

Abstract

Structure-based virtual screening (SBVS) is a cornerstone of modern drug discovery pipelines. However, conventional scoring functions often fail to capture the complexities of protein–ligand binding interactions. To address this limitation, we developed DeepMETTL3, a novel scoring function that integrates 3D convolutional neural networks (CNNs) with multihead attention mechanisms and high-dimensional Structural Protein–Ligand Interaction Fingerprints (SPLIF). This approach enables the model to capture intricate 3D interaction patterns while refining and prioritizing features for precise classification of active and inactive compounds. We validated DeepMETTL3 using METTL3 as a therapeutic target, employing a scaffold-based data-splitting strategy and multiple test sets, including challenging sets with minimal chemical similarity to the training data. Our results demonstrate that DeepMETTL3 outperforms traditional scoring functions, achieving superior accuracy, robustness, and scalability. Key findings include the importance of an active-to-decoy ratio (1:50) in the training set for enhanced performance and the optimal placement of the attention mechanism after CNN1 for improved generalization. DeepMETTL3 represents a significant advancement in target-specific machine learning for SBVS, offering a framework that can be adapted to other biological targets. This work underscores the potential of deep learning in artificial intelligence-based drug design, balancing computational efficiency and predictive power in molecular docking and virtual screening. The scoring function is freely available at https://github.com/juniML/DeepMETTL3.
Introduction
Structure-based virtual screening (SBVS) plays a crucial role in speeding up drug discovery by utilizing the three-dimensional crystal structure of target proteins.1,2 It employs computational techniques to discover new ligands from large molecular libraries.3 A fundamental aspect of SBVS is molecular docking, which combines a scoring function and a search algorithm. The scoring function assesses the binding energy of potential ligands in specific poses, while the search algorithm navigates the conformational space to find the pose with the highest predicted binding affinity.
Despite its utility, SBVS faces notable challenges.4,5 Many docking programs rely on empirical scoring functions with limited parameters, constraining their ability to accurately model the intricate binding modes of protein–ligand interactions.6 Furthermore, the rigid protein structure assumption fails to account for the dynamic conformational changes of proteins and ligands during binding, often resulting in suboptimal predictions. To address these limitations, ongoing research focuses on enhancing scoring function accuracy, incorporating protein flexibility, and employing advanced sampling techniques to thoroughly explore conformational space.7,8 The integration of experimental data, such as binding affinities and mutagenesis findings, also holds promise for refining SBVS predictions. Thus, combining state-of-the-art computational methods with experimental validation remains pivotal in drug discovery. Recent advancements in deep learning and large language models (LLMs) have revolutionized protein–ligand interaction prediction, addressing the limitations of traditional methods. Zhou et al. proposed a global-local framework using pretrained models and advanced algorithms to enhance drug–protein interaction prediction.9 Similarly, Wei et al. introduced DrugReAlign, an LLM-based framework that leverages spatial interaction data for drug repurposing.10,11 These advancements collectively demonstrate the transformative potential of integrating computational techniques to improve the virtual screening precision and scalability.
The past decade in artificial intelligence (AI)-based SBVS has made great strides, especially due to the developments of machine-learning scoring functions that replace conventional empirical scoring functions.12−15 MLSFs use the latest models like neural networks, which take on huge parameter sets in modeling complex binding patterns, which remains unnoticed in conventional scoring functions such as molecular docking. Their advantages are incorporating nonlinear relationships for interaction energy models, considering nonadditive interaction energy modeling, and implicitly putting receptor flexibility implicitly.16 The training of MLSFs is computationally expensive, and the accuracy and speed are ultrafast for the large-scale virtual screening process and thus expedite the drug discovery. The development of a target-specific MLSF relies on three essential components: high-quality data sets containing accurate binding affinities and structural data for diverse protein–ligand complexes, effective representation of protein–ligand interactions using features that have been selected with care, and selection of suitable ML algorithms according to the data set and the desired outcome. This progress in MLSFs is not only enhancing drug candidate identification targeting accuracy and efficiency but also driving a reconsideration of scoring functions in molecular docking.
In initial generations, early ML scoring functions like RFscores17 (random forest) and NNscore13,18 (neural network) were also based on expertly designed features that had been thoroughly designed with significant manual input. However, in recent years, deep convolutional neural networks (CNNs) have gained prominence for predicting binding affinities and performing virtual screening. AtomNet was a pioneering example of a CNN model that showcased the potential for this approach to predict the activity of small molecules.19 Successive CNN-based models, such as KDEEP(20) and Pafnucy,21 leveraged vectorized grid representations of the protein–ligand complex that greatly improved scoring power in comparison to conventional approaches. Other significant methods such as deep learning in molecular docking have also made notable strides with integration of nonbinding data into training for an improvement in pose selection and affinity prediction. Other descriptors, such as protein–ligand topological fingerprints, have been integrated into the ML and CNN models to substantiate their predictive strength. Continuous enhancement in ML-based scoring functions marks a significant step forward in molecular docking, giving way to a more accurate and efficient design of novel drugs. Additionally, several machine learning methods use Structural Interaction Fingerprints (SIFts), which convert structural information into machine-readable one-dimensional vectors, enabling similarity calculations, modeling, and machine learning applications.22 SIFts, introduced by Deng et al., represent protein–ligand interactions as binary strings, capturing specific molecular interactions between receptor residues and ligands.23 They have been widely used in drug discovery for tasks such as selectivity profiling, target prediction, binding mechanism analysis, and scoring function development. SIFts are also effective in filtering virtual libraries and analyzing RNA–ligand complexes, including clustering docking poses and identifying ligands with similar interaction networks.24
METTL3 (methyltransferase-like 3) is a key enzyme involved in the regulation of RNA methylation, specifically catalyzing the addition of a methyl group to the N6 position of adenosine (m6A) in RNA molecules.25 This modification is one of the most prevalent and dynamic post-transcriptional RNA modifications, playing a critical role in regulating RNA stability, splicing, translation, and decay. METTL3, often functioning in a complex with METTL14 and other cofactors, is essential for various biological processes, including embryonic development, cellular differentiation, and immune response.26 Dysregulation of METTL3 has been implicated in numerous diseases, including cancer, where its overexpression is associated with tumor progression and poor prognosis. As a result, METTL3 has emerged as a promising therapeutic target, with ongoing research focused on developing inhibitors to modulate its activity and explore its potential in treating diseases linked to aberrant RNA methylation.26 Virtual screening studies targeting METTL3 have employed molecular docking techniques to identify inhibitors.27−29 These studies have led to the discovery of potential inhibitors, but traditional docking methods are limited by their reliance on empirical scoring functions. In contrast, our study introduces a deep learning-based model trained on high-dimensional Structural Protein–Ligand Interaction Fingerprints (SPLIF) and integrated with multihead attention, enabling more accurate and predictive virtual screening. Unlike conventional docking approaches, our model captures complex binding patterns and dynamic interactions, offering significant advancement in identifying METTL3 inhibitors and accelerating drug discovery.
In this study, we implemented a more streamlined method of featurization for protein–ligand complexes, leading to the development of a new scoring function model designed to predict class of the molecules (active or inactive) after training with a deep three-dimensional (3D) CNN coupled with multihead attention. The representation of features for the protein–ligand complex in our model utilized a 3D grid or a 4D tensor. Unlike the KDeep and Pafnucy models, our approach presents the SPLIF featurization scheme. The CNN subsequently learned the high-dimensional characteristics of protein–ligand complexes from SPLIF features.30 Given that the current scoring methods frequently fall short, improving the likelihood of identifying a substantial number of confirmed actives from a limited set of virtual hits is crucial. SPLIF addresses these shortcomings by leveraging known ligand-bound protein structures, thereby enriching the evaluation of protein–ligand interactions. Unlike previous techniques such as SIFt, which classify interactions into predefined types and may misinterpret bond types, SPLIF captures three-dimensional structures of interacting fragments, allowing for a more comprehensive representation of all possible interaction types, including nuanced interactions like cation–π. The novelty of the present study lies in leveraging SPLIF features, a high-dimensional, knowledge-enriched featurization scheme. Additionally, a second innovative aspect is the integration of multihead attention within the CNN architecture. This mechanism takes the features learned by the CNN layers, assigns attention-based weights to them, and feeds the weighted features back into the CNN, enabling the network to learn more effectively from the emphasized voxel representations. We have successfully applied this method to the METTL3 protein and developed a METTL3-specific target scoring function, which is available at https://github.com/juniML/DeepMETTL3. We anticipate that our model can be extended to other biological targets, thereby making a significant contribution to the field of AI-driven drug discovery.
Material and Methods
Preparation of Actives
Three databases were curated for biochemical studies against METTL3, including bindingDB, PubChem, and ChEMBL.31−34 These databases recorded the experimental activities of compounds against certain targets in the form of IC50, Ki, and Kd. 1015 molecules were extracted from these databases. Molecules with missing IC50 or Ki values were discarded. In this study, only compounds having IC50 were considered. After cleaning the data set, molecules with missing activity, and duplicate removal based on SMILES notation among the three data sets, the final data set contains 736 molecules. According to the PubChem classification, which categorizes a molecule as active if its activity is below 10 μM and inactive if above, 736 molecules were identified as active, while no molecule was classified as inactive.
Preparation of Inactive Molecules
Inactive molecules were generated using the DeepCoy algorithm.35 which leverages graph-generative neural networks to create decoys with matched physicochemical properties. The SMILES of active molecules were input into DeepCoy, which produced structurally distinct molecules sharing similar physicochemical properties.35 For each active molecule, 100 decoys were initially generated, and subsequently reduced to 50 decoys per active based on their DOE and doppelganger scores to balance the data set.35 The DOE score evaluates how well an active molecule is embedded within chemical space using ROC curves based on physicochemical properties. A score of zero indicates optimal embedding, while a score of 0.5 reflects complete separation of actives and decoys. The doppelganger score assesses the structural similarity between active molecules and their most closely related decoys.
Molecular Docking
Protein–ligand complexes were generated through molecular docking using SMINA,36 a fork of Autodock Vina designed to improve minimization and scoring.37 SMINA is available under a GPL2 license at http://smina.sf.net. The crystal structure of the receptor, bound to the substrate SAH (PDB ID 5IL2), was used for docking.38 The ligand search space was defined based on the coordinates of the bound substrate. Docking parameters included an exhaustiveness of 8 and a binding mode set to 1, generating one docked pose per ligand. SMINA’s built-in scoring function, which combines gradient and Monte Carlo steps to search and rank poses, was used to evaluate and rank the docked poses, which were saved in SDF format.
Model Architecture
Each docked target–ligand complex must be numerically encoded as features to enable supervised learning.15,39−41 We select 3D grid-based features from RDKitGridFeaturizer.2,42 The data for this study included protein–ligand complexes expressed in 3D space and enclosed in a cubic grid of 24 Å × 24 Å × 24 Å centered upon the binding pocket of the protein. To characterize the features of protein–ligand interactions, we used SPLIF features,30 which capture atomic-level interactions between the proteins and ligands. The features were encoded as binary fingerprints, creating an input with shape = (batch_size, 1536, 4, 4, 4). The proposed architecture consists of several sections: 3D convolutional layers to extract and refine hierarchical features, multihead attention mechanism, and fully connected layers to perform binary classification. The features extraction begins with the first layer of 3D convolution, with 32 filters, kernel_size 3, stride 1 and padding 1, followed by Batch Normalization and ReLU to stabilize learning and enhance convergence. Two additional convolutional layers, one with 64 filters and one with 128 filters, each of them followed by Batch Normalization and ReLU activation. To capture hierarchical representations while reducing computational complexity, a MaxPooling layer with a kernel size of 2 and a stride of 2 is applied after the final convolutional layer. Six stacked multihead attention layers were added after the first convolutional layer to fine-tune the extracted features and model long-range dependencies. The individual attention layers contain a multihead attention block, which allows the model to consider multiple interaction regions at once and a layer normalization block to stabilize training and promote feature exploration. By combining convolutional layers with attention mechanisms, the model can learn to focus on relevant regions while still retaining spatial context and generalizing across diverse protein–ligand complexes. The classification module is composed of three fully connected layers for processing the features that are filtered through the feature selection module and returns the active/inactive classification of the compound. A dense layer of 256 neurons is followed by 128 neurons and then the output layer with final activation. This is followed by a single neuron with a Sigmoid activation function in the last layer to predict the probability that a compound is active. To prevent overfitting and increase generalization, dropout regularization with a rate of 0.5 was performed between the fully connected layers.
We used the Adam optimizer to train the model with a learning rate of 0.001, and the model was implemented in PyTorch. To optimize the classification of active and inactive compounds, Binary Cross-Entropy Loss was employed as the loss function. We trained the model with a batch size of 32 for 100 epochs.
Binary Cross-Entropy Loss
Binary cross-entropy loss (BCELoss) is a commonly used loss function for binary classification tasks. It calculates the difference between the predicted probabilities and the actual binary labels (0 or 1). BCELoss measures how well the predicted probability distribution matches the actual distribution, penalizing large deviations. The formula for BCELoss is
where yi is the
actual binary label (0 or 1) for the ith sample,  is the predicted probability for the ith sample, and N is the total number of
samples.
is the predicted probability for the ith sample, and N is the total number of
samples.
Multihead Self-Attention Mechanism
Since some features are redundant or less important, the multihead self-attention mechanism can help the model select significant features and give these important features with higher weights. Therefore, the multihead self-attention mechanism can help the model to identify which features are crucial for prediction. The multihead attention is calculated by the following formulas
where X is the drug feature
vector, 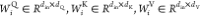 are the parameter matrices. Qi, Ki, and Vi are the Q(query), K(key), and V(value) matrices derived from the linear transformation of X, respectively.
are the parameter matrices. Qi, Ki, and Vi are the Q(query), K(key), and V(value) matrices derived from the linear transformation of X, respectively.
Evaluating ML Models
We trained the classification variants of each algorithm. In the case of classification, a molecule is classified as active if its probability score is above 0.5 and is otherwise classified as inactive. For the classification task, four metrics were utilized; PR-AUC, precision, recall, and F1. During 5-fold CV, and testing, the PR-AUC, Precision, Recall, and F1 were used and imported from the “metrics” module of the “sklearn” Python library (v.1.0.2)): “roc_auc_score”, ”precision_score”, ”auc”, ”recall_score”, ”precision_recall_curve”.
These metrics can be mathematically represented as
where TP represents true positive, FP represents false positive, and FN represents false negative.
Results and Discussion
Model Architecture and Workflow
In this study, we developed a novel DeepMETTL3 architecture integrated with multihead attention layers to classify protein–ligand complexes as active or inactive. The model leverages the strengths of convolutional layers for spatial feature extraction and the attention mechanism for refining and prioritizing critical interactions. The architecture combines hierarchical feature extraction with effective dimensionality reduction and classification, providing robust performance across diverse data sets (Figure 1).
Figure 1.

Architecture of the DeepMETTL3 model. The input consists of 3D grid of protein–ligand complexes encoded with SPLIF features (dimension: 24 Å × 24 Å × 24 Å). The stacked 3D convolutional layers and multihead attention in the feature extraction module are used to focus on refined interactions which are crucial. The output of the attention mechanism is then passed through several fully connected layers of the classification module to predict whether each compound is active or inactive.
The DeepMETTL3 architecture begins with a 3D convolutional layer capable of processing high-dimensional inputs representing protein–ligand complexes. The input is structured as (batch_size, 1536, 4, 4, 4) to represent the spatial grid of protein–ligand interaction features. Key components of the architecture include:
Convolutional layers: Three convolutional layers progressively extract hierarchical spatial features with kernel sizes of 3 × 3 × 3 and appropriate strides and padding to retain the dimensional integrity of the input. Each convolutional layer is followed by Batch Normalization and ReLU activation, ensuring stable learning and faster convergence. The MaxPooling layer further reduces the spatial dimensions to capture abstract representations while avoiding overfitting.
Multihead attention layers: To capture long-range dependencies and refine extracted features, six stacked multihead attention layers are incorporated after the first convolutional layer. Each attention module computes feature relationships across spatial dimensions, allowing the model to focus on crucial protein–ligand interactions. Multihead attention enables the model to simultaneously prioritize multiple features, improving generalization and reducing noise from irrelevant interactions.
Dropout regularization: Dropout layers are added after convolutional and fully connected layers to mitigate overfitting by randomly deactivating neurons during training. This ensures the model remains robust across unseen data.
Fully connected layers: The extracted features are flattened and passed through three fully connected layers with decreasing dimensions (256, 128, and 1) to classify active and inactive compounds. ReLU activation is applied after the first two layers, and the final layer uses a Sigmoid function for binary classification.
The workflow begins with a 3D spatial representation of the protein–ligand complex. The convolutional layers extract hierarchical spatial features, which are further refined by multihead attention layers. This combination enables the model to focus on critical interaction regions while maintaining a broader spatial context. The flattened output is processed through fully connected layers to generate the final classification.
The integration of multi head attention layers with 3D convolutions is a key innovation in this architecture, enabling the model to capture long-range dependencies and focus on critical regions within high-dimensional protein–ligand data. This attention mechanism refines features extracted by convolutional layers, reducing noise and improving the distinction between actives and inactives, especially in challenging data sets. Dropout layers and a fully connected architecture ensure robustness and smooth classification, with Sigmoid activation providing probabilistic outputs. In conclusion, the DeepMETTL3 model effectively combines 3D convolutions and attention mechanisms to accurately classify protein–ligand complexes, making it highly suitable for SBVS. Future work could expand its utility through multitask learning, additional biological features, and testing on diverse drug discovery targets.
Scaffold-Based Data Splitting and Molecular Diversity Analysis
To evaluate the generalizability of our novel approach, we leveraged a Bemis–Murcko (BM) scaffold partitioning strategy to divide the data set for training and testing.43,44 This methodology ensures compounds in the test set contain unique scaffolds absent from training, resembling real-world situations in which models must predict the activity of chemicals with novel architectural scaffolds.
Following BM scaffold splitting, the training subset contained 651 actives, while the initial test set held 82 actives. To push the model further, two extra test sets were developed, termed Hard Test Set 1 and Hard Test Set 2. Hard Test Set 1 originated by removing molecules from the original test set displaying a Tanimoto similarity surpassing 0.80 to any molecule in training. This filtering process reduced the number of actives to 57, keeping the decoys as the initial test set. Similarly, Hard Test Set 2 was formed by enforcing a stricter threshold, excluding compounds with a Tanimoto similarity greater than 0.75 to any molecule in training. As a result, this test set retained only 29 actives, again with an equal number of decoys as for the original test set. The active-to-decoy ratio was calibrated in each test set as follows.
Original test set: 1:50 (active/decoy).
Hard test set 1:1:72, due to the reduction in active molecules while retaining the same number of decoys.
Hard test set 2:1:141, reflecting a further reduction in active molecules while keeping the decoy count constant.
The purpose of constructing these hard test sets was to create increasingly challenging scenarios for the model, thereby simulating situations where the active molecules share minimal chemical similarity with the training data. This ensures a robust evaluation of the model’s ability to identify active molecules accurately in diverse and chemically distinct data sets.
We computed pairwise Tanimoto similarity scores between the Morgan fingerprints (radius = 2) of molecules in the training and test sets. The similarity scores were visualized as heatmaps (Figure 2a–c), with the color intensity reflecting the level of similarity. The heatmaps demonstrate the low similarity between the training and test sets, as indicated by the predominantly blue regions. Specifically, Test Set 1 shows moderate diversity, whereas Hard Test Set 1 and Hard Test Set 2 represent increasingly challenging scenarios with minimal overlap. The similarity between molecules in Hard Test Set 2 and the training set is especially low, highlighting the difficulty of predicting their activities. The cumulative distribution plot (Figure 2d) provides a quantitative overview of scaffold diversity. Approximately 100% of test molecules exhibit Tanimoto similarity scores below 0.8 when compared to the training molecules, suggesting a high degree of structural dissimilarity at the molecular fingerprint level. This highlights the need for the model to demonstrate robustness in accurately classifying molecules with diverse and novel chemical features.
Figure 2.

Analysis of Tanimoto similarity between active scaffolds for training and test sets. Heatmaps show pairwise Tanimoto coefficients for (a) training actives (651 molecules) vs actives from Test Set (82 molecules), (b) Hard Test Set 1 (52 molecules), and (c) Hard Test Set 2 (28 molecules), illustrating the distribution of chemical similarity. (d) Distribution of Tanimoto coefficients, showing structural diversity. Most values are below 0.3, indicating low similarity.
Overall, the BM scaffold-based splitting strategy effectively created diverse and challenging data sets, providing a stringent evaluation of the model’s ability to generalize beyond the training data.
Hyper Parameter Optimization
To evaluate the effect of different hyper parameters on model performance, we systematically tuned the learning rate, number of attention heads, number of layers, and dropout rate. The performance was measured by using several metrics: PR-AUC, ROC-AUC, Precision, Recall, F1 Score, and MCC. The results are summarized in Figure 3. We used 5-fold cross validation with 50 epochs each.
Figure 3.

Hyperparameter optimization results for DeepMETTL3. (a) Learning rate of 0.001 produces the best results. (b) Eight attention heads offer the most balanced performance across metrics. (c) The best PR-AUC and ROC-AUC are with six attention layers. (d) High dropout rate (0.3) used to achieve optimal generalization and robustness. Metrics: PR-AUC, ROC-AUC, Precision, Recall, F1-Score, MCC.
Model performance will be influenced by the learning rate.45 A greater learning rate (0.1) meant that the model would exhibit unstable behavior, which led to poor performance in training. As the learning rate stepped down to 0.01 and 0.001, all results improved significantly, ultimately reaching maximum values for learning rates set at 0.001. The model effectively worked out a stable operation at 0.001, as can be seen by its stable performance. This shows the importance of choosing an appropriate value for the learn_rate to achieve both convergence and good performance. A higher learning rate causes large weight updates, leading to overshooting the optimal solution and unstable training, which results in poor performance.45,46 On the other hand, a reduced learning rate makes it possible to make accurate weight adjustments gradually, which helps the model converge steadily and generalize better to new data. However, training can be slowed down, and there is a chance of becoming trapped in less-than-ideal local minima if learning rates are too low.
The number of attention heads was varied from 1 to 16 to examine the impact on model performance. All metrics demonstrated stable performance across the range, with marginal improvements at 8 attention heads. This indicates that the model can effectively capture complex interactions with multiple attention heads, although increasing beyond 8 did not yield significant benefits.47 The stability suggests that the model is not overly sensitive to the number of attention heads within the tested range.
The effect of increasing the number of layers in the model was evaluated. PR-AUC showed a slight increase as the number of layers increased from 1 to 6, achieving the highest value at 6 layers. Further increases in the layer do not increase the model performance. All metrics ensured that 6 layers provided an optimal trade-off between model depth and performance. Different values for dropouts were also evaluated. Dropout is used to prevent overfitting in the model. The values ranging from 0.1 to 0.5 were evaluated.48,49 The metrics indicated a stable performance at dropout rates of 0.5. The results suggest that a moderate dropout rate of 0.5 is optimal for maintaining a balance between regularization and model expressiveness.
Hyper-Parameter Optimization results showed that the highest accuracy for the model is with learning rate = 0.001, attention heads = 8, layer = 6, dropout rate = 0.5. These provided the best performance (across all metrics) of a considerably robust and reliable model for classifying molecules as being active or inactive. This highlights the requirement of systematic hyper-parameter tuning, in improving the predictive power of deep learning networks in molecular classification tasks, similar to previous studies.45,46,48,49
Effect of Active: Decoys Ratio on Model Performance
Previous research has demonstrated that model performance is positively influenced by incorporating decoys into the training data set.2,41,50−52 Specifically, utilizing a large number of decoys in the training set has been shown to enhance the SBVS effectiveness of MLSFs.17 However, this approach has not yet been explored for METTL3. The ratio of Active/Decoys in the training set was evaluated and the performance of our model on the standard test set when trained with varying ratios of actives to decoys (from 1:1 up to 1:50) was examined. It is worth mentioning that during hyperparameter tuning we used a 1:50 decoy ratio, as this ratio is commonly adopted in the literature and has shown strong performance. In this study, we also hypothesized and explored the impact of using lower decoy ratios. Several performance metrics are plotted, including PR-AUC, ROC-AUC, Precision, Recall, F1-Score, and MCC. Overall, the model achieves near-optimal performance (especially in ROC-AUC) once the training set contains at least a 1:10 decoy ratio. At 1:1, several metrics, particularly Precision and MCC are relatively low, reflecting the difficulty in learning robust decision boundaries without enough negative examples. However, by increasing the proportion of decoys to around 1:10, there is substantial improvement across all metrics. Beyond that point (moving to 1:20, 1:30, and so on), performance remains stable, suggesting that adding more decoys yields diminishing returns in predictive power for this “standard” test set (Figure 4a).
Figure 4.

Test results with DeepMETTL3 with different training decoy ratios on different test sets. (a) Test Set, (b) Hard Test Set 1, and (c) Hard Test Set 2. This is measured using PR-AUC, ROC-AUC, Precision, Recall, F1-Score, and MCC. A training decoy ratio of 1:50 provides optimal performance; and the model generalization and robustness are enhanced by increasing the decoy ratio.
Figure 4b,c shows the same set of metrics on two “hard” test sets; these sets are designed to be more challenging by containing examples that are more difficult to classify, even though the test-training ratio remains the same across experiments. The same general pattern appears: switching from 1:1 to 1:10 produces a pronounced boost in all metrics, and then performance levels off. Compared to the standard test set (Figure 3a), the metrics for these harder test sets are slightly lower overall (e.g., PR-AUC is not as high), which is expected given the increased difficulty. Nevertheless, in both hard test sets, training with at least a 1:10 (or higher) decoy ratio consistently leads to a strong performance across ROC-AUC, Precision, and F1-Score.
In other words, the model strongly benefits from having an ample number of decoys in the training set to learn a robust decision boundary between actives and inactives.17 Once this balance has been achieved (generally at 1:10 or greater), there is no substantial gain from adding even more decoys (e.g., 1:20 or 1:50), indicating that beyond a certain point the model is saturated in terms of negative-class information. To replicate real-world scenarios, we selected a 1:50 decoy ratio to closely align with natural conditions, as in practical applications the proportion of active compounds is significantly smaller compared to the vast size of chemical databases.
Optimizing Multihead Attention Placement in CNNs for Enhanced Virtual Screening
To assess the impact of the placement of a multihead attention block within the CNN architecture, we tested its integration at four different locations: before the first CNN layer, after the first CNN layer (CNN1), after the second CNN layer (CNN2), and after the third CNN layer (CNN3). The results are presented for three test sets: (a) a standard test set, (b) Hard Test Set 1, and (c) Hard Test Set 2. The metrics used for evaluation include PR-AUC, ROC-AUC, Precision, Recall, F1-Score, and MCC (Figure 5).
Figure 5.

Impact of multihead attention placement on model performance across different test sets. (a) Test Set, (b) Hard Test Set 1, and (c) Hard Test Set 2. Metrics include PR-AUC, ROC-AUC, Precision, Recall, F1-Score, and MCC. Optimal performance is observed when the multihead attention block is placed after the first convolutional layer (CNN1), highlighting its effectiveness in refining features and enhancing model generalization.
The results on the standard test set indicate that placing the multihead attention block after CNN1 yields the best overall performance across the majority of metrics. In particular, ROC-AUC and Precision, Recall, and MCC reach their peak values in this configuration, demonstrating the effectiveness of refining low-level spatial features early in the architecture. On the other hand, placing the attention block before the first CNN layer results in suboptimal performance for all metrics, likely due to the lack of extracted features for the attention mechanism to refine.
Interestingly, when the attention block is placed after CNN2, there is a noticeable dip in all the metrics except PR-AUC, suggesting that attention at this stage may disrupt the flow of intermediate feature representations. However, the performance improves when the attention block is placed after CNN3, with metrics recovering to near-optimal levels. This suggests that attention can still be effective in refining high-level abstract features but is less impactful compared with earlier placements.
The performance trends observed in Hard Test Sets 1 and 2 are well aligned with those seen in the standard test set. The optimal performance under all metrics always occurs when the attention block is placed after the CNN1. This suggests that this attention layer enables the model to be able to utilize meaningful features captured from the previous convoluted layers, which is important for distinguishing actives from inactives in a difficult data set. Similar to the standard test set, placing the attention block after CNN2 resulted in declines of all metrics except for PR-AUC and Precision, indicating that the model is sensitive to attention placement. Performance does get back up after CNN3, though. This goes to show that the integration of attention mechanisms into the network must balance how many features are abstracted or refined.
The results demonstrate that the placement of the multihead attention or cross attention block can have a significant impact on performance of the model.47 On the basis of the network architecture and data sets we evaluated, we found that the most appropriate location for the attention unit is after CNN1, where low-level features have been abstracted by the attention mechanism with the aim to enhance generalization. While attention at other stages can still provide benefits, the results highlight the need for careful consideration of attention integration to maximize performance, particularly for more challenging data sets.
Developing METTL3-specific Classification-Based SFs for SBVS
During the tuning phase, we tuned hyper-parameters; based on that we selected the parameters that described the best model. The model was trained using these optimized parameters over 100 epochs, with results repeated over 5 independent runs to ensure robustness and reliability of the results (Figure 6). PR-AUC values as an overall measure of model performance across the test set are illustrated in the figure.
Figure 6.

Precision–Recall (PR) curve analysis and PR-AUC distribution across five independent runs across test sets. (a) Test Set: PR-AUC: 0.92–0.95. (b) PR curves for Hard Test Set 1, reflecting PR-AUC: 0.87–0.97. (c) PR curves for Hard Test Set 2, PR-AUC: 0.80–0.93. (d) Boxplot of PR-AUC values for all test sets, highlighting higher PR-AUC for the Test Set compared to the Hard Test Sets, with lower values and greater variability in Hard Test Set 2.
This visualization illustrates both the consistency and variability of the model’s performance over repeated training and testing with the optimized hyper-parameters. PR-AUC values have been shown as interquartile range (IQR), which indicates the spread between the 25th and 75th percentile. If the IQR is a narrow number, that means that the runs had similar performance, while a wider IQR number would mean that the runs were inconsistent. A narrow spread (small IQR) of PR-AUC values and a high median value indicate not only an effective model but also a reliable one.
PR-AUC focuses on the relationship between Precision (how many of the predicted active molecules are true active molecules) and Recall (how many actual active molecules are identified). It is especially useful for imbalanced data sets that have a heavy skew between active molecules and inactive ones. A high PR-AUC indicates that the model can efficiently recognize active molecules with few false active molecules at any decision threshold. The distribution of PR-AUC values across the 5 independent runs using a boxplot was visualized (Figure 6d). This visualization illustrates both the consistency and variability of the model’s performance over repeated training and testing with the optimized hyper-parameters. PR-AUC values have been shown as IQR, which indicates the spread between the 25th and 75th percentile. If the IQR is narrow, that means that the model had similar performance while a wider IQR would mean that the models were inconsistent. A tight distribution of PR-AUC values (small IQR) and a high median indicate that the model is not only effective but also reliable.
In order to get a holistic view of model performance, we computed various metrics apart from PR-AUC such as Precision, Recall, F1-Score and MCC (as shown in Figure 7). They offer other interesting insights into the model’s capability to balance its false positives and false negatives in order to predict both active and inactive compounds correctly. The following results illustrate the spread of metrics among the three test data. Each metric is represented as a separate boxplot, showing the performance across 5 independent runs. This visualization allows us to assess the variability and consistency of the model’s predictions for each metric. The combination of high PR-AUC, Precision, Recall, F1 Score, and MCC demonstrates the effectiveness of the optimized model across different evaluation metrics. The consistency across multiple independent runs indicates the robustness of the model and its suitability for real-world applications, such as virtual screening or activity prediction.
Figure 7.

Evaluation of Precision, Recall, F1-Score, and MCC across the Test Set, Hard Test Set 1, and Hard Test Set 2. Boxplots demonstrate the performance distribution across multiple runs, with the Test Set showing the highest and most consistent scores. Hard Test Set 1 maintains strong but slightly variable performance, while Hard Test Set 2 exhibits the lowest scores and highest variability, reflecting the increased difficulty of this data set.
Comparison with Other Scoring Functions
The performance comparison of the different models or scoring functions DeepMETTL3, CNN-Score, RF-ScoreVS, SCORCH and SMINA, are performed using three data sets, Test Set, Hard Test Set 1, and Hard Test Set 2 (Figure 8). We measure performance metrics as PR-AUC, ROC-AUC, Precision, Recall, MCC, and F1-Score. The integration of these metrics provides an overview of the performance of each scoring function in distinguishing the active vs inactive compound in the different conditions.
Figure 8.

Comparison of model performance across metrics (PR-AUC, ROC-AUC, Precision, Recall, MCC, and F1-Score) for (a) Test Set, (b) Hard Test Set 2, and (c) Hard Test Set 3. DeepMETTL3 consistently outperforms other methods (CNN-Score, RF-ScoreVS, SCORCH, and SMINA) across all test sets, demonstrating robust and reliable predictions. SCORCH shows high ROC-AUC but poor performance in other metrics, while SMINA exhibits the lowest overall scores. Hard Test Sets highlight the superior generalization capability of DeepMETTL3.
The DeepMETTL3 achieves near-perfect values across all metrics, including PR-AUC, ROC-AUC, Precision, Recall, MCC, and F1-Score, consistently performing well across all three test sets. This highlights its ability to significantly outperform other methods, demonstrating high robustness even in challenging hard test sets. The consistent high performance across all evaluation metrics emphasizes the model’s effectiveness in capturing both low-level and high-level features, enabling it to reliably distinguish actives from inactives. The integration of attention mechanisms into the 4D-CNN framework plays a critical role in this superior performance by allowing the model to focus on the most relevant features in protein–ligand interactions. This targeted focus not only enhances predictive accuracy but also ensures that the model generalizes effectively across diverse and complex data sets, making it a highly reliable approach for SBVS.
The moderate performance of CNN-Score,53 with Recall emerging as its strongest metric followed by ROC-AUC, could be explained by its ability to correctly identify a larger proportion of true positives (actives) while struggling to maintain precision and balance between false positives and false negatives. This suggests that the model is biased toward sensitivity (high Recall) but lacks the capacity to refine predictions to ensure specificity (high Precision). The reliance on convolutional layers for feature extraction enables CNN-Score to capture spatial patterns effectively, which may contribute to its decent ROC-AUC performance, reflecting a good overall ranking ability. However, the absence of mechanisms like attention could hinder the model’s capacity to focus on critical interaction features, leading to poor performance in metrics such as PR-AUC, MCC, and F1-Score. These metrics are more sensitive to a model’s ability to balance the trade-off between true positives and false positives, and CNN-Score’s lower values here indicate inefficiencies in identifying inactive compounds while maintaining a robust decision boundary.
RF-ScoreVS17 relatively strong performance in ROC-AUC can be attributed to its ability to rank compounds effectively, as this metric measures the overall distinction between actives and inactives across thresholds. However, its weaker performance in PR-AUC, Precision, Recall, MCC, and F1-Score stems from its inability to optimize predictions for specific thresholds, which are crucial for balancing true positives and false positives. The noticeable decline in harder test sets highlights its limited generalization capability, likely due to its reliance on handcrafted features and lack of adaptability to complex or noisy data sets. RF-ScoreVS struggles to capture subtle patterns in challenging scenarios, resulting in lower Recall and MCC, which reduces its effectiveness for robust, real-world applications.
The unusual pattern exhibited by SCORCH,54 with extremely high Recall and ROC-AUC values but poor performance in other metrics such as PR-AUC, Precision, MCC, and F1-Score, can be attributed to its tendency to predict a large number of positives, leading to a bias toward high sensitivity. The high Recall indicates that SCORCH captures nearly all true actives, but this comes at the expense of an increased number of false positives, which significantly lowers Precision. The high ROC-AUC reflects its ability to rank actives higher than inactives across thresholds, but this metric does not penalize the model for imbalanced predictions at specific thresholds. As a result, while SCORCH may excel in ranking tasks, its poor performance in PR-AUC, MCC, and F1-Score suggests that it struggles to balance the trade-off between true positives and false positives effectively. This behavior likely stems from SCORCH’s design or scoring function prioritizing recall and ranking over precision and classification balance. While this may be suitable for applications where identifying all actives is critical, it reduces its utility for tasks requiring a more balanced and precise prediction approach.
As a traditional scoring function used in docking software, SMINA36 is likely constrained by its simplistic scoring approach, which may not capture the complexities of protein–ligand interactions effectively. This makes it unsuitable for tasks requiring high predictive accuracy.
The results highlight the superiority of DeepMETTL3, achieving consistently high performance across all metrics and test sets, including hard ones, due to its ability to focus on critical features via attention mechanisms. In contrast, traditional methods like SMINA, RF-ScoreVS, and standard CNNs struggle, especially on challenging data sets. These findings underscore the importance of advanced architectures such as DeepMETTL3 for SBVS.
Conclusions
This study highlights the effectiveness of combining a CNN architecture with multihead attention and SPLIF features to develop a robust machine-learning-based scoring function for SBVS. Our model, DeepMETTL3, consistently outperformed traditional scoring methods such as RF-ScoreVS, CNN-Score, SCORCH, and SMINA across all test sets, including the challenging Hard Test Sets. By leveraging SPLIF features and the integration of attention mechanisms, the model successfully captured both low- and high-level features, enabling precise and reliable classification of active and inactive compounds.
Key findings demonstrated that incorporating an optimal active-to-decoy ratio in the training data set significantly enhances model performance, with a 1:10 ratio providing a substantial improvement in metrics such as PR-AUC, ROC-AUC, Precision, Recall, and F1-Score. The placement of multihead attention also played a critical role, with the optimal position after CNN1 resulting in improved feature refinement and generalization across diverse data sets.
While the DeepMETTL3 method demonstrates significant advancements over traditional scoring functions, it is not without limitations. One key limitation is the reliance on high-quality, target-specific data sets for training, which may not always be available for other biological targets. Additionally, the computational cost of training deep learning models, especially with multihead attention mechanisms, can be substantial, potentially limiting scalability for large-scale virtual screening campaigns. Furthermore, the model’s performance, while robust, is still dependent on the quality of the molecular docking poses generated by tools like SMINA, which may not always accurately reflect the true binding conformations. The use of decoys generated by DeepCoy, while effective, may also introduce biases if the decoys do not fully represent the chemical space of true inactives. In comparison, traditional physics-based scoring functions like AutoDock Vina are limited by their simplistic energy terms and inability to capture complex interactions, while machine learning methods like RF-ScoreVS and CNN-Score often struggle with generalizability and feature representation. Deep learning models such as Pafnucy, though powerful, require large data sets and are computationally intensive, with their black-box nature making interpretability a challenge. Overall, while DeepMETTL3 addresses many of these limitations by leveraging SPLIF and attention mechanisms, it still faces challenges related to data set availability, computational cost, and dependence on docking accuracy, which are common across SBVS methodologies. Future work could focus on reducing computational demands, improving interpretability, and enhancing generalizability to other targets with limited data.
Another potential concern is the risk of overfitting, particularly when dealing with specific compound types or data sets with limited chemical diversity. To mitigate this, we employed techniques such as dropout regularization and scaffold-based splitting, which have proven to be effective in improving generalization. Nevertheless, further validation on larger and more diverse data sets is necessary to fully assess the model’s robustness. Additionally, the model’s performance may be influenced by the size and diversity of the training data set. While scaffold-based splitting ensures chemical diversity in the test sets, the model may still struggle with highly novel scaffolds or underrepresented compound classes. Future work could explore data augmentation techniques or transfer learning to address these challenges. During training, we encountered challenges related to balancing the active-to-decoy ratio and optimizing the hyperparameters. While a 1:50 ratio provided optimal performance, this may not generalize to all data sets, and further tuning may be required for specific applications. Additionally, the computational cost of training deep learning models with large data sets remains a practical limitation, which could be mitigated by leveraging more efficient architectures or distributed computing resources in future work.
The results underscore the importance of advanced architectures such as DeepMETTL3 in addressing the limitations of traditional scoring functions, such as the inability to model complex interaction patterns or generalize effectively to novel scaffolds. Our METTL3-specific scoring function highlights the potential for target-specific machine learning models to enhance the identification of active compounds, offering a scalable and adaptable solution for future SBVS applications. These findings strongly support the hypothesis that integrating multihead attention and SPLIF features within a deep-learning framework can significantly improve prediction accuracy and robustness in drug discovery.
Future studies can expand on this work by testing the model on additional targets, exploring hybrid attention mechanisms, or incorporating dynamic protein flexibility to further enhance its predictive power and utility in real-world drug design scenarios.
Acknowledgments
We would like to express our gratitude to the Natural Science Foundation of Guangdong Province and the Shenzhen Science and Technology Innovation Commission for funding.
Data Availability Statement
The data used and all codes to reproduce this study are freely available on https://github.com/JuniML/DeepMETTL3. The DeepCoy decoys are generated through the DeepCoy algorithm and the code is available on https://github.com/fimrie/DeepCoy/tree/master.
Author Contributions
M.J. designed the study and performed the experiments. M.Z., F.M.A., and A.K. helped in proof reading the manuscript and codes. W.L. supervised the project. All authors analyzed the data, drew conclusions and wrote, read, and approved the final manuscript.
This work was supported by the Natural Science Foundation of Guangdong Province, China (grant no. 2023A1515010471) and the Shenzhen Science and Technology Innovation Commission (grant no. JCYJ20240813142512017).
The authors declare no competing financial interest.
Notes
Ethics statement: No experiment in this study involved animals or human patients.
References
- Chen P.; Ke Y.; Lu Y.; Du Y.; Li J.; Yan H.; Zhao H.; Zhou Y.; Yang Y. DLIGAND2: an improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. J. Cheminf. 2019, 11, 52. 10.1186/s13321-019-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sacristán P.; Simeon S.; Tran-Nguyen V.-K.; Patil S.; Ballester P. J. Inactive-enriched machine-learning models exploiting patent data improve structure-based virtual screening for PDL1 dimerizers. J. Adv. Res. 2025, 67, 185–196. 10.1016/j.jare.2024.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. K.; Maiti N. J.; Mishra M.; Raj S.; Rakshit G.; Ghosh R.; Roy S.. Virtual Screening and Lead Discovery. Computational Methods for Rational Drug Design; John Wiley & Sons, 2025; pp 97–121. [Google Scholar]
- Shen C.; Hu Y.; Wang Z.; Zhang X.; Pang J.; Wang G.; Zhong H.; Xu L.; Cao D.; Hou T. Beware of the generic machine learning-based scoring functions in structure-based virtual screening. Briefings Bioinf. 2021, 22 (3), bbaa070. 10.1093/bib/bbaa070. [DOI] [PubMed] [Google Scholar]
- Tran-Nguyen V.-K.; Ballester P. J. Beware of simple methods for structure-based virtual screening: the critical importance of broader comparisons. J. Chem. Inf. Model. 2023, 63 (5), 1401–1405. 10.1021/acs.jcim.3c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Leung K. S.; Wong M. H.; Ballester P. J. Improving AutoDock Vina using random forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol. Inf. 2015, 34 (2–3), 115–126. 10.1002/minf.201400132. [DOI] [PubMed] [Google Scholar]
- Ricci-Lopez J.; Aguila S. A.; Gilson M. K.; Brizuela C. A. Improving structure-based virtual screening with ensemble docking and machine learning. J. Chem. Inf. Model. 2021, 61 (11), 5362–5376. 10.1021/acs.jcim.1c00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacio-Rodríguez K.; Lans I.; Cavasotto C. N.; Cossio P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9 (1), 5142. 10.1038/s41598-019-41594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Liao Q.; Wei J.; Zhuo L.; Wu X.; Fu X.; Zou Q. Revisiting drug–protein interaction prediction: a novel global–local perspective. Bioinformatics 2024, 40 (5), btae271. 10.1093/bioinformatics/btae271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Zhuo L.; Fu X.; Zeng X.; Wang L.; Zou Q.; Cao D. DrugReAlign: a multisource prompt framework for drug repurposing based on large language models. BMC Biol. 2024, 22 (1), 226. 10.1186/s12915-024-02028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Zhu Y.; Zhuo L.; Liu Y.; Fu X.; Li F. Efficient deep model ensemble framework for drug-target interaction prediction. J. Phys. Chem. Lett. 2024, 15 (30), 7681–7693. 10.1021/acs.jpclett.4c01509. [DOI] [PubMed] [Google Scholar]
- McNutt A. T.; Francoeur P.; Aggarwal R.; Masuda T.; Meli R.; Ragoza M.; Sunseri J.; Koes D. R. GNINA 1.0: molecular docking with deep learning. J. Cheminf. 2021, 13 (1), 43. 10.1186/s13321-021-00522-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. NNScore: a neural-network-based scoring function for the characterization of protein– ligand complexes. J. Chem. Inf. Model. 2010, 50 (10), 1865–1871. 10.1021/ci100244v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francoeur P. G.; Masuda T.; Sunseri J.; Jia A.; Iovanisci R. B.; Snyder I.; Koes D. R. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 2020, 60 (9), 4200–4215. 10.1021/acs.jcim.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junaid M.; Wang B.; Li W. Data-augmented machine learning scoring functions for virtual screening of YTHDF1 m6A reader protein. Comput. Biol. Med. 2024, 183, 109268. 10.1016/j.compbiomed.2024.109268. [DOI] [PubMed] [Google Scholar]
- Masters M. R.; Mahmoud A. H.; Wei Y.; Lill M. A. Deep learning model for efficient protein–ligand docking with implicit side-chain flexibility. J. Chem. Inf. Model. 2023, 63 (6), 1695–1707. 10.1021/acs.jcim.2c01436. [DOI] [PubMed] [Google Scholar]
- Wójcikowski M.; Ballester P. J.; Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7 (1), 46710. 10.1038/srep46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. NNScore 2.0: a neural-network receptor–ligand scoring function. J. Chem. Inf. Model. 2011, 51 (11), 2897–2903. 10.1021/ci2003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach I.; Dzamba M.; Heifets A.. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv preprint. 2015. arXiv:1510.02855, 10.48550/arXiv.1510.02855. [DOI] [Google Scholar]
- Jiménez J.; Skalic M.; Martinez-Rosell G.; De Fabritiis G. K deep: protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. J. Chem. Inf. Model. 2018, 58 (2), 287–296. 10.1021/acs.jcim.7b00650. [DOI] [PubMed] [Google Scholar]
- Stepniewska-Dziubinska M. M.; Zielenkiewicz P.; Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34 (21), 3666–3674. 10.1093/bioinformatics/bty374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witek J.; Smusz S.; Rataj K.; Mordalski S.; Bojarski A. J. An application of machine learning methods to structural interaction fingerprints—a case study of kinase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24 (2), 580–585. 10.1016/j.bmcl.2013.12.017. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein– ligand binding interactions. J. Med. Chem. 2004, 47 (2), 337–344. 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- Szulc N. A.; Mackiewicz Z.; Bujnicki J. M.; Stefaniak F. Structural interaction fingerprints and machine learning for predicting and explaining binding of small molecule ligands to RNA. Briefings Bioinf. 2023, 24 (4), bbad187. 10.1093/bib/bbad187. [DOI] [PubMed] [Google Scholar]
- Bedi R. K.; Huang D.; Eberle S. A.; Wiedmer L.; Śledź P.; Caflisch A. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem 2020, 15 (9), 744–748. 10.1002/cmdc.202000011. [DOI] [PubMed] [Google Scholar]
- Li N.; Wei X.; Dai J.; Yang J.; Xiong S. METTL3: a multifunctional regulator in diseases. Mol. Cell. Biochem. 2025, 480, 1–26. 10.1007/s11010-025-05208-z. [DOI] [PubMed] [Google Scholar]
- Ganguly M.; Gupta R.; Roychowdhury A.; Hazra D. De novo drug designing coupled with brute force screening and structure guided lead optimization gives highly specific inhibitor of METTL3: a potential cure for Acute Myeloid Leukaemia. J. Biomol. Struct. Dyn. 2025, 43 (2), 1038–1051. 10.1080/07391102.2023.2291162. [DOI] [PubMed] [Google Scholar]
- Abo-Dya N. E.; Issahaku A. R. Leveraging Flavonoids as Potential Inhibitors of METTL3 in Combating Cancer: A Combined Structure-Based Drug Design and DFT Approach. ChemistrySelect 2023, 8 (45), e202303481 10.1002/slct.202303481. [DOI] [Google Scholar]
- Du Y.; Yuan Y.; Xu L.; Zhao F.; Wang W.; Xu Y.; Tian X. Discovery of METTL3 small molecule inhibitors by virtual screening of natural products. Front. Pharmacol 2022, 13, 878135. 10.3389/fphar.2022.878135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da C.; Kireev D. Structural protein–ligand interaction fingerprints (SPLIF) for structure-based virtual screening: method and benchmark study. J. Chem. Inf. Model. 2014, 54 (9), 2555–2561. 10.1021/ci500319f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40 (D1), D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento A. P.; Gaulton A.; Hersey A.; Bellis L. J.; Chambers J.; Davies M.; Krüger F. A.; Light Y.; Mak L.; McGlinchey S.; et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014, 42 (D1), D1083–D1090. 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44 (D1), D1202–D1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44 (D1), D1045–D1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrie F.; Bradley A. R.; Deane C. M. Generating property-matched decoy molecules using deep learning. Bioinformatics 2021, 37 (15), 2134–2141. 10.1093/bioinformatics/btab080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D. R.; Baumgartner M. P.; Camacho C. J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53 (8), 1893–1904. 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Feng J.; Xue Y.; Guan Z.; Zhang D.; Liu Z.; Gong Z.; Wang Q.; Huang J.; Tang C.; et al. Structural basis of N 6-adenosine methylation by the METTL3–METTL14 complex. Nature 2016, 534 (7608), 575–578. 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Ballester P. J.; Schreyer A.; Blundell T. L. Does a more precise chemical description of protein–ligand complexes lead to more accurate prediction of binding affinity?. J. Chem. Inf. Model. 2014, 54 (3), 944–955. 10.1021/ci500091r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong G.; Shen C.; Yang Z.; Jiang D.; Liu S.; Lu A.; Chen X.; Hou T.; Cao D. Featurization strategies for protein–ligand interactions and their applications in scoring function development. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12 (2), e1567 10.1002/wcms.1567. [DOI] [Google Scholar]
- Tran-Nguyen V.-K.; Junaid M.; Simeon S.; Ballester P. J. A practical guide to machine-learning scoring for structure-based virtual screening. Nat. Protoc. 2023, 18 (11), 3460–3511. 10.1038/s41596-023-00885-w. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 2018, 9 (2), 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitulescu G. M. Quantitative and qualitative analysis of the anti-proliferative potential of the pyrazole scaffold in the design of anticancer agents. Molecules 2022, 27 (10), 3300. 10.3390/molecules27103300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveja J. J.; Vogt M. Automatic identification of analogue series from large compound data sets: methods and applications. Molecules 2021, 26 (17), 5291. 10.3390/molecules26175291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Chen W.; Chen G.; Zhou S.; Wei D.-Q.; Xiong Y. MDDI-SCL: predicting multi-type drug-drug interactions via supervised contrastive learning. J. Cheminf. 2022, 14 (1), 81. 10.1186/s13321-022-00659-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wei Z.; Xi L. Sfcnn: a novel scoring function based on 3D convolutional neural network for accurate and stable protein–ligand affinity prediction. BMC Bioinf. 2022, 23 (1), 222. 10.1186/s12859-022-04762-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani A.Attention is all you need. Advances in Neural Information Processing Systems, 2017.
- Garbin C.; Zhu X.; Marques O.. Dropout vs. batch normalization: an empirical study of their impact to deep learning. Multimedia Tools and Applications; Springer Science and Business Media LLC, 2020; Vol. 79( (19), ), pp 12777–12815. [Google Scholar]
- Li Z.; Gong B.; Yang T.. Improved dropout for shallow and deep learning. Advances in Neural Information Processing Systems, 2017, Vol. 29. [Google Scholar]
- Caba K.; Tran-Nguyen V.-K.; Rahman T.; Ballester P. J. Comprehensive machine learning boosts structure-based virtual screening for PARP1 inhibitors. J. Cheminf. 2024, 16 (1), 40. 10.1186/s13321-024-00832-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Cui C.; Ding X.; Xiong Z.; Zheng M.; Luo X.; Jiang H.; Chen K. Improving the virtual screening ability of target-specific scoring functions using deep learning methods. Front. Pharmacol. 2019, 10, 924. 10.3389/fphar.2019.00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira M. S.; Koch O. The development of target-specific machine learning models as scoring functions for docking-based target prediction. J. Chem. Inf. Model. 2019, 59 (3), 1238–1252. 10.1021/acs.jcim.8b00773. [DOI] [PubMed] [Google Scholar]
- Ragoza M.; Hochuli J.; Idrobo E.; Sunseri J.; Koes D. R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57 (4), 942–957. 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon M.; Money-Kyrle S.; Blay V.; Houston D. R. SCORCH: Improving structure-based virtual screening with machine learning classifiers, data augmentation, and uncertainty estimation. J. Adv. Res. 2023, 46, 135–147. 10.1016/j.jare.2022.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used and all codes to reproduce this study are freely available on https://github.com/JuniML/DeepMETTL3. The DeepCoy decoys are generated through the DeepCoy algorithm and the code is available on https://github.com/fimrie/DeepCoy/tree/master.