THR221. The remaining six ligands demonstrated minimal variations in binding energy, ranging from − 8.905 to − 8.724 kcal/mol. ARG236 was recognized as a frequent hydrogen bond partner for the majority of ligands, except for norzoanthamine. Notably, kuroshine F demonstrated interactions with four critical residues, forming hydrogen bonds with CYS220, THR221, and LYS222, while engaging in alkyl bonding with ARG224. A similar binding pattern was observed for kuroshine C. In contrast, norzoanthaminone formed a hydrogen bond with ARG224 and an additional hydrogen bond with LYS222. Other notable interactions included LYS211 and VAL219 forming hydrogen bonds with epioxyzoanthamine and zoanthenamine, with LYS211 also acting as a hydrogen bond partner in zoanthamine. Kuroshine B and 5α-methoxykuroshine E exhibited comparable binding interactions with DKK1, engaging THR221, LYS222, and ARG224. Similarly, 18β-hydroxykuroshine J stabilized the receptor by forming a hydrogen bond with ARG236 and CYS220, along with an alkyl bond with ARG224. Although not located within the identified protein hotspot, HIS223, GLU232, HIS261, GLU185, CYS237, and LYS226 consistently formed hydrogen bonds and hydrophobic interactions with the ligand across ten compounds. Consequently, these interactions highlight the significant contribution of these residues to defining the DKK1 active site and to ligand binding affinity, with increased interaction correlating to enhanced complex stability24,25,58.
Table 1.
Docking results of the top ten ligands with their interactions with DKK1.
| Compound | Docking score (kcal/mol) | Interaction residue | |
|---|---|---|---|
| Hydrogen bond | Other bonds | ||
| Kuroshine F | − 8.952 | HIS261, LYS222, THR221, CYS220, ARG236, GLU232, HIS223 | ARG224 |
| Kuroshine C | − 8.95 | GLU232, ARG236, CYS220, LYS222, THR221, LEU260, HIS261 | ARG224 |
| Epioxyzoanthamine | − 8.921 | VAL219, ARG236, LYS211, GLU185 | HIS204 |
| Kuroshine B | − 8.905 | HIS261, LYS222, THR221, ARG236 | ARG224, LYS222 |
| Zoanthenamine | − 8.894 | VAL219, HIS204, LYS211, ARG236, CYS237 | ARG236, VAL219 |
| Norzoanthaminone | − 8.829 | ARG236, HIS261, ARG224, LYS222 | LYS222, HIS223, HIS261 |
| 5α-methoxykuroshine E | − 8.808 | ARG236, GLU232, THR221, CYS220, LYS222, HIS261 | ARG224 |
| 18β-hydroxykuroshine J | − 8.774 | ARG236, CYS220, HIS261 | ARG224 |
| Zoanthamine | − 8.724 | HIS204, LYS211, CYS237, ARG236 | |
| Norzoanthamine | − 8.718 | THR221 | LYS226, HIS223 |
For protein GSK-3β, all ten top-ranked compounds exhibited binding energies lower than that of the reference ligand, TMU (− 7.036 kcal/mol), as summarized in Table 2. Among these, kuroshine E exhibited the lowest binding energy (− 12.807 kcal/mol), followed by 7α-hydroxykuroshine E (− 12.337 kcal/mol), while the remaining eight ligands displayed binding energies ranging from − 11.598 to − 10.603 kcal/mol. Most ligands formed hydrogen bonds with GSK-3β, although 7α-hydroxykuroshine E interacted with TYR134 through pi-alkyl bonds. Notably, TYR134 frequently appeared as a pi-alkyl interaction partner, except in the cases of epioxyzoanthamine, 2-hydroxy-11-ketonorzoanthamide B, 27-hydroxykuroshine A, and 18β-hydroxykuroshine J, which instead exhibited unique interactions with ASN186. Kuroshine E and 3α-hydroxyzoanthenamine formed notable alkyl interactions with LEU188 and hydrogen bonds with LYS85, a pattern also observed in 22-epi-28-deoxyzoanthenamine. Additionally, the latter compound formed further interactions with THR138 and LEU188 via hydrogen and alkyl bonds. In contrast, LYS85 engaged in alkyl bonding with 2-hydroxy-11-ketonorzoanthamide B. Interestingly, 27-hydroxykuroshine A and TMU both interacted with VAL135 through hydrogen bonds and shared interactions with ASP200, CYS199, and LEU188. These residues are critical components of the ATP-binding pocket of GSK-3β59. Conversely, epioxyzoanthamine did not exhibit any interactions with the key residues.
Table 2.
Docking results of the top ten ligands and the control ligand (TMU), along with their interactions with GSK-3β.
| Compound | Docking score (kcal/mol) | Interaction residue | |
|---|---|---|---|
| Hydrogen bond | Other bonds | ||
| Kuroshine E | − 12.807 | LYS85 | ILE62, LEU188, ALA83, TYR134 |
| 7α-hydroxykuroshine E | − 12.337 | – | ILE62, ALA83, TYR140, TYR134 |
| 11β-chloro-11-deoxykuroshine A | − 11.598 | ILE62 | ALA83, ARG141, TYR140, TYR134 |
| 22-epi-28-deoxyzoanthenamine | − 11.131 | LYS85, ILE62 | ILE62, TYR134, TYR140 |
| 18β-hydroxykuroshine J | − 11.008 | ASN186, GLN185 | ARG141, TYR140 |
| Epioxyzoanthamine | − 10.86 | GLY63 | ILE62 |
| 7β-hydroxykuroshine A | − 10.685 | ILE62 | TYR134, ARG141, TYR140 |
| 2-hydroxyl-11-ketonorzoanthamide B | − 10.635 | LYS183, ASN64 | LYS85, VAL70, LEU132 |
| 3α-hydroxyzoanthenamine | − 10.615 | LYS85, GLN185, GLY65, THR138 | ILE62, LEU188, TYR134 |
| 27-hydroxykuroshine A | − 10.603 | VAL135, ILE62 | ARG141, TYR140 |
| TMU* | − 7.036 | VAL135, ASP200, PRO136 | ILE62, LEU188, ALA83, VAL70, CYS199 |
*Control ligand, –: no interaction.
Molecular dynamics simulations
MD simulations lasting 100 ns were performed on ten docked ligand-DKK1 complexes, along with the apoprotein form, to examine conformational alterations and binding stability. The apoprotein and the ten protein-ligand complexes were designated as D0 through D10, as illustrated in Fig. 3, with the corresponding parameters provided in Table S2. An analysis of RMSD across triplicate simulations indicated differing levels of stability among the ten DKK1 complexes (see Fig. S2). The apoprotein (D0) exhibited reproducible RMSD profiles in two of the replicates, with minor fluctuations (maximum peak of 4.5 Å) detected in the final 15 ns of the third replicate. Complexes D3, D6, and D9 displayed comparable RMSD profiles in two of the three replicates; however, the third replicate exhibited increased fluctuations (ranging from 1.5 to 3 Å) over the final 10–20 ns, reflecting a similar, albeit more pronounced fluctuation (maximum peak of 4.2 Å at 45 ns) noted in one replicate of complex D10. Complexes D2 and D5 maintained consistent RMSD trajectories throughout all three replicates, while complex D7 experienced greater fluctuation (with an amplitude of 3 Å) compared to D2 and D5. In complexes D1 and D4, one replicate exhibited stability after 20 and 40 ns, respectively, while the other replicates displayed significant fluctuations (amplitude between 1 and 2.5 Å) post-20 ns. Complex D8 sustained consistent RMSD values up to 60 ns, after which trajectory divergence was observed, leading to stabilization in each replicate. With the exception of zoanthamine (complex D10), all nine ligand-protein complexes demonstrated stable binding, with RMSD values for the ligands remaining below 1.5 Å and minimal fluctuations across the triplicate simulations (see Fig. S3). In contrast, zoanthamine manifested considerable and persistent RMSD fluctuations (amplitude exceeding 1.5 Å) from 10 ns onward across all replicates.
Fig. 3.

Backbone atom RMSD of DKK1 triplicate in apoprotein form and in complexes with 10 ligands. (D0) Apo-protein. (D1) 5α-methoxykuroshine E. (D2) Norzoanthamine. (D3) Kuroshine B. (D4) Kuroshine C. (D5) 18β-hydroxykuroshine J. (D6) Norzoanthaminone. (D7) Kuroshine F. (D8) Epioxyzoanthamine. (D9) Zoanthenamine. (D10) Zoanthamine.
Protein dynamics and ligand-induced effects on DKK1 were assessed by analyzing RMSF, Rg, SASA, and hydrogen bonding interactions from a single replicate of each triplicate MD simulation. RMSF analysis (Fig. 5A) revealed that key DKK1 residues (VAL219, CYS200, LYS211, THR221) exhibited fluctuations below 2 Å throughout the simulation, with similar patterns observed across all complexes except D7, which initially showed elevated RMSF (10 Å) before aligning with the others. Rg values remained within a consistent range (12.44–14.91 Å; Fig. 5B), suggesting maintenance of native protein conformation. SASA analysis (Fig. 5C) indicated that ligands were largely sequestered within the DKK1 binding pocket, resulting in minimal solvent exposure and stable binding interactions (54.72–78.35 nm2 for all complexes except D7, which reached a peak of 78 nm2 at 53 ns). This suggests that the ligands are effectively shielded from solvent, promoting robust binding affinity and stability60.
Fig. 5.

RMSF (A,D), Rg (B,E), SASA (C,F) values for the complexes involving the DKK1 and GSK-3β proteins with ligands.
During the 100 ns molecular dynamics simulation of the DKK1 protein, hydrogen bond occupancy data, as shown in Table S5, indicated that only five out of ten compounds had occupancy exceeding 50%. Norzoanthamine interacted with four out of seven key residues—CYS220, THR221, LYS222, and ARG236—with THR221 exhibiting the highest distribution percentage among them. Moreover, 18β-hydroxykuroshine J and epioxyzoanthamine interacted with three distinct amino acids, with LYS222 (120%) in the case of the former and ARG236 (229.51%) for the latter showing the highest occupancy. Additionally, kuroshine B engaged with ARG224 and THR221 through hydrogen bonds, with occupancy values of 129.17% and 86.48%, respectively, while zoanthamine only interacted with ARG236, registering an occupancy of 92.37%. In contrast, the five remaining ligands interacted with key residues; however, their occupancy percentages for individual amino acids were below 75%, indicating weaker hydrogen bond interactions and less robust binding45.
For the GSK-3β protein, the apoprotein form and eleven docked complexes were categorized as G0 to G11, as depicted in Fig. 4. During the first and second simulation runs involving the apoprotein, the RMSD values exhibited significant fluctuations, with prominent peaks observed at approximately 70 ns and 30 ns. In contrast, the complexes formed between the protein and various ligands—specifically complexes G1, G2, G3, G5, G6, and G11—reached equilibrium conditions relatively quickly and maintained stability throughout the duration of the simulation, with average RMSD values consistently remaining below 3 Å. This observation indicates that these ligands may contribute to reducing the flexibility of the protein in comparison to its apo-form. Furthermore, the GSK-3β complex G4 displayed an RMSD peak nearing 4 Å during the first run, while the third run recorded a minimum RMSD of 1.5 Å, with the second run demonstrating moderate stability between 40 ns and 100 ns. Additionally, the RMSD plots for the protein in complexes G7 and G9 exhibited similar fluctuation patterns until approximately 60 ns across the three simulations, after which significant divergence occurred, accompanied by an increased amplitude around 3.5 Å. The RMSD plots for GSK-3β in complexes G8 (first and second runs) and G10 (second run) revealed two distinct phases of stability, specifically from 10 to 40 ns and from 60 to 100 ns, with a peak RMSD value of 3.5 Å observed during the intermediate period of 40 to 60 ns. The plots of average RMSD for backbone atoms, along with their standard deviations for the twelve protein-ligand complexes, are illustrated in Fig. S4. An analysis of the RMSD trajectories of the protein-ligand complexes revealed that, with the exception of epioxyzoanthamine and 18β-hydroxykuroshine J, all ten ligands exhibited stable binding (RMSD < 1.0 Å) with significantly reduced fluctuations across triplicate simulations when compared to the control (Fig. S5). Epioxyzoanthamine (complex G5) exhibited distinct RMSD profiles across replicates: two rapidly stabilized near 1.2 Å, while the third showed initial stabilization at 0.8 Å before increasing to 1.2 Å. Similarly, 18β-hydroxykuroshine J (complex G10) demonstrated replicate-dependent stability; two replicates remained stable at approximately 1 Å until 90 ns, transitioning to a second equilibrium state around 2 Å thereafter, while the third replicate maintained stability throughout.
Fig. 4.

Backbone atom RMSD of GSK-3β triplicate in apoprotein form and in complexes wih 10 ligands and one control. (G0) Apo-protein. (G1) Kuroshine E. (G2) 3α-hydroxyzoanthenamine. (G3) 7α-hydroxykuroshine E. (G4) 11β-chloro-11-deoxykuroshine A (G5) Epioxyzoanthamine. (G6) 2-hydroxy-11-ketonorzoanthamide B (G7) 22-epi-28-deoxyzoanthenamine. (G8) 7β-hydroxykuroshine A. (G9) 27-hydroxykuroshine A. (G10) 18β-hydroxykuroshine J. (G11) TMU.
Additionally, RMSF plot of the GSK-3β backbone and its complexes, which displayed similar fluctuation patterns, was utilized to characterize the spatial variations of the ligand molecules within the protein structure. As shown in Fig. 5D, key residues at the active site of GSK-3β, including LYS85, TYR134, VAL135, CYS199 and ASP20059, exhibited stable fluctuations with RMSF values below 2 Å throughout the entire simulation. The Rg for the 12 docked complexes, as depicted in Fig. 5E, ranged from 21.11 to 22.35 Å with minimal fluctuations, indicating the absence of any abnormal conformational changes throughout the trajectory. For all ten compounds and the control ligand (TMU), the SASA profiles exhibited relative stability throughout the simulation, with values fluctuating around 185 nm2 and ranging from 170.75 to 199.18 nm2, similar to the apo-protein (Fig. 5F). The consistently low and stable SASA values suggest that the ligands are effectively encapsulated within the protein binding pocket, minimizing solvent exposure and promoting robust, stable binding interactions that enhance both stability and affinity.
Among the compounds assessed, TMU demonstrated the highest frequency of interactions, establishing strong hydrogen bonds with three critical residues: TYR134 (97.67%), VAL135 (173.66%), and LEU188 (104.25%), as detailed in Table S7. Overall, all ligands exhibited significant occupancy of hydrogen bonds with essential amino acids, surpassing 75%, with the exception of 18β-hydroxykuroshine J, which displayed minimal hydrogen bond interactions with all principal amino acids. Notably, 2-hydroxy-11-ketonorzoanthamide B interacted with five out of eight key residues, exhibiting a very high occupancy of hydrogen bonds, specifically with LYS85, TYR134, ASN186, LEU188, and ASP200, with ASN186 (213.60%) ranking highest among the ligands. In contrast, 7β-hydroxykuroshine A and kuroshine E exhibited reduced interaction with key amino acids, forming only one hydrogen bond with ASN186 (186.70%) and two hydrogen bonds with THR138 (61.20%) and ASN186 (98.40%), respectively. Additionally, THR138 was identified as having the highest hydrogen bond occupancy in 3α-hydroxyzoanthenamine, at 276.27%. Furthermore, 7α-hydroxykuroshine E engaged with GSK-3β through four key amino acids, with ASP200 (212.27%) showing the greatest percentage of hydrogen bond acceptor participation. It is noteworthy that epioxyzoanthamine did not form hydrogen bonds with key residues of GSK-3β during docking; however, it established hydrogen bonds with TYR134 (140.75%) and VAL135 (169.29%) during the simulation, acting as both a hydrogen bond donor and acceptor, with TYR134 exhibiting the highest occupancy. This observation underscores the limitations of docking simulations in accurately representing dynamic ligand-receptor interactions, which are more effectively elucidated through molecular dynamics analysis61. Additionally, 27-hydroxykuroshine A interacted with LYS85 with an occupancy value of 251.02%, ranking first among the ligands. Overall, the majority of ligands displayed strong and stable interactions with key residues within the binding pocket throughout the 100 ns molecular dynamics simulation, with the notable exception of 18β-hydroxykuroshine J.
Principle component and free energy landscape analysis
This study employed PCA62, a well-established dimensionality reduction technique, to investigate the functional dynamics of the DKK1 and GSK-3β ligand complexes, focusing on dominant conformational changes and their relationship to binding stability. PCA of the DKK1 complexes revealed distinct patterns in the distribution of principal components (PC1 and PC2). Complexes D2 and D8 exhibited significantly more tightly clustered conformations in the PC1-PC2 space when compared to the apoprotein (D0), strongly suggesting the presence of distinct, stable conformers in these two complexes, as illustrated in Fig. S6. In contrast, the remaining complexes displayed a substantially more dispersed and less dense distribution of conformations, indicating that these complexes underwent significantly greater conformational changes throughout the simulation and, consequently, may exhibit lower stability compared to complexes D2 and D8. Further analysis of the conformational dynamics was conducted using FEL, generated from the first two principal components (PC1 and PC2). These FELs, depicted as three-dimensional surfaces where color intensity corresponds to Gibbs free energy (dark blue-purple: 0–2 kJ/mol; dark red: up to 18 kJ/mol), provided a comprehensive visualization of the range of conformational freedom and stability across all twelve DKK1 complexes (Fig. 6). The apoprotein (D0) displayed a large, central minimum, indicative of a wide range of accessible low-energy conformations. Complexes D2, characterized by a relatively smaller central minimum compared to D0, and D8, exhibiting two distinct minima separated by a high-energy barrier, suggest that these complexes maintained relatively stable equilibrium conditions during the simulation. Conversely, the remaining complexes displayed markedly smaller minimum energy regions and numerous high-energy transitions (red regions), indicating significantly greater conformational flexibility and potentially lower stability relative to complexes D2 and D8.
Fig. 6.

3D FEL plots of DKK1 in apoprotein form (D0) and in complexes with 10 ligands. (D1) 5α-methoxykuroshine E. (D2) Norzoanthamine. (D3) Kuroshine B. (D4) Kuroshine C. (D5) 18β-hydroxykuroshine J. (D6) Norzoanthaminone. (D7) Kuroshine F. (D8) Epioxyzoanthamine. (D9) Zoanthenamine. (D10) Zoanthamine.
A similar PCA analysis was performed on the GSK-3β simulations. This analysis revealed that the majority of the GSK-3β complexes, excluding G10, exhibited relatively tightly clustered conformations, indicating stable binding and low-energy states. However, complex G10 displayed a significantly more dispersed conformational distribution, suggesting increased flexibility and potentially higher energy (Fig. S7). To further investigate these observations, FEL analysis was also conducted on these GSK-3β complexes, as illustrated in Fig. 7. This analysis revealed that complex G10 exhibited the smallest minimum energy region. The apoprotein (G0) displayed four low-barrier minima, suggestive of high conformational flexibility, while the control (G11) displayed two low-barrier minima, indicating instability. Complexes G4, G8, and G9 exhibited frequent fluctuations between distinct conformational states with low energy barriers, further indicative of high flexibility. In contrast, complex G5 demonstrated a single, broad, low-energy minimum separated by high energy barriers, suggesting greater stability compared to the control. Finally, complexes G1, G2, and G3 exhibited similar, centrally located minima, whereas complexes G6 and G7, characterized by smaller, bi-modal minima and significant energy barriers, demonstrated reduced stability relative to complexes G1–G3.
Fig. 7.

3D FEL plots of GSK-3β in apoprotein form (G0) and in complexes with 10 ligands and one control. (G1) Kuroshine E. (G2) 3α-hydroxyzoanthenamine. (G3) 7α-hydroxykuroshine E. (G4) 11β-chloro-11-deoxykuroshine A (G5) Epioxyzoanthamine. (G6) 2-hydroxy-11-ketonorzoanthamide B (G7) 22-epi-28-deoxyzoanthenamine. (G8) 7β-hydroxykuroshine A. (G9) 27-hydroxykuroshine A. (G10) 18β-hydroxykuroshine J. (G11) TMU.
MM-PBSA analysis for binding free energy calculation
Highly accurate binding free energy estimates for the top ten DKK1 and GSK-3β ligand complexes were obtained using MM-PBSA calculations incorporating interaction entropy (Tables 3 and 4). For DKK1, complex D2 exhibited the most favorable binding free energy (− 8.57 kcal/mol), followed by complex D6 (− 2.15 kcal/mol). Similarly, for GSK-3β, complex G2 showed the most favorable binding free energy (− 26.42 kcal/mol), followed by complexes G5 (− 24.85 kcal/mol) and G4 (− 21.20 kcal/mol). Notably, the control GSK-3β complex (− 14.43 kcal/mol) showed less favorable binding than complexes G3 and G8 (− 16.90 kcal/mol). Elevated binding free energies observed in the remaining complexes for both proteins are attributable to a combination of high interaction entropy and low MM-PBSA terms, indicative of conformational instability.
Table 3.
The calculation of binding free energy results of 10 top hit ligands with DKK1.
| Complex with ligand | kcal/mol | |||||||
|---|---|---|---|---|---|---|---|---|
| ΔEvdW | ΔEelec | ΔGPB | ΔGSA | ΔGgas | ΔGsol | − TΔS | ΔGbind | |
| 5α-methoxykuroshine E | − 7.07 ± 5.78 | − 2.51 ± 6.44 | 6.10 ± 7.81 | − 0.90 ± 0.71 | − 9.58 ± 9.47 | 5.20 ± 7.42 | 9.08 ± 3.12 | 4.70 ± 4.93 |
| Norzoanthamine | − 36.15 ± 3.21 | − 15.02 ± 3.52 | 35.10 ± 4.31 | − 3.28 ± 0.21 | − 51.17 ± 5.06 | 31.81 ± 4.22 | 10.78 ± 0.99 | − 8.57 ± 3.38 |
| Kuroshine B | − 15.67 ± 9.88 | − 7.47 ± 8.40 | 16.56 ± 13.36 | − 1.86 ± 1.10 | − 23.14 ± 16.35 | 14.70 ± 12.43 | 15.39 ± 0.12 | 6.95 ± 6.06 |
| Kuroshine C | − 22.22 ± 7.10 | − 11.04 ± 8.55 | 23.19 ± 8.69 | − 2.57 ± 0.73 | − 33.26 ± 10.57 | 20.62 ± 8.23 | 11.79 ± 2.40 | − 0.85 ± 4.90 |
| 18β-hydroxykuroshine J | − 8.63 ± 7.80 | − 1.91 ± 8.80 | 6.34 ± 8.51 | − 1.02 ± 0.84 | − 10.54 ± 10.59 | 5.32 ± 8.29 | 16.42 ± 0.12 | 11.20 ± 4.97 |
| Norzoanthaminone | − 34.16 ± 3.99 | − 16.15 ± 6.41 | 32.58 ± 6.90 | − 3.33 ± 0.33 | − 50.31 ± 9.18 | 29.25 ± 6.63 | 18.90 ± 0.78 | − 2.15 ± 3.50 |
| Kuroshine F | − 12.19 ± 6.00 | − 6.51 ± 10.03 | 11.74 ± 11.38 | − 1.44 ± 0.63 | − 18.71 ± 13.12 | 10.30 ± 11.08 | 23.07 ± 0.11 | 14.66 ± 4.64 |
| Epioxyzoanthamine | − 28.39 ± 2.35 | − 22.41 ± 5.29 | 36.94 ± 4.08 | − 3.15 ± 0.12 | − 50.80 ± 5.95 | 33.79 ± 4.01 | 15.13 ± 0.12 | − 1.88 ± 4.23 |
| Zoanthenamine | − 12.25 ± 5.84 | − 5.32 ± 14.72 | 10.30 ± 14.43 | − 1.41 ± 0.61 | − 17.57 ± 16.52 | 8.89 ± 14.27 | 14.35 ± 1.45 | 5.67 ± 5.36 |
| Zoanthamine | − 14.64 ± 6.79 | − 7.29 ± 13.22 | 13.62 ± 14.06 | − 1.75 ± 0.68 | − 21.93 ± 15.64 | 11.87 ± 13.90 | 17.51 ± 1.33 | 7.45 ± 5.22 |
Table 4.
The calculation of binding free energy results of 10 top hit ligands and control with GSK-3β.
| Complex with ligand | kcal/mol | |||||||
|---|---|---|---|---|---|---|---|---|
| ΔEvdW | ΔEelec | ΔGPB | ΔGSA | ΔGgas | ΔGsol | − TΔS | ΔGbind | |
| Kuroshine E | − 48.44 ± 2.72 | − 15.56 ± 3.18 | 46.46 ± 4.84 | − 4.89 ± 0.11 | − 64.00 ± 4.57 | 41.56 ± 4.80 | 12.66 ± 0.12 | − 9.78 ± 3.85 |
| 3α-hydroxyzoanthenamine | − 53.13 ± 2.52 | − 18.83 ± 4.98 | 45.03 ± 4.33 | − 4.89 ± 0.10 | − 71.96 ± 5.07 | 40.13 ± 4.31 | 5.40 ± 0.09 | − 26.42 ± 3.59 |
| 7α-hydroxykuroshine E | − 50.45 ± 3.73 | − 31.51 ± 5.69 | 60.09 ± 5.20 | − 4.88 ± 0.16 | − 81.96 ± 7.07 | 55.21 ± 5.13 | 9.90 ± 0.12 | − 16.85 ± 3.85 |
| 11β-chloro-11-deoxykuroshineA | − 45.74 ± 2.67 | − 26.19 ± 7.78 | 48.11 ± 4.86 | − 4.42 ± 0.09 | − 71.93 ± 7.52 | 43.69 ± 4.84 | 7.04 ± 1.33 | − 21.20 ± 4.90 |
| Epioxyzoanthamine | − 45.13 ± 3.58 | − 19.09 ± 4.50 | 38.22 ± 5.71 | − 4.46 ± 0.28 | − 64.22 ± 5.36 | 33.75 ± 5.53 | 5.62 ± 0.09 | − 24.85 ± 2.50 |
| 2-hydroxy-11-ketonorzoanthamide B | − 44.68 ± 3.49 | − 17.11 ± 7.78 | 45.72 ± 6.45 | − 4.57 ± 0.19 | − 61.80 ± 7.59 | 41.15 ± 6.48 | 16.30 ± 0.12 | − 4.34 ± 6.25 |
| 22-epi-28-deoxyzoanthenamine | − 38.30 ± 2.54 | − 2.91 ± 7.70 | 23.37 ± 7.39 | − 3.86 ± 0.24 | − 41.21 ± 8.60 | 19.51 ± 7.29 | 21.09 ± 0.12 | − 0.61 ± 4.12 |
| 7β-hydroxykuroshine A | − 47.86 ± 2.37 | − 14.47 ± 5.96 | 45.09 ± 5.36 | − 4.85 ± 0.12 | − 62.33 ± 5.67 | 40.24 ± 5.35 | 5.15 ± 0.10 | − 16.94 ± 3.84 |
| 27-hydroxykuroshine A | − 45.25 ± 2.85 | − 29.05 ± 6.57 | 57.63 ± 5.61 | − 4.61 ± 0.12 | − 74.30 ± 6.69 | 53.02 ± 5.60 | 9.48 ± 0.08 | − 11.79 ± 4.03 |
| 18β-hydroxykuroshine J | − 24.74 ± 6.87 | − 19.56 ± 14.49 | 31.33 ± 14.86 | − 2.65 ± 0.67 | − 44.30 ± 18.48 | 28.68 ± 14.39 | 24.62 ± 6.58 | 9.00 ± 8.39 |
| TMU* | − 35.54 ± 2.94 | − 31.59 ± 4.71 | 52.24 ± 4.57 | − 3.46 ± 0.16 | − 67.13 ± 5.14 | 48.77 ± 4.49 | 3.93 ± 1.28 | − 14.43 ± 3.41 |
*Control ligand.
Following initial identification of high-affinity zoanthamine alkaloid binders to DKK1 and GSK-3β via molecular docking, extensive MD simulations were performed to characterize dynamic binding interactions not fully resolved by static docking. This comprehensive analysis, integrating MD-derived RMSD, ligand interaction occupancy, PCA, FEL, MM-PBSA binding free energies incorporating interaction entropy, identified norzoanthamine as a promising DKK1 inhibitor and 3α-hydroxyzoanthenamine, 7α-hydroxykuroshine E, and epioxyzoanthamine as promising GSK-3β inhibitors. Furthermore, superimposition between the docked complex and post-MD docked complex after 100 ns MD simulation revealed that both structures had a similar binding position in the active points as the four complexes, including D2, G2, G3 and G5. We also took a snapshot (Fig. 8) from 0, 50 and 100 ns from MD simulation trajectory for the aforementioned complexes but no drastic change was observed for their binding pose. This integrated approach focused on identifying zoanthamine alkaloids exhibiting specific and robust binding to key residues within the active sites of DKK1 and GSK-3β. The in vivo anti-osteoporotic activity of norzoanthamine hydrochloride20,21, which alleviates bone loss and enhances cortical bone thickening, supports the potential of these compounds.
Fig. 8.

The surface representation and the binding pockets of DKK1 and GSK-3β complexes with ligands analyzed using snapshots taken at 0, 50, and 100 ns. (D2) Norzoanthamine. (G2) 3α-hydroxyzoanthenamine. (G3) 7α-hydroxykuroshine E. (G5) Epioxyzoanthamine.
Evaluation of cell viability and ALP activity
Based on the availability of zoanthamine alkaloids, we selected three compounds, 3-hydroxynorzoanthamine, 3-acetoxynorzoanthamine, and norzoanthaminone, along with rutin as a positive control for bioassays aimed at assessing osteoporosis, with a specific emphasis on its effects on osteoblasts. In vitro bioassays were conducted to evaluate the potential toxic effects of these three compounds on the growth of MG-63 cells using an MTT-based cell viability assay. As shown in Fig. 9A, none of the compounds inhibited MG-63 cell proliferation at a concentration of 10 µg/mL. Following this, the compounds were analyzed for their effects on ALP activity. Results indicated that 3-hydroxynorzoanthamine significantly increased ALP levels to 197.15 ± 23.47% relative to the control group, while the other compounds did not demonstrate a significant impact on ALP activity (Fig. 9B). Given the activity of these compounds on ALP, further analysis of the top selected zoanthamine alkaloids, guided by docking and molecular dynamics studies, alongside the three tested in vitro compounds, is warranted. This will include DFT analysis of their molecular descriptors to thoroughly elucidate their potential therapeutic applications for osteoporosis treatment.
Fig. 9.

Results of three compounds on cell viability and ALP activity in MG-63 cells. (A) Cell viability was evaluated utilizing the MTT assay. (B) ALP activity was assessed in MG-63 cells treated with 10 µg/mL of each compounds.
Conceptual DFT
The configurations presented in Table S9 represent the geometries corresponding to the lowest energy states. Furthermore, geometry optimizations for four selected compounds satisfied the convergence criteria of the Gaussian 16 software package (see Table S8), indicating that these optimized structures represent minima on the potential energy surface. The electronic energy values for 3α-hydroxyzoanthenamine, epioxyzoanthamine, 7α-hydroxykuroshine E, norzoanthamine, 3-hydroxynorzoanthamine, 3-acetoxynorzoanthamine and norzoanthaminone were determined to be − 47,577.32, − 45,557.34, − 49,624.90, − 42,445.44, − 44,489.00, − 48,644.42, and − 44,456.15 eV, respectively. Notably, these compounds exhibited dipole moment values ranging from 3.06 to 8.68 Debye, indicating a substantial potential for dipole-dipole interaction phenomena within their molecular structures.
The MEP surface provides insights into the reactivity, size, shape, and charge distribution of compounds, aiding in identifying regions prone to electrophilic or nucleophilic attacks63. MEP surfaces, correlated with electron density, are commonly represented graphically using a color-coded scheme. Regions depicted in blue correspond to electrophilic sites, characterized by electron deficiency and an attraction to nucleophiles. Conversely, red regions indicate nucleophilic sites, possessing an excess of electron density and exhibiting an affinity for electrophiles. Green areas represent regions of zero potential. Analysis of the three-dimensional MEP surfaces, computed at the DFT/B3LYP/6-311++G(d, p) level (Fig. 10), reveals regions of pronounced negative (red) and positive (blue) electrostatic potential, indicative of chemically reactive zones. The four zoanthamine alkaloids consistently displayed a significant distribution of red regions, primarily localized around oxygen atoms within ketone and ester functionalities. Specifically, in DKK1, these nucleophilic regions likely interact with basic residues such as LYS—exemplified by LYS211, LYS222—and ARG—particularly ARG236—due to the strong positive charge of their side chains. The formation of hydrogen bonds between these residues and the alkaloids is further supported by the observed hydrogen bond occupancy. Similarly, in GSK-3β, the positively charged Lysine side chain of Lys85 demonstrates a notable interaction with 3α-hydroxyzoanthenamine via hydrogen bonding. Conversely, the light blue zones, indicative of highly positive electrostatic potential, were more pronounced in all compounds, particularly around the methyl groups of the cyclohexanone ring and the central structure, compared to other ligands. These observations confirm that the red regions, being nucleophilic in character, are well-suited for interactions with electrophilic groups and hydrogen bond formation with positively charged residues.
Fig. 10.

The MEP map of four compounds applying DFT calculation.
Molecules with a narrow energy gap demonstrate enhanced biological activity, increased chemical reactivity, and reduced stability relative to those with a wider energy gap57. Hence, the band energy gap of phytochemicals and quantum chemical reactivity metrics are derived from EHOMO and ELUMO were calculated and shown in Table 5. This study investigated the correlation between the HOMO-LUMO energy gap (ΔE) and biological activity in four zoanthamine alkaloids, in addition to three in vitro compounds that were tested57,63. Among these compounds, 3α-hydroxyzoanthenamine exhibited the smallest ΔE of 3.90 eV (Table 5), followed by epioxyzoanthamine at 4.20 eV and 7α-hydroxykuroshine E at 4.44 eV, all of which are smaller than that of norzoanthamine (4.53 eV). Notably, 3-hydroxynorzoanthamine closely mirrored norzoanthamine’s value of 4.54 eV and demonstrated ALP activity, while 3-acetoxynorzoanthamine (4.63 eV) and norzoanthaminone (4.73 eV) displayed higher energy gaps and did not show ALP activity. Furthermore, the reduced ΔE in 3α-hydroxyzoanthenamine corresponds with its superior in silico GSK-3β inhibitory activity, as evidenced by docking and molecular dynamics simulations. This finding reinforces the established relationship between a diminished HOMO-LUMO gap and enhanced biological activity, likely attributable to improved target interactions and binding affinity. Additionally, while this inverse correlation aligns with the in vivo anti-osteoporotic effects of norzoanthamine20, experimental validation is essential, considering that norzoanthamine possesses proven in vivo activity, and 3-hydroxynorzoanthamine exhibited in vitro ALP activity in MG-63 cells despite having a larger ΔE than some computationally predicted higher-potential compounds. Thus, although a smaller ΔE indicates increased potential, both in vitro and in vivo studies are necessary to confirm biological activity and comprehensively elucidate the structure-activity relationships of these zoanthamine alkaloids64.
Table 5.
Quantum reactivity parameters of four selected potential compounds in conjunction with three in vitro tested compounds.
| Compound | Total energy (eV) | Molecular dipole moment (Debye) | EHOMO (eV) | ELUMO (eV) | Energy gap (eV) | Absolute hardness (η) | Global softness (σ) | Electro negativity (χ) | Chemical potential (Pi) | Global electrophilicity (ω) |
|---|---|---|---|---|---|---|---|---|---|---|
| 3α-hydroxyzoanthenamine | − 47,577.32 | 7.74 | − 5.66 | − 1.76 | 3.90 | 1.95 | 0.51 | 3.71 | − 3.71 | 3.53 |
| Epioxyzoanthamine | − 45,557.34 | 8.68 | − 6.26 | − 2.06 | 4.20 | 2.10 | 0.48 | 4.16 | − 4.16 | 4.13 |
| 7α-hydroxykuroshine E | − 49,624.90 | 7.77 | − 6.46 | − 2.02 | 4.44 | 2.22 | 0.45 | 4.24 | − 4.24 | 4.05 |
| Norzoanthamine | − 42,445.44 | 6.25 | − 6.45 | − 1.92 | 4.53 | 2.26 | 0.44 | 4.18 | − 4.18 | 3.87 |
| 3-hydroxynorzoanthamine | − 44,489.00 | 4.77 | − 6.33 | − 1.79 | 4.54 | 2.27 | 0.44 | 4.06 | − 4.06 | 3.63 |
| 3-acetoxynorzoanthamine | − 48,644.42 | 4.93 | − 6.40 | − 1.77 | 4.63 | 2.31 | 0.43 | 4.09 | − 4.09 | 3.61 |
| Norzoanthaminone | − 44,456.15 | 3.06 | − 6.45 | − 1.72 | 4.73 | 2.36 | 0.42 | 4.08 | − 4.08 | 3.53 |
The examination of the HOMO-LUMO distributions for the compounds reveals that the electron density of the HOMO surface is predominantly localized on the azepane ring, with extension into the spirolactone ring, as demonstrated in Fig. 11. In contrast, the LUMO electronic cloud is predominantly dispersed across the cyclohexenone ring. The absolute hardness of seven compounds didn’t show much differentiation with values ranging from 1.95 to 2.36 eV, in which 3α-hydroxyzoanthenamine demonstrated the lowest absolute hardness, suggesting more heightened reactivity. Furthermore, the global electrophilicity (ω) values of these compounds, ranging from 3.53 to 4.13 eV, suggest their potential to engage in multiple interactions with biological macromolecules such as proteins65. The occurrence of a negative Pi implies that the appointed compounds are in a stable condition57.
Fig. 11.

Electron density maps of HOMO and LUMO of four selected compounds.
This study’s computational findings support and extend current understanding of Wnt signaling pathway modulation in osteoporosis, where DKK1 and GSK-3β are established key regulators of bone metabolism. Our results demonstrate the therapeutic potential of targeting these proteins and identify several zoanthamine alkaloids—including norzoanthamine, 3α-hydroxyzoanthenamine, epioxyzoanthamine, and 7α-hydroxykuroshine E. Computational analyses integrating MD and MEP surface data revealed favorable protein-ligand interactions, suggesting high-affinity binding driven by electrostatic interactions, crucial for both binding affinity and biological activity. However, experimental validation is necessary, and a tiered, resource-efficient approach was employed to maximize efficiency. In silico screening prioritized lead compounds for subsequent in vitro (MG-63 cell-based bone formation assays66) and in vivo (ovariectomized mouse model67) testing. This phased approach, focusing resources on computationally promising candidates, optimizes the efficiency and feasibility of this research program.
Conclusion
The Wnt signaling pathway plays a pivotal role in various biological processes, including cell maturation, reproduction, embryogenesis, bone modeling, and organogenesis. All known zoanthamine-type alkaloids isolated from Zoanthus sp. were screened for their ability to regulate fundamental markers within the Wnt signaling pathway. Specifically, inhibition of DKK1 and GSK-β, critical regulators of the pathway, was targeted to evaluate binding affinities and interactions of these phytochemicals. The co-crystallized ligand of GSK-3β, a proven inhibitor, was used as a reference to compare docking scores with the screened alkaloids. Notably, 69 alkaloids exhibited lower binding energies than the reference ligand (TMU). For DKK1, all tested ligands achieved docking scores below − 5 kcal/mol, indicative of strong protein binding. The ten highest-scoring ligands were further analyzed through molecular dynamics simulations. Detailed evaluations incorporating RMSD, hydrogen bond occupation percentages, PCA, FEL and binding free energy with entropy calculations identified 3α-hydroxyzoanthenamine, epioxyzoanthamine, 7α-hydroxyxkuroshine E and norzoanthamine as the most promising inhibitors of GSK-3β and DKK1. Density functional theory analyses revealed the presence of pronounced dipole-dipole interactions within the optimized geometries of these compounds. HOMO-LUMO analysis highlighted 3α-hydroxyzoanthenamine as the most biologically active and chemically reactive compound due to its lower ΔE value, indicating its capability to form multiple interactions with biological macromolecules. Future research should focus on structural modifications to enhance selectivity and anti-osteoporotic efficacy, complemented by in vitro and in vivo studies to validate therapeutic potential. Additionally, broadening molecular docking to other pertinent targets within the pathway may unveil broader implications for these alkaloid derivatives in treating osteoporosis and associated disorders.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
N.-T. Pham: conceptualization, investigation, and writing original draft ; H.-G. Le: validation, writing original draft, and visualization; B.-R. Peng: software, validation, and formal analysis; L.-Y. Chen: methodology and formal analysis; M. El-Shazly: formal analysis, and validation; J.-H. Su: formal analysis, and validation; M.-H. Lee: validation, resources, writing review & editing, and supervision; K.-H. Lai: conceptualization, validation, resources, writing original draft, writing review & editing, visualization, supervision, and funding acquisition.
Funding
The grants that supported this work were from the National Science and Technology Council of Taiwan (NSTC 109-2622-B-038-004, 110-2320-B038-038-MY3, 111-2320-B-038-040-MY3, 113-2321-B-255-001, 113-2628-B-038-009-MY3).
Data availability
Data is provided within the manuscript or supplementary information files.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kamycheva, E. & Goto, T. Camargo Celiac disease is associated with reduced bone mineral density and increased FRAX scores in the US National health and nutrition examination survey. Osteoporos. Int.28 (3), 781–790. 10.1007/s00198-016-3791-4 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Rizzoli, R. & Biver, E. Brennan-Speranza nutritional intake and bone health. Lancet Diabetes Endocrinol.9 (9), 606–621. 10.1016/s2213-8587(21)00119-4 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Song, S., Guo, Y. & Yang, Y. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharmacol. Ther.237, 108168. 10.1016/j.pharmthera.2022.108168 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Pasqualetti, S., Congiu, T. & Banfi, G. Mariotti alendronate rescued osteoporotic phenotype in a model of glucocorticoid-induced osteoporosis in adult zebrafish scale. Int. J. Exp. Pathol.96 (1), 11–20. 10.1111/iep.12106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennel, K. A. & Drake, M. T. Adverse effects of bisphosphonates: Implications for osteoporosis management. Mayo Clin. Proc.84 (7), 632–637. quiz 638. 10.1016/s0025-6196(11)60752-0 (2009). [DOI] [PMC free article] [PubMed]
- 6.Vuong, L. T. & Mlodzik, M. Different strategies by distinct Wnt-signaling pathways in activating a nuclear transcriptional response. Curr. Top. Dev. Biol.149, 59–89. 10.1016/bs.ctdb.2022.02.008 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang, Z. et al. Associations between WNT signaling pathway-related gene polymorphisms and risks of osteoporosis development in Chinese postmenopausal women: A case-control study. Climacteric25 (3), 257–263. 10.1080/13697137.2021.1941848 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Gao, Y., Chen, N. & Fu, Z. Zhang progress of Wnt signaling pathway in osteoporosis. Biomolecules13 (3). 10.3390/biom13030483 (2023). [DOI] [PMC free article] [PubMed]
- 9.Noh, T. et al. Lef1 haploinsufficient mice display a low turnover and low bone mass phenotype in a gender- and age-specific manner. PLoS One4 (5), e5438. 10.1371/journal.pone.0005438 (2009). [DOI] [PMC free article] [PubMed]
- 10.Glass, D. A. 2 et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell8 (5), 751–764. 10.1016/j.devcel.2005.02.017 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Ahamad, S., Saquib, M. & Hussain, M. K. Bhat targeting Wnt signaling pathway with small-molecule therapeutics for treating osteoporosis. Bioorg. Chem.156, 108195. 10.1016/j.bioorg.2025.108195 (2025). [DOI] [PubMed] [Google Scholar]
- 12.Dehghanbanadaki, N. & Taghdir, M. & Naderi-Manesh, H. Structural dynamic investigation of Wnt signalling activation through co-receptor LRP6. J. Biomol. Struct. Dyn., 1–14. 10.1080/07391102.2024.2446667 [DOI] [PubMed]
- 13.Wen, B., Hu, S., Yin, J. & Wu, J. Guo molecular evolution and protein structure variation of Dkk family. Genes14 (10), 1863 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, S. et al. Structural and functional studies of LRP6 ectodomain reveal a platform for Wnt signaling. Dev. Cell21 (5), 848–861. 10.1016/j.devcel.2011.09.007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westendorf, J. J. & Kahler, R. A. Schroeder Wnt signaling in osteoblasts and bone diseases. Gene341, 19–39. 10.1016/j.gene.2004.06.044 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Guillen, P. O., Jaramillo, K. B., Genta-Jouve, & Thomas, G. O. P. Marine natural products from zoantharians: bioactivity, biosynthesis, systematics, and ecological roles. Nat. Prod. Rep.37 (4), 515–540. 10.1039/C9NP00043G (2020). [DOI] [PubMed]
- 17.Villar, R. M. et al. Evaluation of the effects of several zoanthamine-type alkaloids on the aggregation of human platelets. Bioorg. Med. Chem.11 (10), 2301–2306. 10.1016/s0968-0896(03)00107-x (2003). [DOI] [PubMed] [Google Scholar]
- 18.Rao, C. B. et al. Alkaloids from a marine zoanthid. J. Org. Chem.50 (20), 3757–3760. 10.1021/jo00220a016 (1985). [Google Scholar]
- 19.Venkateswarlu, Y., Reddy, N. S., Ramesh, P. & Reddy, P. S. Chemical reduction of Zoanthamine and evaluation of antibacterial activity. Heterocycl. Commun.4, 575–580 (1998). [Google Scholar]
- 20.Kuramoto, M. et al. Structure-activity relationship of norzoanthamine exhibiting significant inhibition of osteoporosis. Bull. Chem. Soc. Jpn. 71 (4), 771–779. 10.1246/bcsj.71.771 (2006). [Google Scholar]
- 21.Yamaguchi, K., Yada, M., Tsuji, T. & Kuramoto, M. Uemura suppressive effect of norzoanthamine hydrochloride on experimental osteoporosis in ovariectomized mice. Biol. Pharm. Bull.22 (9), 920–924. 10.1248/bpb.22.920 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Ahn, V. E. et al. Structural basis of Wnt signaling Inhibition by Dickkopf binding to LRP5/6. Dev. Cell21 (5), 862–873. 10.1016/j.devcel.2011.09.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhat, R. et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418*. J. Biol. Chem.278 (46), 45937–45945. 10.1074/jbc.M306268200 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Rismani, E. et al. Computationally design of inhibitory peptides against Wnt signaling pathway: In silico insight on complex of DKK1 and LRP6. Int. J. Pept. Res. Ther.24 (1), 49–60. 10.1007/s10989-017-9589-1 (2018). [Google Scholar]
- 25.Gregory, C. A. et al. Dkk-1-derived synthetic peptides and lithium chloride for the control and recovery of adult stem cells from bone marrow*. J. Biol. Chem.280 (3), 2309–2323. 10.1074/jbc.M406275200 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Che, X. & Liu, Q. Zhang an accurate and universal protein-small molecule batch Docking solution using Autodock Vina. Results Eng.19, 101335. 10.1016/j.rineng.2023.101335 (2023). [Google Scholar]
- 27.Chen, S. R., Wang, S. W., Chang, F. R. & Cheng, Y. B. Anti-lymphangiogenic alkaloids from the zoanthid Zoanthus vietnamensis collected in Taiwan. J. Nat. Prod.82 (10), 2790–2799. 10.1021/acs.jnatprod.9b00451 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Chen, S. R., Wang, S. W., Sheu, J. H. & Chang, T. H. Zoanthamine alkaloid derivatives from the Zoantharian Zoanthus vietnamensis with antimetastatic activity. J. Org. Chem.85 (19), 12553–12560. 10.1021/acs.joc.0c01731 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Chen, S. R. et al. Additional alkaloids from Zoanthus vietnamensis with neuroprotective and anti-angiogenic effects. Bioorg. Chem.109, 104700. 10.1016/j.bioorg.2021.104700 (2021). [DOI] [PubMed] [Google Scholar]
- 30.O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminf. 3 (1), 33. 10.1186/1758-2946-3-33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated Docking with selective receptor flexibility. J. Comput. Chem.30 (16), 2785–2791. 10.1002/jcc.21256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. 10.1016/j.softx.2015.06.001 (2015).
- 33.Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide, Vol. 21 (2010).
- 34.Park, S. J., Kern, N., Brown, T. & Lee, J. CHARMM-GUI PDB manipulator: Various PDB structural modifications for biomolecular modeling and simulation. J. Mol. Biol.435 (14), 167995. 10.1016/j.jmb.2023.167995 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zoete, V., Cuendet, M. A. & Grosdidier, A. Michielin SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem.32 (11), 2359–2368. 10.1002/jcc.21816 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Huang, J. et al. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods14 (1), 71–73. 10.1038/nmeth.4067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKerell, A. D. Jr. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102 (18), 3586–3616. 10.1021/jp973084f (1998). [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys.79 (2), 926–935. 10.1063/1.445869 (1983). [Google Scholar]
- 39.Ganai, S. A. et al. Delineating binding potential, stability of Sulforaphane-N-acetyl-cysteine in the active site of histone deacetylase 2 and testing its cytotoxicity against distinct cancer lines through stringent molecular dynamics, DFT and cell-based assays. Chem. Biol. Drug Des.98 (3), 363–376. 10.1111/cbdd.13854 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Ibrahim, Z. Y., Uzairu, G. A., Shallangwa, S. E. & Abechi Isyaku. Virtual screening and molecular dynamic simulations of the antimalarial derivatives of 2-anilino 4-amino substituted Quinazolines docked against a Pf-DHODH protein target. Egypt. J. Med. Hum. Genet.23 (1), 119. 10.1186/s43042-022-00329-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Godara, P. et al. Structure-based virtual screening against multiple Plasmodium falciparum kinases reveals antimalarial compounds. Mol. Divers.28 (6), 3661–3681. 10.1007/s11030-023-10770-z (2024). [DOI] [PubMed] [Google Scholar]
- 42.Rout, A. K. et al. Insights into structure and dynamics of extracellular domain of Toll-like receptor 5 in Cirrhinus mrigala (mrigala): A molecular dynamics simulation approach. PLoS One16 (1), e0245358. 10.1371/journal.pone.0245358 (2021). [DOI] [PMC free article] [PubMed]
- 43.Humphrey, W., Dalke, A. & Schulten, K. Visual molecular dynamics. J. Mol. Graph. 14 (1), 33–38. 10.1016/0263-7855(96)00018-5 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Mai, T. T. et al. Discovery of novel flavonoid derivatives as potential dual inhibitors against α-glucosidase and α-amylase: virtual screening, synthesis, and biological evaluation. Mol. Divers.28 (3), 1629–1650. 10.1007/s11030-023-10680-0 (2024). [DOI] [PubMed] [Google Scholar]
- 45.Tran, Q. H. et al. Structure-based 3D-Pharmacophore modeling to discover novel Interleukin 6 inhibitors: an in Silico screening, molecular dynamics simulations and binding free energy calculations. PLoS One. 17 (4), e0266632. 10.1371/journal.pone.0266632 (2022). [DOI] [PMC free article] [PubMed]
- 46.Al-Khafaji, K., Taskin, T. & Tok Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput. Methods Programs Biomed.195, 105660. 10.1016/j.cmpb.2020.105660 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Pan, F. et al. Prediction and evaluation of the 3D structure of Macadamia integrifolia antimicrobial protein 2 (MiAMP2) and its interaction with palmitoleic acid or oleic acid: An integrated computational approach. Food Chem.367, 130677. 10.1016/j.foodchem.2021.130677 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Valdés-Tresanco, M. S., Valdés-Tresanco, M. E. & Valiente, P. A. Moreno Gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput.17 (10), 6281–6291. 10.1021/acs.jctc.1c00645 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Wang, E. et al. End-Point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev.119 (16), 9478–9508. 10.1021/acs.chemrev.9b00055 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Duan, L., Liu, X. & Zhang, J. Z. H. Interaction entropy: A new paradigm for highly efficient and reliable computation of protein–ligand binding free energy. J. Am. Chem. Soc.138 (17), 5722–5728. 10.1021/jacs.6b02682 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Homeyer, N. & Gohlke, H. Free energy calculations by the molecular mechanics Poisson–Boltzmann surface area method. Mol. Inf.31 (2), 114–122. 10.1002/minf.201100135 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Scudiero, D. A. et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res.48 (17), 4827–4833 (1988). [PubMed] [Google Scholar]
- 53.Mao, Y. W., Lin, R. D., Hung, H. C. & Lee, M. H. Stimulation of osteogenic activity in human osteoblast cells by edible Uraria crinita. J. Agric. Food Chem.62 (24), 5581–5588. 10.1021/jf5012177 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Geerlings, P. et al. Conceptual density functional theory: status, prospects, issues. Theor. Chem. Acc.139 (2), 36. 10.1007/s00214-020-2546-7 (2020). [Google Scholar]
- 55.Sahni, V. The Hohenberg–Kohn theorems and Kohn–Sham density functional theory. In Quantal Density Functional Theory (ed Sahni, V.) 99–123 (Springer, 2004).
- 56.Domingo, L. R. & Ríos-Gutiérrez, M. Pérez. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules21 (6), 748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Canakdag, M. et al. Comprehensive evaluation of purine analogues: Cytotoxic and antioxidant activities, enzyme Inhibition, DFT insights, and molecular Docking analysis. J. Mol. Struct.1323, 140798. 10.1016/j.molstruc.2024.140798 (2025). [Google Scholar]
- 58.Zhou, W., Yan, H. & Hao, Q. Analysis of surface structures of hydrogen bonding in protein–ligand interactions using the alpha shape model. Chem. Phys. Lett.545, 125–131. 10.1016/j.cplett.2012.07.016 (2012). [Google Scholar]
- 59.Palomo, V. et al. Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J. Med. Chem.54 (24), 8461–8470. 10.1021/jm200996g (2011). [DOI] [PubMed] [Google Scholar]
- 60.Hassan, A. M. et al. Evaluating the binding potential and stability of drug-like compounds with the Monkeypox virus VP39 protein using molecular dynamics simulations and free energy analysis. Pharmaceuticals17 (12), 1617 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Karmalawy, A. A. et al. Molecular Docking and dynamics simulation revealed the potential inhibitory activity of aceis against SARS-CoV-2 targeting the hACE2 receptor. Front. Chem.10.3389/fchem.2021.661230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dubey, A., Alanazi, A. M., Bhardwaj, R. & Ragusa, A. Identification of potential NUDT5 inhibitors from marine bacterial natural compounds via molecular dynamics and free energy landscape analysis. Mol. Divers.10.1007/s11030-024-10950-5 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abu-Dief, A. M. et al. Synthesize, structural inspection, stoichiometry in solution and DFT calculation of some novel mixed ligand complexes: DNA binding, biomedical applications and molecular Docking approach. J. Mol. Liq. 399, 124422. 10.1016/j.molliq.2024.124422 (2024). [Google Scholar]
- 64.Bhattacharya, S. et al. Computational screening of T-Muurolol for an alternative antibacterial solution against Staphylococcus aureus infections: an in Silico approach for phytochemical-based drug discovery. Int. J. Mol. Sci.25 (17), 9650 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rossi, A. et al. Anticancer activity and morphological analysis of Pt (II) complexes: Their DFT approach, Docking simulation, and ADME-Tox profiling. Appl. Organomet. Chem.38 (5), e7403. 10.1002/aoc.7403 (2024). [Google Scholar]
- 66.Abdel-Naim, A. B. et al. Rutin isolated from Chrozophora tinctoria enhances bone cell proliferation and ossification markers. Oxid. Med. Cell Longev. 2018, 5106469. 10.1155/2018/5106469 (2018). [DOI] [PMC free article] [PubMed]
- 67.Wang, Y., Wang, X., Wang, K. & Qin, W. Li extract of curculigo capitulata ameliorates postmenopausal osteoporosis by promoting osteoblast proliferation and differentiation. Cells13 (23). 10.3390/cells13232028 (2024). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is provided within the manuscript or supplementary information files.