

Abstract
Gene transcripts with invariant abundance during development and in the face of environmental stimuli are essential reference points for accurate gene expression analyses, such as RNA gel-blot analysis or quantitative reverse transcription-polymerase chain reaction (PCR). An exceptionally large set of data from Affymetrix ATH1 whole-genome GeneChip studies provided the means to identify a new generation of reference genes with very stable expression levels in the model plant species Arabidopsis (Arabidopsis thaliana). Hundreds of Arabidopsis genes were found that outperform traditional reference genes in terms of expression stability throughout development and under a range of environmental conditions. Most of these were expressed at much lower levels than traditional reference genes, making them very suitable for normalization of gene expression over a wide range of transcript levels. Specific and efficient primers were developed for 22 genes and tested on a diverse set of 20 cDNA samples. Quantitative reverse transcription-PCR confirmed superior expression stability and lower absolute expression levels for many of these genes, including genes encoding a protein phosphatase 2A subunit, a coatomer subunit, and an ubiquitin-conjugating enzyme. The developed PCR primers or hybridization probes for the novel reference genes will enable better normalization and quantification of transcript levels in Arabidopsis in the future.
Transcripts of stably expressed genes are crucial internal references for normalization of gene expression data. This is especially the case for quantitative reverse transcription (qRT)-PCR studies, which are growing in importance as a means to validate data from whole-genome oligonucleotide arrays and as a primary source of expression data for smaller sets of genes (Czechowski et al., 2004; Gachon et al., 2004). Some of the best known and most frequently used reference transcripts for qRT-PCR in plants and animals include those of 18S rRNA, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), elongation factor-1α (EF-1α), polyubiquitin (UBQ), actin (ACT), and α-tubulin and β-tubulin (TUA and TUB, respectively) genes (Goidin et al., 2001; Bustin, 2002; Kim et al., 2003; Andersen et al., 2004; Brunner et al., 2004; Dheda et al., 2004; Radonić et al., 2004). These genes were chosen in the pre-genomic era because of their known or suspected housekeeping roles in basic cellular processes, such as cell structure maintenance or primary metabolism. Hence, they are often referred to as housekeeping control genes, although we refer to them here simply as reference genes. Unfortunately, the transcript levels of these reference genes are not always stable (Thellin et al., 1999; Suzuki et al., 2000; Lee et al., 2001; this study), and their use for normalization purposes can compromise gene expression studies. Nonetheless, in the absence of better choices, many researchers continue to rely on one or two of these genes, regardless of the developmental or physiological state of the materials being analyzed.
To determine which reference gene(s) is best suited for transcript normalization in a given subset of biological samples, statistical algorithms like geNORM or BestKeeper have been developed (Vandesompele et al., 2002; Pfaffl et al., 2004). For example, geNORM uses pair-wise comparisons and geometric averaging across a matrix of reference genes and biological samples to determine the best reference gene(s) for a given set of samples. Using single-factor ANOVA and linear regression analyses, Brunner et al. (2004) determined the stability of gene expression for 10 reference genes in eight poplar (Populus spp.) tissues by qRT-PCR. A UBQ and a TUA gene were most stably expressed in the set of samples tested. Due to the limited set of samples and genes tested, however, it remains unclear whether these and other traditionally used reference genes are in fact the best possible choices for this species.
To investigate the stability of expression of commonly used reference genes in more depth and to identify novel and superior reference genes, it is necessary to have gene expression data for many (ideally all) expressed genes of an organism from as many different organs and experimental conditions as possible. Tomato (Lycopersicon esculentum) expressed sequence tag (EST) libraries have previously been used for this purpose (Coker and Davies, 2003), but no genes were identified that showed stable expression, as judged by relative EST abundance, across a wide range of developmental and environmental conditions. For Arabidopsis (Arabidopsis thaliana), the prerequisites to identify excellent reference genes for transcript normalization have recently been met in the form of a huge data set, obtained using Affymetrix ATH1 GeneChips representing >23,500 genes (Redman et al., 2004), which includes transcript levels for most Arabidopsis genes over a wide range of developmental and environmental conditions (Altmann et al., 2004; Schmid et al., 2005; http://web.uni-frankfurt.de/fb15/botanik/mcb/AFGN/atgenex.htm). These conditions include an extensive developmental series (79 different tissues, organs, developmental stages, and genotypes), a shoot and a root abiotic stress series (cold, osmotic, salt, drought, genotoxic, oxidative, UV-B, wounding, and heat stress time courses), a biotic stress series (time courses with different virulent and avirulent Pseudomonas syringae strains, Phytophthora infestans, and elicitors), a photomorphogenic light series (time courses of etiolated seedlings treated with different light qualities and durations), and a hormone series (time courses of wild-type seedlings treated with seven different phytohormones).
In this study, we complemented the publicly available AtGenExpress data (http://web.uni-frankfurt.de/fb15/botanik/mcb/AFGN/atgenex.htm) with our own ATH1 data from a nutrient stress series (nitrogen, carbon, sulfur, and phosphorus deprivation of wild-type seedlings and time courses after readdition of the respective nutrient; Scheible et al., 2004; W.-R. Scheible, R. Morcuende, R. Bari, M. Udvardi, Y. Gibon, M. Höhne, B. Pant, O. Thimm, R. Trethewey, and M. Stitt, unpublished data) and a diurnal series (O. Bläsing, Y. Gibon, M. Günther, M. Höhne, R. Morcuende, W.-R. Scheible, and M. Stitt, unpublished data), to assess expression stability of traditional reference genes and to identify hundreds of novel reference genes with very stable expression levels, which together cover a wide range of absolute expression levels. As a result of this analysis, we have validated and recommend 18 new genes as references for superior transcript normalization in Arabidopsis gene expression studies and provide primer sequences for these genes for use in qRT-PCR experiments.
RESULTS
Identification of Very Stably Expressed Arabidopsis Genes from ATH1 Array Data
ATH1 gene probe sets showing very stable hybridization signals were extracted from the different experimental series represented in the AtGenExpress database (http://web.uni-frankfurt.de/fb15/botanik/mcb/AFGN/atgenex.htm; http://arabidopsis.org/info/expression/ATGenExpress.jsp) and from our own nutrient stress and diurnal cycle series in the following way. First, the mean expression value (MV), sd, and the ratio sd/MV (i.e. coefficient of variation, CV) were calculated for each gene in each given experimental series (e.g. developmental series or shoot abiotic stress series; see Supplemental Table I). Second, gene probe sets with at least 80% of “Present” calls, as attributed by Affymetrix Microarray Suite 5 (MAS5) software, within an experimental series were sorted according to their CV value. The 100 genes with the lowest CV values in each series are presented in Supplemental Table I, while Table I displays a selection of five stably expressed genes for the different series.
Table I.
A selection of stably expressed genes for different experimental series
% P Calls, Percentage of Present calls within the experimental series, as attributed by MAS5 software. NA, Not available. *, This low CV value arose due to signal saturation as described in the text. AtGE, AtGenExpress.
| Affymetrix Probe Set | Arabidopsis Gene ID | Annotation | Mean Expression | sd | CV | % P Calls |
|---|---|---|---|---|---|---|
| AtGE developmental series (237 arrays) | ||||||
| 253826_s_at | At4g27960 | UBC9 | 9,979 | 1,940 | 0.194 | 100 |
| At5g53300 | UBC10 | |||||
| 259407_at | At1g13320 | PP2A | 713 | 106 | 0.148 | 100 |
| 262909_at | At1g59830 | PP2A | 454 | 67 | 0.147 | 100 |
| 245863_s_at | At1g58050 | Helicase | 179 | 30 | 0.167 | 97 |
| 248024_at | At5g55840 | PPR protein | 25 | 4 | 0.171 | 99 |
| AtGE shoot abiotic stress series (136 arrays) | ||||||
| 245006_at | psaB | PSI/PSII D1 subunit | 21,284 | 1,364 | 0.064 | 100 |
| 246006_at | At5g08290 | Yellow-leaf-specific protein 8 | 2,228 | 210 | 0.094 | 100 |
| 259407_at | At1g13320 | PP2A | 813 | 70 | 0.086 | 100 |
| 251998_at | At3g53090 | Ubiquitin-transferase | 204 | 19 | 0.093 | 100 |
| 253023_at | At4g38070 | bHLH family protein | 17 | 1 | 0.082 | 81 |
| AtGE root abiotic stress series (136 arrays) | ||||||
| 247644_s_at | At5g60390 | EF-1α | ||||
| At1g07920 | EF-1α | 16,024 | 1,204 | 0.075 | 100 | |
| At1g07930 | EF-1α | |||||
| At1g07940 | EF-1α | |||||
| 248858_at | At5g46630 | Clathrin adaptor complex subunit | 1,438 | 11 | 0.083 | 100 |
| 259407_at | At1g13320 | PP2A | 888 | 67 | 0.075 | 100 |
| 246515_at | At5g15710 | F-box family protein | 223 | 16 | 0.073 | 100 |
| 250311_at | At5g12240 | Expressed protein | 33 | 2 | 0.072 | 96 |
| RIKEN hormone series (48 arrays) | ||||||
| 247644_s_at | At5g60390 | 4× EF-1α (see above) | 44,825 | 111 | 0.002* | NA |
| 259408_at | At3g25800 | PP2A | 6,724 | 274 | 0.041 | NA |
| 254172_at | At4g24550 | Clathrin adaptor complex subunit | 6,255 | 235 | 0.038 | NA |
| 256280_at | At3g12590 | Expressed Protein | 1,390 | 51 | 0.037 | NA |
| 247217_s_at | At5g65050 | MADS-box protein AGL31 | 735 | 24 | 0.032 | NA |
| MPI-MPP nutrient stress series (28 arrays) | ||||||
| 253826_s_at | At4g27960 | UBC9 | 12,150 | 983 | 0.081 | 100 |
| At5g53300 | UBC10 | |||||
| 265697_at | At2g32170 | Expressed protein | 771 | 45 | 0.058 | 100 |
| 261095_at | At1g62930 | PPR protein | 169 | 13 | 0.080 | 100 |
| 248024_at | At5g55840 | PPR protein | 60 | 5 | 0.081 | 100 |
| 246731_at | At5g27630 | RING-finger protein | 29 | 1 | 0.050 | 100 |
| GARNET light series (48 arrays) | ||||||
| 257313_at | At3g26520 | TIP2 | 13,558 | 425 | 0.031 | NA |
| 248573_at | At5g49720 | KORRIGAN | 5,378 | 185 | 0.034 | NA |
| 259407_at | At1g13320 | PP2A | 581 | 25 | 0.043 | NA |
| 265256_at | At2g28390 | SAND family protein | 340 | 15 | 0.045 | NA |
| 261095_at | At1g62930 | PPR protein | 80 | 4 | 0.052 | NA |
| GARNET biotic stress series (90 arrays) | ||||||
| 245006_at | psaB | PSI/PSII D1 subunit | 11,002 | 1,021 | 0.093 | NA |
| 253959_at | At4g26410 | Expressed protein | 748 | 77 | 0.102 | NA |
| 259280_at | At3g01150 | PTB protein | 288 | 30 | 0.103 | NA |
| 256631_at | At3g28320 | Hypothetical protein | 96 | 7 | 0.068 | NA |
| 261095_at | At1g62930 | PPR protein | 74 | 6 | 0.080 | NA |
Within the diurnal series, 96 of the 100 genes with the lowest CV values had MVs <30, putting them among the 20% of lowest-expressed genes. Since the CV was very low (<0.060) for >700 genes in this particular experimental series, 100 of these genes with the highest MVs were chosen to generate a list of both high- and low-expressed, diurnally invariable genes (Supplemental Table I). MAS5-processed files with Present/Absent calls were not available to us for the light, hormone, and biotic stress arrays at the time of the analysis (but they are now available from the Nottingham Arabidopsis Stock Centre; http://affymetrix.arabidopsis.info). To avoid identifying genes with Absent calls as potential reference genes, we considered only the 14,000 genes (approximately 60%) with the highest MVs. This percentage is slightly lower than the percentage of genes typically expressed (called Present by MAS5 software) in an entire Arabidopsis seedling or its major organs (F. Wagner, RZPD Berlin, personal communication; Czechowski et al., 2004; Redman et al., 2004). Moreover, scanning of the hybridized ATH1 arrays from the hormone series was performed at high sensitivity to focus on rare transcripts (Y. Shimada, RIKEN, personal communication), leading to signal saturation and hence feigning stable expression for strongly expressed genes. Following statistical analysis of this series, we therefore disregarded 1,701 probe sets with hybridization signals >7.500 (Supplemental Fig. 1). Still, the results of the hormone series are also presented for the highly expressed genes in Supplemental Figure 6 and for one example in Table I.
Comparison of the CV values for the chosen Top100, i.e. the most stably expressed genes from the different experimental series (Table I; Supplemental Table I), revealed that they were highest for the complete developmental series (0.141–0.200; 237 ATH1 arrays), which presumably reflects the diversity of plant organs, ages, and genotypes included (see http://www.weigelworld.org/resources/microarray/AtGenExpress). In contrast, all other experimental series were obtained with plant material that was more homogenous with respect to tissue type, developmental stage, and genetic background (see Supplemental Fig. 6). The developmental series was analyzed further to identify whether specific types of samples were responsible for the higher CV (see supplemental text and Supplemental Figs. 2–4). Pollen samples and, to a lesser extent, seed samples had a marked influence on the CV, whereas root and flower samples did not. This indicates that pollen and seeds have transcriptomes that are very different from other tissues.
Some of the Affymetrix probe sets (labeled s_at in Table I) are not gene specific, but rather have perfect matches to two or more related genes (Redman et al., 2004). For example, probe set 247644_s_at is representative for four EF-1α genes, transcripts of which will all contribute to the hybridization signal. Therefore, it is possible that individual EF-1α genes have more or less stable expression than the expression pattern mirrored by this probe set. Such uncertainties can be clarified by independent verification by other means (see below). Information on which gene transcripts cross-hybridize to a given probe set (compare e.g. Supplemental Table I) can be obtained from the Affymetrix Web page at http://www.affymetrix.com/analysis/index.affx.
Comparison of Traditional and Novel Arabidopsis Reference Genes
After the identification of stably expressed genes from the ATH1 data, an important aim was to compare the traditional and novel reference genes. Figure 1A shows the developmental expression patterns of five traditional reference genes: ACT2, TUB6, EF-1α, UBQ10, and cytosolic GAPDH. Based on the ATH1 data, TUB6 (red line) had the least stable expression among these five: expression in pollen sample (no. 73), maturing seeds (nos. 81–84), flower sepals (no. 41), stems (nos. 27 and 28), and senescing leaves (no. 25) was >4-fold different than the developmental MV. Accordingly, the CV (0.899) of TUB6 was higher than the CVs of the other four genes (CVACT2 = 0.642; CVGAPDH = 0.233; CVEF-1α = 0.228; CVUBQ10 = 0.205). Like TUB6, EF-1α, ACT2, and GAPDH showed strongly reduced expression in pollen and ACT2 and GAPDH in seed samples. ACT2 also displayed deviant expression in root samples (nos. 3, 9, 94, 95, 98, and 99). In contrast, UBQ10 had remarkably stable expression (<2-fold deviation from the mean value) across all developmental samples. Nevertheless, UBQ10 was found in the developmental Top100 list (Supplemental Table I) only after omission of the seed samples. Similarly, GAPDH was ranked among the 100 most stably expressed genes only after omission of seed and pollen samples. The other three genes were never represented in the Top100.
Figure 1.

Relative expression level of traditional and novel reference genes during development. A, Relative expression levels for five traditional genes, i.e. ACT2 (At3g18780, black), TUB6 (At5g12250, red), EF-1α (At5g60390, green), UBQ10 (At4g05320, cyan), and GAPDH (At1g13440, blue). B, Expression levels for five novel genes, i.e. At4g34270 (black), At1g13320 (red), At1g59830 (green), At4g33380 (cyan), and At2g28390 (blue). The RMA-normalized ATH1 expression level for each gene in every sample (1–101) is expressed relative to the average expression level over the entire developmental series. The descriptions of the developmental samples 1 to 101 are available at http://www.weigelworld.org/resources/microarray/AtGenExpress/. The Affymetrix probe set for At5g60390 is not specific but detects transcripts of four EF-1α genes (see Table I).
The developmental expression patterns and CV values of additional TUB, ACT, and UBQ genes (with >80% Present calls) are shown in Supplemental Figure 5. Whereas UBQ10 appears to be the most constitutively expressed polyubiquitin gene, neither ACT2 nor TUB6 appeared to be the most stably expressed gene in its family. Other proposed reference genes and their homologs were also investigated with respect to their CV values, including TUA, histone, phosphoglyceratekinase, cyclophilin, and EF-Tu genes, and eIF4a (AT1G54270). Only eIF4a and two of the 30 cyclophilin genes (At2g16600, At4g33060) had developmental CVs (0.268, 0.266, and 0.233, respectively) that were close to those of EF-1α, UBQ10, or GAPDH.
Figure 1B depicts the expression patterns of the five “novel” genes with the lowest CVs in the developmental series (compare Supplemental Table I). All five genes are clearly more stably expressed than the traditional reference genes in Figure 1A. The TIP41-like gene At4g34270 (black line) had the lowest CV (0.141) among the five genes in Figure 1B, and the value further decreased to 0.116 after omission of the pollen sample (no. 73; compare Supplemental Table I). Interestingly, two of the five genes shown in Figure 1B encode subunits of Ser/Thr protein phosphatase 2A (PP2A). At1g13320 encodes a 65-kD regulatory subunit, and At1g59830 encodes a catalytic subunit for which two splice variants are annotated at The Arabidopsis Information Resource (TAIR; www.arabidopsis.org) yielding 30- and 35-kD proteins. Moreover, At1g10430 (encoding another catalytic subunit of PP2A) and At2g39840 (encoding a catalytic subunit of protein phosphatase 1) are also represented among the Top100 genes of the developmental series, and At3g25800 (encoding another regulatory PP2A subunit) is represented among the Top100 genes of the root abiotic stress and hormone series (Supplemental Table I). Besides its appearance in the developmental Top100 list(s), At1g13320 is also represented in the Top100 lists of the shoot and the root abiotic stress series, as well as the light series (Table I; Supplemental Tables I and II), and was placed within the Top500 in the hormone, biotic, and nutrient stress series (see Supplemental Table II). In the diurnal series, the gene was not placed among the Top500, although the CV was reasonably low (0.134). In this case, it should be noted that many genes had very low CV values in this series. Altogether, At1g13320 appears to be a very stably expressed gene in the Arabidopsis genome (see Supplemental Fig. 6).
Another important difference between the novel reference genes identified by our analysis and more traditional controls are their absolute expression levels. While EF-1α, UBQ10, and GAPDH usually belong to the first percentile of highly expressed genes in Arabidopsis, the novel genes shown in Figure 1B have expression levels at least 10- to 20-fold lower (Supplemental Fig. 6). Other genes covering an extended range of mean expression levels can be found in Supplemental Table I.
Complete expression profiles, including all available experimental series, for the genes shown in Figure 1, for At5g09810 (ACT7, formerly called ACT2), two other reference genes (i.e. At5g44200/CBP20; At5g25760/UBC) qPCR primers that are marketed by Sigma-Aldrich, and for several other genes selected from Supplemental Table I are presented in Supplemental Figure 6. These genes were selected because they appeared on Top100 lists or extended Top500 lists of two or more experimental series (see selection criteria in Supplemental Table II) and because most displayed much lower absolute expression on ATH1 arrays than traditional reference genes (Table II). Overall selection was biased toward genes that are stably expressed in the developmental series because this series is the most complex one, as mentioned above.
Table II.
qRT-PCR and ATH1 expression levels of traditional and novel reference genes
Arabidopsis Gene IDs of traditional reference genes are in bold. % P Calls, Percentage of Present calls within the experimental series, as attributed by MAS5 software.
| Arabidopsis Gene ID | Annotation | Mean ΔCTa | Mean 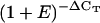
|
Relative Expression | se (n = 20) | ATH1 Absolute/Relative Mean Expressionb | % P Callsb |
|---|---|---|---|---|---|---|---|
| ×106 | |||||||
| AT4G05320 | UBQ10 | 8.76 | 4,270 | 1 | 6.9 E-04 | 6,811/0.412 | 100 |
| AT5G60390 | EF-1α | 9.07 | 3,660 | 0.858 | 6.7 E-04 | 16,528/1 | 100 |
| AT1G13440 | GAPDH | 9.50 | 2,488 | 0.583 | 3.5 E-04 | 8,220/0.497 | 100 |
| AT4G27960 | UBC9 | 10.98 | 1,160 | 0.272 | 1.5 E-04 | 9,979/0.604 | 100 |
| AT3G18780 | ACT2 | 11.74 | 917 | 0.215 | 1.9 E-04 | 5,347/0.323 | 99 |
| AT5G46630 | Clathrin adaptor complex subunit | 12.71 | 421 | 0.099 | 3.9 E-05 | 1,115/0.068 | 100 |
| AT5G08290 | YLS8 | 13.60 | 343 | 0.081 | 3.9 E-05 | 2,924/0.177 | 100 |
| AT4G33380 | Expressed | 13.45 | 342 | 0.080 | 3.2 E-05 | 310/0.019 | 100 |
| AT2G32170 | Expressed | 14.47 | 246 | 0.058 | 3.3 E-05 | 337/0.020 | 100 |
| AT4G34270 | TIP41-like | 13.68 | 243 | 0.057 | 2.9 E-05 | 353/0.021 | 90 |
| AT5G25760 | UBC | 13.82 | 190 | 0.044 | 2.6 E-05 | 422/0.026 | 100 |
| AT1G13320 | PDF2 | 13.41 | 189 | 0.044 | 2.7 E-05 | 713/0.043 | 100 |
| AT2G28390 | SAND family | 14.69 | 167 | 0.039 | 1.9 E-05 | 415/0.025 | 99 |
| AT4G26410 | Expressed | 14.11 | 132 | 0.031 | 1.7 E-05 | 617/0.037 | 100 |
| AT3G01150 | PTB | 18.56 | 40 | 0.009 | 4.7 E-06 | 247/0.015 | 99 |
| AT5G55840 | PPR repeat | 18.59 | 38 | 0.009 | 4.4 E-06 | 24.8/0.001 | 99 |
| AT1G58050 | Helicase | 16.81 | 36 | 0.008 | 3.9 E-06 | 179/0.011 | 97 |
| AT3G53090 | UPL7 | 19.70 | 22 | 0.005 | 2.1 E-06 | 112/0.007 | 100 |
| AT5G15710 | F-box family | 17.53 | 15 | 0.003 | 1.9 E-06 | 99.8/0.006 | 98 |
| AT4G38070 | bHLH | 22.46 | 6 | 0.001 | 1.1 E-06 | 14.1/0.0009 | 84 |
| AT5G12240 | Expressed | 21.91 | 1.6 | 0.00038 | 1.8 E-07 | 5.3/0.0003 | 90 |
| AT1G62930 | PPR repeat | 24.02 | 0.4 | 0.00009 | 6.3 E-08 | 95.1/0.006 | 99 |
| AT3G32260 | Hypothetical | NA | NA | NA | NA | 14.0/0.0008 | 88 |
| AT2G07190 | Hypothetical | NA | NA | NA | NA | 10.7/0.0006 | 89 |
| AT1G47770 | Hypothetical | NA | NA | NA | NA | 5.6/0.0003 | 59* |
Difference in threshold cycle number relative to LjLb2.
Based on the developmental series; NA, data not available since no PCR amplification could be obtained. *, Chosen based on its presence in the Top100 lists of the shoot and root abiotic stress and nutrient stress series and because it showed >80% Present calls in all three.
Gene-Stability Measure and Ranking of Reference Genes Using geNORM
Vandesompele et al. (2002) developed an algorithm named geNORM that determines the expression stability of reference genes. The calculated gene-stability measure (M) relies on the principle that the expression ratio of two ideal internal reference genes is identical in all samples, regardless of the experimental condition or cell type. Accordingly, variation of the expression ratios of two reference genes shows that one or both genes are not stably expressed, hence M > 0. Within a set of reference genes, expression stability for each gene can thus be determined by pair-wise comparison with all other reference genes across all experimental conditions and subsequent calculation of the geometrically averaged sd of log-transformed reference gene ratios.
We used this method as an alternative approach to validate and rank the traditional and novel reference genes, using ATH1 data from the developmental series (Fig. 2; Supplemental Fig. 6). The average expression stability value (M) for most traditional genes (black bars in Fig. 2) was considerably higher (i.e. expression is less stable) than for most of the novel reference genes identified by our approach (white bars). Once again, the polyubiquitin genes At5g25760 (UBC) and At4g05320 (UBQ10) showed the most stable expression within the subset of traditional genes with their M values being comparable to those of most of the novel reference genes (Fig. 2). The novel gene with the highest M value, At1g62930, was chosen because it was represented in three Top100 lists (see Supplemental Table II), although none of these were related to development. Hence, it is not really surprising that it performed less well than other genes as a reference for normalization of developmental expression data. There was good agreement between the average expression stability (M) value and the calculated CV value for this set of genes: Linear regression analysis yielded a correlation coefficient (R2) of 0.91. When the geNORM algorithm was used on combined ATH1 data from all experimental series (Supplemental Fig. 7), the picture remained the same: Novel genes generally had a lower M value, i.e. more stable expression, than traditional genes, with the exception of UBQ10 (At4g05320).
Figure 2.

geNORM ranking of 27 reference genes based on ATH1 data from the developmental series. A low value for the average expression stability M, as calculated by geNORM software, indicates a more stable expression. Seventy-nine samples including different developmental stages, organs, tissues, and genotypes were included in this analysis. Vertical numbers at the top represent the CV values of the genes for the same set of samples.
Validation of Traditional and Novel Reference Genes by qRT-PCR
Affymetrix ATH1 chips have become a “gold standard” for Arabidopsis transcriptome analysis. However, for hybridization-based technologies, like Affymetrix chips, there is not a strict linear relationship between signal strength and transcript amount for different genes, as there is for qRT-PCR (Holland, 2002; Czechowski et al., 2004). Therefore, we used quantitative real-time SYBR Green RT-PCR to assess independently expression stabilities and absolute expression levels of 20 novel and five traditional reference genes.
A diverse set of 20 cDNA pools was synthesized as templates for RT-PCR. Total RNA from the different plant samples (Supplemental Table III) was isolated, DNase I digested, and reverse transcribed, with various quality controls (Fig. 3). To enable quantitative and qualitative assessment of the RT reaction, an equal amount of DNase I-digested total RNA from each sample was spiked with 30 pg cRNA of a foreign gene Lotus japonicus leghemoglobin-2 (LjLb2) before the RT reaction. Subsequently, first-strand cDNAs were analyzed for (1) equal amplification of LjLb2 cDNA and (2) the 5′/3′ amplification ratio of GAPDH [(1 + EGAPDH5′)CT GAPDH5′/(1 + EGAPDH3′)CT GAPDH3′; approximately 2ΔCT(GAPDH5′-GAPDH3′), with E = amplification efficiency], which is an indicator of RNA integrity and processivity of reverse transcriptase. We consider this an important control, since different 5′/3′ ratios for different cDNA samples to be compared can result in erroneous expression ratios for amplicons that are not located immediately upstream or close to the RT initiation site.
Figure 3.

Flowchart of the qRT-PCR methodology. Total RNA was prepared from 20 diverse Arabidopsis plant samples. Total RNA (pre) was quality checked spectrophotometrically (A260/A280 ratio), using an Agilent Bioanalyzer (inset 1) and by gel electrophoresis (inset 2) prior to (lane 1) and after DNase I digest (lane 3; note the absence of the high Mr genomic DNA band). The presence and absence of genomic DNA contamination in total RNA samples before and after DNase I treatment, respectively, was also confirmed by PCR, using primers designed to amplify a 633-bp genomic fragment of the ACT2 gene (inset 2, compare lanes 2 and 4). Prior to RT, equal amounts of DNase I-treated RNA were spiked with a defined amount of LjLb2 cRNA as internal standard. First-strand cDNA pools were checked by (1) gel electrophoresis prior to and after PCR (inset 2, lanes 5 and 6, respectively) with the same ACT2 primers as above, which amplify a 190-bp fragment from cDNA of both annotated ACT2 splice variants; (2) determination of the 5′/3′ ratio of GAPDH cDNA amplification using two primer pairs that amplify DNA from either the 5′ or 3′ region (see Supplemental Table II and “Materials and Methods”), providing a further estimate of RNA integrity and/or processivity of reverse transcriptase in each sample; and (3) quantification of cDNALjLb2 as an independent check of cDNA synthesis efficiency and to normalize cDNA pools. Various cDNA pools were then used for qRT-PCR with primer pairs for a series of novel reference genes extracted from the AtGenExpress database as well as a few traditional reference genes. Primer pairs were all designed to work in the same PCR conditions and were checked for specificity of amplification by gel electrophoresis (inset 3), melting-curve analysis (inset 4), and sequencing of amplicons, before use.
PCR primers for 20 novel and five traditional reference genes were designed and tested in qRT-PCR reactions (see Supplemental Table II). To ensure maximum specificity and efficiency during PCR amplification of cDNA under a standard set of reaction conditions, primers were required to have melting temperatures (TM) of 60°C ± 1°C and to amplify short products, usually around 60 to 70 bp (see Supplemental Table II). Typically primers were also designed to give an amplicon located close to the 3′ end of the transcript of interest (see Supplemental Table II), which should allow better amplification especially for cDNAs with GAPDH 5′/3′ ratios ≪1. In addition, when possible at least one primer of a pair was also designed to cover an exon-exon junction. Care was taken that the primers encompass all known transcript splice variants.
The specificity of PCR primers was tested using the 20 first-strand cDNAs described above (see Supplemental Table III and “Materials and Methods”). Twenty-two of the 25 primer pairs yielded unique PCR amplicons from our cDNA samples. The primers for UBQ10 did not produce amplicons from the two hormone-treatment cDNA pools, which were produced from Arabidopsis accession C24 (see Supplemental Table III). The primer pairs for three hypothetical genes, At1g47770, At2g07190, and At3g32260, yielded no amplicons from any cDNA sample, (Table II; Supplemental Table II). The 22 remaining primer pairs all amplified single PCR products of the expected size from the various cDNA pools, as shown by gel electrophoresis and melting-curve analyses performed by the PCR machine after 40 amplification cycles (Fig. 4, A–D; Supplemental Figs. 8 and 9). A more stringent test of the specificity of PCR was performed by sequencing the products of four novel constitutive genes (Supplemental Fig. 10) and three products from two traditional reference genes (UBQ10, 5′ and 3′ amplicons of GAPDH; data not shown). In all seven cases, the sequence of the PCR product matched that of the intended target cDNA, thereby confirming the exquisite PCR specificity of the developed primer pairs (see also Czechowski et al., 2004).
Figure 4.

Specificity and efficiency of PCR primers. A to D, Specificity of PCR amplification was confirmed by the presence of unique amplicons of the expected size following electrophoresis on agarose gels (A; compare with standard bands and Supplemental Table II), and by dissociation curves with single peaks (B–D). In each section, 54 to 60 individual melting curves are shown obtained with three technical replicates from 18 to 20 different cDNA pools. Dissociation curves and gel pictures for additional amplicons are shown in Supplemental Figures 7 and 8. E to J, Real-time qPCR amplification plots for UBQ10 (E), EF-1α gene At5g60390 (F), At5g25760 (UBC; G), At5g46630 (H), At1g58050 (I), and At1g62930 (J). Each section depicts 54 to 60 amplification curves obtained from 18 (UBQ10) or 20 different cDNA pools. The y axis of each plot shows the logarithmic fluorescence value, while the x axis represents the PCR cycle number (0–40). The gray horizontal line (red line in the online version) represents the threshold fluorescence at which the CT is determined. The slope of each plot in the linear (log) phase is used to determine amplification efficiency (E) for each reaction.
The number of cycles needed to reach a given fluorescence intensity during qPCR depends not only on the amount of cDNA in the extract but also on the amplification efficiency (E). In the ideal case, when the amount of cDNA is doubled in each reaction cycle, E = 1. PCR efficiency can be estimated with various methods (see http://www.gene-quantification.info/). The classical method uses threshold cycle (CT) values obtained from a series of template dilutions (Pfaffl, 2001). An alternative method utilizes absolute fluorescence data captured during the exponential phase of amplification of each real-time PCR reaction (Ramakers et al., 2003). Here, the E value is derived from the slope of the linear portion of the plot of log fluorescence versus cycle number for each primer pair, using the equation (1 + E) = 10slope. Our own previous comparison of the two methods yielded very similar amplification efficiencies for a set of 46 transcription factor primer pairs (Czechowski et al., 2004). Hence, we again used the latter method to establish amplification efficiencies for all reference gene primer pairs, since it does not require standard curves for every primer pair and because it allows estimation of the efficiency for each individual PCR reaction. Supplemental Table II shows the amplification efficiencies for the primer pairs we studied. Each given efficiency value represents an average ± sd calculated from 60 (54 for UBQ10) amplification plots (Fig. 4, E–J; i.e. three technical replicates of 20 different cDNAs). All 22 primer pairs had efficiencies higher than 0.7, 11 between 0.8 and 0.9 and seven higher than 0.9. sds of primer efficiencies were very low, indicating comparable amplification efficiencies in the 20 diverse cDNA samples tested (Fig. 4, compare log slopes in E–J). Mean primer efficiency values (see Supplemental Table II) were taken into account in all subsequent calculations, including calculations of the relative expression level of the constitutive genes (Table II).
Figure 4, E to J, displays the 60 real-time PCR amplification plots for LjLb2 (Fig. 4E) and each of five reference genes. They were obtained with specific primers (see Supplemental Table II) for two traditional genes, UBQ10 and EF-1α (Fig. 4, F and G), and three novel genes, At5g46630, At1g58050, and At1g62930 (Fig. 4, H–J). The 60 samples were measured in parallel for each primer pair. For UBQ10 and EF-1α, most of the amplification curves reached the threshold fluorescence value (0.1), indicated by the gray horizontal line (red line in the online version), after approximately 18 amplification cycles, whereas the amplification curves of the three novel genes (i.e. At5g46630, At1g58050, At1g62930) crossed the threshold fluorescence after approximately 23, approximately 27, and approximately 32 cycles, showing that they are expressed at levels from 20- to >1,000-fold lower than UBQ10 and EF-1α (Table II). The calculated qRT-PCR expression levels were in remarkably good agreement with the developmental ATH1 expression levels (Table II). The linear correlation coefficient of both data sets for all genes in Table II was R2 = 0.88.
The amplification curves for each gene were generally grouped closely together across all treatments and almost as close as those for LjLb2, the external reference. The stable expression of these genes inferred from the ATH1 data was therefore verified by our qRT-PCR experiments. An exception was EF-1α (At5g60390), where the CT values obtained with cDNA from sugar-starved seedlings were larger by about three cycles. Two possible explanations for this shift in CT could be either a lower degree of RNA integrity and/or a less efficient RT reaction (for example, due to reduced processivity of reverse transcriptase) of this batch of RNA, as the cDNA pool prepared from sugar-starved seedlings had the lowest 5′/3′ GAPDH amplification ratio (see Supplemental Table III). The primer pair for At5g60390 was the only one in our set that amplified >1 kb upstream of the transcript 3′ end (see Supplemental Table II), making it the most susceptible to errors generated by differences in RNA integrity and/or RT efficiency. However, lower expression of EF-1α in carbohydrate-starved seedlings might also reflect biological reality since the same behavior was observed with Affymetrix technology (Supplemental Fig. 6). This conclusion is further strengthened by reduced EF-1α expression during an extended night as well as in Arabidopsis pgm mutants at the end of a normal night (Supplemental Fig. 6; Thimm et al., 2004), two additional conditions in which carbohydrate shortage occurs.
To analyze the expression stability M for the genes investigated by qRT-PCR, we used geNORM v.3.4 software (Vandesompele et al., 2002). When expression levels in the form 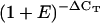 (see “Materials and Methods” for more details) from all 20 cDNA pools were considered, the inferred transcript stability of all novel reference genes was superior to that of EF-1α and ACT2. However, GAPDH and UBC transcripts had M values similar to most of the novel reference genes (Fig. 5). Data for UBQ10 were excluded in this analysis because primers for this gene did not amplify cDNA of wild-type accession C24 for the two hormone treatments. When these two cDNA samples were excluded and UBQ10 was included, the result remained very similar (Supplemental Fig. 11).
(see “Materials and Methods” for more details) from all 20 cDNA pools were considered, the inferred transcript stability of all novel reference genes was superior to that of EF-1α and ACT2. However, GAPDH and UBC transcripts had M values similar to most of the novel reference genes (Fig. 5). Data for UBQ10 were excluded in this analysis because primers for this gene did not amplify cDNA of wild-type accession C24 for the two hormone treatments. When these two cDNA samples were excluded and UBQ10 was included, the result remained very similar (Supplemental Fig. 11).
Figure 5.

geNORM ranking of reference genes based on qRT-PCR data. Data from all 20 cDNA pools were considered for 21 genes (UBQ10 was omitted). Traditional and novel reference genes are shown as black and white bars, respectively. A low value for the average expression stability, M, as calculated by geNORM software, indicates more stable expression throughout the various conditions.
DISCUSSION
Gene expression databases for Arabidopsis and other organisms are important resources to investigate the expression pattern of a given gene or gene families, to identify genes that respond to specific stimuli, and to search for coexpressed genes (http://www.ncbi.nlm.nih.gov/geo/; http://www.ebi.ac.uk/arrayexpress/; Steinhauser et al., 2004; Zimmermann et al., 2004; Shen et al., 2005). AtGenExpress is a very recent gene expression database for Arabidopsis, and possibly the most standardized and comprehensive gene expression resource for any multicellular organism (Altmann et al., 2004). We complemented this dataset with our own data (see introduction) and used the resulting set of Affymetrix Arabidopsis ATH1 whole-genome GeneChip data (approximately 16 million data points i.e. 721 arrays or 323 conditions) to investigate the expression stability of traditional reference genes and to identify novel Arabidopsis reference genes (see Supplemental Table I) that outperform the traditional ones in terms of expression stability, as determined by their CV. Furthermore, the novel genes cover a wide range of absolute expression levels, which makes transcripts of these genes valuable reference points for normalizing transcript levels of other genes that are expressed at high, moderate, or low levels in Arabidopsis. We developed high-efficiency qPCR primers for a set of 20 novel and five traditional reference genes and were able to confirm superior expression stability of most of the novel genes, using a set of cDNA samples from 20 different plant samples that included different organs, developmental stages, and abiotic stresses.
Although the scope of the AtGenExpress database is great, it is certainly not all encompassing. Hence, it is possible that genes other than those identified here may exist that are better references for normalizing gene expression under special conditions. For instance, the developmental series contains data from various organs at different stages of development, but information about specific tissue or cell types is lacking. It is likely that the transcriptome of each cell type is very different from that of other cell types, and tissues and organs as a whole. Therefore, care is required in selecting reference genes for specific comparisons. As pointed out by Radonić et al. (2004), it seems unreasonable to expect that the transcription of any gene in a living cell is absolutely resistant to cell cycle fluctuations or nutrient status. Nevertheless, the identification and validation of genes like the PP2A catalytic subunit gene At1g13320 or the TIP41-like gene At4g34270 among others clearly show that traditional reference genes can be easily outperformed, especially when samples originating from different tissues, organs, and developmental stages are being compared (Supplemental Fig. 6).
Traditional reference genes were chosen in the pre-genomic era based on their known or suspected housekeeping roles in basic cellular processes (e.g. protein translation, ubiquitin-dependent protein degradation), cell structure maintenance (e.g. cytoskeleton), or primary metabolism (e.g. glycolysis), and comprise a small number of gene families (see “Results”). Our study revealed that the polyubiquitin gene family contains very stably expressed members, and several stably expressed genes involved in the ubiquitin/26S proteasome pathway (Smalle and Vierstra, 2004) were also found, including genes encoding an ubiquitin-conjugating enzyme (At4g27960), several E3-ubiquitin protein ligases (e.g. At1g75950, and other F-box, RING-finger, and HECT-domain proteins; see Supplemental Table I), an ubiquitin-specific protease (At5g06600), and proteasome subunits. Genes involved in other cellular processes were also found to be very stably expressed. These included PP2A subunit genes; several members of the large family of pentatricopeptide (PPR) repeat-containing protein-encoding genes (Lurin et al., 2004); and many genes encoding proteins involved in protein secretion and intracellular protein transport (Sanderfoot and Raikhel, 2002), such as SNAREs (At1g28490, At3g24350, At1g32270, At5g39510, At3g58170), a SNARE effector (At1g77140, VPS45), adaptor complex subunits of clathrin coated vesicles (At5g46630, At1g48760, At2g19790, At2g20790, At3g55480, At1g31730, At5g11490, At4g24550, At4g14160, At3g01340, At3g51310), and additional genes involved in the process of protein transport (At1g32050, At3g10380, At3g01340). It is intriguing to notice that genes encoding proteins with potential regulatory functions (such as F-box proteins, protein phosphatase subunits, and others; see Supplemental Table I) display very stable transcript levels, and it is therefore tempting to speculate that mechanisms downstream of transcription control their expression.
All genes with moderate or higher ATH1 mean expression in our chosen subset (Table II) were successfully validated and confirmed by qRT-PCR with respect to their high expression stability (Fig. 5) as well as their expression level. As pointed out before, there was good agreement between expression values calculated from the qRT-PCR data and the developmental ATH1 data with a linear correlation coefficient R2 = 0.88. This indicates that it should be straightforward to successfully validate by qRT-PCR also others of the many potential reference genes with moderate or higher ATH1 expression listed in Supplemental Table I. In contrast, for three hypothetical genes (Table II) that showed stable expression but had very low ATH1 MVs, no transcripts could be detected using qRT-PCR, even when a second primer pair was tested for each (data not shown). One possible explanation for this failure could be incorrect electronic gene model predictions, which make it difficult to design functional PCR primers. Another possibility is that these are pseudogenes and are not expressed, which is supported by the lack of any reported cDNAs, ESTs, or Multiple Parallel Signature Sequencing signatures in public databases. Thus, it is probable that the ATH1 expression values for these genes and the high percentage (80%) of Present calls initially used to filter the data arose by unspecific hybridization of the probe set. Another two genes in our chosen subset (At1g62390 and At4g38070) also had very low expression (Table II) and turned out to yield M values similar to those of traditional reference genes (Fig. 5). Again, this might indicate that low ATH1 expression values are not always reliable indicators for true gene expression, and shows that validation of stable gene expression for such genes (Supplemental Table I) is critical before they are used as references for sensitive and accurate gene expression by qRT-PCR. Still, these two genes and, in particular, AT5G12240 and AT5G15710 (Table II) demonstrate that it is perfectly possible to extract from ATH1 data and validate by qRT-PCR genes that have high expression stability but expression levels 1,000- to 10,000-fold lower than UBQ10, EF-1α, or GAPDH. Such stably but lowly expressed genes are the internal references of choice in qRT-PCR experiments where low abundance transcripts, for example, those of many transcription factor genes (Czechowski et al., 2004), are being investigated. Because the difference in threshold cycle number (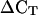 ) between the reference transcript and the gene under investigation (e.g. a transcription factor transcript) will then be much smaller, the calculation of the results will be less influenced by variations in amplification efficiencies and hence more accurate.
) between the reference transcript and the gene under investigation (e.g. a transcription factor transcript) will then be much smaller, the calculation of the results will be less influenced by variations in amplification efficiencies and hence more accurate.
We have shown here that it is possible to identify excellent reference genes from large collections of comprehensive transcriptome data obtained from DNA-array hybridization studies. However, care must be taken that hybridization data is truly gene-specific for the genes of interest. While this appears to be the case for most probe sets on the Arabidopsis ATH1 arrays, it is not true for all sets. For instance, the probe set 247644_s_at detects four EF-1α genes (At5g60390, At1g07920, At1g07930, At1g07940; Table I). All four of these genes are expressed at similar levels in Arabidopsis, as judged by the number of ESTs for each from different cDNA libraries (see gene locus pages at www.arabidopsis.org). The EF-1α probe set (labeled with At5g60390) had an average expression stability M = 0.5 to 0.6, not much higher than those of the best Affymetrix probe sets we selected (Fig. 2; Supplemental Fig. 7). However, when primers specific for At5g60390 were used for qRT-PCR validation, the gene was found to be one of the least stably expressed of those tested (Fig. 5). One way to utilize the expression stability of this group of genes for normalization of qRT-PCR could be to generate a primer pair that amplifies all four transcripts simultaneously. Such an approach was used for AtACT2 and AtACT8, which display complementary patterns of expression, making their combined expression profile quasi-constitutive (Charrier et al., 2002).
In conclusion, this analysis revealed that hundreds of genes in the Arabidopsis genome are more stably expressed and at lower levels than traditional reference genes. This was confirmed for a subset of genes by qRT-PCR analysis. The gene-specific primer pairs developed here for novel reference genes will enable more accurate normalization and quantification of small- and medium-scale gene expression studies in Arabidopsis by qRT-PCR in the future. Probes to these reference genes will also aid normalization of transcripts using other methods, such as RNA gel-blot analysis. Finally, orthologs of these novel reference genes could serve the same purposes in other species.
MATERIALS AND METHODS
ATH1 Data Mining for Stably Expressed Genes
Affymetrix CEL files from each experimental series (see Table I; 721 ATH1 CEL files in total representing 323 experimental conditions) were processed using RMA software (Bolstad et al., 2003). The normalized data were delogged and biological replicate values (n = 3 for all developmental conditions; n = 2 for all others) averaged. MVs, sds, and CVs were subsequently calculated for each of the 22,750 probe sets in each experimental series, giving equal weighting of each condition. When available, the Present calls attributed by MAS5 software were used to flag the probe sets (typically around 12,000 to 13,000) that had Present calls in at least 80% of the arrays of a given experimental series. The genes represented by these probe sets were regarded as expressed and sorted by their CVs in ascending order. The top 100 genes (probe sets) of each experimental series were extracted into Supplemental Table I.
RT-PCR Primer Design and Test
Gene models, including information on exon/intron structure, 3′ and 5′ untranslated regions, and known splice variants for all investigated reference genes were downloaded from sequence viewer (SeqViewer) at TAIR (http://www.arabidopsis.org/). To facilitate RT-PCR measurement of transcripts of all investigated genes under a standard set of reaction conditions, qPCR primers were designed on these sequences using PrimerExpress 2.0 software (Applied Biosystems) and the following criteria: TM of 60°C ± 1°C and PCR amplicon lengths of 60 to 150 bp, yielding primer sequences with lengths of 20 to 30 nucleotides and guanine-cytosine contents of 35% to 55%. Primers were also designed to amplify close to the annotated 3′ end of the transcripts, to encompass all known splice variants, and at least one primer of a pair was designed to cover an exon-exon junction if possible (see Supplemental Table II). The specificity of the resulting primer pair sequences was checked against the Arabidopsis (Arabidopsis thaliana) transcript database using TAIR BLAST (http://www.arabidopsis.org/Blast/). Specificity of the primer amplicons was checked by melting-curve analysis performed by the PCR machine after 40 amplification cycles and by gel-electrophoretic analysis. To that effect, primer amplicons were resolved on 4% (w/v) agarose gels (3:1 HR agarose; Amresco) run at 4 V cm−1 in Tris-borate/EDTA buffer, along with a 50-bp DNA-standard ladder (Invitrogen GmbH). Identity of the short PCR products was checked by direct sequencing at AGOWA.
Plant Materials
Arabidopsis (Col-0) wild-type plants were grown under long-day conditions (16-h day/8-h night) on GS90 soil (Gebr. Patzer). The following tissue samples/organs were harvested at the given time after sowing: rosette leaves and shoot apices (4 weeks); cauline leaves and stems (5 weeks); flowers, green siliques, and old rosette leaves (6 weeks); and mature seeds (8 weeks). Shoot and root samples were also harvested from 14-d-old Arabidopsis (Col-0) wild-type plants that were grown vertically on half-strength Murashige and Skoog medium (Murashige and Skoog, 1962), supplemented with 0.5% (w/v) Suc and solidified with 0.7% agar, at 22°C under a 16-h-day (140 μmol m−2 s−1)/8-h-night regime. Abiotically stressed Arabidopsis (Col-0) wild-type seedling materials were produced in sterile liquid culture essentially as described by Scheible et al. (2004), and unstressed “control” seedlings were harvested after 9 d (full nutrition). Osmotically stressed seedlings were harvested after adding 100 mm NaCl or 200 mm mannitol during 3 h. Sulfur-, sugar-, or phosphorus-deprived seedlings were grown in sterile liquid cultures for 7 d in essentially the same full nutrition medium (Scheible et al., 2004) but with a concentration of the respective element that prevents accumulation in the seedlings (i.e. 200 μm phosphate, 200 μm sulfate, 0.5% Suc). After 7 d, the medium was replaced with medium lacking the respective nutrient and seedlings were allowed to grow for another 2 d, thereby developing molecular, metabolic, and visible phenotypes characteristic of sulfur, phosphorus, or sugar starvation. Harvesting was performed as described (Scheible et al., 2004). Leaf samples after cold-acclimation were harvested from Arabidopsis (Col-0) wild-type plants that were first grown for 6 weeks in ambient conditions (see above) and then transferred to a +4°C phytotron (16-h day/8-h night, light intensity 60 to 80 μE) for 2 weeks. Hormone-treated Arabidopsis (C24) wild-type plants were grown in liquid culture (0.5× Murashige and Skoog medium supplemented with 1% [w/v] Suc) with continuous shaking under a 16-h-day (140 μmol m−2 s−1, 22°C)/8-h-night (22°C) regime. After 7 d of culture, the medium was replaced for fresh one. One micromolar 2,4-dichlorophenoxyacetic acid (Duchefa) was added 3 d later, and samples were harvested 3 h after hormone addition. Control plants (without hormone treatment) were harvested in parallel.
RNA Extraction
Total RNA from most of the samples was isolated using TRIZOL reagent (Invitrogen GmbH) as described (http://www.Arabidopsis.org/info/2010_projects/comp_proj/AFGC/RevisedAFGC/site2RnaL). RNA from hormone-treated plant materials was prepared using the Invisorb Spin Plant RNA mini kit (Invitek GmbH), according to the manufacturer's protocol. RNA samples from green siliques and seeds were prepared using a “hot borate” method (Wan and Wilkins, 1994).
LjLb2 cRNA Template Preparation
One microgram of pSPORT1 plasmid containing the Lotus japonicus LjLb2 full-length cDNA sequence (GenBank accession no. BI416412) was linearized by BamHI digestion, phenol/chloroform treated, ethanol precipitated, and in vitro transcribed using T7 RNA polymerase with Ambion's mMESSAGE kit (catalog no. 1340), according to the manufacturer's instructions. Concentration of the cRNA was measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies), and the presence of a discrete 700-bp LjLb2 cRNA band was checked on an Agilent-2100 Bioanalyzer using RNA 6000 NanoChips (Agilent Technologies).
DNase I Digest, cDNA Synthesis, and Quality Control
RNA concentration and integrity were measured before and after DNase I digestion with a NanoDrop ND-1000 UV-Vis spectrophotometer (NanoDrop Technologies) and an Agilent-2100 Bioanalyzer using RNA 6000 NanoChips (Agilent Technologies). Altogether 150 μg of total RNA were digested with Turbo DNA-free DNase I (Ambion) according to the manufacturer's instructions. Absence of genomic DNA contamination in DNase I-treated samples was tested by PCR using primers (5′-TTTTTTGCCCCCTTCGAATC-3′ and 5′-ATCTTCCGCCACCACATTGTAC-3′) designed to amplify an intron sequence of a reference gene (At5g65080), and primers (5′-ACTTTCATCAGCCGTTTTGA-3′ and 5′-ACGATTGGTTGAATATCATCAG-3′) designed to amplify a 633-bp genomic fragment of the ACT2 gene (At3g18780). DNase I-treated RNA was subsequently spiked with 30 pg of LjLb2 cRNA template, and RT reactions were performed with SuperScript III reverse transcriptase (Invitrogen GmbH), according to the manufacturer's instructions. Amplification from LjLb2 cDNA was achieved with primers 5′-TTCGCGGTGGTTAAAGAAGC-3′ and 5′-TCCATTTGTCCCCAACTGCT-3′ (primer efficiency 108% ± 2%, amplicon length 61 bp, distance from annotated transcript 3′ end 212 bp). The 5′/3′ ratio of GAPDH cDNA [(1 + EGAPDH5′)CT GAPDH5′/(1 + EGAPDH3′)CT GAPDH3′; approximately 2ΔCT(GAPDH5′-GAPDH3′)] was determined by qRT-PCR with two primer pairs that amplify in the 5′ region (5′-TCTCGATCTCAATTTCGCAAAA-3′ and 5′-CGAAACCGTTGATTCCGATTC-3′, primer efficiency 104% ± 5%, amplicon length 61 bp, distance from annotated transcript 3′ end 1,279 bp) or 3′ region (primer specifications are given in Supplemental Table II).
Real-Time qRT-PCR Conditions and Analysis
PCR reactions were performed in an optical 384-well plate with an ABI PRISM 7900 HT sequence detection system (Applied Biosystems), using SYBR Green to monitor dsDNA synthesis. Reactions contained 3 μL 2× SYBR Green Master Mix reagent (Applied Biosystems), 1 μL of cDNA (1 ng/μL), and 200 nm of each gene-specific primer in a final volume of 6 μL. A master mix of sufficient cDNA and 2× SYBR Green reagent was prepared prior to dispensing into individual wells, to reduce pipetting errors and to ensure that each reaction contained an equal amount of cDNA. The following standard thermal profile was used for all PCR reactions: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. Amplicon dissociation curves, i.e. melting curves, were recorded after cycle 40 by heating from 60°C to 95°C with a ramp speed of 1.9°C min−1. Data were analyzed using the SDS 2.2.1 software (Applied Biosystems). To generate a baseline-subtracted plot of the logarithmic increase in fluorescence signal (ΔRn) versus cycle number, baseline data were collected between cycles 3 and 15. All amplification plots were analyzed with an Rn threshold of 0.1 to obtain CT values. PCR efficiency (E) was estimated from the data obtained from the exponential phase of each individual amplification plot and the equation (1 + E) = 10slope (Ramakers et al., 2003). The expression level of each gene of interest (GOI) is presented as 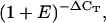 where
where 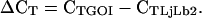
geNORM Analysis of Gene Expression Stability
To analyze gene expression stability, we used geNORM v.3.4 software (Vandesompele et al., 2002). Data imported to the software were RMA-normalized expression levels from ATH1 arrays or absolute expression levels calculated from qRT-PCR data by  for each of the tested reference genes in 18 to 20 different cDNA samples, where Emean is the value shown in Supplemental Table II and
for each of the tested reference genes in 18 to 20 different cDNA samples, where Emean is the value shown in Supplemental Table II and 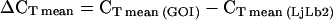 for each of three technical replicates. Results obtained from hormone-treated samples were excluded from the analysis when UBQ10 was considered and vice versa UBQ10 was omitted when the hormone-samples were included, because the UBQ10 primers do not work on UBQ10 cDNA from the Arabidopsis C24 ecotype.
for each of three technical replicates. Results obtained from hormone-treated samples were excluded from the analysis when UBQ10 was considered and vice versa UBQ10 was omitted when the hormone-samples were included, because the UBQ10 primers do not work on UBQ10 cDNA from the Arabidopsis C24 ecotype.
Acknowledgments
We thank Lutz Nover, Detlef Weigel, and all other researchers that contributed to the Deutsche Forschungsgemeinschaft-, GARNET-, RIKEN-, and National Science Foundation-funded AtGenExpress dataset, and who agreed to make the data freely available to the research community prior to publication. We are also grateful to Thomas Ott at Max-Planck Institute for Molecular Plant Physiology (MPI-MPP) for providing the LjLb2 cDNA clone, to Florian Wagner and his team at RZPD Berlin (German Resource Center for Genome Research, Berlin) for expert Affymetrix array service, including all steps from total RNA to data acquisition, and to Rajendra Bari, Monika Bielecka, Dirk Hincha, Tomasz Kobylko, Janina Lisso, Rosa-Maria Morcuende, Ana-Silvia Nita, Daniel Osuna, Armin Schlereth, and Wenming Zheng at MPI-MPP for donations of total RNA samples.
This work was supported by the Max-Planck Society and the Bundesministerium für Bildung und Forschung-funded project GABI Verbund Arabidopsis III Gauntlets (Carbon and Nutrient Signaling: Test Systems, and Metabolite and Transcript Profiles; 0312277A).
The online version of this article contains Web-only data.
References
- Altmann T, Weigel D, Nover L (2004) AtGenExpress—Ein multinational koordiniertes Programm zur Erforschung des Arabidopsis Transkriptoms. GenomXpress 3: 13–14 [Google Scholar]
- Andersen CL, Jensen JK, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64: 5245–5250 [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Brunner AM, Yakovlev IA, Strauss SH (2004) Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol 4: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR RT-PCR: trends and problems. J Mol Endocrinol 29: 23–29 [DOI] [PubMed] [Google Scholar]
- Charrier B, Champion A, Henry Y, Kreis M (2002) Expression profiling of the whole Arabidopsis shaggy-like kinase multigene family by real-time reverse transcriptase-polymerase chain reaction. Plant Physiol 130: 577–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker JS, Davies E (2003) Selection of candidate housekeeping controls in tomato plants using EST data. Biotechniques 35: 740–748 [DOI] [PubMed] [Google Scholar]
- Czechowski T, Bari RP, Stitt M, Scheible W-R, Udvardi MK (2004) Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J 38: 366–379 [DOI] [PubMed] [Google Scholar]
- Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A (2004) Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 37: 112–119 [DOI] [PubMed] [Google Scholar]
- Gachon C, Mingam A, Charrier B (2004) Real-time PCR: what relevance to plant studies? J Exp Bot 55: 1445–1454 [DOI] [PubMed] [Google Scholar]
- Goidin D, Mamessier A, Staquet M-J, Schmitt D, Berthier-Vergnes O (2001) Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and β-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and non-invasive human melanoma cell subpopulations. Anal Biochem 295: 17–21 [DOI] [PubMed] [Google Scholar]
- Holland MJ (2002) Transcript abundance in yeast varies over six orders of magnitude. J Biol Chem 277: 14363–14366 [DOI] [PubMed] [Google Scholar]
- Kim B-R, Nam H-Y, Kim S-U, Kim S-I, Chang Y-J (2003) Normalization of reverse transcription quantitative-PCR with housekeeping genes in rice. Biotechnol Lett 25: 1869–1872 [DOI] [PubMed] [Google Scholar]
- Lee PD, Sladek R, Greenwood CMT, Hudson TJ (2001) Control genes and variability: absence of ubiquitous reference transcripts in diverse mammalian expression studies. Genome Res 12: 292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurin C, Andres C, Aubourg S, Bellaoui M, Bitton F, Bruyere C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16: 2089–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15: 473–497 [Google Scholar]
- Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol Lett 26: 509–515 [DOI] [PubMed] [Google Scholar]
- Radonić A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A (2004) Guideline for reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun 313: 856–862 [DOI] [PubMed] [Google Scholar]
- Ramakers C, Ruijter JM, Deprez RH, Moorman AF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 13: 62–66 [DOI] [PubMed] [Google Scholar]
- Redman JC, Haas BJ, Tanimoto G, Town CD (2004) Development and evaluation of an Arabidopsis whole genome Affymetrix probe array. Plant J 38: 545–561 [DOI] [PubMed] [Google Scholar]
- Sanderfoot AA, Raikhel NV (2002) The secretory system of Arabidopsis. In CR Somerville, EM Meyerowitz, eds, The Arabidopsis Book. American Society of Plant Biologists, Rockville, MD, doi/10.1199/tab.0098, http://www.aspb.org/publications/arabidopsis/
- Scheible W-R, Morcuende R, Czechowski T, Fritz C, Osuna D, Palacios-Rojas N, Schindelasch D, Thimm O, Udvardi MK, Stitt M (2004) Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol 136: 2483–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann J (2005) A gene expression map of Arabidopsis development. Nat Genet 37: 501–506 [DOI] [PubMed] [Google Scholar]
- Shen L, Gong J, Caldo RA, Nettleton D, Cook D, Wise RP, Dickerson JA (2005) BarleyBase—an expression profiling database for plant genomics. Nucleic Acids Res 33: D614–D618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalle J, Vierstra RD (2004) The ubiquitin 26s proteasome proteolytic pathway. Annu Rev Plant Biol 55: 555–590 [DOI] [PubMed] [Google Scholar]
- Steinhauser D, Usadel B, Luedemann A, Thimm O, Kopka J (2004) CSB.DB: a comprehensive systems-biology database. Bioinformatics 20: 3647–3651 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Higgins PJ, Crawford DR (2000) Control selection for RNA quantitation. Biotechniques 29: 332–337 [DOI] [PubMed] [Google Scholar]
- Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Henne G, Grisar T, Igout A, Heinen E (1999) Housekeeping genes as internal standards: use and limits. J Biotechnol 75: 197–200 [DOI] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939 [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 37: RESEARCH0034 [DOI] [PMC free article] [PubMed]
- Wan C-Y, Wilkins TA (1994) A modified hot-borate method significantly enhances the yield of high-quality RNA from cotton Gossypium hirsutum L. Anal Biochem 223: 7–12 [DOI] [PubMed] [Google Scholar]
- Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol 136: 2621–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]