

Abstract
The intrinsic conformational biases of individual amino acids and their interstrand side-chain–side-chain (SC–SC) interactions both contribute to the stability of β-sheets. The relative magnitudes of these effects have been difficult to assess in the context of folded proteins, where tertiary contacts complicate the quantitative analysis of local effects. We now report the results of such an analysis in a much simpler system, a short, stabilized β-hairpin structure where intrastrand (conformational) and interstrand (SC–SC) influences can be distinguished in the absence of competing protein tertiary interactions. A comprehensive comparison of all pairwise combinations of 11 N-terminal and 7 C-terminal amino acids within an 8-residue, @-tide-stabilized [in which @ denotes the 1,2-dihydro-3(6H)-pyridinyl unit] β-hairpin reveals distinct differences between the various pairings and shows that the intrastrand and interstrand effects are of comparable magnitude in contributing to the stability of the folded forms over the unfolded forms.
Keywords: β-strand conformation, peptide conformation, peptidomimetic
Although the β-sheet conformation is a firmly established component of protein secondary structure, the factors that stabilize the interaction of one peptide β-strand with another are not well understood (1). The recognition that β-sheet motifs and β-sheet interactions are key elements in protein structure (2) and function (3) and in a number of disease states (4) heightens the need for a more quantitative understanding of the roles that individual amino acids play in their assembly. The statistical prevalence of individual amino acids and of pairwise combinations within β-sheet and α-helical regions of known protein structures has provided a general foundation for understanding their relative stabilizing effects (5, 6). However, these predilections are not easily related to energetic differences, and, moreover, they reflect the context of folded proteins where tertiary as well as secondary interactions are important (3, 7). As a consequence, small mimics of protein secondary structure have played a key role in evaluating local influences on backbone conformation and side-chain–side-chain (SC–SC) interactions, with fewer complications from remote effects (2, 7, 8). For example, the stabilization of short segments of α-helices has made it possible to determine the α-helical propensities of different amino acids quantitatively, which has facilitated the rational design of α-helical peptides (9). In contrast, model systems have been notably less successful in providing the “rules” governing β-sheet formation and stability (7, 8, 10).
β-Sheet models have included both small proteins (3, 11–18), in which the impact of specific mutations on stability is reflected in melting behavior or on ligand binding, as well as minimalist, two-stranded β-hairpins (7, 8, 19). The contextual issues are reduced to different degrees in these models, with the result that there is similarity, but not quantitative correlation, among the results observed. The discrepancies between β-sheet model systems may arise from the fact that contributions from individual amino acids in the β-sheet region are affected by more remote tertiary contacts within the folded proteins. As a consequence, the influence of specific local interactions on β-structure may be best measured in the context of β-hairpins, where amino acids pair in the absence of competing protein contacts. Although the problems with conformational heterogeneity and aggregation of early β-sheet mimics have been solved with recent β-hairpin models, small variations in their structure can still result in a dramatic reduction in conformational stability (20, 21). Moreover, the folding equilibria of these hairpins is assessed by NMR, which is of modest precision and not readily applied to the large number of analogs required for an understanding of the individual and pairwise effects of different amino acids (22, 23). The disulfide-cyclized hairpins of Cochran and colleagues (19), in which the folding equilibrium is deduced from the redox behavior, represent a departure from NMR-based approaches; however, the need remains for a truly versatile β-sheet model system that tolerates a wide range of amino acid replacements and provides a straightforward spectroscopic indication of their effect on structure.
We have recently reported a greatly simplified β-hairpin system, illustrated in Fig. 1A, that not only is significantly structured in water but also provides a spectroscopic signature that allows rapid determination of the conformational equilibrium (24). The key component in this β-hairpin model is the 1,2-dihydro-3(6H)-pyridinyl unit, a cyclic amino acid surrogate that favors the extended conformation (25). [We use the @ symbol, pronounced “at,” as the one-letter code for the 1,2-dihydro-3(6H)-pyridinyl unit and refer to oligomers containing @-units and amino acids as “@-tides.”] Because the @-unit lacks an NH group, it reduces the hydrogen bond-mediated aggregation often observed with organized peptides (26). And, fortuitously, the conformational sensitivity of the CD signal of the @-unit provides an indication of the equilibrium between structured and unstructured forms (24). Using this model system, we analyzed 77 analogs and determined 16 individual and 60 pairwise effects of different amino acids on β-sheet stability.
Fig. 1.

The design and synthesis of @-tide-stabilized β-hairpins. (A) The templating effect of the @-unit combined with the reverse-turn conformation favored by the dPro-Ala dipeptide (8) results in a significant proportion of β-hairpin structure at equilibrium. The specific sequences shown, with Val residues flanking the reverse turn and Thr opposite the @-unit, are sufficiently stabilized that a broad range of β-hairpin populations can be sampled, depending on the identity of the N- and C-terminal residues. (B) Peptide analogs containing the @-unit were synthesized in high yield and purity on solid phase by using semiautomated techniques (27). After the Fmocprotected @-unit is coupled to an amino acid by direct condensation, the product is incorporated into the growing chain by using standard conditions for peptide synthesis.
Materials and Methods
Solid-Phase Synthesis of @-Tide-Peptide Hybrids. General procedures for the preparation of protected @-amino acids, including 9-fluorenylmethoxycarbonyl (Fmoc)-@-Val, and their incorporation in oligomeric @-tides and @-tide-peptide hybrids are described in ref. 27. @-Tide-peptide hybrids were synthesized on Wang polystyrene resin by using a Quest parallel synthesizer with automatic washings (Argonaut Technologies, Foster City, CA). The resin was washed between the coupling and deprotection steps with dimethylformamide (DMF) (three times) and then with CH2Cl2 (three times), agitating for 2 min before the solvent was removed. Deprotection of the Fmoc group was accomplished by shaking the resin in 20% piperidine in DMF for 15 min. The initial amino acid was added to resin by using the 1-(mesitylene-2-sulfonyl)-3-nitro-1H-1,2,4-triazole procedure (28, 29).
Fmoc-Amino Acid Addition. The resin-bound peptide (0.1 g, 1.1 mmol/g) was suspended in 2 ml of DMF. The desired Fmocprotected amino acid (2.5 eq in relation to the resin) was added to the resin, followed by 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate (2.5 eq), 1-hydroxy-7-benzotriazole (2.5 eq), and diisopropylethylamine (DIEA) (5 eq). The reaction mixture was agitated at room temperature for 1 h, the resin was washed, and the Fmoc protecting group was removed.
Coupling to the Secondary Amine of the @-Unit or Proline. The resin-bound peptide (0.1 g, 1.1 mmol/g) was suspended in 2 ml of DMF. The desired Fmoc-amino acid or di-@-tide (2.5 eq in relation to the resin) was added to the resin, followed by N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (2.5 eq) and DIEA (5 eq). The reaction mixture was agitated at room temperature for 16 h, the resin was washed, and the Fmoc protecting group was removed.
Cleavage from Resin. To cleave the product from the solid support, the resin was dried in vacuo for at least 16 h and then suspended in 1:1 CH2Cl2:trifluoroacetic acid (3 ml) and rotated in a glass vial for 2 h. The solvent was removed under reduced pressure, and the resin was dried for 16 h under high vacuum. The resulting resin was resuspended in methanol, the slurry was filtered, and the resin was washed (three times each in 2 ml of MeOH and CH2Cl2). The combined filtrates were concentrated under reduced pressure, and the crude product was immediately purified by using preparative HPLC. Isolated yields ranged from ≈50% to 80% with a typical yield of ≈55%. The yields varied depending on the hydrophobicity of the sequence: Higher yields were obtained for nonpolar @-tide-peptide hybrids; lower yields were observed for polar hybrids.
Purification and Characterization. The @-tide-peptide hybrids were purified by reverse-phase HPLC and characterized by analytical HPLC (purity) and high-resolution mass spectrometry (identity). Detailed procedures and characterization data are provided in the supporting information, which is published on the PNAS web site.
CD Methods. Samples were prepared in aqueous buffer (10 mM sodium phosphate, pH 7). Compound concentrations were determined at 284 nm (extinction coefficient = 22,200 M–1·cm–1) by using a UV spectrometer. Each compound was measured at three different concentrations, ranging from 15 to 60 μM. CD spectra were obtained on a 62DS spectropolarimeter (Aviv Associates, Lakewood, NJ) using a scan speed of 20 nm/min with a band width of 2 nm. The data were imported into excel (Microsoft), corrected for solvent contributions, and converted to molar ellipticities: [Θ] (104 degrees·cm2·dmol–1) = 100ψ/lcn, where ψ is the observable signal in millidegrees, l is the path length in cm, c is concentration in millimolar, and n is the number of @-units. The CD results and the calculated values for the β-hairpin equilibrium constants (Keq), folding energies (ΔG°XY), and SC–SC contributions (ΔΔG°XY) are tabulated in the supporting information.
Results and Discussion
Conformational analysis poses a significant challenge in the evaluation of peptides and peptidomimetics (10). The traditional spectroscopic tool, CD of the peptide bond, is useful for assessing relative proportions of different elements of secondary structure in a folded protein, but it is poorly parameterized for short peptide sequences (30). Most critically, in the context of a β-hairpin, the signal at <220 nm due to the peptide unit is a complex combination of β-turn, β-sheet, and random-coil signals, precluding the deconvolution of the β-hairpin population (31). For the @-tide derivatives, the analysis is further complicated by the strong absorption in this region of the @-unit itself. However, the vinylogous amide moiety of the @-unit also absorbs strongly at 284 nm (ε = 22,200 M–1cm–1), providing a CD signal that is outside the envelope of the peptide backbone and dependent only on the asymmetric environment of this chromophore. The CD signal of the @-unit was parameterized by determining the molar ellipticity [Θ] for a number of control compounds of 0% and 100% β-sheet populations (Fig. 2) (24). Similar ellipticity for the various compounds within each of these classes suggests that the CD signal of the @-unit not only reflects the local backbone conformation but also, like that of a peptide, is relatively insensitive to the identity of the flanking amino acids.
Fig. 2.

Mean @-unit ellipticities [Θ] (104 degrees·cm2·dmol–1) at 282 nm for benchmark @-tides representing 0 and 100% β-hairpin conformations. Ellipticities were determined in 10 mM phosphate buffer (pH 7) at 25°C; each value is the average of three independent measurements over the concentration range of 15–45 μM.
From the limiting values of [Θ]100 and [Θ]0, the mole fraction xβ of β-hairpin conformation for the @-tide-templated molecules can be derived from Eq. 1:
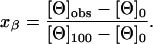 |
[1] |
Using this rapid assay for determining the extent of β-hairpin folding, we analyzed all combinations of the N- and C-terminal amino acids indicated in Fig. 1 A as X and Y.‡ Although this library does not include all amino acid pairs, it encompasses a large portion of chemical space by including large and small hydrophobic, charged, and polar amino acids.
We have shown previously that @-tide-β-hairpins adopt the expected β-hairpin conformation in water (24). In the library presented here, the @-unit template and the residues around the β-turn were kept as an invariant element among the analogs in this study to ensure a common hairpin conformation. Fig. 3 shows the superimposed CD curves of all of the hairpins studied, plus those of the macrocyclic benchmark compounds and some additional hairpins reported previously. Although there are a few analogs for which the minimum is shifted to longer wave-lengths, the similarities among the curves and the appearance of isodichroic areas at ≈320, 298, and 246 nm are consistent with the view that the folded conformations are also similar. Our analysis assumes that the templated hairpins exist in a two-state folded–unfolded equilibrium, as demonstrated for a number of much longer peptidic β-hairpins (19, 20, 23, 32–37). Although this model has not been proved rigorously, it is consistent with the observation of cooperative effects from alterations at the terminal positions and at the positions flanking the β-turn in our earlier study on hairpin stability (24).
Fig. 3.

Superposition of CD spectra for templated hairpins and macrocyclic benchmark analogs.
Within this library, the mole fraction β-hairpin ranges from a low of 0.14 (Keq = 0.16) for S@VDPAVTE (designated as the SE analog) to a high of 0.56 (Keq = 1.26) for I@VDPAVTI (the II derivative). Their relative stabilities are depicted graphically in Fig. 4A, in which the β-hairpin populations have been converted into free-energy differences. This bar graph has been ordered along each axis according to the stability of the XG and GY analogs (white bars), which divides the compounds roughly into two groups: toward the back are β-hairpins that are more stable than the GG analog; at the front are those that are less stable than GG.
Fig. 4.

Experimental free-energy components of β-hairpin folding. (A) Free energies of β-hairpin folding for X@VDPAVTY: ΔG°XY = –RTlnKeq(XY) = –RTln[xβ/(1 – xβ)]; the dividing plane of the bar graph is set at the free energy of folding for G@VDPAVTG (Keq = 0.38; ΔG°GG = +633 cal/mol). β-Hairpin populations are determined in triplicate, with standard deviations ranging from 3% to 5%. (B) Components of free energies of β-hairpin folding for X@VDPAVTY that can be attributed to the intrinsic effects of the terminal amino acids: ΔG°XY(intr) = ΔG°GG + (ΔG°XG – ΔG°GG) + (ΔG°GY – ΔG°GG) = ΔG°XG + ΔG°GY – ΔG°GG; the dividing plane is set as in A. (C) The difference between A and B. Shown are energetic contributions from SC–SC interactions across the ends of the X@VDPAVTY β-hairpins: ΔΔG°XY =ΔG°XY –ΔG°XY(intr).
Intrinsic Effects. The general characteristics of these two groups are not unexpected: Hydrophobic side chains are found in the more stable hairpins, and charged and polar side chains are found in the less stable ones (38). However, much more information can be gleaned from this data set. For example, the XG and GY analogs (the white bars in Fig. 4A) reveal the intrinsic contributions of the varied amino acids (that is, their effect on the peptide backbone conformation in the absence of interstrand SC–SC interaction). However, it is important to note that the choice of reference state has an influence on how the intrinsic effects due to the side chains are calculated. By comparing the XG and GY hairpins with the GG hairpin, the intrinsic effects that are measured for each side chain include those due to conformational constraint and those arising from interaction with functional groups common to all of the hairpins.
Because the two termini of the β-hairpin represent different environments, it is not surprising that location also influences the measured intrinsic effects, especially for those with charged and polar side chains. Whereas the hydrophobic amino acids V and I show similar favorable intrinsic effects at both positions, striking differences are observed for the charged amino acids D, E, and K. When the negative side chains of D and E are at the C terminus, they augment the charge and favor a salt bridge with the N terminus; in contrast, when these residues are located at the N terminus, they neutralize the charge and diminish β-hairpin formation (Fig. 5). The opposite preference is observed for the positively charged lysine. With those caveats, these intrinsic effects, (ΔG°XG or ΔG°GY) – ΔG°GG, correlate generally with the statistical propensities of the amino acids to occur in β-sheet structures within proteins (5) (Fig. 5).
Fig. 5.

Relationship between intrinsic effects at the N termini (filled symbols) and C termini (open symbols) and statistical β-sheet propensities from ref. 5. Blue, neutral side chains; green, charged side chains at their reinforcing positions in the hairpin (i.e., D and E at the C terminus and K at the N terminus); red, charged side chains at their destabilizing positions.
Interstrand SC–SC Interactions. Interstrand SC–SC interactions are potentially of greatest interest in understanding the energetics of β-sheet formation, yet they have been the most difficult to quantify by other methods (14). Although the effects of different amino acids on β-sheet and β-hairpin stability have been investigated in several contexts (7, 14, 19, 39), few studies in simple model β-sheets have distinguished the specific contributions due to SC–SC contacts from their intrinsic effects. In the absence of SC–SC interaction across the ends of the β-hairpin, the intrinsic effects of the individual amino acids would be additive, resulting in the free-energy pattern illustrated in Fig. 4B. The difference, ΔΔG°XY, between the observed free energy and the intrinsic free energy of folding reveals the effects of SC–SC interactions between the N- and C-terminal amino acids (Table 1 and Fig. 4C).
Table 1.
ΔΔG°XY = ΔG°XY–ΔG°XY(intr): Energy of SC–SC interactions at terminal positions of β-hairpins in cal·mol–1

Positive or negative values >200 cal·mol–1 are shown in red or blue, respectively. *ΔΔG° = 0 for analogs containing Gly at either terminus.
Table 1 shows some clear and intuitive trends for the energies of SC–SC interaction. Pairing hydrophobic amino acids (I, L, and V) with other hydrophobes (I and V) or with side chains with substantial hydrophobic regions (e.g., K and E) affords the most stable hairpins among those we explored. Conversely, in hairpins where side-chain charges are mismatched (DD, DE, ED, EE, and KK), the interactions are uniformly destabilizing. Table 1 reveals some less obvious relationships as well. Aspartic acid, regardless of location, interacts unfavorably with almost all partners, even the oppositely charged lysine; glutamate shows similar tendencies but is able to take advantage of the extra methylene group to interact more favorably with hydrophobic side chains and with lysine. Serine and threonine are perhaps the most enigmatic as interaction partners. For threonine, there are significant differences for the same pairs, depending on their location [e.g., IT (ΔΔG°IT = +281 cal·mol–1) vs. TI (–182); ET (–75) vs. TE (+216)]. And, although serine is a favorable partner for C-terminal residues of all types, it discriminates between isoleucine and valine [SI (–269) vs. SV (+93)] and aspartate and glutamate [SD (–79) vs. SE (+198)]. The ability of the threonine and serine side chains to hydrogen-bond both within and between strands may be responsible for the subtle effects manifested in these β-hairpins.
Because the intrinsic effects, ΔG°XY(intr), and the SC–SC interactions, ΔΔG°XY, are comparable in magnitude, both must be favorable for X@VDPAVTY to fold into a β-hairpin that is more stable than the baseline G@VDPAVTG sequence (xβ = 0.26; Keq = 0.34) (see Fig. 4). The two effects are stabilizing for amino acids with hydrophobic side chains, which explains the behavior of combinations of I, L, and V. In contrast, when these effects are opposed, as for EK and VE (xβ = 0.26 for both), no advantage over baseline is seen. In EK, the favorable interaction energy is offset by the poor intrinsic effects from Glu at the N terminus and Lys at the C terminus; in VE, the highly favorable intrinsic effects are negated by the unfavorable interaction of the side chains. For the polar amino acids in general, either ΔG°XY(intr) or ΔΔG°XY is frequently destabilizing, with the result that the sequences with combinations of polar residues are typically less stable than G@VDPAVTG.
One of the most extensive previous studies that dissected β-sheet folding energies into the intrinsic effects and SC–SC interactions is that of Smith and Regan (14), who investigated changes in the thermal stability of the B1 domain of streptococcal protein G from substitutions at two hydrogen-bonded positions in the β-sheet. Although the model systems and methods of analysis differ, there is a good correlation between the side-chain interaction energies calculated in the β-hairpins (ΔΔG°XY) and the GB1 protein [Δ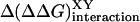 (14)] if our X is matched with their Y, and vice versa (Fig. 6). However, the interaction energies are smaller in magnitude at the ends of the β-hairpin structure, and the KE pairing, which is strongly favored in the B1 protein, is reduced in significance in the presence of the terminal charges of the β-hairpin. As Smith and Regan noted in their system, the side-chain interaction energies are strongly dependent on location, and the reverse comparison (i.e., X matched with X and Y matched with Y) shows no correlation at all (graph not shown). However, the basis for this comparison has been called into question by Distefano et al. (18), whose analysis of the effect of mutations on phage-expressed GB1 domain mutants indicates that specific cross-strand side-chain interactions play only a minor role in stabilizing mutants of this protein. Their results suggest that the correlation observed in Fig. 5 is coincidental and that SC–SC interactions are more significant in the context of flexible hairpins than in the folded GB1 protein.
(14)] if our X is matched with their Y, and vice versa (Fig. 6). However, the interaction energies are smaller in magnitude at the ends of the β-hairpin structure, and the KE pairing, which is strongly favored in the B1 protein, is reduced in significance in the presence of the terminal charges of the β-hairpin. As Smith and Regan noted in their system, the side-chain interaction energies are strongly dependent on location, and the reverse comparison (i.e., X matched with X and Y matched with Y) shows no correlation at all (graph not shown). However, the basis for this comparison has been called into question by Distefano et al. (18), whose analysis of the effect of mutations on phage-expressed GB1 domain mutants indicates that specific cross-strand side-chain interactions play only a minor role in stabilizing mutants of this protein. Their results suggest that the correlation observed in Fig. 5 is coincidental and that SC–SC interactions are more significant in the context of flexible hairpins than in the folded GB1 protein.
Fig. 6.

Correlation between side-chain interaction energies observed in streptococcal protein G B1 domain [Δ(ΔΔG)XYinteraction from ref. 14] and in @-tide-stabilized β-hairpins (ΔΔG°XY).
Conclusion
The quantitative and rapid determination of the contributions of individual amino acids to the stability of peptide β-structure will further the mechanistic understanding of general effects that have been observed in diverse and more complex systems. The approach using @-tide-templated β-hairpins can be readily expanded to encompass additional amino acid combinations and applied to other analogs to probe interactions in different contexts, including longer hairpins in which the varied sites are removed from the N and C termini. Key advantages of this strategy derive from the spectroscopic and structural characteristics of the @-unit itself: This amino acid surrogate is integral to the oligomer structure, stabilizes the hairpin conformation, and, through its CD signature, provides a general and rapid method for probing peptide conformation that is independent of amino acid composition. Quantitative insights from this work should contribute to an understanding of protein folding, the design of unnatural proteins, and the interpretation of protein aggregation processes.
Supplementary Material
Acknowledgments
We thank Prof. Susan Marqusee and her laboratory for assistance with CD measurements. This work was supported by a grant from the National Institutes of Health and by graduate fellowships from Eli Lilly (to S.T.P.), Boehringer Ingelheim (to S.T.P.), Ministero dell'Istruzione, dell'Università e della Ricerca: XVI Ciclo-Dottorato di Ricerca, and Università Degli Studi di Urbino “Carlo Bo” (to G.P.). The Center for New Directions in Organic Synthesis is supported by Bristol-Myers Squibb as a sponsoring member and Novartis Pharma as a supporting member.
Author contributions: S.T.P. and P.A.B. designed research; and S.T.P. and G.P. performed research.
Abbreviations: SC–SC, side chain–side chain; @, 1,2-dihydro-3(6H)-pyridinyl; Fmoc, 9-fluorenylmethoxycarbonyl.
Footnotes
Because the CD signal of @-tide β-hairpins can be affected by exciton coupling with aromatic side chains located at certain positions on the opposing strand (26), these amino acids were omitted from the current investigation.
References
- 1.Baltzer, L., Nilsson, H. & Nilsson, J. (2001) Chem. Rev. 101, 3153–3163. [DOI] [PubMed] [Google Scholar]
- 2.Lacroix, E., Kortemme, T., de la Paz, M. L. & Serrano, L. (1999) Curr. Opin. Struct. Biol. 9, 487–493. [DOI] [PubMed] [Google Scholar]
- 3.Smith, C. K. & Regan, L. (1997) Acc. Chem. Res. 30, 153–161. [Google Scholar]
- 4.Cohen, F. E. & Kelly, J. W. (2003) Nature 426, 905–909. [DOI] [PubMed] [Google Scholar]
- 5.Zhu, H. & Braun, W. (1999) Protein Sci. 8, 326–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutchinson, E. G., Sessions, R. B., Thornton, J. M. & Woolfson, D. N. (1998) Protein Sci. 7, 2287–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Searle, M. S. (2001) J. Chem. Soc. Perkin Trans. 2, 1011–1020.
- 8.Gellman, S. H. (1998) Curr. Opin. Chem. Biol. 2, 717–725. [DOI] [PubMed] [Google Scholar]
- 9.Venkatraman, J., Shankaramma, S. C. & Balaram, P. (2001) Chem. Rev. 101, 3131–3152. [DOI] [PubMed] [Google Scholar]
- 10.Nesloney, C. L. & Kelly, J. W. (1996) Bioorg. Med. Chem. 4, 739–766. [DOI] [PubMed] [Google Scholar]
- 11.Otzen, D. E. & Fersht, A. R. (1995) Biochemistry 34, 5718–5724. [DOI] [PubMed] [Google Scholar]
- 12.Minor, D. L., Jr., & Kim, P. S. (1994) Nature 367, 660–663. [DOI] [PubMed] [Google Scholar]
- 13.Minor, D. L., Jr., & Kim, P. S. (1994) Nature 371, 264–267. [DOI] [PubMed] [Google Scholar]
- 14.Smith, C. K. & Regan, L. (1995) Science 270, 980–982. [DOI] [PubMed] [Google Scholar]
- 15.Kim, C. A. & Berg, J. M. (1993) Nature 362, 267–270. [DOI] [PubMed] [Google Scholar]
- 16.Smith, C. K., Withka, J. M. & Regan, L. (1994) Biochemistry 33, 5510–5517. [DOI] [PubMed] [Google Scholar]
- 17.Kotz, J. D., Bond, C. J. & Cochran, A. G. (2004) Eur. J. Biochem. 271, 1623–1629. [DOI] [PubMed] [Google Scholar]
- 18.Distefano, M. D., Zhong, A. & Cochran, A. G. (2002) J. Mol. Biol. 322, 179–188. [DOI] [PubMed] [Google Scholar]
- 19.Russell, S. J., Blandl, T., Skelton, N. J. & Cochran, A. G. (2003) J. Am. Chem. Soc. 125, 388–395. [DOI] [PubMed] [Google Scholar]
- 20.Espinosa, J. F., Syud, F. A. & Gellman, S. H. (2002) Protein Sci. 11, 1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santiveri, C. M., Rico, M. & Jiménez, M. A. (2000) Protein Sci. 9, 2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramírez-Alvarado, M., Kortemme, T., Blanco, F. J. & Serrano, L. (1999) Bioorg. Med. Chem. 7, 93–103. [DOI] [PubMed] [Google Scholar]
- 23.Syud, F. A., Espinosa, J. F. & Gellman, S. H. (1999) J. Am. Chem. Soc. 121, 11577–11578. [Google Scholar]
- 24.Phillips, S. T., Blasdel, L. K. & Bartlett, P. A. (2005) J. Am. Chem. Soc. 127, 4193–4198. [DOI] [PubMed] [Google Scholar]
- 25.Phillips, S. T., Rezac, M., Abel, U., Kossenjans, M. & Bartlett, P. A. (2002) J. Am. Chem. Soc. 124, 58–66. [DOI] [PubMed] [Google Scholar]
- 26.Phillips, S. T., Blasdel, L. K. & Bartlett, P. A. (2005) J. Org. Chem. 70, 1865–1871. [DOI] [PubMed] [Google Scholar]
- 27.Phillips, S. T., Piersanti, G., Rüth, M., Gubernator, N., van Lengerich, B. & Bartlett, P. A. (2004) Org. Lett. 6, 4483–4485. [DOI] [PubMed] [Google Scholar]
- 28.Blankenmeyer-Menge, B., Nimtz, M. & Frank, R. (1990) Tetrahedron Lett. 31, 1701–1704. [Google Scholar]
- 29.Nielsen, J. & Lyngsø, L. O. (1996) Tetrahedron Lett. 37, 8439–8442. [Google Scholar]
- 30.Perczel, A., Park, K. & Fasman, G. D. (1992) Proteins Struct. Funct. Genet. 13, 57–69. [DOI] [PubMed] [Google Scholar]
- 31.Bush, C. A., Sarkar, S. K. & Kopple, K. D. (1978) Biochemistry 17, 4951–4954. [DOI] [PubMed] [Google Scholar]
- 32.Maynard, A. J., Sharman, G. J. & Searle, M. S. (1998) J. Am. Chem. Soc. 120, 1996–2007. [Google Scholar]
- 33.Searle, M. S., Griffiths-Jones, S. R. & Skinner-Smith, H. (1999) J. Am. Chem. Soc. 121, 11615–11620. [Google Scholar]
- 34.Honda, S., Kobayashi, N. & Eisuke Munekata, E. (2000) J. Mol. Biol. 295, 269–278. [DOI] [PubMed] [Google Scholar]
- 35.Espinosa, J. F. & Gellman, S. H. (2000) Angew. Chem. Int. Ed. 39, 2330–2332. [DOI] [PubMed] [Google Scholar]
- 36.Cochran, A. G., Skelton, N. J. & Starovasnik, M. A. (2001) Proc. Natl. Acad. Sci. USA 98, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciani, B., Jourdan, M. & Searle, M. S. (2003) J. Am. Chem. Soc. 125, 9038–9047. [DOI] [PubMed] [Google Scholar]
- 38.Street, A. G. & Mayo, S. L. (1999) Proc. Natl. Acad. Sci. USA 96, 9074–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanger, H. E., Syud, F. A., Espinosa, J. F., Giriat, I., Muir, T. & Gellman, S. H. (2001) Proc. Natl. Acad. Sci. USA 98, 12015–12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}