

Abstract
Inflammatory bowel disease (IBD) is a chronic gastrointestinal condition that encompasses ulcerative colitis (UC) and Crohn’s disease (CD). Targeting both inflammation and the epithelial barrier simultaneously can significantly improve symptom management in IBD, as a promising strategy. In this study, we focused on addressing both inflammation and the epithelial barrier. Until now, each therapeutic target including phosphodiesterase 4 (PDE4) and prolyl hydroxylase domain enzymes 1 and 2 (PHD1/2) have been studied separately. PDE4 plays a key role in the inflammatory process by converting cyclic AMP (cAMP) to AMP and its inhibition can suppress the production of inflammatory cytokines. Research has shown that inhibiting PHD1 and PHD2 increases levels of hypoxia-inducible factor-alpha (HIF-α), which in turn strengthens the epithelial barrier by promoting the expression of protective factors such as mucins and β-defensins. Through virtual screening, molecular docking, and molecular dynamics simulations, we identified five compounds—Cassiamin C, Ginkgetin, Hinokiflavone, Sciadopitysin, and Sojagol—as promising new drug candidates for IBD treatment. All compounds demonstrated superior free binding energy for the three targets compared to reference ligands, except Sojagol concerning PDE4B. Among these compounds, Ginkgetin was the best compound with potential ability of targeting multiple drug target proteins. Future experimental studies are warranted to validate these findings.
Keywords: Inflammatory bowel disease, MD simulation, Bioactive compounds, Ginkgetin, Multi-target drugs
Subject terms: Virtual drug screening, Gastrointestinal diseases
Introduction
Inflammatory bowel disease (IBD) is a chronic gastrointestinal disease that includes ulcerative colitis (UC) and Crohn’s disease (CD)1–3. The main cause of this disease is still unknown; however, it is known that genetics and the environment play a role in its development. Symptoms of this disease include abdominal pain, diarrhea, and weight loss4. In general, this disease is associated with dysbiosis, inflammation and loss of the epithelial barrier. According to studies, the incidence of inflammatory bowel disease is increasing worldwide5–7. This issue highlights the need to discover and develop new drugs for this disease. Following the loss of mucus and increased permeability of the epithelial barrier, due to the loss of tight junctions, immune system cells come into excessive contact with antigens in the intestinal lumen and produce and secrete inflammatory cytokines IL-12, IL-23, and IL-6. In addition, antigen presentation to T cells causes their differentiation into Th1 and Th17. These cells also produce inflammatory cytokines IL-17 A, IL-17 F, and IFN-γ. In this way, the immune system is activated and causes the onset of inflammation8–12. Inflammatory cytokines secreted by immune cells affect the epithelial barrier by reducing the expression of claudin-1 and occludin proteins, further increasing the permeability of the epithelial barrier and increasing the interaction of immune cells with antigens, which contributes to the chronicity of inflammation in the intestine. For this reason, epithelial barrier regeneration, along with the suppression of inflammation, is of particular importance for the treatment of this disease13. Furthermore targeting multiple pathways associated with inflammatory processes and regenerating the epithelial can lead to a more effective therapeutic strategy in IBD. The multi-target approach in drug discovery and development has gained recognition as a more effective strategy compared to the traditional single-target approach for several reasons14,15. Reducing drug resistance is a very important outcome of using multi target approach. Multi-target drugs can be designed to balance the effects on different targets, which may reduce side effects compared to high-dose single-target drugs that can lead to toxicity. Additionally, IBD is characterized by a multifactorial etiology involving genetic, environmental, and immunological factors. By simultaneously targeting PDE4B, PHD1, and PHD2, we aim to address the diverse pathways involved in the inflammatory response, and epithelial regeneration, which may result in enhanced efficacy compared to single-target therapies14,15.
The enzyme phosphodiesterase 4 (PDE4) plays an important role in inflammation which is involved in the production of inflammatory cytokines by converting cAMP to AMP16,17. The inhibition of this enzyme is associated with inhibition of nuclear factor kappa B (NF-κB) and B-cell lymphoma 6 / Early growth response 1(Bcl6/Egr1) and activation of Ras-related protein 1 (Rap1), which leads to inhibition of inflammatory cytokines production and also causes the activation of cAMP-response element binding protein / activating transcription factor 1 (CREB/ATF-1), resulting in the production of anti-inflammatory cytokines16. Due to the effective role of this enzyme in inflammation, its inhibition has always been of interest to pharmaceutical companies for the treatment of inflammatory diseases. Roflumilast, Oglemilast, GSK256066, Crisaborole, Apremilast, Tetomilast, and Revamilast are among the PDE4 inhibitors that have entered clinical trials. Among them, Roflumilast, Crisaborole and Apremilast have been approved by the Food and Drug Administration (FDA). After Amgen acquired Apremilast for $13.4 billion, Amgen conducted a Phase II clinical trial of Apremilast for IBD, which reported improvements in clinical and endoscopic features in patients. In addition, a Phase III clinical trial of Tetomilast has been completed in CD and UC. These cases demonstrate the effective role of PDE4 inhibitors in suppressing inflammation16,18–21. Research indicates that the potential side effects of PDE4B inhibitors are significantly lower compared to those associated with PDE4D, another member of the same family. Therefore, this study utilized the three-dimensional structure of the PDE4B protein. Additionally, Apremilast, a well-established PDE4 inhibitor, served as a reference ligand in this research.
Regarding the pathogenesis of inflammatory bowel disease, the reconstruction and repair of the epithelial barrier by inhibiting epithelial cell apoptosis, in addition to inhibiting the production and secretion of inflammatory cytokines, could be one of the potential therapeutic approaches for IBD. PHD1, a member of the PHD family of enzymes, plays a role in regulating epithelial cell apoptosis, as apoptosis in epithelial cells was reduced in PHD1−/− knockout mice, resulting in reduced disruption of the epithelial barrier22. In addition, deletion of PHD1 in the RAW264.7 macrophage cell line resulted in reduced production of inflammatory cytokines. Deletion of PHD2, another member of the PHD family of enzymes, in macrophages was associated with a decrease in the expression of the inflammatory factor TNF23. It has also been shown that PHDs play a role in the degradation of HIF-a by the proteasome through hydroxylation24–28. Thus, inhibition of PHD1/2 increases HIF-α levels and improves the epithelial barrier following the expression of protective factors such as mucin and β-defensin. So far, PHD inhibitors such as Vadadustat, AKB-6899, Roxadustat, and Daprodustat have been investigated in different diseases by Akebia Therapeutics, Fibrogen, and Glaxo-Smith-Kline, respectively24–27. Additionally, the drug AKB-4924 has entered clinical trials by Aerpio Therapeutics for IBD29. One of the companies currently investigating PHD1/2 inhibitors in IBD is Insilico Medicine, which has announced that its inhibitor has good efficacy in preclinical studies, and colitis models30. Therefore, inhibition of PHD1/2 can be a potential therapeutic target for the treatment of IBD and the improvement of pathological symptoms associated with it.
The fascination with natural and herbal remedies has been a part of human history for centuries. In recent times, as awareness grows about the side effects associated with chemical and synthetic medications, there has been a notable shift towards using natural alternatives31. According to projections, the global herbal medicine market is anticipated to reach a value of $437.59 billion by 2031, up from $168.86 billion in 202332. This surge in interest has prompted numerous studies to explore the therapeutic effects of herbal compounds on various diseases. For instance, curcumin, a compound derived from turmeric, is recognized for its anti-inflammatory properties, which have been studied in the context of inflammatory diseases such as IBD33. Additionally, fennel (Foeniculum vulgare) is commonly used as a digestive aid, and a 2022 study in the United States highlighted its potential as a complementary treatment for IBD34. Furthermore, research has indicated that cinnamon extract may play a role in preventing the relapse of chronic inflammatory conditions35.
Today, the use of in silico methods has garnered significant attention from pharmaceutical companies due to their ability to dramatically reduce both time and costs in drug development. Identifying the suitable herbal compounds is crucial for treating diseases; however, traditional methods for finding these compounds can be incredibly time-consuming and expensive. This is where in silico tools prove to be immensely valuable. By utilizing these tools, we can avoid the lengthy processes of screening herbal compounds and performing numerous tests, allowing us to identify effective treatments fastly36. In this study, we performed a virtual screening of 2299 herbal compounds. We evaluated their physicochemical properties, absorption, distribution, metabolism, excretion, and toxicity (ADMET) characteristics, and affinity scores for various compounds. Additionally, we analyzed molecular dynamics, binding free energy, and intermolecular interactions within the complexes to identify potential bioactive agents for the treatment of IBD.
Materials and methods
Structure-based drug design using virtual screening
The wild-type variants of human PDE4B (5OHJ), PHD1 (5V1B), and PHD2 (5OX6) three dimensional structures, were obtained from the Protein Data Bank (PDB) (https://www.rcsb.org/)22,27,40. Apremilast and Vadadustat were chosen as reference ligands for PDE4B and PHD1/2, respectively, as they are approved inhibitors for these targets37,38 AutoDock Tools (ADT) is a GUI for preparing input and analyzing output files for the AutoDock suite, used in molecular docking studies crucial for drug design. AutoDockFR (ADFR) enhances this by enabling flexible receptor docking, improved sampling methods, and user-friendly interfaces, facilitating accurate ligand-binding predictions in drug discovery. The AutoDock FR software was utilized to prepare the proteins for virtual screening and molecular docking39. The ligands, which consisted of herbal bioactive compounds, were processed using the OpenBabel program to add hydrogen atoms and to generate their 3D structures in PDB format. The dimensions of the selected grid box and the binding sites are detailed in Table 1. Docking studies were conducted using the AutoDock Vina tool40, which employed the PDBQT files of the PDE4B, PHD1, and PHD2 structures along with a library of 2299 ligands in PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The properties of the herbal bioactive compounds with respect to ADMET as well as Lipinski’s Rules were assessed using the OSIRIS Property Explorer and pkCSM websites41,42. Each 3D structure of the docked complexes was analyzed with the PLIP (protein–ligand interaction profiler), and PyMOL visualization tool43 (The PyMOL Molecular Graphics System, Version 3.0, Schrödinger, LLC) (projects.biotec.tu-dresden.de/plip-web) (Table 2).
Table 1.
Grid box size surrounding binding site of PDE4B, PHD1, and PHD2.
| Protein | Grid box size | Binding site |
|---|---|---|
| PDE4B (5OHJ) | 22.50 Å × 18.00 Å × 16.50 Å |
Ser454 Thr517 Met519 Asn567 Tyr575 Thr579 Il582 Fhe586 Gln589 Pro602 Met603 Cys604 Ser614 Gln615 Phe618 Il622 |
| PHD1 (5V1B) | 21.00 Å × 21.00 Å × 30.75 Å |
Tyr287 Tyr294 His297 Asp299 Tyr313 Asn315 Arg367 His358 |
| PHD2 (5OX6) | 15.75 Å × 26.25 Å × 26.25 Å |
Asp254 Met299 Tyr310 His313 Asp315 Trp389 |
Table 2.
Affinity of candidate compounds in complex with PDE4B, PHD1, and PHD2. 3D structures were analyzed with the PLIP, and PyMOL visualization tool.

Physicochemical characteristics and ADMET properties
In this research, we employed the OSIRIS Property Explorer (https://www.cheminfo.org/flavor/cheminformatics/Utility/Property_explorer/index.html) and the pkCSM platforms (https://biosig.lab.uq.edu.au/pkcsm/) to assess a range of parameters for our selected candidates. We examined various physicochemical characteristics, including the number of rotatable bonds, the octanol-water partition coefficient, molecular weight, water solubility, and the counts of hydrogen bond donors and acceptors, as well as the topological polar surface area. We also evaluated pharmacokinetic parameters such as Caco-2 permeability, P-glycoprotein inhibition, and human intestinal absorption, alongside distribution metrics like central nervous system permeability, volume of distribution, and blood-brain barrier penetration. Furthermore, we investigated metabolic parameters, particularly the ability to inhibit different cytochrome P450 subtypes. We assessed excretion factors, including total clearance and renal Organic Cation Transporter 2 (OCT2) substrate potential, along with toxicity indicators such as AMES toxicity, inhibition of human ether-related gene channels, and both acute and chronic toxicity in oral rats, as well as skin sensitization44.
Molecular dynamics (MD) simulations
Molecular dynamics (MD) simulations were conducted on the docked complexes using GROMACS-2022 to explore how ligands interact with receptors under physiological conditions45,46. The protein topology was created using the OPLS-AA force field47. Ligand topology files were generated via the LigParGen server (https://traken.chem.yale.edu/ligpargen/). Each system was placed in a cubic box filled with the TIP3P water model48, and neutralization was achieved by adding counter ions (Na· and Cl−) at a concentration of 0.15 M. The energy of the neutralized systems was minimized using both steepest descent and conjugate gradient methods, with 50,000 steps allocated to each. Equilibration of the systems involved regulating the volume (NVT) and adjusting the pressure (NPT). The temperature was gradually raised to 310 K in the NVT ensemble, followed by pressure and density equilibration in the NPT ensemble. The SHAKE algorithm was utilized to keep hydrogen atoms at their equilibrium distances. Throughout the MD simulations, periodic boundary conditions were applied, and long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) method49. Coulombic and van der Waals forces were truncated at 1.2 nm, while bond lengths and angles were constrained with the LINC algorithm50. Following the model setup, we performed equilibration phases with position restraints on the protein and ligand molecules for 1 ns, utilizing the NVT and NPT ensembles through standard coupling techniques. The production phase was then conducted for 150 ns without any restraints on the molecules. Hydrogen bond (H-bond), Root Mean Square Fluctuation (RMSF), Root Mean Square Deviation (RMSD), Radius of gyration (Rg), and Radial Distribution Function (RDF) analysis were conducted using the GROMACS tool gmx hbond, gmx rmsf, gmx rms, gmx gyrate, and gmx rdf, respectively.
Clustering analysis
We performed clustering on bound compounds to identify variations in their binding poses. For each compound, we found significant clusters and chose the central structure as the representative. Additionally, we analyzed MD simulation trajectories using GROMACS with the GROMOS method and an RMSD cutoff of 0.2 nm. We aligned the trajectories to the protein backbone to minimize noise and focused on binding site residues for clustering. This approach helps us study key interactions between ligands and proteins effectively.
Binding free energy
To investigate the energy components that influence the binding of proteins to ligands, we utilized the MM-PBSA method, implemented in the g_mmpbsa program. This approach calculates the binding free energy (ΔGbind) of the receptor-ligand complex by assessing the difference between the free energy of the complex (ΔGRL) and the free energies of the individual components—the receptor (ΔGR) and the ligand (ΔGL). Mathematically, this relationship is represented as:
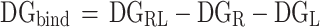 |
1 |
The free energies are derived from several energy components, as outlined in the following equations:
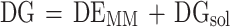 |
2 |
In this equation, the molecular mechanical energy in the gas phase (ΔEMM) is determined by adding the van der Waals interaction energy (ΔEvdW) to the electrostatic energy (ΔEele):
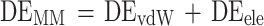 |
3 |
Furthermore, the solvation free energy (ΔGsol) is divided into polar (ΔGsol−pol) and non-polar (ΔGsol−np) components:
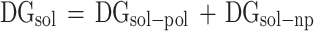 |
4 |
This methodology allows for a comprehensive exploration of the energetic dynamics involved in receptor-ligand interactions51,52.
Results
ADMET analysis and toxicity profiling
A total of 2299 herbal compounds from the PubChem database were analyzed, and five compounds—Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, and Ginkgetin—were selected based on their affinity scores, ADMET properties, and adherence to Lipinski’s Rule. The Vina scores and 2D chemical structures of these compounds are detailed in Table 2. Generally, an intestinal absorption threshold of over 30% and a Caco-2 cell permeability greater than 0.9 are considered acceptable. These compounds demonstrate a longer half-life within the human body, likely due to their low renal clearance rates. The intestinal absorption rates for Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, and Ginkgetin are 95.903%, 80.035%, 79.914%, 98.322%, and 95.376%, respectively (Table 3). Their permeability across the blood–brain barrier is recorded at − 2.29, − 0.96, − 1.78, − 1.85, and − 1.88, and central nervous system (CNS) permeability values are − 1.64, − 2.97, − 3.03, − 3.24, and − 3.27, indicating restricted access to the CNS (Table 3). These compounds could not be toxic or mutagenic (Table 3), leading to their selection for MD simulation.
Table 3.
ADME analysis and toxicity profiling of candidate compounds, received from OSIRIS property explorer and PkCSM. (TPSA, H bond acceptor, H bond donor, Nb rotatable bonds, and molecular weight predicted by OSIRIS, ADMET properties, toxicity profiles, solubility, and permeability metrics predicted by PkCSM.
| Sojagol (5281809) | Cassiamin C (442728) | Hinokiflavone (5281627) | Sciadopitysin (5281696) | Ginkgetin (5271805) | Apremilast | Vadadustat | |
|---|---|---|---|---|---|---|---|
| Water solubility | − 3.53 | − 2.97 | − 2.89 | − 3.02 | − 2.94 | − 4.46 | − 2.61 |
| TPSA | 68.90 | 149.20 | 162.98 | 140.98 | 151.98 | 127.46 | 99.52 |
| H bond acceptor | 5 | 8 | 10 | 10 | 10 | 7 | 4 |
| H bond donor | 1 | 4 | 5 | 3 | 4 | 1 | 3 |
| Nb rotatable bonds | 0 | 1 | 4 | 6 | 5 | 8 | 4 |
| Molecular weight | 336.34 | 506.46 | 538.46 | 580.54 | 566.52 | 460.51 | 306.71 |
| Caco2 permeability | 1.18 | 0.17 | 0.24 | 0.23 | − 0.08 | 0.21 | 0.65 |
| P-glycoprotein I inhibitor | No | Yes | Yes | Yes | Yes | Yes | No |
| P-glycoprotein II inhibitor | No | Yes | Yes | Yes | Yes | Yes | No |
| Intestinal absorption (human) | 95.90 | 80.04 | 79.91 | 98.32 | 95.38 | 83.79 | 46.50 |
| BBB permeability | − 2.29 | − 0.96 | − 1.78 | − 1.85 | − 1.88 | − 0.57 | − 1 |
| CNS permeability | − 1.64 | − 2.97 | − 3.03 | − 3.24 | − 3.27 | − 3.51 | − 2.64 |
| Total Clearance | 0.56 | − 0.13 | 0.51 | 0.83 | 0.65 | 0.23 | 0.02 |
| Renal OCT2 substrate | No | No | No | No | No | No | No |
| AMES toxicity | No | No | No | No | No | No | No |
| Oral rat acute toxicity (LD50) | 2.44 | 2.68 | 2.65 | 3.06 | 2.73 | 2 | 2.70 |
| Oral rat chronic toxicity (LOAEL) | 0.79 | 2.11 | 3.57 | 2.08 | 2.48 | 2.06 | 3.23 |
| Skin sensitisation | No | No | No | No | No | No | No |
Complex stability and compactness
The RMSD analysis of the PDE4B protein systems revealed a consistent trend throughout the simulation (Fig. 1A). In all the complexes that were tested, the RMSD for the PDE4B protein exhibited a significant rise, reaching a maximum between 0.5 and 0.64 nm. After this peak, the fluctuations began to decrease and eventually leveled off, settling into a stable plateau as the simulation progressed. This stabilization indicates that the PDE4B protein maintained sufficient stability for subsequent analyses (Table 4).
Fig. 1.

Molecular dynamic results of PDE4B complexes. The RMSD (A), RMSDligand (B), of the Cα atoms, Rg (C), RDF (D), RMSF (E), and H-bond (F).The black, red, green, blue, brown, yellow, and cyan colors are related to the PDE4B-Cassiamin C, PDE4B-Ginkgetin, PDE4B-Hinokiflavone, PDE4B-Sciadopitysin, PDE4B-Sojagol, PDE4B-Apremilast complexes, and PDE4B-free form (apo) respectively.
Table 4.
Molecular dynamic result of PDE4B complexes.
| RMSF (Avg-nm) | RMSD (Avg-nm) | RMSDligand (Avg-nm) | RDF (max) | Rg (Avg-nm) | |
|---|---|---|---|---|---|
| Sojagol | 0.19 | 0.51 | 0.05 | 48.73 | 2.28 |
| Cassiamin C | 0.21 | 0.64 | 0.05 | 415.16 | 2.29 |
| Hinokiflavone | 0.20 | 0.64 | 0.08 | 192.12 | 2.25 |
| Sciadopitysin | 0.24 | 0.52 | 0.08 | 340.08 | 2.30 |
| Ginkgetin | 0.17 | 0.50 | 0.09 | 289.21 | 2.28 |
| Apremilast | 0.15 | 0.55 | 0.15 | 473.90 | 2.23 |
The average RMSD values for the PDE4B complexes with Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Apremilast were recorded as 0.51, 0.64, 0.64, 0.52, and 0.50 nm, respectively. In comparison, the average RMSD for the reference ligand, Apremilast, was 0.55 nm. To assess the local fluctuations of individual amino acids in the PDE4B protein as influenced by the bioactive compounds, we calculated the RMSF (Fig. 1E). The average RMSF for the complexes were measured at 0.19, 0.21, 0.20, 0.24, and 0.17 nm, respectively. These values suggest that there are minimal local fluctuations within these systems. For comparison, the RMSF for Apremilast was recorded at 0.15 nm (Table 4). Additionally, we utilized the radius of gyration (Rg) to evaluate the shape and compactness of the protein’s conformation. Our findings showed average Rg values of 2.28, 2.29, 2.25, 2.30, 2.28, and 2.23 nm for the complexes with Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Apremilast, respectively (Table 4). The Rg analysis showed that all complexes, including the reference ligand, displayed higher values at the start of the simulation, which gradually decreased over time, revealing a consistent trend. In general, only slight changes were noted throughout the simulations, suggesting that the protein retained its structural stability while maintaining a reasonable level of compactness (Fig. 1C).
To further understand the distribution of compounds within the protein binding site and their behavior during simulations, we analyzed the RDF. This analytical tool helped identify patterns, with peaks suggesting preferred distances where ligands are likely positioned relative to specific amino acids, particularly F342 in the binding site. As shown in Fig. 1D; Table 4, the simulation revealed variations in the RDF. All compounds, including the reference ligand Apremilast, exhibited their highest peaks at a distance of 0.4 nm from the reference residue F342, with the exception of Sojagol. This indicates that, except for Sojagol, the compounds remained stable with only slight fluctuations, successfully maintaining their conformations within their binding sites.
We also examined the conformational changes of the compounds at the PDE4B protein’s binding site by measuring the RMSD values of the heavy atoms in the compounds. The recorded values were 0.05, 0.05, 0.08, 0.08, 0.09, and 0.15 for Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Apremilast, respectively. These results indicate that the compounds interact effectively with the binding site while maintaining suitable conformations. This suggests that there is favorable stability within the complex structures, comparable to the reference ligand (Fig. 1B).
Similarly, the RMSD analysis of the PHD1 protein systems displayed comparable patterns throughout the simulation (Fig. 2A). In all the complexes studied, the RMSD of the PHD1 protein varied between 0.28 and 0.41 nm. Following this initial variation, the fluctuation rates decreased and eventually stabilized, reaching a plateau by the end of the simulation.This indicates that PHD1 exhibited acceptable stability for further analysis (Table 5).The average RMSD values for PHD1 when bound to Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, and Ginkgetin were 0.4 nm, 0.38 nm, 0.41 nm, 0.31 nm, and 0.28 nm, respectively. In comparison, the reference ligand, Vadadustat, had an RMSD value of 0.34 nm. The mean RMSF values for PHD1 complexes were 0.22, 0.15, 0.13, 0.15, and 0.14 nm, respectively, and 0.14 nm for Vadadustat (Fig. 2E), indicating low local fluctuations within these complex systems as the reference ligand (Table 5).
Fig. 2.

Molecular dynamic results of PHD1 complexes. The RMSD (A), and RMSDligand (B), of the Cα atoms, Rg (C), RDF (D), RMSF (E), and H-bond (F). The black, red, green, blue, brown, yellow, and cyan colors are related to the PHD1-Cassiamin C, PHD1-Ginkgetin, PHD1-Hinokiflavone, PHD1-Sciadopitysin, PHD1-Sojagol, PHD1-Vadadustat complexes, and PHD1-free form (apo), respectively.
Table 5.
Molecular dynamic result of PHD1complexes.
| RMSF (Avg-nm) | RMSD (Avg-nm) | RMSDligand (Avg-nm) | RDF (Max) | Rg (Avg-nm) | |
|---|---|---|---|---|---|
| Sojagol | 0.22 | 0.40 | 0.04 | 134.48 | 1.80 |
| Cassiamin C | 0.15 | 0.38 | 0.05 | 155.42 | 1.79 |
| Hinokiflavone | 0.13 | 0.41 | 0.17 | 186.58 | 1.80 |
| Sciadopitysin | 0.15 | 0.31 | 0.10 | 173.33 | 1.78 |
| Ginkgetin | 0.14 | 0.28 | 0.10 | 148.11 | 1.72 |
| Vadadustat | 0.14 | 0.34 | 0.15 | 163.41 | 1.80 |
The average Rg values for PHD1 complexes with Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Vadadustat were found to be 1.80, 1.79, 1.80, 1.78, 1.72, and 1.80 nm, respectively (Table 5). The Rg analysis revealed that all complexes, like the reference ligand, showed high values at the beginning of the simulation. However, these values gradually declined towards the end, following a trend similar to that observed in PDE4B. Overall, minimal changes were noted during the simulations, suggesting that PHD1 also maintained its structural stability and exhibited an acceptable level of compactness (Fig. 2C). Figure 2D shows the changes in the RDF over the course of the simulation. Notably, all compounds reached their highest peaks at a distance of 0.4 nm from the reference residue Y294. This suggests that the compounds remained stable with only minor fluctuations, effectively maintaining their conformations at the binding sites. It is worth mentioning that Hinokiflavone and Sciadopitysin exhibited higher peaks compared to Vadadustat.
The RMSD values for the heavy atoms of the compounds were recorded as follows: 0.04, 0.05, 0.17, 0.10, 0.10, and 0.15 for Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Vadadustat, respectively. These values suggest that the compounds effectively interacted with the binding site, maintaining their proper conformations. All candidate compounds, similar to the reference ligand, exhibited stability within their complex structures, as shown in Fig. 2B.
The RMSD analysis of the PHD2 protein systems exhibited a consistent pattern throughout the simulation period (Fig. 3A). The RMSD values for the PHD2 protein across all the complexes analyzed varied between 0.77 and 0.99 nm. Over the course of the simulation, these values showed a decline in fluctuation rates, eventually leveling off into a stable plateau by the end. This indicates that PHD2 maintained acceptable stability for further analyses (Table 6). The RMSD values for PHD2 when complexed with Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Vadadustat were 0.95, 0.99, 0.77, 0.84, 0.91, and 0.96 nm, respectively. The RMSD values indicate that the complexes are quite stable. Similarly, the RMSF values for PHD2 in association with these compounds were 0.36, 0.16, 0.19, 0.22, 0.18, and 0.33, respectively. These figures suggest that there are minimal local fluctuations within these complex systems, comparable to those observed with Vadadustat, which serves as the reference ligand (Fig. 3E; Table 6). On average, the Rg values for the protein complexes with Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Vadadustat were recorded as 1.91, 1.84, 1.86, 1.81, 1.84, and 1.96 nm, respectively (Table 6). Similar to the previously analyzed complexes, the Rg analysis for PHD2 revealed higher values at the beginning of the simulation, which decreased over time, following a similar final pattern.Overall, similar to the reference ligand, only minor changes were observed throughout the simulations. This suggests that PHD2 preserved its structural stability and demonstrated an appropriate level of compactness (Fig. 3C).
Fig. 3.

Molecular dynamic results of PHD2 complexes. The RMSD (A), and RMSDligand (B), of the Cα atoms, Rg (C), RDF (D), RMSF (E), and H-bond (F). The black, red, green, blue, brown, yellow, and cyan colors are related to the PHD2-Cassiamin C, PHD2-Ginkgetin, PHD2-Hinokiflavone, PHD2-Sciadopitysin, PHD2-sojagol, PHD2-Vadadustat complexes, and PHD2-free form (apo), respectively.
Table 6.
Molecular dynamic result of PHD2 complexes.
| RMSF (Avg-nm) | RMSD (Avg-nm) | RMSDligand (Avg-nm) | RDF (Max) | Rg (Avg-nm) | |
|---|---|---|---|---|---|
| Sojagol | 0.36 | 0.95 | 0.04 | 54.58 | 1.91 |
| Cassiamin C | 0.16 | 0.99 | 0.05 | 46.44 | 1.84 |
| Hinokiflavone | 0.19 | 0.77 | 0.10 | 41.97 | 1.86 |
| Sciadopitysin | 0.22 | 0.84 | 0.10 | 255.11 | 1.81 |
| Ginkgetin | 0.18 | 0.91 | 0.11 | 20.87 | 1.84 |
| Vadadustat | 0.33 | 0.96 | 0.14 | 117.34 | 1.96 |
The findings presented in Fig. 3D; Table 6 reveal differences in the RDF throughout the simulation. Notably, Sojagol and Ginkgetin demonstrated the highest peaks at 0.8 nm. In contrast, Cassiamin C, Hinokiflavone, Sciadopitysin, and Vadadustat displayed peaks at 0.7 nm, 1.0 nm, 0.4 nm, and 0.5 nm, respectively, when compared to the reference residue Y310. This data suggests significant interactions among the compounds (Fig. 3D). To evaluate the conformational changes of compounds at the binding site of the PHD2 protein, we measured the mean RMSD values of the heavy atoms in the compounds. The recorded values were 0.04, 0.05, 0.10, 0.10, 0.11, and 0.14 nm for Sojagol, Cassiamin C, Hinokiflavone, Sciadopitysin, Ginkgetin, and Vadadustat, respectively. The results demonstrate that the compounds interact effectively with the binding site while maintaining their proper conformations. This suggests that the compounds are likely stable within the complex structures, much like the reference ligand (Fig. 3B).
Consequently, the analysis reveals that both PDE4B and PHD proteins exhibited stable conformations throughout the simulations, with low local fluctuations and acceptable compactness, as indicated by RMSD, RMSF, and Rg measurements. The compounds analyzed demonstrated effective interactions with their respective binding sites, maintaining stable conformations, which suggests their potential for further study in therapeutic contexts. The RDF analysis further supports the stability of these complexes, highlighting the significant interactions among the ligands and their target residues.
H-bond analysis was performed for all complexes (Figs. 1F, 2F and 3F). Furthermore, interactions, in all proteins had no negative effect on protein stability compared to the apo state, in RMSF, RMSD, and Rg analysis. Overall, these findings suggest that the studied compounds may serve as promising candidates for targeting PDE4B and PHD proteins in future therapeutic applications compared to reference ligands.
Free binding energy and interactions analysis using MMPBSA
The binding free energy (ΔGbind) values, along with the van der Waals (ΔEvdw) and electrostatic energy (ΔEele) components for the PDE4B, PHD1, and PHD2 complexes during molecular dynamics (MD) simulations, are detailed in Tables 7 and 8, and 9. The MMPBSA method was employed to evaluate the energetic interactions of each compound within the binding landscape and to calculate the binding free energy. The calculated binding free energy for Ginkgetin in complex with PDE4B was − 231.24 ± 2.69 kJ/mol (Table 7), outperforming other candidate compounds associated with PDE4B. This complex exhibited five hydrophobic interactions, and four hydrogen bonds (Fig. 4B). In contrast, the complex of PDE4B with Sojagol displayed five hydrophobic interaction, no hydrogen bond, and one salt bridge. (Fig. 4E), resulting in a ΔGbind of − 24.61 ± 14.78 kJ/mol (Table 7). PDE4B in complex with Cassiamin C showed eight hydrophobic interactions, four hydrogen bonds, (Fig. 4A) and a ΔGbind of − 116.57 ± 14.48 kJ/mol (Table 7). The complex with Hinokiflavone had four hydrophobic interactions, seven hydrogen bonds, (Fig. 4C), and a ΔGbind of − 165.87 ± 9.33 kJ/mol (Table 7), while the complex with Sciadopitysin had ten hydrophobic interactions, three hydrogen bonds (Fig. 4D), and a ΔGbind of − 170.28 ± 6.64 kJ/mol (Table 7). In these complexes, non-polar interactions were the main factors affecting the ΔGbind values except for PDE4B-Hinokiflavone. For comparison, the ΔGbind of Apremilast when bound to PDE4B was − 75.28 ± 13.32 kJ/mol (Table 7), and it had four hydrophobic interactions, and one hydrogen bond (Fig. 4F). This suggests that our compounds generally outperformed the reference ligand, with the exception of Sojagol.
Table 7.
Free binding energy and the individual energy contributions result of PDE4B complexes (kJ/mol).
| Sojagol | Cassiamin C | Hinokiflavone | Sciadopitysin | Ginkgetin | Apremilast | |
|---|---|---|---|---|---|---|
| ΔEele | − 37.14 ± 2.59 | − 14.49±3.64 | − 86.93 ± 8.48 | − 56.72 ± 4.48 | − 42.46 ± 3.70 | − 67.61 ± 3.03 |
| ΔEvdW | − 162.04 ± 2.86 | − 279.23±7.29 | − 287.71 ± 7.88 | − 316.93 ± 6.20 | − 303.25 ± 2.52 | − 230.83 ± 2.92 |
| ΔGGB | 192.22±17.28 | 200.47±7.21 | 237.70 ± 9.42 | 233.06 ± 7.12 | 143.30±4.06 | 249.05±14.76 |
| ΔGSA | − 16.97 ± 0.37 | − 24.03 ± 0.27 | − 28.50 ± 0.62 | − 29.47 ± 0.29 | − 28.84 ± 0.44 | − 26.13±0.53 |
| ΔEnon−polara | − 179.01 ± 2.89 | − 303.26 ± 7.29 | − 316.20 ± 7.90 | − 346.40±6.21 | − 332.09 ± 2.56 | − 256.96±2.97 |
| ΔEpolarb | 155.08 ± 17.47 | 185.99 ± 8.08 | 150.76 ± 12.67 | 176.34 ± 8.41 | 100.83 ± 5.49 | 181.44±15.07 |
| ΔGbind | − 24.61 ± 14.78 | − 116.57 ± 14.48 | − 165.87 ± 9.33 | − 170.28 ± 6.64 | − 231.24 ± 2.69 | − 75.28±13.32 |
Table 8.
Free binding energy and the individual energy contributions result of PHD1 complexes (kJ/mol).
| Sojagol | Cassiamin C | Hinokiflavone | Sciadopitysin | Ginkgetin | Vadadustat | |
|---|---|---|---|---|---|---|
| ΔEele | − 15.33 ± 1.63 | − 30.12 ± 4.09 | − 31.75 ± 6.06 | − 31.46 ± 5.81 | − 21.68 ± 2.55 | − 36.84±4.91 |
| ΔEvdW | − 158.19 ± 4.06 | − 183.25 ± 8.47 | − 174.07 ± 5.40 | − 214.87 ± 6.86 | − 242.20 ± 4.11 | − 145.39 ± 1.35 |
| ΔGGB | 102.23 ± 2.64 | 121.58 ± 7.48 | 100.77 ± 6.08 | 141.22 ± 3.59 | 157.10 ± 2.28 | 114.32±4.89 |
| ΔGSA | − 18.11 ± 0.24 | − 20.55 ± 0.51 | − 20.25 ± 0.94 | − 27.71 ± 0.36 | − 29.56 ± 0.41 | − 17.43±0.26 |
| ΔEnon-polara | − 176.29 ± 4.07 | − 203.80 ± 8.48 | − 194.31 ± 5.48 | − 242.58 ± 6.87 | − 271.76 ± 4.13 | − 162.82±1.38 |
| ΔEpolarb | 86.90 ± 3.10 | 91.46 ± 8.53 | 69.01 ± 8.58 | 109.77 ± 6.83 | 135.42 ± 4.13 | 77.47 ± 6.93 |
| ΔGbind | − 89.25 ± 4.88 | − 112.20 ± 6.79 | − 125.03 ± 5.34 | − 133.15 ± 5.31 | − 135.99 ± 4.73 | − 84.90±2.61 |
Table 9.
Free binding energy and the individual energy contributions result of PHD2 complexes (kJ/mol).
| Sojagol | Cassiamin C | Hinokiflavone | Sciadopitysin | Ginkgetin | Vadadustat | |
|---|---|---|---|---|---|---|
| ΔEele | − 41.83 ± 2.27 | − 3.09 ± 1.47 | − 112.08 ± 5.97 | − 19.60 ± 2.52 | − 70.54 ± 8.19 | 5.32±2.19 |
| ΔEvdW | − 128.28 ± 2.84 | − 162.92 ± 2.01 | − 240.95 ± 4.21 | − 198.66 ± 8.46 | − 221.26 ± 3.27 | − 101.79±1.71 |
| ΔGGB | 95.18 ± 3.04 | 95.66 ± 5.65 | 209.14 ± 7.51 | 126.68 ± 6.83 | 186.93 ± 6.74 | 43.73±1.95 |
| ΔGSA | − 15.28 ± 0.23 | − 17.69 ± 0.36 | − 26.35 ± 0.32 | − 24.00 ± 0.64 | − 27.16 ± 0.39 | − 11.14±0.33 |
| ΔEnon-polara | − 143.56 ± 2.85 | − 180.62 ± 2.04 | − 267.30 ± 4.22 | − 222.66± 8.48 | − 248.41 ± 3.29 | − 112.93±2.26 |
| ΔEpolarb | 53.35 ± 3.79 | 92.57 ± 5.84 | 97.06 ± 9.59 | 107.08 ± 7.28 | 116.39±10.60 | 54.36±2.94 |
| ΔGbind | − 90.15 ± 2.91 | − 88.22 ± 5.50 | − 169.99 ± 3.83 | − 115.22 ± 6.90 | − 132.04 ± 5.12 | − 63.83 ± 1.79 |
a ΔEnonpolar = ΔGsol-np + ΔEvdw. bΔEpolar = ΔGsol-pol + Δee.
Fig. 4.

PDE4B complex with (A) Cassiamin C, (B) Ginkgetin, (C) Hinokiflavone, (D) Sciadopitysin (E) sojagol, and (F) Apremilast. The highlighted residues are marked with light blue sticks. Blue lines, gray and yellow dashed lines indicate hydrogen bonds, hydrophobic interactions and salt bridges respectively.
Similar to the findings for PDE4B, Ginkgetin also emerged as the most effective inhibitor for PHD1, with a ΔGbind of − 135.99 ± 4.73 kJ/mol (Table 8) and four hydrophobic interactions, and tow hydrogen bonds (Fig. 5B). Sciadopitysin displayed a ΔGbind of − 133.15 ± 5.31 kJ/mol (Table 8) with five hydrophobic interactions, and five hydrogen bonds (Fig. 5D). The Hinokiflavone-PHD1 complex had four hydrophobic interactions, and four hydrogen bonds (Fig. 5C), with a ΔGbind of − 125.03 ± 5.34 kJ/mol (Table 8) while Cassiamin C showed four hydrophobic interactions, and four hydrogen bonds (Fig. 5A) with a ΔGbind of − 112.20 ± 6.79 kJ/mol (Table 8). Sojagol in complex with PHD1 resulted in a ΔGbind of − 89.25 ± 4.88 kJ/mol (Table 8) and exhibited four hydrophobic interactions, one hydrogen bond, and one salt bridge (Fig. 5E). The analysis indicated that non-polar contributions, particularly hydrophobic interactions, were the most significant factors influencing the binding energies of the PHD1-Ginkgetin, and PHD1-Sojagol complexes. The ΔGbind for Vadadustat in complex with PHD1 was − 84.90 ± 2.61 kJ/mol with seven hydrophobic interactions, and two hydrogen bonds (Fig. 5F). Overall, our findings indicate that all four compounds show significant potential as inhibitors of PHD1 when compared to the reference ligand.
Fig. 5.

PHD1 complex with (A) Cassiamin C, (B) Ginkgetin, (C) Hinokiflavone, (D) Sciadopitysin (E) sojagol, and (F) Vadadustat. The highlighted residues are marked with light blue sticks. Blue lines, gray and yellow dashed lines indicate hydrogen bonds, hydrophobic interactions and salt bridges respectively.
In contrast to the previous complexes, Hinokiflavone demonstrated superior inhibition of PHD2, with a ΔGbind of − 169.99 ± 3.83 kJ/mol (Table 9). This complex comprised five hydrophobic interactions, and seven hydrogen bonds (Fig. 6C). The other compounds also showed potential as inhibitors of PHD2, with ΔGbind values of − 88.22 ± 5.50 kJ/mol for Cassiamin C, − 89.25 ± 4.88 kJ/mol for Sojagol, − 115.22 ± 6.90 kJ/mol for Sciadopitysin, and − 132.04 ± 5.12 kJ/mol for Ginkgetin (Table 9). The interactions in these complexes included six hydrophobic interactions, and one hydrogen bond for Cassiamin C-PHD2 (Fig. 6A), four hydrophobic interactions, two hydrogen bonds, and one π–π stacking for Sojagol-PHD2 (Fig. 6E), five hydrophobic interactions and one hydrogen bond for Sciadopitysin-PHD2 (Fig. 6D), and four hydrophobic interactions, one hydrogen bond, and one π–π stacking for Ginkgetin-PHD2 (Fig. 6B). Ultimately, the binding free energy (ΔGbind) for Vadadustat when complexed with PHD2 was measured at − 63.83 ± 1.79 kJ/mol with only four hydrophobic interactions (Fig. 6F). This indicates that all of our compounds performed better than the reference ligand.
Fig. 6.

PHD2 complex with (A) Cassiamin C, (B) Ginkgetin, (C) Hinokiflavone, (D) Sciadopitysin (E) sojagol, and (F) Vadadustat. The highlighted residues are marked with light blue sticks. Blue lines, gray and green dashed lines indicate hydrogen bonds, hydrophobic interactions and π–π stacking respectively.
Discussion
The development of IBD suggests that addressing both inflammation and the integrity of the epithelial barrier could help reduce symptoms53. Our study explores this combined strategy, setting it apart from previous research that focused on single therapeutic targets such as PDE4B and PHD1/2. Finding small molecules that can inhibit multiple targets is crucial for the successful treatment of complex conditions like IBD.
PDE4, member of the phosphodiesterase family, is crucial in inflammatory cytokine production and a promising target for inflammatory disease treatment16. In a recent study conducted by Abdullah R Alanzi et al. in 2024, several PDE4 inhibitors were identified, with the most effective compounds being D356-2630, C700-2058, G842-0420, and F403-0203, which achieved glide scores of − 8.06, − 7.85, − 7.32, and − 7.07 kcal/mol, respectively54. In separate investigations of various PDE4B structures (PDB IDs: 3O0J, 4KP6, 3W5E), binding energies for HTS04529 as their best compound were reported at − 83.68, − 84.93, and − 97.90 kJ/mol, respectively, with the reference ligand Rolipram displaying values of − 67.78, − 69.87, and − 83.26 kJ/mol55. In contrast, our candidate compounds exhibited superior results, with docking scores for Sojagol, Cassiamin C, Hinokiflavone, Ginkgetin, Sciadopitysin, and Apremilast recorded at − 10.3, − 10.5, − 9.8, − 10.8, − 10.7 kcal/mol, respectively. Correspondingly, their binding energies were − 24.61, − 116.57, − 165.88, − 231.24, − 170.28, and − 75.28 kJ/mol. Therefore, inhibiting PDE4B using these compounds based on the established role of PDE4B in regulating inflammatory pathways, specifically its influence on NF-κB and Bcl6/Egr1, as well as the activation of Rap1, may suppress NF-κB and Bcl6/Egr1, activate Rap1, and reduce inflammatory cytokine production.
PHD1 and PHD2 hydroxylate HIF-α for proteasomal degradation; inhibiting them offers a potential therapeutic target for IBD treatment24. A study investigating PHD2 inhibitors derived from Cistanche tubulosa identified top compounds with docking scores including acteoside (− 11.184 kcal/mol), 8-epi-loganic acid (− 10.718 kcal/mol), castanoside B (− 10.419 kcal/mol), succinic acid (− 10.415 kcal/mol), and echinacoside-qt (− 9.271 kcal/mol)56. Our candidate compounds yielded comparable results, with docking scores for Sojagol, Cassiamin C, Hinokiflavone, Ginkgetin, and Sciadopitysin of − 9.9, − 10.1, − 10.8, − 8.6, and − 8 kcal/mol, respectively. Inhibiting PHD1 and PHD2 can elevate HIF-α levels, enhancing the epithelial barrier and promoting protective factors like mucin and β-defensin24–27. Sojagol is a natural compound found in Glycine max or soybeans. It was predicted to have structural similarity to (+)-rutamarin alcohol. Rutamarin alcohol is reported to target topoisomerase II that is critical for viral replication. Sojagol was reported to have inhibitory activity of both topoisomerase I and II57. An other study found that sojagol could be a mineralocorticoid receptor (MR) antagonist, which could be a potential herbal compound for the treatment of heart disease58. Additionally, Cassiamin C exhibited significant inhibitory effects on all three targets, with docking scores of − 10.5, − 9.6, and − 10.1 kcal/mol for PDE4B, PHD1, and PHD2, respectively. The binding free energies recorded were − 116.57 ± 14.48, − 112.20 ± 6.79, and − 88.22 ± 5.50 kJ/mol. Our research supports the notion that Cassiamin C shows great potential as a candidate for future therapies aimed at IBD.
Hinokiflavone, a biflavonoid compound, has been recognized for its anti-proliferative and anti-metastatic properties59. Studies have shown that it inhibits the migration and invasion of breast cancer cells through the epithelial-mesenchymal transition (EMT) signaling pathway60. Hinokiflavone may serve as a potential antitumor agent for colorectal cancer by suppressing tumor cell proliferation, migration, and invasion59. In our study, Hinokiflavone exhibited favorable results, with docking scores of − 9.8, − 10.3, and − 10.8 kcal/mol against PDE4B, PHD1, and PHD2, respectively, and binding energies of − 165.87 ± 9.33, − 125.03 ± 5.34, and − 169.99 ± 3.83 kJ/mol. Sciadopitysin, a flavonoid, significantly enhanced alkaline phosphatase, collagen synthesis, osteocalcin, mineralization, and glutathione levels in MC3T3-E1 cells, while reducing TNF-α production induced by antimycin A (P < 0.05)61. Our study found that Sciadopitysin effectively inhibited PDE4B, PHD1, and PHD2, with docking scores of − 10.7, − 11, and − 8 kcal/mol, and binding energies of − 170.28 ± 6.64, − 133.15 ± 5.31, and − 115.22 ± 6.90 kJ/mol, respectively.
Additionally, a wealth of research has underscored the diverse benefits of Ginkgetin. This compound is known for its anti-inflammatory properties, which it achieves by inhibiting various inflammatory mediators. Furthermore, it has demonstrated potential in combating cancer, exhibiting antimicrobial effects, and providing neuroprotection against cell death caused by oxidative stress62. One study identified Ginkgetin as a promising candidate for ovarian cancer treatment by inhibiting the JAK2/STAT3 and Mitogen-activated protein kinase (MAPK) pathways, in addition to modulating the Sirtuin (SIRT1) protein63 Additionally, Ginkgetin has been recognized as a potential treatment for atherosclerosis64. In our study, Ginkgetin exhibited promising results, inhibiting PDE4B, PHD1, and PHD2, with docking scores of − 10.8, − 11.0, and − 8.6 kcal/mol, accompanied by binding energies of − 231.24 ± 2.69, − 135.99 ± 4.73, and − 132.04 ± 5.12 kJ/mol. The candidate compounds identified—Sojagol, Cassiamin C, Hinokiflavone, Ginkgetin, and Sciadopitysin—demonstrated significant inhibitory activity against PDE4B, PHD1, and PHD2. These compounds exhibited favorable docking scores and binding energies, indicating their potential to modulate the pathways involved in inflammation and epithelial integrity. Specifically, our results suggest that inhibiting PDE4B may reduce inflammatory cytokine production, while inhibiting PHD1 and PHD2 may enhance HIF-α levels and support the epithelial barrier through the expression of protective factors.
Future research should prioritize in vitro and in vivo studies to confirm the efficacy and safety of candidate compounds for IBD in animal models. Exploring their synergistic effects and specific molecular mechanisms will enhance understanding and clinical application. Additionally, expanding the search for novel multi-target inhibitors could improve treatment options for complex diseases like IBD.
Conclusion
In recent years, the field of computer-aided drug design has gained significant momentum, leading to the rapid and cost-effective development of novel inhibitors for a variety of diseases. One area that has garnered considerable attention is IBD, a condition characterized by a complex and multifaceted inflammatory pathology. Researchers have increasingly focused on designing and producing new pharmacological agents aimed at alleviating the symptoms and underlying mechanisms of IBD.
Despite the identification of numerous drugs that have therapeutic potential for IBD, a notable gap remains in the literature regarding the discovery of compounds capable of simultaneously targeting multiple proteins implicated in the disease’s pathogenesis. Specifically, proteins such as PDE4B, PHD1 and PHD2 play crucial roles at various levels of IBD progression. In this research, we adopted a multi-target strategy to evaluate the inhibitory potential of specific compounds: Sojagol, Cassiamin C, Hinokiflavone, Ginkgetin, and Sciadopitysin. Our research showed that Cassiamin C, Hinokiflavone, Ginkgetin, and Sciadopitysin were predicted to have potential inhibitory effects on all three targets. These compounds demonstrated both high affinity and stability in their interactions when compared to the reference ligands.
We are optimistic that the outcomes of this study, coupled with future experimental investigations of the identified candidate compounds, will pave the way for a novel therapeutic approach to managing IBD. These compounds can be used in vivo and in vitro studies. For example, conducting molecular, and cellular assessments, like examining the effect of these compounds on activated inflammatory cells as well as evaluating in DSS-induced colitis mouse model. Also, investigating their effects in clinical trials, in the following, can be suitable for advancing drug discovery and development.
This research not only highlights the potential of multi-target drug design but also underscores the importance of a more integrated strategy in the treatment of complex diseases like IBD.
Acknowledgements
We appreciate Fasa University of Medical Sciences for kind supports of this work. This article is derived from a master’s thesis completed at Fasa University of Medical Sciences.
Abbreviations
- IBD
Inflammatory bowel disease
- IL
Interleukin
- JAK/STAT
Janus kinase/signal transducers and activators of transcription
- PDE4
Phosphodiesterase 4
- PHD1/2
Prolyl hydroxylase domain enzymes 1 and 2
- AMP
Adenosine monophosphate
- cAMP
Cyclic AMP
- HIF-α
Hypoxia-inducible factor-alpha
- Th
T helper
- INF-γ
Interferon-γ
- NF-κB
Nuclear factor kappa B
- Bcl6
B-cell lymphoma 6
- Egr1
Early Growth Response 1
- Rap1
Ras-related protein 1
- CREB/ATF-1
cAMP-response element binding protein/activating transcription factor 1
- FDA
Food and drug administration
- UC
Ulcerative colitis
- CD
Crohn’s disease
- TNF
Tumor necrosis factor
- ADMET
Absorption, distribution, metabolism, excretion, and toxicity
- PDB
Protein Data Bank
- OCT2
Organic Cation Transporter 2
- MD
Molecular dynamics
- PME
Particle mesh Ewald
- ΔGbind
Binding free energy
- ΔGRL
Free energy of the complex
- ΔGR
Free energy of the receptor
- ΔGL
Free energy of the ligand
- ΔEMM
Molecular mechanical energy in the gas phase
- ΔEvdW
van der Waals interaction energy
- ΔEele
Electrostatic energy
- ΔGsol
Solvation free energy
- CNS
Central nervous system
- Rg
Radius of gyration
- RDF
Radial distribution function
- RMSF
Root mean square fluctuation
- RMSD
Root mean square deviation
- MR
Mineralocorticoid receptor
- EMT
Epithelial–mesenchymal transition
- MAPK
Mitogen-activated protein kinase
- SIRT1
Sirtuin
- PLIP
Protein–ligand interaction profiler
Author contributions
P.M.: Conceptualization, methodology, and writing-original draft. P.M.: Conceptualization, methodology, and writing original draft. S.A.K.: Conceptualization, and writing original draft. S.N.: Conceptualization, and writing original draft. A.F.: Supervision, writing-review & editing, and conceptualization. E.B.: Supervision, writing-review and editing, and methodology.
Funding
This work was supported by Fasa University of Medical Sciences [Grant number: 402125].
Data availability
All the data generated in this article are available from the first author on request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Akbar Farjadfar, Email: farjadbio@gmail.com.
Esmaeil Behmard, Email: ebehmard@yahoo.com.
References
- 1.Dai, C., Huang, Y. H. & Jiang, M. Combination therapy in inflammatory bowel disease: current evidence and perspectives. Int. Immunopharmacol.114, 109545 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Cui, X. et al. Therapeutic potential of a synthetic dual JAK1/TYK2 inhibitor in inflammatory bowel disease. Int. Immunopharmacol.126, 111238 (2024). [DOI] [PubMed] [Google Scholar]
- 3.Xu, G., Xu, Y., Zheng, T. & Liu, T. Type 2 diabetes and inflammatory bowel disease: a bidirectional two-sample Mendelian randomization study. Sci. Rep.14 (1), 5149 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaddoura, R. et al. Identification of specific biomarkers and pathways in the treatment response of Infliximab for inflammatory bowel disease: in-silico analysis. Life13 (3), 680 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caviglia, G. P. et al. Epidemiology of inflammatory bowel diseases: a population study in a healthcare district of North-West Italy. J. Clin. Med.12 (2), 641 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang, Y. et al. Biomarkers prediction and immune landscape in ulcerative colitis: findings based on bioinformatics and machine learning. Comput. Biol. Med.168, 107778 (2024). [DOI] [PubMed] [Google Scholar]
- 7.Vaga, S. et al. Compositional and functional differences of the mucosal microbiota along the intestine of healthy individuals. Sci. Rep.10 (1), 14977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, J. T. Pathophysiology of inflammatory bowel diseases. Longo DL, editor. N. Engl. J. Med.383(27), 2652–2664 (2020). [DOI] [PubMed]
- 9.Liang, B. et al. New insights into bacterial mechanisms and potential intestinal epithelial cell therapeutic targets of inflammatory bowel disease. Front. Microbiol.13, 1065608 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedrich, M., Pohin, M. & Powrie, F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity50 (4), 992–1006 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Lee, S. H., Kwon, J. E. & Cho, M. L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res.16 (1), 26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sewell, G. W. & Kaser, A. Interleukin-23 in the pathogenesis of inflammatory bowel disease and implications for therapeutic intervention. J. Crohn’s Colitis. 16 (Supplement_2), ii3–19 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrews, C., McLean, M. H. & Durum, S. K. Cytokine tuning of intestinal epithelial function. Front. Immunol.9, 368738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makhoba, X. H., Viegas, C. Jr, Mosa, R. A., Viegas, F. P. & Pooe, O. J. Potential impact of the multi-target drug approach in the treatment of some complex diseases. Drug Des. Dev. Ther. 3235–3249 (2020). [DOI] [PMC free article] [PubMed]
- 15.Löscher, W. Single-target versus multi-target drugs versus combinations of drugs with multiple targets: preclinical and clinical evidence for the treatment or prevention of epilepsy. Front. Pharmacol.12, 730257 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crocetti, L., Floresta, G., Cilibrizzi, A. & Giovannoni, M. P. An overview of PDE4 inhibitors in clinical trials: 2010 to early 2022. Molecules27 (15), 4964 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong, X. L., Wang, Y. H., Xu, J. & Zhang, N. The protective effect of the PDE-4 inhibitor rolipram on intracerebral haemorrhage is associated with the cAMP/AMPK/SIRT1 pathway. Sci. Rep.11 (1), 19737 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.https://www.amgen.com/newsroom/press-releases/2019/08/amgen-to-acquire-otezla-for-134-billion-in-cash-or-approximately-112-billion-net-of-anticipated-future-cash-tax-benefits [Internet]. [cited 2024 Apr 14].
- 19.Danese, S. et al. Effects of Apremilast, an oral inhibitor of phosphodiesterase 4, in a randomized trial of patients with active ulcerative colitis. Clin. Gastroenterol. Hepatol.18 (11), 2526–2534 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Otsuka, P. D. & Commercialization, Inc. FACTS II: A phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-arm study of the efficacy and safety of OPC-6535 tablets in the treatment of subjects with active ulcerative colitis [Internet]. clinicaltrials.gov; 2012 May [cited 2024 Jan 1]. Report No.: NCT00064454. https://clinicaltrials.gov/study/NCT00064454.
- 21.Otsuka Pharmaceutical, D., Commercialization, I. P. & Double-Blind, M. C. R. 3, Parallel-Arm, 52-week dose comparison study of the efficacy and safety of 25 mg QD and 50 mg QD of OPC-6335 oral tablets and 800 mg BID of Asacol® in the maintenance of ulcerative colitis remission [Internet]. clinicaltrials.gov; 2007 Jun [cited 2024 Jan 1]. Report No.: NCT00092508. https://clinicaltrials.gov/study/NCT00092508.
- 22.Tambuwala, M. M. et al. Loss of Prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology139 (6), 2093–2101 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Takeda, K. et al. Inhibition of Prolyl hydroxylase domain-containing protein suppressed lipopolysaccharide-induced TNF-α expression. Arterioscler. Thromb. Vasc. Biol.29 (12), 2132–2137 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Pergola, P. E., Spinowitz, B. S., Hartman, C. S., Maroni, B. J. & Haase, V. H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int.90 (5), 1115–1122 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Yan, Z. & Xu, G. A novel choice to correct inflammation-induced anemia in CKD: oral hypoxia-inducible factor Prolyl hydroxylase inhibitor Roxadustat. Front. Med.7, 393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brigandi, R. A. et al. A novel hypoxia-inducible factor – prolyl hydroxylase inhibitor (GSK1278863) for anemia in CKD: a 28-day, phase 2A randomized trial. Am. J. Kidney Dis.67 (6), 861–871 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Roda, J. M. et al. Stabilization of HIF-2α induces sVEGFR-1 production from tumor-associated macrophages and decreases tumor growth in a murine melanoma model. J. Immunol.189 (6), 3168–3177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu, X. et al. Prolyl hydroxylase domain inhibitor is an effective pre-hospital pharmaceutical intervention for trauma and hemorrhagic shock. Sci. Rep.14 (1), 3874 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aerpio Therapeutics. Phase 1a Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose Study to Assess the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamic Effects of AKB 4924 in Healthy Male Volunteers [Internet]. clinicaltrials.gov; 2017 Mar [cited 2024 Jan 1]. Report No.: NCT02914262. Available from: https://clinicaltrials.gov/study/NCT02914262
- 30.Fu, Y. et al. Intestinal mucosal barrier repair and immune regulation with an AI-developed gut-restricted PHD inhibitor. Nat. Biotechnol.10.1038/s41587-024-02503-w (2024). [DOI] [PubMed]
- 31.Qin, Z., Tang, R., Liang, J. & Jia, X. Berberine, a natural alkaloid: advances in its pharmacological effects and mechanisms in the treatment of autoimmune diseases. Int. Immunopharmacol.137, 112422 (2024). [DOI] [PubMed] [Google Scholar]
- 32.Herbal Medicine Market Size, Share, G., Analysis, B. & Form Application, Product Type, Source, Distribution Channel-Industry Forecast 2022–2028 [Internet]. [cited 2024 Apr 23]. https://www.skyquestt.com/report/herbal-medicine-market.
- 33.Peng, Y. et al. Anti-inflammatory effects of curcumin in the inflammatory diseases: Status, limitations and countermeasures. Drug Des. Dev. Ther. 4503–4525 (2021). [DOI] [PMC free article] [PubMed]
- 34.Das, B. et al. The effect of a fennel seed extract on the STAT signaling and intestinal barrier function. PLoS One. 17 (7), e0271045 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lonati, E. et al. Digested cinnamon (Cinnamomum verum J. Presl) bark extract modulates claudin-2 gene expression and protein levels under TNFα/IL-1β inflammatory stimulus. Int. J. Mol. Sci.24 (11), 9201 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaker, B., Ahmad, S., Lee, J., Jung, C. & Na, D. Silico methods and tools for drug discovery. Comput. Biol. Med.137, 104851 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Perez-Aso, M. et al. Apremilast, a novel phosphodiesterase 4 (PDE4) inhibitor, regulates inflammation through multiple cAMP downstream effectors. Arthritis Res. Ther. 17, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markham, A. Vadadustat: first approval. Drugs80 (13), 1365–1371 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Ravindranath, P. A., Forli, S., Goodsell, D. S., Olson, A. J. & Sanner, M. F. AutoDockFR: advances in protein-ligand Docking with explicitly specified binding site flexibility. PLoS Comput. Biol.11 (12), e1004586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of Docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.31 (2), 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pires, D. E., Blundell, T. L. & Ascher, D. B. PkCSM: predicting small-molecule Pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.58 (9), 4066–4072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sander, T. Actelion’s Property Explorer (Actelion’s Pharmaceuticals Ltd: Allschwil, 2001).
- 43.Behmard, E. et al. Advanced simulations and screening to repurposing a 3 C protease inhibitor against the rupintrivir-resistant human norovirus-induced gastroenteritis. J. Mol. Graph. Model.118, 108345 (2023). [DOI] [PubMed] [Google Scholar]
- 44.Ebrahimi, M., Karami, L. & Alijanianzadeh, M. Computational repurposing approach for targeting the critical Spike mutations in B. 1.617. 2 (delta), AY. 1 (delta plus) and C. 37 (lambda) SARS-CoV-2 variants using exhaustive structure-based virtual screening, molecular dynamic simulations and MM-PBSA methods. Comput. Biol. Med.147, 105709 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX1, 19–25 (2015). [Google Scholar]
- 46.Hess, B., van der Spoel, D., Abraham, M. J. & Lindahl, E. On the importance of accurate algorithms for reliable molecular dynamics simulations. (2019).
- 47.Smith, M. D., Rao, J. S., Segelken, E. & Cruz, L. Force-field induced bias in the structure of Aβ21–30: A comparison of OPLS, AMBER, CHARMM, and GROMOS force fields. J. Chem. Inf. Model.55 (12), 2587–2595 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A. 105 (43), 9954–9960 (2001). [Google Scholar]
- 49.Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys.103 (19), 8577–8593 (1995). [Google Scholar]
- 50.Hess, B., Bekker, H., Berendsen, H. J. & Fraaije, J. G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem.18 (12), 1463–1472 (1997). [Google Scholar]
- 51.Cournia, Z., Allen, B. & Sherman, W. Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J. Chem. Inf. Model.57 (12), 2911–2937 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Federico, L. B. et al. Potential Colchicine binding site inhibitors unraveled by virtual screening, molecular dynamics and MM/PBSA. Comput. Biol. Med.137, 104817 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Mansouri, P. et al. Novel targets for mucosal healing in inflammatory bowel disease therapy. Int. Immunopharmacol.144, 113544 (2025). [DOI] [PubMed] [Google Scholar]
- 54.Alanzi, A. R., Alsalhi, M. S., Mothana, R. A., Alqahtani, J. H. & Alqahtani, M. J. Insilico discovery of novel phosphodiesterase 4 (PDE4) inhibitors for the treatment of psoriasis: insights from computer aided drug design approaches. PLoS One. 19 (11), e0305934 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Nema, M., Gaurav, A. & Lee, V. S. Docking based screening and molecular dynamics simulations to identify potential selective PDE4B inhibitor. Heliyon. 6(9). (2020). [DOI] [PMC free article] [PubMed]
- 56.Zhang, S. et al. A novel PHD2 inhibitor acteoside from Cistanche tubulosa induces skeletal muscle mitophagy to improve cancer-related fatigue. Biomed. Pharmacother.150, 113004 (2022). [DOI] [PubMed] [Google Scholar]
- 57.Ashley, C. N. et al. Identifying potential Monkeypox virus inhibitors: an in Silico study targeting the A42R protein. Front. Cell. Infect. Microbiol.14, 1351737 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kusumawati, R., Sulastomo, H., Setyawan, A. N. & Dewi, R. Sojagol from mung beans as A potential antagonist of mineralocorticoid receptor. Hiroshima J. Med. Sci.67, 185–190 (2018). [Google Scholar]
- 59.Zhou, J. et al. Antitumor activity in colorectal cancer induced by Hinokiflavone. J. Gastroenterol. Hepatol.34 (9), 1571–1580 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Huang, W. et al. Hinokiflavone induces apoptosis and inhibits migration of breast cancer cells via EMT signalling pathway. Cell Biochem. Funct.38 (3), 249–256 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suh, K. S., Lee, Y. S., Kim, Y. S. & Choi, E. M. Sciadopitysin protects osteoblast function via its antioxidant activity in MC3T3-E1 cells. Food Chem. Toxicol.58, 220–227 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Adnan, M. et al. Ginkgetin: A natural biflavone with versatile pharmacological activities. Food Chem. Toxicol.145, 111642 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Wu, L. et al. Ginkgetin suppresses ovarian cancer growth through Inhibition of JAK2/STAT3 and MAPKs signaling pathways. Phytomedicine. 116, 154846 (2023). [DOI] [PubMed] [Google Scholar]
- 64.Zhang, Y., Niu, Y. & Weng, Q. Ginkgetin promotes proliferation and migration of Schwann cells via PIGF/p38 MAPK signaling pathway. Tissue Cell.79, 101967 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data generated in this article are available from the first author on request.