

Abstract
Background
Genetic resources are essential components of biodiversity. As national strategy, the conservation of genetic resources is crucial not only for biodiversity but also for sustainable agriculture and cultural heritage. However, the exact origin of most local breeds remains unclear at the genomic level. The conservation efforts are becoming more challenging as local breeds are currently experiencing genetic drift and admixture, which may be further complicated by historical hybridizations. A typical example is the Beijing-You chicken, a local breed renowned for its excellent meat flavor and unique appearance. With a relatively recent history (~ 300 years), it displays mixed phenotypes which may have resulted from genomic admixture, with its exact origin yet to be determined.
Results
Through comprehensive genomic similarity analysis, we identified 12 genetic donor breeds for the Beijing-You chicken and quantified their genetic contributions, with the highest ancestry proportion coming from Henan chickens. The local ancestry components and genomic structure analyses of the Beijing-You chicken suggest recent hybridization in the formation of this breed. Furthermore, we innovatively used ancestry components as new material for genetic evaluation and selection signature detection, demonstrating that conservation efforts over the past decade have been effective. Analysis of selection signatures revealed genes and regions associated with polydactyly, egg production, intramuscular fat, and spermatogenesis.
Conclusions
By integrating various analytical strategies, we developed a novel framework for genetic traceability and evaluation. Our results highlight the effectiveness of ancestry components in genetic assessment and offer valuable insights for the conservation, improvement, and sustainable utilization of local breeds.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-025-11563-4.
Keywords: Ancestry analysis, Genetic traceability, Conservation effectiveness, Selection signatures, Beijing-You chicken
Background
Domesticated animals play a crucial role in human society. They are not only an important part of the human diet [1], but also contribute to various fields such as the economy, culture, ecology, and scientific research [2–5]. Due to the impact of foreign breeds on commercial performance, local breeds face issues such as decline in population size, reduced diversity, and genetic admixture or pollution [6–11]. The genetic survey, conservation, development and utilization of local breeds are urgently needed. However, these tasks cannot be effectively carried out without a good understanding of the genetic background of the breeds. As the breed origins are often not well-documented and frequent hybridization among breeds reduces phenotypic distinctiveness, genetic traceability thus become crucial in characterizing and evaluating the genetic resources of local breeds [12, 13]. Breed origin tracing can not only reveal its genetic background but also promote the improvement and utilization of local breeds by identifying breed-specific genes [14, 15]. However, when the genetic relationships between breeds became complex, resolution power of a single analytical method may be limited [16, 17]. Integrating multiple analytical strategies may provide confidence in breed origin tracing.
Domestic chickens were derived from the Red Junglefowl subspecies (G. g. spadiceus) and spread northward across China after initial domestication [18]. China has a rich genetic resource of domestic chickens, with local chicken breeds often embodying the cultural and ecological characteristics of their specific regions [19, 20]. As a typical representative of local chicken breeds, Beijing-You chicken (BY) is famous for excellent meat flavor as well as unique appearance [21–23]. BY has a medium-sized body, and its feather color is reddish-brown or yellow and distinctive morphological features, including a large and fluffy comb. Majority of BY individuals have beard feathers, some individuals also have toe feathers, and a few have five toes [22, 24]. Overall, BY exhibits mixed phenotypes of several Chinese local chicken (CLC) breeds, likely resulting from genetic admixture; however, certain phenotypes have not yet been stabilized. BY is recorded to have appeared around the mid-Qing Dynasty in Beijing, approximately 300 years ago [22], but its exact origin remains unknown.
Here, based on the whole-genome sequencing of 39 CLC breeds (both newly sequenced and previously published) and 2 wild junglefowl breeds, we address the genetic component, conservation effectiveness, and selection signatures of BY. Using various analytical strategies, we comprehensively determine the genetic component of BY, which requires no prior information of breed formation history. We innovatively use ancestral components for genetic evaluation and selection signature detection, expanding the research scope in relevant domains. Our established suite of methodologies provides both analytical standards and practical example for breed origin tracing and genetic assessment of local breeds.
Methods
Samples and sequencing
The resequencing data used in this study includes a total of 1,097 samples, comprising 39 CLCs (both newly sequenced and previously published) and 2 wild junglefowl breeds (Table S1) [18, 25–36]. BY consist of 5 subpopulations, totaling 177 samples: the 2010 random conservation population (BY_R1, 40 samples), the 2019 random conservation population (BY_R2, 37 samples), the 2019 phenotypic selection line (BY_P, 40 samples), the 2019 intramuscular fat selection line (BY_F, 40 samples), and the 2023 BY samples high-depth sequenced in this study (BY_H, 20 samples) [26]. The year number represents the year of sample collection. The distribution of the geographical origin of the CLC is shown in Fig. 1a. Based on the origins of these CLCs, they are divided into five groups: Southwest China, South China, Northwest China, Central China, and North China.
Fig. 1.

Distribution Map and Genetic Characterization of Beijing-You chicken. (a) Distribution map of the CLC breeds used in this study. (b) PCA results for 39 CLC breeds. We selected eight samples for each breed to constitute a balanced dataset for PCA. (c) Distribution of genetic probability γ from HyDe analysis according to the geographic regions
BY_H was sampled from the National Conservation Farm of Beijing You Chicken. No animal was anesthetized or sacrificed in this study. Blood samples were obtained from the brachial vein underneath the wing by venipuncture and stored in EDTA tubes at -80 °C before DNA extraction. DNA was extracted from each sample using DNeasy Blood & Tissue Kit (Qiagen, Shanghai, China), including an RNase A treatment. DNA integrity was checked on an agarose gel, and the concentration was quantified using a Qubit 2.0 fluorometer (Life Sciences, CA). The paired-end (PE) sequencing libraries were then constructed using the MGIEasy Universal DNA Library Prep Kit and sequenced on the MGISEQ-2000 platform (BGI Genomics, Shenzhen, China) with PE150 whole-genome sequencing. The average sequencing depth of the 20 (BY_H) samples was approximately 35×, while the sequencing depth of other samples ranged from 3× to 30×.
Quality control and variant calling
In this study, the GTX-One platform by Genetalks company, a commercially available FPGA-based hardware accelerator, was used for building the reference genome index (bGalGal1.mat.broiler.GRCg7b), reads alignment, and variant calling, resulting in a total of 68,625,088 variants [37]. GTX-One accelerates the Smith–Waterman algorithm for alignment process, and the variant calling is accelerated according to the HaplotypeCaller (Pair-HMM) from the Genome Analysis Toolkit (GATK 3.7). The variants quality was filtered using GATK VariantFiltration (QD < 2.0, QUAL < 30.0, SOR > 3.0, MQ < 40.0, MQRankSum < -12.5, ReadPosRankSum < -20.0), retaining 48,504,105 variants. Subsequently, the PLINK (v1.9) [38] was used with the parameters ‘--geno 0.1’ and ‘--mind 0.2’ to filter out variants and individuals with high missing rates, leaving 34,903,658 variants. Minor allele frequency (MAF) and linkage disequilibrium (LD) filtering were applied also by PLINK (v1.9) using the parameters ‘--maf 0.05’ and ‘--indep-pairwise 50 10 0.2’ for MAF and LD filtering, a total of 1,479,867 variants from 1,097 samples were finally retained.
Population structure analysis and hybridization detection
We use principal component analysis (PCA) to assess population structure. Since PCA can be affected by imbalanced sample sizes [39, 40], we randomly selected eight samples from each CLC breed to avoid biases. PCA was performed by SMARTPCA (v18140) from EIGENSOFT (v8.0.0) [41] on 312 samples from 39 CLC breeds.
We used HyDe software [42] to detect genomic hybridization signals. Similar to the ABBA-BABA test, HyDe considers a rooted, four-taxon network consisting of an outgroup and three ingroup populations. The distribution of site patterns was used to infer a hybrid ingroup population, with probability γ being sister to one population (P1) and with probability 1 − γ being sister to another population (P2). The null hypothesis is that when no hybridization occurs, the expected value of γ should be 0. In this study, we assumed that BY is a hybrid population, with the remaining 38 CLC breeds as potential parental populations, and Green Junglefowl as outgroup. We tested all possible combinations and filtered the results based on Z-scores and P-values.
Genomic similarity analysis
IBS Neighbor-Joining tree
The identity by state (IBS) distance between samples was calculated by PLINK (v1.9) with the parameter “--genome”. The distances between samples were then converted into an average IBS distance matrix between populations. A Neighbor-Joining tree was constructed by FastMe (v2.1.6.1) [43], and the tree was visualized by iTOL (v4) [44].
Fst Neighbor-Joining tree
The pairwise population Fst values on each individual SNP were calculated by VCFtools (v0.1.13) [45] with the parameter “--weir-fst-pop”. These values were then converted into an average Fst distance matrix across the entire genome.
Outgroup f3
The outgroup f3 statistic measures the genetic drift shared between populations, and was calculated using the qp3pop program of AdmixTools (v7.0.2) [46] with default parameters. The Green Junglefowl was set as outgroup, with BY as the fixed population (pop1) and the other CLC breeds as the variable population (pop2). The f3 statistic was computed in the form of f3 (BY, CLC; Green Junglefowl). A higher f3 value indicates a closer genetic relationship between BY and the CLC breed compared to the Green Junglefowl.
The normalized IBD (nIBD)
The identity by descent (IBD) between BY and other CLC breeds were calculated on a per-chromosome base using the RefinedIBD (v17Jan20.102) with default parameters [47]. The nIBD was then calculated by normalizing the total IBD length (tIBD) across chromosomes through division by the sample sizes of the two populations (n1 and n2).
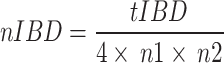 |
Allelic frequency modeling based on whole-genome SNPs
To make the downstream modeling more computationally efficient and statistically sound, the genomic SNPs were firstly thinned by LD-pruning using the PLINK (v1.9) parameter “--indep-pairwise 50 10 0.1” to generate a subset of relatively independent sites. The frequency of the reference allele at each site in BY was set as the dependent variable (Y), while the frequency of corresponding reference allele in other CLC breeds were included as the independent variable (X). LASSO (least absolute shrinkage and selection operator) regression, which shrinks certain coefficients to exactly zero, was employed to model relationships between multiple X variables (CLC breeds except BY) and the single Y variable (BY). The objective can be reflected with formula.
 |
Where 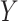 represents the frequency of the reference allele at each site in BY,
represents the frequency of the reference allele at each site in BY, 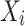 represents the frequencies of the reference alleles at the corresponding sites in other CLC breeds,
represents the frequencies of the reference alleles at the corresponding sites in other CLC breeds, 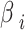 represents the regression coefficients for the respective independent variables, and
represents the regression coefficients for the respective independent variables, and 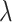 is the regularization parameter that controls the strength of the penalty. Breeds with large non-zero coefficients after LASSO fitting were considered as significant genetic donors to BY.
is the regularization parameter that controls the strength of the penalty. Breeds with large non-zero coefficients after LASSO fitting were considered as significant genetic donors to BY.
Ancestry inference and analysis
RFMix (v2.03-r0) [48] is a program that infers the ancestry of haplotype segments of admixed individuals by employing a conditional random field (CRF) approach based on reference panels. The genetic donor breeds for BY were used as reference ancestral groups to analyze both the global and local ancestry of BY genome. For each sample, RFMix outputs ancestry proportions as well as the ancestry assignment for haplotype segments of varying lengths across the genome. The ratio of ancestry heterozygote for each sample was calculated by comparing whether the ancestry assignments between the two haplotype alleles was identical at each locus. To evaluate the ancestry heterogeneity among BY individuals, we computed the Hamming distance among BY samples based on the ancestry assignment of haplotype segments across the genome.
We designed the ancestry H1 and H12 statistics, according to the concepts of the haplotype H1 and H12 statistics [49] as follows:
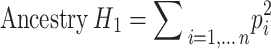 |
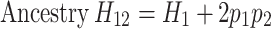 |
Where 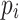 represents the frequency of the i-th ranked ancestry for the haplotype segment, as outputted by RFMix. Higher values of ancestry H1 and H12 indicate a more homogeneous ancestry background.
represents the frequency of the i-th ranked ancestry for the haplotype segment, as outputted by RFMix. Higher values of ancestry H1 and H12 indicate a more homogeneous ancestry background.
We defined ancestry H12 peaks as regions where H12 value of haplotype segments exceeded 0.9 for at least three consecutive segments. These ancestry H12 peak segments represent genomic regions with relatively homogeneous ancestry, suggesting selection through the purification of ancestry components. We used the ancestry H1 statistic to evaluate the degree of ancestry admixture, while ancestry H12 peaks were used to identify selection sweeps across the genome.
Phylogenetic weighting analysis
Twisst (topology weighting by iterative sampling of sub-trees) [50] infers the topological relationships among populations by sampling sub-trees and quantifies the weighting of each topology. To better understand the phylogenies, we first performed hierarchical clustering on the IBS tree of BY’s genetic donor breeds. Subsequently, we selected one representative genetic donor breed from each lineage for the Twisst analysis. Using the phyml_sliding_windows.py script, neighbor-joining trees were constructed for genome-wide windows with a block size of 10 SNPs. These trees were then used as input for Twisst to compute topology weightings using default parameters.
Estimation of effective population size and divergence times
To estimate the divergence time between BY and its genetic donor, we first calculated the effective population sizes of them using the MSMC2 (v.2.1.4) [51] with default parameters, assuming a mutation rate of 1.91e-9 per generation and a generation length of one year [18]. We then inferred the divergence time between BY and its genetic donor using the cross-population test implemented in the MSMC2. The relative cross-population coalescence rate (CCR) value ranges between 0 and 1. A CCR value close to 1 indicates that the two populations are part of a connected population, while a value of 0 suggests complete separation between populations. We considered a CCR value of 0.5 as indicative of population split [52].
Given the relatively short history of BY, we used the GONE [53] to estimate the effective population size over the past 50 to 200 generations for the two BY conservation subpopulations. This method employs genetic algorithms to infer population history and it is highly effective in detecting recent population changes. The GONE program was executed using default parameters with unphased data, utilizing an average recombination rate of 0.01 cm/Mb and applying genetic distance correction according to Haldane’s function.
Linkage disequilibrium (LD) analysis
PopLDdecay [54] is a fast and efficient tool for calculating linkage disequilibrium (LD) based on VCF files. Using the “-SubPop” parameter, we calculated the LD decay between BY and its genetic donor breeds. The LD decay plots for multiple populations were generated using the Plot_MultiPop.pl script come with the tool.
Genetic diversity assessment
To evaluate the conservation effectiveness, we calculated nucleotide diversity (π) and inbreeding coefficient (F) of two conservation BY subpopulations. The nucleotide diversity for each subpopulation was calculated based on whole-genome SNPs using the VCFtools (v0.1.13). The inbreeding coefficient for each subpopulation was calculated by PLINK (v1.9) with the parameter “-- het”.
Detection of runs of homozygosity (ROH)
ROH are continuous homozygous genotypes in an individual’s genome, resulting from the inheritance of identical haplotypes from both parents. Long ROH is often attributed to recent inbreeding or population bottlenecks [55]. ROH segments were detected using PLINK (v1.9) with the parameters “--homozyg-kb 500” and “--homozyg-window-snp 50,” where the window size was set to 50 SNPs, and ROH segments longer than 500 kb were reported.
Analysis of selection signatures
Using VCFtools (v0.1.13), we calculated fixation index Fst [56] and nucleotide diversity (π) using a sliding window approach with a window size of 50 kb and a step size of 25 kb. The π ratio was determined by dividing the π value for each window in the control population (BY_R2) by the π value for each window in the selected populations (BY_P, BY_F) (π ratio = π (control) / π (selection)). We also detected signatures of positive selection in populations by comparing the extent of haplotype homozygosity using XP-EHH [57] with the “-xpehh” parameter in Selscan (v2.0.2) [58], normalizing the results with the “norm” command (v1.3.0). The top 5% of values for Fst, π ratio, and XP-EHH were considered as evidence of selection. Selection signatures were annotated using SnpEff (v5.2c) [59] to retrieve the functional effects of variants, and genes overlapping with these selection signatures were extracted. Functional enrichment analysis for genes was performed based on Gene Ontology (GO) terms using the R package “clusterProfiler” [60].
Results
Population structure analysis and hybridization detection
To retrieve the genomic relationship between CLC breeds, we conducted PCA projection on whole-genome SNPs for 39 breeds. It showed that most of the CLCs clustered by breed, indicating a relatively clear population structure (Fig. 1b). Populations such as Yunnan chickens (CH, DL, DWS, NX, XG), Tibetan chickens (TB), and Game chickens (TG, XG, LXG, HNG) were located on the periphery in the PC space, while the remaining CLC breeds were more concentrated. BY was found to be closest to CLC breeds from North China, followed by those from Central China. This pattern aligns with their geographical distribution, suggesting that BY is more genetically close to CLC breeds from North and Central China.
HyDe analysis detected hybridization signals in BY, with multiple CLC breeds identified as potential parental sources (Table S2). The genetic probability γ, calculated by grouping CLCs according to geographic regions, supported the PCA findings. Chickens from North China showed the highest genetic probability γ to BY, with a median value of 0.75, followed by chickens from Central China with a median of 0.60 (Fig. 1c).
These results suggest that the formation of BY likely involves multiple ancestral breeds, with a more significant genetic contribution from CLC breeds in North and Central China.
Potential genetic donors for Beijing-You chicken
We further explored the potential genetic donors for BY using multiple analytical strategies on genomic data. We applied five different analyses to identify potential genetic donors for BY from various analytical perspectives (Table 1). The IBS neighbor-joining tree shows that BY clusters with the Zhengyang-Yellow chickens (ZYY), Huaibei-Partridge chicken (HBP), Gushi chicken (GS), Silkies (SK), Langshan chicken (LS), Shouguang chicken (SG), Dagong chicken (DG), and Bian chicken (BIAN) in the same branch (Fig. 2a). The Fst neighbor-joining tree exhibits a similar topology to the IBS tree, with the same eight CLC breeds clustered in the same branch (Fig. 2b). We calculated the nIBD between BY and other CLCs. The results indicate that the nIBD values for Hetian-Black chicken (HTB), Aijiao chicken (AJ), Liyang chicken (LY), ZYY and BIAN were higher, suggesting that these breeds may share a closer genetic relationship to BY in specific genomic regions (Fig. 2a). In the allelic frequency modeling based on whole-genome SNPs, we found that HBP, ZYY, GS, LY, LS, Xichuan-Black-Bone chicken (XCB), and HTB had large non-zero coefficients in the final LASSO regression model. This suggests that, from a global perspective, these breeds make stronger contributions to the genetic component of BY (Fig. 2b). Finally, we calculated the outgroup f3 using the Green Junglefowl as outgroup. It showed that SG, BIAN, DG, LS, HBP, GS, ZYY, SK, Lushi chicken (LUS), and XCB had higher outgroup f3 values, indicating higher genetic similarity to BY (Fig. 2c).
Table 1.
Summary of similarity indicators and Top-Ranking breeds across different analyses
| Analysis | Similarity Indicators from Different Analytical Perspectives | Top-Ranking Breeds |
|---|---|---|
| IBS Neighbor-Joining Tree | Small IBS distance between populations | BIAN, DG, GS, HBP, LS, SG, SK, ZYY |
| Fst Neighbor-Joining Tree | Low genetic differentiation between populations | BIAN, DG, GS, HBP, LS, SG, SK, ZYY |
| nIBD | Significant inherited DNA segments between populations | AJ, BIAN, HTB, LY, ZYY |
| Allelic Frequency Modeling | Strong influence on the genome allele frequency spectrum | GS, HBP, HTB, LS, LY, XCB, ZYY |
| Outgroup f3 | Highly shared genetic drift between populations | SG, BIAN, DG, LS, HBP, GS, ZYY, SK, LUS, XCB |
| - | - | Potential genetic donors: AJ, BIAN, DG, GS, HBP, HTB, LS, LY, SG, SK, XCB, ZYY |
Fig. 2.

Searching Potential Genetic Donors for BY. (a) The IBS neighbor-joining tree, with the result from nIBD shown in the outer circle. (b) The Fst neighbor-joining tree, with the result from allelic frequency modeling shown in the outer circle. (c) Outgroup f3 values using the Green Junglefowl as outgroup. Blue represents BY, red represents the top-rankings from different analyses, and gray represents the remaining CLC breeds
Based on the results above, there was low consistency in the outcomes across different analytical strategies, suggesting that the genetic structure and formation of BY are more complex than previously anticipated. We therefore retained the top-ranking breeds from each analysis that appeared among the top ranks at least twice across all analyses as potential genetic donors for BY. Specifically, for the IBS and Fst neighbor-joining trees, we selected breeds that grouped with BY in the same branch as top-ranking; for allelic frequency modeling and outgroup f3, we selected the top 25% of breeds as top-ranking. The nIBD results exhibited clear stratification, so CLC breeds with high nIBD values were directly chosen as top-ranking. Ultimately, the potential genetic donors for BY were determined from the 39 CLC breeds, including 12 breeds: BIAN, DG, GS, HBP, LS, LY, HTB, SG, SK, XCB, AJ and ZYY (Table 1).
Global and local ancestry of Beijing-You chicken
To quantify the genetic contribution of the 12 donor breeds to BY, we used RFMix to infer the ancestry component for each sample. From the perspective of global ancestry proportions, the proportions of different genetic donor breeds were relatively stable across individuals of BY (Fig. 3a). Among them, the highest ancestry proportion was from ZYY (about 30%), followed by XCB (about 20%). The ancestry proportions of HTB, LY, and GS were similar, each around 10%, while HBP and AJ had close proportions, both approximately 5%. The ancestry proportions of LS and SK were lower, at around 2%.
Fig. 3.

Global and Local Ancestry of BY. (a) Global ancestry proportions of BY individuals. Each column represents a sample. (b) Heatmap of the ancestry H1 statistic along the chromosomes. The ancestry H1 statistic quantifies the degree of admixture in local ancestry. (c) The heatmap of the Hamming distance matrix for local ancestry of haplotypes in BY individuals. Sample clustering results shown above the heatmap, colors corresponding to e. (d) Most frequent topologies from Twisst analysis (complete results are shown in Figure S2). (e) Violin plot of the heterozygosity of haplotype ancestry proportions in different BY subpopulations. (f) LD decay from BY and all its genetic donor breeds
We used the ancestry H1 statistic to measure the degree of genetic admixture in BY. A lower value of ancestry H1 suggests more diverse ancestry components and higher levels of admixture. The results show that most regions across the genome exhibit high levels of admixture, indicating that the genetic structure of BY is highly heterogeneous (Fig. 3b). To investigate ancestry heterogeneity in BY, we calculated Hamming distances based on local ancestry assignments for haplotype alleles among BY individuals along the genomic blocks. Figure 3c reveals substantial overall Hamming distances between samples, indicating diverse ancestry components along chromosome segments among BY individuals, even within the same subpopulation. However, Hamming distances among individuals within BY_P and BY_F subpopulations were comparatively smaller, suggesting reduced ancestry heterogeneity in subpopulations under selection (Fig. 3c). In addition, we used Twisst to evaluate the topological discrepancies across genomic blocks. The results revealed no dominant topology among the various topologies across these blocks (Fig. 3d, Figure S1-S2), providing further evidence of the heterogeneous genetic architecture of BY.
Altogether, we found that the global ancestry proportions of BY are stable, but the local ancestry components exhibit high heterogeneity. The ancestry components of individuals from the BY_P and BY_F subpopulations are relatively less variable, which may be attributable to the selective pressures acting on these two BY subpopulations (discuss later).
Genomic structure of Beijing-You chicken exhibits characteristics of recent hybridization
By utilizing MSMC2, we estimated the divergence time between BY and its genetic donors, revealing that BY and most of its genetic donors diverged approximately 5,000 years ago (Figure S3). Given that historical records show that BY has been a stable breed for only about 300 years, this suggests that BY did not gradually evolve from these genetic donor breeds. Instead, it is more likely that BY was recently formed through hybridization of them.
We also discovered genomic evidence that supports the recent hybridization of BY. First, we assessed whether the two haplotypes from a locus in each sample shared the same ancestral background. The results showed that the median ratio of ancestry heterozygote for genomic blocks in all BY subpopulations exceeded 50% (Fig. 3e). Furthermore, we analyzed the LD decay for BY and its genetic donors, finding that the genomic LD level of BY was significantly higher than that of its genetic donors (Fig. 3f). Since LD levels typically increase in hybrid populations during the early stages of hybridization [61], our findings align with this expectation.
Conservation effectiveness of Beijing-You chicken
As a stable breed existing for 300 years, BY had become quite rare by the end of the 20th century. The Chinese Academy of Agricultural Sciences and Beijing Academy of Agriculture and Forestry Sciences have since collected and protected the breed, and BY is currently in a state of maintenance [22]. To evaluate the conservation effectiveness over the past decade, we compared two random conservation subpopulations: BY_R1 and BY_R2.
We first compared the recent effective population size of the two random conservation subpopulations. The results showed that, over the past 50 to 200 generations, the effective population size of BY_R2 was comparable to that of BY_R1, suggesting that the effective population size has remained stable over the past 10 years (Figure S4). Furthermore, the global ancestry proportions of BY_R1 and BY_R2 remained stable too, with no obvious loss of ancestral diversity (Fig. 4a).
Fig. 4.

Conservation Effectiveness Evaluation for BY. (a) Global ancestry proportions of BY_R1 and BY_R2. (b) Distribution of Hamming distances between haplotypes in BY_R1 and BY_R2. Wilcoxon test p = 3.7 × 10⁻⁴. (c) The distribution of ROH fragment lengths in BY_R1 and BY_R2 individuals
The nucleotide diversity (π) of BY_R1 was measured at 0.0022, while the inbreeding coefficient (F) was 0.2804. In contrast, the π for BY_R2 decreased by 10% to 0.0020, and the F decreased by 17% to 0.3284. To assess changes in local ancestry heterogeneity among individuals within the two subpopulations, we compared the Hamming distances between haplotypes in BY_R1 and BY_R2. Our analysis revealed a statistically significant difference in Hamming distances between the two, with BY_R2 showing a loss of some higher values in the distance distribution (Fig. 4b). This suggests a slight decrease in local ancestry heterogeneity in BY_R2, which aligns with the observed increase in the inbreeding coefficient. Additionally, the lengths of the ROH segments in BY subpopulations further support this finding. Compared to BY_R1, individuals in BY_R2 had significantly longer ROH segments, indicating that BY_R2 has experienced a certain degree of inbreeding (Fig. 4c), consistent with earlier reports [26].
In summary, our analysis indicates that the past decade of conservation efforts has resulted in a decline in both genomic diversity and ancestry heterogeneity. However, given the challenges associated with pure conservation practices in preventing breed extinction and keeping breed purity, the conservation effectiveness of BY seems to be relatively optimal.
Selection signatures in two Beijing-You subpopulations
Besides conservation efforts, selective breeding has been applied to BY in recent years. Currently, two BY selection lines have been established: i.e., BY_P, primarily selected for polydactyly, and BY_F, selected for high intramuscular fat content.
To identify genomic signatures of selection, we first compared the two selected lines with BY_R2 using Fst, π ratio and XP-EHH. For BY_P, π ratio and XP-EHH analysis revealed a prominent signal on chromosome 2: 8.03–8.58 Mb (Fig. 5a), covering the Lmbr1 gene, which has been reported being associated with polydactyly in chickens [62, 63]. By intersecting the top 5% of selection signatures identified through Fst, π ratio, and XP-EHH, a total of 33 regions containing 40 genes were identified. In contrast, 317 regions containing 330 genes were identified in BY_F (Fig. 5a-b, Tables S3-S6). The significantly larger number of selected regions in BY_F indicates a much stronger selection intensity for intramuscular fat content. GO enrichment analysis of the encompassed genes in BY_F revealed that they were primarily enriched in pathways related to dephosphorylation, coagulation, mitochondrial membrane, and peptidase activity (Figure S5).
Fig. 5.

Analysis of Selection Signatures in BY_P and BY_F. (a and b) show the circos Manhattan plots of Fst, π ratio, and XP-EHH results for BY_P and BY_F, respectively. (c and d) display the distribution of selected regions in BY_P and BY_F subpopulations that meet the top 5% criteria for Fst, π ratio, and XP-EHH, as well as the ancestry H12 peaks along the chromosomes (complete results are shown in Figures S6-S7). The heatmap represents the ancestry H12 statistic. (e) The dominant haplotype of the HENMT1 gene in BY and its genetic donor breeds
Due to the hybrid background of BY, we introduced a new ancestry H12 statistic to identify selection sweeps. Like selection would reduce levels of polymorphism, a higher H12 value and the presence of H12 peaks indicate a more homogeneous ancestry background resulting from the purifying selection that eliminates less favorable ancestry components. As shown in Fig. 5c-d and Figures S6-S7, we identified a total of 79 regions containing 197 genes in BY_P, and 581 regions containing 1,066 genes were found in BY_F (Tables S7-S10). Notably, eight shared H12 peaks were identified in BY_R1, BY_R2, BY_F, and BY_P (Figure S8), spanning the 12.21–13.89 Mb region on chromosome Z. This region shows a striking 88% ancestry contribution from HTB, which is substantially higher than the genome-wide average HTB ancestry proportion of 10%. Previous research has identified multiple SNPs and genes within the 10.81–13.05 Mb region of chromosome Z that are associated with egg production traits [64]. These findings suggest that excellent egg-laying trait of BY may have been inherited from HTB through this specific genomic region.
For BY_P and BY_F, we found that the top selected regions identified by Fst, π ratio, and XP-EHH were overlapped with the ancestry H12 peaks, indicating that some alleles under selection may be donor-ancestry specific (Fig. 5c-d, Figures S6-S7). By intersecting the selected genes identified by both approaches (Figure S9a), we found four genes on chromosome 8 in BY_P: EEIG2, FNDC7, HENMT1, and PRPF38B. The HENMT1 gene has been shown to be associated with spermatogenesis in humans and mice [65, 66]. By comparing the dominant haplotypes of the HENMT1 gene between BY and its genetic donor breeds, we found that the dominant haplotype of BY exhibited high similarity to the dominant haplotype found in SG (Fig. 5e). Due to the relatively low hatching rate observed in SG (personal communication), the disadvantageous haplotype may have been transmitted from SG to BY and reached high frequency in the BY, potentially as a pleiotropic consequence of selection on this haplotype for other traits. Similarly, in BY_F, 64 genes were identified as being selected by both approaches (Figure S9b, Table S11). Among these, the ACOX3 gene, which is involved in fatty acid degradation, plays a role in regulating fatty acid oxidation and affects the deposition of intramuscular fat in the pectoralis major of chicks [67]. The KLHL4 gene, on the other hand, is known to be associated with fat deposition in chickens [68, 69].
Discussion
In this study, we developed an analytical framework that uses whole-genome SNPs to determine genetic traceability, and assess conservation effectiveness and selection signatures based on ancestral information. Our method, which does not require any prior information on breed formation history, narrows down potential genetic donors by examining genomic similarity. When searching for potential genetic donors of BY, we observed low consistency among the results obtained from different analytical strategies. This inconsistency may stem from the varying analytical perspectives adopted by different analyses, leading to the capture of distinct genomic features and, consequently, discrepancies in results. Moreover, such inconsistencies are common in genetic studies, as multiple factors in natural environments—such as genetic drift, natural selection, incomplete lineage sorting, gene flow, and complex evolutionary events (e.g., ancient hybridization)—may interact and obscure the true phylogenetic history of the studied populations [16, 70, 71]. Given the potential complexity of the genomic structure and formation of BY, integrating multiple analytical strategies is an effective approach to enhancing the robustness and reliability of the study. By integrating multiple analytical results, we identified 12 genetic donors for BY. Notably, the genetic contribution from Henan chickens, including GS, ZYY, and XCB, accounts for around 60% of its genetic makeup. We therefore consider Henan chickens to be the primary genetic contributor to BY bloodline. During the Ming and Qing dynasties, Henan had convenient water and land transportation, and its communication and connectivity played a significant role [72]. The Gushi County Annals records that Gushi chicken was offered as a tribute to the imperial court during the Qianlong period of the Qing Dynasty [22]. We hypothesize that BY was primarily developed using Henan chickens as the genetic backbone, while being continuously interbred with other CLC breeds possessing excellent traits from across the country. BY was ultimately formed through selective breeding and cultivation following hybridization. It should be noted that our analysis assumes that the lineage of the genetic donor breeds is relatively pure or phenotypically stable. Although the donor breeds identified in this study all standard breeds listed in the Animal Genetic Resources in China: Poultry encyclopedia, the historical genetic admixture among chicken populations [11, 73, 74], complicates the assurance of perfect genetic purity for these donor breeds. Even if a breed was initially pure, subsequent genetic admixture may have led to a dilution of its original characteristics. Therefore, the genetic donors of BY proposed in this study do not refer to current breeds that are direct ancestors of BY, but rather to the ancient genetic components present in the genomes of the hypothetical purebred genetic donors that are included in the current breed’s genome.
Traditional studies primarily rely on genomic variations as the material for genetic evaluation and selection signature detection [75]. In this study, we innovatively used ancestry component as the new material for these purposes. The inclusion of ancestry component broadens the scope of evaluation, offering new insights that conventional genomic analyses cannot provide and resulting in a more comprehensive assessment. The selected regions identified by the traditional methods overlapped with the ancestry H12 peaks, demonstrating the validity of our new approach. Although BY, as a 300-year-old breed, has a relatively stable phenotypic characteristics and overall genetic components, Hamming distances based local ancestry among BY individuals showed substantial ancestry heterogeneity. This suggests that the genetic components have not fully stabilized since the formation of BY. Based on the results of the ancestry analysis, the conservation of BY over the past decade has been successful, with no significant external genetic contamination. We successfully used the peak of ancestry H12, which represents the purification of ancestry components, to identify selection signatures. Our findings show that in four BY subpopulations, the genomic region on chromosome Z (12.21–13.89 Mb) is almost completely fixed with genetic material from the HTB, suggesting that the breeding of BY has incorporated the desirable traits of the genetic donor breed. We further compared the distribution of the top 5% of shared selection regions, based on Fst, π ratio, and XP-EHH, with the distribution of ancestry H12 peaks across BY genome. The significant overlaps confirm that certain traits from the genetic donor breeds were further selected during the breeding of BY. Interestingly, the overlap was significantly higher in BY_F (24.1%) compared to BY_P (11.11%), suggesting that, while the unique appearance of BY was a consideration, the selection for intramuscular fat may have been more closely aligned with the breed’s breeding objectives. This implies that the breeding of BY was likely driven more by a focus on meat quality and flavor than on ornamental characteristics. Additionally, this study identified several key genes in BY, and further functional studies may help confirm the mechanisms by which these selected genes influence target traits. By combining CRISPR/Cas9 with primordial germ cell (PGC) culture technology, gene knockout or knock-in chicken models can be established to validate the functions of key genes [76, 77]. Furthermore, tissue-specific recombinant viral vectors can be used for in situ injection, enabling gene overexpression or gene regulation in live animals to verify their biological functions [78].
As BY is preserved as a purebred population, a certain degree of nucleotide diversity (π) reduction and inbreeding coefficient (F) increase is inevitable. To maintain breed purity while preserving genetic diversity and mitigating inbreeding effects, optimizing breeding strategies is essential. Approaches such as adjusting the male-to-female ratio, implementing rotational mating, applying minimum kinship mating, and conducting regular genomic monitoring can help alleviate these issues. In fact, local chicken breeds are facing similar genetic conservation challenges. A study on Silkie chickens found that, compared to seven other CLC breeds, Silkie chickens exhibited lower genetic diversity and a higher level of inbreeding [79]. The study also detected a prolonged population bottleneck and strong artificial selection in Silkie chickens, with potential gene introgression from other CLC breeds. A conservation study on Henan chickens (Gushi chickens and Xichuan-Black-Bone chickens) revealed that with an increasing number of conservation generations, the Gushi chicken population showed a declining trend in genetic diversity [80]. Some individuals also showed signs of admixture between the two breeds. Similarly, a study on southern CLC breeds detected extensive hybridization signals, suggesting a certain level of genetic exchange among these breeds [81]. In addition, CLCs are also facing challenges from genetic introgression and contamination due to foreign commercial breeds [11]. These studies suggest that some CLC breeds exhibit genetic characteristics similar to BY during conservation, including a decline in genetic diversity, an increase in inbreeding levels, and, in some cases, gene introgression or admixture. In fact, this issue is not limited to China but is a global phenomenon [8, 82]. The rapid development of intensive livestock production systems worldwide may pose challenges to the genetic conservation of local chicken breeds. This highlights the importance of strengthening genetic diversity management and optimizing conservation strategies for local chicken breeds.
Conclusions
In this study, we developed a novel analytical framework that integrates whole-genome SNPs and ancestry components for breed origin tracing, genetic evaluation, conservation assessment, and detection of selection signatures. Using this framework, we identified 12 genetic donor breeds for the Beijing-You chicken, with Henan chickens forming its primary genetic backbone. Based on ancestral information, we demonstrated that conservation efforts over the past decade have been effective. Our analysis of selection signatures revealed genes and regions associated with polydactyly, egg production, intramuscular fat, and spermatogenesis in selection lines. These results highlight the utility of ancestry components in genetic evaluation and provide valuable insights for the conservation, improvement, and sustainable utilization of local breeds.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the support of the Xihe high-performance computing platform of the National Research Facility for Phenotypic and Genotypic Analysis of Model Animals (Beijing).
Abbreviations
- BY
Beijing-You chicken
- BY_R1
BY random conservation population from 2010
- BY_R2
BY random conservation population from 2019
- BY_P
BY phenotypic selection line from 2019
- BY_F
BY intramuscular fat selection line from 2019
- BY_H
BY samples high-depth sequenced in this study in 2023
- CLC
Chinese local chicken
- MAF
Minor allele frequency
- LD
Linkage disequilibrium
- PCA
Principal component analysis
- IBS
Identity by state
- IBD
Identity by descent
- nIBD
The normalized IBD
- LASSO
Least absolute shrinkage and selection operator
- CRF
Conditional random field
- Twisst
Topology weighting by iterative sampling of sub-trees
- CCR
Cross-population coalescence rate
- π
Nucleotide diversity
- F
Inbreeding coefficient
- ROH
Runs of homozygosity
- GO
Gene Ontology
- ZYY
Zhengyang-Yellow chicken
- HBP
Huaibei-Partridge chicken
- GS
Gushi chicken
- SK
Silkies
- LS
Langshan chicken
- SG
Shouguang chicken
- DG
Dagong chicken
- BIAN
Bian chicken
- HTB
Hetian-Black chicken
- AJ
Aijiao chicken
- LY
Liyang chicken
- XCB
Xichuan-Black-Bone chicken
- LUS
Lushi chicken
Author contributions
YK performed the data analysis and prepared the manuscript. ZLW, XYC, LZT, and ZXL contributed to the study design. QW and QHL raised and sampled the chickens, and collected the data. YQZ and YZW revised the manuscript. YQZ received the funding. YQZ, YZW, and NY supervised the study. All authors read and approved the final manuscript.
Funding
This work was funded by the National Key Research and Development Program of China (2021YFD1200803 and 2022YFF1000204).
Data availability
The clean DNA sequencing data of 20 Beijing-You chickens reported in this study were deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA022028).
Declarations
Ethics approval and consent to participate
All procedures involving animals in this study were carried out in accordance with the Guidelines for the Care and Use of Experimental Animals established by the Ministry of Science and Technology of the People’s Republic of China (approval number: 2006 − 398). Furthermore, all the animal experiment protocols were approved by the Animal Welfare Committee of China Agricultural University (permission number: SKLAB-2014-04-02).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yuzhan Wang, Email: yuzhanwang@cau.edu.cn.
Yiqiang Zhao, Email: yiqiangz@cau.edu.cn.
References
- 1.Bradford GE. Contributions of animal agriculture to meeting global human food demand. Livest Prod Sci. 1999;59(2):95–112. [Google Scholar]
- 2.Bahr A, Wolf E. Domestic animal models for biomedical research. Reprod Domest Anim. 2012;47(Suppl 4):59–71. [DOI] [PubMed] [Google Scholar]
- 3.Andersson L, Georges M. Domestic-animal genomics: Deciphering the genetics of complex traits. Nat Rev Genet. 2004;5(3):202–12. [DOI] [PubMed] [Google Scholar]
- 4.Herrero M, Grace D, Njuki J, Johnson N, Enahoro D, Silvestri S, et al. The roles of livestock in developing countries. Animal. 2013;7(Suppl 1):3–18. [DOI] [PubMed] [Google Scholar]
- 5.Kayabasi ET, Yılmaz O. Contribution of domestic animals to human economic and social life. Acta Scientiarum Polonorum Zootechnica. 2022;21(1):29–34. [Google Scholar]
- 6.Mathew E, Mathew L. Conservation of landraces and Indigenous breeds: an investment for the future. Conservation and sustainable utilization of bioresources. Springer; 2023. pp. 291–321.
- 7.Berthouly-Salazar C, Thevenon S, Van TN, Nguyen BT, Pham LD, Chi CV, et al. Uncontrolled admixture and loss of genetic diversity in a local Vietnamese pig breed. Ecol Evol. 2012;2(5):962–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ali TM. A review of genetic diversity erosion in Ethiopia’s local chicken gene pool: implications on determination of suitable breeding and conservation strategies. World’s Poult Sci J. 2024;80(2):371–85. [Google Scholar]
- 9.Mongke T, Budsuren U, Tiemuqier A, Bozlak E, Wallner B, Dulamsuren S et al. Genomic conservation of Mongolian horses promoted by preservation of the intangible cultural heritage of Naadam in Mongolia. Conserv Lett. 2024:e13019.
- 10.Tisdell C. Socioeconomic causes of loss of animal genetic diversity: analysis and assessment. Ecol Econ. 2003;45(3):365–76. [Google Scholar]
- 11.Zhang C, Lin D, Wang Y, Peng D, Li H, Fei J, et al. Widespread introgression in Chinese Indigenous chicken breeds from commercial broiler. Evol Appl. 2019;12(3):610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Supple MA, Shapiro B. Conservation of biodiversity in the genomics era. Genome Biol. 2018;19(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogg CJ. Translating genomic advances into biodiversity conservation. Nat Rev Genet. 2024;25(5):362–73. [DOI] [PubMed] [Google Scholar]
- 14.Dalvit C, De Marchi M, Cassandro M. Genetic traceability of livestock products: A review. Meat Sci. 2007;77(4):437–49. [DOI] [PubMed] [Google Scholar]
- 15.Dadousis C, Munoz M, Ovilo C, Fabbri MC, Araujo JP, Bovo S, et al. Admixture and breed traceability in European Indigenous pig breeds and wild Boar using genome-wide SNP data. Sci Rep. 2022;12(1):7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scherz MD, Masonick P, Meyer A, Hulsey CD. Between a rock and a hard polytomy: phylogenomics of the rock-Dwelling Mbuna cichlids of lake Malaŵi. Syst Biol. 2022;71(3):741–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bravo GA, Antonelli A, Bacon CD, Bartoszek K, Blom MPK, Huynh S, et al. Embracing heterogeneity: coalescing the tree of life and the future of phylogenomics. PeerJ. 2019;7:e6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang MS, Thakur M, Peng MS, Jiang Y, Frantz LAF, Li M, et al. 863 Genomes reveal the origin and domestication of chicken. Cell Res. 2020;30(8):693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Y, Cui H, Yuan X, Liu L, Liu X, Wang Y, et al. Identification of the main aroma compounds in Chinese local chicken high-quality meat. Food Chem. 2021;359:129930. [DOI] [PubMed] [Google Scholar]
- 20.Xiao L, Qi L, Fu R, Nie Q, Zhang X, Luo W. A large-scale comparison of the meat quality characteristics of different chicken breeds in South China. Poult Sci. 2024;103(6):103740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge Y, Gai K, Li Z, Chen Y, Wang L, Qi X, et al. HPLC-QTRAP-MS-based metabolomics approach investigates the formation mechanisms of meat quality and flavor of Beijing you chicken. Food Chem X. 2023;17:100550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Resources CNCoAG. Animal genetic resources in China: poultry. Beijing: Chinese Agricultural; 2011. [Google Scholar]
- 23.Meng L, Mao P, Guo Q, Tian X. Evaluation of meat and egg traits of Beijing-you chickens rotationally grazing on Chicory pasture in a chestnut forest. Brazilian J Poult Sci. 2016;18:1–6. [Google Scholar]
- 24.Chu Q, Yan Z, Zhang J, Usman T, Zhang Y, Liu H, et al. Association of SNP rs80659072 in the ZRS with polydactyly in Beijing you chickens. PLoS ONE. 2017;12(10):e0185953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Bai X, Liu H, Zhao B, Yan Z, Hou Y, et al. Population genomic sequencing delineates global landscape of copy number variations that drive domestication and breed formation of in chicken. Front Genet. 2022;13:830393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q, Zhang J, Wang H, Wang Z, Li Q, Zhao G, et al. Estimates of genomic inbreeding and identification of candidate regions in Beijing-You chicken populations. Anim Genet. 2023;54(2):155–65. [DOI] [PubMed] [Google Scholar]
- 27.Ulfah M, Kawahara-Miki R, Farajalllah A, Muladno M, Dorshorst B, Martin A, et al. Genetic features of red and green junglefowls and relationship with Indonesian native chickens Sumatera and Kedu Hitam. BMC Genomics. 2016;17:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qanbari S, Rubin CJ, Maqbool K, Weigend S, Weigend A, Geibel J, et al. Genetics of adaptation in modern chicken. PLoS Genet. 2019;15(4):e1007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li D, Che T, Chen B, Tian S, Zhou X, Zhang G, et al. Genomic data for 78 chickens from 14 populations. Gigascience. 2017;6(6):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang X, Otecko NO, Peng M, Weng Z, Li W, Chen J, et al. Genome-wide genetic structure and selection signatures for color in 10 traditional Chinese yellow-feathered chicken breeds. BMC Genomics. 2020;21(1):316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo Y, Ou JH, Zan Y, Wang Y, Li H, Zhu C, et al. Researching on the fine structure and admixture of the worldwide chicken population reveal connections between populations and important events in breeding history. Evol Appl. 2022;15(4):553–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu J, Liang Q, Liu C, Li S. Genomic analyses reveal adaptation to hot arid and harsh environments in native chickens of China. Front Genet. 2020;11:582355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Li D, Guo A, Li M, Li L, Zhou J, et al. Whole-genome resequencing of Dulong chicken reveal signatures of selection. Br Poult Sci. 2020;61(6):624–31. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Sun G, Zhang M, Cao Y, Zhang C, Fu Y, et al. Breeding history and candidate genes responsible for black skin of Xichuan black-bone chicken. BMC Genomics. 2020;21(1):511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun J, Chen T, Zhu M, Wang R, Huang Y, Wei Q, et al. Whole-genome sequencing revealed genetic diversity and selection of Guangxi Indigenous chickens. PLoS ONE. 2022;17(3):e0250392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhi Y, Wang D, Zhang K, Wang Y, Geng W, Chen B, et al. Genome-Wide genetic structure of Henan Indigenous chicken breeds. Animals. 2023;13(4):753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bu L, Wang Q, Gu W, Yang R, Zhu D, Song Z, et al. Improving read alignment through the generation of alternative reference via iterative strategy. Sci Rep. 2020;10(1):18712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maruthi Padmaja T, Raju BS, Hota RN, Krishna PR. Class imbalance and its effect on PCA preprocessing. Int J Knowl Eng Soft Data Paradigms. 2014;4(3):272–94. [Google Scholar]
- 40.Elhaik E. Principal component analyses (PCA)-based findings in population genetic studies are highly biased and must be reevaluated. Sci Rep. 2022;12(1):14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blischak PD, Chifman J, Wolfe AD, Kubatko LS. HyDe: A python package for Genome-Scale hybridization detection. Syst Biol. 2018;67(5):821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lefort V, Desper R, Gascuel O. FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol. 2015;32(10):2798–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and vcftools. Bioinformatics. 2011;27(15):2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192(3):1065–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics. 2013;194(2):459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maples BK, Gravel S, Kenny EE, Bustamante CD. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet. 2013;93(2):278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015;11(2):e1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin SH, Van Belleghem SM. Exploring evolutionary relationships across the genome using topology weighting. Genetics. 2017;206(1):429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schiffels S, Wang K. MSMC and MSMC2: the multiple sequentially Markovian coalescent. Statistical population genomics: Humana; 2020. pp. 147–65. [DOI] [PubMed] [Google Scholar]
- 52.Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences. Nat Genet. 2014;46(8):919–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santiago E, Novo I, Pardiñas AF, Saura M, Wang J, Caballero A. Recent demographic history inferred by high-resolution analysis of linkage disequilibrium. Mol Biol Evol. 2020;37(12):3642–53. [DOI] [PubMed] [Google Scholar]
- 54.Zhang C, Dong SS, Xu JY, He WM, Yang TL. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics. 2019;35(10):1786–8. [DOI] [PubMed] [Google Scholar]
- 55.Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF. Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet. 2018;19(4):220–34. [DOI] [PubMed] [Google Scholar]
- 56.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984:1358–70. [DOI] [PubMed]
- 57.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szpiech ZA, Hernandez RD. Selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. fly. 2012;6(2):80–92. [DOI] [PMC free article] [PubMed]
- 60.Yu G, Wang L-G, Han Y, He Q-Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson JF, Goldstein DB. Consistent long-range linkage disequilibrium generated by admixture in a Bantu-Semitic hybrid population. Am J Hum Genet. 2000;67(4):926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang YQ, Deng XM, Du ZQ, Qiu X, Du X, Chen W, et al. Single nucleotide polymorphisms in the chicken Lmbr1 gene are associated with chicken polydactyly. Gene. 2006;374:10–8. [DOI] [PubMed] [Google Scholar]
- 63.He C, Chen Y, Yang K, Zhai Z, Zhao W, Liu S, et al. Genetic pattern and gene localization of polydactyly in Beijing fatty chicken. PLoS ONE. 2017;12(5):e0176113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding J, Ying F, Li Q, Zhang G, Zhang J, Liu R, et al. A significant quantitative trait locus on chromosome Z and its impact on egg production traits in seven maternal lines of meat-type chicken. J Anim Sci Biotechnol. 2022;13(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim SL, Qu ZP, Kortschak RD, Lawrence DM, Geoghegan J, Hempfling A-L, et al. HENMT1 and PiRNA stability are required for adult male germ cell transposon repression and to define the spermatogenic program in the mouse. PLoS Genet. 2015;11(10):e1005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hempfling A, Lim S, Adelson D, Evans J, O’Connor A, Qu Z, et al. Expression patterns of HENMT1 and PIWIL1 in human testis: implications for transposon expression. Reproduction. 2017;154(4):363–74. [DOI] [PubMed] [Google Scholar]
- 67.Liu L, Cui H, Fu R, Zheng M, Liu R, Zhao G, et al. The regulation of IMF deposition in pectoralis major of fast-and slow-growing chickens at hatching. J Anim Sci Biotechnol. 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ikeobi CO, Woolliams JA, Morrice DR, Law A, Windsor D, Burt DW, et al. Quantitative trait loci affecting fatness in the chicken. Anim Genet. 2002;33(6):428–35. [DOI] [PubMed] [Google Scholar]
- 69.Zhu F, Cui QQ, Yang YZ, Hao JP, Yang FX, Hou ZC. Genome-wide association study of the level of blood components in Pekin ducks. Genomics. 2020;112(1):379–87. [DOI] [PubMed] [Google Scholar]
- 70.Stiller J, Feng S, Chowdhury AA, Rivas-Gonzalez I, Duchene DA, Fang Q, et al. Complexity of avian evolution revealed by family-level genomes. Nature. 2024;629(8013):851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pezzi PH, Wheeler LC, Freitas LB, Smith SD. Incomplete lineage sorting and hybridization underlie tree discordance in Petunia and related genera (Petunieae, Solanaceae). Mol Phylogenet Evol. 2024;198:108136. [DOI] [PubMed] [Google Scholar]
- 72.Wang C, Ducruet C, Wang W. Evolution, accessibility and dynamics of road networks in China from 1600 BC to 1900 AD. J Geog Sci. 2015;25:451–84. [Google Scholar]
- 73.Zhang J, Nie C, Li X, Ning Z, Chen Y, Jia Y, et al. Genome-wide population genetic analysis of commercial, Indigenous, game, and wild chickens using 600K SNP microarray data. Front Genet. 2020;11:543294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ren X, Guan Z, Li H, Wen J, Zhao X, Wang G, et al. Extensive intra-and inter-genetic admixture of Chinese Gamecock and other Indigenous chicken breeds revealed by genomic data. Poult Sci. 2023;102(7):102766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vitti JJ, Grossman SR, Sabeti PC. Detecting natural selection in genomic data. Annu Rev Genet. 2013;47(1):97–120. [DOI] [PubMed] [Google Scholar]
- 76.Ballantyne M, Woodcock M, Doddamani D, Hu T, Taylor L, Hawken RJ, et al. Direct allele introgression into pure chicken breeds using sire dam surrogate (SDS) mating. Nat Commun. 2021;12(1):659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hellmich R, Sid H, Lengyel K, Flisikowski K, Schlickenrieder A, Bartsch D, et al. Acquiring resistance against a retroviral infection via CRISPR/Cas9 targeted genome editing in a commercial chicken line. Front Genome Ed. 2020;2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang D, Tan L, Zhi Y, Bu L, Wang Y, Wang Z, et al. Genome-wide variation study and inter-tissue communication analysis unveil regulatory mechanisms of egg-laying performance in chickens. Nat Commun. 2024;15(1):7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang R, Zhu C, Zhen Y. Genetic diversity, demographic history, and selective signatures of Silkie chicken. BMC Genomics. 2024;25(1):754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wenting L, Chaoqun G, Zhao C, Sensen Y, Yanru L, Mengya W, et al. Assessing the conservation impact of Chinese Indigenous chicken populations between ex-situ and in-situ using genome-wide SNPs. J Integr Agric. 2024;23(3):975–87. [Google Scholar]
- 81.Lin Q, Liangchao X, Rong F, Qinghua N, Xiquan Z, Wen L. Genetic characteristics and selection signatures between Southern Chinese local and commercial chickens. Poult Sci. 2024;103(7):103863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arora R, Kumar H, Sharma U, Ahlawat S, Sharma R, Chhabra P, et al. Mapping genome-wide diversity and population dynamics in Indian chicken breeds for targeted conservation and breeding. Br Poult Sci. 2024;65(6):665–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The clean DNA sequencing data of 20 Beijing-You chickens reported in this study were deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA022028).