

Abstract
Hepatocellular carcinoma (HCC) poses a significant therapeutic challenge, driving the need for novel treatment strategies. This study investigates the combination of photothermal therapy (PTT) and metalloimmunotherapy for HCC treatment using Co + diABZI@J-dICG nanoparticles. Indocyanine green (ICG), an FDA-approved near-infrared (NIR) dye, is dimerized into J-aggregates to enhance PTT by improving light absorption and photothermal efficiency. The cGAS-STING pathway, a key mediator of innate immunity, is activated by the STING agonist diABZI, while cobalt ions (Co2+) further enhance immune responses. The Co + diABZI@J-dICG nanoparticles take advantage of ICG’s hepatotropic properties for sustained tumor accumulation and immune activation, resulting in significant tumor growth inhibition and reduced HCC recurrence following hepatectomy.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s12951-025-03353-7.
Keywords: Hepatocellular carcinoma, Photothermal therapy, Metalloimmunotherapy, Indocyanine green, Nanoparticles
Introduction
Hepatocellular carcinoma (HCC) presents a significant global health challenge, with treatment options ranging from systemic therapies to localized interventions [1]. While targeted therapies and immunotherapies have expanded the therapeutic arsenal, there is an unmet need for more effective and precise treatment modalities [2]. Localized treatments, such as transarterial chemoembolization (TACE) and radiofrequency ablation (RFA), are critical for managing early-stage HCC but are limited by their invasive nature, which can lead to complications and patient discomfort [3, 4].
Photothermal therapy (PTT) is an innovative, non-invasive cancer treatment approach that converts light energy into heat to induce apoptosis in cancer cells [5, 6]. Indocyanine green (ICG), the only FDA-approved near-infrared (NIR) dye, plays a pivotal role in PTT due to its heat-generating properties upon NIR irradiation [7]. The photothermal efficacy of ICG can be enhanced through dimerization into dimeric ICG (dICG), which self-assembles into J-aggregates [8]. These J-aggregates exhibit a narrow, red-shifted absorption spectrum, improving light absorption and increasing photothermal conversion efficiency [9]. Additionally, J-dICG outperforms monomeric ICG in terms of photostability and photodegradation, making it an ideal agent for PTT. This advancement also enhances its drug-loading capacity, enabling precise tumor targeting and minimizing side effects on healthy tissues [10, 11].
The cGAS-STING pathway, a key mediator of innate immunity, is activated when cytosolic double-stranded DNA (dsDNA) is detected by cGAS, leading to the formation of the secondary messenger 2’3’-cyclic-GMP-AMP (cGAMP). This molecule then binds to the STING receptor, initiating a signaling cascade that produces type I interferons and other immune mediators, enhancing the anti-tumor immune response [12]. diABZI, a STING agonist, mimics the action of cGAMP by directly activating STING, thereby boosting the immune response [13]. By incorporating diABZI into J-dICG, the tumor-targeting capabilities of ICG are leveraged [14], while the photothermal effects of J-dICG enhance the intracellular delivery of diABZI and create a hyperthermic tumor environment that releases dsDNA, further activating the cGAS-STING pathway [15]. This synergistic approach utilizes the immunostimulatory effects of diABZI and the photothermal effects of J-dICG to potentiate a robust immune response against HCC.
Metalloimmunotherapy, which utilizes metal ions such as manganese (Mn2+), zinc (Zn2+), and cobalt (Co2+), is an emerging field that activates the cGAS-STING pathway and amplifies the immune response against tumors [16–18]. These ions increase the sensitivity of cGAS and STING, boosting their binding affinity for cGAMP and significantly enhancing the production of type I interferons (IFNs) [19, 20]. This results in the activation of dendritic cells (DCs), the initiation of tumor antigen-specific T cells, and the activation of natural killer (NK) cells. Cobalt ions, in particular, help modulate the tumor microenvironment [21], enhancing the efficacy of immunotherapy. When combined with PTT, which induces immunogenic cell death (ICD) through the heat generated by J-dICG upon NIR irradiation, the synergistic effect of cobalt ions and PTT can significantly improve therapeutic outcomes [22].
Herein, we developed Co + diABZI@J-dICG nanoparticles (NPs), designed to harness the power of photothermal therapy and immunomodulation. These nanoparticles integrate the photothermal properties of J-dICG and the immunostimulatory effects of diABZI, while cobalt ions may further enhance their immunogenicity [23, 24]. We demonstrated that Co + diABZI@J-dICG NPs accumulated in tumor tissue and underwent lysosomal escape upon internalization by HCC cells. Under NIR irradiation, Co + diABZI@J-dICG NPs generated heat, causing cellular and mitochondrial damage, apoptosis, and activation of the cGAS-STING-IFN-β pathway. The subsequent release of damage-associated molecular patterns (DAMPs) promoted ICD and activated DCs within both the tumor and spleen, leading to the activation of CD8 + T cells, polarization of M1 macrophages, and activation of NK cells. This also reduced the suppressive function of regulatory T cells (Tregs), resulting in a potent anti-tumor immune response. Furthermore, Co + diABZI@J-dICG NPs induced interleukin-6 (IL-6) expression in HCC cells, upregulating prostaglandin dehydrogenase (PGDH), an enzyme that degrades prostaglandin E2 (PGE2), a molecule known to promote tumor growth and immune evasion [25]. The upregulation of PGDH induced by the nanoparticles could inhibit HCC growth by disrupting the pro-tumorigenic effects of PGE2, potentially reducing HCC recurrence after surgery (Fig. 1). In summary, Co + diABZI@J-dICG NPs represent a significant advancement in nanomedicine and immunotherapy, offering a promising strategy for the effective treatment of HCC.
Fig. 1.

Schematic illustration of Co+diABZI@J-dICG nanoparticles synergistically combining photothermal therapy and metalloimmunotherapy to activate cGAS-STING signaling, enhance IL-6/PGDH-mediated tumor suppression, and inhibit hepatocellular carcinoma progression and recurrence through immunogenic cell death and immune microenvironment remodeling
Results
Preparation and characterization of Co + diABZI@J-dICG nanoparticles
The preparation and structural properties of Co + diABZI@J-dICG NPs were thoroughly characterized using scanning electron microscopy (SEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). As shown in Fig. 2A, the SEM and HAADF-STEM images, along with elemental mapping for carbon (C), nitrogen (N), oxygen (O), chlorine (Cl), and cobalt (Co), confirmed the composition and uniform distribution of elements within the nanoparticles. Transmission electron microscopy (TEM) images further validated the structural integrity of the nanoparticles (Fig. 2B). The hydrodynamic size of J-dICG was approximately 90 nm, while the Co + diABZI@J-dICG NPs were slightly larger, measuring around 110 nm as determined by nanoparticle tracking analysis (NTA) (Fig. 1C). Zeta potential measurements, shown in Fig. 2D, revealed that J-dICG exhibited a positive charge of approximately + 30 mV (Fig. 1D). Upon incorporation of the drug components, the zeta potential of Co + diABZI@J-dICG NPs decreased to near-neutral values, approaching 0 mV, suggesting enhanced colloidal stability and improved circulation characteristics, as well as an increased affinity for tumor cells [26]. As depicted in Fig. 2E, the temperature of Co + diABZI@J-dICG NPs increased significantly compared to saline, with a temperature change (Δ℃) of 24.7 ℃ observed at a concentration of 80 µg/mL after 6 min of 808 nm NIR irradiation (1 W/cm2). Furthermore, the temperature change exhibited a linear relationship with the laser power (Fig. 2F), which is essential for controlled PTT [27]. The photothermal stability of Co + diABZI@J-dICG NPs was further confirmed through repeated heating and cooling cycles. As shown in Fig. 2G, the temperature rise remained consistent across five heating and natural cooling cycles, demonstrating excellent photothermal stability and reliability for therapeutic applications. Infrared thermograms showed a concentration-dependent photothermal effect of Co + diABZI@J-dICG solutions (Fig. 2H), with distinct temperature responses observed at different nanoparticle concentrations (0, 2.5, 5, 10, 20 µg/mL) and various times (0–6 min) under NIR irradiation (1 W/cm2). The cytotoxicity of Co + diABZI@J-dICG NPs was assessed by live/dead staining (Fig. 2I), which revealed an increasing proportion of dead cells in the Co + diABZI@J-dICG + NIR group compared to the control group, correlating with the progression of treatment. Apoptosis induction was quantified by flow cytometry (Fig. 2J), where the percentage of Annexin V-FITC+ & PI + cells increased from 0.51% in the control group to 37.7% in the Co + diABZI@J-dICG + NIR group, indicating significant apoptosis and enhanced cell death upon treatment with the nanoparticles and NIR irradiation. These results demonstrate that Co + diABZI@J-dICG NPs exhibit excellent photothermal properties, stability, and cytotoxicity, supporting their potential as an effective platform for targeted therapy.
Fig. 2.

Characterization of Co + diABZI@J-dICG NPs. (A) SEM and HAADF-STEM images of Co + diABZI@J-dICG NPs with uniform size and morphology and EDS energy spectra confirming the elemental composition of Co + diABZI@J-dICG NPs. (B) TEM image of Co + diABZI@J-dICG NPs (scale bar: 200 nm). (C) Hydrodynamic size distributions of J-dICG and Co + diABZI@J-dICG NPs solution. (D) Zeta potential distributions of J-dICG and different drug-loaded J-dICG NPs. (E) The temperature curves with different concentrations of Co + diABZI@J-dICG NPs under NIR irradiation (1 W/cm2). (F) The temperature curves of the same concentration of Co + diABZI@J-dICG NPs (40 µg/mL) under NIR irradiation with different power densities (0.5 W/cm2, 1 W/cm2, 1.5 W/cm2). (G) Photothermal stability evaluation of Co + diABZI@J-dICG NPs solution (80 µg/mL) under laser administration (1 W/cm2) for five lasers on/off cycles. (H) Infrared thermal images of different concentrations of Co + diABZI@J-dICG NPs under NIR irradiation (1 W/cm2). (I) Live/dead cell staining of Hepa1-6 after J-dICG or Co + diABZI@J-dICG treatment with or without laser irradiation (1 W/cm2, 5 min), scale bar: 200 μm. (J) Flow cytometry analysis of Hepa1-6 apoptosis after treatment with various formulations
Combination of Cobalt ions and DiABZI enhances ICD and activates the cGAS-STING pathway in HCC cells
The potential formation of a coordination complex between cobalt ions and diABZI at multiple coordination sites may significantly enhance the photothermal effect of J-dICG, which further potentiates its anti-tumor effects [28]. These findings emphasize the importance of understanding the synergistic anti-tumor mechanisms of cobalt ions and diABZI in augmenting the therapeutic potential of J-dICG. Confocal laser scanning microscopy (CLSM) images shown in Fig. 3A and Fig. S2A illustrated the active cellular uptake and intracellular trafficking of Co + diABZI@J-dICG NPs, with notable nanoparticle accumulation observed within 2 h. A decrease in lysosomal co-localization, indicated by Pearson’s correlation coefficients (Rpearson), suggests lysosomal escape. To evaluate the potential synergy of this drug combination, we conducted synergy studies using the Chou-Talalay method [29, 30]. The IC50 values, which represent the concentration required to inhibit 50% of cell growth, were found to be 129.7 µg/mL for diABZI in PLC/PRF/5 cell, and 127.3 µg/mL for cobalt ions in the same cell line (Fig. S1A). Additionally, we generated novel dose-response curves by incorporating a set quantity of diABZI along with diverse concentrations of cobalt ions (Fig. S1B). The Combination Index (CI) values, determined by these curves, were used to evaluate the drug interaction. The findings indicated that the combination of diABZI and cobalt ions produced synergistic effects in PLC/PRF/5 cell, as shown by CI values and isobolograms (Fig. S1C, D). Further assays in Fig. 3B-D and Fig. S2B-D revealed that cobalt ions and diABZI inhibited cell migration, proliferation, and wound closure in Hepa1-6 and PLC/PRF/5 cells, demonstrating the nanoparticles’ ability to suppress key processes associated with cancer progression. Flow cytometry results showed a significant increase in Annexin V-FITC+ & PI + cells in the Co + diABZI group, indicative of enhanced apoptosis (Fig. 3E, F). The apoptosis rate of cells treated with a combination of cobalt ions and diABZI was higher than the linear addition of apoptosis in cells co-incubated with cobalt ions or diABZI alone [31, 32]. Additionally, intracellular reactive oxygen species (ROS) levels were significantly elevated in the Co + diABZI group (Fig. 3G and Fig. S2E). This increase is attributed to redox reactions triggered by cobalt ions and the activation of NADPH oxidase induced by diABZI, indicating a synergistic pro-oxidative effect [33, 34]. Live/dead staining in Fig. 3H and Fig. S2F further corroborated the cytotoxic effects of cobalt ions and diABZI, with an increased proportion of dead cells in the Co + diABZI group. The release of DAMPs confirmed the induction of ICD, evidenced by increased HMGB1 release, calreticulin (CRT) expression, and ATP secretion in the Co + diABZI group (Fig. 3I-M and Fig. S2G, H). The proportion of CRT-positive cells was greater when treated with a combination of cobalt ions and diABZI than the linear addition of ICD in cells co-incubated with cobalt ions or diABZI alone. Western blot analysis demonstrated the activation of the cGAS-STING pathway (Fig. 3N, O and Fig. S2I, J), with increased phosphorylation of TBK1, IRF3, STING, and NF-κB in the Co + diABZI group, indicative of enhanced immune signaling. Furthermore, as shown in Fig. 3P and Fig. S2K, the upregulation of pro-apoptotic proteins (Bax, Cleaved Caspase-9, Cytochrome C) and the downregulation of anti-apoptotic proteins (Bcl-2, Survivin) confirmed the induction of apoptosis by cobalt ions and diABZI.
Fig. 3.

Cobalt ions and diABZI synergistically promote ICD and activate the cGAS-STING pathway in HCC cells, inhibiting growth and invasion, as well as inducing apoptosis. (A) CLSM images demonstrating the internalization and lysosomal release of Co + diABZI@J-dICG NPs in Hepa1-6 cells over various time intervals (green: Co + diABZI@J-dICG, red: Lysotracker Red), scale bar 50 μm. (B) Invasion analysis in Hepa1-6 cells following various drug treatments, scale bar: 100 μm. (C) Clone formation assay demonstrating the synergistic inhibition of Hepa1-6 cell growth by cobalt ions and diABZI. (D) Wound healing assay evaluating the migratory response of PLC/PRF/5 cells to various drug treatments, scale bar: 400 μm. (E) Flow cytometry analysis of apoptosis in PLC/PRF/5 and Hepa1-6 cells following various drug treatments. (F) Quantification of early and late apoptotic events in PLC/PRF/5 and Hepa1-6 cells following various drug treatments. (G) Visualization of ROS induction in Hepa1-6 cells after various drug treatments (24-hour), scale bar: 50 μm. (H) Live/dead staining of Hepa1-6 cells after various drug treatments (24-hour), scale bar: 100 μm. (I) Detection of CRT translocation to the cell surface in Hepa1-6 cells after various drug treatments (24-hour, green: CRT, blue: DAPI), scale bar: 50 μm, and (K) the quantification of CRT-positive cells. (J) Co-localization analysis of HMGB1 with the nucleus in Hepa1-6 cells after various drug treatments (24-hour) (green: HMGB1, blue: DAPI), scale bar: 100 μm, and (L) the quantification of HMGB1-positive cells. (M) Measurement of ATP release in Hepa1-6 cells following various drug treatments (24-hour). (O) Examination of cGAS-STING pathway activation in Hepa1-6 cells post 48-hour drugs treatments (TBK1, p-TBK1, IRF3, p-IRF3, STING, p-STING and NF-κB) and (N) the quantification of protein expression levels. (P) Apoptosis-associated proteins (Bax, Cleaved Caspase-9, Cytochrome C, Bcl-2, and Survivin) in Hepa1-6 cells after different drug treatments (48-hour)
Activation of BMDCs maturation and antigen-presenting capacity by Co + diABZI@J-dICG NPs enhances immune response
The combination of cobalt ions and diABZI in Co + diABZI@J-dICG nanoparticles enhances the maturation of bone marrow-derived dendritic cells (BMDCs) and their antigen-presenting function, thereby activating splenic lymphocytes. The activation of the cGAS-STING pathway, initiated by the recognition of DNA double-strand breaks (DSBs) by cGAS, was evident from γ-H2AX immunofluorescence (Fig. 4A and Fig. S2L), highlighting DNA damage-induced by treatment with cobalt ions and diABZI [35]. Additionally, mitochondrial membrane potential was disrupted, as shown in Fig. 4B and Fig. S2M, where JC-1 staining transitioned from red aggregates to green monomers, indicating increased mitochondrial depolarization caused by the drug combination. The release of dsDNA into the cytoplasm, a consequence of cellular and mitochondrial damage, was visualized in Fig. 4C and Fig. S2N. TOMM20 staining delineated the mitochondrial membrane, while Picogreen highlighted the dsDNA released from mitochondria into the cytoplasm following treatment with the drug combination, emphasizing the cellular stress induced by the combination therapy.
Fig. 4.

Stimulation of BMDCs maturation and antigen-presenting capabilities by drugs, leading to the subsequent activation of splenic lymphocytes. (A) Evaluation of nuclear damage in Hepa1-6 cells after various drug treatments (48-hour), as indicated by γ-H2AX staining (blue: nuclei, green: γ-H2AX), scale bar: 200 μm. (B) Assessment of mitochondrial membrane potential decline in Hepa1-6 cells after various drug treatments (48-hour, blue: nuclei, green: JC-1 monomer, red: JC-1 polymer), scale bar: 50 μm. (C) Assessment of mitochondrial-to-cytoplasmic dsDNA release in Hepa1-6 cells following various drug treatments (48-hour, blue: nuclei, red: TOMM20, green: dsDNA probe), scale bar: 20 μm. (D) BMDCs induction is characterized by CD11c and MHC-II expression after stimulation with GM-CSF and IL-4 (1, 3, 5, and 7 days). (E) CD80 and CD86 expression levels on BMDCs following various drug treatments. (F) MHC II and CD40 expression levels on BMDCs after different drug treatments. (G) Schematic overview of the interaction between splenic lymphocytes and supernatants from HCC cells treated with different drugs (The intensity of NIR: 1 W/cm²). (H) CD11c+ & MHC II + cells in splenic lymphocytes following incubation with various culture supernatants, with (L) MFI quantification for MHC II expression. (I) CD3+ & CD4 + T cell populations following various supernatants incubation, and (M) MFI assessment for CD4 expression. (J) CD45+ & SIINFEKL/H-2Kb + cells following incubation of splenic lymphocytes with supernatants, and (N) MFI quantification for SIINFEKL/H-2Kb expression. (K) CD3+ & CD8 + cells after incubating splenic lymphocytes with various supernatants, and (O) MFI quantification for CD8 expression
The maturation and functional differentiation of DCs are critical for metalloimmunotherapy [36]. BMDCs were successfully induced, with over 70% of cells expressing CD11c and MHC II by day 7 (Fig. 4D). Treatment with cobalt ions and diABZI led to a significant increase in the proportion of mature DCs, as indicated by the upregulation of CD11c+ & CD80+ & CD86 + cells, with the Co + diABZI group showing a more than 30% enhancement in comparison to the control group (Fig. 4E). Additionally, the Co + diABZI group exhibited approximately 40% increase in CD11c+ & CD40+ & MHC II + cells, suggesting a substantial elevation in antigen-presenting function (Fig. 4F). To explore the indirect effects of Co + diABZI@J-dICG nanoparticles on immune cell function, supernatants from treated tumor cells were used to culture splenic lymphocytes (Fig. 4G). Flow cytometry analysis revealed a 15% increase in CD11c+ & MHC II + cells in splenic lymphocytes from the Co + diABZI@J-dICG + NIR group compared to the control group (Fig. 3H). As a result, the proportion of CD3+ & CD4 + cells increased by approximately 10% (Fig. 4I) indicating a robust activation of CD4 + T cells that play a regulatory role in the immune response. This activation also led to an increase in “licensed” immune cells [37], such as CD11b+ & CD86 + M1 macrophages (Fig. S3A, B), as evidenced by the significant rise in CD45+ & SNIINFEKL-H-2Kb + cells to 30% (Fig. 4J). The overall activation of the immune microenvironment was reflected in the increased numbers and activation of CD3+ & CD8 + cells (Fig. 4K) and CD45+ & NK1.1 + cells (Fig. S3C, D). The MFI of MHC II, CD4, SNIINFEKL, and CD8 (Fig. 4L-O) further confirmed the enhanced immune response induced by the Co + diABZI@J-dICG NPs’ PTT.
Evaluation of anti-tumor activity of drug-loaded J-dICG NPs in HCC models
In our study, we established an in-situ HCC model in mice using a Hepa1-6-luc cell line. Figure 5 A outlines the experimental design for the in-situ liver tumor model, which included the administration of drug-loaded J-dICG nanoparticles combined with NIR irradiation. The mice were randomly divided into four treatment groups: Control, J-dICG + NIR, diABZI@J-dICG + NIR, and Co + diABZI@J-dICG + NIR (n = 6). As shown in Fig. 5B, in vivo bioluminescence imaging performed on days 5, 10, 15, and 20 post-treatment revealed a significant reduction in bioluminescence signals, an indicator of tumor burden, in the Co + diABZI@J-dICG + NIR group compared to the control group, particularly on day 20 (Fig. S4A). This reduction was attributed to the excellent photothermal conversion efficiency of the Co + diABZI@J-dICG NPs under NIR irradiation, which transformed absorbed light energy into heat, triggering PTT and inducing tumor cell death. Figure 5 C quantified the average bioluminescence intensity of each group at different time points, demonstrating a time-dependent decrease in signal intensity in the treated groups. By day 20, statistical analysis revealed the Co + diABZI@J-dICG + NIR group had the lowest bioluminescence intensity (Fig. 5D), indicating the most effective suppression of tumor growth. No significant weight loss was observed in any treatment group compared to the control group (Fig. 5E, F), suggesting minimal systemic toxicity. Hematoxylin and eosin (H&E) staining of organs, including the heart, lungs, spleen, kidneys, and liver, from both treated and control groups, indicated good biocompatibility and low toxicity of the nanoparticles (Fig. S5A). Additionally, serum biomarker levels, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CREA), urea (UREA), creatine kinase (CK), and lactate dehydrogenase (LDH), in the different groups (Fig. S5B), further supported the biosafety of the drug-loaded Co + diABZI@J-dICG nanoparticles. Macroscopic liver examination on day 20 revealed distinct tumor nodules in the control group (Fig. 5G), whereas tumors were less conspicuous in the treated groups, especially in the Co + diABZI@J-dICG + NIR group, where the smallest tumor lesions were observed.
Fig. 5.

Assessment of anti-tumor activity of drug-loaded J-dICG nanoparticles in HCC models. (A) Schematic overview of the experimental protocol, depicting HCC model establishment, treatment timing and methodology, and tissue collection points (The intensity of NIR: 2 W/cm²). (B) Live imaging of mice at multiple time points post-administration of various drug-loaded J-dICG nanoparticles and NIR irradiation demonstrating in vivo effects. (C) Mean bioluminescence intensity of tumors in various groups at multiple time points (n = 6). (D) Comparison of average bioluminescence intensity of tumors in different groups on day 20 (n = 6). (E) Chart of mice body weight changes during the treatment period across various groups (n = 6). (F) Comparison of mice body weight changes among various groups on day 20 (n = 6). (G) Photographs of liver specimens from mice untreated and treated with various drug-loaded J-dICG nanoparticles and NIR irradiation (red dashed area: in-situ tumor, n = 3). (H) Live and NIR imaging of the front aspects of liver specimens from various groups of mice (n = 3). (I) Flow cytometric analysis of DCs maturation (CD11c+ & CD86+ & MHC II+) in tumor and spleen samples. (J) CD11b+ & CD86 + and CD11b+ & CD206 + cells in tumors and spleens, with determination of the M1/M2 macrophage phenotype ratio. (K) Analysis of CD3+ & CD8 + cell populations in tumors and spleens by flow cytometry. (L) NK1.1+ & CD107a + cells within tumor and spleen samples. (M) CD3+ & CD8 + cells infiltration in tumor tissues (green: CD8, red: CD3), scale bar: 100 μm. (N) CD11c+ & CD8a + cells infiltration in tumor tissues (green: CD8a, red: CD11c), scale bar: 100 μm. (O) CD4+ & FOXP3 + cells infiltration within tumor tissues (green: FOXP3, red: CD4), scale bar: 100 μm. (P) cGAS-STING pathway activation in tumor tissues following treatment with various drug-loaded J-dICG nanoparticles and NIR irradiation, profiling TBK1, p-TBK1, IRF3, p-IRF3, STING, p-STING, and NF-κB expression
In vivo imaging of the excised livers confirmed the presence of ICG fluorescence (Fig. 5H and Fig. S4B), indicating the accumulation of J-dICG nanoparticles in the liver. The highest fluorescence was observed in the Co + diABZI@J-dICG + NIR group, which likely resulted from the enhanced permeability and retention (EPR) effect, a characteristic of nanomedicine-based PTT. Flow cytometry analysis revealed increased proportions of mature DCs (CD11c+ & MHC II+ & CD86+), M1 macrophages (CD11b+ & CD86+), CD8 + T cells (CD3+ & CD8+), functional NK cells (NK1.1+ & CD107a+), and a reduction in Tregs (CD25+ & FOXP3+) and M2 macrophages (CD11b+ & CD206+) in the tumor tissues and spleen tissues of the treated groups compared to the control group, suggesting an enhanced immune response (Fig. 5I-L and Fig. S4C-H). Immunofluorescence analysis further confirmed increased infiltration of mature DCs (CD11c+ & CD8a+), CD8 + T cells (CD3+ & CD8+), and a reduction in Tregs (CD4+ & FOXP3+) in the tumor tissues of the treated groups, supporting the activation of immune responses (Fig. 5M-O and Fig. S4I-K). Western blotting analysis revealed upregulation of key proteins involved in the cGAS-STING pathway (p-TBK1, TBK1, p-IRF3, IRF3, p-STING, STING, NF-κB) in the treated groups, with the Co + diABZI@J-dICG + NIR group showing the highest expression levels, indicative of robust immune signaling activation (Fig. 5P and Fig. S6A). Histological examination, including H&E staining, Ki67, TUNEL, CRT, and HMGB1 immunofluorescence staining, demonstrated significant tumor cell necrosis, reduced proliferation, enhanced apoptosis, and increased ICD markers in the treated groups, with the Co + diABZI@J-dICG + NIR group exhibiting the most pronounced effects (Fig. S6B-G). Western blot analysis of apoptosis-related proteins revealed increase expression of pro-apoptotic markers (Bax, Cleaved PARP, Cytochrome C, Cleaved Caspase9, Cleaved Caspase3) and decreased expression of anti-apoptotic markers (Bcl, Survivin) in the treated groups, suggesting effective induction of apoptosis in tumor cells (Fig. S6H, I). Overall, these results demonstrated the therapeutic efficacy of Co + diABZI@J-dICG NPs in suppressing tumor growth through the activation of the cGAS-STING pathway and the induction of ICD, resulting in a robust anti-tumor immune response in an in-situ HCC model.
Genomic expression profiling reveals the immune-modulating impact of Co + diABZI@J-dICG NPs and their mechanisms in tumor suppression
To explore the immunomodulatory effects of Co + diABZI@J-dICG + NIR and understand the underlying mechanisms behind the suppression of tumor growth, we conducted RNA sequencing analysis on tumor tissues collected from both the control and Co + diABZI@J-dICG + NIR groups. Principal component analysis (PCA) of the transcriptomic profiles (Fig. 6A), highlighted the distinct impact of Co + diABZI@J-dICG + NIR therapy. Figure 6B displays volcano plots, revealing significant differential gene expression between the Control and Co + diABZI@J-dICG + NIR groups. Gene set enrichment analysis (GSEA) confirmed that Co + diABZI@J-dICG + NIR treatment, which combines metalloimmunotherapy with PTT, enhanced T cell proliferation, activation, and migration (Fig. 5C). The heatmap further emphasized the upregulation of genes associated with apoptosis, inflammation, immunity, and PTT (Fig. 6D and Fig. S7A), suggesting an impact on the cGAS-STING signaling pathway, indicative of the synergistic effects of metalloimmunotherapy and PTT. Additionally, gene ontology (GO) enrichment analysis revealed that Co + diABZI@J-dICG + NIR therapy upregulated immune cell functions such as proliferation, activation, and effector roles (Fig. 6E), while also promoting tumor cell apoptosis (as annotated by the blue solid-line box). Notably, the Co + diABZI@J-dICG + NIR treatment group exhibited a marked enrichment of regulatory pathways associated with IL-6 (Fig. 6E, as highlighted by the red solid-line box). Further analysis revealed a positive association between IL-6 expression and PGDH level within the HCC cohort (Fig. 6F), where PGDH is involved in the serine biosynthesis pathway and correlates with tumor cell growth and metastasis [38]. We subsequently explored the effect of Co + diABZI@J-dICG on PGDH expression and its role in HCC treatment. Western blotting of various HCC cell lines revealed differing PGDH expression levels (Fig. S7B), leading to the selection of PLC/PRF/5 (high PGDH expression) and HepG2 (low PGDH expression) for further study. To investigate the role of PGDH in HCC cell proliferation, PGDH expression was manipulated in these cells: PGDH was knocked down in PLC/PRF/5 cells using lentivirus (sh-PGDH) and overexpressed in HepG2 cells (PGDH-OE) through viral transduction (Fig. 6G, Fig. S7C, E). Clonal expansion assays revealed that PGDH knockdown in PLC/PRF/5 cells enhanced their proliferation, while PGDH overexpression in HepG2 cells suppressed growth (Fig. 6H). EDU staining further confirmed these findings (Fig. 6I), showing an increased EDU + cell proportion in sh-PGDH PLC/PRF/5 cells and a decreased EDU + cell proportion in PGDH-OE HepG2 cells. Cell viability assays corroborated the results observed in clonal expansion and EDU assays (Fig. S7D, F).
Fig. 6.

Transcriptome analysis reveals the immunomodulatory effects of Co + diABZI@J-dICG NPs and the underlying mechanisms by which Co + diABZI@J-dICG inhibits tumor growth. (A) Principal component analysis revealing sample clustering according to treatment conditions (n = 3 independent samples). (B) Comparative analysis of differentially expressed genes between Co + diABZI@J-dICG + NIR and Control groups, demonstrating notable transcriptional shifts (n = 3 independent samples). (C) GSEA indicates the enrichment of lymphocyte immune effector functions in the Co + diABZI@J-dICG group relative to the Control group. (D) Gene expression profiles of key inflammatory and apoptotic pathway genes, revealing the molecular basis of therapeutic outcomes (n = 3 independent samples). (E) GO enrichment of differentially expressed genes involved in immune cell functions (marked by the blue solid-line box) and signaling pathways, with alterations induced by Co + diABZI@J-dICG therapy relative to the Control group, and the IL-6-associated pathways (highlighted by the red solid-line box, n = 3 independent samples). (F) A significant positive correlation between the expression of IL-6 and the expression of PGDH in the liver hepatocellular carcinoma cohort. (G) Western blotting analysis reveals the knockdown of PGDH in PLC/PRF/5 cells and the overexpression of PGDH in HepaG2 cells through viral transduction. (H) Monoclonal assays demonstrating enhanced proliferation of PGDH-knockdown PLC/PRF/5 cells and attenuated proliferation of PGDH-overexpressing HepG2 cells, with quantitative analysis. (I) EDU staining demonstrating increased proliferation in PGDH-knockdown PLC/PRF/5 cells and decreased proliferation in PGDH-overexpressing HepaG2 cells, scale bar: 100. (J) Live imaging of nude mice bearing subcutaneous tumors derived from sh-PGDH-PLC/PRF/5 cells via viral transduction, captured at various time points across different experimental groups (n = 3, The intensity of NIR: 2 W/cm²). (K) Live imaging of nude mice with PGDH-OE-HepG2 cell-derived subcutaneous tumors following viral transduction, with multiple time point assessments within each experimental group (n = 3, The intensity of NIR: 2 W/cm²). (L) Average tumor bioluminescence intensity across different groups at various time points and the analysis of mean tumor bioluminescence intensity among various groups on day 28 (Tumors derived from PLC/PRF/5 and sh-PGDH-PLC/PRF/5 cells, n = 3). (M) Average tumor bioluminescence intensity across different groups at various time points and the analysis of mean tumor bioluminescence intensity among various groups on day 28 (Tumors derived from HepaG2 and PGDH-OE-HepaG2 cells, n = 3). (N) Photographs of subcutaneous tumor specimens from mice across different treatment groups (Tumors derived from PLC/PRF/5 and sh-PGDH-PLC/PRF/5 cells, n = 3). (O) Photographs of subcutaneous tumor specimens from mice across different treatment groups (Tumors derived from HepaG2 and PGDH-OE-HepaG2 cells, n = 3). (P) Western blotting analysis of PGDH protein expression levels in subcutaneous tumor tissues from mice across various treatment groups (Tumors derived from PLC/PRF/5 and sh-PGDH-PLC/PRF/5 cells, n = 3). (Q) Western blotting analysis of PGDH protein expression levels in subcutaneous tumor tissues from mice across various treatment groups (Tumors derived from HepaG2 and PGDH-OE-HepaG2 cells, n = 3)
To evaluate the effect of Co + diABZI@J-dICG on PGDH and its role in HCC growth, PLC/PRF/5 and sh-PGDH PLC/PRF/5 cells were subcutaneously implanted in nude mice (control and sh-PGDH groups, respectively), with Co + diABZI@J-dICG + NIR treatment applied to the sh-PGDH group (sh-PGDH + Co + diABZI@J-dICG + NIR). By day 28, tumors in the sh-PGDH group were significantly larger than in the control group, highlighting PGDH’s suppressive effect on tumor growth. Conversely, the sh-PGDH + Co + diABZI@J-dICG + NIR group showed significantly reduced tumor volume compared to the sh-PGDH group, suggesting that Co + diABZI@J-dICG + NIR induced PGDH expression, counteracting the effects of PGDH knockdown. In vivo imaging and tumor pictures also showed that the tumor volume in the sh-PGDH + Co + diABZI@J-dICG + NIR + Tocilizumab (IL-6 receptor monoclonal antibody) group was larger than in the sh-PGDH + Co + diABZI@J-dICG + NIR group but smaller than in the sh-PGDH group. This suggested that Co + diABZI@J-dICG + NIR-mediated tumor reduction might be through activation of the IL-6-PGDH pathway (Fig. 6J, L, N, P, and Fig. S7G, I). To further confirm the role of IL-6-PGDH in tumor growth, HepG2, and PGDH-OE HepG2 cells were implanted subcutaneously in nude mice (control and PGDH-OE groups, respectively), with Tocilizumab applied to the PGDH-OE group (PGDH-OE + Tocilizumab). By day 28, tumors in the PGDH-OE group were significantly smaller than in the Control group, confirming PGDH’s tumor-suppressive effect. In contrast, the PGDH-OE + Tocilizumab group showed significantly larger tumors than the PGDH-OE group, indicating that Tocilizumab inhibition of IL-6 downregulated PGDH expression, thus reversing its tumor-suppressive effect. Moreover, tumor volume in the PGDH-OE + Tocilizumab + Co + diABZI@J-dICG + NIR group was reduced compared to the PGDH-OE + Tocilizumab group but still larger than in the PGDH-OE group, reinforcing the idea that Co + diABZI@J-dICG + NIR inhibits tumor growth by upregulating the IL-6-PGDH pathway (Fig. 5K, M, O, Q, and Fig. S7H, J). As shown in Fig. S8A-C, the levels of PGE2 in liver cancer tissues were negatively correlated with PGDH levels. In combination with the aforementioned experimental results, it is suggested that Co + diABZI@J-dICG NPs may inhibit the growth of liver cancer by targeting the IL-6-PGDH-PGE2 signaling axis [39]. Overall, our findings demonstrate that Co + diABZI@J-dICG + NIR treatment effectively upregulates IL-6 expression, promoting PGDH expression and subsequently inhibiting HCC growth.
The combination of Co + diABZI@J-dICG NPs with NIR irradiation effectively prevents HCC recurrence post-resection
Building on our previous findings in Fig. 6, the administration of Co + diABZI@J-dICG NPs, in combination with NIR irradiation, led to the upregulation of IL-6 and subsequent increase in PGDH expression. HCC is known for its high recurrence rate following surgical resection, with a reduced level of PGDH being linked to this recurrence pattern [39]. We further investigated whether Co + diABZI@J-dICG NPs could inhibit tumor recurrence in a mouse model following hepatectomy. Figure 7 A presents the schematic workflow, involving in-situ liver tumor-bearing mice models that underwent hepatic resection followed by treatment with drug-loaded J-dICG NPs + NIR. Figure 7B depicted the in vivo imaging and subsequent ex vivo organ imaging in mice injected with Co + diABZI@J-dICG NPs or free ICG. Both Co + diABZI@J-dICG and free ICG accumulated in the liver. However, free ICG was rapidly metabolized, showing a significant reduction by hour 1 and being nearly absent by hour 4. In contrast, Co + diABZI@J-dICG NPs exhibited a slower metabolic rate, with substantial accumulation persisting through day 8 and a notable presence even by day 16. The changes in the fluorescence intensity of ICG in mouse serum also indicated that the metabolic rate of Co + diABZI@J-dICG nanoparticles was significantly slower than that of free ICG [40] (Fig. S9A). This prolonged drug retention in the liver suggested a potential for sustained therapeutic effects [41]. To explore the post-hepatectomy therapeutic potential, we injected Co + diABZI@J-dICG NPs into an in-situ HCC mouse model and performed tumor resection on day 6, followed by NIR irradiation. Figure 7 C demonstrates a significant elevation in the temperature at the liver resection site after NIR irradiation, confirming the PTT effect of Co + diABZI@J-dICG NPs in the liver. The ability for sustained liver retention and PTT effects positions Co + diABZI@J-dICG as a promising candidate for preventing tumor recurrence post-hepatectomy. We used an incomplete resection model of in-situ HCC in mice (n = 8) and randomly assigned them to four groups: Control, J-dICG + NIR, diABZI@J-dICG + NIR, and Co + diABZI@J-dICG + NIR. Different drug-loaded NPs were administered two days before tumor resection, and NIR irradiation was applied on the day of resection and days 3, 6, 9, and 12 post-resection. Tumor recurrence was monitored using in vivo bioluminescence imaging (Fig. 7D and Fig. S7A). Furthermore, in vivo imaging revealed that while tumors in the J-dICG + NIR group recurred, their growth was slower than that of the control group, confirming the photothermal effect of J-dICG. The diABZI@J-dICG + NIR group exhibited a 50% recurrence rate, with slower tumor growth compared to the J-dICG + NIR group, highlighting the immunoactivating effect of diABZI. The Co + diABZI@J-dICG + NIR group showed the best outcomes, with 75% of mice exhibiting no recurrence, and those that did showed significantly slower tumor growth (Fig. 7D-F and Fig. S10A). This demonstrates the synergistic effect of cobalt ions in enhancing the immunoactivation of diABZI. Additionally, the body weight of mice in the Co + diABZI@J-dICG + NIR group remained consistently higher than in the other groups, indicating not only significant inhibition of tumor recurrence but also the absence of overt systemic toxicity (Fig. 7G). The superior performance of Co + diABZI@J-dICG NPs combined with NIR irradiation in preventing HCC recurrence can be attributed to their prolonged liver retention, enhanced PTT effects, and the metalloimmunotherapy of cobalt ions and diABZI.
Fig. 7.

Co + diABZI@J-dICG NPs in combination with NIR irradiation can effectively inhibit the recurrence of hepatocellular carcinoma after resection. (A) Schematic illustrating the treatment modality combining Co + diABZI@J-dICG NPs with NIR irradiation in an incomplete resection model of in-situ HCC in mice (The intensity of NIR: 2 W/cm²). (B) NIR fluorescence imaging of Co + diABZI@J-dICG NPs and free ICG in mice at different time points following intravenous tail injection. (C) Photographs of murine in-situ hepatocellular carcinoma sample and incompletely resected HCC specimen, and thermal imaging of the resection surface after NIR irradiation. (D) In vivo bioluminescence imaging of tumor recurrence post-incomplete tumor resection (n = 8). (E) Recurrence and growth dynamics of HCC after incomplete resection in mice across different treatment groups (n = 8). (F) Mean bioluminescence intensity of recurred tumors at various time points in different treatment groups (n = 8). (G) Body weight changes in mice subjected to various treatment approaches (n = 8)
Discussion
This study highlights the therapeutic potential of Co + diABZI@J-dICG NPs in HCC treatment, demonstrating the synergistic effects of PTT and metalloimmunotherapy. The PTT benefits of J-dICG are notable, as its strong absorption in the NIR region allows for the efficient conversion of light into heat. This localized heat generation elevates the temperature within the tumor microenvironment while enhancing drug release from the nanoparticles. Furthermore, the induced heat activates immune responses and increases the permeability of tumor cells to the therapeutic agents, which facilitates enhanced cellular uptake and lysosomal escape. The drug-loading capacity of the nanoparticles further bolsters their therapeutic potential by enabling the co-delivery of multiple agents, including cobalt ions and diABZI, which work synergistically to enhance the anti-tumor effects. Co + diABZI@J-dICG NPs also capitalize on the inherent hepatotropism of ICG [42], and when combined with the enhanced EPR effect [43, 44], ensure targeted accumulation within the tumor tissue.
Metalloimmunotherapy plays a pivotal role in the activation of the immune system. Cobalt ions can lead to DNA double-strand breaks in tumor cells through multiple mechanisms, including the induction of reactive oxygen species (ROS), oxidative stress responses, and the inhibition of DNA repair. The incorporation of cobalt ions within the nanoparticles activates the cGAS-STING pathway, which amplifies IFN production, thereby enhancing the maturation and migration of DCs. This activation in turn triggers tumor antigen-specific T cells, and NK cells, which work together to mount a robust immune response against the tumor. In addition to direct activation, Co + diABZI@J-dICG NPs also indirectly activate various lymphocytes in the spleen, further modulating the systemic immune response [45].
Our study also reveals that Co + diABZI@J-dICG NPs induce the expression of interleukin-6 (IL-6) in HCC cells, which in turn upregulates PGDH. PGDH plays a crucial role in the degradation of PGE2, a molecule known to promote tumor growth and immune evasion. By boosting PGDH expression, the drug-loaded nanoparticles effectively disrupt the pro-tumorigenic effects of PGE2 [46], potentially inhibiting HCC growth and recurrence after surgery. The extended retention of the nanoparticles in the liver, exceeding 10 days, provides a significant advantage over free ICG, allowing for effective monitoring of tumor recurrence and enabling early therapeutic intervention post-hepatectomy.
However, despite the promising results, there are limitations to this study. Firstly, the research is currently confined to specific cancer models, and additional studies are needed to evaluate the nanoparticles’ effectiveness across a broader range of HCC subtypes [47]. Furthermore, optimizing the structural modification of the J-dICG NPs is essential to improve drug loading and release profiles, which are critical for maintaining sustained therapeutic effects over time. Exploring the potential synergy between Co + diABZI@J-dICG NPs and other immunotherapeutic agents, such as checkpoint inhibitors, may also provide opportunities to amplify the anti-tumor immune response. In addition, further exploration of the synergistic effects of PTT with other therapeutic approaches is needed to enhance the anticancer efficacy. The focus of this study is to utilize the excellent photothermal effect and liver targeting of J-dICG, and the photodynamic effect of ICG under NIR irradiation provides new possibilities for its application in tumor treatment. In the future, we will further explore the application of ICG in multimodal imaging and combined therapy [48]. By focusing on these areas of improvement, we can advance the integration of PTT and metalloimmunotherapy via Co + diABZI@J-dICG NPs, marking a significant step forward in the field of nanomedicine for HCC treatment.
Conclusion
This study highlights the promising therapeutic potential of Co + diABZI@J-dICG nanoparticles in the treatment of HCC by synergistically combining PTT and immunomodulatory approaches. These nanoparticles effectively induce tumor cell death through the photothermal effect of J-dICG, while simultaneously activating the immune response by enhancing the cGAS-STING pathway through the inclusion of cobalt ions and the STING agonist diABZI. The ability of Co + diABZI@J-dICG NPs to induce apoptosis and stimulate anti-tumor immune responses, as demonstrated in both in vitro and in vivo models, offers a novel strategy for targeting HCC. By increasing the expression of prostaglandin dehydrogenase (PGDH), which degrades prostaglandin E2 (PGE2), these nanoparticles help inhibit HCC growth and recurrence. Thus, Co + diABZI@J-dICG NPs represent an innovative advancement in nanomedicine, leveraging the dual therapeutic powers of PTT and metalloimmunotherapy for effective HCC treatment.
Materials and methods
Materials
Indocyanine Green (ICG) and 1, 2, 3-Tri-n-Octanoylglycerol were purchased from MACKLIN. CoCl2•6H2O (C434161) was purchased from Aladdin. diABZI STING agonist-1 (A1250006) was purchased from AmBeed. Human Prostaglandin E2, PGE2 ELISA Kit was purchased from CUSABIO. TUNEL assay (T2195), and anti-Ki67 (K010075P) were purchased from Solarbio. Crystal violet (C805210) was purchased from Macklin. 808 nm point laser was purchased from KERONG. A handheld infrared thermal imager was purchased from Uni-Trend Technology. sh-PGDH lentivirus (sequence: ACTCATAACAACACAGACATA) was provided by GeneChem Co., Ltd., Shanghai, China. The lentivirus which overexpresses PGDH was generated by Genechem Co., Ltd., Shanghai, China. Lyso-tracker Red (C1046), DCFH-DA (S0033M), Calcein/PI Live/Dead Cytotoxicity Kit (C2015M), Goat Anti-Mouse IgG H&L (Sulfo-Cyanine3) (A0521), DNA Damage Assay Kit by γ-H2AX Immunofluorescence (C2035S) and Enhanced mitochondrial membrane potential assay kit with JC-1 (C2003S) were purchased from Beyotime. Anti-Bax, anti-cleaved Casp3, anti-cleaved Casp9, anti-cytochorme C, anti-cleaved PARP, anti-Survivin, and anti-Bcl-2, as well as anti-TBK1, anti-phospho-TBK1 (Ser172), anti-IRF3, anti-phospho-IRF3 (Ser396), anti-STING, anti-phospho-STING (Ser366), and anti-NF-κB p65 were purchased from Cell Signaling Technology (CST). FITC anti-mouse CD11c, APC anti-mouse CD86, APC/Cyanine7 anti-mouse MHC II, PE anti-mouse CD40, FITC anti-mouse CD3, APC anti-mouse CD8, PE anti-mouse CD45, APC anti-mouse NK1.1, PE anti-mouse CD107a, APC anti-mouse SIINFEKL/H-2Kb, PerCP/Cyanine5.5 anti-mouse CD11b, PE anti-mouse CD86, APC anti-mouse CD206, and FITC anti-mouse CD4, APC anti-mouse CD25, PE anti-mouse FOXP3 were purchased from BioLegend. Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 488) (ab150073), Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 647) (ab150075), Goat Anti-Mouse IgG H&L (Alexa Fluor® 568) (ab175473), Goat Anti-Rabbit IgG H&L (HRP) (ab6721), anti-GAPDH (ab8245), anti-TOMM20 (ab186735), were purchased from Abcam. All flow cytometry antibodies were purchased from BioLegend. The other reagents in this study were of analytical purity and used without further purification. All the water employed in the study was ultrapure, with an electrical resistivity not exceeding 18.2 MΩ cm–1, prepared using a laboratory water purification system (Milli-Q Integral 3, Merck Millipore, Germany).
Cell culture
Mouse HCC cell lines Hepa1-6 and Hepa1-6-luc, along with human tumor cell lines PLC/PRF/5, HCCLM3, SK-HEP-1, HUH-7, HepG2, and Hep3B were sourced from the National Center for Cell Culture Identification and Repository (Shanghai, China). These cells were cultured in DMEM (Gibco, USA) with high glucose and pyruvate, supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin-streptomycin (Thermo Fisher Scientific, USA).
Preparation of nanoemulsion precursor
Dissolve 10 mg of CoCl2·6H2O and 5 mg of diABZI (for Co + diABZI@J-dICG) in 3 mL of chloroform. Concurrently, dissolve 40 µL of glyceryl trioctanoate in 3 mL of chloroform. Mix the two solutions and stir for 3 h to ensure uniformity. Afterward, dry the mixture under a gentle stream of nitrogen gas to form a thin lipid film. For the synthesis of Co@J-dICG, only CoCl2·6H2O was used, and for diABZI@J-dICG, only diABZI was incorporated in the initial step.
Hydration and sonication
Hydrate the lipid film with 4 mL of deionized water and subject it to sonication using a sonicator (BioRuptor) at 25 °C for 60 cycles, with each cycle consisting of 30 s of sonication followed by a 30-second rest.
Storage and maturation of nanoemulsion
Store the resulting nanoemulsion at 4 °C to facilitate the dimerization and J-aggregation of ICG at the nanoemulsion interface. Centrifuge the nanoemulsion at 15,000 rpm for 15 min to remove any large aggregates. Wash the nanoemulsion with deionized water (ddH2O) and recentrifuge to ensure the removal of any unwanted byproducts and to achieve a stable dispersion of Co + diABZI@J-dICG NPs.
Characterization of nanoparticle morphology and composition
The morphology and structure of the drug-loaded nanoparticles were characterized using a 300 kV Transmission Electron Microscope (TEM), while Energy Dispersive Spectroscopy (EDS) operating voltage was employed to analyze elemental distribution on prepared Co + diABZI@J-dICG samples mounted on copper grids. Particle size measurements of J-dICG and Co + diABZI@J-dICG were conducted using nanoparticle tracking analysis (NanoSight NS300, Malvern). Additionally, the Zeta potential of nanoparticles (J-dICG, Co@J-dICG, diABZI@J-dICG, Co + diABZI@J-dICG) was determined using a nanoparticle analyzer (Litesizer 500, Anton Paar) to assess their surface charge.
Photothermal effect characterization
To assess the photothermal properties of J-dICG, diABZI@J-dICG, and Co + diABZI@J-dICG nanoparticles, we created solutions of different concentrations for each nanoparticle type using deionized water. The temporal variation of infrared thermal imaging was monitored under 808 nm laser irradiation at various power densities to assess the photothermal response and stability of the nanoparticles. The laser irradiation was applied in a controlled manner, with power densities ranging from 0.5 to 2 W/cm², to simulate different clinical treatment scenarios. The temperature changes of the nanoparticle solutions were recorded using an infrared thermal imaging camera at 30-second intervals during a 6-minute irradiation period. After each irradiation, the nanoparticles were subjected to an “on/off” cycle, where the laser was turned off to mimic the intermittent treatment that might be used in photothermal therapy. This process was repeated multiple times to evaluate the photothermal stability of the nanoparticles under cyclic laser irradiation conditions.
Cellular uptake and lysosomal co-localization
For cellular uptake, Mouse HCC cell line Hepa1-6 and human tumor cell line PLC/PRF/5 were incubated with Co + diABZI@J-dICG for different times, followed by Lysotracker Red staining and imaging using a confocal laser scanning microscope (TCS SP8, Leica). Fiji software was used for fluorescence quantification.
Invasion assay of drug combinations treatment in HCC cells
For the invasion assay of HCC cells, matrigel was diluted in a serum-free medium and applied to the upper chamber of Transwell inserts then polymerized for 3 h at 37 °C. PLC/PRF/5 cells (5 × 105 cells/well) were seeded in the upper chamber and pre-incubated for 6 h. After 24 h of co-culture with different drug combinations, cells from the following groups were fixed and stained with crystal violet to visualize cell nuclei and assess invasion: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (diABZI, 5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM and diABZI, 5 µM). The invaded cells were counted in four random fields of view per well under a microscope, and the invasion was quantified by comparing the number of invaded cells across different treatment groups. Hepa1-6 cells were processed in the same manner.
Wound healing assay for drug combinations in HCC cells
PLC/PRF/5 cells (5 × 105 cells/well) were cultured to confluence in 24-well plates and then serum-starved in DMEM without serum for 24 h to synchronize the cell cycle. The cell monolayer was wounded with a pipette tip, and the wells were gently washed to remove cellular debris. The cells were then treated with different drug combinations: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM and diABZI, 5 µM). Following treatment, cells were incubated with DMEM containing 10% FBS. Images of the wound area were captured at 0 and 24 h post-wounding to track cell migration. The cell migration rate was calculated using the following formula:
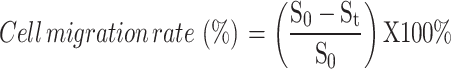
where 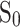 is the initial scratch area, and
is the initial scratch area, and  is the area at 24 h. The assay was repeated with Hepa1-6 cells.
is the area at 24 h. The assay was repeated with Hepa1-6 cells.
Clonogenic assay for drug combinations in HCC cells
For the proliferation assay of HCC cells, the logarithmic growth phase PLC/PRF/5 (2 × 103 cells/well) cells were seeded evenly in 6-well plates. After adherence of the cells, they were treated with the following drug combinations respectively: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (diABZI, 5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM and diABZI, 5 µM). The drug-containing culture medium was replaced every 3 days, and the cells were cultured for about 2 weeks. After the formation of cell clones, cells were fixed with 4% paraformaldehyde for 15–30 min and then washed three times with PBS. After fixation, the cells were stained with crystal violet for 20 min, and then washed three times with PBS before being counted and statistically analyzed under a microscope. The assay was repeated with Hepa1-6 cells.
Apoptosis induction by drug combinations in HCC cells
PLC/PRF/5 cells (5 × 105 cells/well) were cultured in 6-well plates and allowed to adhere for 12 h. The cells were then treated with the following drug combinations: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM + diABZI, 5 µM). Each treatment was applied for 24 h. After treatment, cells were stained with Annexin V-FITC/PI to detect early apoptotic (Annexin V+, PI−) and late apoptotic (Annexin V+, PI+) cells. Apoptosis was quantified using flow cytometry. This protocol was repeated for Hepa1-6 cells to evaluate the apoptotic effects of the drug combinations.
Intracellular ROS assessment post-drug combinations treatment
PLC/PRF/5 cells (2 × 105 cells/well) were seeded in 12-well plates and allowed to adhere overnight. The cells were then treated with the following drug combinations: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM + diABZI, 5 µM). After treatment, cells were incubated with the ROS-sensitive dye DCFH-DA (5 µM) for a specified period to allow for ROS detection. The cells were then imaged using fluorescence microscopy, and the average ROS fluorescence intensity was measured and compared across different treatment groups. This protocol was also applied to Hepa1-6 cells to assess the effect of the drug combinations on intracellular ROS levels.
Live/Dead cell assessment after exposure to drug combinations
PLC/PRF/5 cells (5 × 104 cells/well) were seeded in 48-well plates and allowed to adhere for 24 h. The cells were then exposed to the following drug combinations: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM and diABZI, 5 µM). After the exposure period, live/dead staining was performed using Calcein-AM (5 µM) to label live cells and Propidium Iodide (PI, 5 µM) to label dead cells. The percentage of dead cells was calculated using the formula:
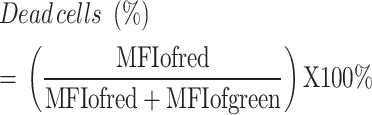
This analysis was also conducted on Hepa1-6 cells.
γ-H2AX foci analysis for nuclear damage induced by drug combinations
PLC/PRF/5 cells (5 × 104 cells/well) were pre-cultured in confocal dishes and then exposed to the following drug treatments: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM and diABZI, 5 µM) for 48 h. γ-H2AX foci, an indication of DNA double-strand breaks, were visualized using immunofluorescence with Alexa Fluor® 488-conjugated secondary antibodies. The fluorescence intensity and number of γ-H2AX foci were quantified to assess the level of nuclear damage in response to the drug treatments. The same procedure was applied to Hepa1-6 cells.
Induction of Immunogenic cell death (ICD) by drug combinations
PLC/PRF/5 cells (2 × 104 cells/well) were cultured in confocal dishes to adherence and treated with Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), or a combination of Cobalt + diABZI (CoCl2·6H2O, 10 µM with diABZI, 5 µM) for 24 h. The induction of ICD was assessed by immunofluorescence staining for the release of Calreticulin (CRT) and High Mobility Group Box 1 Protein (HMGB1), which are indications of ICD, and quantification of extracellular ATP secretion using a luminescent ATP detection assay kit (Beyotime). The same procedure was applied to Hepa1-6 cells.
Assessment of mitochondrial membrane potential disruption in HCC cells
PLC/PRF/5 cells (5 × 104 cells/well) were seeded in confocal Petri dishes and incubated for 12 h. The cells were then treated with Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM + diABZI, 5 µM) for 48 h. The mitochondrial membrane potential was assessed using the JC-1 kit (Beyotime). The ratio of green to red mean fluorescence intensity (MFI) was calculated to determine the extent of mitochondrial membrane potential disruption:
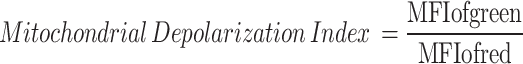
The same procedure was applied to Hepa1-6 cells.
Cytoplasmic double-stranded DNA (dsDNA) release induced by drug treatments in HCC cells
PLC/PRF/5 cells (5 × 104 cells/well) were cultured in confocal petri dishes for 12 h before treatment with Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM + diABZI, 5 µM) for 48 h. To quantify cytoplasmic dsDNA release, cells were incubated with an anti-TOMM20 antibody (1:100, Abcam) to label mitochondria and a dsDNA-specific marker (1:100, Abcam), followed by incubation with Goat Anti-Mouse IgG H&L (Alexa Fluor® 568) for immunofluorescence staining. Nuclei were counterstained with DAPI using an anti-fade mounting medium. Images were acquired using a confocal laser scanning microscope (CLSM). The same procedure was applied to Hepa1-6 cells.
Western blotting analysis of cGAS-STING pathway and apoptosis in HCC cells and tissues
Cell Western blotting
PLC/PRF/5 cells (4 × 105 cells/well) were seeded in 6-well plates and treated with Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM + diABZI, 5µM) for 48 h. Protein extraction and Western blotting were performed as previously described. Primary antibodies targeting the cGAS-STING pathway (anti-GAPDH, anti-TBK1, anti-phospho-TBK1, anti-IRF3, anti-phospho-IRF3, anti-STING, anti-phospho-STING, anti-NF-κB) and apoptosis-related proteins (anti-Bcl-2, anti-Survivin, anti-Bax, anti-Cleaved PARP, anti-Cytochorme C, anti-Cleaved Casp9, anti-Cleaved Casp3) were used with β-tubulin as a loading control. Hepa1-6 cells were processed similarly.
Tissue Western blotting
Tumor tissues from mice with in-situ HCC treated with the above drug regimens were minced and homogenized in ice-cold RIPA lysis buffer with protease and phosphatase inhibitors. The homogenate was centrifuged, and the supernatant was collected for protein concentration determination using the BCA assay kit. Equal amounts of total protein were mixed with Laemmli sample buffer, denatured, and resolved by SDS-PAGE. Proteins were transferred onto nitrocellulose membranes, blocked, and incubated with primary antibodies against cGAS-STING pathway-related proteins (with GAPDH as a loading control) and apoptosis-related proteins (with β-tubulin as a loading control). After incubation with HRP-conjugated secondary antibodies, proteins were visualized using an ECL detection system, and band intensities were quantified using Fiji software to assess the relative expression levels of the proteins of interest.
Induction and characterization of bone marrow-derived dendritic cells (BMDCs)
BMDCs were induced from 6-week-old C57BL/6 mice. Bone marrow was extracted from the femurs and tibiae, flushed with phosphate-buffered saline (PBS), and the cells were suspended in RPMI 1640 medium containing IL-4 (10 ng/mL) and GM-CSF (20 ng/mL). Cultures were maintained with partial medium changes every 48 h. On day 7, mature BMDCs were harvested by gently pipetting the culture medium to collect both suspended and semi-adherent cells. The maturation of BMDCs was confirmed by flow cytometry, evaluating the expression of CD11c and MHC II.
Maturation and activation of BMDCs by drug combinations
Induced BMDCs were seeded in 12-well plates at a density of 2 × 105 cells/well and exposed to the following treatments: Control, Cobalt (CoCl2·6H2O, 10 µM), diABZI (5 µM), and Cobalt + diABZI (CoCl2·6H2O, 10 µM + diABZI, 5 µM) for 24 h. Post-treatment, BMDCs were harvested and stained with FITC-conjugated anti-mouse CD11c, PE-conjugated anti-mouse CD40, APC-conjugated anti-mouse CD86, FITC-conjugated anti-mouse CD80, and APC/Cyanine7-conjugated anti-mouse MHC-II antibodies to assess the expression of maturation markers. After a 30-minute incubation at 4 °C, cells were washed and suspended in PBS, then analyzed using a CytoFLEX flow cytometer. This analysis was conducted to evaluate the functional activation of BMDCs in response to the drug combinations.
Isolation and culture of splenic lymphocytes from C57BL/6 mice
Splenic lymphocytes were obtained from 6-week-old C57BL/6 mice. Under ethical guidelines, the mice were euthanized via cervical dislocation, and the splenic area was prepared by sanitizing with 75% alcohol. A midline incision facilitated spleen exposure and extraction. The spleen was rinsed in RPMI 1640 medium and processed to obtain a single-cell suspension. Lymphocyte isolation was performed using a lymphocyte isolation solution (Dakewe Biotech), followed by centrifugation to collect the mononuclear cell layer. The cells were then washed with PBS to complete the isolation process.
Indirect activation of splenic lymphocytes by supernatants from hepa1-6 cells treated with drug combinations
Hepa1-6 cells were incubated with the following treatments for 24 h: Control, J-dICG + NIR, diABZI@J-dICG + NIR, and Co + diABZI@J-dICG + NIR (CoCl2·6H2O, 10 µM + diABZI, 5µM). The supernatants were collected and used to culture splenic lymphocytes isolated from C57BL/6 mice for an additional 24 h. The lymphocytes were divided into groups corresponding to the incubation conditions: Control-Sup, J-dICG + NIR-Sup, diABZI@J-dICG + NIR-Sup, and Co + diABZI@J-dICG + NIR-Sup. Post-incubation, the lymphocytes were stained with FITC anti-mouse CD11c, APC/Cyanine7 anti-mouse MHC II, APC anti-mouse CD3, FITC anti-mouse CD4, PE anti-mouse CD8, PE anti-mouse CD45, APC/Cyanine7 anti-mouse NK1.1, and APC anti-mouse SIINFEKL/H-2Kb, PerCP/Cyanine5.5 anti-mouse CD11b, PE anti-mouse CD86, and incubated for 30 min at 4 °C. Following staining, cells were washed, resuspended in PBS, and analyzed using a CytoFLEX flow cytometer to evaluate the indirect activation of splenic lymphocytes induced by the supernatants from Hepa1-6 cells treated with the specified drug combinations.
Animal studies
This study aimed to assess the therapeutic efficacy of drug combinations in HCC models. We hypothesized that these treatments would elicit tumor regression and modulate the immune response. The experiments utilized in-situ and subcutaneous HCC mouse models, including a syngeneic model in C57BL/6 mice and a human cell-derived subcutaneous model in nude mice, as well as a model of HCC recurrence post-hepatectomy in C57BL/6 mice. Sample sizes were determined based on prior studies to ensure adequate statistical power to detect significant differences, with each experiment including at least three replicates for reproducibility and reliability.
Animals were randomly allocated to treatment groups to minimize bias, though the study was not blinded. Monitoring of tumor volume and body weight served as primary endpoints, while immune cell analysis and mRNA sequencing were considered secondary endpoints. Data collection ceased upon reaching predefined endpoints, such as tumor size thresholds or the conclusion of the study period. Outliers were identified using statistical criteria and were reported if excluded from the analysis. All animal experiments adhered to the Guidelines for the Care and Use of Laboratory Animals at Sun Yat-sen University and were approved by the Guangzhou GENNIO Biological Laboratory Animal Management and Use Committee (Ethics No: JENNIO-IACUC-2024-A039). The study was conducted in strict compliance with ethical guidelines, and all procedures were reviewed and approved by the relevant institutional animal care committees.
HCC mouse models
In situ HCC model
Hepa1-6-luc cells (5 × 105 per mouse), mixed with Matrigel, were injected into the sub-pericardial region of the left liver lobe. Tumor establishment and growth were monitored non-invasively by live imaging following the intraperitoneal injection of D-Luciferin (MCE). Mice were imaged at regular intervals to track tumor progression (p/s/cm2/sr).
Post-hepatectomy recurrence model
For the model of HCC recurrence post-hepatectomy, in-situ HCC was established by injecting Hepa1-6-luc cells (5 × 105 per mouse) mixed with Matrigel into the sub-pericardial region of the left liver lobe of mice. Tumor establishment was confirmed using bioluminescence imaging. On day 6 post-implantation, when the tumors were well-established, mice underwent a surgical procedure to remove 95% tumor mass. Following surgery, mice were monitored closely for signs of tumor recurrence. Notably, live imaging was performed every 3 days post-hepatectomy to assess tumor recurrence by detecting any re-emergence of luciferase-expressing tumor cells at the resection site or in the liver.
Nude mouse subcutaneous HCC model
Nude mice were used to establish subcutaneous tumor models by injecting 100 µL of cell suspension containing 5 × 106 cells per mouse, derived from sh-PGDH-PLC/PRF/5 and PGDH-OE-HepG2 cell lines, which were generated through viral transduction to modulate PGDH expression. These injections were administered subcutaneously in the dorsal region, and tumor growth was monitored until the tumors reached approximately 50 mm3, as calculated using the formula.
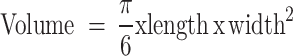
Tumor size was tracked using live imaging.
In vivo distribution and metabolism of drug-loaded nanoparticles
To investigate the in vivo distribution and metabolism of drug-loaded nanoparticles, 8 normal C57BL/6 mice were injected with Co + diABZI@J-dICG or ICG at a concentration of 2 mg/mL (100 µl/mouse) via tail vein, and the drug concentration and metabolism in the liver were monitored in real time using in vivo imaging.
Efficacy of different drug-loaded nanoparticles in treating HCC
To evaluate the therapeutic effects of various drug-loaded nanoparticles on HCC, 24 mice with the in-situ HCC model were randomly assigned to four groups (Control, J-dICG + NIR, diABZI@J-dICG + NIR, Co + diABZI@J-dICG + NIR) and treated with saline or nanoparticles at a concentration of 2 mg/mL (100 µl/mouse) via tail vial tail vein injection followed by NIR irradiation. Tumor size was monitored using in vivo imaging every 5 days, and body weight was recorded every 3 days for a 21-day observation period. At the end of the experiment, blood and tumor tissues were collected for mRNA extraction using the TRIzol reagent. Sequencing libraries were prepared, and 2 × 150 bp double-end sequencing was conducted using Illumina Novaseq™ 6000. Data analysis was performed using R-packages provided by Lc-Bio Technology.
Investigating the anti-tumor mechanism of Co + diABZI@J-dICG + NIR
To explore the anti-tumor mechanism of Co + diABZI@J-dICG + NIR, 24 mice with the subcutaneous HCC model were generated through viral transduction to create sh-PGDH-PLC/PRF/5 and PGDH-OE-HepG2 cell lines. Mice were divided into following groups (Control, sh-PGDH, sh-PGDH + Co + diABZI@J-dICG + NIR, sh-PGDH + Co + diABZI@J-dICG + NIR + Tocilizumab, Control, PGDH-OE, PGDH-OE + Tocilizumab, PGDH-OE + Tocilizumab + Co + diABZI@J-dICG + NIR) and treated with Co + diABZI@J-dICG or Tocilizumab at a concentration of 2 mg/mL (100 µl/mouse) followed by NIR irradiation. Tumor progression was assessed every 7 days for 28 days, and at the study’s conclusion, blood and tumor samples were collected for immunofluorescence and other analyses.
Efficacy of Co + diABZI@J-dICG NPs in preventing recurrence post-hepatectomy
To assess the efficacy of Co + diABZI@J-dICG NPs in preventing HCC recurrence post-hepatectomy, 32 mice in the post-hepatectomy recurrence model were divided into four groups (Control, J-dICG + NIR, diABZI@J-dICG + NIR, Co + diABZI@J-dICG + NIR). These mice were treated with the respective solutions two days before tumor resection at a concentration of 2 mg/mL (100 µl/mouse). After tumor resection, these mice were treated with NIR irradiation on days 0, 3, 6, 9, and 12. Recurrence was monitored every 3 days, and body weight was recorded every 3 days for 12 days. Upon completion, blood and tumor tissues were harvested for further examination.
Flow cytometry analysis of tumor and spleen tissues
Post-treatment, tumor, and spleen tissues were harvested and processed into single-cell suspensions via mechanical dissociation and enzymatic digestion with collagenase and DNase. The cell suspensions were filtered to obtain a homogeneous single-cell population. Phenotypic characterization of immune cells, including DCs, T cells, and NK cells, was performed using a panel of fluorophore-conjugated antibodies: FITC anti-mouse CD11c, APC anti-mouse CD86, APC/Cyanine7 anti-mouse MHC II, FITC anti-mouse CD3, APC anti-mouse CD8, FITC anti-mouse CD4, APC anti-mouse CD25, PE anti-mouse FOXP3, APC anti-mouse NK1.1, and PE anti-mouse CD107a. Macrophage polarization was assessed with PerCP/Cyanine5.5 anti-mouse CD11b, PE anti-mouse CD86, and APC anti-mouse CD206. Samples were analyzed using a CytoFLEX flow cytometer to evaluate immune cell populations and functions post-treatment.
Immunofluorescence staining and infiltration analysis
For both cultured HCC cells and tumor tissue sections, samples were fixed with paraformaldehyde and methanol, permeabilized with Triton X-100, and blocked with BSA before incubation with primary antibodies at 4 °C overnight. Secondary antibodies were applied at 37 °C for 1 h, and nuclei were counterstained with DAPI using an anti-fade mounting medium. Fluorescence was visualized using a microscope. For immune cell infiltration, tumor tissue sections were incubated with primary antibodies specific to immune cell markers (anti-CD11c and anti-CD8a for DCs, anti-CD3 and anti-CD8 for CD8 + T cells, anti-CD4 and anti-FOXP3 for Tregs), followed by treatment with secondary antibodies (Goat Anti-Mouse IgG H&L conjugated with Alexa Fluor® 488 or Sulfo-Cyanine3) and counterstaining with DAPI. Images were captured using fluorescence microscopy and analyzed with Fiji software to quantify immune cell infiltration.
Statistical analysis
The data from this study have been validated through triplicate replication to ensure the consistency of the findings. Results are reported as the mean ± standard deviation (mean ± SD). We employed SPSS 24.0 (IBM) and GraphPad Prism 10 for data analysis and visualization. For pairwise comparisons, a Student’s t-test was utilized, while one-way ANOVA was applied to assess differences across three or more groups. Subsequent pairwise comparisons were made using two-tailed t-tests or one-way ANOVA where necessary. And p < 0.05 was considered statistically significant, suggesting that any observed group differences were unlikely to be attributed to random variation. The abbreviation “ns” was used to indicate statistical non-significance. Additionally, asterisks (*) denote the level of statistical significance as follows: * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, and **** indicates p < 0.0001.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We appreciate Dr. WIN TOPATANA for polishing our manuscript.
Author contributions
Kaiming He, Desheng Chen, Dongzi Zhu, Wenjie Zheng contributed equally to this work. Kaiming He: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Visualization, Writing - Original Draft. Desheng Chen: Methodology, Visualization, Formal analysis. Dongzi Zhu and Wenjie Zheng: Conceptualization, Formal analysis, Investigation, Visualization. Lei Lyu and Mingshen Zhang: Investigation, Software. Zeping Chen: Investigation. Xiaowen Wang: Writing - Review &Editing, Supervision. Yongwei Hu: Resources, Supervision, Funding acquisition, Project administration, Writing - Review & Editing. Binsheng Fu: Writing - Review & Editing, Conceptualization, Resources, Supervision, Project administration, and Funding acquisition.
Funding statement
This work was supported by the following grants: Guangdong Basic and Applied Basic Research Foundation, 2020A1515010302. Postdoctoral Fellowship Program of CPSF under Grant Number GZC20242089.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval
All animal experiments adhered to the Guidelines for the Care and Use of Laboratory Animals at Sun Yat-sen University and were approved by the Guangzhou GENNIO Biological Laboratory Animal Management and Use Committee (Ethics No: JENNIO-IACUC-2024-A039).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kaiming He, Desheng Chen, Dongzi Zhu and Wenjie Zheng contributed equally to this work.
Contributor Information
Xiaowen Wang, Email: wangxw68@mail3.sysu.edu.cn.
Yongwei Hu, Email: huyongwei123456@126.com.
Binsheng Fu, Email: fubinsh@mail.sysu.edu.cn.
References
- 1.Yang X, Yang C, Zhang S, Geng H, Zhu AX, Bernards R, Qin W, Fan J, Wang C, Gao Q. Precision treatment in advanced hepatocellular carcinoma. Cancer Cell. 2024;42:180–97. [DOI] [PubMed] [Google Scholar]
- 2.Sankar K, Gong J, Osipov A, Miles SA, Kosari K, Nissen NN, Hendifar AE, Koltsova EK, Yang JD. Recent advances in the management of hepatocellular carcinoma. Clin Mol Hepatol. 2024;30:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee M, Shin HP. Efficacy of transarterial chemoembolization (TACE) for Early-Stage hepatocellular carcinoma. Med (Kaunas). 2023; 59. [DOI] [PMC free article] [PubMed]
- 4.Zhang Y, Qin Y, Dong P, Ning H, Wang G. Liver resection, radiofrequency ablation, and radiofrequency ablation combined with transcatheter arterial chemoembolization for very-early- and early-stage hepatocellular carcinoma: A systematic review and bayesian network meta-analysis for comparison of efficacy. Front Oncol. 2022;12:991944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li B, Luo Y, Liu G, Gou M, Feng L, Ye X, Xu J, Fan Y, You Z. NIR-II-Absorbing NDI polymer with superior penetration depth for enhanced photothermal therapy efficiency of hepatocellular carcinoma. Int J Nanomed. 2024;19:6577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu GL, Liu F, Li N, Fu Q, Wang CK, Yang S, Xiao H, Tang L, Wang F, Zhou W, et al. Trisulfide Bond-Mediated molecular phototheranostic platform for activatable NIR-II Imaging-Guided enhanced Gas/Chemo-Hypothermal photothermal therapy. Adv Sci (Weinh). 2023;10:e2304104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prince Y, Hiremath N, Vankayala R. Near-infrared light activatable niosomes loaded with indocyanine green and plasmonic gold nanorods for theranostic applications. Biomater Sci. 2023;11:7759–67. [DOI] [PubMed] [Google Scholar]
- 8.Kwon N, Jasinevicius GO, Kassab G, Ding L, Bu J, Martinelli LP, Ferreira VG, Dhaliwal A, Chan HHL, Mo Y, et al. Nanostructure-Driven indocyanine green dimerization generates Ultra-Stable phototheranostics nanoparticles. Angew Chem Int Ed Engl. 2023;62:e202305564. [DOI] [PubMed] [Google Scholar]
- 9.Millard M, Bernhard Y, Canilho N, Grandemange S, Parant S, Mourer M, Lassalle HP, Pasc A. Enhanced stability and photothermal efficiency of indocyanine green J-aggregates by nanoformulation with Calix[4]arene for photothermal therapy of cancers. Colloids Surf B Biointerfaces. 2023;230:113516. [DOI] [PubMed] [Google Scholar]
- 10.Alotaibi H, Hatahet T, Al-Jamal WT. Indocyanine green J-aggregate (IJA) theranostics: challenges and opportunities. Int J Pharm. 2024;661:124456. [DOI] [PubMed] [Google Scholar]
- 11.Gowsalya K, Yasothamani V, Vivek R. Emerging indocyanine green-integrated nanocarriers for multimodal cancer therapy: a review. Nanoscale Adv. 2021;3:3332–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gan Y, Li X, Han S, Liang Q, Ma X, Rong P, Wang W, Li W. The cGAS/STING Pathway: A novel target for cancer therapy. Front Immunol. 2021;12:795401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aishajiang R, Liu Z, Liang Y, Du P, Wei Y, Zhuo X, Liu S, Lei P, Wang T, Yu D. Concurrent amplification of ferroptosis and immune system activation via Nanomedicine-Mediated radiosensitization for Triple-Negative breast cancer therapy. Adv Sci (Weinh) 2024:e2407833. [DOI] [PMC free article] [PubMed]
- 14.Le JQ, Song XH, Tong LW, Lin YQ, Feng KK, Tu YF, Hu YS, Shao JW. Dual-drug controllable co-assembly nanosystem for targeted and synergistic treatment of hepatocellular carcinoma. J Colloid Interface Sci. 2024;656:177–88. [DOI] [PubMed] [Google Scholar]
- 15.Li LS, Chen PW, Zhao XJ, Cheng D, Liu BB, Tang XJ, Zhu WQ, Yang X, Zhao MX. Nuclear-targeted smart nanoplatforms featuring double-shell Hollow mesoporous copper sulfide coated with manganese dioxide synergistically potentiate chemotherapy and immunotherapy in hepatocellular carcinoma cells. J Colloid Interface Sci. 2025;680:202–14. [DOI] [PubMed] [Google Scholar]
- 16.Lei J, Zhang W, Ma L, He Y, Liang H, Zhang X, Li G, Feng X, Tan L, Yang C. Sonodynamic amplification of cGAS-STING activation by cobalt-based nanoagonist against bone and metastatic tumor. Biomaterials. 2023;302:122295. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Tian H, Chen Y, Liang B, Nice EC, Huang C, Xie N, Zheng S. A metal-organic nanoframework for efficient colorectal cancer immunotherapy by the cGAS-STING pathway activation and immune checkpoint Blockade. J Nanobiotechnol. 2024;22:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng SJ, Yang M, Luo JQ, Liu R, Song J, Chen Y, Du JZ. Manganese-Based immunostimulatory Metal-Organic framework activates the cGAS-STING pathway for cancer metalloimmunotherapy. ACS Nano. 2023;17:15905–17. [DOI] [PubMed] [Google Scholar]
- 19.Sun X, Zhang Y, Li J, Park KS, Han K, Zhou X, Xu Y, Nam J, Xu J, Shi X, et al. Amplifying STING activation by Cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy. Nat Nanotechnol. 2021;16:1260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Zhang R, Wei X, Lv M, Jiang Z. Metalloimmunology: the metal ion-controlled immunity. Adv Immunol. 2020;145:187–241. [DOI] [PubMed] [Google Scholar]
- 21.Shang K, Montesdeoca N, Zhang H, Efanova E, Liang G, Ochs J, Karges J, Song H, Zhang L. Cobalt(III) prodrug-based nanomedicine for inducing Immunogenic cell death and enhancing chemo-immunotherapy. J Control Release. 2024;373:493–506. [DOI] [PubMed] [Google Scholar]
- 22.Hu H, Yao S, Xu Q, Cai X, Mo Z, Yang Z, Chen W, He Q, Dai X, Xu Z. Protein-coated Cobalt oxide-hydroxide nanospheres deliver photosensitizer IR780 iodide for near-infrared light-triggered photodynamic/photothermal/chemodynamic therapy against colon cancer. J Mater Chem B. 2023;11:9185–200. [DOI] [PubMed] [Google Scholar]
- 23.Wu YT, Fang Y, Wei Q, Shi H, Tan H, Deng Y, Zeng Z, Qiu J, Chen C, Sun L, Chen ZJ. Tumor-targeted delivery of a STING agonist improvescancer immunotherapy. Proc Natl Acad Sci U S A. 2022;119:e2214278119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Q, Luo Y, Xu L, Du C, Zhang W, Xu J, Liu Y, Liu B, Chen S, Wang Y, et al. Smart responsive Fe/Mn nanovaccine triggers liver cancer immunotherapy via pyroptosis and pyroptosis-boosted cGAS-STING activation. J Nanobiotechnol. 2024;22:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.So JY, Skrypek N, Yang HH, Merchant AS, Nelson GW, Chen WD, Ishii H, Chen JM, Hu G, Achyut BR, et al. Induction of DNMT3B by PGE2 and IL6 at distant metastatic sites promotes epigenetic modification and breast cancer colonization. Cancer Res. 2020;80:2612–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boggio E, Gigliotti CL, Stoppa I, Pantham D, Sacchetti S, Rolla R, Grattarola M, Monge C, Pizzimenti S, Dianzani U et al. Exploiting nanomedicine for cancer polychemotherapy: recent advances and clinical applications. Pharmaceutics. 2023; 15. [DOI] [PMC free article] [PubMed]
- 27.Ma W, Sun R, Tang L, Li Z, Lin L, Mai Z, Chen G, Yu Z. Bioactivable STING nanoagonists to synergize NIR-II mild photothermal therapy primed robust and Long-Term anticancer immunity. Adv Mater. 2023;35:e2303149. [DOI] [PubMed] [Google Scholar]
- 28.Wang S, Zhang J, Chu L, Xiao H, Miao C, Pan Z, Qiao Y, Wang Z, Zhou B. Crown-ether threaded covalent organic Polyrotaxane framework (COPF) towards synergistic crown/Zn(2+)/photothermal/photodynamic antibacterial and infected wound healing therapy. Biomater Adv. 2024;159:213814. [DOI] [PubMed] [Google Scholar]
- 29.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Xu Y, Mintz RL, Luo X, Fang Y, Lao YH, Chan HF, Li K, Lv S, Chen G, et al. Self-intensified synergy of a versatile biomimetic nanozyme and doxorubicin on electrospun fibers to inhibit postsurgical tumor recurrence and metastasis. Biomaterials. 2023;293:121942. [DOI] [PubMed] [Google Scholar]
- 32.Yang B, Ding L, Yao H, Chen Y, Shi J. A Metal-Organic framework (MOF) Fenton Nanoagent-Enabled nanocatalytic cancer therapy in synergy with autophagy Inhibition. Adv Mater. 2020;32:e1907152. [DOI] [PubMed] [Google Scholar]
- 33.Angelé-Martínez C, Murray J, Stewart PA, Haines J, Gaertner AAE, Brumaghim JL. Cobalt-mediated oxidative DNA damage and its prevention by polyphenol antioxidants. J Inorg Biochem. 2023;238:112024. [DOI] [PubMed] [Google Scholar]
- 34.Hayman TJ, Baro M, MacNeil T, Phoomak C, Aung TN, Cui W, Leach K, Iyer R, Challa S, Sandoval-Schaefer T, et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat Commun. 2021;12:2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ba S, Yu M. Ultrasound-stimulated microbubbles enhances radiosensitivity of ovarian cancer. Acta Radiol. 2022;63:1433–40. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Zhang C, Chen Y, Xu Y, Yao W, Fan W. Biodegradable Long-Circulating nanoagonists optimize Tumor-Tropism Chemo-Metalloimmunotherapy for boosted antitumor immunity by cascade cGAS-STING pathway activation. ACS Nano. 2024;18:23711–26. [DOI] [PubMed] [Google Scholar]
- 37.Veatch JR, Lee SM, Shasha C, Singhi N, Szeto JL, Moshiri AS, Kim TS, Smythe K, Kong P, Fitzgibbon M, et al. Neoantigen-specific CD4(+) T cells in human melanoma have diverse differentiation States and correlate with CD8(+) T cell, macrophage, and B cell function. Cancer Cell. 2022;40:393–e409399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu X, Yasuda T, Yasuda-Yosihara N, Yonemura A, Umemoto T, Nakachi Y, Yamashita K, Semba T, Arima K, Uchihara T, et al. Downregulation of 15-PGDH enhances MASH-HCC development via fatty acid-induced T-cell exhaustion. JHEP Rep. 2023;5:100892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun L, Suo C, Zhang T, Shen S, Gu X, Qiu S, Zhang P, Wei H, Ma W, Yan R, et al. ENO1 promotes liver carcinogenesis through YAP1-dependent arachidonic acid metabolism. Nat Chem Biol. 2023;19:1492–503. [DOI] [PubMed] [Google Scholar]
- 40.Zhu HJ, Sun YY, Du Y, Zhou SY, Qu Y, Pang SY, Zhu S, Yang Y, Guo ZN. Albumin-seeking near-infrared-II probe evaluating blood-brain barrier disruption in stroke. J Nanobiotechnol. 2024;22:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Yang T, Yu Z, Liu T, Jin R, Weng L, Bai Y, Gooding JJ, Zhang Y, Chen X. Intelligent gold nanoparticles with oncogenic MicroRNA-Dependent activities to manipulate tumorigenic environments for synergistic tumor therapy. Adv Mater. 2022;34:e2110219. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Li X, Shang H, Sun Y, Wang C, Wang X, Tian H, Yang H, Zhang L, Deng L, et al. Mechanism exploration of synergistic photo-immunotherapy strategy based on a novel exosome-like nanosystem for remodeling the immune microenvironment of HCC. Nano Converg. 2024;11:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu PC, Guo LZ, Yu S, Zeng N, Liu YC, Yu J, Zhang Z, Lu K, Sun L, Wang C, et al. Noninvasive assessment of liver function reserve with fluorescent dosimetry of indocyanine green. Biomed Opt Express. 2022;13:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okamoto Y, Ishizuka M, Sumiyama F, Kosaka H, Suganami A, Tamura Y, Sekimoto M, Kaibori M. Inhibitory effects and gene expression analysis of chemotherapeutic photodynamic therapy by using a liposomally formulated indocyanine green derivative. Photodiagnosis Photodyn Ther. 2022;39:102961. [DOI] [PubMed] [Google Scholar]
- 45.Song J, Wang H, Meng X, Li W, Qi J. A hypoxia-activated and microenvironment-remodeling nanoplatform for multifunctional imaging and potentiated immunotherapy of cancer. Nat Commun. 2024;15:10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morotti M, Grimm AJ, Hope HC, Arnaud M, Desbuisson M, Rayroux N, Barras D, Masid M, Murgues B, Chap BS, et al. PGE(2) inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature. 2024;629:426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen S, Liao C, Hu H, Liao J, Chen Z, Li S, Zeng X, Peng B, Shen S, Li D, et al. Hypoxia-driven tumor stromal remodeling and immunosuppressive microenvironment in scirrhous HCC. Hepatology. 2024;79:780–97. [DOI] [PubMed] [Google Scholar]
- 48.Wu GLSB, He YX, Tan XF, Pan Q, Yang S, Li N, Wang MH, Wu PX, Liu F, et al. Rational design of molecular phototheranostic platform for NIR-II fluorescence imaging guided chemodynamic-photothermal combined therapy. Chem Eng J. 2023;5:463. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.