

Abstract
AIM
To determine the therapeutic benefits of fenofibrate (Feno) on the dysfunction of high glucose (HG)-induced human retinal microvascular endothelial cells (HRMECs) and to elucidate the underlying molecular mechanism.
METHODS
HRMEC dysfunction model was established by 48h glucose (30 mmol/L) treatment and treated with Feno/NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activator (Nigericin). Cell viability/apoptosis were assessed by cell counting kit-8 (CCK-8)/terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay (TUNEL) staining and flow cytometry assays. Levels of apoptosis- (Bcl-2-associated X protein, Bax/B-cell lymphoma 2, Bcl-2), vascular permeability-(vascular endothelial growth factor, VEGF) and inflammasome activation-related proteins (NLRP3/cleaved caspase-1/apoptosis-associated speck-like protein containing a CARD, ASC), as well as inflammatory factors (interleukin, IL-6/IL-1β/tumor necrosis factor, TNF-α/IL-18) were determined with Western blot/enzyme linked immunosorbent assay (ELISA). Cell permeability/reactive oxygen species (ROS) level/superoxide dismutase (SOD) activity/malondialdehyde (MDA) content were assessed by Evans blue staining/2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe/SOD kit/MDA kit.
RESULTS
HRMEC dysfunction was successfully induced by HG, evidenced by decreased viability (P<0.001), increased apoptosis (P<0.001), permeability (P<0.001), and inflammatory factor levels (P<0.001). Feno treatment significantly ameliorated HG-induced HRMEC dysfunction (P<0.01). Meanwhile, HG induction increased ROS production (P<0.001) and MDA content (P<0.001) in HRMECs, while reducing SOD activity (P<0.001), indicative of oxidative stress. This was, however, abolished by Feno (P<0.05). Moreover, Feno eliminated activation of NLRP3 inflammasomes (P<0.05) in HG-induced HRMECs. Strikingly, activation of NLRP3 inflammasomes partially averted the inhibition of Feno on HG-induced HRMEC dysfunction (P<0.05).
CONCLUSION
Feno represses oxidative stress and NLRP3 inflammasome activation, consequently alleviating HG-induced HRMEC dysfunction.
Keywords: fenofibrate, human retinal microvascular endothelial cells, high glucose, NOD-like receptor thermal protein domain associated protein 3 inflammasomes, oxidative stress
INTRODUCTION
Diabetic retinopathy (DR) is referred to as mild, moderate, or proliferative retinopathy in at least one eye; it is a common microvascular complication of diabetes, and continues to be a predominant cause of preventable blindness among working-aged populations[1]. Microvascular dysfunction, breakdown of the blood-retinal barrier in the retina, and retinal neovascularization are the main pathological changes of DR[2]. A former study has shown that due to a long-term contact with glucose-rich blood, human retinal microvascular endothelial cells (HRMECs) are damaged by high glucose (HG), characterized by an increased production of reactive oxygen species (ROS), and activated inflammatory reactions as a consequence of activation of NOD-like receptor thermal protein domain associated protein 3 (NLRP3)-mediated inflammatory factors. This reaction facilitates cell apoptosis, and increases vascular endothelial growth factor (VEGF) expression and endothelial permeability, ultimately triggering DR[3]–[4]. As the dysfunction of HRMECs caused by hyperglycemia is a critical event in the early DR vascular leakage and late DR angiogenesis, inhibition of their dysfunction may be a therapeutic avenue to alleviate DR.
NLRP3 inflammasomes are intracellular multimolecular complexes that are emerging as key regulators of innate immune response, consisting of sensor molecule NLRP3 protein, effector protease caspase-1, and apoptosis-associated speck-like protein containing a CARD (ASC)[5]–[6]. It is reported that HG environment is a potential mechanism underlying the increase of ROS production and oxidative stress, along with activation of NLRP3; the activated NLRP3 can recruit ASC and pro-caspase-1 to assemble the inflammasome complex, driving activation of caspase-1, followed by generation of pro-inflammatory factors, such as interleukin (IL)-1β and IL-18. This process further induces inflammatory reactions in HRMECs and provokes cell apoptosis[7]–[8]. Of note, existing studies have identified the intimate association of the activation of NLRP3 inflammasome with DR progression; specifically, nuclear factor-kappa B/NLRP3 inflammasome pathway activation can cause oxidative damage in human retinal pigment epithelial cells treated by HG, consequently leading to DR[8]. Although the activation of NLRP3 inflammasomes engaged in the pathological process of DR, its possible participation in the dysfunction of HRMECs remains largely unknown. Notably, fenofibrate (Feno) is an agonist of peroxisome proliferator-activated receptor-α (PPAR)-α, which has demonstrated therapeutic potentials in alleviating diabetes-associated vascular diseases, especially DR[9]–[10]. Previous research has established definitively that Feno is able to reduce retinal inflammation, ameliorate hypertriglyceridemia and dyslipidemia, and decrease macular degeneration, thus providing protection against DR[11]. Moreover, there is experimental evidence that Feno can limit NLRP3 inflammasome activation and alleviate DR[12]. Accordingly, in this research, we aimed to elucidate whether Feno prevented the HG-induced dysfunction of HRMECs by suppressing the NLRP3 inflammasome activation, and to examine the possible clinical implications of this phenomenon. Through these aspects, we hope to provide new ideas and interventional targets for DR treatment.
MATERIALS AND METHODS
Cell Culture and Treatment
HRMEC line acquired from ATCC (Rockville, MD, USA) underwent culture in DMEM (10564011, Gibco, Carlsbad, CA, USA) that contains glucose (5.5 mmol/L), fetal bovine serum (10%, 10099, Gibco) and penicillin-streptomycin (1%) in an incubator with 5% CO2 and 95% humidity at 37°C. Upon reaching confluence (around 80%-90%), cells were subjected to two washes with phosphate buffer saline (PBS) and incubation with DMEM containing 30 mmol/L glucose for 48h[13] to establish an HRMEC dysfunction model.
Then the cells were assigned as follows: normal glucose (NG) group (cells were incubated with the 5.5 mmol/L glucose-containing medium for 48h), osmotic pressure (OSM) group (cells underwent incubation with the medium containing 25 mmol/L mannitol and 5 mmol/L glucose for 48h), HG+dimethyl sulphoxide (DMSO) group (cells underwent incubation with the 30 mmol/L glucose-containing medium for 48h or treated with the same volume of DMSO as Feno/Nigericin treatment), HG+Feno group [cells were incubated with the 30 mmol/L glucose-containing medium and simultaneously treated with 100 µmol/L Feno (F6020, Sigma-Aldrich, St. Louis, MO, USA) for 48h][14], and HG+Feno+Nigericin group (cells were incubated with the 30 mmol/L glucose-containing and simultaneously treated with 100 µmol/L Feno and 10 µmol/L nigericin for 48h[15]. Among them, nigericin (481990, Sigma-Aldrich) is an activator of the NLRP3 inflammasomes.
Cell Viability Assessment
Viability of HRMECs was evaluated by Enhanced cell counting kit-8 (CCK-8; C0041, Beyotime, Shanghai, China). Cells collected from each group were seeded into 96-well plates and subjected to incubation with CCK-8 reagent (volume of 10 µL) in the dark at ambient temperature for 2h. Absorbance value at 450 nm was then measured by means of a plate-reading luminometer (acquired from Varioskan™; Thermo Scientific, Rockford, IL, USA).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick End Labeling Assay Staining
Cells underwent 1h fixation at ambient temperature by means of 4% paraformaldehyde (P0099, Beyotime). After washing with PBS, cells underwent incubation on ice with 1% Triton X-10 (P0096, Beyotime) for 2min and then with terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay (TUNEL) reaction mixture (C1086, Beyotime) at 37°C in the dark for 1h. After counterstaining by virtue of DAPI (C1005, Beyotime), the cells were observed and imaged by virtue of fluorescence microscope (acquired from Olympus Life-Sciences, Tokyo, Japan).
Flow Cytometry
Cells were detached with trypsin, centrifuged and resuspended in PBS to make cell suspension and counted. After centrifugation (105 cells), the cells underwent resuspension with Annexin V-fluorescein isothiocyanate (FITC, 195 µL) binding buffer, and incubation with Annexin V-FITC (5 µL, C1062S, Beyotime) and PI staining solution for 10-20min in the dark. Finally, the cells were subjected to analysis by virtue of flow cytometer (acquired from BD Biosciences, San Jose, CA, USA).
Evans Blue Staining
Cell permeability was examined by Evans blue (E2129, Sigma-Aldrich) staining. Cells underwent staining with Evans blue staining solution for 2h. After washing, cells were extracted with formamide (47671, Sigma-Aldrich) for 18h at 70°C and centrifuged at low temperature, lasting 30min. Absorbance of the supernatant was measured at 620 nm.
Enzyme Linked Immunosorbent Assay
Tumor necrosis factor (TNF)-α, IL-6, IL-18 and IL-1β levels in cell supernatants were assayed following instructions in enzyme linked immunosorbent assay (ELISA) kits (all acquired from R&D Systems, Minneapolis, MN, USA).
Western Blot
Cells were treated by virtue of RIPA (P0013B, Beyotime) which contains protease inhibitors. Afterwards, protein concentration measurement was followed by virtue of bicinchoninic acid (BCA) protein quantification kit (P0012, Beyotime). Different concentrations of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel were selected on the basis of the molecular weight of the target protein. After the separation gel and concentration gel were prepared, the protein was separated by electrophoresis using an electrophoresis instrument. Afterwards, the protein was transferred onto polyvinylidene fluoride (PVDF) membranes for a duration of 1.5h at a low temperature of 4°C. Subsequently, the PVDF membranes underwent 5% skim milk powder blocking at ambient temperature for a duration of 1.5h, which was followed by overnight incubation at 4°C with rabbit anti-human primary antibodies against B-cell lymphoma 2 (Bcl-2; 1:2000, ab182858, Abcam, Cambridge, UK), Bcl-2-associated X protein (Bax; 1:1000-1:10000, ab32503, Abcam), NLRP3 (1:1000, ab263899, Abcam), ASC (1:1000, ab283684, Abcam), cleaved caspase-1 (1:500, ab32042, Abcam), and VEGF (1:1000-1:5000, ab32152, Abcam), with glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:10000, ab181602, Abcam) as the internal reference. After 4 rinses with tris-buffered saline-tween 20 (TBST; 10min/time), the membranes underwent incubation with goat anti-rabbit immunoglobulin G (1:5000, ab205718, Abcam) labelled with horseradish peroxidase at ambient temperature for 2h. Enhanced chemiluminescence (acquired from EMD Millipore, Billerica, MA, USA) was utilized for PVDF development. Grayscale analysis was followed using Media Cybernetics' Image Pro Plus 6.0 (Silver Spring, MD, USA).
ROS Content Determination
DCFH-DA fluorescent probe (acquired from Beyotime) was adopted for intracellular ROS measurement. In short, cells were cultured with DCFH-DA (10 µmol/L) for 20min at 37°C and rinsed three times with serum-free medium. A multimode microplate reader (Varioskan LUX 3020, Thermo Scientific) or FACSCalibur flow cytometer (acquired from BD Biosciences) was adopted to determine the fluorescence (525 nm emission wavelength, 488 nm excitation wavelength).
Superoxide Dismutase Activity Evaluation
Superoxide dismutase (SOD) activity was assessed by SOD Kit (S0101S, Beyotime). In short, cells were lysed by virtue of sonication, whereupon the lysate was centrifuged at 3000 rpm for 20min. BCA kit was applied for measuring protein concentration. The double-distilled water and supernatant were placed to a 96-well plate, and the samples underwent incubation with reaction solution at temperature of 37°C for 20min. Then, a VICTOR X5 Multilabel Plate Reader (acquired from PerkinElmer, Waltham, MA) was used to determine SOD activity at 450 nm.
Malondialdehyde Content Detection
Malondialdehyde (MDA) Assay Kit (S0131S, Beyotime) was applied for measuring MDA content. Cells were ultrasonically lysed. Subsequent to centrifugation at 3000 rpm, lasting 20min, the supernatant was mixed with MDA precipitating solution, and allowed to stand. The supernatant was discarded by means of centrifugation, and the samples were mixed with distilled water. Then, the absorbance value at 532 nm was evaluated and the MDA content was calculated.
Statistical Analysis
All analyses were done by means of GraphPad Prism software (version 8.0.1, from GraphPad Software Inc., San Diego, CA, USA). Measurement data were summarized as mean±standard deviation (SD). Two-group data were compared using t-test, and multi-group data using one-way ANOVA with Tukey's test. P<0.05 reflects significance.
RESULTS
HG-Induced Dysfunction of HRMECs
HRMECs were cultured in vitro and treated with 30 mmol/L HG to establish a HRMEC dysfunction model, which were simultaneously cultured in a medium with the addition of 5 mmol/L glucose and 25 mmol/L mannitol to exclude the OSM effect[13]–[14]. CCK-8 manifested that the viability of HRMECs was prominently reduced in HG group versus NG group (P<0.001; Figure 1A). TUNEL staining and flow cytometry (Figure 1B-1C) showed augmented HRMEC apoptosis in HG group versus NG group (P<0.001). As reflected by Western blot, in comparison with NG group, Bax protein was elevated, and Bcl-2 protein was lowered in HG group (all P<0.001; Figure 1D). Subsequently, the permeability of HRMECs was measured by Evans blue staining. It was noted that cell permeability was remarkably increased after HG induction (P<0.001; Figure 1E). Western blot was employed to assess the level of VEGF, and revealed that its protein level was up-regulated in cells after HG induction (all P<0.001; Figure 1D). Further, ELISA was used to determine levels of inflammatory factors, namely TNF-α and IL-6. As displayed in Figure 1F, their levels were higher in HG group versus NG group (all P<0.001). Besides, all of these indicators had no apparent changes between OSM and NG groups (all P>0.05; Figure 1A-1F), indicating that high OSM due to HG did not lead to HRMEC dysfunction. Overall, these results confirmed the successful induction of HRMEC dysfunction by HG.
Figure 1. The influence of HG on apoptosis of HRMECs.

A: CCK-8 was conducted to evaluate cell viability; B: TUNEL staining was conducted to assess apoptosis; C: Flow cytometry was adopted to evaluate apoptosis; D: Western blot was conducted to measure the protein levels of apoptosis-associated proteins Bax and Bcl-2, as well as vascular permeability-related protein VEGF; E: Cell permeability was assayed by Evans blue staining; F: ELISA was conducted to measure TNF-α and IL-6 levels. The cell-based in vitro experiments were run three times in an independent manner, with the data represented as mean±SD. Multi-group data were compared by one-way ANOVA, with Tukey's test. cP<0.001. HRMECs: Human retinal microvascular endothelial cells; NG: Normal glucose; HG: High glucose; OSM: Osmotic pressure; CCK-8: Cell counting kit-8; DMSO: Dimethyl sulphoxide; TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling; TNF-α: Tumor necrosis factor-α; IL-6: Interleukin-6; ELISA: Enzyme-linked immunosorbent assay; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; VEGF: Vascular endothelial growth factor; OD: Optical density; DAPI: 4′,6-diamidino-2-phenylindole; SD: Standard deviation; ANOVA: Analysis of variance.
Feno's Amelioration of HG-Induced Dysfunction of HRMECs
In order to investigate whether Feno had an ameliorative effect on HG-induced HRMEC dysfunction, we treated the HG-induced HRMECs with 100 µmol/L Feno. Relative to the HG+DMSO group, the HG+Feno group showed an observable increase in cell viability and Bcl-2 protein level while cell apoptosis and Bax protein level exhibited a tendency to decrease (all P<0.01; Figure 2A-2D). Furthermore, after Feno treatment, VEGF protein level and cell permeability were significantly reduced, and the TNF-α and IL-6 levels were decreased (all P<0.01; Figure 2D-2F). Taken together, the above-revealed results indicated that Feno could alleviate HG-induced HRMEC dysfunction.
Figure 2. The role of Feno in HG-induced dysfunction of HRMECs.

A: CCK-8 was performed to determine cell viability; B: TUNEL staining was performed to evaluate cell apoptosis; C: The cell apoptosis was assessed by flow cytometry; D: Determination of Bax, Bcl-2, and VEGF protein levels by Western blot; E: Evaluation of cell permeability by Evans blue staining; F: ELISA assay was performed to assess the levels of IL-6 and TNF-α. The cell-based in vitro experiments were run three times in an independent manner. The data were represented as mean±SD, with independent sample t-test adopted for two-group data comparisons. bP<0.01. CCK-8: Cell counting kit-8; HG: High glucose; Feno: Fenofibrate; HRMECs: Human retinal microvascular endothelial cells; DAPI: 4′,6-diamidino-2-phenylindole; DMSO: Dimethyl sulphoxide; TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling; Bax: Bcl-2-associated X protein; TNF-α: Tumor necrosis factor-α; IL-6: Interleukin-6; OD: Optical density; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; VEGF: Vascular endothelial growth factor; Bcl-2: B-cell lymphoma 2; SD: Standard deviation; ELISA: Enzyme-linked immunosorbent assay.
Feno's Suppression of HG-Induced Oxidative Stress
HG can exacerbate production of ROS, which can induce oxidative stress, and damage cells[8]. To this end, in this study, we used fluorescent probe DCFH-DA to detect intracellular ROS. As shown in Figure 3A, a higher content of ROS was found in HG+DMSO group versus NG group (P<0.001); by contrast, the content of ROS was decreased in HG+Feno group versus HG+DMSO group (all P<0.05). SOD activity (Figure 3B) and MDA content (Figure 3C) were subsequently measured by corresponding kits. Versus NG group, SOD activity was down-regulated and MDA content was elevated in HG+DMSO group (all P<0.001). Strikingly, these changes were annulled by Feno treatment (all P<0.05). Thus, it was plausible to conclude that Feno could arrest HG-induced oxidative stress.
Figure 3. Influence of Feno on HG-induced oxidative stress.

A: Fluorescent probe DCFH-DA was adopted to detect intracellular ROS; B: SOD activity measurement was done by the SOD kit; C: The MDA content measurement was done by an MDA kit. The cell-based in vitro experiments were run three times in an independent manner, and the data were represented as mean±SD. Data were analyzed by virtue of one-way ANOVA with Tukey's test. aP<0.05, bP<0.01, cP<0.001. Feno: Fenofibrate; SOD: Superoxide dismutase; NG: Normal glucose; DMSO: Dimethyl sulphoxide; ROS: Reactive oxygen species; MDA: Malondialdehyde; HG: High glucose; ANOVA: Analysis of variance; DCFH-DA: 2′,7′-Dichlorodihydrofluorescein diacetate.
Feno Inhibited the Activation of NLRP3 Inflammasomes
We extended our mechanistic findings to determine the inhibiting effect of Feno on HG-induced dysfunction of HRMECs and the resultant oxidative stress. HG-induced HRMECs were treated with 100 µmol/L Feno. Western blot was performed to detect the levels of NLRP3 inflammasome-associated proteins (ASC, NLRP3, and cleaved caspase-1). The results in Figure 4A revealed that the protein levels of ASC, NLRP3, and cleaved caspase-1 in HG+DMSO group were evidently higher than in NG group (all P<0.001); conversely, these changes were all reversed after Feno treatment (all P<0.05). Subsequently, we examined the levels of inflammatory factors IL-1β and IL-18, major pro-inflammatory factors participating in activation of NLRP3 inflammasome, by ELISA. HG+DMSO group exhibited notably higher IL-18 and IL-1β levels than NG group (all P<0.001; Figure 4B), whereas Feno treatment abolished this trend (all P<0.01). These results emanated that Feno curbed of NLRP3 inflammasome activation.
Figure 4. Influence of Feno on the activation of NLRP3 inflammasomes.

A: Western blot was performed to measure the protein levels of ASC, NLRP3 and cleaved caspase-1; B: ELISA was performed to analyze the levels of IL-18 and IL-1β. The cell-based in vitro experiments were run three times in an independent manner, and the data were represented as the mean±SD. Data were analyzed by one-way ANOVA with Tukey's test. aP<0.05, bP<0.01, cP<0.001. Feno: Fenofibrate; NLRP3: NOD-like receptor thermal protein domain-associated protein 3; ASC: Apoptosis-associated speck-like protein containing a CARD; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; HG: High glucose; IL: Interleukin; SD: Standard deviation; DMSO: Dimethyl sulphoxide; ELISA: Enzyme-linked immunosorbent assay; NG: Normal glucose; ANOVA: Analysis of variance.
Activation of NLRP3 inflammasomes partially reversed the alleviative effect of Feno on HG-induced HRMEC dysfunction. To further explore whether Feno prevented HG-induced dysfunction in HRMECs via inactivation of NLRP3 inflammasomes, we processed HG-induced HRMECs with 10 µmol/L NLRP3 inflammasome activator Nigericin and 100 µmol/L Feno concomitantly. ASC, NLRP3, and cleaved caspase-1 protein levels, the levels of IL-18 and IL-1β, cell apoptosis and permeability, as well as the VEGF protein level were apparently increased in HG+Feno+Nigericin group versus HG+Feno group (all P<0.05; Figure 5A-5F). Moreover, the Bax protein level and the levels of inflammatory factors TNF-α and IL-6 were increased but the protein level of Bcl-2 was diminished (all P<0.05; Figure 5F-5G). Remarkably, the results were reproduced in HG-induced mouse retinal microvascular endothelial cells (mRMECs) treated with Feno or combined with Nigericin. The above diagrams served to illustrate that activation of the NLRP3 inflammasomes partially averted the suppressing effect induced by Feno on HG-induced dysfunction in HRMECs.
Figure 5. The effect of activating NLRP3 inflammasomes on the prevention of Feno on HRMEC dysfunction.

A: Detection of ASC, NLRP3, and cleaved caspase-1 protein levels were done by virtue of Western blot; B: ELISA was conducted to analyze IL-1β and IL-18 levels; C: TUNEL assay was conducted to assess HRMEC apoptosis; D: Flow cytometry was conducted to evaluate HRMEC apoptosis; E: Cell permeability was tested by Evans blue staining; F: Determination of Bax, Bcl-2 and VEGF protein levels by Western blot; G: ELISA was conducted to measure the levels of IL-6 and TNF-α. The cell-based in vitro experiments were run three times in an independent manner, with the data represented as mean±SD. Independent sample t-test was performed to analyze. aP<0.05, bP<0.01. Feno: Fenofibrate; DAPI: 4′,6-diamidino-2-phenylindole; NLRP3: NOD-like receptor thermal protein domain-associated protein 3; HRMEC: Human retinal microvascular endothelial cell; HG: High glucose; ASC: Apoptosis-associated speck-like protein containing a CARD; DMSO: Dimethyl sulphoxide; TNF-α: Tumor necrosis factor-α; IL: Interleukin; ELISA: Enzyme-linked immunosorbent assay; Bcl-2: B-cell lymphoma 2; VEGF: Vascular endothelial growth factor; OD: Optical density; Bax: Bcl-2-associated X protein; SD: Standard deviation.
DISCUSSION
Feno has been extensively studied in a variety of contexts, especially in diabetes; Feno exerts profound therapeutic effects on diabetes, by concurrently ameliorating hypertriglyceridaemia and dyslipidaemia, and attenuating inflammation in the retina[11]. It has been also proven effective in combating DR; mechanistically, it can alleviate HG-induced HRMEC dysfunction due to the regulation on lipid metabolism and retinal inflammation reduction[16]. Accordingly, this study explored the mechanism whereby Feno alleviates DR, and the findings highlighted that Feno relieved the dysfunction of HG-induced HRMECs by disrupting activation of NLRP3 inflammasomes. These data provide new insights into the pathogenesis of DR and points towards novel therapeutic targets.
The permeability through interfering the blood-retinal barrier cell junctions has been well established as a signature of DR[17]. Increasing studies have found that inflammation is also a core event in the progression of diabetes and metabolic syndrome[18]. Notably, accumulating evidence has demonstrated that Feno is an effective modality for treating DR[9]–[10]. Suppression of the ANGPTL3 pathway may be one of the underlying mechanisms responsible for Feno's protection against DR[14]. In addition, another study has documented that Feno impedes HG cellular metabolic memory in DR via a sirtuin 1-dependent pathway[19]. Not surprisingly, HG could trigger dysfunction of HRMECs, which is manifested by reduced viability, amplified apoptosis and permeability, as well as elevated levels of inflammatory cytokines IL-6 and TNF-α in HRMECs after HG induction in vitro. By contrast, Feno treatment yielded opposite results. Partially in agreement, Feno is capable of protecting retinal-choroidal vascular endothelial cells against apoptosis induced by oxidative stress[20]. Besides, Feno has the potency to hinder apoptotic cell death due to serum deprivation through the PPARalpha-independent, but AMPK-dependent pathway[21]. There is also evidence that Feno improves systemic and retinal inflammation and regulates gut microbiota in mice caused by high-fat diet[22]. In conclusion, Feno could alleviate HG-induced HRMEC dysfunction.
HG leads to overwhelming ROS production and sustained oxidative stress, which damages cells and exacerbates disease via triggering inflammation[8]. ROS and MDA contents as well as SOD activity are often used to evaluate the oxidative stress levels; increase of SOD activity and decrease of ROS and MDA contents are indicatives of promoted oxidative stress[23]–[24]. Our results demonstrated that HG induced oxidative stress manifested by stimulated ROS and MDA contents, and suppressed SOD activity, whereas Feno treatment produced opposite trends. This implied that Feno could attenuate HG-induced oxidative stress. In line with our findings, previous work has signified that Feno can ameliorate oxidative stress-driven retinal microvascular dysfunction in diabetic rats[25]. Mechanistic investigations indicated that the mitigation of Feno on renal function was attributed to the attenuation of oxidative stress, inflammation and cell apoptosis[26]. What's more, PPAR-α induced by Feno can strengthen anti-oxidant defense systems and improve oxidative stress caused by diabetes[27]. Furthermore, another previous study indicated that Feno prominently declines the levels of inflammatory chemokines and decreases oxidative products in diabetic retina, thus preventing DR progression[28]. Altogether, our findings preliminarily uncovered that Feno could inhibit oxidative stress induced by HG.
It is also noteworthy that high level of ROS is capable of activating NLRP3 inflammasomes[3]. NLRP3 inflammasome has the potency to sense soluble pathogen and danger-associated molecular patterns, which is the central hub of the inflammatory response system that triggers pro-inflammatory responses[29]. When inflammatory stimuli are sensed, the activated NLRP3 then binds adaptor protein ASC to recruit cleaved caspase-1, which provokes the downstream reaction, causing IL-1β release[30]. Importantly, the NLRP3 inflammasomes are activated in DR models both in vivo and in vitro[31]. Studies have shown that inactivating the NLRP3 inflammasomes can ameliorate DR[32]–[33]. Based on prior studies and the results from our own analysis, we have evidence supporting that Feno impeded NLRP3 inflammasome activation. Thus, we processed the HG-induced HRMECs with Feno, and identified suppression in HG-activated NLRP3 inflammasomes, manifested by the decreased ASC, NLRP3, and cleaved caspase-1 protein levels and IL-18 and IL-1β levels. Feno, by inhibiting NLRP3 inflammasome activation, reduces the production of downstream inflammatory factors (namely IL-18 and IL-1β), thereby alleviating HG-induced HRMEC apoptosis and dysfunction (Figure 6).
Figure 6. Schematic representation summarizing the role of Feno in HG-induced dysfunction of HRMECs.
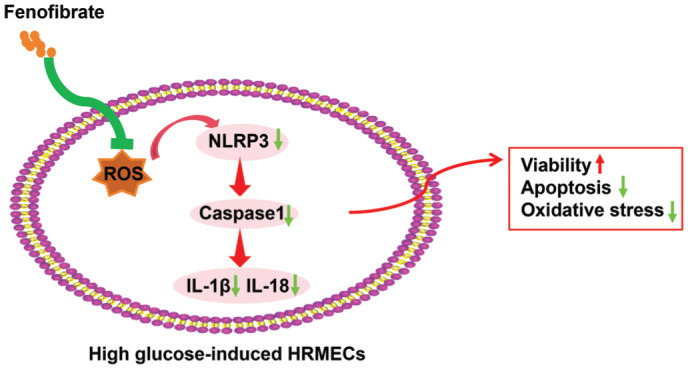
Feno reduces ROS generation, increases SOD activity, and suppresses NLRP3 inflammasome activation, which in turn decreases caspase-1 activation and pro-inflammatory cytokine release. This process plays a key role in reducing inflammation, oxidative stress, and cellular damage under HG conditions. SOD: Superoxide dismutase; NLRP3: NOD-like receptor protein 3; Caspase-1: Cysteine-dependent aspartate-specific protease 1; ROS: Reactive oxygen species; IL-18: Interleukin-18; IL-1β: Interleukin-1β; HRMEC: Human retinal microvascular endothelial cells.
Upon exposure to HG environment, NLRP3 inflammasomes are activated, driving the cleavage of caspase-1 and generation of pro-inflammatory cytokines, a process that exacerbates inflammation and oxidative stress in HRMECs[34]. By regulating the NLRP3-ASC-caspase-1 signaling pathway, Feno significantly suppresses these inflammatory responses, contributing to reduction of cell apoptosis and increase of vascular permeability. Furthermore, Feno also decreases the generation of ROS, which further attenuates the activation of NLRP3 inflammasomes, as high levels of oxidative stress can promote NLRP3 activation[35]. Overall, Feno inhibits the activation of NLRP3 inflammasomes through multiple mechanisms, thereby protecting HRMECs from damage induced by a HG environment. Next, to clarify the role of NLRP3 inflammasomes in Feno-mediated effects on HRMEC dysfunction, we activated NLRP3 inflammasomes in HG and Feno-treated HRMECs, and uncovered notable increases in cell apoptosis and permeability, as well as in the levels of IL-6 and TNF-α. Likewise, during hyperglycemia, the NLRP3 inflammasome dysfunction and activation are discovered in HRMECs[36]. Feno mitigates DR via regulating Nrf2 signaling and the inactivation of NLRP3 inflammasomes[12]. NLRP3 leads to HRMEC apoptosis and reduced NLRP3 weakens the levels of NLRP3 inflammasome components[37]. These suggested that Feno inactivated the NLRP3 inflammasomes, and conversely, activated NLRP3 inflammasomes partially counteracted the inhibition effect of Feno on the HG-induced dysfunction of HRMECs.
In summary, this study supported that Feno could reduce HRMEC dysfunction by curbing NLRP3 inflammasome activation. However, besides oxidative stress, NLRP3 activation can also be triggered by other factors, such as extracellular ATP and bacterial infections[38]–[39]. In this study, we didn't explore whether Feno exerted its effect by inactivating NLRP3 inflammasomes via these pathways and their mechanisms. Therefore, it is necessary to further study whether the mitigation of Feno on the dysfunction of HRMECs is due to other mechanisms associated with NLRP3 inflammasomes.
Footnotes
Foundation: Supported by grants from the Tianjin Key Medical Discipline (Specialty) Construction Project (No.TJYXZDXK-037A).
Conflicts of Interest: Shi Y, None; Chen HM, None; Liu AH, None; Li XR, None.
REFERENCES
- 1.Roy S, Kim D. Retinal capillary basement membrane thickening: Role in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res. 2021;82:100903. doi: 10.1016/j.preteyeres.2020.100903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonetti DA, Silva PS, Stitt AW. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol. 2021;17(4):195–206. doi: 10.1038/s41574-020-00451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang XL, Zhang Q, Xue WW, et al. Suppression of cAMP/PKA/CREB signaling ameliorates retinal injury in diabetic retinopathy. Kaohsiung J Med Sci. 2023;39(9):916–926. doi: 10.1002/kjm2.12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Li HY, Shao J, et al. GRP75 modulates endoplasmic reticulum–mitochondria coupling and accelerates Ca2+-dependent endothelial cell apoptosis in diabetic retinopathy. Biomolecules. 2022;12(12):1778. doi: 10.3390/biom12121778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magupalli VG, Negro R, Tian YZ, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369(6510):eaas8995. doi: 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Y, Xu W, Zhou RB. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–2127. doi: 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Wang Y, Xie TH, et al. Prostaglandin E2/EP2 receptor signalling pathway promotes diabetic retinopathy in a rat model of diabetes. Diabetologia. 2019;62(2):335–348. doi: 10.1007/s00125-018-4755-3. [DOI] [PubMed] [Google Scholar]
- 8.Choi YH. Reduction of high glucose-induced oxidative injury in human retinal pigment epithelial cells by sarsasapogenin through inhibition of ROS generation and inactivation of NF-κB/NLRP3 inflammasome pathway. Genes Genomics. 2023;45(9):1153–1163. doi: 10.1007/s13258-023-01417-2. [DOI] [PubMed] [Google Scholar]
- 9.Moran EP, Ma J-X. Therapeutic effects of PPARαon neuronal death and microvascular impairment. PPAR Res. 2015;2015:595426. doi: 10.1155/2015/595426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin L, Hua H, Ji Y, et al. Anti-inflammatory role of fenofibrate in treating diseases. Biomol Biomed. 2023;23(3):376–391. doi: 10.17305/bb.2022.8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kataoka SY, Lois N, Kawano S, et al. Fenofibrate for diabetic retinopathy. Cochrane Database Syst Rev. 2023;6(6):CD013318. doi: 10.1002/14651858.CD013318.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu QP, Zhang FJ, Zhang X, et al. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol Cell Biochem. 2018;445(1-2):105–115. doi: 10.1007/s11010-017-3256-x. [DOI] [PubMed] [Google Scholar]
- 13.He FT, Fu XL, Li MH, et al. USP14 regulates ATF2/PIK3CD axis to promote microvascular endothelial cell proliferation, migration, and angiogenesis in diabetic retinopathy. Biochem Genet. 2023;61(5):2076–2091. doi: 10.1007/s10528-023-10358-0. [DOI] [PubMed] [Google Scholar]
- 14.Wang N, Zou C, Zhao SZ, et al. Fenofibrate exerts protective effects in diabetic retinopathy via inhibition of the ANGPTL3 pathway. Invest Ophthalmol Vis Sci. 2018;59(10):4210–4217. doi: 10.1167/iovs.18-24155. [DOI] [PubMed] [Google Scholar]
- 15.Cai BS, Zhao J, Zhang YL, et al. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy. 2022;18(5):990–1004. doi: 10.1080/15548627.2021.1965426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomita Y, Lee D, Tsubota K, et al. Updates on the current treatments for diabetic retinopathy and possibility of future oral therapy. J Clin Med. 2021;10(20):4666. doi: 10.3390/jcm10204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu SH, Lai MC, Zheng YY, et al. miR-195 inhibits the ubiquitination and degradation of YY1 by Smurf2, and induces EMT and cell permeability of retinal pigment epithelial cells. Cell Death Dis. 2021;12(7):708. doi: 10.1038/s41419-021-03956-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forrester JV, Kuffova L, Delibegovic M. The role of inflammation in diabetic retinopathy. Front Immunol. 2020;11:583687. doi: 10.3389/fimmu.2020.583687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao SZ, Li J, Wang N, et al. Fenofibrate suppresses cellular metabolic memory of high glucose in diabetic retinopathy via a sirtuin 1-dependent signalling pathway. Mol Med Rep. 2015;12(4):6112–6118. doi: 10.3892/mmr.2015.4164. [DOI] [PubMed] [Google Scholar]
- 20.Hsu YJ, Lin CW, Cho SL, et al. Protective effect of fenofibrate on oxidative stress-induced apoptosis in retinal-choroidal vascular endothelial cells: implication for diabetic retinopathy treatment. Antioxidants (Basel) 2020;9(8):712. doi: 10.3390/antiox9080712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oshitari T. The pathogenesis and therapeutic approaches of diabetic neuropathy in the retina. Int J Mol Sci. 2021;22(16):9050. doi: 10.3390/ijms22169050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Yu CF, Liu XM, et al. Fenofibrate ameliorated systemic and retinal inflammation and modulated gut microbiota in high-fat diet-induced mice. Front Cell Infect Microbiol. 2022;12:839592. doi: 10.3389/fcimb.2022.839592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng WJ, Wang H, Wu BJ, et al. Selenium-layered nanoparticles serving for oral delivery of phytomedicines with hypoglycemic activity to synergistically potentiate the antidiabetic effect. Acta Pharm Sin B. 2019;9(1):74–86. doi: 10.1016/j.apsb.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Luo JS, Liu YJ, et al. Brain-targeted biomimetic nanodecoys with neuroprotective effects for precise therapy of Parkinson's disease. ACS Cent Sci. 2022;8(9):1336–1349. doi: 10.1021/acscentsci.2c00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Wang PP, Chen Z, et al. Fenofibrate ameliorates oxidative stress-induced retinal microvascular dysfunction in diabetic rats. Curr Eye Res. 2018;43(11):1395–1403. doi: 10.1080/02713683.2018.1501072. [DOI] [PubMed] [Google Scholar]
- 26.Yaribeygi H, Mohammadi MT, Rezaee R, et al. Fenofibrate improves renal function by amelioration of NOX-4, IL-18, and p53 expression in an experimental model of diabetic nephropathy. J Cell Biochem. 2018;119(9):7458–7469. doi: 10.1002/jcb.27055. [DOI] [PubMed] [Google Scholar]
- 27.Yaribeygi H, Mohammadi MT, Sahebkar A. PPAR-α agonist improves hyperglycemia-induced oxidative stress in pancreatic cells by potentiating antioxidant defense system. Drug Res (Stuttg) 2018;68(6):355–360. doi: 10.1055/s-0043-121143. [DOI] [PubMed] [Google Scholar]
- 28.Yeh PT, Wang LC, Chang SW, et al. Effect of fenofibrate on the expression of inflammatory mediators in a diabetic rat model. Curr Eye Res. 2019;44(10):1121–1132. doi: 10.1080/02713683.2019.1622020. [DOI] [PubMed] [Google Scholar]
- 29.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15(8):738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 30.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim RR, Wieser ME, Ganga RR, et al. NOD-like receptors in the eye: uncovering its role in diabetic retinopathy. Int J Mol Sci. 2020;21(3):899. doi: 10.3390/ijms21030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen W, Zhao MJ, Zhao SZ, et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res. 2017;66(2):157–166. doi: 10.1007/s00011-016-1002-6. [DOI] [PubMed] [Google Scholar]
- 33.Yin YZ, Chen F, Wang WY, et al. Resolvin D1 inhibits inflammatory response in STZ-induced diabetic retinopathy rats: Possible involvement of NLRP3 inflammasome and NF-κB signaling pathway. Mol Vis. 2017;23:242–250. [PMC free article] [PubMed] [Google Scholar]
- 34.Bai BC, Yang YY, Wang Q, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11(9):776. doi: 10.1038/s41419-020-02985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, Qian KL, Sun YL, et al. Omarigliptin ameliorated high glucose-induced nucleotide oligomerization domain-like receptor protein 3 (NLRP3) inflammasome activation through activating adenosine monophosphate-activated protein kinase α (AMPKα) in renal glomerular endothelial cells. Bioengineered. 2021;12(1):4805–4815. doi: 10.1080/21655979.2021.1957748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang K, Liu JN, Zhang XH, et al. H3 relaxin alleviates migration, apoptosis and pyroptosis through P2X7R-mediated nucleotide binding oligomerization domain-like receptor protein 3 inflammasome activation in retinopathy induced by hyperglycemia. Front Pharmacol. 2020;11:603689. doi: 10.3389/fphar.2020.603689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang YX, Tao JX, Yao Y. Prostaglandin E2 activates NLRP3 inflammasome in endothelial cells to promote diabetic retinopathy. Horm Metab. 2018;50(9):704–710. doi: 10.1055/a-0664-0699. [DOI] [PubMed] [Google Scholar]
- 38.Lee Y, Schulte DJ, Shimada K, et al. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–1550. doi: 10.1161/CIRCULATIONAHA.111.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong XW, Zheng ZH, Lin P, et al. ACPAs promote IL-1β production in rheumatoid arthritis by activating the NLRP3 inflammasome. Cell Mol Immunol. 2020;17(3):261–271. doi: 10.1038/s41423-019-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]