

Abstract
Background
Keratinopathic ichthyoses are a group of hereditary skin disorders caused by pathogenic variants in keratin genes such as KRT1, KRT2 and KRT10, resulting in conditions such as epidermolytic ichthyosis (EI), autosomal‐recessive EI, superficial EI and epidermal nevus. Case reports highlight the diversity of clinical manifestations, but only limited information exists regarding the quality of life and burden of disease.
Objectives
The objective of this study was to assess the clinical spectrum, genotype–phenotype correlations and burden of disease in patients with epidermolytic ichthyosis in Germany.
Methods
We conducted an observational study involving 48 patients diagnosed with EI. Evaluations included the severity of skin involvement using the Investigator's Global Assessment (IGA), the modified Ichthyosis Area Severity Index (mIASI) and complications. The burden of disease was evaluated using the Dermatology Life Quality Index (DLQI) or the Children's Dermatology Life Quality Index (cDLQI).
Results
Based on clinical features, mIASI and IGA, EI can be categorized into localized, intermediate and severe forms. Patients with keratin 1 mutations tended to have severe EI, while the three forms were evenly distributed in those with keratin 10 mutations. The study highlights that around half of the patients with EI experienced itch and severe pain. Quality of life was affected, with daily life restrictions of 78% due to care and therapies. Reimbursement for moisturizing ointments by health insurance was insufficient for one‐quarter of cases.
Conclusions
The results emphasize the need for targeted interventions and comprehensive care strategies to enhance the quality of life for affected individuals.
INTRODUCTION
Keratinopathic ichthyoses (KPI) encompass hereditary keratinization disorders caused by pathogenic variants in genes encoding keratins expressed in the suprabasal layers of the skin, KRT1, KRT10 or KRT2. 1 The main entity is epidermolytic ichthyosis (EI, syn. bullous ichthyosis) which is characterized by epidermolytic hyperkeratosis on histopathology. It is inherited in an autosomal‐dominant (KRT1 or KRT10), or rarely in a recessive (KRT10) manner. Superficial EI is linked to anomalies of keratin 2. Postzygotic mutations in any of these genes cause epidermolytic nevi, which represent a mosaic manifestation.2, 3, 4, 5, 6
There are only case series and small cohort studies available7, 8, 9, 10, 11, 12 describing the clinical impact and the overall burden of EI. In a large series of kindreds with ichthyosis, patients with EI suffered significantly more often from skin pain, odour and infections, while ocular involvement, hypertrichosis or collodion membrane were less common compared to other ichthyosis subtypes. Moreover, patients with KRT1 mutations were often affected by functionally limiting palmoplantar keratoderma (PPK), while in patients with KRT10 mutations, mild and smooth PPK with focal accentuation has been described. 12 In a Danish cohort of 16 EI patients, the severity of the condition with manifestations such as hyperkeratosis (particularly on joint regions), PPK, blister formation and erosions exhibited a spectrum ranging from mild to severe. 7 In an Austrian cohort, genotype–phenotype correlations were described based on the typical clinical presentation. 11 Quality of life studies in congenital ichthyosis revealed that general cutaneous manifestations often lead to considerable discomfort, including unpleasant odour, itching and pain.9, 10
By elucidating the genetic underpinnings, clinical spectrum and impact on patients' lives affected by EI in Germany, this research contributes to a broader understanding of this rare skin disorder and the gaps in medical care.
MATERIALS AND METHODS
Study cohort
Patients were recruited from the genodermatoses clinics at the University Hospitals of Freiburg, Munich, Cologne, and the support group for ichthyosis in Germany (https://www.ichthyose.de/). Only patients with a molecularly and histologically confirmed diagnosis were included. Forty‐eight cases with pathogenic variants in KRT1, KRT10 and KRT2, either in the germ line or in mosaic form who were born between 1958 and 2021 throughout Germany were included in this study. Ethical approvals were obtained from the University of Freiburg (EK Freiburg‐ 92/2020) and the University of Munich (LMU Nr. 21‐1254). The current mean age is 20.4 years (range: 2–65 years), among the cases were 30 females and 18 males. Consanguinity was not reported in any family.
Data collection
A structured questionnaire, focusing on the course of pregnancy, childbirth, child development (including physical development), symptoms, previous and current therapies and complications was utilized to collect data (Table S1). The questionnaire data were obtained from medical records or during the patients' visits or contacts. The questionnaire was fully filled out for 43 out of the 48 patients, and partially for 5 patients. Only genetic information was available for case 29. The Dermatology Life Quality Index (DLQI) or the Children's Dermatology Life Quality Index (cDLQI) was determined to assess the impact of skin disease on various aspects of an individual's life, including daily activities, emotions, symptoms and social interactions. It provides a quantitative measure, with a higher score indicating a greater impact of the disease on the quality of life.13, 14
Scoring of skin involvement
The skin findings were documented photographically at each visit, and the severity was standardized using the Ichthyosis Area Severity Index (IASI) score.15, 16, 17 To assess the palmoplantar involvement in these patients, the modified IASI (mIASI) score was employed. 18 Patients were categorized into three severity levels, localized, intermediate or severe, based on the mIASI calculations and the Investigator's Global Assessment (IGA 0–4; 0: clear, 1: almost clear, 2: mild, 3: moderate and 4: severe).
Laboratory investigations
Laboratory parameters were assessed in the clinical context.
Statistical analysis
GraphPad Prism version 9.5.1 (GraphPad Software, La Jolla, CA) was utilized for statistical analysis and visualization of the results. Statistical analysis was conducted using the Mann–Whitney test and Fisher's exact test. The significance level was set at p < 0.05.
RESULTS
Epidemiology and genetics of epidermolytic ichthyosis
Based on the records of the main clinical and diagnostic centres in Germany, 130 patients born between 1941 and 2022 with KRT1, KRT10 or KRT2 pathogenic variants in germline or mosaic were identified. However, data on the survival rate is unavailable. This figure yields an estimated prevalence of around 1.7 per 1 million, or 1 in 0.59 million inhabitants in Germany (84.36 million inhabitants in 2022). Assuming that mild and mosaic cases were not fully captured, we suppose having included most moderate and severe cases.
The genetic basis was disclosed in all 48 cases included in this study (Table 1). Nineteen patients had pathogenic variants in KRT1, 25 in KRT10, and one family with 4 affected members had a pathogenic variant in KRT2. Most patients were affected by de novo mutations (27 out of 48) and in 19 cases from 7 unrelated families, the inheritance was autosomal‐dominant. Two cases (20, 32) had an autosomal‐recessive EI due to biallelic pathogenic variants in KRT10. In 13 of 32 cases (40.6%), a clinical diagnosis could not be established immediately after birth, or epidermolysis bullosa was suspected. Genetic diagnosis was performed in adulthood in cases with a late onset, or in familial cases.
TABLE 1.
Overview of the genetic basis, inheritance patterns and severity spectrum of the EI cohort.
| Patient ID | Gender | Current age (years) | Affected gene | Position of mutation | Mutation on protein level | Inheritance | EI subtype | IASI‐E | IASI‐S | IASI‐ES | mIASI | Additional gene variant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | M | 6 | KRT1 | Coil 1A | p.lle184Thr | De novo | Severe | 9.6 | 14.4 | 25.4 | 28.4 | |
| 12 | M | 18 | KRT1 | Coil 1A | p.Ser186Pro | De novo | Intermediate | 0 | 7.4 | 7.4 | 10.4 | |
| 10 | M | 9 | KRT1 | Coil 1A | p.Leu187Phe | De novo | Severe | 9.4 | 14.5 | 23.9 | 26.9 | |
| 14 | M | 2 | KRT1 | Coil 1A | p.Asn188Asp | De novo | Severe | 5.8 | 10.3 | 16.1 | 19.1 | |
| 37 | F | 2 | KRT1 | Coil 1A | p.Asn188Ser | De novo | Severe | 5.8 | 10.8 | 16.6 | 19.6 | |
| 44 | F | 8 | KRT1 | Coil 1A | p.Asn188Ser | De novo | Severe | 17.1 | 12.5 | 29.6 | 32.6 | |
| 34 | F | 26 | KRT1 | Coil 1A | p.Asn188Ser | De novo | Severe | 5.2 | 20.7 | 25.9 | 29.9 | |
| 6 | M | 8 | KRT1 | Coil 1A | p.Ser193Pro (15%) mosaic (Blood) | De novo | Severe | 23.1 | 12.1 | 35.2 | 39.2 | |
| 17 | M | 9 | KRT1 | Linker 12 | p.Leu337His | AD | Localized | 0 | 1.8 | 1.8 | 2.8 | |
| 18 | M | 9 | KRT1 | Linker 12 | p.Leu337His | AD | Localized | 0 | 1.8 | 1.8 | 2.8 | |
| 16 | F | 37 | KRT1 | Linker 12 | p.Leu337His | AD | Localized | 0 | 2.4 | 2.4 | 3.4 | |
| 33 | F | 64 | KRT1 | Linker 12 | p.Leu337His | AD | Localized | 0 | 2.4 | 2.4 | 2.4 | |
| 7 | F | 13 | KRT1 | Coil 2 | p.Glu478Lys | De novo | Severe | 21.9 | 22.8 | 44.7 | 48.7 | |
| 2 | F | 43 | KRT1 | Coil 2 | p.Leu485Pro | De novo | Severe | 9.3 | 8.6 | 17.9 | 20.9 | |
| 8 | F | 6 | KRT1 | Coil 2 | p.Leu486Gln | De novo | Severe | 9.7 | 12.1 | 21.8 | 24.8 | |
| 21 | F | 6 | KRT1 | Tail | p.Tyr587Leufs*63 | De novo | Intermediate | 3.2 | 6.2 | 9.4 | 13.4 | ALOX12B : p.Pro620Gln |
| 41 | M | 2 | KRT1 | Tail | p.Gly589Trpfs*65 | AD | Intermediate | 2.6 | 7 | 9.6 | 10.6 | FLG : p.Arg501* |
| 42 | F | 9 | KRT1 | Tail | p.Gly589Trpfs*65 | AD | Intermediate | 2.9 | 6.2 | 9.1 | 12.1 | |
| 43 | F | 46 | KRT1 | Tail | p.Gly589Trpfs*65 | De novo | Severe | 8.5 | 6.6 | 15.1 | 19.1 | FLG : p.Arg501* |
| 3 | M | 22 | KRT10 | Coil 1A | p.Leu153Val | De novo | Severe | 7.3 | 15.6 | 22.9 | 23.9 | |
| 24 | M | 3 | KRT10 | Coil 1A | p.Arg156Leu | AD | Severe | 5.6 | 19.3 | 24.9 | 26.9 | |
| 15 | F | 3 | KRT10 | Coil 1A | p.Arg156Cys | De novo | Intermediate | 3 | 9.4 | 12.4 | 12.4 | |
| 38 | M | 3 | KRT10 | Coil 1A | p.Arg156His | AD | Intermediate | 4.8 | 5.2 | 10 | 11 | |
| 39 | F | 6 | KRT10 | Coil 1A | p.Arg156His | AD | Intermediate | 5 | 7.6 | 12.6 | 13.6 | |
| 23 | F | 8 | KRT10 | Coil 1A | p.Arg156Leu | AD | Severe | 6.4 | 16.2 | 22.6 | 24.6 | |
| 9 | F | 9 | KRT10 | Coil 1A | p.Arg156His | De novo | Severe | 10.9 | 6.2 | 17.1 | 17.1 | |
| 19 | M | 9 | KRT10 | Coil 1A | p.Arg156Cys | De novo | Intermediate | 4.2 | 4.6 | 8.8 | 8.8 | |
| 30 | F | 11 | KRT10 | Coil 1A | p.Arg156His | AD | Severe | 4.8 | 12.4 | 17.2 | 17.2 | |
| 28 | M | 25 | KRT10 | Coil 1A | p.Arg156=/Cys (24%) mosaic (Skin) | De novo | Localized EN | |||||
| 40 | F | 31 | KRT10 | Coil 1A | p.Arg156His | De novo | Intermediate | 5.8 | 7.6 | 13.4 | 14.4 | |
| 5 | F | 38 | KRT10 | Coil 1A | p.Arg156His | De novo | Severe | 9.2 | 11 | 20.2 | 20.2 | |
| 13 | F | 40 | KRT10 | Coil 1A | p.Arg156Leu | De novo | Intermediate | 0 | 12.8 | 12.8 | 13.8 | |
| 31 | F | 43 | KRT10 | Coil 1A | p.Arg156His | De novo | Intermediate | 4.8 | 6.2 | 11 | 11 | |
| 36 | M | 43 | KRT10 | Coil 1A | p.Arg156His | De novo | Severe | 9.4 | 8.1 | 17.5 | 18.5 | |
| 11 | M | 52 | KRT10 | Coil 1A | p.Arg156=/Cys (11%) mosaic (Skin) | De novo | Localized EN | 2.2 | 4.2 | 6.4 | 6.4 | TGM1 : p.Arg554Gln |
| 1 | F | 2 | KRT10 | Coil 1A | p.Arg156Cys | De novo | Severe | 9.6 | 14.4 | 18.6 | 19.6 | |
| 29 | M | 29 | KRT10 | Coil 1A | p.Arg156=/Cys (24%) mosaic | De novo | Localized EN | |||||
| (Skin) | ||||||||||||
| 20 | F | 6 | KRT10 | Coil 2 | p.Glu289=;Cys401* | AR | Localized | 5.2 | 5.2 | |||
| 32 | F | 21 | KRT10 | Coil 2 | p.Lys439Glyfs*3 | AR | Intermediate | 4.2 | 5 | 9.2 | 9.2 | |
| 27 | F | 5 | KRT10 | Coil 2 | p.Tyr449Ser | AD | Localized | |||||
| 26 | M | 7 | KRT10 | Coil 2 | p.Tyr449Ser | AD | Localized | |||||
| 25 | F | 33 | KRT10 | Coil 2 | p.Tyr449Ser | AD | Localized | |||||
| 22 | F | 62 | KRT10 | Coil 2 | p.Tyr449Ser | AD | Localized | 3.2 | 3 | 6.2 | 7.2 | GJB3: p.Gln26His |
| 35 | F | 34 | KRT10 | Coil 2 | p.Leu453Gln | de novo | Severe | 9.4 | 19.5 | 28.9 | 29.9 | |
| 46 | M | 4 | KRT2 | Coil 2 | p.Ile477Asn | AD | Intermediate SEI | 3.6 | 5.7 | 9.3 | 10.3 | |
| 47 | F | 6 | KRT2 | Coil 2 | p.Ile477Asn | AD | Intermediate SEI | 3.5 | 5.5 | 9 | 10 | |
| 45 | F | 37 | KRT2 | Coil 2 | p.Ile477Asn | AD | Intermediate SEI | 0 | 10 | 10 | 11 | |
| 48 | F | 65 | KRT2 | Coil 2 | p.Ile477Asn | AD | Localized SEI | 0 | 2.5 | 2.5 | 3.5 |
Note: Mutations not previously reported are highlighted in bold.
Abbreviations: AD, autosomal‐dominant; AR, autosomal‐recessive; f, female; m, male; EN, epidermolytic nevus; SEI, superficial epidermolytic ichthyosis; if one pathogenic variant, then monoallelic. * inidicates a stop codon.
To the best of our knowledge, five KRT1 and two KRT10 variants have not been reported before (Genome Aggregation Database, ClinVar, or LODV) (Table 1). The substitution of Arg156 in keratin 10 was recurrent in 16 cases of which 11 were de novo. When present in the germline, it led to severe or intermediate phenotypes. In 4 patients, postzygotic mosaicism was genetically identified and corresponded to clinically extensive mosaic lesions in three of them (11, 28 and 29), all carrying the recurrent substitution of Arg156 in keratin 10 in the skin. Patient 6 however, had the keratin 1 substitution p.Ser193Pro as a mosaic in the DNA from the blood and demonstrated the clinical picture of full‐blown severe EI. Additional monoallelic variants in genes associated with cornification disorders, FLG, ALOX12B, GJB3 and TGM1 were found in 5 patients, but their contribution to the phenotype remains unclear.
The course of epidermolytic ichthyosis in infancy
All cases except for nine (18, 33, 41–43 and 45–48), were born with skin manifestations, particularly erythroderma, blisters and erosions. Cases 18 and 33 developed blisters, erosions and keratoses a few weeks after birth. Two families (41–43 and 45–48) developed keratoses on the main joints, and only rarely blisters in infancy. Most patients (30 out of 43; 69.8%) were born through vaginal delivery, while 13 cases (13 out of 43; 30.2%) through a caesarean section. Six patients (1, 2, 6, 14, 36 and 37; 13.3%) were born preterm before the 37th week of gestation. Shortly after birth, complications included bacterial skin infections, dehydration, electrolyte imbalances and temperature regulation problems. As a result, in more than half (25 out of 41) of the patients, immediate care in an incubator or a neonatal intensive care unit was needed. More than one‐third of the cases (17 out of 44; 38.6%) were underweight or below the fifth growth percentile at birth or during early childhood. Among these 17 individuals, 11 (3, 4, 6–8, 10, 14, 30, 34, 35 and 37) exhibited a severe phenotype, while 5 (12, 13, 15, 19 and 32) showed an intermediate phenotype.
Clinical spectrum of epidermolytic ichthyosis
Different severity levels were observed, and were categorized into three groups based on localized, intermediate or severe cutaneous involvement. Localized EI cases were characterized by limited affected skin areas that were exposed to mechanical stress and by a mIASI score of up to 7 and IGA 1. Intermediate EI cases exhibited disseminated skin involvement, mIASI scores ranging between 7 and 15, and IGA 2–3, while skin involvement in severe EI was generalized with mIASI scores exceeding 15 and IGA 3–4. Using this classification, 13 patients had localized EI (mean mIASI: 4.3, range: 2.4–7.2), 15 intermediate (mean mIASI: 11.7, range: 8.8–14.4) and 20 severe (mean mIASI: 25.4, range: 17.1–32.6; Figure 1a, Table 1).
FIGURE 1.

Distribution of severity grades in EI. (a) mIASI scores in the respective groups (mean mIASI: localized, 4.3; intermediate, 11.7; severe, 25.4). (b) KRT1 is associated with a higher prevalence of severe cases. Distribution of mIASI scores was based on genotype.
While the patients with KRT10 germline mutations showed a relatively even distribution across different severity levels, 15 out of 19 patients with KRT1 germline mutations exhibited intermediate or severe EI, and higher mIASI (KRT1 vs. KRT10 median 19.10, range: 2.4–48.70 vs. 14.40, range: 5.2–29.9) and IASI‐E (KRT1 vs. KRT10 median 5.8, range: 0–23.10 vs. 5.0 range: 0–10.9) scores compared to KRT10 (Figure 1b, Figure 2). Amino acid substitutions in the coil regions of keratin 1 lead to more severe phenotypes achieving the top three highest mIASI (48.7, 39.2 and 32.6, maximum possible 52) and IASI‐E (17.1, 21.9 and 23.1, maximum possible 24, at an age range between 8 and 13 years) scores. The maximal mIASI scores in patients with KRT10 mutations were below 30.
FIGURE 2.

Clinical spectrum of epidermolytic ichthyosis. (a) Mild manifestation of EI due to KRT1 defect (mIASI: 3.4). (b) Severe case with generalized keratosis and erythema (mIASI: 26.9). (c) Intermediate case with erosions and keratoses (mIASI 9.2). (d) Generalized keratosis, erythema (mIASI: 19.6).
Eight of 39 patients (20.5%) suffered from recurrent skin infections, particularly caused by gram‐positive bacteria. Among them, six were severely, while two were intermediately affected.
Palmoplantar involvement was present in 38 patients. The severity varied from hyperlinearity to extensive diffuse involvement of the entire area with pronounced keratosis and fissures (Figure 3). All 19 patients with KRT1 mutations had palmoplantar keratoderma (PPK). Among the 20 patients displaying a severe phenotype, 17 presented with palmoplantar keratoderma (PPK) (85.0%). Two severe cases with KRT10 and 10 severe cases with KRT1 mutations showed pronounced PPK, causing an increase of at least 2 points in the mIASI score. In some severe cases with KRT1 mutations (e.g. 6 and 43), extremely pronounced PPK was observed, with thick keratoderma and painful fissures across the entire palm and sole surfaces, resulting in a 4‐point increase in the mIASI score despite otherwise relatively limited skin involvement. Although PPK is more commonly associated with KRT1 mutations, 13 patients with KRT10 mutations had mild keratosis or localized blisters, or in 8 cases only hyperlinearity. One of these patients exhibited a variant in filaggrin, while three cases demonstrated an elevation of total IgE levels.
FIGURE 3.
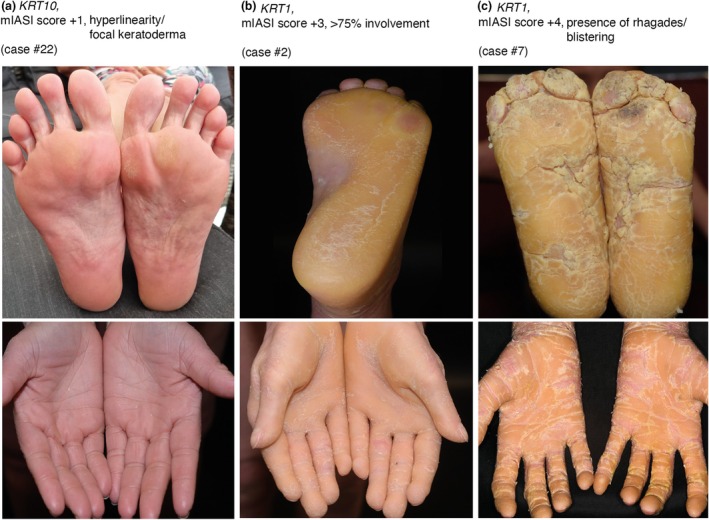
Palmoplantar keratoderma. (a) Palmoplantar hyperlinear patterns and focal keratoses due to a pathogenic variant in KRT10. mIASI score + 1. (b) Marked diffuse hyperkeratosis palmoplantar involving up to 100% of the area. mIASI score + 3. (c) Pronounced hyperkeratosis with the presence of fissures. mIASI score + 4.
Patients 43 and 21, with premature termination codons in the tail region of keratin 1, Tyr587Leufs* and Gly589Trpfs*, had a predominant involvement of palms and soles, which is in agreement with a previous report. 19
The two patients with autosomal‐recessive EI (Cases 20 and 32) exhibited localized and intermediate skin lesions, respectively. In contrast to the rest of the cohort, these patients experienced severe itching (VAS 9/10), along with elevated total IgE levels (>3000 U/mL). An allergy to cat hair was identified in Patient 20. Clinically, both cases exhibited erosive scratch excoriations on the skin with eczematous features.
The family with superficial EI (45–48) and mutation in KRT2 demonstrated an improvement of the skin lesions with age, across three generations. While the two children, aged 4 and 6, still experienced blisters on the extremities, the adults exhibited hyperkeratosis on the joints and acral regions. PPK was present in all these patients. The mean mIASI score of these patients was 8.7, lower than 17.7, the mean mIASI for the rest of the cohort.
The body mass index (BMI) was in the normal range in 11 of the 16 adults examined, (BMI range: 17.7–27.8, age range: 18–65), 2 were underweight (BMI: 16.6, 16.9; age: 22 Case 2, 43 Case 3) and 2 were overweight (BMI: 33.3, 35.4; age: 37, 38).
Vitamin D deficiency (range: 6.7–16.1 ng/mL, normal values >20.0 ng/mL) was found in 8 out of 15 patients (53.3%) for which data were available. Among the patients with normal levels, 5 were receiving vitamin D supplementation. Elevated total IgE were present in 10 out of 16 patients with values up to 4116 U/mL (range: 177–4116 U/mL, normal values <100 U/mL). To the best of our knowledge, only patient 20 had a manifest allergy (see above). All patients with elevated IgE reported itching.
Burden of disease and treatment in epidermolytic ichthyosis
Itch was present in 31 of 45 patients, the majority (16 out of 31; 51.6%) with severe EI. In comparison with patients with a localized phenotype, a significantly higher number of patients with a severe phenotype reported experiencing itching (p = 0.0214). Twenty‐one (21 out of 46, 45.7%) patients reported experiencing severe pain regularly (numeric analogue scale 8–9/10), particularly during blister formation and the occurrence of fissures on the hands and feet. A greater number of patients with a severe phenotype experienced pain compared to those with a localized condition (p = 0.0028). Six patients stated that they rarely experience pain (Figure 4a, Table S2). More than half of the patients (25 out of 42, 59.5%) reported that their skin had an unpleasant odour due to pronounced hyperkeratosis and bacterial colonization.
FIGURE 4.
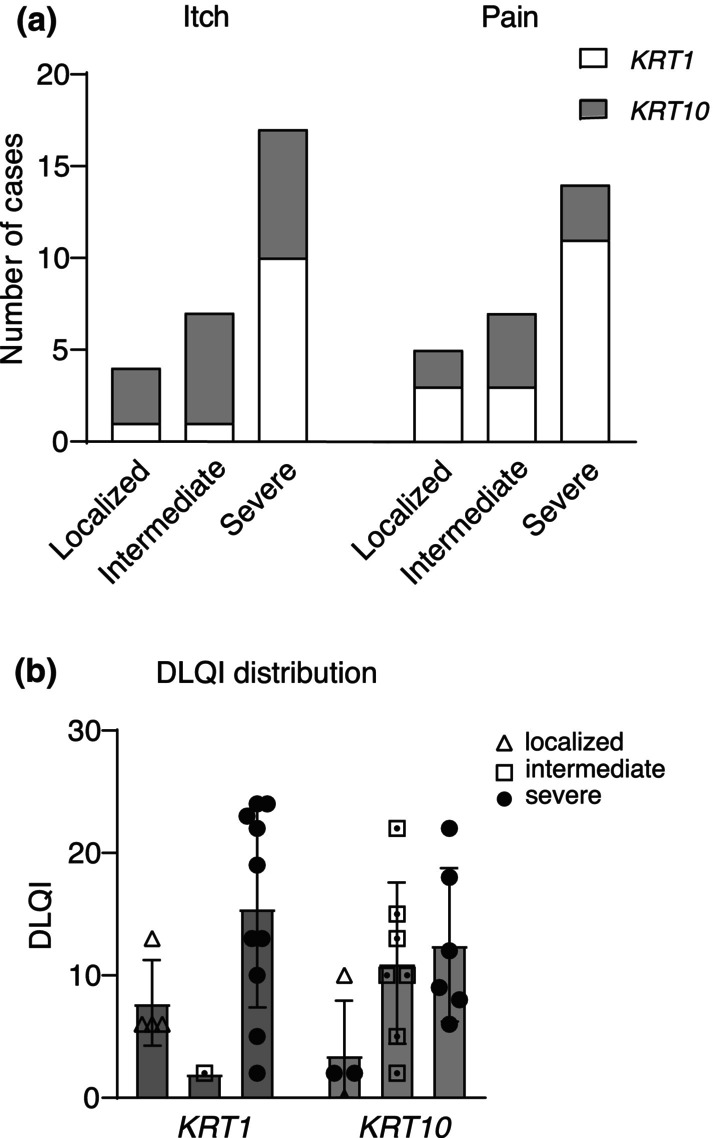
Symptoms of the disease and impairment of quality of life. (a) Depending on the severity, patients experience itching and pain (more common in the KRT1 group overall). (b) Distribution of DLQI values based on genotype and phenotype.
In KRT1‐EI, the DLQI was slightly higher (mean: 12.5, range: 2–24) compared to KRT10 (mean: 10.3, range: 0–22). The group with a localized phenotype exhibited an average DLQI score of 5.3 (range: 0–13), whereas the intermediately affected group scored 10.3 (range: 2–22) and the severely affected group scored 13.9 (range: 2–24) (Figure 4b). Patients with a severe phenotype had a significantly higher DLQI score compared to the localized group (p = 0.0027). Surprisingly, 31.3% of the severely affected individuals had low DLQI scores of under 10 (5 out of 16).
Seventy‐eight per cent (32 out of 41) of the patients reported being permanently restricted in their quality of life due to the high time commitment required for care and additional therapies such as occupational therapy, physiotherapy or psychotherapy for psychological distress caused by factors like bullying or social withdrawal related to their skin condition. All severely affected patients reported significant mobility limitations, particularly manifested as painful blistering after walking and because of the pronounced palmoplantar keratosis, the patients also experienced reduced participation in everyday life activities. In the group of severely affected patients, eight reported regularly relying on a wheelchair due to their intensity of pain. A need for care level up to Grade 4 (classification system in Germany: Grade 1, minimal care; Grade 5, most intensive care) was present in 45.0% of the cases (18 out of 40) due to their skin condition. Twenty‐four of 39 patients (61.5%) for whom the information was available were members of the German support group for ichthyosis.
Most patients were treated solely with local therapy, including frequent application of panthenol, urea‐containing creams and glycerine, in combination with baths utilizing additives such as oil or sodium bicarbonate. In 8 patients, systemic therapy with acitretin was administered (8 out of 40, 20.0% doses: from 0.13 mg/kg to 0.31 mg/kg body weight per day). They were aged between 8 and 62 years, six having KRT10 and two KRT1 mutations, with severity levels severe (4 out of 8) and intermediate (3 out of 8). Under therapy, clinical improvements were observed in these cases. The removal of palmoplantar keratosis was primarily carried out by the patients themselves or their parents using nonprofessional tools such as abrasive devices or callus shavers. Only a few cases were treated by medical podiatrists. Monthly to annual consultations with ear, nose and throat doctors for cleaning and removal of scales were required in 11 patients.
In 25.6% (10 out of 39) of the cases for which this information was available, moisturizing ointments were either not, or only partially reimbursed by the health insurance company.
DISCUSSION
This study underscores the impact of EI, an ultra‐rare disease, on health and quality of life. We identified a minimum of 130 patients, which yields an estimated prevalence of at least 1.7 cases per million people in Germany. This is similar to France, where the estimated prevalence of all keratinopathic ichthyoses is 1.1 per Million. 20 Our cohort, represents approximately one‐third of the individuals with this disease living in Germany. The results suggest that due to its extremely low prevalence and missing causal therapy or cure, this disease is challenging in terms of diagnosis, management and patient care.
From a practical point of view, patients were categorized according to disease severity into three groups: localized, intermediate and severe, akin to the classification used for epidermolysis bullosa. 21
Analysis of the present cohort indicates that individuals with EI, particularly those falling into the intermediate and severe disease subtypes, experience severe cutaneous features including erythroderma, blistering, itch and invalidating palmoplantar keratoderma that limits mobility. The disrupted epidermal barrier may contribute to an increased susceptibility to infections and elevated IgE. However, to the best of our knowledge, classical atopy with specific IgE or manifest allergies was described in only one patient. These patients also suffer from low vitamin D levels which is why supplementation should be considered in most cases. Although patients often adapt and become accustomed to their living conditions, this study demonstrates that the patients' quality of life remains consistently compromised. As a result of pain and itching, patients experienced disruptions in their ability to achieve uninterrupted sleep at night. The presence of odour and reduced mobility can cause social withdrawal and reduced self‐sufficiency. This study revealed a considerable prevalence of psychological distress among patients with EI and an expressed need for psychological support.
The management of EI poses significant challenges for patients and healthcare providers alike. The nature of the condition requires long‐term care, involving regular follow‐up appointments, specialized treatments and extensive skincare routines. For scaling, patients need to bathe more frequently, in many cases daily or even multiple times a day. Moreover, emollient baseline skin therapy needs to be applied up to four times a day. In severe cases, additional dressings have to be applied daily due to blister formation or erosions. This demands a considerable investment of time and resources for both patients and their families. Proper podiatric care is crucial to maintain mobility and prevent complications, but the costs are not covered by the healthcare system. Moreover, limited reimbursement options for treatments and supportive care create financial burdens for both patients and their families. In Germany, health insurance covers expenses exclusively for systemic retinoids and formulations containing urea. As an orphan disease, EI seems to be ignored by the health system, leaving patients and their families without essential resources. A detailed analysis of these aspects requires a separate study on the socio‐economic burden of the disease in a larger, international cohort.
The results of the current study also underline the urgent need for an interdisciplinary treatment approach to children and adults with EI facilitating comprehensive care of this potentially severe skin disease associated with significant extracutaneous symptoms such as chronic pain, restricted mobility and psychosocial impairment (Table 2).
TABLE 2.
Key recommendations for addressing the needs of patients with epidermolytic ichthyosis.
| 1. Diagnosis | Histopathology of the skin and early genetic testing for suspected cases of EI |
| 2. Treatment | Implement multidisciplinary approach involving dermatologists, geneticists, podiatrists and mental health professionals |
|
Regular skincare routines including frequent bathing, moisturizing, antiseptics and wound care to manage symptoms effectively Low‐dose retinoids may be indicated and helpful in cases with prominent hyperkeratosis | |
| 3. Additional support | Provide psychological support to address the emotional impact of chronic symptoms, including anxiety, depression and social withdrawal |
| Employ strategies for pain management, considering the severity of symptoms, such as topical analgesics or systemic therapy as appropriate | |
|
Offer mobility support and adaptive aids to improve independence and quality of life, particularly for patients with severe palmoplantar involvement Patients support groups provide advice and assistance |
Although some molecular players in the pathogenesis of EI have been identified,22, 23 translation to the bedside has not taken place yet. Further research is required to address the complex interplay between epidermal fragility, abnormal keratinization and inflammation in EI, hopefully leading to causal, gene‐directed treatments.
AUTHOR CONTRIBUTIONS
LF collected and analysed the data and drafted the manuscript together with CH; CH planned the study, analysed the data and drafted the manuscript; JH, SH, KG, KS, ITP, KK, HO, SA, JF and TS collected data, reviewed them and the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors state no conflict of interest.
ETHICS STATEMENT
The patients in this manuscript have given written informed consent to the publication of their case details.
Supporting information
Table S1.
Table S2.
ACKNOWLEDGEMENTS
We thank the patients, their families and ‘Kinderforschungsallianz’ for their involvement, support and funding. L.F. is supported by the Munich Medical & Clinician Scientist Program (MCSP) of the LMU Munich. C.H. is supported by EADV (PPRC‐2018‐65). C.H. and K.G. are members of the ERN skin. I.T.P. is supported by a grant from FOR2722 ‘Musculoskeletal Extracellular Matrix’ (‘Clinician Scientist’). Open Access funding enabled and organized by Projekt DEAL.
Frommherz L, Giehl K, Hofmann J, Huebner S, Kiekbusch K, Sabkova T, et al. Epidermolytic ichthyosis: Clinical spectrum and burden of disease in a large German cohort. J Eur Acad Dermatol Venereol. 2025;39:1028–1037. 10.1111/jdv.20096
Linked article: J. Mazereeuw‐Hautier. J Eur Acad Dermatol Venereol. 2025;39:893–894. https://doi.org/10.1111/jdv.20636.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, et al. Revised nomenclature and classification of inherited ichthyoses: results of the first ichthyosis consensus conference in Soreze 2009. J Am Acad Dermatol. 2010;63:607–641. [DOI] [PubMed] [Google Scholar]
- 2. Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, Robinson‐Bostom L. Histopathologic characterization of epidermolytic hyperkeratosis: a systematic review of histology from the National Registry for ichthyosis and related skin disorders. J Am Acad Dermatol. 2008;59:86–90. 10.1016/j.jaad.2008.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hotz A, Oji V, Bourrat E, Jonca N, Mazereeuw‐Hautier J, Betz RC, et al. Expanding the clinical and genetic Spectrum of KRT1, KRT2 and KRT10 mutations in Keratinopathic ichthyosis. Acta Derm Venereol. 2016;96:473–478. 10.2340/00015555-2299 [DOI] [PubMed] [Google Scholar]
- 4. Covaciu C, Castori M, de Luca N, Ghirri P, Nannipieri A, Ragone G, et al. Lethal autosomal recessive epidermolytic ichthyosis due to a novel donor splice‐site mutation in KRT10. Br J Dermatol. 2010;162:1384–1387. 10.1111/j.1365-2133.2010.09665.x [DOI] [PubMed] [Google Scholar]
- 5. Gutierrez JA, Hannoush ZC, Vargas LG, Momany A, Garcia CC, Murray JC, et al. A novel non‐sense mutation in keratin 10 causes a familial case of recessive Epidermolytic ichthyosis. Mol Genet Genomic Med. 2013;1:108–112. 10.1002/mgg3.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vodo D, Sarig O, Peled A, Samuelov L, Malchin N, Grafi‐Cohen M, et al. Recessive epidermolytic ichthyosis results from loss of keratin 10 expression, regardless of the mutation location. Clin Exp Dermatol. 2018;43:187–190. 10.1111/ced.13324 [DOI] [PubMed] [Google Scholar]
- 7. Bygum A, Virtanen M, Brandrup F, Gånemo A, Sommerlund M, Strauss G, et al. Generalized and naevoid epidermolytic ichthyosis in Denmark: clinical and mutational findings. Acta Derm Venereol. 2013;93:309–313. 10.2340/00015555-1447 [DOI] [PubMed] [Google Scholar]
- 8. Diociaiuti A, Castiglia D, Corbeddu M, Rotunno R, Rossi S, Pisaneschi E, et al. First case of KRT2 Epidermolytic nevus and novel clinical and genetic findings in 26 Italian patients with Keratinopathic ichthyoses. Int J Mol Sci. 2020;21. 10.3390/ijms21207707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gånemo A, Sjöden PO, Johansson E, Vahlquist A, Lindberg M. Health‐related quality of life among patients with ichthyosis. Eur J Dermatol. 2004;14:61–66. [PubMed] [Google Scholar]
- 10. Mazereeuw‐Hautier J, Dreyfus I, Barbarot S, Serrentino L, Bourdon‐Lanoy E, Ezzedine K, et al. Factors influencing quality of life in patients with inherited ichthyosis: a qualitative study in adults using focus groups. Br J Dermatol. 2012;166:646–648. 10.1111/j.1365-2133.2011.10701.x [DOI] [PubMed] [Google Scholar]
- 11. Seidl‐Philipp M, Schatz UA, Gasslitter I, Moosbrugger‐Martinz V, Blunder S, Schossig AS, et al. Spectrum of ichthyoses in an Austrian ichthyosis cohort from 2004 to 2017. J Dtsch Dermatol Ges. 2020;18:17–25. [DOI] [PubMed] [Google Scholar]
- 12. Sun Q, Burgren NM, Cheraghlou S, Paller AS, Larralde M, Bercovitch L, et al. The genomic and phenotypic landscape of ichthyosis: an analysis of 1000 kindreds. JAMA Dermatol. 2022;158:16–25. 10.1001/jamadermatol.2021.4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lewis‐Jones MS, Finlay AY. The Children's dermatology life quality index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132:942–949. 10.1111/j.1365-2133.1995.tb16953.x [DOI] [PubMed] [Google Scholar]
- 14. Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)–a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210–216. 10.1111/j.1365-2230.1994.tb01167.x [DOI] [PubMed] [Google Scholar]
- 15. Kamalpour L, Rice ZP, Pavlis M, Veledar E, Chen SC. Reliable methods to evaluate the clinical severity of ichthyosis. Pediatr Dermatol. 2010;27:148–153. 10.1111/j.1525-1470.2010.01114.x [DOI] [PubMed] [Google Scholar]
- 16. Marukian NV, Deng Y, Gan G, Ren I, Thermidor W, Craiglow BG, et al. Establishing and validating an ichthyosis severity index. J Invest Dermatol. 2017;137:1834–1841. [DOI] [PubMed] [Google Scholar]
- 17. Paller AS, Renert‐Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, et al. An IL‐17‐dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol. 2017;139:152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frommherz L, Krause A, Kopp J, Hotz A, Hübner S, Reimer‐Taschenbrecker A, et al. High rate of self‐improving phenotypes in children with non‐syndromic congenital ichthyosis: case series from south‐western Germany. J Eur Acad Dermatol Venereol. 2021;35:2293–2299. [DOI] [PubMed] [Google Scholar]
- 19. Sprecher E, Yosipovitch G, Bergman R, Ciubutaro D, Indelman M, Pfendner E, et al. Epidermolytic hyperkeratosis and epidermolysis bullosa simplex caused by frameshift mutations altering the v2 tail domains of keratin 1 and keratin 5. J Invest Dermatol. 2003;120:623–626. [DOI] [PubMed] [Google Scholar]
- 20. Dreyfus I, Chouquet C, Ezzedine K, Henner S, Chiavérini C, Maza A, et al. Prevalence of inherited ichthyosis in France: a study using capture‐recapture method. Orphanet J Rare Dis. 2014;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner‐Tuderman L, Diem A, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614–627. 10.1111/bjd.18921 [DOI] [PubMed] [Google Scholar]
- 22. Ansai O, Miyauchi T, Hayashi R, Katsumi T, Nishiguchi T, Hasegawa A, et al. Interleukin‐18 as a severity marker and novel potential therapeutic target for epidermolytic ichthyosis. Clin Exp Dermatol. 2023;48:199–210. 10.1093/ced/llac069 [DOI] [PubMed] [Google Scholar]
- 23. Malik K, He H, Huynh TN, Tran G, Mueller K, Doytcheva K, et al. Ichthyosis molecular fingerprinting shows profound T(H)17 skewing and a unique barrier genomic signature. J Allergy Clin Immunol. 2019;143:604–618. 10.1016/j.jaci.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Table S2.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.