

Abstract
As obesity and diabetes reach epidemic proportions in the developed world, the role of insulin resistance and its consequences are gaining prominence. Understanding the role of insulin in wide-ranging physiological processes and the influences on its synthesis and secretion, alongside its actions from the molecular to the whole body level, has significant implications for much chronic disease seen in Westernised populations today. This review provides an overview of insulin, its history, structure, synthesis, secretion, actions and interactions followed by a discussion of insulin resistance and its associated clinical manifestations. Specific areas of focus include the actions of insulin and manifestations of insulin resistance in specific organs and tissues, physiological, environmental and pharmacological influences on insulin action and insulin resistance as well as clinical syndromes associated with insulin resistance. Clinical and functional measures of insulin resistance are also covered. Despite our incomplete understanding of the complex biological mechanisms of insulin action and insulin resistance, we need to consider the dramatic social changes of the past century with respect to physical activity, diet, work, socialisation and sleep patterns. Rapid globalisation, urbanisation and industrialisation have spawned epidemics of obesity, diabetes and their attendant co-morbidities, as physical inactivity and dietary imbalance unmask latent predisposing genetic traits.
Introduction
With obesity and diabetes reaching epidemic proportions in the developed world,1 the role of insulin resistance and its sequelae is gaining prominence. Understanding the role of insulin across a wide range of physiological processes and the influences on its synthesis and secretion, alongside its actions from the molecular to the whole body level, has significant implications for much chronic disease seen in Westernised populations today. Consequently, more than a century after scientists began to elucidate the role of the pancreas in diabetes, the study of insulin and insulin resistance remain in the forefront of medical research, relevant at all levels from bench to bedside and to public health policy.
Scope of this Review
This review will include an overview of insulin, its history, structure, synthesis, secretion, actions and interactions followed by a discussion of insulin resistance and its associated clinical manifestations. This latter section will discuss the mechanisms and contexts, physiological and pathological, of insulin resistance, and its assessment in the clinical laboratory. Given the breadth of the topic, this review will not be exhaustive, but will set the scene, bringing together the diverse areas of current understanding of this continuously emerging field.
Definitions and Concepts
Insulin is a peptide hormone secreted by the β cells of the pancreatic islets of Langerhans and maintains normal blood glucose levels by facilitating cellular glucose uptake, regulating carbohydrate, lipid and protein metabolism and promoting cell division and growth through its mitogenic effects.
Insulin resistance is defined where a normal or elevated insulin level produces an attenuated biological response;2 classically this refers to impaired sensitivity to insulin mediated glucose disposal.3
Compensatory hyperinsulinaemia occurs when pancreatic β cell secretion increases to maintain normal blood glucose levels in the setting of peripheral insulin resistance in muscle and adipose tissue.
Insulin resistance syndrome refers to the cluster of abnormalities and related physical outcomes that occur more commonly in insulin resistant individuals. Given tissue differences in insulin dependence and sensitivity, manifestations of the insulin resistance syndrome are likely to reflect the composite effects of excess insulin and variable resistance to its actions.3
Metabolic syndrome represents the clinical diagnostic entity identifying those individuals at high risk with respect to the (cardiovascular) morbidity associated with insulin resistance.3
The Discovery of Insulin
In 1889 German scientists Minkowski and von Mering noted, from their experimental work with animals, that total pancreatectomy led to the development of severe diabetes.4 They hypothesised that a substance secreted by the pancreas was responsible for metabolic control. Others later refined this hypothesis, noting diabetes to be associated with destruction of the islets of Langerhans. While Minkowski, as well as Zuelzer in Germany and Scott in the USA attempted, with inconsistent results, to isolate and administer the missing pancreatic islet substance, Belgian investigator de Meyer in 1909 proposed the name “insuline”, as did British researcher Schaefer in 1916.
Finally in 1921, a decade later, insulin was finally isolated, purified and available in a form capable of therapeutic administration. In May 1921, Toronto surgeon Banting, assisted by medical student Best, and under the supervision of McLeod, Professor of Carbohydrate Metabolism, began experiments in dogs. They administered chilled saline extracts of pancreas intravenously to dogs rendered diabetic by pancreatectomy and observed lowering of blood glucose. In December 1921 this work was presented to the American Physiological Association, and biochemist Collip, who had joined the team, further demonstrated that this extract also restored hepatic glycogen mobilisation and the capacity to clear ketones. One month later, in January 1922 the first human experiments began on a 14 year old diabetic boy whose clinical symptoms and biochemical abnormalities were essentially reversed by administration of the pancreatic isolate. In May 1922, the active component had been named insulin, and the results of these experiments presented to the Association of American Physicians. Eli Lilly subsequently began production of porcine insulin, enhancing purification through iso-electric precipitation, making commercial quantities by early 1923. The Nobel Prize was awarded in 1923 to Banting and McLeod.4
Structure and Chemical Properties of Insulin
Insulin was found to be a polypeptide in 1928 with its amino acid sequence identified in 1952. It is in fact a dipeptide, containing A and B chains respectively, linked by disulphide bridges, and containing 51 amino acids, with a molecular weight of 5802. Its iso-electric point is pH 5.5.5 The A chain comprises 21 amino acids and the B chain 30 amino acids. The A chain has an N-terminal helix linked to an anti-parallel C-terminal helix; the B chain has a central helical segment. The two chains are joined by 2 disulphide bonds, which join the N- and C-terminal helices of the A chain to the central helix of the B chain. In pro-insulin, a connecting peptide links the N-terminus of the A chain to the C-terminus of the B chain.6
Synthesis and Release of Insulin
Insulin is coded on the short arm of chromosome 117 and synthesised in the β cells of the pancreatic islets of Langherhans as its precursor, proinsulin. Proinsulin is synthesised in the ribosomes of the rough endoplasmic reticulum (RER) from mRNA as pre-proinsulin. Pre-proinsulin is formed by sequential synthesis of a signal peptide, the B chain, the connecting (C) peptide and then the A chain comprising a single chain of 100 amino acids. Removal of the signal peptide forms proinsulin, which acquires its characteristic 3 dimensional structure in the endoplasmic reticulum. Secretory vesicles transfer proinsulin from the RER to the Golgi apparatus, whose aqueous zinc and calcium rich environment favours formation of soluble zinc-containing proinsulin hexamers.6 As immature storage vesicles form from the Golgi, enzymes acting outside the Golgi convert proinsulin to insulin and C-peptide.8 Insulin forms zinc-containing hexamers which are insoluble, precipitating as chemically stable crystals at pH 5.5. When mature granules are secreted into the circulation by exocytosis, insulin, and an equimolar ratio of C-peptide are released. Proinsulin and zinc typically comprise no more than 6% of the islet cell secretion.6
Insulin secretion from the islet cells into the portal veins is characteristically pulsatile, reflecting the summation of coordinate secretory bursts from millions of islet cells. An ultradian oscillatory pattern of insulin release, in addition to post meal variation, has been reported.9 In response to a stimulus such as glucose, insulin secretion is characteristically biphasic, with an initial rapid phase of insulin secretion, followed by a less intense but more sustained release of the hormone.10
Factors Influencing Insulin Biosynthesis and Release
Insulin secretion may be influenced by alterations in synthesis at the level of gene transcription, translation, and post-translational modification in the Golgi as well as by factors influencing insulin release from secretory granules. Longer-term modification may occur via influences on β cell mass and differentiation.11 Given insulin’s pivotal role in glucose utilisation and metabolism, it is not surprising that glucose has multiple influences on insulin biosynthesis and secretion. However, other factors such as amino acids, fatty acids, acetylcholine, pituitary adenylate cyclase-activating polypeptide (PACAP), glucose-dependent insulinotropic polypeptide (GIP), glucagon-like peptide-1 (GLP-1), and several other agonists, together in combination, also influence these processes.10
Mechanisms of Insulin Secretion
Increased levels of glucose induce the “first phase” of glucose-mediated insulin secretion by release of insulin from secretory granules in the β cell. Glucose entry into the β cell is sensed by glucokinase, which phosphorylates glucose to glucose-6-phosphate (G6P), generating ATP.12 Closure of K+-ATP-dependent channels results in membrane depolarization and activation of voltage dependent calcium channels leading to an increase in intracellular calcium concentration; this triggers pulsatile insulin secretion.13 Augmentation of this response occurs by both a K+-ATP channel-independent Ca2+-dependent pathway and K+-ATP channel-independent Ca2+-independent pathways of glucose action.10 Other mediators of insulin release include activation of phospholipases and protein kinase C (e.g. by acetycholine) and by stimulation of adenylyl cyclase activity and activation of β cell protein kinase A, which potentiates insulin secretion. This latter mechanism may be activated by hormones, such as vasoactive intestinal peptide (VIP), PACAP, GLP-1, and GIP. These factors appear to play a significant role in the second phase of glucose mediated insulin secretion, after refilling of secretory granules translocated from reserve pools.10
Regulation and Mechanisms of Insulin Secretion at the Cellular Level
Synthesis and secretion of insulin is regulated by both nutrient and non-nutrient secretagogues, in the context of environmental stimuli and the interplay of other hormones.8 Nutrient secretagogues such as glucose appear to trigger insulin secretion from the β cell by increasing intracellular ATP and closing of K+-ATP channels as outlined above. Generation of cyclic AMP and other cellular energy intermediates is also augmented, further enhancing insulin release. Glucose does not require insulin action to enter the β cell (nor do fructose, mannose or galactose).8 Non-nutrient secretagogues may act via neural stimuli such as cholinergic and adrenergic pathways, or through peptide hormones and cationic amino acids.
Neural Stimuli
1. Cholinergic Transmission
It has been well recognised that vagus nerve stimulation results in pancreatic insulin secretion. This is thought to mediate the so-called “cephalic phase” of insulin secretion, occurring when food is seen, smelled or acutely ingested. Islet cell cholinergic muscarinic receptors activate phospholipase C, with subsequent intracellular events activating protein kinase C, phospholipase A2 and mobilizing intracellular calcium. Insulin secretion by these mechanisms does not occur in the fasting state or if blood glucose levels are low, but may augment the anabolic response to feeding.8
2. Adrenergic Pathway
Catecholamines, through α2-adrenoceptors, typically inhibit insulin release during stress and exercise. The pancreas also has β-adrenoceptors, which enhance insulin secretion via cAMP, but this effect appears to be minor. The α2-mediated effects appear to act downstream to those of nutrient secretogogues.8
Peptide Hormones
A number of peptide hormones influence insulin secretion. The gut hormones GLP-114 and somatostatin enhance and inhibit insulin action respectively. Their secretion is stimulated by nutrients in the gut. Their mechanisms of action appear to be, at least in part, mediated by cAMP generation and activation of a cAMP-responsive protein kinase.8 Leptin and adiponectin appear to act in a similar fashion whereas acylation-stimulating protein appears to work via glucose phosphorylation, calcium influx and protein kinase C.15
Amino Acids
The insulinotropic effect of the cationic amino acid arginine is well established. L-ornithine has similar effects. Arginine’s action is associated with an increase in potassium permeability without any effect on proinsulin biosynthesis. Uptake of arginine into the β cell is associated with depolarization of the cell membrane and gating of voltage dependent calcium channels.8 Leucine, an essential branched chain amino acid, is a potent enhancer of insulin secretion, allosterically stimulating glutamate dehydrogenase and forming α-ketoglutarate via the Krebs cycle to generate ATP.12 The major pathways and influences on insulin secretion are presented schematically in Figure 1.
Figure 1.
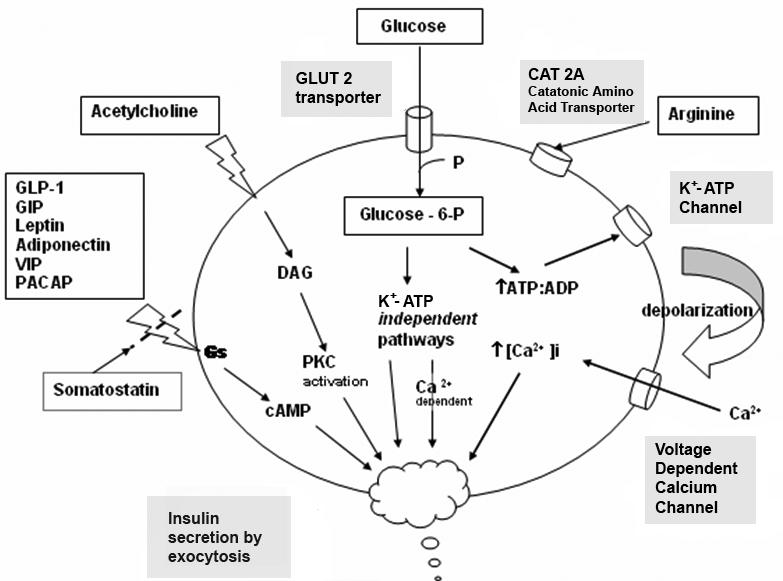
Schematic presentation of insulin secretory pathways. Adapted from references: 10 & 14.
Footnotes: Figure Abbreviations
Akt/PKB = protein kinase B
GLP-1 = glucagon-like peptide 1
ATP = adenosine triphosphate
IRS = insulin receptor substrate
ADP = adenosine diphosphate
MAP kinase = mitogen activated protein kinase
cAMP= cyclic adenosine monophosphate
PACAP = pituitary adenylate cyclase-activating polypeptide
DAG = diacylglycerol
PI3K = phosphatidylinositol 3-kinase
GIP = gastric inhibitory peptide /
PIPD1 & 2 = phosphatidylinositol dependent protein kinases 1 & 2
glucose-dependent insulinotropic polypeptide
PKC = protein kinase C
Glucose- 6- P = glucose 6 phosphate
RAS = rat sarcoma protein
GLUT 2 = glucose transport protein 2
SHC =adaptor protein with src-homology
GLUT 4 = glucose transport protein 4
VIP= vasoactive intestinal peptide
Factors Influencing β Cell Mass
Longer term regulation of insulin secretion may be mediated via effects on β cell mass. Growth hormone, prolactin, placental lactogen and GLP-1 not only increase glucose-stimulated insulin release and insulin gene expression, but also increase β cell proliferation.11,14 For a comprehensive review on this topic see Nielsen and Serup.16
Physiology of Insulin Secretion
Glucose is the principal stimulus for insulin secretion, though other macronutrients, hormones, humoral factors and neural input may modify this response. Insulin, together with its principal counter-regulatory hormone glucagon, regulates blood glucose concentrations.17 Pancreatic β cells secrete 0.25–1.5 units of insulin per hour during the fasting (or basal) state, sufficient to enable glucose insulin-dependent entry into cells. This level prevents uncontrolled hydrolysis of triglycerides and limits gluconeogenesis, thereby maintaining normal fasting blood glucose levels. Basal insulin secretion accounts for over 50% of total 24 hour insulin secretion. Following secretion of insulin into the portal venous system, 60% is subsequently removed by the liver; so portal vein insulin concentrations reaching the liver approach triple that of the peripheral circulation. In healthy lean individuals circulating venous (or arterial) fasting insulin concentrations are about 3–15 mIU/L or 18–90 pmol/L.17 Meal-related insulin secretion accounts for the remaining fraction of the total daily output.
Insulin Secretion in Response to Stimuli
Response to Glucose
In healthy individuals glucose stimulated pancreatic secretion is biphasic. Intravenous administration of glucose is associated with a rapid “first phase” of insulin release within 1 minute, peaking at 3–5 minutes, and lasting about 10 minutes; the slower onset “second phase” of insulin secretion begins shortly after the glucose bolus but is not apparent until 10 minutes later, lasts the duration of the hyperglycaemia and is proportional to the glucose concentration immediately prior to the glucose administration.17 The first phase of insulin secretion represents release of insulin already synthesised and stored in secretory granules; the second phase represents secretion of both stored and newly synthesised insulin. Overall insulin secretion relates to the total dose of glucose and its rate of administration; maximal pancreatic response occurs with 20 g of glucose given intravenously over 3 minutes in humans.18
In contrast to the reproducible pattern of insulin secretion in response to intravenous glucose, insulin secretion following oral glucose is much more variable. With an oral glucose load, gastric emptying and gastrointestinal motility affect glucose absorption, gastro-intestinal hormones and neural input associated with glucose ingestion modify the insulin response, and insulin secretion continues some time after glucose ingestion.17
Response to Arginine
The response to arginine is well characterised. Arginine-induced insulin secretion is proportional to the basal glucose level and occurs 2–10 minutes after IV injection.17 Oral ingestion of amino acids stimulates the secretion of insulin, as well as growth hormone and glucagon.19
Effects of Lipids
In the circulation, non-esterified fatty acids (NEFA) may be derived from dietary lipids and released by the liver, or synthesised in adipocytes, liver, skin and small intestine from excess carbohydrate. NEFA not only increase hepatic glucose output and reduce peripheral insulin sensitivity, but may also modify glucose stimulated insulin secretion (GSIS). Acute elevation of plasma NEFA is associated with an increase in GSIS, dependent on the prevailing glucose concentration. Chronic elevation of NEFA is associated with a decrease in GSIS, and reduced insulin synthesis.20 Ingestion of lipid may also modify the insulin response to glucose by effects on gastrointestinal hormones and gastric emptying.21
Response to Mixed Meal
This is a much more physiological stimulus for insulin secretion. The pattern of insulin secretion will depend on the relative proportions of various nutrients, the physical form in which the component foods are ingested and those factors influencing response to oral glucose alone.22,23
Incretin Hormones
Nutrients in the GI tract stimulate the secretion of hormones known as incretins which amplify glucose-induced insulin release. These account for the greater insulin response to oral, as opposed to intravenous, glucose. GIP and GLP-1 are the two most important incretin hormones. GLP-1 also inhibits glucagon release, delays gastric emptying and reduces appetite.24
Effects of Neural and Hormonal Stimuli
See Table 1.
Table 1.
Mediators of insulin secretion.
| Stimulus | Nutrient | Hormone | Neural |
|---|---|---|---|
| Stimulatory | Glucose | Growth hormone | β-adrenergic |
| Amino acids | Glucagon | Vagal | |
| (Ketones) | GLP-1 | (parasympathetic) | |
| GIP | |||
| Secretin | |||
| Cholecystokinin | |||
| Gastrin | |||
| VIP | |||
| Gastrin releasing peptide | |||
| Inhibitory | Adrenocorticosteroids | α-adrenergic | |
| Somatostatin | |||
| Adrenalin | |||
| Noradrenalin | |||
| Galanin | |||
| Neuropeptide Y | |||
| Calcitonin gene-related peptide | |||
| (CGRP) | |||
| Prostaglandin E |
Reference: Adapted from reference 17.
Insulin Receptors and Insulin Binding
Insulin mediates its actions through binding to insulin receptors. The insulin receptor was first characterised in 1971. It consists of a heterotetramer consisting of 2 α and 2 β glycoprotein subunits linked by disulphide bonds and is located on the cell membrane.25 The gene coding for the insulin receptor is located on the short arm of chromosome 19.17 Insulin binds to the extracellular α subunit, resulting in conformational change enabling ATP to bind to the intracellular component of the β subunit.23 ATP binding in turn triggers phosphorylation of the β subunit conferring tyrosine kinase activity. This enables tyrosine phosphorylation of intracellular substrate proteins known as insulin responsive substrates (IRS). The IRS can then bind other signalling molecules which mediate further cellular actions of insulin.25
There are four known specifically-named IRS proteins. IRS 1 and 2 have widely overlapping tissue distribution. IRS 1 is phosphorylated by both the insulin receptor and insulin-like growth factor 1 (IGF-1 see below) receptor, mediates the mitogenic effects of insulin and couples glucose sensing to insulin secretion with IRS 1 proposed to be the major IRS in skeletal muscle. IRS 2, proposed to be the main IRS in liver, mediates peripheral actions of insulin and growth of pancreatic β cells.25 IRS 3 and 4 are less well characterised. IRS 3 is found only in adipose tissue, β cells and liver and IRS 4 in thymus, brain and kidney.26,27 Phosphorylated IRS proteins bind specific src-homology-2 domain proteins (SH2), which include important enzymes such as phosphatidylinositol 3-kinase (PI 3-kinase) and phosphotyrosine phosphatase SHPTP2 (or Syp), and other proteins that lack enzymatic activity but which link IRS-1 and other intracellular signalling systems, e.g. the adaptor protein Grb2 which connects with the RAS (rat sarcoma protein) pathway.
PI 3-kinase promotes the translocation of glucose transporter proteins, glycogen, lipid and protein synthesis, anti-lipolysis and the control of hepatic gluconeogenesis.27 PI 3-kinase acts via serine and threonine kinases such as Akt/protein kinase B (PKB), protein kinase C (PKC) and PI dependent protein kinases1& 2 (PIPD 1&2). The RAS pathway activates transcription factors and stimulates the growth promoting actions of insulin.25 Thus broadly, PI 3-kinase mediates insulin’s metabolic effects, e.g. cellular glucose uptake, while RAS significantly mediates insulin’s mitogenic effects17,25 together with other less well described actions. These pathways are presented schematically in Figure 2.
Figure 2.
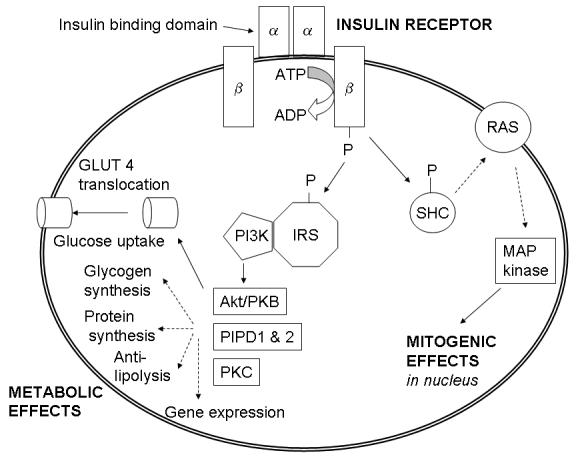
Schematic presentation of insulin signalling pathways. Adapted from references: 25, 28 & 35.
Footnotes: Figure Abbreviations
Akt/PKB = protein kinase B
GLP-1 = glucagon-like peptide 1
ATP = adenosine triphosphate
IRS = insulin receptor substrate
ADP = adenosine diphosphate
MAP kinase = mitogen activated protein kinase
cAMP= cyclic adenosine monophosphate
PACAP = pituitary adenylate cyclase-activating polypeptide
DAG = diacylglycerol
PI3K = phosphatidylinositol 3-kinase
GIP = gastric inhibitory peptide /
PIPD1 & 2 = phosphatidylinositol dependent protein kinases 1 & 2
glucose-dependent insulinotropic polypeptide
PKC = protein kinase C
Glucose- 6- P = glucose 6 phosphate
RAS = rat sarcoma protein
GLUT 2 = glucose transport protein 2
SHC =adaptor protein with src-homology
GLUT 4 = glucose transport protein 4
VIP= vasoactive intestinal peptide
Glucose Transporter Proteins
Glucose enters cells in an ATP-independent manner by means of glucose transporter proteins (GLUT), of which at least 5 subtypes have been identified28,29 (Table 2). Differing in characteristics such as Km for maximal glucose transport and insulin dependency, they enable different cell types to utilise glucose according to their specific functions. For example, most brain cells, having GLUT 1 as the principal transporter protein, are able to move glucose intracellularly at very low blood glucose blood concentrations without the need for insulin. Thus these neurons, which are principally dependent on glucose for intracellular energy, are able to extract it from the circulation and function despite the low glucose and insulin levels seen during the fasting state. On the other hand, adipose cells and muscle cells have GLUT 4 as the major glucose transporter protein, which requires insulin for its action and has a much higher Km for glucose. This enables adipose tissue cells, whose function is to store excess energy, to respond to the higher glucose levels characteristic of the fed state, and allows glucose to enter the cells where fatty acid and glycerol synthesis is stimulated and lipolysis suppressed. However, where glucose and insulin levels fall to fasting values, glucose no longer enters the cells, promoting lipolysis. In muscle cells, intracellular glucose transport facilitates glycogen synthesis in the fed state.27 PI 3-kinase appears to be essential for the translocation of GLUT 4 to the cell membrane in muscle cells and adipocytes; this facilitates the downstream actions of this key intracellular enzyme.25
Table 2.
| ISOFORM | Tissue Distribution | Affinity for Glucose | Km | Characteristics | Gene location |
|---|---|---|---|---|---|
| GLUT 1 | Brain microvessels, | Ubiquitous | |||
| Red blood cells | High | 1 mmol/L | Chr 1 | ||
| Placenta | Basal transporter | ||||
| Kidney | |||||
| All tissues | |||||
| GLUT 2 | Liver | High Km transporter | |||
| Kidney | Low | 15–20 mmol/L | Chr 3 | ||
| β cell | Insulin-independent | ||||
| Small intestine | |||||
| GLUT 3 | Brain neurons | Low Km transporter | |||
| Placenta | High | <1 mmol/L | Chr 12 | ||
| Foetal muscle | Found in glucose- dependent tissues | ||||
| All tissues | |||||
| GLUT 4 | Muscle cells | Sequestered intracellularly and translocates to cell surface in response to insulin | |||
| Fat cells | Medium | 2.5–5 mmol/L | Chr 17 | ||
| Heart | |||||
| GLUT 5 | Small intestine | High affinity for fructose | |||
| Testes | Medium | 6 mmol/L | Chr 1 |
Actions of Insulin at the Cellular Level
Insulin’s actions at the cellular level encompass carbohydrate, lipid and amino acid metabolism and mRNA transcription and translation.
Carbohydrate Metabolism
Insulin acts at multiple steps in carbohydrate metabolism. Its effect on facilitated diffusion of glucose into fat and muscle cells via modulation of GLUT 4 translocation has been discussed. Glycogen synthesis is increased, and glycogen breakdown decreased, by dephosphorylation of glycogen synthase and glycogen phosphorylase kinase respectively. Glycolysis is stimulated and gluconeogenesis inhibited by dephosphorylation of pyruvate kinase (PK) and 2,6 biphosphate kinase. Signalling intracellular energy abundance, insulin enhances the irreversible conversion of pyruvate to Acetyl Co-A by activation of the intra-mitochondrial enzyme complex pyruvate dehydrogenase. Acetyl-CoA may then be directly oxidised via the Krebs’ cycle, or used for fatty acid synthesis.30
Lipid Metabolism
Insulin stimulates fatty acid synthesis in adipose tissue, liver and lactating mammary glands along with formation and storage of triglycerides in adipose tissue and liver. Fatty acid synthesis is increased by activation and increased phosphorylation of acetyl-CoA carboxylase, while fat oxidation is suppressed by inhibition of carnitine acyltransferase. Triglyceride synthesis is stimulated by esterification of glycerol phosphate, while triglyceride breakdown is suppressed by dephosphorylation of hormone sensitive lipase. Cholesterol synthesis is increased by activation and dephosphorylation of HMG Co-A reductase while cholesterol ester breakdown appears to be inhibited by dephosphorylation of cholesterol esterase. Phospholipid metabolism is also influenced by insulin.28
Protein Synthesis
Insulin promotes protein synthesis in a range of tissues. There are effects on transcription of specific mRNA, as well as translation of mRNA into proteins in the ribosomes. Examples of enhanced mRNA transcription include the mRNA for glucokinase, PK, fatty acid synthase and albumin in the liver, pyruvate carboxylase in the adipose tissue, casein in the mammary gland and amylase in the pancreas. Insulin action decreases mRNA for liver enzymes such as carbamoyl phosphate synthetase, a key enzyme in the urea cycle. Effects on translation are widespread and influenced by both insulin itself and by various growth factors, e.g. IGF-1.19,28
Other Ligands for the Insulin Receptor
Insulin-like growth factors (IGF) are so-called because they have significant structural homology with proinsulin but mainly mitogenic effects, significantly regulated by growth hormone.31 IGF-1 and 2 are coded for on the long arm of chromosome 12 and short arm of chromosome 11 respectively.32 They have specific receptors and bind with different affinities to the various IGF binding proteins. Insulin can bind to the receptors for IGF-1 and 2 but with much lower affinity (10−2 and 5x10−3) respectively. IGF-1 binds weakly to the insulin receptor, with only 1.25x10−3 the affinity for the IGF-1 receptor; it binds the IGF-2 receptor with 1/4 the affinity for the IGF-2 receptor. IGF-2 does not bind to the insulin receptor; it does bind the IGF-1 receptor but with 1/3 the affinity for the IGF-2 receptor.29 Therefore overlap in physiological functions is more limited in vivo.
Physiological Role of Insulin
Insulin is the pivotal hormone regulating cellular energy supply and macronutrient balance, directing anabolic processes of the fed state.27 Insulin is essential for the intra-cellular transport of glucose into insulin-dependent tissues such as muscle and adipose tissue. Signalling abundance of exogenous energy, adipose tissue fat breakdown is suppressed and its synthesis promoted. In muscle cells, glucose entry enables glycogen to be synthesised and stored, and for carbohydrates, rather than fatty acids (or amino acids) to be utilised as the immediately available energy source for muscle contraction. Insulin therefore promotes glycogen and lipid synthesis in muscle cells, while suppressing lipolysis and gluconeogenesis from muscle amino acids. In the presence of an adequate supply of amino acids, insulin is anabolic in muscle.29
Mechanisms of Insulin Resistance
Physiologically, at the whole body level, the actions of insulin are influenced by the interplay of other hormones. Insulin, though the dominant hormone driving metabolic processes in the fed state, acts in concert with growth hormone and IGF-1; growth hormone is secreted in response to insulin, among other stimuli, preventing insulin-induced hypoglycaemia. Other counter-regulatory hormones include glucagon, glucocorticoids and catecholamines. These hormones drive metabolic processes in the fasting state. Glucagon promotes glycogenolysis, gluconeogenesis and ketogenesis. The ratio of insulin to glucagons determines the degree of phosphorylation or dephosphorylation of the relevant enzymes.29 Catecholamines promote lipolysis and glycogenolysis; glucocorticoids promote muscle catabolism, gluconeogenesis and lipolysis. Excess secretion of these hormones may contribute to insulin resistance in particular settings, but does not account for the vast majority of insulin resistant states.
Insulin resistance in most cases is believed to be manifest at the cellular level via post-receptor defects in insulin signalling. Despite promising findings in experimental animals with respect to a range of insulin signalling defects, their relevance to human insulin resistance is presently unclear. Possible mechanisms include down-regulation, deficiencies or genetic polymorphisms of tyrosine phosphorylation of the insulin receptor, IRS proteins or PIP-3 kinase, or may involve abnormalities of GLUT 4 function.33
Sites of Insulin Action and Manifestations of Insulin Resistance
The effects of insulin, insulin deficiency and insulin resistance vary according to the physiological function of the tissues and organs concerned, and their dependence on insulin for metabolic processes. Those tissues defined as insulin dependent, based on intracellular glucose transport, are principally adipose tissue and muscle. However, insulin’s actions are pleotropic and widespread, as are the manifestations of insulin resistance and the associated compensatory hyperinsulinaemia.3
Muscle
Glucose uptake into muscle is essentially insulin dependent via GLUT 4, and muscle accounts for about 60–70% of whole-body insulin mediated uptake.34 In the fed state insulin promotes glycogen synthesis via activation of glycogen synthase. This enables energy to be released anaerobically via glycolysis, e.g. during intense muscular activity. Muscle cells do not rely on glucose (or glycogen) for energy during the basal state, when insulin levels are low. Insulin suppresses protein catabolism while insulin deficiency promotes it, releasing amino acids for gluconeogenesis. In starvation, protein synthesis is reduced by 50%.35 Whilst data regarding a direct anabolic effect of insulin are inconsistent, it is clearly permissive, modulating the phosphorylation status of intermediates in the protein synthetic pathway. In experimental studies, the insulin dose promoting protein synthesis is significantly greater than the dose required to suppress proteolysis. Insulin is anabolic in conjunction with growth hormone, IGF-1 and sufficient amino acids.35 In insulin resistance, muscle glycogen synthesis is impaired; this appears largely mediated by reduced intracellular glucose translocation.28 In regard to protein turnover, one study reported no difference between insulin resistant type 2 diabetics and controls, though this was at the expense of hyperinsulinaemia in this hyperinsulinemic euglycemic clamp study.36
Adipose Tissue
Intracellular glucose transport into adipocytes in the postprandial state is insulin-dependent via GLUT 4; it is estimated that adipose tissue accounts for about 10% of insulin stimulated whole body glucose uptake.34 Insulin stimulates glucose uptake, promotes lipogenesis while suppressing lipolysis, and hence free fatty acid flux into the bloodstream. As adipocytes are not dependent on glucose in the basal state, intracellular energy may be supplied by fatty acid oxidation in insulin-deficient states, whilst liberating free fatty acids into the circulation for direct utilization by other organs e.g. the heart, or in the liver where they are converted to ketone bodies. Ketone bodies provide an alternative energy substrate for the brain during prolonged starvation.30
In insulin resistance the effects on adipose tissue are similar, but in the liver the increased free fatty acid flux tends to promote hepatic very low density lipoprotein (VLDL) production37 whilst ketogenesis typically remains suppressed by the compensatory hyperinsulinaemia. Furthermore, since lipoprotein lipase activity is insulin-dependent and impaired by insulin resistance, peripheral uptake of triglycerides from VLDL is also diminished. These mechanisms contribute to the observed hypertriglyceridaemia of insulin resistance.38 In addition to free fatty acids, adipose tissue secretes a number of cytokines which have systemic effects on insulin resistance. These include IL-6, TNFα, plasminogen activator inhibitor 1 (PAI-1), angiotensinogen and leptin which are associated with increased insulin resistance, and adiponectin with reduced insulin resistance.39 TNFα and IL-6 impair insulin signalling, lipolysis and endothelial function. IL-6 production is enhanced by sympathetic nervous system activation, e.g. stress.39 Adipose tissue depots differ in their response to insulin.35 Adipocytes from diabetic and insulin resistant individuals have reduced GLUT 4 translocation, impaired intracellular signalling via reduced IRS-1 gene and protein expression, impaired insulin-stimulated PIP-3 kinase and Akt (protein kinase B).34
Liver
While glucose uptake into the liver is not insulin-dependent, it accounts for about 30% of whole body insulin-mediated glucose disposal,34 with insulin being needed to facilitate key metabolic processes. Through intracellular signalling described above, glycogen synthesis is stimulated while protein synthesis and lipoprotein metabolism are modulated.30 Gluconeogenesis and ketone body production are inhibited. Mitogenic effects of insulin (and growth hormone) are mediated via hepatic production of insulin-like growth factors and potentially via suppression of sex-hormone binding globulin (SHBG) production.28
Whilst in insulin deficiency, e.g. starvation, these processes are more uniformly affected, this is not necessarily the case with insulin resistance. Compensatory hyperinsulinaemia, differential insulin resistance and differential tissue requirements may dissociate these processes.3 Resistance to insulin’s metabolic effects results in increased glucose output via increased gluconeogenesis (as in starvation), however, unlike starvation, compensatory hyperinsulinaemia depresses SHBG production and promotes insulin’s mitogenic effects. Alterations in lipoprotein metabolism represent a major hepatic manifestation of insulin resistance. Increased free fatty acid delivery, and reduced VLDL catabolism by insulin resistant adipocytes, results in increased hepatic triglyceride content and VLDL secretion.38 Hepatic synthesis of C-reactive protein, fibrinogen and PAI-1 is induced in response to adipocyte-derived pro-inflammatory cytokines such as TNFα and IL-6. Insulin may also increase factor VII gene expression.39
Endothelium and Vasculature
Insulin and its actions play an important role in various aspects of endothelial function, e.g. nitric oxide production, while insulin resistance is strongly associated with endothelial dysfunction. Whether these associations are causal, or mediated by common mechanisms, awaits clarification. The functions of vascular endothelial cells are critical to many aspects of cardiovascular biology, with endothelial dysfunction being seen at a very early stage of atherosclerosis and its associated clinical risk factors. Endothelial cells not only provide the physical lining of the blood vessels but secrete various factors influencing vessel tone, platelet function, coagulation and fibrinolysis. Clinical problems develop when these processes are in imbalance.
Nitric oxide (NO) is the major factor in large arteries mediating endothelial dependent relaxation. It also inhibits platelet aggregation, cell adhesion and smooth muscle cell proliferation. NO is synthesised from L-arginine, molecular oxygen and NADPH, via the activity of endothelial enzyme nitric oxide synthase (eNOS), and its cofactors tetrahydrobiopterin, flavin adenine dinucleotide and flavin mononucleotide. Interestingly, arginine is a potent secretatogue for insulin and there is a final common pathway for the intracellular signalling of both eNOS and insulin. Insulin enhances tetrahydrobiopterin production by stimulating its biosynthetic enzyme GTP cyclohydrolase, and stimulates eNOS by calcium-independent phosphorylation of eNOS at serine and threonine residues via PIP-3 kinase and Akt (protein kinase B). Thus nitric oxide production is enhanced. Insulin also promotes release of the vasoconstrictor endothelin while TNFα decreases eNOS expression and induces von Willebrand Factor release. Ininsulin resistance tetrahydrobiopterin levels are reduced, the pathways for eNOS stimulation are down-regulated, and vasodilator responses to insulin and cholinergic agonists are impaired. Insulin’s ability to counteract the TNFα-mediated Akt dephosphorylation in endothelial cells is also lost. Free fatty acids, elevated in insulin resistant states, also inhibit eNOS activity, decreasing NO production.
The compensatory hyperinsulinaemia that accompanies insulin resistance is associated with increased levels of pro-coagulant factors such as PAI-1. These factors are thought to contribute to the enhanced platelet aggregation seen in insulin resistant states. Endothelin 1 secretion is stimulated by insulin and elevated in insulin resistant states. Endothelin 1, a potent vasoconstrictor also inhibits insulin signalling via PIP-3 kinase and competes with NO resulting in endothelial dysfunction. Whilst the metabolic effects of insulin are variably affected in insulin resistance, the mitogenic properties, mediated via the MAP (mitogen activated protein) kinase pathway, remain intact. These mitogenic effects of insulin on endothelial smooth muscle cell proliferation probably contribute to atherosclerosis.33
Brain
While the brain is not insulin-dependent so far as intracellular glucose uptake is concerned, insulin receptors have been located in the brain and are concentrated in the olfactory bulb, hypothalamus, hippocampus,40 retina and vessels of the choroid plexus, as well as in regions of the striatum and cerebral cortex e.g. medial temporal lobes. Insulin is believed to act as a neuropeptide, involved in satiety, appetite regulation, olfaction, memory and cognition.41 It may be actively transported from the bloodstream or locally synthesised. Insulin also appears to act through other appetite regulating neurotransmitters and peptides. Leptin and insulin appear to share a common signalling pathway in the hypothalamus.39 There is suggestion of a potential link to Alzheimer’s disease, given insulin’s role in normal cognitive functioning and in the regulation of amyloid precursor protein and beta-amyloid itself.42 The possibility that syndromes associated with insulin resistance in the periphery, such as obesity and type 2 diabetes, may also be associated with insulin resistance in the brain, with dysregulation of appetite and body weight, is intriguing.41
Pancreas
Pancreatic β cells possess receptors for both insulin and IGF-1. Insulin may have a role in regulating glucose-stimulated insulin secretion, possibly via glucose sensing or β cell growth.43
Pituitary
Insulin receptors are found in the anterior pituitary gland44 and are found in association with β endorphin, consistent with a role in food intake regulation.45 Insulin acts in concert with other hormones. Insulin stimulates growth hormone production from the pituitary gland, which in turn promotes IGF-1 production by the liver.
Kidney
The kidney does not require insulin for glucose transport. Insulin receptors are found in the proximal tubules; insulin regulates mineral transport and gluconeogenesis in the kidney. Insulin is degraded in the kidney.46 In vivo, physiological concentrations of insulin reduce urinary sodium excretion.47
Gonads
Insulin receptors are found in the ovary and appear to have a role in enhancing steroidogenesis.48 Insulin and IGF-1 act synergistically with FSH to enhance oestrogen production by granulosa cells and with LH to enhance androgen production by thecal stromal cells.49 They have also been found in the rat testis where insulin may act synergistically with LH on steroidogenesis.50
Bone
Insulin appears to be anabolic in bone. Insulin receptors are found in osteoblasts and osteoclasts. Insulin stimulates bone formation by osteoblasts and is also reported to suppress osteoclast function.51
Physiological Influences on Insulin Action and Insulin Resistance
Diet and Starvation
Insulin levels are low in the basal state, as during starvation, facilitating mobilisation of fatty acids and glycerol from adipose tissue and amino acids from muscle.29 While insulin levels are typically higher in the fed state, the degree of insulin sensitivity may be influenced by the composition of the diet. Chronic excess energy consumption promotes hyperinsulinaemia and insulin resistance through stimulation of insulin secretion, triglyceride synthesis and fat accumulation with down-regulation of insulin receptors and post receptor signalling.17
High fat diets tend to be associated with insulin resistance, particularly with respect to saturated fat52 and trans-fatty acids.53 Fatty acid composition is thought to play a role in the long term development of insulin resistance, via effects on the composition of membrane lipids. Long chain polyunsaturated fatty acids play a physiological role in maintaining cell membrane fluidity and cell signalling; they also influence gene expression and are endogenous ligands for peroxisome proliferator receptors.54 Epidemiological studies report an association between the ratio of omega 6 to omega 3 fatty acids and the prevalence of type 2 diabetes.55 There is some experimental work to support this hypothesis; though long chain omega-3 fatty acids in pharmacological doses have been thought to impair glucose tolerance,56 meta-analyses do not confirm this.57
High carbohydrate diets are generally associated with improved insulin sensitivity in the short term. Insulin secretion depends on the type and physical form of the carbohydrates consumed. The glycaemic index (GI) of a food refers to the area under the curve for blood glucose concentration versus time, relative to an equivalent dose of glucose. The GI is lower where digestion, absorption and/or conversion to glucose occur more slowly. The rate of insulin secretion tends to be lower when low GI foods are consumed. Not all carbohydrates (in their purified form) are equivalent in their insulin response.58 For example, fructose tends not to elicit as marked an insulin response as glucose. Chronic overfeeding with sucrose has been reported to increase visceral adipose tissue deposition in rodents, which may have long-term implications for insulin resistance.59 Whilst there is concern that high carbohydrate diets may exacerbate the clinical manifestations of the insulin resistance syndrome, this likely depends on the type of carbohydrate and intake of dietary fibre which has a favourable effect.60
Dietary fibre has indirect effects on insulin secretion and action. Effects on gut motility and transit time, gastrointestinal hormone secretion e.g. GIP,61 GLP-1,62 and colonic fermentation yielding short chain fatty acids which suppress hepatic gluconeogenesis, have all been reported.63 Resistant starch, which is not digested in the small intestine but passes to the large bowel acts as a substrate for colonic fermentation (like dietary fibre) and has been reported to decrease post-prandial glucose and insulin responses, improve whole body insulin sensitivity, lower triglycerides and HDL, increase satiety and reduce fat storage.64
Protein ingestion stimulates insulin secretion, favouring anabolism; it possibly mediates its effect on satiety this way. It also stimulates glucagon secretion from the α cells of the pancreas and promotes gluconeogenesis.65
Moderate alcohol consumption has been reported to augment the postprandial increment in insulin and reduce the postprandial increment in glucose following a low carbohydrate meal.66 Population studies report a U-shaped curve with respect to level of alcohol consumption and insulin resistance.67
Micronutrients of potential importance include zinc, chromium and possibly iron. Zinc appears to be important in insulin biosynthesis and secretion, and is concentrated in the pancreas.6 Chromium deficiency has been associated with glucose intolerance and insulin resistance in patients on long-term parenteral nutrition, though work in this area has been hampered by difficulties in accurate measurement of the trace concentrations of this element in body fluids.68 The active form of chromium is an oligopeptide called chromomodulin which enhances the tyrosine kinase activity of the occupied insulin receptor. The chromium ion is transported in the bloodstream on transferrin.69 Iron accumulation is associated with impaired insulin sensitivity and type 2 diabetes, and features of the metabolic syndrome.70 Whether the adverse effects of iron accumulation are compounded by the potential resultant deleterious effects on chromium metabolism has not been addressed. In addition to the effects of nutrient components of the diet, individual foods and their component phytochemicals may also influence insulin secretion and action.71–73.
Exercise & Physical Activity
Since Chauveu & Kaufman’s remarkable observation in 1887 that “When a horse chews on hay the concentration of glucose in the blood draining its masseter muscle substantially decreases”74 a large body of evidence supports the role of exercise in improving insulin sensitivity and its beneficial outcomes in insulin resistant states. Epidemiological studies such as the US Physicians Health Study have reported substantial decreases in the relative risk of type 2 diabetes with lifelong regular physical activity.75 Large scale randomised controlled clinical trials such as the Diabetes Prevention Program76 and the Finnish Prevention Study77 demonstrate a 58% reduction in progression of impaired glucose tolerance to type 2 diabetes by intensive lifestyle modification which included a minimum of 20–30 minutes of exercise per day. Acute exercise increases GLUT 4 translocation to sarcolemmal membrane, whereas chronic exercise training increases Glut 4 mRNA expression.78 In addition to this insulin-dependent mechanism, enhanced glucose uptake into exercising muscle occurs by multiple insulinin dependent mechanisms.79 Exercise training appears to enhance insulin sensitivity by increased post-receptor insulin signalling;80 increased insulin-mediated glucose transport appears to be related to enhanced signal transduction at the level of IRS proteins and PI 3-kinase.79
Stress
Insulin resistance is typically seen in the catabolic stress of severe illness with implications for morbidity and mortality. Mechanisms include activation of the hypothalamo-pituitary adrenal (HPA) axis resulting in marked elevation of counter-regulatory hormones, as well as the effects of inflammatory cytokines.81 The latter impair insulin receptor signalling in skeletal muscle, liver and adipose tissue; intermediates such as IkB kinase, MAP kinase, ceramides and ganglioside GM3 affect several key signal transduction pathways. Important examples include the inhibition of insulin receptor kinase activity via serine phosphorylation of IRS-1, with downstream effects on PI-3 kinase, and thereby GLUT 4 translocation, as well as direct effects on PI-3 kinase, Akt/protein kinase B and protein kinase C.82 The potential significance of chronic psychosocial stress in the development of the metabolic syndrome has recently been reviewed.83
Sleep and Sleep Deprivation
Acute sleep deprivation in healthy young adults has been reported to raise fasting blood glucose concentrations in association with altered diurnal cortisol secretion and reduced heart rate variability.84 These effects suggest increased counter-regulatory hormone secretion via hyper-arousal with activation of the hypothalamo-pituitary adrenal axis.85 There is also accumulating evidence that chronic sleep deprivation may impact on insulin and insulin resistance. Recent epidemiological studies report that reduced sleep duration is associated with increased BMI.86 Sleep deprivation is associated with decreased plasma concentrations of leptin, the adipocyte peptide hormone regulating fat mass and appetite, and increased concentrations of ghrelin, which increases appetite.87 Growth hormone is secreted during slow wave sleep, sleep declines with age88 and growth hormone deficiency in adults has been associated with central adiposity and insulin resistance,89 but whether sleep deprivation acts through these mechanisms is not clearly established. Obstructive sleep apnoea (OSA), where sleep disturbance results from obstruction to breathing during sleep, is associated with impaired glucose tolerance independent of adiposity,90 and improves with continuous positive airway pressure treatment91 but whether this is due to resolution of hypoxia and hypercapnia, or to effects on sleep quality, is unclear.
Pregnancy
Normal pregnancy is characterised by insulin resistance which is greatest in the third trimester. This appears to be an adaptive response, diverting glucose and lipids to the developing foetus92 and thought due to the combined effects of human placental lactogen, progesterone, oestradiol and cortisol, which act as counter-regulatory hormones to insulin. Exaggeration of the insulin resistance normally seen in pregnancy is associated with gestational diabetes mellitus and gestational hypertension.93
Obesity
Increased adipose tissue, especially that in an upper body or “android” deposition, was first associated with diabetes and vascular disease by French endocrinologist Jean Vague in 1956.94 Insulin resistance increases with increasing body mass index, waist circumference and in particular waist-hip ratio.95 These reflect increased adiposity especially increased levels of visceral adipose tissue. Visceral adipose tissue refers to intra-abdominal fat around the intestines and correlates with liver fat. Visceral adipose tissue has metabolic characteristics which differ from that of subcutaneous fat. It is more metabolically active with regard to free fatty acid turnover; the increased flux of free fatty acids promotes insulin resistance at a cellular level and increases hepatic VLDL production.35 Omental pre-adipocytes have increased 11-β hydroxysteroid dehydrogenase type 1 activity, initially thought to promote insulin resistance by the effects of locally produced active glucocorticoid, via conversion of cortisone to cortisol, though the significance of these findings in vivo is debated.96
Adipose tissue produces a number of cytokines which have been associated with insulin resistance, including those with pro-inflammatory activity e.g. TNFα, interleukins, and PAI-1. There are regional differences in adipocyte cytokine production. Interestingly recent reports suggest improvements in insulin resistance by high dose aspirin, supporting the authors’ hypothesis regarding the role of the serine kinase IkB kinase-β (IKK-Eβ) as an intracellular mediator of insulin resistance in muscle. Muscle insulin resistance is associated with increased intramyocellular triglyceride, derived from adipose tissue lipolysis.97
The insulin resistance seen in obesity is believed to involve primarily muscle and liver, with increased adipocyte-derived free fatty acids promoting triglyceride accumulation in these tissues.97 This is more likely where adipocytes are insulin resistant.32 Free fatty acid flux is greater from visceral adipose tissue and more likely in those individuals with genetically mediated adipocyte insulin resistance.98 Whilst individual differences in the effects of increasing adiposity exist, weight gain worsens and weight loss improves insulin resistance in those so predisposed.99
Pharmacological Influences on Insulin Action and Insulin Resistance
A wide range of pharmacological agents have been associated with impaired glucose tolerance. Antihypertensive agents such as diuretics and β-blockers, corticosteroids, oral contraceptives, nicotinic acid and antipsychotic agents have been reported to impair glucose tolerance100,101 as have the anti-retroviral protease inhibitors used to treat human immunodeficiency virus infection.102 The mechanisms vary; β-blockers impair insulin secretion from the pancreas by blockade of β-adrenoceptors, thiazide diuretics are thought to act by depleting potassium levels, corticosteroids and oral contraceptives have counter-regulatory hormonal activity,101 and the HIV-1 protease inhibitors result in partial lipodystrophy with loss of peripheral subcutaneous fat and accumulation of truncal adipose tissue leading to insulin resistance.102
Aside from insulin itself, a number of agents are used therapeutically to modulate insulin action. These include sulphonylureas, which stimulate insulin secretion from the β-cell by activation of the sulphylurea receptor adjoining the K+-ATP channel at the cell membrane.12 They are used in type 2 diabetes where residual β cell function exists. Other agents act by reducing insulin resistance. Metformin, a biguanide, reduces hepatic glucose output and to a lesser extent peripheral insulin resistance and is used in type 2 diabetes and polycystic ovarian syndrome (PCOS). Its cellular actions are less well characterised. Recently, a class of agonists for peroxisome proliferator gamma receptors has been available and these agents are more potent than metformin in reducing peripheral insulin resistance. They promote the differentiation of subcutaneous adipocytes and favour redistribution of triglycerides from hepatic and visceral adipose fat depots to the periphery. They improve insulin resistance in adipose tissue but not in muscle. Thiazolidinediones such as rosiglitazone and pioglitazone are used in type 2 diabetes103 and have been effective in PCOS.28 However, there are concerns regarding the safety of thiazolidinediones in pregnancy; moreover the long term pregnancy outcome of all these insulin-sensitizing agents, including metformin, is unknown.104
The Insulin Resistance Syndrome
The insulin resistance syndrome describes the cluster of abnormalities which occur more frequently in insulin resistant individuals. These include glucose intolerance, dyslipidaemia, endothelial dysfunction and elevated procoagulant factors, haemodynamic changes, elevated inflammatory markers, abnormal uric acid metabolism, increased ovarian testosterone secretion and sleep-disordered breathing.3 Clinical syndromes associated with insulin resistance include type 2 diabetes, cardiovascular disease, essential hypertension, polycystic ovary syndrome, non-alcoholic fatty liver disease, certain forms of cancer and sleep apnoea.3
Clinical Syndromes Associated with Insulin Resistance
Type 2 diabetes and the Metabolic Syndrome would be the most common clinical syndromes associated with insulin resistance. Others include hypertension, PCOS, non-alcoholic fatty liver disease, certain forms of cancer and OSA,3 which some authors consider a component of the metabolic syndrome per se. There are also relatively common conditions where insulin resistance is a secondary phenomenon; these include acute illness, hepatic cirrhosis, renal failure, pregnancy, hyperthyroidism, Cushing’s disease and Cushing’s syndrome as well as acromegaly and phaeochromocytoma which are less common.26 In many of these, the insulin resistance is due to increased production of counter-regulatory hormones.
However there are also a large number of generally rare disorders where insulin resistance is a major clinical feature.28,105 (Table 3) Though individually rare, these conditions may provide insight into the mechanisms of insulin resistance in other settings. Typically they are characterised by disturbances in organ systems where insulin action plays a critical role.
Table 3.
| Down’s Syndrome |
| Turner’s Syndrome |
| Klinefelter’s Syndrome |
| Thalassaemia |
| Haemochromatosis |
| Lipodystrophy |
| Progeria |
| Huntington’s Chorea |
| Myotonic dystrophy |
| Friedrich’s ataxia |
| Laurence-Moon-Biedel syndrome |
| Glycogen storage diseases type I & III |
| Mitochondrial disorders (?) |
Uncommon Conditions Associated with Insulin Resistance The lipodystrophies represent a group of disorders typified by lack of subcutaneous adipose tissue, which may be inherited or acquired (e.g. secondary to treatment with protease inhibitors for HIV). They are associated with fat accumulation in liver and muscle and insulin resistance. Transplanting deposits of subcutaneous adipose tissue has been shown to improve insulin resistance in animal models of these disorders. For a recent review on this topic see Garg & Misra.106
Inherited metabolic disorders such as mitochondrial disorders and glycogen storage diseases affect muscle and liver in particular, and are associated with diabetes and insulin resistance respectively. In mitochondrial disorders there is frequently pancreatic failure reflecting failure of β cell energy generation which contributes to the development of diabetes;107 however impaired fatty acid oxidation may contribute to defects in insulin action at the cellular level.97 Interestingly diabetic patients with MELAS (Myoclonus, Elipepsy, Lactic Acidosis and Stroke-like episodes) seem to do better on a high fat diet,108 but also therefore, a low carbohydrate diet, which minimises accumulation of lactate resulting from incomplete metabolism of pyruvate in the Krebs Cycle. Recently increased intramyocellular lipid and reduced mitochondrial phosphorylation has been reported in lean insulin-resistant offspring of patients with Type 2 diabetes, suggesting subtle mitochondrial dysfunction may be more widespread in this disorder.109 In glycogen storage disease type I the adult manifestations are striking clinically for their overlap with the Metabolic Syndrome.105 Insulin resistance in this group has recently been documented,110 and likely relates to the hepatic fat accumulation that occurs in this condition. The topic of genetic syndromes causing diabetes, including those resulting in insulin resistance, is extensively reviewed by Scheuner et al.111
Common Conditions Associated with Insulin Resistance
Type 2 Diabetes (T2DM)
Following pioneering work by Bornstein112 and the Nobel Prize-winning work of Yalow and Berson,113 the first insulin assays became widely available in the late 1960s;28 it was subsequently confirmed that diabetic patients with so-called or maturity onset or type 2 diabetes had normal or increased plasma insulin levels. Insulin resistance was reported to be a characteristic feature of T2DM in the early 1970s.3 A progressive inability of the β cells to compensate for the prevailing insulin resistance by sufficient hyperinsulinaemia, heralds the clinical onset of this disorder.3 While twin studies and linkage analyses are consistent with a strong genetic component in the development of type 2 diabetes, several decades of research have failed to identify a predominant genetic abnormality in the majority of cases.26 The aetiology of T2DM is thought to be polygenic, with environmental factors being superimposed upon this basic predisposition.
Insulin resistance typically predates the development of diabetes and is commonly found in unaffected first-degree relatives.28 The morbidity of the disorder relates both to the severity of hyperglycaemia and the metabolic consequences of insulin resistance itself. The primary defects in insulin action appear to be in muscle cells and adipocytes, with impaired GLUT 4 translocation resulting in impaired insulin-mediated glucose transport.28
Compensatory hyperinsulinaemia develops initially, but the first phase of insulin secretion is lost early in the disorder. Additional environmental and physiological stresses such as pregnancy, weight gain, physical inactivity and medications may worsen the insulin resistance. As the β cells fail to compensate for the prevailing insulin resistance, impaired glucose tolerance and diabetes develops. As glucose levels rise, β cell function deteriorates further, with diminishing sensitivity to glucose and worsening hyperglycaemia. The pancreatic islet cell mass is reported to be reduced in size in diabetic patients; humoral and endocrine factors may be important in maintaining islet cell mass.11 In contrast to most forms of type 2 diabetes, the genetic basis of Maturity Onset diabetes of the Young (MODY) has been well characterised and relates to defects in glucokinase, hence intracellular glucose transport.26
Metabolic Syndrome
Jean Vague’s early observations in the 1950s were rediscovered some three decades later, when in 1988 it was proposed that individuals with glucose intolerance, elevated triglycerides, low HDL cholesterol and essential hypertension were at greatly increased cardiovascular risk.3 The underlying connection with insulin resistance was not fully appreciated at the time and it was initially referred to as Syndrome X, but later as the Metabolic Syndrome. The Adult Treatment Panel III of the National Cholesterol Advisory Panel defines Metabolic syndrome as the “constellation of lipid and non-lipid risk factors of metabolic origin”. Their diagnostic criteria for metabolic syndrome require three of the following:
-
Abdominal obesity
Men: waist circumference >40 inches (102 cm )
Women: waist circumference >35 inches (88 cm)
Fasting glucose ≥ 110 <126 mg/dL (6.1–7.0 mmol/L)
Blood pressure ≥ 130/80 mmHg
Triglycerides >150 mg/dL (>1.7 mmol/L)
-
HDL cholesterol
Men <40 mg/dL (<1.0 mmol/L)
Women <50 mg/dL (<1.3 mmol/L)
It has been estimated that in the United States some 25% of adults have the syndrome.3 In addition to an underlying genetic predisposition to insulin resistance, physical inactivity and excess energy intake are considered the main risk factors, with an unknown contribution from other factors such as chronic stress114 and sleep deprivation.115
Dyslipidaemia
The lipid abnormalities associated with insulin resistance affect all lipid fractions. They are characterised by elevated fasting triglyceride levels, elevated postprandial triglyceride-rich remnant lipoproteins, low HDL cholesterol, and small dense LDL particles. This pattern correlates strongly with cardiovascular risk, and treatment decreases this risk.3 Apolipoprotein B (apo B) is typically associated with LDL cholesterol; however, in insulin resistant atherogenic dyslipidaemia, in response to increased delivery of free fatty acids to the liver, there is an overproduction of apo B and increased VLDL triglyceride synthesis. This includes VLDL particles which contain apo B. Low HDL cholesterol levels are an independent risk factor for cardiovascular disease; the low HDL seen in atherogenic dyslipidaemia relates to a reduced HDL particle size. The low HDL particle size relates to exchange of VLDL triglycerides for cholesterol esters in LDL and HDL via cholesterol ester transfer protein. When the triglycerides in LDL and HDL undergo hydrolysis, small, cholesterol depleted LDL and HDL remain.116 Small dense LDL particles are reported to be more atherogenic possibly because of their increased propensity to oxidation and greater proportion of apo B. Compared with non-insulin resistant states, a given LDL cholesterol level represents a greater number of apo B containing small dense LDL particles and this confers increased risk.107
Hypertension
Essential hypertension has been associated with insulin resistance in up to 50% of cases.117 There is a strong correlation of blood pressure with body weight. The significance of insulin resistance in hypertension is subject to some controversy, as 50% of essential hypertensives appear not to have insulin resistance. Proposed mechanisms have included increased renal sodium retention and increased sympathetic nervous system activity from compensatory hyperinsulinaemia.3 However endothelial dysfunction from resistance to insulin-mediated nitric oxide formation is thought to be of clearer significance.118,119
PCOS
Convincing evidence links insulin resistance to the pathogenesis of this, the most common endocrine disorder of premenopausal women.3 The severity of genetic defects in insulin sensitivity may perhaps determine the probability of developing PCOS for a given body mass index. Genetically determined defects in transduction of the insulin signal have been described in PCOS, with insulin resistance preceding the clinical manifestations. Further deterioration in insulin sensitivity occurs with the adolescent growth spurt and the development of obesity.120 The ovarian dysfunction relates to the effects of compensatory hyperinsulinaemia increasing pituitary LH secretion and androgen production by the theca cells of the ovary. Aromatisation of androgens in the setting of obesity increases production of oestrogens, further impairing the function of the hypothalamo-pituitary gonadal axis. Hyperinsulinaemia also suppresses SHBG production by the liver, further elevating free androgens. Elevated androgens in turn further aggravate insulin resistance.120 Therapeutic modalities which reduce insulin resistance typically result in improvements in ovarian function. These include weight loss, exercise and insulin-sensitizing pharmacological agents such as metformin and troglitazone.28
Non-alcoholic Fatty Liver Disease (NAFLD)
NAFLD is becoming increasingly recognised with different countries reporting prevalence rates 10–24% of the general population; however amongst obese individuals, 50–75% are affected. 25–50% of obese children are affected. NAFLD may progress to non-alcoholic steatohepatosis, steatohepatitis, fibrosis or cirrhosis, representing increasing liver damage. Over a follow-up period of 3.5–11 years, 28% of patients with NAFLD had progressive liver damage.121 Peripheral insulin resistance in fat and muscle lead to increased delivery of free fatty acids to the liver, increasing triglyceride synthesis. Fatty liver develops when hepatic triglyceride synthesis exceeds hepatic synthesis and export of VLDL triglyceride.3
Cancer
Insulin resistance with compensatory hyperinsulinaemia has been implicated in the aetiology of certain cancers, including colon, endometrial, possibly pancreatic and renal-cell cancers122 and breast cancer.3 In the case of colonic cancer it is proposed that insulin resistance leads to hyperinsulinaemia with elevated IGF-1 levels promoting intestinal epithelial cell proliferation; alterations in NF- B and peroxisome proliferator activated receptor signalling may also influence colonocyte kinetics.123 In addition to mitogenic effects of insulin itself, the effects of insulin resistance on ovarian function and sex hormone metabolism, along with low levels of SHBG potentially elevating bio-available hormone levels, may also contribute to carcinogenesis.124
OSA
OSA is generally regarded as a complication of obesity manifested by mechanical obstruction to the upper respiratory tract during sleep. However there is some evidence suggesting that OSA may be a systemic condition related to insulin resistance.3 However the resultant sleep fragmentation and hyperarousal may promote insulin resistance via activation of the HPA axis.125 Sympathetic nervous system hyperactivity has also been reported in OSA.126 Treatment of OSA by nasal continuous positive airway pressure preferentially decreases visceral adipose tissue.127 Whilst these studies do not preclude insulin resistance as a contributor to OSA, there are several mechanisms, described above, by which OSA might contribute to insulin resistance.
Measurement of Insulin and Insulin Resistance
There are a variety of approaches to the laboratory assessment of insulin resistance. Over the years the limited specificity of older radio-immunoassays that cross-react with proinsulin have reduced the credibility of measuring insulin resistance in clinical settings. Current assays have improved specificity and precision. A comprehensive review of insulin assays is beyond the scope of this review and the reader is encouraged to consult Sapin128 in this regard. Insulin resistance may be measured by looking directly at insulin mediated glucose uptake in the basal or post-stimulated state, by inference from the relative concentrations of glucose and insulin, or by looking at surrogate markers of insulin action.
Research Methods
The most accepted methods for measuring insulin resistance are limited to research settings due to their invasiveness and complexity. The clamp studies specifically measure insulin mediated glucose uptake under controlled conditions. These include the euglycaemic clamp (considered the gold standard) and the hyperinsulinaemic clamp. Less invasive are the insulin sensitivity test and short insulin tolerance test. Less direct methods include assessments of insulin resistance based on mathematical modelling include the Homeostasis Assessment Model (HOMA)129 and the Quantitative Insulin Sensitivity Check Index (QUICKI), Continuous Infusion of Glucose with Model Assessment (CIGMA), Frequently Sampled Intravenous Glucose Tolerance Test and minimal modelling. These methods have been recently reviewed.130
Clinical Methods
Most of the methods outlined above have limited applicability in clinical settings. The most widely used approach would be to measure fasting plasma insulin. While additional information may be obtained with HOMA and QUICKI, by incorporating the simultaneous fasting glucose into the equation, it has been argued that most of the variance in insulin resistance is reflected in the insulin level alone.131 Belfiore et al have developed Insulin Sensitivity Indices based on basal and OGTT-induced insulin, glucose and free fatty acid levels; these are a practical measure of whole body insulin resistance in the clinical setting and are unique in measuring sensitivity to insulin-mediated suppression of free fatty acid levels. They can be applied to either basal or post OGTT glucose and insulin levels. Although this method is performed under physiological conditions, unlike the euglycaemic clamp technique, the authors report values for r of 0.93 to 0.99 for the correlations between the methods.132
Functional Measures of Insulin Resistance
Another approach is to identify insulin resistant patients, based on functional markers of insulin resistance. McLoughlin et al were able to identify insulin resistant individuals from an overweight-obese cohort by looking at plasma triglyceride concentration, ratio of triglyceride to high-density lipoprotein cholesterol concentrations and insulin concentration. Using cut points of 1.47 mmol/L for triglyceride, 1.8 mmol/L for the triglyceride-high-density lipoprotein cholesterol ratio and 109 pmol/L (16 mIU/L) for insulin, they achieved comparable sensitivity and specificity to the Adult Treatment Panel III to diagnose the metabolic syndrome.133
Concluding Comments
A century or more since research into this field began in earnest neither the significance nor the medical and scientific interest in this area has waned. Instead, rapid globalisation, urbanisation and industrialisation have spawned epidemics of obesity, diabetes and their attendant co-morbidities, as physical inactivity and “convenience” foods unmask latent predisposing genetic traits. The biological mechanisms are intricate and complex and incompletely understood. However, taking a step back, we may need to consider the dramatic social changes of the past century with respect to physical activity, diet, work, socialisation and sleep patterns. Aside from the challenges that remain in unravelling the genetic and mechanistic factors, perhaps a greater challenge, is creatively, to adapt contemporary lifestyles to our genetic make-up and physiological requirements.
The contents of articles or advertisements in The Clinical Biochemist – Reviews are not to be construed as official statements, evaluations or endorsements by the AACB, its official bodies or its agents. Statements of opinion in AACB publications are those of the contributors. Print Post Approved - PP255003/01665.
No literary matter in The Clinical Biochemist – Reviews is to be reproduced, stored in a retrieval system or transmitted in any form by electronic or mechanical means, photocopying or recording, without permission. Requests to do so should be addressed to the Editor. ISSN 0159 – 8090.
Footnotes
Competing interests: none declared.
References
- 1.World Health Organization. Obesity: Preventing and Managing the Global Epidemic Report of a WHO Consultation Technical Report Series. World Health Organization, Geneva 2000. [PubMed]
- 2.Cefalu WT. Insulin resistance: cellular and clinical concepts. Exp Biol Med (Maywood) 2001;226:13–26. doi: 10.1177/153537020122600103. [DOI] [PubMed] [Google Scholar]
- 3.Reaven G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin North Am. 2004;33:283–303. doi: 10.1016/j.ecl.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Bliss M. The history of insulin Diabetes Care. 1993;16(Suppl 3):4–7. doi: 10.2337/diacare.16.3.4. [DOI] [PubMed] [Google Scholar]
- 5.Home PD. Insulin therapy. In: Alberti KGMM, Zimmet P, Defronzo RA editors & Keen H (Hon) editor International Textbook of Diabetes Mellitus (2nd ed) John Wiley & Sons, New York; 1997 p. 899–928.
- 6.Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol. 1998;8:189–94. doi: 10.1016/s0959-440x(98)80037-7. [DOI] [PubMed] [Google Scholar]
- 7.Schroder D, Zuhlke H. Genetechnology, characterization of insulin gene and the relationship to diabetes research. Endokrinologie. 1982;79:197–209. [PubMed] [Google Scholar]
- 8.Malaisse WJ. Insulin biosynthesis and secretion in vitro. In: Alberti KGMM, Zimmet P, Defronzo RA & Keen H (Hon) editors. International Textbook of Diabetes Mellitus (2nd ed) John Wiley & Sons, New York; 1997 p. 315–36.
- 9.Porksen N, Hollingdal M, Juhl C, Butler P, Veldhuis JD, Schmitz O. Pulsatile insulin secretion: detection, regulation, and role in diabetes. Diabetes. 2002;51 (Suppl 1):S245–54. doi: 10.2337/diabetes.51.2007.s245. [DOI] [PubMed] [Google Scholar]
- 10.Bratanova-Tochkova TK, Cheng H, Daniel S, et al. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes. 2002;51 (Suppl. 1):S83–90. doi: 10.2337/diabetes.51.2007.s83. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen JH, Galsgaard ED, Moldrup A, et al. Regulation of beta-cell mass by hormones and growth factors. Diabetes. 2001;50 (Suppl 1):S25–9. doi: 10.2337/diabetes.50.2007.s25. [DOI] [PubMed] [Google Scholar]
- 12.De Lonlay, Saudubray J-M. Persistent hyperinsulinaemic hypoglycaemia. In: Fernandes J, Sudubray J-M, van den Berghe editors Inborn Metabolic Diseases: Diagnosis and treatment. (3rd ed): Springer, Heidelberg Germany; 2000 p.117–26.
- 13.Soria B, Quesada I, Ropero AB, Pertusa JA, Martin F, Nadal A. Novel players in pancreatic islet signaling: from membrane receptors to nuclear channels. Diabetes. 2004;53 (Suppl 1):S86–91. doi: 10.2337/diabetes.53.2007.s86. [DOI] [PubMed] [Google Scholar]
- 14.MacDonald PE, El-Kholy W, Riedel MJ, Salapatek AM, Light PE, Wheeler MB. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes. 2002;51 (Suppl 3):S434–42. doi: 10.2337/diabetes.51.2007.s434. [DOI] [PubMed] [Google Scholar]
- 15.Havel PJ. Peripheral signals conveying metabolic information to the brain: short-term and long-term regulation of food intake and energy homeostasis. Exp Biol Med (Maywood) 2001;226:963–77. doi: 10.1177/153537020122601102. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen JH, Serup P. Molecular basis of islet development, growth and regeneration. Current Opinion in Endocrinology & Diabetes. 1998;5:97–107. [Google Scholar]
- 17.Kahn SE, McCulloch DK, Porte D. Insulin secretion in the normal and diabetic human. In: Alberti KGMM, Zimmet P, Defronzo RA, editors & Keen H, (hon) editor. International Textbook of Diabetes Mellitus. (2nd ed) John Wiley & Sons, New York; 1997 p. 337–54.
- 18.Chen M, Porte D., Jr The effect of rate and dose of glucose infusion on the acute insulin response in man. J Clin Endocrinol Metab. 1976;42:1168–75. doi: 10.1210/jcem-42-6-1168. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Barrett EJ. Human protein metabolism: its measurement and regulation. Am J Physiol Endocrinol Metab. 2002;283:E1105–12. doi: 10.1152/ajpendo.00337.2002. [DOI] [PubMed] [Google Scholar]
- 20.Amery CM, Nattrass M. Fatty acids and insulin secretion. Diabetes Obes Metab. 2000;2:213–21. doi: 10.1046/j.1463-1326.2000.00059.x. [DOI] [PubMed] [Google Scholar]
- 21.Frost GS, Brynes AE, Dhillo WS, Bloom SR, McBurney MI. The effects of fiber enrichment of pasta and fat content on gastric emptying, GLP-1, glucose, and insulin responses to a meal. Eur J Clin Nutr. 2003;57:293–8. doi: 10.1038/sj.ejcn.1601520. [DOI] [PubMed] [Google Scholar]
- 22.Reaven GM. Effects of differences in amount and kind of dietary carbohydrate on plasma glucose and insulin responses in man. Am J Clin Nutr. 1979;32:2568–78. doi: 10.1093/ajcn/32.12.2568. [DOI] [PubMed] [Google Scholar]
- 23.Wolever TM. The glycemic index. World Rev Nutr Diet. 1990;62:120–85. [PubMed] [Google Scholar]
- 24.Efendic S, Portwood N. Overview of incretin hormones. Horm Metab Res. 2004;36:742–6. doi: 10.1055/s-2004-826157. [DOI] [PubMed] [Google Scholar]
- 25.Kido Y, Nakae J, Accili D. The insulin receptor and its cellular targets. J Clin Endocrinol Metab. 2001;86:972–9. doi: 10.1210/jcem.86.3.7306. [DOI] [PubMed] [Google Scholar]
- 26.Withers DJ, White M. Perspective: The insulin signaling system--a common link in the pathogenesis of type 2 diabetes. Endocrinology. 2000;141:1917–21. doi: 10.1210/endo.141.6.7584. [DOI] [PubMed] [Google Scholar]
- 27.Burks DJ, White MF. IRS proteins and beta-cell function. Diabetes. 2001;50 (Suppl 1):S140–5. doi: 10.2337/diabetes.50.2007.s140. [DOI] [PubMed] [Google Scholar]
- 28.Hunter SJ, Garvey WT. Insulin action and insulin resistance: diseases involving defects in insulin receptors, signal transduction, and the glucose transport effector system. Am J Med. 1998;105:331–45. doi: 10.1016/s0002-9343(98)00300-3. [DOI] [PubMed] [Google Scholar]
- 29.Karam JH. Pancreatic Hormones and Diabetes Mellitus. In: Greenspan FS, Strewler GJ, editors. Basic and Clinical Endocrinology. Appleton & Lange, Stamford CT USA; 1997 p. 601–2.
- 30.Denton RM, Tavaré JM. Molecular basis of insulin action on intracellular metabolism. In: Alberti KGMM, Zimmet P, Defronzo RA, Keen H (Hon), editors. International Textbook of Diabetes Mellitus (2nd ed) John Wiley & Sons, New York; 1997 p. 469–88.
- 31.Clemmons DR. Structural and functional analysis of insulin-like growth factors. Br Med Bull. 1989;45:465–80. doi: 10.1093/oxfordjournals.bmb.a072335. [DOI] [PubMed] [Google Scholar]
- 32.Frystyk J, Ørskof H. IGFI, IGFII, IGF-binding proteins and diabetes. In: Alberti KGMM, Zimmet P, Defronzo RA, Keen H (hon), editors. International Textbook of Diabetes Mellitus (2nd ed) John Wiley & Sons, New York; 1997 p. 417–36.
- 33.Wheatcroft SB, Williams IL, Shah AM, Kearney MT. Pathophysiological implications of insulin resistance on vascular endothelial function. Diabet Med. 2003;20:255–68. doi: 10.1046/j.1464-5491.2003.00869.x. [DOI] [PubMed] [Google Scholar]
- 34.Smith U. Impaired (‘diabetic’) insulin signaling and action occur in fat cells long before glucose intolerance--is insulin resistance initiated in the adipose tissue? Int J Obes Relat Metab Disord. 2002;26:897–904. doi: 10.1038/sj.ijo.0802028. [DOI] [PubMed] [Google Scholar]
- 35.Giorgino F, Laviola L, Eriksson JW. Regional differences of insulin action in adipose tissue: insights from in vivo and in vitro studies. Acta Physiol Scand. 2005;183:13–30. doi: 10.1111/j.1365-201X.2004.01385.x. [DOI] [PubMed] [Google Scholar]
- 36.Halvatsiotis PG, Turk D, Alzaid A, Dinneen S, Rizza RA, Nair KS. Insulin effect on leucine kinetics in type 2 diabetes mellitus. Diabetes Nutr Metab. 2002;15:136–42. [PubMed] [Google Scholar]
- 37.Grundy SM. What is the contribution of obesity to the metabolic syndrome? Endocrinol Metab Clin North Am. 2004;33:267–82. doi: 10.1016/j.ecl.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Krauss RM, Siri PW. Metabolic abnormalities: triglyceride and low-density lipoprotein. Endocrinol Metab Clin North Am. 2004;33:405–15. doi: 10.1016/j.ecl.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 39.Devaraj S, Rosenson RS, Jialal I. Metabolic syndrome: an appraisal of the pro-inflammatory and procoagulant status. Endocrinol Metab Clin North Am. 2004;33:431–53. doi: 10.1016/j.ecl.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 40.Stockhorst U, de Fries D, Steingrueber HJ, Scherbaum WA. Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration to humans. Physiol Behav. 2004;83:47–54. doi: 10.1016/j.physbeh.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Gerozissis K. Brain insulin and feeding: a bi-directional communication. Eur J Pharmacol. 2004;490:59–70. doi: 10.1016/j.ejphar.2004.02.044. [DOI] [PubMed] [Google Scholar]
- 42.Watson GS, Craft S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs. 2003;17:27–45. doi: 10.2165/00023210-200317010-00003. [DOI] [PubMed] [Google Scholar]
- 43.Kulkarni RN. The islet betal-cell. Int J Biochem Cell Biol. 2004;36:365–71. doi: 10.1016/j.biocel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Unger JW, Betz M. Insulin receptors and signal transduction proteins in the hypothalamo-hypophyseal system: are view on morphological findings and functional implications. Histol Histopathol. 1998;13:1215–24. doi: 10.14670/HH-13.1215. [DOI] [PubMed] [Google Scholar]
- 45.Unger JW, Lange W. Insulin receptors in the pituitary gland: morphological evidence for influence on opioid peptide-synthesizing cells. Cell Tissue Res. 1997;288:471–83. doi: 10.1007/s004410050833. [DOI] [PubMed] [Google Scholar]
- 46.Nagasaka Y, Kaneko T. [Molecular biology of regulation of renal function-structure, function and distribution of the receptor-insulin, glucagon] Nippon Rinsho. 1992;50:2921–4. [PubMed] [Google Scholar]
- 47.Sechi LA, Bartoli E. Molecular mechanisms of insulin resistance in arterial hypertension. Blood Pres Suppl. 1996;1:47–54. [PubMed] [Google Scholar]
- 48.Poretsky L, Kalin MF. The gonadotropic function of insulin. Endocr Rev. 1987;8:132–41. doi: 10.1210/edrv-8-2-132. [DOI] [PubMed] [Google Scholar]
- 49.Samoto T, Maruo T, Katayama K, Barnea ER, Mochizuk M. Altered expression of insulin and insulin-like growth factor-1 receptors in follicular and stromal compartments of polycystic ovaries. Endocr J. 1993;40:413–24. doi: 10.1507/endocrj.40.413. [DOI] [PubMed] [Google Scholar]
- 50.Abele V, Pelletier G, Tremblay RR. Radioautographic localization and regulation of the insulin receptors in rat testis. J Recept Res. 1986;6:461–73. doi: 10.3109/10799898609074825. [DOI] [PubMed] [Google Scholar]
- 51.Thomas DM, Udagawa N, Hards DK, et al. Insulin receptor expression in primary and cultured oseoclast-like cells. Bone. 1998;23:181–6. doi: 10.1016/s8756-3282(98)00095-7. [DOI] [PubMed] [Google Scholar]
- 52.Lichtenstein AH, Schwab US. Relationship of dietary fat to glucose metabolism. Atherosclerosis. 2000;150:227–43. doi: 10.1016/s0021-9150(99)00504-3. [DOI] [PubMed] [Google Scholar]
- 53.Bray GA, Lovejoy JC, Smith SR, et al. The Influence of Different Fats and Fatty Acids on Obesity, Insulin Resistance and Inflammation. J Nutr. 2002;132:2488–91. doi: 10.1093/jn/132.9.2488. [DOI] [PubMed] [Google Scholar]
- 54.Sampath H, Ntambi JM. Polyunsaturated fatty acid regulation of gene expression. Nutr Rev. 2004;62:333–9. doi: 10.1111/j.1753-4887.2004.tb00058.x. [DOI] [PubMed] [Google Scholar]
- 55.Simopoulos AP. Essential fatty acids in health and chronic disease. Am J Clin Nutr. 1999;70(3 Suppl):560S–569S. doi: 10.1093/ajcn/70.3.560s. [DOI] [PubMed] [Google Scholar]
- 56.Borkman M, Chisholm DJ, Furler SM, et al. Effects of fish oil supplementation on glucose and lipid metabolism in NIDDM. Diabetes. 1989;38:1314–9. doi: 10.2337/diab.38.10.1314. [DOI] [PubMed] [Google Scholar]
- 57.Friedberg CE, Janssen MJ, Heine RJ, Grobbee DE. Fish oil and glycemic control in diabetes. A meta-analysis. Diabetes Care. 1998;21:494–500. doi: 10.2337/diacare.21.4.494. [DOI] [PubMed] [Google Scholar]
- 58.Wolever TM. Dietary carbohydrates and insulin action in humans. Br J Nutr. 2000;83 (Suppl 1):S97–102. doi: 10.1017/s0007114500001021. [DOI] [PubMed] [Google Scholar]
- 59.Toida S, Takahashi M, Shimizu H, Sato N, Shimomura Y, Kobayashi I. Effect of high sucrose feeding on fat accumulation in the male Wistar rat. Obes Res. 1996;4:561–8. doi: 10.1002/j.1550-8528.1996.tb00270.x. [DOI] [PubMed] [Google Scholar]
- 60.Davy BM, Melby CL. The effect of fiber-rich carbohydrates on features of Syndrome X. J Am Diet Assoc. 2003;103:86–96. doi: 10.1053/jada.2003.50005. [DOI] [PubMed] [Google Scholar]
- 61.Beck B, Villaume C, Bau HM, et al. Long-term influence of a wheat-bran supplemented diet on secretion of gastrointestinal hormones and on nutrient absorption in healthy man. Hum Nutr Clin Nutr. 1986;40:25–33. [PubMed] [Google Scholar]
- 62.Cani PD, Dewever C, Delzenne NM. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr. 2004;92:521–6. doi: 10.1079/bjn20041225. [DOI] [PubMed] [Google Scholar]
- 63.Roberfroid M. Dietary fiber, inulin, and oligofructose: a review comparing their physiological effects. Crit Rev Food Sci Nutr. 1993;33:103–48. doi: 10.1080/10408399309527616. Erratum in: Crit Rev Food Sci Nutr 1993;33:553. [DOI] [PubMed] [Google Scholar]
- 64.Higgins JA. Resistant starch: metabolic effects and potential health benefits. J AOAC Int. 2004;87:761–8. [PubMed] [Google Scholar]
- 65.Linn T, Santosa B, Gronemeyer D, et al. Effect of long-term dietary protein intake on glucose metabolism in humans. Diabetologia. 2000;43:1257–65. doi: 10.1007/s001250051521. [DOI] [PubMed] [Google Scholar]
- 66.Greenfield JR, Samaras K, Hayward CS, Chisholm DJ, Campbell LV. Beneficial postprandial effect of a small amount of alcohol on diabetes and cardiovascular risk factors: Modification by Insulin Resistance. J Clin Endocrinol Metab. 2004 Nov 2 [Epub ahead of print] [DOI] [PubMed]
- 67.Magis DC, Jandrain BJ, Scheen AJ. Alcohol, insulin sensitivity and diabetes. Rev Med Liege. 2003;58:501–7. [PubMed] [Google Scholar]
- 68.Anderson RA. Chromium, glucose intolerance and diabetes. J Am Coll Nutr. 1998;17:548–55. doi: 10.1080/07315724.1998.10718802. [DOI] [PubMed] [Google Scholar]
- 69.Vincent JB. The biochemistry of chromium. J Nutr. 2000;130:715–8. doi: 10.1093/jn/130.4.715. [DOI] [PubMed] [Google Scholar]
- 70.Fernandez-Real JM, Lopez-Bermejo A, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes. 2002;51:2348–54. doi: 10.2337/diabetes.51.8.2348. [DOI] [PubMed] [Google Scholar]
- 71.Bhathena SJ, Velasquez MT. Beneficial role of dietary phytoestrogens in obesity and diabetes. Am J Clin Nutr. 2002;76:1191–201. doi: 10.1093/ajcn/76.6.1191. [DOI] [PubMed] [Google Scholar]
- 72.Broadhurst CL, Polansky MM, Anderson RA. Insulin-like biological activity of culinary and medicinal plant aqueous extracts in vitro. J Agric Food Chem. 2000;48:849–52. doi: 10.1021/jf9904517. [DOI] [PubMed] [Google Scholar]
- 73.Grassi D, Lippi C, Necozione, Desideri, Ferri C. Short-term administration of dark chocolate is followed by a significant increase in insulin sensitivity and a decrease in blood pressure in healthy persons. Am J Clin Nutr. 2005;81:611–4. doi: 10.1093/ajcn/81.3.611. [DOI] [PubMed] [Google Scholar]
- 74.Tomás E, Zorzano A, Ruderman NB. Exercise and insulin signaling: a historical perspective. J Appl Physiol. 2002;93:765–72. doi: 10.1152/japplphysiol.00267.2002. [DOI] [PubMed] [Google Scholar]
- 75.Manson JE, Nathan DM, Krolewski AS, Stampfer MJ, Willett WC, Hennekens CH. A prospective study of exercise and incidence of diabetes among US male physicians. JAMA. 1992;268:63–7. [PubMed] [Google Scholar]
- 76.Knowler WC, Barrett-Connor E, Fowler SE, et al. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. NEJM. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tuomilehto J, Lindstrom J, Eriksson JG, et al. Finnish Diabetes Prevention Study Group.. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–50. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 78.Dohm GL. Regulation of skeletal muscle GLUT-4 expression by exercise. J Appl Physiol. 2002;93:782–7. doi: 10.1152/japplphysiol.01266.2001. [DOI] [PubMed] [Google Scholar]
- 79.Henriksen EJ. Effects of acute exercise and exercise training on insulin resistance. J Appl Physiol. 2002;93:788–96. doi: 10.1152/japplphysiol.01219.2001. [DOI] [PubMed] [Google Scholar]
- 80.Juleen R. Zierath. Exercise training-induced changes in insulin signaling in skeletal muscle. J Appl Physiol. 2002;93:773–81. doi: 10.1152/japplphysiol.00126.2002. [DOI] [PubMed] [Google Scholar]
- 81.Van den Berghe G. How does blood glucose control with insulin save lives in intensive care? J Clin Invest. 2004;114:1187–95. doi: 10.1172/JCI23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marette A. Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care. 2002;5:377–83. doi: 10.1097/00075197-200207000-00005. [DOI] [PubMed] [Google Scholar]
- 83.Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30:1–10. doi: 10.1016/j.psyneuen.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 84.Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–9. doi: 10.1016/S0140-6736(99)01376-8. [DOI] [PubMed] [Google Scholar]
- 85.Vgontzas AN, Mastorakos G, Bixler EO, Kales A, Gold PW, Chrousos GP. Sleep deprivation effects on the activity of the hypothalamic-pituitary-adrenal and growth axes: potential clinical implications. Clin Endocrinol (Oxf) 1999;51:205–15. doi: 10.1046/j.1365-2265.1999.00763.x. [DOI] [PubMed] [Google Scholar]
- 86.Vorona RD, Winn MP, Babineau TW, Eng BP, Feldman HR, Ware JC. Overweight and obese patients in a primary care population report less sleep than patients with a normal body mass index. Arch Intern Med. 2005;165:25–30. doi: 10.1001/archinte.165.1.25. [DOI] [PubMed] [Google Scholar]
- 87.Spiegel K, Tasali E, Penev P, Van Cauter E. Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med. 2004;141:846–50. doi: 10.7326/0003-4819-141-11-200412070-00008. [DOI] [PubMed] [Google Scholar]
- 88.Mullington J, Hermann D, Holsboer F, Pollmacher T. Age-dependent suppression of nocturnal growth hormone levels during sleep deprivation. Neuroendocrinology. 1996;64:233–41. doi: 10.1159/000127122. [DOI] [PubMed] [Google Scholar]
- 89.Hew FL, O’Neal D, Kamarudin N, Alford FP, Best JD. Growth hormone deficiency and cardiovascular risk. Baillieres Clin Endocrinol Metab. 1998;12:199–216. doi: 10.1016/s0950-351x(98)80018-9. [DOI] [PubMed] [Google Scholar]
- 90.Tassone F, Lanfranco F, Gianotti L, et al. Obstructive sleep apnoea syndrome impairs insulin sensitivity independently of anthropometric variables. Clin Endocrinol (Oxf) 2003;59:374–9. doi: 10.1046/j.1365-2265.2003.01859.x. [DOI] [PubMed] [Google Scholar]
- 91.Yee B, Liu P, Philips C, Grunstein R. Neuroendocrine changes in sleep apnea. Curr Opin Pulm Med. 2004;10:475–81. doi: 10.1097/01.mcp.0000143967.34079.27. [DOI] [PubMed] [Google Scholar]
- 92.Butte NF. Carbohydrate and lipid metabolism in pregnancy:normal compared with gestational diabetes mellitus. Am J Clin Nutr. 2000;71(suppl):1256S–61S. doi: 10.1093/ajcn/71.5.1256s. [DOI] [PubMed] [Google Scholar]
- 93.Seely EW, Solomon CG. Insulin resistance and its potential role in pregnancy-induced hypertension. J Clin Endocrinol Metab. 2003;88:2393–8. doi: 10.1210/jc.2003-030241. [DOI] [PubMed] [Google Scholar]
- 94.Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. 1956. Obes Res. 1996;4:204–12. doi: 10.1002/j.1550-8528.1996.tb00536.x. [DOI] [PubMed] [Google Scholar]
- 95.Aronne LJ, Segal KR. Adiposity and fat distribution outcome measures: assessment and clinical implications. Obes Res. 2002;10 (Suppl 1):14S–21S. doi: 10.1038/oby.2002.184. [DOI] [PubMed] [Google Scholar]
- 96.Tomlinson JW, Sinha B, Bujalska I, Hewison M, Stewart PM. Expression of 11 beta-hydroxysteroid dehydrogenase type 1 in adipose tissue is not increased in human obesity. J Clin Endocrinol Metab. 2002;87:5630–5. doi: 10.1210/jc.2002-020687. [DOI] [PubMed] [Google Scholar]
- 97.Perseghin G, Petersen K, Shulman GI. Cellular mechanism of insulin resistance: potential links with inflammation. Int J Obes Relat Metab Disord. 2003;27 (Suppl 3):S6–11. doi: 10.1038/sj.ijo.0802491. [DOI] [PubMed] [Google Scholar]
- 98.Jansson PA, Pellme F, Hammarstedt A, et al. A novel cellular marker of insulin resistance and early atherosclerosis in humans is related to impaired fat cell differentiation and low adiponectin. FASEB J. 2003;17:1434–40. doi: 10.1096/fj.02-1132com. [DOI] [PubMed] [Google Scholar]
- 99.Reaven GM. Importance of identifying the overweight patient who will benefit the most by losing weight. Ann Intern Med. 2003;138:420–3. doi: 10.7326/0003-4819-138-5-200303040-00012. [DOI] [PubMed] [Google Scholar]
- 100.Bressler P, De Fronzo RA. In: Alberti KGMM, Zimmet P, Defronzo RA, Keen H (hon), editors. International Textbook of Diabetes Mellitus (2nd ed) John Wiley & Sons, New York; 1997 p. 213–54.
- 101.Ananth J, Parameswaran S, Gunatilake S. Side effects of atypical antipsychotic drugs. Curr Pharm Des. 2004;10:2219–29. doi: 10.2174/1381612043384088. [DOI] [PubMed] [Google Scholar]
- 102.Chen D, Misra A, Garg A. Lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab. 2002;87:4845–56. doi: 10.1210/jc.2002-020794. [DOI] [PubMed] [Google Scholar]
- 103.Lebovitz HE. Oral antidiabetic agents. Med Clin North Am. 2004;88:847–63. doi: 10.1016/j.mcna.2004.05.002. ix-x. [DOI] [PubMed] [Google Scholar]
- 104.Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223–36. doi: 10.1056/NEJMra041536. [DOI] [PubMed] [Google Scholar]
- 105.Fernandez J, Smit GPA. The glycogen storage diseases. In: Fernandes J, Sudubray J-M, van den Berghe, editors. Inborn Metabolic Diseases: Diagnosis and treatment. (3rd ed). Springer, Heidelberg Germany; 2000 p. 86–101.
- 106.Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome. Endocrinol Metab Clin North Am. 2004;33:305–31. doi: 10.1016/j.ecl.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 107.Maassen JA, Hart LM, Van Essen E, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53 (Suppl 1):S103–9. doi: 10.2337/diabetes.53.2007.s103. [DOI] [PubMed] [Google Scholar]
- 108.Panetta J, Smith LJ, Boneh A. Effect of high-dose vitamins, coenzyme Q and high-fat diet in paediatric patients with mitochondrial diseases. J Inherit Metab Dis. 2004;27:487–98. doi: 10.1023/B:BOLI.0000037354.66587.38. [DOI] [PubMed] [Google Scholar]
- 109.Pedersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:660–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Giacchero R, Fiorir L, Scaglioni S, Sala M, Giovanni M. Insuline resistance and pancreatic function in glycogen storage disease Type 1. J Inherit Metab Dis. 2003;26(suppl 2):124. [Google Scholar]
- 111.Scheuner MT, Raffel LJ, Rotter JI. Genetics of Diabetes. In: Alberti KGMM, Zimmet P, Defronzo RA, Keen H (hon), editors. International Textbook of Diabetes Mellitus (2nd ed) John Wiley & Sons, New York; 1997 p. 37–88.
- 112.Bornstein J. A technique for the assay of small quantities of insulin using alloxan diabetic, hypophysectomized, adrenalectomized rats. Aust J Exp Biol Med Sci. 1950;28:87–91. doi: 10.1038/icb.1950.7. [DOI] [PubMed] [Google Scholar]
- 113.Yalow, RS, Berson SA. Immunoassay of endogenous plasma insulin in man. J Clin Invest. 1960;39:1157–75. doi: 10.1172/JCI104130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bjorntorp P. Do stress reactions cause abdominal obesity and comorbidities? Obes Rev. 2001;2:73–86. doi: 10.1046/j.1467-789x.2001.00027.x. [DOI] [PubMed] [Google Scholar]
- 115.Bass J, Turek FW. Sleepless in America: a pathway to obesity and the metabolic syndrome? Arch Intern Med. 2005;165:15–6. doi: 10.1001/archinte.165.1.15. [DOI] [PubMed] [Google Scholar]
- 116.Grundy SM. What is the contribution of obesity to the metabolic syndrome? Endocrinol Metab Clin North Am. 2004;33:267–82. doi: 10.1016/j.ecl.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 117.Reaven GM. Insulin resistance/compensatory hyperinsulinemia, essential hypertension, and cardiovascular disease. J Clin Endocrinol Metab. 2003;88:2399–403. doi: 10.1210/jc.2003-030087. [DOI] [PubMed] [Google Scholar]
- 118.Wang CC, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004;53:2735–40. doi: 10.2337/diabetes.53.11.2735. [DOI] [PubMed] [Google Scholar]
- 119.Shinozaki K, Kashiwagi A, Masada M, Okamura T. Molecular mechanisms of impaired endothelial function associated with insulin resistance. Curr Drug Targets Cardiovasc Haematol Disord. 2004;4:1–11. doi: 10.2174/1568006043481248. [DOI] [PubMed] [Google Scholar]
- 120.Jacobs HS, Conway GS. Leptin, polycystic ovaries and polycystic ovary syndrome. Hum Reprod Update. 1999;5:166–71. doi: 10.1093/humupd/5.2.166. [DOI] [PubMed] [Google Scholar]
- 121.Angulo P, Lindor KD. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2002;17 (Suppl):S186–90. doi: 10.1046/j.1440-1746.17.s1.10.x. [DOI] [PubMed] [Google Scholar]
- 122.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 123.Komninou D, Ayonote A, Richie JP, Jr, Rigas B. Insulin resistance and its contribution to colon carcinogenesis. Exp Biol Med (Maywood) 2003;228:396–405. doi: 10.1177/153537020322800410. [DOI] [PubMed] [Google Scholar]
- 124.Kaaks R. Nutrition, hormones, and breast cancer: is insulin the missing link? Cancer Causes Control. 1996;7:605–25. doi: 10.1007/BF00051703. [DOI] [PubMed] [Google Scholar]
- 125.Lanfranco F, Gianotti L, Pivetti S, et al. Obese patients with obstructive sleep apnoea syndrome show a peculiar alteration of the corticotroph but not of the thyrotroph and lactotroph function. Clin Endocrinol (Oxf) 2004;60:41–8. doi: 10.1111/j.1365-2265.2004.01938.x. [DOI] [PubMed] [Google Scholar]
- 126.Wilcox I, McNamara SG, Collins FL, Grunstein RR, Sullivan CE. “Syndrome Z”: the interaction of sleep apnoea, vascular risk factors and heart disease. Thorax. 1998;53 (Suppl 3):S25–8. [PMC free article] [PubMed] [Google Scholar]
- 127.Chin K, Shimizu K, Nakamura T, et al. Changes in intra-abdominal visceral fat and serum leptin levels in patients with obstructive sleep apnea syndrome following nasal continuous positive airway pressure therapy. Circulation. 1999;100:706–12. doi: 10.1161/01.cir.100.7.706. [DOI] [PubMed] [Google Scholar]
- 128.Sapin R. Insulin assays: previously known and new analytical features. Clin Lab. 2003;49:113–21. [PubMed] [Google Scholar]
- 129.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 130.Wallace TM, Matthews DR. The assessment of insulin resistance in man. Diabet Med. 2002;19:527–34. doi: 10.1046/j.1464-5491.2002.00745.x. [DOI] [PubMed] [Google Scholar]
- 131.Sikaris KA. The clinical biochemistry of obesity. Clin Biochem Rev. 2004;25:165–82. [PMC free article] [PubMed] [Google Scholar]
- 132.Belfiore F, Ianello S, Volpicelli G. Insulin sensitivity indices calculated from basal and OGTT-induced insulin, glucose and FFA levels. Mol Genet Metab. 1998;63:134–41. doi: 10.1006/mgme.1997.2658. [DOI] [PubMed] [Google Scholar]
- 133.McLaughlin T, Abbasi F, Cheal K, Chu J, Lamendola C, Reaven G. Use of metabolic markers to identify overweight individuals who are insulin resistant. Ann Intern Med. 2003;139:802–9. doi: 10.7326/0003-4819-139-10-200311180-00007. [DOI] [PubMed] [Google Scholar]