

Abstract
In this study, two series of kojic acid triazole hybrids, namely 6a–6p and 13a–13t, were designed and synthesized. Subsequently, their biological activities including anti-tyrosinase, antioxidant, and as anti-browning effects were investigated. The results showed that most of compounds demonstrated excellent inhibitory effect against mushroom tyrosinase compared with standard reference drug (kojic acid, IC50 = 26.090 µM). Of particular note, 13t proved to be the most potent tyrosinase inhibitor with an IC50 value as low as 1.363 µM. Further kinetic inhibition studies suggested that 13t presented such powerful anti-tyrosinase efficacy by functioning as a mixed-type inhibitor (Ki = 0.3647 µM, Kis = 0.8492 µM). Moreover, the results from molecular docking and fluorescence quenching studies revealed that 13t’s inhibitory effect on tyrosinase stemmed from its ability to directly bind to the active site of mushroom tyrosinase. Besides, the antioxidant activity, anti-browning effect, and cytotoxicity of 13t were accordingly investigated, all yielding highly satisfactory results. Collectively, these findings position 13t as a highly promising candidate, providing a valuable molecular framework for the development of novel, efficient, and safe tyrosinase inhibitors endowed with potent antioxidant and anti-browning capabilities.
Keywords: Kojic acid-triazole hybrids, Tyrosinase inhibitory activity, Inhibition mechanism, Antioxidant activity, Anti-browning effect
Subject terms: Biochemistry, Drug discovery
Introduction
Tyrosinase, a metalloenzyme incorporating two active copper ions, is also referred to as polyphenol oxidase1. The crystal structures of mushroom tyrosinase manifest an H2L2 tetramer structure, with a molecular mass of approximately 120 kDa. Notably, the H subunit constitutes the tyrosinase domain, which encompasses multiple loops, α-helices, and β-strands. In contrast, the L subunit features a lectin-like fold composed of β-strands. The active site of tyrosinase is situated within two antiparallel α-helices of the H subunit, where the two copper ions are housed. These copper ions engage in robust interaction with three histidine residues2–4. It is widely acknowledged that tyrosinase plays a pivotal role in melanin pigment biosynthesis, catalyzing two distinct reactions. Firstly, it facilitates the conversion of monophenols into o-diphenols (a process mediated by monophenolase activity), and secondly, it enables the transformation of o-diphenols into o-quinones (driven by diphenolase activity). In mammals, melanin serves as the pigment dictating the color for skin and hair5,6. Appropriate melanin production not only shields human skin from the harmful effects of ultraviolet rays but also mitigates the damage inflicted by numerous toxic and noxious substances. However, abnormal levels of melanin production can lead to aesthetic concerns in humans, as well as hyperpigmentary disorders, such as post-inflammatory hyperpigmentation, maturational dyschromia, periorbital hyperpigmentation, melasma, age spots, freckles, and even melanoma7,8. Moreover, in the context of neurological processes, tyrosinase catalyzes the production of neuromelanin through the oxidation of dopamine to dopaquinone species. Excessive generation of dopaquinones, unfortunately, results in neuronal damage and cell death, thereby implicating tyrosinase in the neurodegenerative processes associated with Parkinson’s and Huntington’s diseases9. Shifting the focus to the food industry, enzymatic browning of fruits and vegetables poses a significant challenge during storage and handling. This phenomenon leads to rapid degradation, adversely affecting the storage lifespan, visual appearance, taste, safety, and nutritional value of fresh produce10,11. Finally, recent studies have shown that tyrosinase is also specifically connected to three metabolic processes in insects, namely cuticle sclerotization, protective encapsulation, and melanization of invading organisms as well as wound healing12,13.
Considering the key roles of tyrosinase as shown above, the development of tyrosinase inhibitors should have broad potential for utilization in multiple fields, such as being used as insecticides in the agricultural sector, anti-browning agents for vegetables and fruits, drugs for the treatment of hyper-pigmentation disorders, as well as whitening agents in cosmetics. In addition, UV irradiation results in the overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which can stimulate the biosynthesis of melanin7. Antioxidant systems are capable of removing free radicals from the body and may also limit excessive melanin production14. Up to now, many tyrosinase inhibitors from both natural and synthetic sources have been reported; however, their application is very limited due to several drawbacks including toxicity, lack of stability, off-odours, and economic unsustainability15,16. For example, kojic acid, an organic acid with an antibacterial effect, is produced by the aerobic fermentation of glucose by Aspergillus and Candida at 30–32 °C. It serves as the key raw material for the preparation of biologically active derivatives. Moreover, it also functions as a tyrosinase inhibitor and is commonly used as a potent whitening agent in the pharmaceutical industry17. However, the use of kojic acid in cosmetics is currently limited due to its cytotoxicity, chemical instability, and low lipophilicity, which may result in skin irritations18. Besides, several adverse effects including genotoxic, hepatocarcinogenic, and allergic dermatitis have also been involved during its clinical use19. Subsequently, another well-known tyrosinase inhibitor, hydroquinone, can lead to erythema, stinging, colloid milium, allergic contact dermatitis, nail discoloration, and paradoxical postinflammatory hypermelanosis20. Based on these severe side effects, recent EU (European Union) cosmetic regulations have restricted or even banned the use of kojic acid and hydroquinone as skin whitening agents21.
To overcome the above-mentioned shortcomings of kojic acid, the hydroxypyranone structure of kojic acid as a good pharmacophoric unit for the design of new tyrosinase inhibitors22,23. In fact, several kojic acid derivatives have been developed as the new generation of anti-browning agents24–26. In addition, 1,2,3-triazole is a highly attractive and privileged five-membered heterocyclic scaffold with several salient features27. For instance, it has been widely applied as a potent hydrogen bond acceptor to dynamically enhance the binding affinity with the corresponding target proteins in the field of new drug discovery28. Besides, 1,2,3-Triazole-based derivatives have therapeutic potential for antioxidant, antitumor, antifungal, anti-HIV, anti-inflammatory, antimicrobial, antitubercular, antiparasitic, tyrosinase inhibitory and antiviral activities29. Given these advantages, and inspired by the principle of active skeleton hybridization in compound design, we reasoned that the proper hybridization of kojic acid and 1,2,3-triazole motif might be feasible for developing a new tyrosinase inhibitor with an improved therapeutic index.
As part of ongoing efforts to discover potent tyrosinase inhibitors with minimum side effects, in the present study, two series of kojic acid-triazole hybrids were designed and synthesized. Subsequently, their structure-activity relationships regarding the inhibition of mushroom tyrosinase were primarily discussed. Besides, the inhibition mechanism, the mode of action, the antioxidant activity, the anti-browning effect as well as the cytotoxicity of the optimized compound were also systematically investigated.
Results and discussion
Chemistry
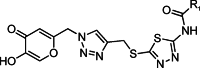
The synthesis route of kojic acid-triazole hybrids is shown in Scheme 1. Briefly, kojic chloride was synthesized by the reaction of kojic acid and thionyl chloride in CH₂Cl₂ at room temperature for 8 h. Then, kojic chloride reacted with sodium azide to give azido kojic acid. Propargyl bromide was reacted with 2-amino-5 -mercapto-1,3,4-thiadiazole in the presence of KOH to provide propargyl thiadiazole. Subsequently, the click reaction of propargyl thiadiazole with azido kojic acid was carried out to yield the key intermediate (kojic acid-triazole hybrid species). Compounds 6a–6p were prepared by stirring the mixture of the corresponding kojic acid-triazole, aromatic acid species, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI), and 1-Hydroxybenzotriazole (HOBT) in N, N-Dimethylformamide (DMF) at room temperature and were purified by preparative chromatography. Subsequently, alkynyl ether-substituted aromatic aldehyde or ketone was readily synthesized through a substitution reaction between hydroxyl-substituted aromatic aldehyde/ketone and propargyl bromide in the presence of K₂CO₃. Then, the click reaction with azido kojic acid under the Cu-catalyzed conditions was performed to produce the final compounds 13a–13t in good yields. The synthesized compounds were characterized by ¹H-NMR, ¹³C-NMR, IR and HR-MS.
Scheme 1.

General synthetic route of kojic acid-triazole hybrids
Tyrosinase inhibitory activities
With the synthetic compounds (6a–6p, 13a–13t) in hand, their tyrosinase inhibitory activities were evaluated using L-DOPA as the substrate, with kojic acid selected as a positive reference for comparison. In the case of the 6 series (Table 1), a remarkable trend was observed. With the exception of compounds 6b–6d, all the remaining compounds demonstrated potent inhibitory activities against mushroom tyrosinase. Their IC50 values ranged from 3.289 ± 0.011 µM to 20.59 ± 0.037 µM, outperforming the positive control, kojic acid, which had an IC50 value of 26.09 ± 0.027 µM. Notably, compound 6 l exhibited the most prominent tyrosinase inhibitory potency, with an IC50 value of 3.289 ± 0.011 µM, which was over eight times more potent than that of kojic acid. Generally speaking, those compounds within the 6 series that bore no substituent on the aryl ring exhibited a moderate level of activity. Furthermore, the introduction of an amide group derived from methyl-substituted phenylacetic acid appeared to significantly enhance the inhibitory activity. This was clearly evidenced by the fact that 6j–6 l exhibited potent inhibitory activities against tyrosinase, while 6b–6d showed no detectable inhibitory effects on the enzyme. A comparative analysis of 6 h and 6k revealed that nearly equivalent inhibition effects were achieved when the benzene ring was substituted with a methoxyl group. In contrast, when one hydrogen atom on the benzene ring was replaced with halogen atoms (–F, –Cl, –Br) as seen in 6e–6 g and 6 m–6p, the inhibitory effect underwent significant and dynamic alterations. Moreover, 6j–6 l demonstrated potent but distinct inhibitory activities, with IC50 values of 17.26 ± 0.038 µM, 13.81 ± 0.066 µM, and 3.289 ± 0.011 µM, respectively. This pattern suggests that the presence of a methyl group at the para-position of the aromatic ring is more favorable compared to its placement at the ortho- and meta-positions. It is worth noting that the compound featuring a chlorine substitution exhibited slightly superior tyrosinase inhibitory potency relative to those compounds with other halogen substituents. This implies that the inhibition effect is likely influenced not only by the electronic properties of the substituents but also by the steric size of the halogen atoms.
Table 1.
Tyrosinase inhibitory activity of the Kojic acid-triazole derivatives.
| |||||
|---|---|---|---|---|---|
| Cpds | R1 | IC50 (µM) | Cpds | R1 | IC50 (µM) |
| 6a | Ph | 20.590 ± 0.037 | 6i | 4-NH2-Ph | 16.310 ± 0.016 |
| 6b | 2–CH3-Ph | > 200 | 6j | 2–CH3-Bn | 17.260 ± 0.038 |
| 6c | 3–CH3-Ph | > 200 | 6k | 3–CH3-Bn | 13.810 ± 0.066 |
| 6d | 4–CH3-Ph | > 200 | 6l | 4–CH3-Bn | 3.289 ± 0.011 |
| 6e | 4-Cl-Ph | 6.073 ± 0.035 | 6m | 4-Cl-Bn | 10.170 ± 0.032 |
| 6f | 4-Br-Ph | 6.701 ± 0024 | 6n | 4-Br-Bn | 12.360 ± 0.039 |
| 6g | 4-F-Ph | 8.240 ± 0.023 | 6o | 4-F-Bn | 11.980 ± 0.022 |
| 6h | 4-OCH3-Ph | 5.559 ± 0.078 | 6p | 4-OCH3-Bn | 3.832 ± 0.005 |
| Kojic acid | – | 26.090 ± 0.027 | |||
With respect to the 13 series (Table 2), all compounds demonstrated more potent inhibitory activities against mushroom tyrosinase, with IC50 values ranging from 1.363 ± 0.003 µM to 9.908 ± 0.044 µM, in contrast to kojic acid (whose IC50 value was 26.09 ± 0.027 µM). Particularly, 13t exhibited the most potent tyrosinase inhibitory activity among the 13 series compounds, possessing an IC50 value of 1.363 ± 0.003 µM. Subsequently, the structure–activity relationships (SARs) of these compounds were rationally discussed and are summarized as follows:
Table 2.
Tyrosinase inhibitory activity of the kojic acid-triazole derivatives.
|
| |||||||
|---|---|---|---|---|---|---|---|
| Cpds | R 2 | R 3 | IC50 (µM) | Cpds | R 2 | R 3 | IC50 (µM) |
| 13a | H | H | 2.474 ± 0.023 | 13k | CH3 | H | 1.517 ± 0.010 |
| 13b | CH3 | H | 4.604 ± 0.046 | 13l | CH3 | 3-tBu | 4.976 ± 0.057 |
| 13c | H | H | 4.546 ± 0.062 | 13m | H | 4-Cl | 4.571 ± 0.004 |
| 13d | CH3 | H | 9.908 ± 0.044 | 13n | CH3 | 4–CH3 | 6.818 ± 0.027 |
| 13e | CH3 | 3–CH3 | 1.737 ± 0.048 | 13o | CH3 | 5–CH3 | 1.831 ± 0.042 |
| 13f | H | 4-F | 5.961 ± 0.036 | 13p | H | 4-Br | 4.231 ± 0.029 |
| 13g | H | H | 2.101 ± 0.078 | 13q | CH3 | 4-Cl | 3.029 ± 0.059 |
| 13h | H | 5–CH3 | 4.676 ± 0.025 | 13r | CH3 | 4-Br | 3.325 ± 0.057 |
| 13i | H | 4–CH3 | 6.895 ± 0.042 | 13s | CH3 | 4-F | 2.582 ± 0.049 |
| 13j | H | 4-OCH3 | 2.817 ± 0.007 | 13t | CH3 | 4-OCH3 | 1.363 ± 0.003 |
| Kojic acid | / | / | 26.090 ± 0.027 | ||||
The position of the carbonyl group on the phenyl ring exerted an obvious effect in determining the tyrosinase inhibitory activity. In the current investigation, the ortho position was found to be optimal. This was evidenced by comparing 13a with 13c and 13g, as well as 13b with 13d and 13k. For para-substituted carbonyl species, the introduction of the 3-methyl substituent was beneficial for enhancing the inhibitory activity (as demonstrated by the comparison between 13d and 13e). This result might be related to the hydrophobicity of the compound.
The introduction of a methyl substituent into the carbonyl group led to a noticeable variation in the inhibitory effect. For ortho-substituted carbonyl species, the introduction of the methyl substituent was advantageous for enhancing the inhibitory activity (as seen in the comparison of 13k with 13g). However, the opposite trend was observed for either meta- or para-substituted carbonyl species (such as the comparisons between 13b and 13a, and 13d and 13c).
By examining the data from 13g to 13j, 13m, and 13p, it was suggested that the introduction of relevant substituent forms, including methyl, methoxyl, and halogen-substituted (e.g., –Cl, –Br) groups, seemed to have little impact on the tyrosinase inhibitory effect. In many cases, no enhancement or even a reduction in inhibitory activities was frequently observed.
Interestingly, when the ortho-substituted keto carbonyl group remained fixed, the introduction of an additional substituent onto the phenyl ring had a significant impact on the inhibitory activity against tyrosinase. This ultimately led to the discovery of the most potent inhibitor within the series, 13t, which boasted an IC50 value of 1.363 ± 0.003 µM. Significantly, this finding implies that an appropriately enlarged size of the substituents on the phenyl ring could enhance the overall interaction between the enzyme and the inhibitor, thereby augmenting the inhibitory potency.
Kinetic studies
Considering that 13t exhibited the highest tyrosinase inhibitory activity among the tested kojic acid-triazole hybrids, we proceeded to further explore its inhibition mechanism. Subsequently, the inhibitory type of 13t on mushroom tyrosinase was identified through the Lineweaver–Burk double reciprocal plots, following treatment with varying concentrations of compound 13t and the substrate.
As illustrated in Fig. 1A, the plots of 1/V against 1/[S] yielded a family of straight lines with diverse slopes. Notably, the slope of the curve exhibited a downward trend as the inhibitor concentration decreased, and all of the straight lines intersected at a single point within the second quadrant. This pattern strongly suggested that compound 13t functions as a mixed-type inhibitor. To determine the inhibition constant (Ki) of the compound, a plot was constructed with the slope values plotted against the concentrations of compound 13t. Additionally, a plot of the vertical intercept (1/Vm) against the concentrations of compound 13t was generated to obtain the Kis value of the compound. As depicted in Fig. 1B,C, the Ki value and the Kis value of the compound were calculated to be 0.3647 µM and 0.8492 µM, respectively.
Fig. 1.

Kinetic Inhibition Measurement of Compound 13t. The inhibition type of 13t was demonstrated through the Lineweavere–Burk plot. (A) In this figure, the concentrations of 13t were 0 µM, 2 µM, and 4 µM, respectively. (B) The secondary replot was constructed by plotting the slope against the concentrations of 13t, which was used to determine the inhibition constant Ki. (C) The secondary replot in this case involved plotting the intercept against the concentrations of 13t, aiming to define the inhibition constant Kis.
Molecular electrostatic potential (MEP)
MEP is an indispensable tool for chemical structure of compound in biological study which used to investigate the biological discovery such as enzyme-substrate, ligand-substrate and drug receptor. The MEP map of compound 13t was figured out in Fig. 2. The yellowish color is present electrophile site, blue color present nucleophilic site. The negative charge is predominantly located on the oxygen atom of carbonyl group, 4,5-oxygen atoms of kojic acid and 1,2-nitrogen atoms of triazole ring.The most positive region of the molecule is the –CH hydrogen on the triazole ring.
Fig. 2.

MEPM of compound 13t.
Frontier molecular orbitals analysis
The FMOs helpful to describe the stability and chemical reactivity of organic compound. The highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) are related to ionization potential and electron affinity, respectively. The HOMO and LUMO of compound 13t are shown in Fig. 3.The larger ΔEgap value means the compound is more stable than those with less ΔEgap value. ΔEgap value of the compound 13t is 4.68e V, and ΔEgap value of the kojic acid is 5.019e V, indicating a higher reactivity of compound 13t than kojic acid.
Fig. 3.

HOMO-LUMO of compound 13t.
Fluorescence quenching
Fluorescence quenching occurs when certain small-molecule compounds bind to proteins, thereby affecting the fluorescence intensity of the fluorophore. Indeed, to date, fluorescence spectroscopy has evolved into a crucial research tool for obtaining valuable information regarding conformational changes of enzymes and probing the possible interaction modes. Consequently, the fluorescence spectra of tyrosinase in the presence of different concentrations of 13t were investigated. As demonstrated in Fig. 4A, the fluorescence intensity of tyrosinase decreased dramatically with increasing concentrations of 13t, clearly indicating the potent quenching effect of 13t on the fluorescence of tyrosinase. Moreover, no significant red shift or blue shift was observed in the fluorescence emission peak of tyrosinase following treatment with 13t. This result implies that the interaction between 13t and tyrosinase did not alter the hydrophobic environment in the vicinity of the chromophore tryptophan residues.
Fig. 4.

(A) This panel illustrates the alterations in tyrosinase fluorescence at varying concentrations of compound 13t (with the excitation wavelength, λex, set at 280 nm and the temperature maintained at 298 K). The final concentration of the enzyme was 0.1 mg/mL. For the curves shown, from top to bottom respectively, the concentrations of compound 13t were 0 µM, 10 µM, 20 µM, 30 µM, 40 µM, 50 µM, 60 µM, 70 µM, 80 µM, and 90 µM (a-j). (B) the Stern-Volmer plots of 13t and tyrosinase are presented at temperatures of 298 K, 303 K, and 310 K.
As depicted in Fig. 4B, the Stern-Volmer plots of 13t with tyrosinase at 298, 303, and 310 K exhibited a favorable linear relationship, signifying a single quenching type of 13t. Moreover, it was also indicated that the predominant quenching mechanism of compound 13t was likely to be dynamic quenching, as the quenching constant increased with a rise in temperature. The quenching of the intrinsic fluorescence furnished direct and unequivocal evidence that the inhibitor 13t was capable of binding to tyrosinase. The binding of 13t to tyrosinase induced a change in the microenvironment surrounding the fluorophore. The detailed values of Ksv and Kq are summarized in Table 3.
Table 3.
Stern–Volmer equation parameters for the interaction between 13t and tyrosinase.
| T/K | R 2 | Ksv/(L/µmol) | Kq/( L/µmol/s) |
|---|---|---|---|
| 298 | 0.98871 | 0.01952 | 1.952 × 106 |
| 303 | 0.99326 | 0.02478 | 2.478 × 106 |
| 310 | 0.99939 | 0.02823 | 2.823 × 106 |
Molecular docking study
To clarify whether 13t binds directly to tyrosinase, we conducted a molecular docking study using Discovery Studio 2019 software (Fig. 5A–D). As demonstrated in Fig. 5A and 13t was identified as a mixed-type inhibitor via kinetic assay. In Fig. 5, the results revealed that the kojic acid fragment within 13t penetrated the bottom of the tyrosinase activity site pocket. Notably, it not only overlapped precisely with the original ligand in the crystal structure but also showed a remarkable overlap with kojic acid itself. The carbonyl group of the kojic acid fragment interacted with copper ions, maintaining a bond length of 2.4 Å. Meanwhile, the 5-hydroxyl substituent on the pyranone ring formed a hydrogen bond with Met280, with a bond length of 2.9 Å. The triazole ring and the acetyl group of the inhibitor 13t likely occupied the spacious cavity surrounding the active site. Moreover, the carbonyl and methoxyl groups on the benzene ring engaged in significant hydrogen bond interactions with Arg268 and Ser282, boasting bond lengths of 2.0 Å and 2.3 Å, respectively. Overall, the docking simulation results strongly suggested that, similar to kojic acid, 13t, being the most potent inhibitor among those studied, could bind to the active site of mushroom tyrosinase, thereby directly suppressing tyrosinase activity.
Fig. 5.

(A) Docking model showing the best binding pose of compound 13t to tyrosinase. (B) Superposition of 13t-bound with kojic acid in mushroom tyrosinase. (C) The simulated docking model depicting the interactions of compound 13t with the active site of mushroom tyrosinase using AutoDock4.2. (D) The schematic representation of the interactions of compound 13t within the binding pocket of mushroom tyrosinase, which is derived from the docking model. Dashed lines indicate the bond distances between the interacting functionalities of the ligand and the receptor. The legends inset display the type of interaction between the ligand atoms and the amino acid residues of the protein.
Anti-browning study
Among the synthesized target compounds, 13t emerged as the candidate with the best tyrosinase inhibitory activity. Thus, we proceeded to evaluate its anti-browning effect on apples, with kojic acid and Vitamin C (Vc) serving as positive controls. Figure 6A illustrates the change in color values of post-cut apples treated with 13t at concentrations of 25 µM, 50 µM, and 100 µM, along with those of the blank control group, the kojic acid-treated group (50 µM), and the Vc-treated group (50 µM) during storage at 5 °C. After being stored at 5 °C for 4 days, the L* values of the 13t groups at 25 µM, 50 µM, and 100 µM, as well as the Vc group, the kojic acid group, and the blank control group were 65.91, 66.59, 67.87, 63.58, 63.04, and 38.44, respectively. Notably, the L* values of the different concentration groups of compound 13t were significantly higher than that of the blank control group and were also slightly greater than those of both the Vc and kojic acid groups. Throughout the measurement period spanning 0 to 4 days, the browning extent of the apple slices in the treatment groups was conspicuously lower than that in the blank control group, the kojic acid group, and the Vc group. Moreover, after 4 days of storage at 5 °C, the color of the apple slices in the compound 13t group was lighter compared to that of the positive control groups (Fig. 6B). These results provide further evidence suggesting that compound 13t holds promise as a potent anti-browning agent.
Fig. 6.

(A) Photographs are presented as follows: fresh-cut apples prior to treatment, control fresh-cut apples after being stored at 5 °C for 4 days, and fresh-cut apples treated with 13t at concentrations of 25 µM, 50 µM, and 100 µM, respectively, after 4 days of storage at 5 °C. (B) Illustrates the effects of compound 13t in suppressing the browning of fresh-cut apples during a 4-day storage period at 5 °C.
Antioxidant activities and cell viability assessments of 13t
Subsequently, the 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging ability and the 2,2’-azino-bis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) free radical scavenging ability of 13t were evaluated, with vitamin C serving as the reference drug. As depicted in Fig. 7A,B and 13t demonstrated potent scavenging activities against both DPPH and ABTS radicals, exhibiting IC50 values of 57.90 ± 0.28 µM and 45.64 ± 0.25 µM, respectively. Remarkably, its efficacy in this regard surpassed that of the positive control drug, vitamin C.
Fig. 7.

(A) The DPPH free radical scavenging ability of 13t and Vitamin C (Vc) was determined across a range of concentrations, from 0.78125 M to 200 µM, and the results are presented on a log10 scale. (B) The ABTS free radical scavenging ability of 13t and Vc was likewise measured at multiple concentrations spanning from 0.78125 to 200 µM and is presented on a log10 scale as well. (C) The cell viability of 13t in relation to human umbilical vein endothelial cells (HUVEC) is depicted.
Encouraged by the foregoing results and with the aim of assessing the safety of 13t, an MTT assay was carried out on human umbilical vein endothelial cells (HUVEC), a normal cell line, following a 24-h treatment with 13t. As illustrated in Fig. 7C, this compound exhibited no discernible toxicity even at a concentration as high as 32 µM, revealing a good safety window of 13t for the treatment of future tyrosinase-mediated disorders.
Prediction of ADMET properties
The ADMET analysis reveals that the compound demonstrates favorable aqueous solubility (logS = − 1.784, categorized as optimal) and moderate absorption potential, meeting the requirements for oral bioavailability (Table 4). Key distribution parameters include moderate lipophilicity (ALogP98 = 0.313) and a polar surface area (PSA = 113.957 Å2), suggesting acceptable membrane permeability. The compound is classified as a non-inhibitor of CYP2D6 and exhibits low plasma protein binding, indicating metabolic stability and a predominance of free form available in circulation. However, concerns arise due to predicted hepatotoxicity and atypical blood-brain barrier (BBB) penetration (undefined classification), which may limit its application in CNS-targeted therapies. These findings highlight the need for further optimization to address hepatotoxic risks and clarify BBB interaction mechanisms.
Table 4.
The ADMET properties of the compound 13t.
| Property | Value | Description | Drug-likeness |
|---|---|---|---|
| Solubility | − 1.784 | The base 10 logarithm of the molar solubility as predicted by the regression | |
| Solubility_Level | 4 | Categorical solubility level | Yes, optimal |
| PSA_2D | 113.957 | Fast polar surface area | |
| AlogP98 | 0.313 | ALogP | |
| Absorption_Level | 0 | Categorical absorption level | Good absorption |
| Blood brain barrier | Base 10 logarithm of (brain concentration)/(blood concentration) as predicted by a robust (least-median-of-squares) regression derived from literature in vivo brain penetration data | ||
| BBB_Level | 4 | Categorical level | Undefined-Outside 99% confidence ellipse |
| EXT_CYP2D6 | − 6.93099 | Bayesian score from the model | |
|
EXT_CYP2D6 #Prediction |
False | The classification whether a compound is an CYP2D6 inhibitor using the cutoff Bayesian score of 0.161 (obtained by minimizing the total number of false positives and false negatives) | |
| EXT_Hepatotoxic | − 0.184948 | Bayesian score from the model | |
|
EXT_Hepatotoxic #Prediction |
True | The classification whether a compound is hepatotoxic using the cutoff Bayesian score of − 4.154 (obtained by minimizing the total number of false positives and false negatives) | |
| EXT_PPB | − 3.61468 | Bayesian score from the model | |
|
EXT_PPB #Prediction |
False | The classification whether a compound is highly bounded ( > = 90% bound) to plasma proteins using the cutoff Bayesian score of − 2.209 (obtained by minimizing the total number of false positives and false negatives) |
Conclusion
In summary, a series of kojic acid-triazole hybrids were meticulously designed, synthesized, and evaluated in terms of their tyrosinase inhibitory activities, antioxidant capabilities, and anti-browning effects. This process led to the discovery and identification of the lead compound, 13t, which demonstrated the most remarkable tyrosinase inhibitory effect, with an IC50 value of 1.363 ± 0.003 µM. Subsequently, the structure–activity relationships (SARs) of these compounds were rationally and comprehensively discussed. Furthermore, additional comprehensive biological evaluations unveiled the following characteristics of 13t: Firstly, 13t functions as a mixed-type inhibitor, with Ki and Kis values of 0.3647 µM and 0.8492 µM, respectively. Secondly, dynamic quenching appears to be the predominant quenching mechanism of 13t. Thirdly, 13t is capable of binding to the active site of mushroom tyrosinase, thereby directly suppressing tyrosinase activity. Fourthly, 13t exhibits a potent anti-browning effect on fresh-cut apples. Lastly, 13t displays no evident toxicity towards human normal cells, even at a relatively high concentration. Collectively, all these findings suggest that compound 13t holds great promise as a candidate for the future development of highly effective and safe tyrosinase inhibitors as well as anti-browning agents.
Experimental
General
All the reactions described below were monitored by TLC (thin layer chromatography), which was carried out with 60F254 silicone plates. Melting points were determined using an uncorrected SGW-X4 melting point apparatus. 1H NMR and13C NMR spectra were obtained on a Bruker 400 MHz at 25 °C in DMSO-d6 using tetramethyl silane (TMS) as an internal standard. The values of coupling constant (J) and chemical shift were measured in hertz (Hz) and parts per million (ppm), respectively. The abbreviations used in 1H NMR as follow: s (singlet), d (doublet), t (triplet), and m (multiplet). FT-IR spectra were recorded on a Nicolet-iS5 spectrophotometer (KBr disks). Mass spectral ESI measurements were executed on Thermo Fisher Scientific Q Exactive Focus LC/MS instrument. UV-2600 spectrophotometer (Shimadzu Corporation, Tokyo, Japan) was used to record UV spectra. The fluorescence spectra of the selected compound were inspected on Cary Eclipse G9800A fluorescence spectrophotometer (Agilent Technologies Co). The colorimeter is CR-400 Minolta chronometer instrument (Konica Minolta, Osaka, Japan). Purity for target compounds was measured by high-performance liquid chromatography (HPLC) with an Thermo Scientific UltiMate 3000. Tyrosinase, L-3,4-dhydroxyphenylalanine (L-DOPA), 1,1-diphenyl-2-picrylhydrazyl radical 2,2-diphenyl-1- (2,4,6-trinitrophenyl) hydrazyl (DPPH), 2, 2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), dimethyl sulfoxide (DMSO), dimethyl sulfoxide-d6 (DMSO-d6), and kojic acid were obtained from Sigma–Aldrich Chemical Co (Shanghai, China). Other chemicals were purchased from commercial suppliers.
Synthesis of Kojic acid-triazole derivatives
Synthesis of 2-(chloromethyl)-5-hydroxy-4 H-pyran-4-one
At room temperature, to a rapidly stirring suspension of kojic acid (8.52 g, 60 mmol) in dichloromethane (CH₂Cl₂, 60 mL), thionyl chloride (10 mL, 1.38 mol) was added dropwise over a period of 30 min. Subsequently, the reaction mixture was continuously stirred at room temperature for 6–8 h. The progress of the reaction was monitored by thin-layer chromatography (TLC) using a solvent system of petroleum ether and ethyl acetate in a volume ratio of 1:1 (v/v). Once the reaction was complete, the precipitates were separated by filtration and then washed with acetone. As a result, kojic chloride was obtained as a white solid with an 85% yield (8.16 g).
Synthesis of 2-(azidomethyl)-5-hydroxy-4 H-pyran-4-one
A mixture of kojic chloride (8.03 g, 50.0 mmol) and sodium azide (3.25 g, 50.0 mmol) in 60 mL of DMF was stirred for 8 h at room temperature. The reaction process was monitored by TLC using a solvent system of petroleum ether and ethyl acetate in a volume ratio of 1:1 (v/v). After the reaction was complete, water (60 mL) was slowly added to the reaction mixture. The mixture was extracted with ethyl acetate (3 × 50 mL), the combined organic phase was washed three times with water, and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure using a rotary evaporator. Azido kojic acid was obtained as a yellow solid in 61.7% yield (5.15 g) .
Synthesis of 5-(prop-2-yn-1-ylthio)-1,3,4-thiadiazol-2-amine
Propargyl bromide (2.476 g, 20.0 mmol) was added to a mixture of 2-amino-5-mercapto-1,3,4-thiadiazole (2.664 g, 20.0 mmol) and KOH (1.124 g, 20.0 mmol) in 60 mL of ethanol. The reaction mixture was stirred under reflux and monitored by TLC using a solvent system of petroleum ether and ethyl acetate in a volume ratio of 1:3 (v/v). After the reaction was complete, the mixture was extracted with dichloromethane (3 × 50 mL). The combined organic phase was washed three times with water, and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure using a rotary evaporator to give the yellow precipitate of crude thiadiazole sulfide, followed by crystallization in 95% ethanol to yield the pure product as a yellow solid in 84.5% yield (2.89 g).
Synthesis of kojic acid-triazole species
A mixture of azido kojic acid (2.51 g, 15.0 mmol), propargyl thiadiazole (2.56 g, 15.0 mmol), copper sulfate pentahydrate (0.297 g, 1.2 mmol), and sodium ascorbate (0.742 g, 3.75 mmol) in a mixed solvent of THF (50 mL) and water (50 mL) was stirred at room temperature and monitored by TLC using a solvent system of dichloromethane and methanol in a volume ratio of 10:1 (v/v). After the reaction was completed, water (50 mL) was added to the reaction mixture. The mixture was extracted with dichloromethane (3 × 50 mL), the combined organic phase was washed three times with water and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure using a rotary evaporator to yield the desired product, kojic acid-triazole as a yellow solid in 72.7% yield (3.69 g).
Synthesis of 6a–6p
A mixture of kojic acid-triazole (0.338 g, 1.0 mmol), substituted aromatic acid (1.2 mmol), EDCI (0.958 g, 5 mmol), and HOBT (0.162 g, 1.2 mmol) in DMF (10 mL) was stirred at room temperature. The reaction progress was monitored by TLC using a solvent system of dichloromethane and methanol in a volume ratio of 10:1 (v/v). After the reaction was completed, water (20 mL) was added to the reaction mixture. The mixture was extracted with ethyl acetate (3 × 10 mL). The organic phase was dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure using a rotary evaporator, and the residue was purified by preparative chromatography to afford the compounds 6a–6p.
Synthesis of 13a–13t
To a solution of hydroxyl-substituted aromatic aldehyde or ketone (2 mmol) in acetone (15 mL), propargyl bromide (0.833 g, 7 mmol), and K2CO3 (0.415 g, 3 mmol) were added, and the solution was heated to 60 °C. The reaction progress was monitored by TLC using a solvent system of petroleum ether and ethyl acetate in a volume ratio of 1:3 (v/v). After the reaction was completed, the precipitates were filtered and washed with acetone. The solvent of the filtrate was removed by vacuum evaporation to afford alkynyl ether-substituted aromatic aldehyde or ketone as a yellow solid and without purifcation. A mixture of azido kojic acid (0.334 g, 2.0 mmol), alkynyl ether-substituted aromatic aldehyde or ketone (1.8 mmol), copper sulfate pentahydrate (0.04 g, 0.16 mmol), and sodium ascorbate (0.099 g, 0.5 mmol) in a mixed solvent of THF (10 mL) and water (10 mL) was stirred and heated to 70 oC. The reaction progress was monitored by TLC, using a solvent system of dichloromethane and methanol in a volume ratio of 10:1 (v/v). After the reaction was completed, water (40 mL) was added to the reaction mixture. The precipitates were filtered to afford the crude product, followed by crystallization in 50% ethanol to yield the corresponding target product.
6aN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)meth-yl)thio)-1,3,4-thiadiazol-2-yl)benzamide
Yellow solid; Yield: 63%; m.p: 170.3–173.8 °C; 97% purity; IR ( KBr, cm− 1) 3410, 3151, 1734, 1659, 1636, 1579, 1492, 788, 705;1H NMR (DMSO-d6, 400 MHz): δ 8.62 (s, 1 H, –OH), 8.13 (s, 1 H, =CH), 8.03 (d, J = 7.7 Hz, 2 H, Ph-H), 7.72 (d, J = 6.9 Hz, 1 H, Ph-H), 7.57 (t, J = 7.5 Hz, 2 H, Ph-H), 7.27 (s, 2 H, =CH), 6.52 (s, 1 H, -NH), 5.65 (s, 2 H, –CH2), 4.35 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 157.6, 156.5, 150.8, 150.2, 140.3, 139.6, 135.2, 133.0, 128.0, 124.4, 123.7, 122.5, 120.2, 112.6, 60.2, 43.5; HR-MS (m/z) calcd. For C18H14N6O4S2 [M + H] + 443.0596, found 443.0591.
6bN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)meth-yl)thio)-1,3,4-thiadiazol-2-yl)-2-methylbenzamide
Yellow solid; Yield: 48%; m.p: 162.1–164.5 °C; 98% purity; IR ( KBr, cm− 1) 3365, 3105, 1741, 1661, 1640, 1514, 1456, 733; 1 H NMR (DMSO-d6, 400 MHz): δ 8.64 (s, 1 H, –OH), 8.17 (s, 1 H, =CH), 8.00 (d, J = 7.8 Hz, 1 H, Ph-H), 7.59 (t, J = 7.5 Hz, 1 H, Ph-H), 7.38–7.44 (m, 2 H, Ph-H), 7.32 (s, 2 H, =CH), 6.57 (s, 1 H, –NH), 5.69 (s, 2 H, –CH2), 4.39 (s, 2 H, –CH2), 2.54 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 157.7, 156.4, 151.3, 150.1, 140.2, 139.6, 135.2, 133.0, 132.7, 127.1, 125.9, 125.1, 122.1, 121.4, 120.1, 112.6, 60.2, 43.5, 37.3; HR-MS (m/z) calcd. For C19H16N6O4S2 [M + H] + 457.0753, found 457.0745.
6cN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)meth-yl)thio)-1,3,4-thiadiazol-2-yl)-3-methylbenzamide
Yellow solid; Yield: 51%; m.p: 162.8–165.8 °C; 98% purity; IR ( KBr, cm− 1) 3367, 3111, 1740, 1665, 1640, 1514, 1468, 788, 740;1H NMR (DMSO-d6, 400 MHz): δ 8.65 (s, 1 H, –OH), 8.17 (s, 1 H, =CH), 7.87 (d, J = 12.3 Hz, 2 H, Ph-H), 7.58 (d, J = 7.1 Hz, 1 H, Ph-H), 7.44–7.53 (m, 1 H, Ph-H), 7.32 (s, 2 H, =CH), 6.55 (s, 1 H, –NH), 5.69 (s, 2 H, –CH2), 4.39 (s, 2 H, –CH2), 2.40 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 157.6, 156.5, 150.9, 150.2, 140.2, 139.6, 135.2, 133.0, 131.3, 128.5, 124.6, 123.6, 122.5, 122.1, 120.2, 112.6, 60.2, 43.5, 37.0; HR-MS (m/z) calcd. For C19H16N6O4S2 [M + H] + 457.0753, found 457.0745.
6dN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)meth-yl)thio)-1,3,4-thiadiazol-2-yl)-4-methylbenzamide
Yellow solid; Yield: 55%; m.p: 182.3–185.8 °C; 98% purity; IR ( KBr, cm− 1) 3387, 3092, 1739, 1660, 1649, 1524, 1468, 824;1H NMR (DMSO-d6, 400 MHz): δ 8.60 (s, 1 H, –OH), 8.13 (s, 1 H, =CH), 7.91 (d, J = 7.6 Hz, 2 H, Ph-H), 7.36 (d, J = 7.8 Hz, 2 H, Ph-H), 7.29 (s, 2 H, =CH), 6.51 (s, 1 H, –NH), 5.61(s, 2 H, –CH2), 4.34 (s, 2 H, –CH2), 2.37 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 157.3, 150.8, 150.1, 140.2, 136.5, 135.2, 133.0, 124.4, 124.1, 120.3, 120.2, 112.6, 60.2, 43.5, 37.4; HR-MS (m/z) calcd. For C19H16N6O4S2 [M + H] + 457.0753, found 457.0746.
6e 4–CHloro-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)benzamide
Yellow solid; Yield: 47%; m.p: 164.8–168.0 °C; 97% purity; IR ( KBr, cm− 1) 3404, 3094, 1744, 1660, 1639, 1591, 1498, 847;1H NMR (DMSO-d6, 400 MHz): δ 8.60 (s, 1 H, –OH), 8.11 (s, 1 H, =CH), 8.03 (d, J = 8.2 Hz, 2 H, Ph-H), 7.64 (d, J = 8.3 Hz, 2 H, Ph-H), 7.19 (s, 2 H, =CH), 6.51 (s, 1 H, –NH), 5.64 (s, 2 H, –CH2), 4.35 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 157.3, 156.7, 151.3, 150.5, 140.2, 139.8, 135.4, 133.5, 124.0, 123.6, 122.6, 122.1, 120.3, 113.1, 62.0, 43.9; HR-MS (m/z) calcd. For C18H13ClN6O4S2 [M + H] + 477.0206, found 477.0211.
6f 4-bromo-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)benzamide
Yellow solid; Yield: 43%; m.p: 168.2–171.4 °C; 98% purity; IR ( KBr, cm− 1) 3400, 3090, 1741, 1661, 1640, 1587, 1498, 845;1H NMR (DMSO-d6, 400 MHz): δ 8.46 (s, 1 H, –OH), 7.65–8.19 (m, 5 H, Ph-H, =CH), 7.24 (s, 2 H, =CH), 6.48 (s, 1 H, –NH), 5.79 (s, 2 H, –CH2), 4.53 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 157.3, 156.6, 151.3, 150.5, 140.5, 139.8, 135.4, 133.5, 124.2, 123.7, 122.6, 121.9, 120.6, 113.1, 61.7, 43.7; HR-MS (m/z) calcd. For C18H13BrN6O4S2 [M + H] + 520.9701, found 520.9704.
6 g 4-fluoro-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)benzamide
Yellow solid; Yield: 46%; m.p: 230.3–231.9 °C; 99% purity; IR ( KBr, cm− 1) 3406, 3093, 1743, 1660, 1627, 1603, 1547, 852;1H NMR (DMSO-d6, 400 MHz): δ 8.48 (s, 1 H, –OH), 8.36 (s, 1 H, =CH), 8.09 (d, J = 8.2 Hz, 2 H, Ph-H), 7.62 (d, J = 8.3 Hz, 2 H, Ph-H), 7.24 (s, 2 H, =CH), 6.31 (s, 1 H, –NH), 5.57 (s, 2 H, –CH2), 4.36 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 157.4, 156.5, 151.5, 150.5, 140.6, 139.6, 135.6, 133.3, 124.2, 123.7, 122.6, 121.9, 121.0, 113.1, 61.6, 44.0; HR-MS (m/z) calcd. For C18H13FN6O4S2 [M + H] + 461.0502, found 461.0502.
6 hN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)meth-yl)thio)-1,3,4-thiadiazol-2-yl)-4-methoxybenzamide
Yellow solid; Yield: 52%; m.p: 169.2–173.4 °C; 99% purity; IR ( KBr, cm− 1) 3493, 3078, 1721, 1662, 1601, 1500, 1464, 848;1H NMR (DMSO-d6, 400 MHz): δ 8.58 (s, 1 H, –OH), 8.14 (s, 1 H, =CH), 8.00 (t, J = 7.8 Hz, 2 H, Ph-H), 7.27 (s, 2 H, =CH), 7.07 (d, J = 8.0 Hz, 2 H, Ph-H), 6.50 (s, 1 H, –NH), 5.64 (s, 2 H, –CH2), 4.34 (s, 2 H, –CH2), 3.80 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 158.1, 156.6, 151.5, 149.2, 140.6, 139.7, 135.6, 133.5, 124.2, 123.3, 122.6, 122.1, 121.0, 112.8, 62.3, 55.8, 44.6; HR-MS (m/z) calcd. For C19H16N6O5S2 [M + H] + 473.0702, found 473.0702.
6i 4-amino-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)benzamide
Yellow solid; Yield: 57%; m.p: 173.7–174.5 °C; 97% purity; IR ( KBr, cm− 1) 3352, 3113, 1705, 1659, 1640, 1602, 1498, 846;1H NMR (DMSO-d6, 400 MHz): δ 8.50 (s, 1 H, –OH), 8.12 (s, 1 H, =CH), 7.67 (d, J = 8.3 Hz, 2 H, Ph-H), 7.29 (s, 2 H, Ph-H), 6.56 (d, J = 8.4 Hz, 2 H, =CH), 6.46 (s, 1 H, -NH), 6.20 (s, 2 H, -NH2), 5.63 (s, 2 H, –CH2), 4.34 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 158.0, 156.4, 150.8, 149.9, 144.0, 140.1, 139.6, 135.1, 133.1, 126.1, 120.1, 112.5, 110.8, 110.6, 60.2, 43.5; HR-MS (m/z) calcd. For C18H15N7O4S2 [M + H] + 458.0705, found 458.0709.
6jN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)met-hyl)thio)-1,3,4-thiadiazol-2-yl)-2-(o-tolyl)acetamide
Yellow solid; Yield: 52%; m.p: 156.0–157.4 °C; 99% purity; IR ( KBr, cm− 1) 3328, 3136, 1760, 1673, 1642, 1548, 1494, 746;1H NMR(DMSO-d6, 400 MHz): δ 8.15 (s, 1 H, –OH), 7.32–7.51 (m, 3 H, =CH), 7.18–7.30 (m, 4 H, Ph-H), 6.69 (s, 1 H, -NH), 5.73 (s, 2 H, –CH2), 4.66 (s, 2 H, –CH2), 3.48 (s, 2 H, –CH2), 2.55 (s, 3 H, –CH3);13C NMR (DMSO- d6, 100 MHz): δ 157.3, 156.6, 151.3, 150.6, 140.2, 139.6, 135.2, 133.2, 132.7, 127.1, 126.1, 125.3, 122.1, 121.4, 120.6, 112.6, 61.3, 43.5, 42.3, 37.6; HR-MS (m/z) calcd. For C20H18N6O4S2 [M + H] + 471.0909, found 471.0908.
6kN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)met-hyl)thio)-1,3,4-thiadiazol-2-yl)-2-(m-tolyl)acetamide
Yellow solid; Yield: 50%; m.p: 150.9–152.1 °C; 98% purity; IR ( KBr, cm− 1) 3410, 3137, 1764, 1674, 1642, 1611, 1547, 796, 713;1H NMR(DMSO-d6, 400 MHz): δ 8.27 (s, 1 H, –OH), 7.29–7.44 (m, 3 H, =CH), 7.12–7.24 (m, 4 H, Ph-H), 6.98 (s, 1 H, -NH), 5.47 (s, 2 H, –CH2), 4.62 (s, 2 H, –CH2), 3.57 (s, 2 H, –CH2), 2.59 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 157.4, 156.0, 151.3, 149.7, 140.4, 139.6, 135.5, 133.2, 132.6, 127.1, 126.2, 125.3, 122.1, 121.7, 120.3, 113.0, 62.0, 42.6, 42.3, 37.4; HR-MS (m/z) calcd. For C20H18N6O4S2 [M + H] + 471.0909, found 471.0907.
6 LN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)met-hyl)thio)-1,3,4-thiadiazol-2-yl)-2-(p-tolyl)acetamide
Yellow solid; Yield: 53%; m.p: 150.1–152.4 °C; 99% purity; IR ( KBr, cm− 1) 3409, 3051, 1762, 1693, 1667, 1645, 1553, 817;1H NMR (DMSO-d6, 400 MHz): δ 8.10 (s, 1 H, –OH), 7.12–7.21 (m, 3 H, =CH), 7.04–7.12 (m, 4 H, Ph-H), 6.41 (s, 1 H, -NH), 5.55 (s, 2 H, –CH2), 4.49 (s, 2 H, –CH2), 3.70 (s, 2 H, –CH2), 2.23 (s, 3 H, –CH3);13C NMR (DMSO- d6, 100 MHz): δ 157.4, 156.5, 151.5, 150.5, 140.6, 139.6, 135.6, 133.3, 124.2, 123.7, 122.6, 121.9, 121.0, 113.1, 61.6, 44.2, 43.5, 37.6; HR-MS (m/z) calcd. For C20H18N6O4S2 [M + H] + 471.0909, found 471.0908.
6 m 2-(4–CHlorophenyl)-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)acetamide
Yellow solid; Yield: 38%; m.p: 173.3–176.9 °C; 99% purity; IR ( KBr, cm− 1) 3345, 3137, 1763, 1679, 1548, 1492, 1409, 858;1H NMR (DMSO-d6, 400 MHz): δ 8.01 (s, 1 H, –OH), 7.09–7.53 (m, 7 H, =CH, Ph-H), 6.36 (s, 1 H, -NH), 5.51 (s, 2 H, –CH2), 4.45 (s, 2 H, –CH2), 3.73 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 158.3, 156.4, 151.3, 150.6, 141.5, 140.2, 135.5, 133.5, 124.2, 123.7, 122.4, 121.9, 121.6, 113.1, 63.7, 43.7, 41.7; HR-MS (m/z) calcd. For C19H15ClN6O4S2 [M + H] + 491.0363, found 491.0362.
6n 2-(4-bromophenyl)-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)acetamide
Yellow solid; Yield: 42%; m.p: 165.6–173.0 °C; 97% purity; IR ( KBr, cm− 1) 3409, 3104, 1764, 1679, 1547, 1488, 1462, 815;1H NMR (DMSO-d6, 400 MHz): δ 8.16 (s, 1 H, –OH), 7.52 (d, J = 4.2 Hz, 4 H, Ph-H), 7.07–7.46 (m, 3 H, =CH), 6.41 (s, 1 H, -NH), 5.60 (s, 2 H, –CH2), 4.54 (s, 2 H, –CH2), 3.81 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 158.7, 156.3, 151.3, 150.6, 141.5, 140.7, 134.5, 133.5, 124.2, 123.4, 122.2, 121.9, 121.5, 112.4, 63.0, 42.4, 41.2; HR-MS (m/z) calcd. For C19H15BrN6O4S2 [M + H] + 534.9858, found 534.9862.
6o 2-(4-fluorophenyl)-N-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-thiadiazol-2-yl)acetamide
Yellow solid; Yield: 45%; m.p: 159.1–160.2 °C; 98% purity; IR ( KBr, cm− 1) 3409, 3138, 1766, 1675, 1640, 1547, 1511, 825;1H NMR (DMSO-d6, 400 MHz): δ 8.39 (s, 1 H, –OH), 7.75 (d, J = 4.2 Hz, 4 H, Ph-H), 7.13–7.56 (m, 3 H, =CH), 6.69 (s, 1 H, -NH), 5.65 (s, 2 H, –CH2), 4.34 (s, 2 H, –CH2), 3.81 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 157.9, 156.5, 152.4, 150.4, 142.0, 140.3, 134.5, 133.4, 124.2, 123.4, 122.3, 121.9, 121.0, 112.4, 63.4, 42.2, 41.6; HR-MS (m/z) calcd. For C19H15FN6O4S2 [M + H] + 475.0658, found 475.0655.
6pN-(5-(((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)met-hyl)thio)-1,3,4-thiadiazol-2-yl)-2-(4-methoxyphenyl)acetamide
Yellow solid; Yield: 53%; m.p: 176.0–181.6 °C; 99% purity; IR ( KBr, cm− 1) 3410, 3052, 1777, 1684, 1661, 1586, 1514, 791;1H NMR (DMSO-d6, 400 MHz): δ 8.09(s, 1 H, –OH), 7.05–7.31 (m, 3 H, =CH), 6.69–6.99 (m, 4 H, Ph-H), 6.33 (s, 1 H, -NH), 5.50 (s, 2 H, –CH2), 4.40 (s, 2 H, –CH2), 3.75( s, 2 H, –CH2), 3.66 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 159.3, 155.9, 151.5, 150.6, 140.4, 139.7, 135.6, 134.5, 124.2, 123.1, 122.8, 122.3, 121.0, 112.5, 64.7, 55.8, 44.6, 43.6; HR-MS (m/z) calcd. For C20H18FN6O5S2 [M + H] + 487.0858, found 487.0858.
13a 3-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-benzaldehyde
Yellow solid; Yield: 60%; m.p: 159.8–162.9 °C; 97% purity; IR ( KBr, cm− 1) 3264, 3137, 2738, 1695, 1655, 1585, 1489, 794, 681;1H NMR (DMSO-d6, 400 MHz): δ 9.94 (s, 1 H, –OH), 9.27 (s, 1 H, =CH), 8.32 (s, 1 H, =CH), 8.02 (s, 1 H, =CH), 7.44–7.58 (m, 3 H, Ph-H), 7.28–7.40 (m, 1 H, Ph-H), 6.36 (s, 1 H, –CHO), 5.58 (s, 2 H, –CH2), 5.22 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 174.7, 159.3, 148.8, 147.2, 137.2, 134.6, 132.4, 130.5, 124.7, 120.8, 118.6, 117.6, 111.7, 110.8, 69.3, 60.4; HR-MS (m/z) calcd. For C16H13N3O5 [M + H] + 328.0933, found 328.0928.
13b 2-((4-((3-acetylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hydroxy-4H-pyran-4-one
Yellow solid; Yield: 75%; m.p: 165.2–167.0 °C; 99% purity; IR ( KBr, cm− 1) 3411, 3152, 1679, 1654, 1615, 1593, 1487, 791, 700;1H NMR (DMSO-d6, 400 MHz): δ 9.28 (s, 1 H, –OH), 8.32 (s, 1 H, =CH), 8.02 (s, 1 H, =CH), 7.52 (s, 2 H, Ph-H), 7.42 (d, J = 7.1 Hz, 1 H, Ph-H), 7.27 (d, J = 6.9 Hz, 1 H, Ph-H), 6.36 (s, 1 H, =CH), 5.58 (s, 2 H, –CH2), 5.21 (s, 2 H, –CH2), 2.53 (s, 3 H, –CH3);13C NMR (DMSO-d6, 100 MHz): δ 178.5, 159.3, 148.8, 146.9, 137.2, 134.7, 132.4, 131.0, 124.3, 120.8, 117.2, 116.2, 111.4, 110.8, 69.3, 60.4, 41.9; HR-MS (m/z) calcd. For C17H15N3O5 [M + H] + 342.1090, found 342.1091.
13c 4-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde
Yellow solid; Yield: 57%; m.p: 174.3–175.1 °C; 98% purity; IR ( KBr, cm− 1) 3403, 3244, 2922, 1697, 1655, 1578, 1453, 849;1H NMR (DMSO-d6, 400 MHz): δ 9.83 (s, 1 H, –OH), 9.28 (s, 1 H, =CH), 8.35 (s, 1 H, =CH), 8.02 (s, 1 H, =CH), 7.84 (d, J = 8.1 Hz, 2 H, Ph-H), 7.20 (d, J = 8.1 Hz, 2 H, Ph-H), 6.37 (s, 1 H, –CHO), 5.58 (s, 2 H, –CH2), 5.26 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 173.5, 159.3, 150.7, 148.8, 137.2, 134.4, 132.4, 125.8, 124.3, 121.0, 112.5, 110.9, 69.4, 60.4; HR-MS (m/z) calcd. For C16H13N3O5 [M + H] + 328.0933, found 328.0931.
13d 2-((4-((4-acetylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hydroxy-4H-pyran-4-one
Yellow solid; Yield: 80%; m.p: 184.1–188.3 °C; 99% purity; IR ( KBr, cm− 1) 3399, 3083, 1677, 1654, 1609, 1579, 1456, 835; 1H NMR (DMSO-d6, 400 MHz): δ 9.28 (s, 1 H, –OH), 8.34 (s, 1 H, =CH), 8.02 (s, 1 H, =CH), 7.89 (d, J = 8.6 Hz, 2 H, Ph-H), 7.11 (d, J = 8.6 Hz, 2 H, Ph-H), 6.37 (s, 1 H, =CH), 5.58 (s, 2 H, –CH2), 5.23 (s, 2 H, –CH2), 2.48 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 177.4, 159.3, 149.8, 148.8, 137.2, 134.5, 132.4, 124.7, 124.5, 120.9, 112.0, 110.9, 69.3, 60.4, 41.5; HR-MS (m/z) calcd. For C17H15N3O5 [M + H] + 342.1090, found 342.1091.
13e 2-((4-((4-acetyl-2-methylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hydr-oxy-4H-pyran-4-one
Yellow solid; Yield: 85%; m.p: 177.2–178.9 °C; 97% purity; IR ( KBr, cm− 1) 3345, 3177, 1669, 1635, 1600, 1578, 1505, 877, 811; 1H NMR (DMSO-d6, 400 MHz): δ 9.28 (s, 1 H, –OH), 8.33 (s, 1 H, =CH), 8.02 (s, 1 H, =CH), 7.79 (d, J = 8.5 Hz, 1 H, Ph-H), 7.73 (s, 1 H, Ph-H), 7.21 (d, J = 8.6 Hz, 1 H, Ph-H), 6.36 (s, 1 H, =CH), 5.58 (s, 1 H, –CH2), 5.25 (s, 2 H, –CH2), 2.46 (s, 3 H, –CH3), 2.12 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 177.6, 159.3, 148.8, 148.4, 137.2, 134.8, 132.4, 124.9, 124.2, 123.1, 121.3, 120.7, 110.8, 109.4, 69.6, 60.4, 41.5, 33.2; HR-MS (m/z) calcd. For C18H17N3O5 [M + H] + 356.1246, found 356.1240.
13f 4-fluoro-2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde
Yellow solid; Yield: 80%; m.p: 172.3–173.1 °C; 97% purity; IR ( KBr, cm− 1) 3405, 3113, 2923, 1682, 1652, 1594, 1496, 875, 802;1H NMR (DMSO-d6, 400 MHz): δ 10.20 (s, 1 H, –OH), 9.31 (s, 1 H, =CH), 8.43 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.75 (t, J = 7.7 Hz, 1 H, Ph-H), 7.37 (d, J = 11.3 Hz, 1 H, Ph-H), 6.94 (d, J = 7.9 Hz, 1 H, Ph-H), 6.39 (s, 1 H, –CHO), 5.61 (s, 2 H, –CH2), 5.37 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 170.6, 159.3, 154.9, 152.9, 150.1, 148.8, 137.2, 132.4, 124.8, 124.7, 121.0, 117.6, 107.3, 102.3, 70.4, 60.4; HR-MS (m/z) calcd. For C16H12FN3O5 [M + H] + 346.0839, found 346.0833.
13 g 2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde
Yellow solid; Yield: 53%; m.p: 179.9–181.2 °C; 97% purity; IR ( KBr, cm− 1) 3417, 3151, 2922, 1687, 1651, 1597, 1483, 770;1H NMR (DMSO-d6, 400 MHz): δ 10.32 (s, 1 H, –OH), 9.29 (s, 1 H, =CH), 8.41 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.66 (dd, J = 16.7, 7.5 Hz, 2 H, Ph-H), 7.42 (d, J = 8.1 Hz, 1 H, Ph-H), 7.09 (t, J = 6.9 Hz, 1 H, Ph-H), 6.39 (s, 1 H, –CHO), 5.60 (s, 2 H, –CH2), 5.35 (s, 2 H, –CH2);13C NMR (DMSO-d6, 100 MHz): δ 171.7, 159.3, 148.8, 148.6, 137.2, 134.7, 132.4, 129.5, 122.5, 120.8, 120.0, 117.4, 111.8, 110.9, 70.1, 60.4; HR-MS (m/z) calcd. For C16H13N3O5 [M + H] + 328.0933, found 328.0928.
13 h 2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-5-methylbenzaldehyde
Yellow solid; Yield: 73%; m.p: 179.8–180.6 °C; 98% purity; IR ( KBr, cm− 1) 3417, 3151, 2922, 1687, 1651, 1597, 1483, 884, 770; 1H NMR (DMSO-d6, 400 MHz): δ 10.24 (s, 1 H, –OH), 9.30 (s, 1 H, =CH), 8.41 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.57 (d, J = 7.2 Hz, 1 H, Ph-H), 7.24 (s, 1 H, Ph-H), 6.90 (d, J = 7.1 Hz, 1 H, Ph-H), 6.39 (s, 1 H, –CHO), 5.61 (s, 2 H, –CH2), 5.32 (s, 2 H, –CH2), 2.36 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 175.1, 163.1, 152.6, 152.5, 142.2, 141.0, 138.5, 136.2, 126.3, 124.6, 122.0, 121.9, 115.8, 114.7, 73.8, 64.2, 41.6; HR-MS (m/z) calcd. For C17H15N3O5 [M + H] + 342.1090, found 342.1092.
13i 2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methylbenzaldehyde
Yellow solid; Yield: 68%; m.p: 151.6–152.1 °C; 97% purity; IR ( KBr, cm− 1) 3321, 2923, 2853, 1687, 1648, 1603, 1497, 882, 803; 1H NMR (DMSO-d6, 400 MHz): δ 10.27 (s, 1 H, –OH), 9.33 (s, 1 H, =CH), 8.37 (s, 1 H, =CH), 8.01 (s, 1 H, =CH), 7.45 (d, J = 7.5 Hz, 2 H, Ph-H), 7.29 (d, J = 8.9 Hz, 1 H, Ph-H), 6.38 (s, 1 H, –CHO), 5.59 (s, 2 H, –CH2), 5.30 (s, 2 H, –CH2), 2.24 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 175.6, 163.1, 152.6, 150.9, 141.0, 138.5, 136.2, 133.7, 128.4, 126.2, 124.6, 123.6, 115.7, 114.6, 73.9, 64.2, 40.0; HR-MS (m/z) calcd. For C17H15N3O5 [M + H] + 342.1090, found 342.1085.
13j 2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methox-y)-4-methoxybenzaldehyde
Yellow solid; Yield: 84%; m.p: 193.3–195.1 °C; 97% purity; IR ( KBr, cm− 1) 3410, 3136, 2836, 1657, 1631, 1604, 1499, 893, 802; 1H NMR (DMSO-d6, 400 MHz): δ 10.14 (s, 1 H, –OH), 9.30 (s, 1 H, =CH), 8.42 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.65 (d, J = 8.0 Hz, 1 H, Ph-H), 6.91 (s, 1 H, Ph-H), 6.65 (d, J = 7.9 Hz, 1 H, Ph-H), 6.39 (s, 1 H, –CHO), 5.61 (s, 2 H, –CH2), 5.35 (s, 2 H, –CH2), 3.85 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 174.0, 163.1, 156.9, 153.9, 152.6, 141.0, 138.4, 136.2, 127.9, 124.6, 118.9, 114.7, 110.1, 103.9, 73.9, 68.9, 64.2; HR-MS (m/z) calcd. For C17H15N3O6 [M + H] + 358.1039, found 358.1033.
13k 2-((4-((2-acetylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hydroxy-4H-pyran-4-one
Yellow solid; Yield: 67%; m.p: 165.6–168.8 °C; 98% purity; IR ( KBr, cm− 1) 3405, 3204, 1671, 1646, 1617, 1593, 1448, 760; 1H NMR (DMSO-d6, 400 MHz): δ 8.43 (s, 1 H, –OH), 7.68 (s, 1 H, =CH), 7.41 (s, 1 H, =CH), 7.01 (dd, J = 13.3, 6.1 Hz, 2 H, Ph-H), 6.85 (d, J = 6.6 Hz, 1 H, Ph-H), 6.60 (t, J = 5.9 Hz, 1 H, Ph-H), 6.07 (s, 1 H, =CH), 5.48 (s, 2 H, –CH2), 5.23 (s, 2 H, –CH2), 2.94 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 179.5, 159.3, 148.9, 146.1, 137.2, 134.7, 132.5, 127.4, 124.0, 123.0, 120.8, 117.1, 111.5, 110.8, 69.8, 60.4, 45.7; HR-MS (m/z) calcd. For C17H15N3O5 [M + H] + 342.1090, found 342.1093.
13 L 3-(tert-butyl)-2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde
Yellow solid; Yield: 53%; m.p: 179.8–180.9 °C; 98% purity; IR ( KBr, cm− 1) 3366, 3154, 2885, 1683, 1654, 1593, 1487, 863, 802; 1H NMR (DMSO-d6, 400 MHz): δ 9.25 (s, 1 H, –OH), 8.43 (s, 1 H, =CH), 7.75 (s, 1 H, =CH), 7.43 (s, 1 H, =CH), 7.11 (t, J = 5.8 Hz, 2 H, Ph-H), 6.78 (t, J = 6.1 Hz, 1 H, Ph-H), 6.11 (s, 1 H, –CHO), 5.50 (s, 2 H, –CH2), 5.05 (s, 2 H, –CH2), 2.07 (s, 9 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 172.7, 159.3, 148.8, 148.7, 137.2, 135.2, 134.5, 132.4, 127.3, 124.2, 122.9, 121.0, 119.9, 110.9, 77.1, 60.4, 48.3, 45.0; HR-MS (m/z) calcd. For C20H21N3O5 [M + H] + 384.1559, found 384.1551.
13 m 4–CHloro-2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde
Yellow solid; Yield: 78%; m.p: 182.7–183.9 °C; 98% purity; IR ( KBr, cm− 1) 3409, 3144, 2923, 1681, 1651, 1592, 1483, 875, 800; 1H NMR (DMSO-d6, 400 MHz): δ 10.99 (s, 1 H, –OH), 9.19 (s, 1 H, =CH), 8.47 (s, 1 H, =CH), 7.74 (s, 1 H, =CH), 7.43 (s, 1 H, Ph-H), 7.15 (s, 1 H, Ph-H), 7.06 (s, 1 H, Ph-H), 6.13 (s, 1 H, –CHO), 5.50 (s, 2 H, –CH2), 5.32 (s, 2 H, –CH2); 13C NMR (DMSO-d6, 100 MHz): δ 171.0, 159.3, 149.0, 148.8, 137.3, 134.3, 133.0, 132.4, 123.9, 120.9, 119.1, 117.6, 112.2, 110.9, 70.4, 60.4; HR-MS (m/z) calcd. For C16H12ClN3O5 [M + H] + 362.0544, found 362.0546.
13n 2-((4-((2-acetyl-5-methylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hy-droxy-4H-pyran-4-one
Yellow solid; Yield: 83%; m.p: 203.7–205.0 °C; 99% purity; IR ( KBr, cm− 1) 3423, 3236, 1667, 1645, 1617, 1512, 1494, 873, 813; 1H NMR (DMSO-d6, 400 MHz): δ 9.30 (s, 1 H, –OH), 8.37 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.49 (d, J = 7.8 Hz, 1 H, Ph-H), 7.17 (s, 1 H, Ph-H), 6.83 (d, J = 7.8 Hz, 1 H, Ph-H), 6.35 (s, 1 H, =CH), 5.62 (s, 2 H, –CH2), 5.29 (s, 2 H, –CH2), 2.41 (s, 3 H, –CH3), 2.33 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 178.7, 159.3, 148.9, 146.4, 137.2, 136.1, 134.7, 132.4, 124.2, 120.8, 120.7, 117.7, 111.8, 110.7, 69.8, 60.4, 45.8, 37.5; HR-MS (m/z) calcd. For C18H17N3O5 [M + H] + 356.1246, found 356.1245.
13o 2-((4-((2-acetyl-4-methylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hy-droxy-4H-pyran-4-one
Yellow solid; Yield: 86%; m.p: 185.5–188.2 °C; 99% purity; IR ( KBr, cm− 1) 3245, 3148, 1668, 1648, 1619, 1586, 1493, 877, 808; 1H NMR (DMSO-d6, 400 MHz): δ 9.24 (s, 1 H, –OH), 8.32 (s, 1 H, =CH), 8.00 (s, 1 H, =CH), 7.31 (d, J = 12.5 Hz, 2 H, Ph-H), 7.21 (d, J = 8.3 Hz, 1 H, Ph-H), 6.32 (s, 1 H, =CH), 5.58 (s, 2 H, –CH2), 5.24 (s, 2 H, –CH2), 2.45 (s, 3 H, –CH3), 2.21 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 179.5, 159.2, 148.9, 144.5, 137.2, 134.7, 132.4, 127.6, 124.2, 124.1, 122.8, 120.7, 111.6, 110.7, 69.9, 60.3, 45.7, 36.3; HR-MS (m/z) calcd. For C18H17N3O5 [M + H] + 356.1246, found 356.1246.
13p 4-bromo-2-((1-((5-hydroxy-4-oxo-4H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde
Yellow solid; Yield: 81%; m.p: 201.3–202.9 °C; 99% purity; IR ( KBr, cm− 1) 3401, 3175, 2922, 1681, 1651, 1588, 1481, 875, 798; 1H NMR (DMSO-d6, 400 MHz): δ 11.21 (s, 1 H, –OH), 9.30 (s, 1 H, =CH), 8.44 (s, 1 H, =CH), 7.69 (s, 1 H, =CH), 7.48 (s, 1 H, Ph-H), 7.21 (s, 1 H, Ph-H), 6.85 (s, 1 H, Ph-H), 6.19 (s, 1 H, –CHO), 5.63 (s, 2 H, –CH2), 5.36 (s, 2 H, –CH2); 13C NMR (DMSO-d6, 100 MHz): δ 172.3, 159.6, 150.2, 148.3, 137.5, 134.7, 134.0, 132.6, 123.9, 121.9, 120.2, 117.6, 111.8, 110.6, 71.0, 61.3; HR-MS (m/z) calcd. For C16H12BrN3O5 [M + H] + 406.0039, found 406.0037.
13q 2-((4-((2-acetyl-5–CHlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hy-droxy-4H-pyran-4-one
Yellow solid; Yield: 90%; m.p: 197.4–199.5 °C; 97% purity; IR ( KBr, cm− 1) 3246, 3098, 1662, 1628, 1590, 1563, 1481, 908, 811; 1H NMR (DMSO-d6, 400 MHz): δ 8.47 (s, 1 H, –OH), 7.74 (s, 1 H, =CH), 7.46 (s, 1 H, =CH), 7.06 (d, J = 27.5 Hz, 2 H, Ph-H), 6.72 (s, 1 H, Ph-H), 6.12 (s, 1 H, =CH), 5.53 (s, 2 H, –CH2), 5.32 (s, 2 H, –CH2), 2.98 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 178.5, 159.3, 148.9, 146.7, 137.2, 134.3, 132.4, 130.9, 125.3, 121.9, 120.9, 117.2, 111.9, 110.8, 70.2, 60.4, 45.7; HR-MS (m/z) calcd. For C17H14ClN3O5 [M + H] + 376.0700, found 376.0695.
13r 2-((4-((2-acetyl-5-bromophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hy-droxy-4H-pyran-4-one
Yellow solid; Yield: 94%; m.p: 194.6–195.7 °C; 98% purity; IR ( KBr, cm− 1) 3265, 3148, 1651, 1627, 1610, 1582, 1468, 893, 834; 1H NMR (DMSO-d6, 400 MHz): δ 9.29 (s, 1 H, –OH), 8.37 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.60 (s, 1 H, Ph-H), 7.49 (d, J = 8.1 Hz, 1 H, Ph-H), 7.23 (d, J = 8.0 Hz, 1 H, Ph-H), 6.36 (s, 1 H, =CH), 5.62 (s, 2 H, –CH2), 5.35 (s, 2 H, –CH2), 2.41 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 146.5, 128.0, 125.9, 124.8, 121.8, 119.8, 119.8, 118.5, 117.0, 117.0, 114.4, 107.2, 105.0, 104.8, 67.2, 61.7, 47.3; HR-MS (m/z) calcd. For C17H14BrN3O5 [M + H] + 420.0195, found 420.0193.
13s 2-((4-((2-acetyl-5-fluorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hy-.
droxy-4H-pyran-4-one.
Yellow solid; Yield: 89%; m.p: 178.5–179.9 °C; 97% purity; IR ( KBr, cm− 1) 3265, 3145, 1648, 1618, 1586, 1526, 1492, 875, 804; 1H NMR (DMSO-d6, 400 MHz): δ 9.31 (s, 1 H, –OH), 8.39 (s, 1 H, =CH), 8.03 (s, 1 H, =CH), 7.66 (t, J = 7.6 Hz, 1 H, Ph-H), 7.29 (d, J = 10.8 Hz, 1 H, Ph-H), 6.87 (t, J = 7.6 Hz, 1 H, Ph-H), 6.35 (s, 1 H, =CH), 5.62 (s, 2 H, –CH2), 5.33 (s, 2 H, –CH2), 2.41 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 178.1, 159.3, 151.7, 147.6, 137.2, 134.3, 133.7, 132.4, 126.1, 121.0, 120.1, 110.8, 106.8, 101.7, 70.2, 60.4, 45.7; HR-MS (m/z) calcd. For C17H14FN3O5 [M + H] + 360.0996, found 360.0990.
13t 2-((4-((2-acetyl-5-methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-hydroxy-4H-pyran-4-one
Yellow solid; Yield: 95%; m.p: 168.1–170.5 °C; 98% purity; IR ( KBr, cm− 1) 3288, 3056, 1667, 1644, 1606, 1500, 1443, 879, 825; 1H NMR (DMSO-d6, 400 MHz): δ 8.42 (s, 1 H, –OH), 7.68 (s, 1 H, =CH), 7.41 (s, 1 H, =CH), 7.09 (d, J = 6.9 Hz, 1 H, Ph-H), 6.46 (s, 1 H, Ph-H), 6.26 (d, J = 7.0 Hz, 1 H, Ph-H), 6.06 (s, 1 H, =CH), 5.48 (s, 2 H, –CH2), 5.24 (s, 2 H, –CH2), 4.04 (s, 3 H, –CH3), 2.89 (s, 3 H, –CH3); 13C NMR (DMSO-d6, 100 MHz): δ 177.5, 159.3, 151.7, 148.9, 148.0, 137.2, 134.6, 132.4, 125.8, 120.8, 116.9, 110.7, 105.6, 100.2, 69.9, 64.9, 60.4, 45.8; HR-MS (m/z) calcd. For C18H17N3O6 [M + H] + 372.1196, found 372.1184.
In vitro tyrosinase Inhibition
The mushroom tyrosinase inhibitory activity was determined according to previous described method30. Briefly, 168 µL of phosphate buffer (0.1 M, pH 6.8), 10 µL of mushroom tyrosinase (0.5 mg/mL, Sigma Chemical, USA) and 2 µL of the inhibitor solution were placed in the wells of a 96-well micro plate. After pre-incubation for 20 min at 37 °C, 20 µL of 2.0 mg/mL L-DOPA (3,4-dihydroxyphenylalanine, Sigma Chemical, USA) was added and the enzyme activity was measured at 475 nm every 60 s for 180 s in a Microplate Reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Kojic acid and phosphate buffer were respectively used as positive and negative control. The extent of inhibition by the test compounds was expressed as the percentage of concentration necessary to achieve 50% inhibition (IC50). The percentage of inhibition was calculated as follows: Inhibitory rate (%) = [Ac − At)/Ac] × 100. Ac is the absorbance of the negative control and At is the absorbance of the test compound. Each concentration was analyzed in three independent experiments run in triplicate31,32. The IC50 values were determined by the data analysis software GraphPad Prism 8.
Tyrosinase kinetic analysis
To study the inhibition type of kojic acid-triazole derivatives, the investigation of inhibitory type of selected compound 13t on mushroom tyrosinase was carried out according to reported protocol33. Compound 13t was used at the concentrations of 0 µM, 2 µM and 4 µM, respectively. Substrate L-DOPA concentration was between 500µM and 1000 µM in the process of all kinetic study. Pre-incubation time and measurement time were the same as described in mushroom tyrosinase inhibition assay procedure. Lineweavere-Burk plots of inverse of velocities (1/V) versus inverse of substrate concentration 1/[S] µM− 1was used to determine the type of enzyme inhibition.
DFT calculations
The structural optimization and single point energy calculations of compound 13t were performed using ORCA 5.0.3 software at the theoretical level of PBE(D3BJ)/Def2SVP and PBE0(D3BJ)/Def2TZVP34, respectively. The surface electrostatic potential of the molecule was plotted by Multiwfn software combined with VMD software.
Measurements of fluorescence spectra
To investigate the fluorescence quenching role of the inhibitor on tyrosinase, fluorescence experiments were carried out according to the reported method30. To determine the linear concentration range of the fluorescence, compound 13t was first dissolved in DMSO. The concentration of the solution was 1.0 mM, which was then diluted with sodium phosphate buffer. The final concentration was ranged from 0 to 90 µM. A series of 2.5 mL solutions containing 0.2 mL of tyrosinase solution were added to a centrifuge tube and accurately mixed with different concentrations of 13t solution ranging from 0 to 90 µM. Cary Eclipse G9800A fluorescence spectrophotometer (Agilent Technologies Co) was employed to detect the fluorescence intensity. The excitation wavelength was set to 280 nm, the bath was set at three temperatures (298 K, 304 K and 310 K) with a scanning wavelength range from 300 to 500 nm. The excitation and emission bandwidths were 5 nm. The final tyrosinase concentration was 0.2 mg/mL.
In Silico Docking simulation of tyrosinase with compound 13t
Molecular docking calculations were performed molecular docking calculations by using the X-ray structure of mushroom tyrosinase in complex with tyrosinase inhibitor (PDB ID: 2Y9X)35. The most potent compound 13t was prepared by conducting Prepare Ligands procedure (Discovery studio 2019) in its neutral form and preferential conformation was obtained in the CHARMm force field. Choosing one of the eight monomers from the PDB entry, and the protein structure was prepared using the Prepare Protein module in Discovery studio 2019 software, the specific steps included adding missing residues and hydrogen atoms as well as removing water molecules and spectator ions. Then their conformation optimized in the CHARMm force field. The search grid of binding site was identified as center_x: − 10.021, center_y: − 28.823, and center_z: − 43.596 with Radius value of 10. Molecular modeling simulations were performed with CDOCKER protocol (Discovery studio 2019), reporting the top 10 poses for each ligand and the graph generation was done by PyMOL (Schrödinger).
Anti-browning experiment
Fuji apples were planted in Yantai of Shandong province, China, and were purchases from a Walmart supermarket in Shaoyang, Hunan province, China. The apples were from the same batch having similar shapes, maturity, and without injuries, rot, diseases or pests. Apples was cut into thin slices with the same size by using a sharp knife. Then, the fresh-cut slices were then randomly divided into different groups: the blank control group was immersed in ethanol for 10 min at room temperature, the treatment groups were dipped in a solution of compound 13t under the same conditions, the positive groups were dipped in 50 µM kojic acid and 50 µM vitamin C (Vc) under the same conditions. The test samples were air-dried naturally, immediately packaged separately in polyethylene clam-shell packs, and stored at 5 °C for the following experimental measurements.
According to the reported protocol36, the color of apples was tested using a CR-400 Minolta chroma meter instrument (Konica Minolta, Osaka, Japan). Three data of L*, a*, and b* were recorded for each sample. The L* values indicate lightness, the a* values show reddish-greenish, the b* values represent yellowish-bluish. The color measurement was carried out in triplicate by random sampling.
DPPH free radical scavenging activity assay
The anti-oxidation effect on DPPH radical was determined using a slightly modified version of a previously described DPPH radical scavenging assay37,38. The reaction mixture consisted of 100 µL DPPH (150 µM) and 20 µL test compound at different concentrations, and then the volume was adjusted to 200 µL with DMSO. The tested solution was incubated for 30 min at room temperature (25 °C) in a dark environment. After incubating, a UV-2600 spectrophotometer was used to measure the absorbance at 517 nm. The experiments were performed in triplicate. DMSO was used as a control, and ascorbic acid was used as an antioxidant standard for comparison. The calculation of DPPH radical scavenging activity was carried out by this formula:
 |
ABTS radical cation scavenging activity assay
According to the reported procedure30, ABTS free radical scavenging activity was assayed. The test solution was prepared by mixing 2.45 mM potassium persulfate and 7 mM ABTS in an 8:12 (volume ratio). The reaction solution was kept for 12 to 18 h at room temperature in the dark. Before the ABTS radical scavenging test, the reaction mixture was diluted with DMSO to the absorbance of 0.7 ± 0.02 at 734 nm. 1 mL diluted ABTS test solution was added to 10 µl DMSO or test sample solution. After mixing, the mixture was kept for 2 to 6 min in the dark at room temperature, and the absorbance at 734 nm was tested. This measurement was performed in triplicate. DMSO was used as a blank control. Ascorbic acid was used as antioxidant standard to compare the ABTS scavenging activity. The ABTS scavenging activity was calculated by following formula:
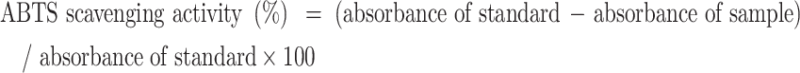 |
MTT assay for cell viability
The cytotoxic activity of 13t against the HUVEC cell line (Shanghai Fusheng Industrial Co., Ltd., China) was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay39. The HUVEC cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco-BRL, Gaithersburg, MD, USA), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were cultured in a standard humidified incubator at 37 °C with a 5% CO2 atmosphere. Cells were plated in 96-well microtiter plates at a density of 5 × 103/well and incubated in a humidified atmosphere with 5% CO2 at 37 °C for 24 h. 13t was added onto triplicate wells with different concentrations and 0.1% DMSO used for the control. After they had been incubated for 48 h, 10 µL of CCK-8 (Cell Counting Kit-8) solution was added to each well, and the plate was incubated for an additional 1 h. The absorbance (OD) was read on a microplate reader at 450 nm.
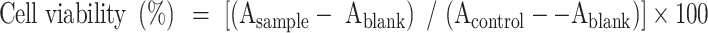 |
Asample denotes the OD450 absorbance of test compound and Ablank is the OD450 absorbance for the blank. Acontrol represents the OD450 absorbance of control.
ADMET analysis
ADMET study is designed to predict ADMET properties (absorption, distribution, metabolism, excretion, and toxicity) of the compound 13t via utilizing the Discovery Studio 2019 and the SwissADME web server40.
Statistical analyses
All data were analyzed with the unpaired study t-test by using GraphPad Prism 8, and the data were presented as the mean ± SEM. All cell culture experiments were performed independently for at least three times and in triplicate each time. P value of < 0.05 or < 0.001 was considered statistically significant.
Acknowledgements
This work was supported by the key Scientific Research Project of Hunan Education Department (22A0536), Hunan Provincial Natural Science Foundation of China (2025JJ70200) and Aid Program for Science and Technology Innovative Research Team in Higher Educational Institutions of Hunan Province(Xiangjiaotong[2023] NO 233).
Author contributions
D.L. and J.T. performed research and drafted the manuscript. D.L. synthesized all kojic acid-triazole derivatives. D.L. and S.T. tested the in vitro bioactivity. D.L., J.T., and D.W. performed the computational work and interpreted the data. J.L., J.T., S.T. and D.W. conceived and supervised the overall project and revised the manuscript. All authors have read and approved the final text and consent to its publication.
Data availability
All data generated or analyzed during this study are included in this published article.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Raza, H. et al. 2-Aminothiazole-oxadiazole bearing N-arylated butanamides: convergent synthesis, tyrosinase Inhibition, kinetics, structureactivity relationship, and binding conformations. Chem. Biodivers.20, e202201019 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Yousefnejad, F. et al. Ugi bis-amide derivatives as tyrosinase inhibitor; synthesis, biology assessment, and in Silico analysis. Chem. Biodivers.20, e202200607 (2023). [DOI] [PubMed] [Google Scholar]
- 3.Sepehri, N. et al. The natural-based optimization of kojic acid conjugated to different thio-quinazolinones as potential anti-melanogenesis agents with tyrosinase inhibitory activity. Bioorg. Med. Chem.36, e116044 (2021). [DOI] [PubMed]
- 4.Ismaya, W. T. et al. Crystallization and preliminary X-ray crystallographic analysis of tyrosinase from the mushroom Agaricus bisporus. Acta Crystallogr. F. 67, 575–578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ielo, L., Deri, B., Germanò, M. P. & Luca, L. D. Exploiting the 1-(4-fluorobenzyl)pi- perazine fragment for the development of novel tyrosinase inhibitors as anti-melanogenic agents: design, synthesis, structural insights and biological profile. Eur. J. Med. Chem.178, 380–389 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Nazir, Y., Saeed, A. & Rafiq, M. Hydroxyl substituted benzoic acid/cinnamic acid derivatives. Tyrosinase inhibitory kinetics, anti-melanogenic activity and molecular Docking studies. Bioorg. Med. Chem. Lett.30, e126722 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Nasab, N. H. et al. Synthesis and discovery of potential tyrosinase inhibitor of new coumarin-based thiophenyl-pyrazolylthiazole nuclei: in vitro evaluation, cytotoxicity, kinetic, and computational studies. Chem. Biol. Drug Des.101, 1262–1272 (2023). [DOI] [PubMed] [Google Scholar]
- 8.Patrycja, L. et al. Rafał, L. Tripeptides conjugated with thiosemicarbazones: new inhibitors of tyrosinase for cosmeceutical use. J. Enzym. Inhib. Med. Chem.38, e2193676 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khoa, N. M., Phong, N. V., Yang, S. Y., Min, B. S. & Kim, J. A. Spectroscopic analysis, kinetic mechanism, computational docking, and molecular dynamics of active metabolites from the aerial parts of Astragalus Membranaceus bunge as tyrosinase inhibitors. Bioorg. Chem.134, e106464 (2023). [DOI] [PubMed] [Google Scholar]
- 10.Chen, Q. et al. Inhibitory mechanism of scutellarein on tyrosinase by kinetics, spectroscopy and molecular simulation. Spectrochim. Acta A. 296, e122644 (2023). [DOI] [PubMed] [Google Scholar]
- 11.He, M., Fan, M., Yang, W., Peng, Z. & Wang, G. Novel Kojic acid-1,2,4- triazine hybrids as anti-tyrosinase agents: synthesis, biological evaluation, mode of action, and anti-browning studies. Food Chem.419, e136047 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Al-Rooqi, M. M. et al. Evaluation of 2,3-dihydro-1,5-benzothiazepine derivatives as potential tyrosinase inhibitors: In vitro and in silico studies. ACS Omega. 8, 17195–17208 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng, Z. et al. A systematic review of synthetic tyrosinase inhibitors and their structure-activity relationship. Crit. Rev. Food Sci. Nutr.62, 4053–4094 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Xue, S. et al. Design, synthesis, and biological evaluation of novel hybrids containing dihydrochalcone as tyrosinase inhibitors to treat skin hyperpigmentation. J. Med. Chem.66, 5099–5117 (2023). [DOI] [PubMed] [Google Scholar]
- 15.Mohammadsadeghi, N., Mahdavi, A., Saadati, F. & Mohammadi, F. In silico and in vitro studies of novel derivatives of tyrosol and raspberry ketone as the mushroom tyrosinase inhibitors. Food Chem.424, 136413 (2023). [DOI] [PubMed] [Google Scholar]
- 16.Muddathir, A. M., Yamauchi, K., Batubara, I., Mohieldin, E. A. M. & Mitsunaga, T. Anti-tyrosinase, total phenolic content and antioxidant activity of selected Sudanese medicinal plants. S. Afr. J. Bot.109, 9–15 (2017). [Google Scholar]
- 17.He, M., Fan, M., Peng, Z. & Wang, G. An overview of hydroxypyranone and hydroxypyridinone as privileged scaffolds for novel drug discovery. Eur. J. Med. Chem.221, e113546 (2021). [DOI] [PubMed] [Google Scholar]
- 18.He, M., Fan, M., Liu, W., Li, Y. & Wang, G. Design, synthesis, molecular modeling, and biological evaluation of novel Kojic acid derivatives containing bioactive heterocycle moiety as inhibitors of tyrosinase and antibrowning agents. Food Chem.362, e130241 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Santi, M. D., Peralta, M. A., Puiatti, M., Cabrera, J. L. & Ortega, M. G. Melanogenic inhibitory effects of triangularin in B16F0 melanoma cells, in vitro and molecular Docking studies. Bioorg. Med. Chem.27, 3722–3728 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Radhakrishnan, S., Shimmon, R., Conn, C. & Baker, A. Design, synthesis and biological evaluation of hydroxy substituted amino chalcone compounds for antityrosinase activity in B16 cells. Bioorg. Chem.62, 117–123 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Lee, S. et al. Inhibitory effects of N-(acryloyl)benzamide derivatives on tyrosinase and melanogenesis. Bioorg. Med. Chem.27, 3929–3937 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Hashemi, S. M. & Emami, S. Kojic acid-derived tyrosinase inhibitors: synthesis and bioactivity. Pharm. Biomed. Res.1 (1), 1–17 (2015). [Google Scholar]
- 23.Emami, S., Ahmadi, R., Ahadi, H. & Ashooriha, M. Diverse therapeutic potential of 3-hydroxy-4-pyranones and related compounds as Kojic acid analogs. Med. Chem. Res.31 (11), 1842–1861 (2022). [Google Scholar]
- 24.Rho, H. S. et al. Synthesis and biological evaluation of Kojyl thioether derivatives as tyrosinase inhibitors. B Korean Chem. Soc.31, 2375–2378 (2010). [Google Scholar]
- 25.Xie, W. et al. Synthesis and biological evaluation of Kojic acid derivatives containing 1,2,4-triazole as potent tyrosinase inhibitors. Chem. Biol. Drug Des.86, 1087–1092 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Wang, G. C., He, M., Huang, Y. & Peng, Z. Y. Synthesis and biological evaluation of new Kojic acid-1,3,4-oxadiazole hybrids as tyrosinase inhibitors and their application in the anti-browning of fresh-cut mushrooms. Food Chem.409, e135275 (2023). [DOI] [PubMed] [Google Scholar]
- 27.Dheer, D., Singh, V. & Shankar, R. Medicinal attributes of 1,2,3– Triazoles: current developments. Bioorg. Chem.71, 30–54 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Badar, A. D. et al. Synthesis, antimicrobial evaluation and Docking study of novel thiosemicarbazone clubbed with 1,2,3-triazoles. Curr. Bioact. Compd.17, e010621185856 (2021). [Google Scholar]
- 29.Singh, G. et al. 1,2,3-Triazole functionalized organosilanes as naked eye sensor of Zr (IV) and a potent tyrosinase inhibitor. J. Mol. Struct.1318, e139309 (2024). [Google Scholar]
- 30.Zhou, W., Tang, J., Zhou, X. & Liu, J. Tyrosinase inhibition by novel benzimidazole-thione schiff base derivatives. Lett. Drug Des. Discov. 19, 782–790 (2022). [Google Scholar]
- 31.Gulcin, I. & Alwasel, S. H. Metal ions, metal chelators and metal chelating assay as antioxidant method. Process10 (1), 132–148 (2022). [Google Scholar]
- 32.Taslimi, P. et al. Anti-Alzheimer, antidiabetic and antioxidant potential of Satureja cuneifolia and analysis of its phenolic contents by LC-MS/MS. Arab. J. Chem.13 (3), 4528–4537 (2020). [Google Scholar]
- 33.Zhou, W., Wu, F. & Liu, J. Biological evaluation and synthesis of thiazole schiff base derivatives. Heterocycles102 (7), 1337–1353 (2021). [Google Scholar]
- 34.Idris, S., Jan, F., Wheed, M., Alam, A. & Khan, M. Multifaceted bioactivity of Thiosemicarbazide derivatives: synthesis, characterization, and DFT investigations on Inhibition of α-amylase, hydroxyl radical scavenging, and iron chelating activities with molecular Docking insights. J. Mol. Struct.1304, e137669 (2024). [Google Scholar]
- 35.Tang, J., Liu, J. & Wu, F. Molecular docking studies and biological evaluation of 1,3,4-thiadiazole derivatives bearing schiff base moieties as tyrosinase inhibitors. Bioorg. Chem.69, 29–36 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Hou, Z. Q., Feng, Y. Y., Wei, S. C. & Wang, Q. G. Effects of curing treatment on the Browning of fresh-cut potatoes. Am. J. Potato Res.91, 655–662 (2014). [Google Scholar]
- 37.Gulcin, I. & Alwasel, S. H. DPPH radical scavenging assay. Processes11 (8), 2248–2268 (2023). [Google Scholar]
- 38.Gulcin, I. Antioxidants and antioxidant methods—An updated overview. Arch. Toxicol.94 (3), 651–715 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Romagnoli, R., Oliva, P., Prencipe, F., Manfredini, S. & Bortolozzi, R. Cinnamic acid derivatives linked to arylpiperazines as novel potent inhibitors of tyrosinase activity and melanin synthesis. Eur. J. Med. Chem.231, e114147 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.7(1), e42717 (2017). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.