

Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is a severe genetic disorder characterized by renal cystogenesis and hepatic fibrosis, primarily associated with PKHD1 mutations. While differential expression analysis (DEG) has identified key genes involved in ARPKD, their network-level interactions remain unclear. Recent studies have implicated WNT signaling in ARPKD pathogenesis, but a topological framework may provide additional insights into gene community structures. This study applied a network-based approach integrating single-sample gene set enrichment analysis (ssGSEA) and topological centrality analysis to investigate gene communities in ARPKD. We identified three key communities: Community 2, centered on IFT22, exhibited stable activation in both ARPKD and healthy samples, suggesting its role in ciliary function. Community 5, predominantly activated in ARPKD, included genes linked to tissue repair and immune regulation. In contrast, Community 3 was suppressed in ARPKD, indicating potential structural instability. Notably, PKHD1 was mathematically isolated, suggesting limited direct involvement in ARPKD-specific transcriptional networks, while the absence of WNT5A, CDH1, and FZD10 from defined communities in ARPKD may indicate potential alterations in their network associations compared to healthy individuals. These findings highlight the advantages of network topology over conventional DEG analysis in elucidating ARPKD pathophysiology. By identifying gene communities and regulatory hubs, this approach offers novel insights into disease mechanisms and potential therapeutic targets.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-99048-y.
Subject terms: Kidney, Kidney diseases, Network topology
Introduction
Polycystic kidney disease (PKD) is a genetic disorder characterized by the formation of multiple cysts in the kidneys, often leading to kidney failure and systemic complications1,2. PKD is classified into autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD), with ARPKD affecting children and causing early-onset nephromegaly and hepatic fibrosis1–4.
PKD1 (polycystic kidney disease 1) is a major regulator of cyst formation and is also involved in cellular processes such as osteoclastogenesis and bone resorption4. In contrast, ARPKD is caused by mutations in PKHD1 (polycystic kidney and hepatic disease 1), which encodes fibrocystin, a membrane-associated protein essential for kidney and bile duct development5. Additionally, the involvement of ciliary genes such as DZIP1L (DAZ interacting zinc finger protein 1-like) and TULP3 (Tubby like protein 3) classifies ARPKD as a ciliopathy6. The variability in clinical phenotypes among patients with the same PKHD1 mutation suggests that genetic modifiers influence disease severity7. Furthermore, dysregulation of signaling pathways, including Hedgehog signaling, planar cell polarity (PCP), WNT signaling, and metabolic pathways, has been implicated in ARPKD progression8,9.
A study by Richards et al. (2019) analyzed ARPKD kidney tissues using whole exome sequencing (WES) and RNA sequencing (RNA-Seq) and demonstrated that ATMIN (ataxia telangiectasia mutated interactor) regulates PKHD1 expression and influences ARPKD pathology through non-canonical WNT/PCP signaling9. Increased expression of ATMIN, WNT5A (Wnt family member 5A), VANGL2 (Van Gogh-like protein 2), and SCRIBBLE (scribbled planar cell polarity protein) was observed in ARPKD kidney tissues, along with a reduction in β-catenin protein levels.
More recently, Richards et al. (2024) confirmed that mutations in PKHD1 are the primary cause of ARPKD1. However, no correlation was found between PKHD1 mutation positions and disease severity. Instead, mutations in PKD1 were associated with severe ARPKD phenotypes, and transcriptomic analysis revealed significant alterations in WNT signaling pathways. These findings suggest that changes in WNT-related gene expression may contribute to ARPKD progression1.
Traditional differential gene expression (DEG) analysis is critical for understanding disease mechanisms but primarily focuses on individual gene expression changes, potentially overlooking gene–gene interactions. To address this limitation, this study employed a network-based approach using graph-theoretic and topological analyses to investigate coordinated gene communities involved in ARPKD progression.
Unlike conventional DEG analysis, which treats genes as independent entities, this study integrates genes from publicly available datasets into functional networks, providing a more comprehensive understanding of disease mechanisms. This approach leverages discrete mathematics to characterize the structural properties of gene communities and identify key regulatory hub genes10–14.
Transcriptomic data from ARPKD patients were reanalyzed using a publicly available dataset from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (accession: GSE242476), initially published by Goggolidou et al.1,9,10. This dataset includes kidney samples from four ARPKD patients and four age-matched healthy controls.
Furthermore, topological centrality and single-sample gene set enrichment analysis (ssGSEA) were applied to identify gene communities and their hub genes. This network-based approach uncovered previously unrecognized functional relationships, providing a comprehensive understanding of the genetic architecture of ARPKD and identifying potential therapeutic targets1,9.
Results
Reanalysis of ARPKD transcriptomic data: beyond simple differential expression
Transcriptomic data from ARPKD patients were reanalyzed using a publicly available dataset from the NCBI Gene Expression Omnibus (GEO) (accession: GSE242476), originally published by Goggolidou et al. This dataset includes kidney samples from four ARPKD patients and four age-matched healthy controls1,9,10. Since PKHD1, the primary causative gene of ARPKD, was not measured, this analysis focused on other genes associated with ARPKD pathology.
RNA-seq data were processed using standard bioinformatics workflows, and differential expression analysis was performed using DESeq215–19, identifying multiple significantly altered genes (Table 1). Among them, PKD1, primarily associated with autosomal dominant polycystic kidney disease (ADPKD), exhibited higher expression in ARPKD samples than in controls (14,028 vs. 7,715, ranked 113th). While PKD1 is not the causative gene for ARPKD, its altered expression suggests a potential regulatory role in disease progression.
Table 1.
Top 10 genes with the most significant differential expression between ARPKD and control groups.
| Gene_ID | Gene_Description | PKD_ expression_avg |
Control_ expression_avg |
p_value |
|---|---|---|---|---|
| ENSG00000121753 | ADGRB2 | 1159.5 | 115.5 | 5.90E-05 |
| ENSG00000134569 | LRP4 | 1541.75 | 463.75 | 0.000621 |
| ENSG00000004838 | ZMYND10 | 340.75 | 87.75 | 0.000769 |
| ENSG00000118513 | MYB | 36.75 | 0 | 0.000819 |
| ENSG00000130294 | KIF1A | 388.25 | 60.25 | 0.000923 |
| ENSG00000129038 | LOXL1 | 1572 | 497.25 | 0.001394 |
| ENSG00000034053 | APBA2 | 182.5 | 51 | 0.001465 |
| ENSG00000135074 | ADAM19 | 1511.5 | 396.75 | 0.001491 |
| ENSG00000135127 | BICDL1 | 3496.5 | 14,724.5 | 0.001496 |
| ENSG00000136378 | ADAMTS7 | 1125.75 | 306 | 0.001612 |
This table lists the top 10 genes with the most significant differential expression between ARPKD and control groups. For each gene, the table includes the Gene ID, gene name, average expression levels in the ARPKD and control groups, and the corresponding p-values.
Previous transcriptomic studies, including the original analysis by Goggolidou et al., have primarily focused on identifying DEGs. While these approaches have contributed to understanding disease mechanisms, they tend to emphasize genes with large expression changes, potentially overlooking interactions among lowly expressed genes.
The following section details the topology of ARPKD-specific gene networks, describing gene interactions, functional clustering, and the positioning of key genes within the network.
Mathematical approach reveals diverse gene communities in ARPKD
In this study, the topology of gene networks in ARPKD patients was analyzed by constructing a correlation-based network. This transformation enabled the identification of meaningful gene communities that might not be evident in conventional linear gene expression analyses.
Given the computational constraints of analyzing all gene pairs in large-scale RNA-seq datasets, a reproducible random subset of 100 genes was selected to ensure consistency across analyses. Correlation coefficients were calculated for each gene pair, and only those with a correlation of 0.6 or higher were incorporated into the network20,21.
The Louvain method was applied to detect communities of closely interacting genes, and the central genes in each community were identified. This analysis revealed key regulatory hub genes in ARPKD, including PKD1, KIF1A (kinesin family member 1A), and LRP4 (low-density lipoprotein receptor-related protein 4). Further details on the network structure and sensitivity analysis of correlation thresholds are provided in Supplementary Figures S1 and S2.
This analysis demonstrated that topology-based network methods are a powerful approach for uncovering hidden gene relationships in ARPKD. Unlike conventional differential expression analysis, which evaluates genes individually, our approach integrated gene associations to identify functional communities, providing novel insights into disease progression.
Mathematical approach identifies central genes in ARPKD communities
Figure 1 visualizes gene connectivity, showing how genes cluster into functional groups. In the gene network analysis for ARPKD patients, key genes with high betweenness centrality were identified within each community. Betweenness centrality quantifies a gene’s role in network connectivity by measuring the fraction of shortest paths passing through it. Genes with high betweenness may act as critical connectors, facilitating interactions across distinct functional modules.
Fig. 1.

Gene network topology in ARPKD patients. This figure illustrates the gene network in ARPKD patients, where each node represents a gene, and each edge indicates a gene pair with a correlation coefficient of 0.6 or higher. To emphasize the distribution of nodes, edges are rendered fully transparent. Node size is proportional to its degree, reflecting the number of connections with other genes. Larger nodes represent highly connected hub genes, which may play key roles in network stability. Clusters were detected using the Louvain method, with each color representing a distinct community: Community 0 (dark purple), Community 1 (green), and Community 2 (yellow). Genes within each community exhibit higher intra-cluster connectivity, visually delineating the structure of correlated gene groups.
Among them, IFT22 (intraflagellar transport protein 22) in Community 2 exhibited the highest betweenness centrality (0.05737)22,23, suggesting its central role in network structure. IFT22 is involved in intraflagellar transport, essential for cilia maintenance. While cilia dysfunction is a hallmark of ARPKD, further studies are needed to determine IFT22's specific contribution.
Other key genes included PMPCB (mitochondrial processing peptidase beta subunit) in Community 0 (0.013874) and TPSD1 (tryptase delta 1) in Community 1 (0.038575)24. PMPCB plays a crucial role in mitochondrial protein processing and overall mitochondrial function, while TPSD1, a tryptase enzyme, is involved in immune response regulation. These findings highlight distinct biological pathways within the network, but their direct relevance to ARPKD pathology requires further investigation.
This network-based approach offers insight into gene connectivity, but functional validation is essential to confirm these genes’ roles in disease mechanisms. Future studies integrating differential expression analysis and experimental validation will help clarify their biological significance.
Gene communities reveal functional connections in ARPKD
Gene communities identified through network analysis reveal the structural organization of the ARPKD genetic network. A community consists of genes with higher internal connectivity, often reflecting shared functions. Some genes appear in multiple communities due to their involvement in different pathways, a known feature of complex gene interaction networks. Table 2 summarizes these communities and their functional relevance.
Table 2.
Gene communities and their associated genes identified in ARPKD patient samples through network analysis.
| Community | Gene symbol (Ensembl ID) |
|---|---|
| 0 | KLF12 (ENSG00000118922), RBM3 (ENSG00000102317), SUMF2 (ENSG00000129103), MCHR1 (ENSG00000128285), RGS13 (ENSG00000127074), HOMER2 (ENSG00000103942), LACTB (ENSG00000103642), PUS7L (ENSG00000129317), PMPCB (ENSG00000105819), SUCO (ENSG00000094975), GSTM1 (ENSG00000086189), GLRB (ENSG00000109738), CREBL2 (ENSG00000111269), IL20RA (ENSG00000016402), AP1S1 (ENSG00000106367), HSDL1 (ENSG00000103160), PRPH2 (ENSG00000112619), RCBTB2 (ENSG00000136161), FGF14 (ENSG00000102466), RRAS (ENSG00000126458), PPP2CA (ENSG00000113575), ASPHD2 (ENSG00000128203), DCBLD2 (ENSG00000057019), HLTF (ENSG00000071794), LRRC4B (ENSG00000131409), CUL2 (ENSG00000108094), SKAP2 (ENSG00000005020), PNO1 (ENSG00000115946), MRPL44 (ENSG00000135900), KDM5A (ENSG00000073614), MAG (ENSG00000105695), SORT1 (ENSG00000134243), CSDE1 (ENSG00000009307), EHD3 (ENSG00000013016) |
| 1 | CEMIP2 (ENSG00000135048), PGLYRP1 (ENSG00000008438), BAIAP3 (ENSG00000007516), SP100 (ENSG00000067066), TSPAN32 (ENSG00000064201), SLC4A11 (ENSG00000088836), BPIFB9P (ENSG00000125997), CLNK (ENSG00000109684), BCKDK (ENSG00000103507), P3H3 (ENSG00000110811), WIPI1 (ENSG00000070540), TPSD1 (ENSG00000095917), SLC7A9 (ENSG00000021488), SLC8A2 (ENSG00000118160), AQP6 (ENSG00000086159), CAP2 (ENSG00000112186), EBF3 (ENSG00000108001), ADGRE2 (ENSG00000127507), BYSL (ENSG00000112578), UTP18 (ENSG00000011260), TSGA10 (ENSG00000135951), TREM1 (ENSG00000124731), GSKIP (ENSG00000100744), PHF20 (ENSG00000025293), ARFGEF2 (ENSG00000124198), MXD1 (ENSG00000059728), NSFL1C (ENSG00000088833), LSR (ENSG00000105699), DDX39A (ENSG00000123136) |
| 2 | APOA1 (ENSG00000118137), KCNIP3 (ENSG00000115041), SLC35F5 (ENSG00000115084), COX6A1 (ENSG00000111775), MCM8 (ENSG00000125885), KHDRBS3 (ENSG00000131773), POLR3H (ENSG00000100413), IFT22 (ENSG00000128581), PLA2G12A (ENSG00000123739), POLQ (ENSG00000051341), CLEC4M (ENSG00000104938), TENM1 (ENSG00000009694), CEP250 (ENSG00000126001), MFSD11 (ENSG00000092931), C22orf23 (ENSG00000128346), GALNT15 (ENSG00000131386), DYNLRB1 (ENSG00000125971), BRD1 (ENSG00000100425), CCNP (ENSG00000105219), PRMT7 (ENSG00000132600), HOXC8 (ENSG00000037965), CDKL5 (ENSG00000008086), G6PC1 (ENSG00000131482), TRHDE (ENSG00000072657), NDC80 (ENSG00000080986), NDUFS7 (ENSG00000115286), TNNT3 (ENSG00000130595), SLC9A2 (ENSG00000115616), DNAAF11 (ENSG00000129295), EHHADH (ENSG00000113790), SMS (ENSG00000102172), CCDC88C (ENSG00000015133), LLGL2 (ENSG00000073350) |
Gene communities identified through network analysis in the ARPKD samples were categorized as communities 0, 1, and 2. Each community comprises distinct genes associated with specific biological functions relevant to ARPKD pathology, including cellular regulation, immune responses, and metabolic pathways. The table lists the gene IDs and corresponding proteins within each community, emphasizing their potential roles in ARPKD progression.
Community 0 includes PMPCB (mitochondrial processing peptidase beta subunit), KLF12 (Kruppel-like factor 12)25, RBM3 (RNA-binding motif protein 3)26, SUMF2 (sulfatase modifying factor 2)27, MCHR1 (melanin-concentrating hormone receptor 1)28, and RGS13 (regulator of G-protein signaling 13)29. This community is linked to transcriptional regulation, cellular stress response, and signal transduction.
Community 1 contains TPSD1 (tryptase delta 1), CEMIP2 (cell migration-inducing hyaluronidase 2)30, TREM1 (triggering receptor expressed on myeloid cells 1), and SLC4A11 (solute carrier family 4 member 11)31, which are involved in immune responses and inflammation. The presence of cytokine signaling and immune receptor genes suggests a role in immune regulation.
Community 2 includes IFT22, CEP250 (centrosomal protein 250 kDa), DYNLRB1 (dynein light chain roadblock-type 1), COX6A1 (cytochrome c oxidase subunit 6A1)32, NDUFS7 (NADH oxidoreductase subunit S7)33, POLQ (DNA polymerase theta), MCM8 (minichromosome maintenance complex component 8), CLEC4M (C-type lectin domain family 4 member M), and PLA2G12A (phospholipase A2 group XIIA)34. These genes are involved in intracellular trafficking, protein modification, and metabolism.
Figure 1 visualizes the network topology, while Table 2 details representative genes in each community. This analysis provides a framework for understanding gene connectivity in ARPKD, though further validation is needed to determine the functional significance of these communities.
Classification of gene communities in the ARPKD network
This study’s method determines whether additional genes belong to Community 0, 1, or 2.
Under the applied correlation threshold, none of the genes listed in Table 1 were assigned to any community. However, when genes with weaker correlations were included, ADGRB2 was assigned to Community 2, while LRP4, ZMYND10, MYB, and KIF1A were classified into Community 1. Similarly, LOXL1, APBA2, and BICDL1 were assigned to Community 0. These genes exhibited weak correlations with other members, suggesting limited but measurable network connectivity. Additionally, ADAM19 and ADAMTS7 were assigned to Community 1, reflecting weak correlations indicative of interactions at the network level.
Gene network analysis was performed using a publicly available dataset from Goggolidou et al. Although this dataset was limited in scope, it included WNT5A, CDH1, and FZD10, which are key genes in ARPKD. These genes were not classified into any community in ARPKD.
Regarding PKD1, which is also important in ARPKD, its expression was increased in ARPKD but did not correlate with genes in Community 0 and showed only weak correlations with some genes in Communities 1 and 235,36.
Further details are provided in Supplementary Table 1.
Gene network analysis and functional organization in control (normal) individuals
The gene interaction network in healthy individuals was analyzed to identify distinct gene communities and their organizational structures in normal kidney tissues. Figure 2 visualizes gene connectivity, showing how genes cluster into functional groups.
Fig. 2.

Gene network topology in healthy control individuals. This figure shows the gene network in healthy control individuals, with each node representing a gene and each edge representing a gene pair with a correlation of 0.6 or higher. The edges are fully transparent to focus on the structure and prominence of individual nodes. The size of each node is proportional to the number of other genes and is strongly correlated with, emphasizing the hub genes that are central to the network. Different colors indicate clusters detected by the Louvain method, with each color representing a distinct community of genes: Community 3 is shown in purple, Community 4 in green, and Community 5 in yellow. Genes within each community exhibit strong mutual interactions, illustrating the structure of gene relationships based on correlations in healthy kidney function. These clusters highlight potential functional groupings of genes, with central genes in each community likely playing key roles in maintaining normal cellular processes in the kidneys.
Each community contained genes with strong correlation-based associations, reflecting coordinated biological functions. Central genes were identified based on their connectivity patterns, indicating their potential role as key regulators in maintaining homeostasis. Notably, genes with high betweenness centrality serve as communication hubs, facilitating interactions between different functional groups.
The identified network structure in healthy individuals serves as a reference for comparison with disease states. Understanding how genes naturally organize within a stable physiological system provides insight into how gene interactions may be disrupted in pathological conditions. Future studies examining network perturbations in ARPKD may help pinpoint key mechanisms underlying disease progression.
In the gene network analysis of normal kidney tissues, key genes with high betweenness centrality were identified within each community. These central genes function as hubs that integrate essential biological processes, contributing to homeostasis and normal kidney function.
The results demonstrated that SMS (spermine synthase) in Community 3 is involved in polyamine biosynthesis, a process essential for cellular growth and stability37. In Community 4, CLEC4M (C-type lectin domain family 4 member M) was identified as a regulator of immune response, facilitating pathogen recognition and contributing to infection defense38. CCNP (cyclin C-like nucleolar protein) in Community 5 was associated with cell cycle regulation, which supports tissue repair and regeneration39.
Through their roles in metabolic, immune, and proliferative pathways, these genes contribute to the functional organization of the normal kidney gene network. Their identification provides a reference for understanding potential alterations in gene interactions in pathological states such as ARPKD40–42.
Based on the network analysis of normal kidney samples, three distinct gene communities (Communities 3, 4, and 5) were identified as functional hubs contributing to kidney homeostasis.
Community 3 includes IFT22, CEP250 (centrosomal protein 250 kDa), COX6A1 (cytochrome c oxidase subunit 6A1), NDUFS7 (NADH oxidoreductase subunit S7), and LLGL2 (lethal giant larvae homolog 2). These genes are involved in intracellular trafficking, mitochondrial function, and protein modification, indicating their roles in maintaining metabolic balance and cellular homeostasis.
Community 4 is characterized by APOA1 (apolipoprotein A1)43, KCNIP3 (potassium voltage-gated channel interacting protein 3)44, CEMIP2 (cell migration-inducing hyaluronidase 2)45, and CLEC4M (C-type lectin domain family 4 member M)46. These genes participate in lipid metabolism, immune regulation, and cellular stress responses, reflecting their involvement in immune balance and renal protection.
Community 5 features POPDC3 (Popeye domain-containing protein 3)47, COX6A1 (cytochrome c oxidase subunit 6A1)33, KLF12 (Kruppel-like factor 12)25, and CCNP (cyclin C-like nucleolar protein)48. These genes are associated with cellular growth, tissue repair, and metabolic regulation, supporting kidney cell function under normal conditions.
In healthy kidney samples, PKD1, ADGRB2, LRP4, and KIF1A did not exhibit strong or weak correlations within the normal gene network. This suggests that these genes are not functionally co-regulated in healthy kidney tissues and that the altered connectivity observed in ARPKD reflects disease-specific interactions.
Additionally, WNT5A, CDH1, and FZD10, which were part of Community 0 in healthy controls, were not classified into any community in ARPKD. This suggests that their network associations may have been disrupted in the disease state, as detailed in Supplementary Table 1. These genes are involved in Wnt/PCP signaling and cell adhesion, both of which play essential roles in maintaining kidney homeostasis and may contribute to ARPKD pathogenesis when dysregulated1,9.
Table 3 provides an overview of gene composition and connectivity within these communities, emphasizing their contributions to normal kidney function.
Table 3.
Gene communities and their associated genes identified in normal kidney samples through network analysis.
| Community | Gene symbol (Ensembl ID) |
|---|---|
| 3 | SLC35F5 (ENSG00000115084), KHDRBS3 (ENSG00000131773), MCHR1 (ENSG00000128285), BAIAP3 (ENSG00000007516), LACTB (ENSG00000103642), IFT22 (ENSG00000128581), PMPCB (ENSG00000105819), SUCO (ENSG00000094975), GLRB (ENSG00000109738), AP1S1 (ENSG00000106367), HSDL1 (ENSG00000103160), POLQ (ENSG00000051341), PRPH2 (ENSG00000112619), MRPL44 (ENSG00000135900), CAP2 (ENSG00000112186), EHHADH (ENSG00000113790), SMS (ENSG00000102172), MXD1 (ENSG00000059728) |
| 4 | APOA1 (ENSG00000118137), KCNIP3 (ENSG00000115041), CEMIP2 (ENSG00000135048), PGLYRP1 (ENSG00000008438), MCM8 (ENSG00000125885), TGM5 (ENSG00000104055), POLR3H (ENSG00000100413), HOMER2 (ENSG00000103942), TSPAN32 (ENSG00000064201), BPIFB9P (ENSG00000125997), IL20RA (ENSG00000016402), CLNK (ENSG00000109684), CLEC4M (ENSG00000104938), TENM1 (ENSG00000009694), BCKDK (ENSG00000103507), CEP250 (ENSG00000126001), MFSD11 (ENSG00000092931), P3H3 (ENSG00000110811), C22orf23 (ENSG00000128346), SLC7A9 (ENSG00000021488), BRD1 (ENSG00000100425), PRMT7 (ENSG00000132600), HOXC8 (ENSG00000037965), MAG (ENSG00000105695), ADGRE2 (ENSG00000127507), G6PC1 (ENSG00000131482), TRHDE (ENSG00000072657), TNNT3 (ENSG00000130595), DNAAF11 (ENSG00000129295), LLGL2 (ENSG00000073350), EHD3 (ENSG00000013016), DDX39A (ENSG00000123136) |
| 5 | POPDC3 (ENSG00000132429), COX6A1 (ENSG00000111775), KLF12 (ENSG00000118922), RBM3 (ENSG00000102317), SUMF2 (ENSG00000129103), SP100 (ENSG00000067066), PUS7L (ENSG00000129317), SLC4A11 (ENSG00000088836), DIMT1 (ENSG00000086189), CREBL2 (ENSG00000111269), PLA2G12A (ENSG00000123739), RCBTB2 (ENSG00000136161), FGF14 (ENSG00000102466), RRAS (ENSG00000126458), GALNT15 (ENSG00000131386), PPP2CA (ENSG00000113575), ASPHD2 (ENSG00000128203), DCBLD2 (ENSG00000057019), DYNLRB1 (ENSG00000125971), HLTF (ENSG00000071794), LRRC4B (ENSG00000131409), CUL2 (ENSG00000108094), SKAP2 (ENSG00000005020), WIPI1 (ENSG00000070540), PNO1 (ENSG00000115946), SLC8A2 (ENSG00000118160), CCNP (ENSG00000105219), AQP6 (ENSG00000086159), KDM5A (ENSG00000073614), CDKL5 (ENSG00000008086), EBF3 (ENSG00000108001), BYSL (ENSG00000112578), UTP18 (ENSG00000011260), NDC80 (ENSG00000080986), SORT1 (ENSG00000134243), NDUFS7 (ENSG00000115286), TSGA10 (ENSG00000135951), SLC9A2 (ENSG00000115616), CSDE1 (ENSG00000009307), TREM1 (ENSG00000124731), GSKIP (ENSG00000100744), PHF20 (ENSG00000025293), ARFGEF2 (ENSG00000124198), CCDC88C (ENSG00000015133), NSFL1C (ENSG00000088833), LSR (ENSG00000105699) |
The gene communities identified through network analysis in normal kidney samples, categorized as Communities 3, 4, and 5. Each community comprises distinct genes associated with specific biological functions that are essential for maintaining normal kidney physiology, including structural integrity, immune balance, and metabolic pathways. This table lists the gene IDs and corresponding proteins within each community, highlighting their potential roles in normal kidney function.
Single sample gene set enrichment analysis
Table 4 presents the application of single-sample Gene Set Enrichment Analysis (ssGSEA) to gene communities identified in ARPKD patients and mapped to healthy samples. The analysis examined Communities 0, 1, and 2 in the control group and Communities 3, 4, and 5 in ARPKD patients. For each community, the mean enrichment score (ES_mean) and normalized enrichment score (NES_mean), along with their standard deviations (ES_std and NES_std), were calculated to assess activation trends in both groups.
Table 4.
Comparison of community activation in control and ARPKD samples via ssGSEA analysis.
| Term | Group | ES_mean | ES_std | NES_mean | NES_std |
|---|---|---|---|---|---|
| Community_0 | Control | 151.67 | 235.40 | 0.170 | 0.264 |
| Community_1 | Control | 182.25 | 361.60 | 0.204 | 0.405 |
| Community_2 | Control | 511.60 | 158.94 | 0.573 | 0.178 |
| Community_3 | ARPKD | − 283.63 | 94.42 | − 0.293 | 0.097 |
| Community_4 | ARPKD | 99.99 | 150.57 | 0.103 | 0.155 |
| Community_5 | ARPKD | 458.04 | 68.47 | 0.473 | 0.071 |
Comparison of community activation between control (healthy) and ARPKD patients. For each community, the mean Enrichment Score (ES_mean) and standard deviation (ES_std) indicate the activation level and variability within the gene set, while the mean Normalized Enrichment Score (NES_mean) and standard deviation (NES_std) were adjusted for gene set size to facilitate community comparisons. Positive scores indicate activation, whereas negative scores indicate suppression. Control community structures (Communities 0, 1, 2) are applied to ARPKD samples, and ARPKD community structures (Communities 3, 4, 5) are applied to control samples.
Community 0 exhibited an ES_mean of 151.67, indicating a slight positive activation in healthy samples, with an NES_mean of 0.170, suggesting a modest activation trend relative to other communities. The relatively large standard deviations (ES_std of 235.40 and NES_std of 0.264) suggest variability across samples, potentially reflecting heterogeneity in activation levels within this community in healthy individuals.
Community 1 exhibited an ES_mean of 182.25, indicating mild activation in healthy samples, with an NES_mean of 0.204. However, the high standard deviations (ES_std of 361.60 and NES_std of 0.405) reflect substantial inter-sample variability, suggesting that some healthy samples exhibit particularly low activation in this community.
Community 2 displayed the highest activation level, with an ES_mean of 511.60 and an NES_mean of 0.573, indicating robust and consistent activation in healthy samples. The lower standard deviations (ES_std of 158.94 and NES_std of 0.178) compared to other communities suggest that this activation trend is stable across samples, highlighting the potential significance of Community 2 in maintaining normal kidney function.
In comparing healthy and ARPKD samples, Community 2 exhibited the highest activation in both groups, suggesting its involvement in fundamental physiological processes or homeostasis. If activation in ARPKD samples is elevated, it may reflect disease-related alterations rather than normal physiological regulation. Conversely, a decrease in activation could indicate disruption in key pathways associated with disease progression.
Community 0 and Community 1 exhibit only mild activation in healthy samples, suggesting that abnormal activation or suppression in ARPKD samples could indicate their involvement in disease-related processes.
Community 3: The mean enrichment score (ES_mean) was -283.63, with a normalized enrichment score mean (NES_mean) of -0.293, suggesting reduced activation in ARPKD patients. The ES standard deviation (ES_std) was 94.42, indicating significant variability across samples and suggesting individual differences in Community 3 activation among ARPKD patients.
Community 4: The ES_mean was 99.99, while the NES_mean was 0.103, indicating mild positive activation. High standard deviations (ES_std of 150.57 and NES_std of 0.155) suggest considerable variation in activation across samples, implying that this community is either not directly involved in ARPKD or only minimally affected.
Community 5: With an ES_mean of 458.04, Community 5 exhibited the highest positive score, and the NES_mean was 0.473, indicating stronger activation in ARPKD samples. The standard deviations were relatively low (ES_std of 68.47 and NES_std of 0.071), suggesting consistent activation across samples.
These results suggest that Community 5 is consistently activated in ARPKD patients, potentially reflecting its involvement in disease-related pathways. Conversely, Community 3 appears to be suppressed, which may indicate a regulatory response to disease or a loss of function in ARPKD.
Discussion
This study proposes a pilot methodological approach for analyzing gene networks in ARPKD using a random subset of 100 genes. By integrating ssGSEA and topological centrality analysis, this approach identifies gene community structures that conventional DEG analysis may overlook. This provides a framework for exploring potential gene interactions and guiding future network-based studies.
First, RNA overexpression was observed in both ARPKD and healthy samples, suggesting the possible involvement of compensatory mechanisms in ARPKD5. Community 2, identified in ARPKD samples, also exhibited high activation in healthy samples, indicating a potential role in kidney function maintenance. The hub gene IFT22 supports ciliary function and may contribute to compensatory responses in ARPKD22,23. Additionally, abnormalities in the Hedgehog, Wnt, and Notch pathways have been linked to ARPKD, with Hedgehog signaling as a proposed therapeutic target9. This community includes APOA1, PLA2G12A, MCM8, POLQ, CEP250, and HOXC8, which may be involved in inflammation regulation, DNA repair, ciliary maintenance, and tissue remodeling35,43,49–51. Conversely, Community 5 was predominantly activated in ARPKD samples and may reflect both disease-related changes and potential compensatory mechanisms. It includes CCNP, TREM1, PLA2G12A, COX6A1, and RRAS, which may be involved in cell cycle regulation, immune responses, and metabolic adaptation30,39,52–54. Further studies are needed to determine whether these genes have a direct role in disease progression or reflect secondary effects.
Second, genes upregulated in ARPKD but not in healthy controls may contribute to disease pathology. Community 0 analysis identified PMPCB and MRPL44, involved in mitochondrial protein processing, whose dysfunction may lead to oxidative stress and impaired ATP production, exacerbating cellular stress responses24,33. KLF12 may be activated under such conditions, potentially influencing cell growth and tissue remodeling25. Overexpression of IL20RA and CREBL2 could sustain chronic inflammation, which may contribute to renal damage55–57. Community 1 analysis highlighted TPSD1 as a hub gene potentially involved in renal tissue damage through inflammatory pathways, along with CEMIP2 and TREM1, which may sustain inflammation and promote kidney remodeling30. Abnormal ion channel genes, including SLC4A11 and SLC8A2, could disrupt osmotic regulation and contribute to cyst formation31. Excessive activation of AQP6 may further accelerate fluid transport abnormalities in ARPKD58,59.
Third, gene communities suppressed in ARPKD may indicate a loss of functions essential for kidney homeostasis. Community 3 was downregulated, suggesting a role in structural integrity. The hub gene SMS, essential for spermine synthesis, is involved in DNA stability and cell growth, and its suppression may weaken kidney tissue and be associated with disease progression2,35. Additionally, PMPCB, MRPL44, LACTB, and IFT22 may contribute to impaired mitochondrial support, disrupted cell growth, and metabolic dysfunction in ARPKD2,22–24,60.
Fourth, to place our findings in a broader context, we compared them with previous analyses by Richards et al. (2019, 2024). Gene communities were identified using a randomly selected set of 100 genes, but additional genes were evaluated based on their correlation with existing communities to infer their potential network associations.
Examining the network positions of WNT5A, CDH1, and FZD10 allowed us to compare our results with prior studies and assess structural changes in gene communities associated with ARPKD pathophysiology.
Our analysis revealed that WNT5A, CDH1, and FZD10 were part of Community 0 in healthy individuals but were not classified into any community in ARPKD. This suggests that these genes were components of a network involved in transcriptional regulation, cellular stress response, and signal transduction. However, in ARPKD patients, their network integration was lost, implying potential changes in their functional roles. Whether these alterations arise due to disease progression or reflect primary pathogenic abnormalities remains uncertain.
WNT5A is a key component of the non-canonical Wnt/PCP pathway. Richards et al. (2019) reported that excessive activation of WNT5A in ARPKD may contribute to aberrant Wnt/PCP signaling, potentially influencing disease progression1. Additionally, Richards et al. (2024) demonstrated that variations in WNT5A expression in ARPKD patients are associated with abnormalities in Wnt signaling. Our findings suggest that while WNT5A plays a role in transcriptional regulation and cellular response in healthy individuals, its functional role may shift in ARPKD.
CDH1 (E-cadherin) is crucial for maintaining cell–cell adhesion. Previous studies reported that increased CDH1 expression in ARPKD kidneys does not necessarily enhance cell adhesion but may instead contribute to abnormal cell polarity and adhesion defects1. Our results indicate that while CDH1 is associated with maintaining adhesion homeostasis in healthy kidneys, its role may change during ARPKD progression.
FZD10 (Frizzled class receptor 10) functions as a Wnt signaling receptor involved in cell proliferation, differentiation, and polarity control. Previous studies have indicated that FZD10 is differentially expressed in ARPKD, potentially contributing to disease progression through altered Wnt signaling9. In our study, FZD10 was part of a transcriptional regulation and cellular response network in healthy individuals, but its function appeared to be altered in ARPKD.
Furthermore, conventional gene expression analysis using DESeq2 identified genes that, despite being highly expressed in ARPKD, exhibited weak correlations (0.3–0.5) with major gene communities. These weak associations may reflect compensatory mechanisms or network reorganization in ARPKD, but further validation is required to determine their biological significance.
A detailed comparison with traditional differential gene expression (DEG) analysis and an expanded evaluation of genes, including PKD1, LOXL1, KIF1A, ADGRB2, LRP4, and APBA2, are provided in Supplementary Information61–63. In particular, while our method does not determine whether PKD1 is mathematically isolated, this finding may carry significant implications35,36.
Fifth, this study has several limitations. An important limitation is the small sample size (n = 4 per group), which restricts the generalizability of our findings and may be influenced by inter-sample variability. The cross-sectional design restricts the ability to determine whether gene expression changes are primary contributors to ARPKD progression or secondary responses to the disease. Longitudinal studies with larger cohorts are needed to clarify the causal relationship between these alterations and disease progression3.
While ssGSEA and topological centrality analysis provide valuable insights into gene community structures, they do not account for other critical aspects of gene regulation, such as epigenetic modifications, post-transcriptional regulation, or protein-level interactions, which may also influence ARPKD pathology11. Functional validation using cellular and animal models is essential to confirm the biological significance of these gene communities and assess their potential as therapeutic targets50.
Due to computational constraints, this study analyzed a reproducible random subset of 100 genes rather than a systematically selected biologically relevant set. Methods such as Deterministic Column Subset Selection or Hierarchical Representative Set Selection, which could improve biological relevance, were not applied. Future studies should incorporate these approaches to refine gene selection and enhance network analysis.
Despite these limitations, this study provides a useful framework for analyzing gene networks associated with ARPKD and establishes a foundation for further research and therapeutic development. Integrating both gene-specific analyses and network-based approaches will be essential to deepen the understanding of ARPKD pathogenesis. Functional validation of gene communities through experimental models and iterative incorporation of newly identified genes into TDA-based analyses will improve the precision of disease modeling. These efforts will contribute to a more comprehensive understanding of ARPKD pathophysiology and support the development of novel therapeutic strategies.
Methods
Ethics
This investigation utilized publicly available data from the National Center for Biotechnology Information (NCBI), and approval from the Yokosuka Urogynecology and Urology Clinic Ethics Committee was not required.
Sample and data usage
In this study, a transcriptomic analysis of ARPKD was conducted using a publicly available dataset from NCBI GEO. This dataset was generated and provided by Goggolidou et al. (GEO accession number: GSE242476), comprising four kidney samples from ARPKD patients and four age-matched healthy kidney samples1,9,10. An independent analytical approach was employed to examine this dataset to provide additional insights and compare the findings with those presented in the original publication1,9.
RNA extraction, library preparation, and sequencing
In this study, no genetic manipulation was conducted, and the protocols for RNA extraction and sequencing adhered to those established in the original dataset by Goggolidou et al. The specimens were obtained from the ARPKD Biobank at University College London, RNA extraction was performed using the Qiagen RNeasy Mini Kit, and sequencing was performed on the Illumina NovaSeq 6000 platform.
Read processing and alignment
Post-sequencing, raw reads were processed for quality control and adapter trimming using CutAdapt. The cleaned reads were then aligned to the GRCh38 reference genome using HISAT2 to ensure accurate mapping of transcriptomic sequences.
Gene quantification and differential expression analysis
Gene-level expression counts were obtained using StringTie. Differential expression analysis was conducted using DESeq214–18, applying an adjusted p-value threshold of < 0.05 to identify significantly differentially expressed genes. Among the identified genes, PKD1, associated with autosomal dominant polycystic kidney disease (ADPKD), exhibited significantly higher expression in ARPKD samples compared to controls. Other differentially expressed genes included ADGRB2, which is involved in cellular adhesion, LRP4, a key regulator of Wnt signaling, and KIF1A, an essential component of intracellular transport.
Functional enrichment analysis
To explore the biological relevance of differentially expressed genes, functional enrichment analysis was performed using GOSeq and Revigo19,20, accessed via Galaxy.org, with the exception of Revigo21. Enrichment results highlighted pathways associated with cystogenesis, cellular adhesion, and immune response, which are known to be implicated in ARPKD pathophysiology.
Data processing and analysis
The sequence data were analyzed in their original form without any genetic manipulation. Using the StringTie and DESeq2 packages, the data were re-analyzed with an independent analytical approach. To enable direct comparison with the original study, the same reference genome (GRCh38) and aligner (HISAT2) were used for differential expression analysis.
Data citation
This dataset was made publicly available as part of the study by Goggolidou et al. and uploaded to the NCBI GEO database for utilization by researchers. In this study, the data provided by the authors were used to generate new insights based on an independent analytical approach, contributing to an enhanced understanding of gene expression in ARPKD1,9,10.
Data preparation
Two datasets were obtained: one representing gene expression in ARPKD (abnormal) samples and the other in control (normal) samples. Both datasets contained gene expression levels across four samples. The column names were standardized to facilitate merging and comparison, and the datasets were subsequently merged based on common gene identifiers (Gene_ID).
Statistical testing
For each gene, a paired t-test was performed between the ARPKD and Control expression levels across the four samples. This test was chosen because it assesses whether there is a statistically significant difference in the expression of each gene between ARPKD and Control groups. A p-value threshold of 0.05 was applied to identify genes with significant differences in expression.
Data loading and preprocessing
Gene expression data are provided in a CSV file, with rows representing gene names and columns representing individual samples. These data were loaded using Python’s `pandas` library. Only numeric data columns were extracted to focus on the gene expression levels. To improve the computational efficiency, a subset of 100 genes was randomly selected for analysis. If the total number of genes was less than 100, all genes were used. Random sampling was performed to retain a representative set of genes for correlation analysis while maintaining manageable computational requirements.
Constructing the gene network
Given the computational constraints associated with analyzing all gene pairs in large-scale RNA-seq datasets, a reproducible random subset of 100 genes was selected to ensure consistency across analyses23,24. This selection was performed using a fixed random seed to allow reproducibility. While this approach reduces computational burden and maintains network integrity, it does not prioritize biological relevance. In future studies, deterministic selection methods such as DSSC (Deterministic Column Subset Selection) or HRSS (Hierarchical Representative Set Selection) may be implemented to improve the biological relevance of selected genes while maintaining computational feasibility20,21,64–66.
A correlation matrix was computed for the selected subset of genes using the transpose of the data to assess gene-to-gene correlations. This matrix displays the correlation coefficient for each gene pair, with higher values indicating stronger co-expression. An edge threshold of 0.6 was applied, meaning that only gene pairs with a correlation of 0.6 or above were considered strongly related and were retained as edges in the network. Using the `NetworkX` library, a network graph was built where each gene was represented as a node and an edge connected to any two genes with a correlation meeting or exceeding the threshold12. Next, to focus on the most interconnected structure, only the largest connected component of the graph is retained, removing any isolated nodes. This refined graph was visualized with `matplotlib,’ using a layout algorithm (`spring_layout`) to place closely correlated genes near one another for clearer identification of clusters11,12.
To ensure statistical rigor, Pearson’s correlation coefficients were used to quantify pairwise gene co-expression.
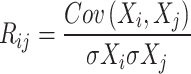 |
where Xi and Xj represent the expression profiles of genes i and j, and σX denotes the standard deviation. Edge retention was determined based on two criteria: ∣Rij∣exceeding a threshold T = 0.6 and statistical significance (pij < 0.05), ensuring that only robust co-expression relationships were included in the network.
Sensitivity analysis method
To evaluate the robustness of the chosen correlation coefficient threshold (0.6), a sensitivity analysis was performed by varying the threshold across a range of values (0.1,0.2,0.3, 0.4, 0.5, 0.6, 0.7, 0.8 and 0.9). For each threshold, a gene correlation network was constructed using only gene pairs with correlations above a given threshold. Network topology was analyzed in terms of the number of nodes, edges, and communities detected using the Louvain method. The results were compared to assess the impact of the threshold on network structure and community detection.
Community detection and centrality calculation
The Louvain method was applied using the Community Louvain module to identify gene clusters within the network12. This method groups genes into communities, where genes within a community have stronger connections with each other than with those in other communities. Next, betweenness centrality was calculated separately for each community to identify genes acting as central 'hubs' within their respective groups. The gene with the highest centrality score in each community was designated as the 'central gene,' suggesting its potential key role in regulating interactions within that community. These central genes are then listed in a summary table, showing their community assignment and centrality scores, providing insights into genes that may significantly influence ARPKD-specific gene expression networks.
Definition of disease-related gene communities
Gene communities were identified from ARPKD patient data based on prior network analyses that grouped genes into distinct functional communities: Community_0, Community_1, and Community_2. Each community consisted of genes with related functions and interaction patterns that were used as gene sets for ssGSEA analysis. The list of genes within each community was curated and labeled as "Community_0," "Community_1," and "Community_2" in the gene set definitions.
Application of ssGSEA analysis to healthy sample data
Gene expression data from the healthy control samples were collected and uploaded. Each column in the dataset was treated as an independent sample, with rows representing the expression levels of different genes11. Predefined gene sets representing the ARPKD communities were used to conduct ssGSEA on healthy sample data11. This analysis produced two scores for each gene community for each sample.
Enrichment Score (ES): A score reflecting the cumulative level of activation or repression of genes within a community.
Normalized Enrichment Score (NES): An adjusted score that accounts for differences in gene set size, enabling comparisons across communities.
Calculation and comparison of activation scores
The results of the ssGSEA were averaged across samples to calculate the mean and standard deviation for both ES and NES within each community. The mean scores indicate the overall activation trend in each community, whereas the standard deviations reflect the variability of activation across different samples11. These baseline values for healthy samples served as a reference point for comparing activation patterns in ARPKD patients.
Interpretation and comparison with disease states
The mean ES and NES values, along with their standard deviations, were analyzed to understand the baseline activation of ARPKD-related gene communities in healthy individuals. Comparing these scores to those obtained from ARPKD patients allowed us to detect deviations in community activation that may be associated with disease pathology. Communities showing significantly altered activation in ARPKD patients ARPKD relative to the baseline observed in healthy controls are considered candidates for further investigation into their roles in disease progression.
Graph theoretical isolation and mathematical isolation
To analyze the mathematical isolation of PKD1, we represented the network as an undirected graph G = (V, E), where V represents nodes (genes) and E represents edges, indicating statistically significant pairwise correlations66,67. The adjacency matrix (A) for this graph is constructed such that:
 |
The connectivity of each node was determined by its degree, which was calculated as
 |
A node is mathematically isolated if its degree is zero
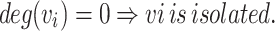 |
For PKD1, represented as node v₀, isolation was verified by checking if
 |
We further analyzed the network’s connectivity by constructing the graph Laplacian matrix (L), defined as
 |
Here, D is the degree matrix, which is a diagonal matrix where
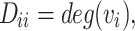 |
and A is the adjacency matrix. The eigenvalues of L, denoted as λ1, λ2, …, λn, reveal the network structure. The number of zero eigenvalues corresponds to the number of connected components in the graph67,68. An isolated node forms its own trivial connected component, defined as.
To construct the graph, pairwise correlations were calculated using Pearson’s correlation coefficient.
 |
where Xi and Xj represent the expression profiles of genes i and j, and σx denotes the standard deviation. Edges were retained if:
where Xi and Xj represent the expression profiles of genes i and j, and σx denotes the standard deviation67,68. Edges were retained if:
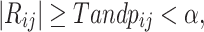 |
where T is the correlation threshold, and pij is the p-value associated with the correlation, with a significance level of α = 0.05.
Statistical analysis
Statistical analyses were performed using Google Colab (Google LLC, Mountain View, CA, USA). The Python code for the analysis was developed with the assistance of ChatGPT (OpenAI, San Francisco, CA, USA) and cross-checked using Claude 3 (Anthropic PBC, San Francisco, CA, USA) and Jimmniy (Grok Ventures Pty Ltd., Sydney, NSW, Australia). After the verification, the code was executed to obtain the results of this study. To ensure reproducibility, detailed Python code is publicly available in the "Code Availability section”. The analysis employed various scientific computing libraries, including pandas, numpy, and matplotlib, for data handling and visualization, and NetworkX for network-based analyses.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to express our sincere gratitude to Dr. P. Goggolidou, Dr. T. Richards, and Dr. P. Wilson for making their valuable ARPKD transcriptomic dataset (GSE242476) publicly available through the NCBI Gene Expression Omnibus, and for their groundbreaking research on WNT signaling in ARPKD published in Biochimica et Biophysica Acta—Molecular Basis of Disease (2024). Their work has provided crucial insights into ARPKD pathogenesis and has been fundamental to our network-based analysis. We also thank Mrs. Karen Okui for her expert assistance in reviewing and editing the English language presentation of this manuscript. Her bilingual expertise has significantly enhanced the clarity and accessibility of our work.
Author contributions
N.O. and S.H. wrote the initial manuscript draft. N.O. and T.H. designed the statistical analysis plan. N.O. developed the code and validated the data. All authors reviewed and approved the final manuscript.
Funding
This research received no specific grants from any funding agency in the public, commercial, or not-for-profit sectors.
Data availability
The transcriptomic data used in this study were obtained from the publicly available NCBI Gene Expression Omnibus (GEO) repository under accession number GSE242476 (Goggolidou et al., 2024). This dataset includes samples from ARPKD patients and age-matched healthy controls.
Code availability
The code used in this study is available at: https://github.com/Okuinobuo/ARPKD2024/.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Richards, T. et al. Atmin modulates Pkhd1 expression and may mediate autosomal recessive polycystic kidney disease (ARPKD) through altered non-canonical Wnt/planar cell polarity (PCP) signaling. Biochim. Biophys. Acta Mol. Basis Dis.1865, 378–390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song, X. et al. Reprogramming of energy metabolism in human PKD1 polycystic kidney disease: A systems biology analysis. Int. J. Mol. Sci.25, 7173 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nobakht, N. et al. Advances in autosomal dominant polycystic kidney disease: A clinical review. Kidney Med.2, 196–208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang, M. et al. Mechanical protein polycystin-1 directly regulates osteoclastogenesis and bone resorption. Sci. Bull.69, 1964–1979 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adamiok-Ostrowska, A. & Piekiełko-Witkowska, A. Ciliary genes in renal cystic diseases. Cells9, 907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devane, J. et al. Progressive liver, kidney, and heart degeneration in children and adults affected by TULP3 mutations. Am. J. Hum. Genet.109, 928–943 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgmaier, K. et al. Refining genotype-phenotype correlations in 304 patients with autosomal recessive polycystic kidney disease and PKHD1 gene variants. Kidney Int.100, 650–659 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Hsieh, C. L., Jerman, S. J. & Sun, Z. Non-cell-autonomous activation of hedgehog signaling contributes to disease progression in a mouse model of renal cystic ciliopathy. Hum. Mol. Genet.31, 4228–4240 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards, T. et al. Atmin modulates Pkhd1 expression and may mediate autosomal recessive polycystic kidney disease (ARPKD) through altered non-canonical Wnt/planar cell polarity (PCP) signalling. Biochim. Biophys. Acta Mol. Basis Dis.1865, 378–390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goggolidou, P., Richards, T., Wilson, P. Next Generation Sequencing Technologies to Investigate Autosomal Recessive Polycystic Kidney Disease (ARPKD). NCBI Gene Expression Omnibus (GSE242476). Available at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242476 (Accessed January 30, 2025).
- 11.Muley, V. Y. Centrality analysis of protein-protein interaction networks using R. Methods Mol. Biol.2690, 445–456 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol.11, R14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okui, N., Ikegami, T. & Okui, M. Topological data analysis of Ninjin’yoeito effects unraveling complex interconnections in patients with frailty: A pilot study. Cureus16, e74855 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okui, N. Innovative decision-making tools using discrete mathematics for stress urinary incontinence treatment. Sci. Rep.14, 9900 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J.17, 10–12 (2011). [Google Scholar]
- 16.Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yates, A. D. et al. Ensembl 2020. Nucleic Acids Res.48, D682–D688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol.20, 278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCurdy, S. R., Ntranos, V. & Pachter, L. Deterministic column subset selection for single-cell RNA-Seq. PLoS ONE14, e0210571 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tung, L. H. & Kingsford, C. Practical selection of representative sets of RNA-seq samples using a hierarchical approach. Bioinformatics37, i334–i341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko, J. Y. et al. Inactivation of max-interacting protein 1 induces renal cilia disassembly through reduction in levels of intraflagellar transport 20 in polycystic kidney. J. Biol. Chem.288, 6488–6497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wachter, S. et al. Binding of IFT22 to the intraflagellar transport complex is essential for flagellum assembly. EMBO J.38, e101251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeo, J. H. C. et al. A role for the mitochondrial protein Mrpl44 in maintaining OXPHOS capacity. PLoS ONE10, e0134326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang, C. X. et al. Krüppel-like factor 12 regulates aging ovarian granulosa cell apoptosis by repressing SPHK1 transcription and sphingosine-1-phosphate (S1P) production. J. Biol. Chem.299, 105126 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, J. et al. Cold-induced RNA-binding protein and RNA-binding motif protein 3: Two RNA molecular chaperones closely related to reproductive development and reproductive system diseases. Protein Pept. Lett.30, 2–12 (2023). [DOI] [PubMed] [Google Scholar]
- 27.Jarenbäck, L. et al. Single-nucleotide polymorphisms in the sulfatase-modifying factor 1 gene are associated with lung function and COPD. ERJ Open Res.8, 00668–02021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada, R., Michimae, M., Hamamoto, A. & Takemori, H. Melanin-concentrating hormone receptor 1 is discarded by exosomes after internalization. Biochem. Biophys. Res. Commun.710, 149917 (2024). [DOI] [PubMed] [Google Scholar]
- 29.Yuan, G. & Yang, S. Effect of regulator of G protein signaling proteins on bone. Front. Endocrinol.13, 842421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tammaro, A. et al. Role of TREM1-DAP12 in renal inflammation during obstructive nephropathy. PLoS ONE8, e82498 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panagopoulos, A. et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) inhibition in atherosclerosis. Pharmacol. Ther.238, 108182 (2022). [DOI] [PubMed] [Google Scholar]
- 32.Schmidt, T. R., Jaradat, S. A., Goodman, M., Lomax, M. I. & Grossman, L. I. Molecular evolution of cytochrome c oxidase: rate variation among subunit VIa isoforms. Mol. Biol. Evol.14, 595–601 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Xu, Y. et al. First-in-class NADH/ubiquinone oxidoreductase core subunit S7 (NDUFS7) antagonist for the treatment of pancreatic cancer. ACS Pharmacol. Transl. Sci.6, 1164–1181 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lutzmann, M. et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell.47, 523–534 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Engelhardt, B. E. & Stephens, M. Analysis of population structure: A unifying framework and novel methods based on sparse factor analysis. PLoS Genet.6, e1001117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olson, R. J. et al. Synergistic genetic interactions between Pkhd1 and Pkd1 result in an ARPKD-like phenotype in murine models. J. Am. Soc. Nephrol.30, 2113–2127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, L. et al. Spermine enhances antiviral and anticancer responses by stabilizing DNA binding with the DNA sensor cGAS. Immunity56, 272–288 (2023). [DOI] [PubMed] [Google Scholar]
- 38.Zeng, S. et al. Malassezia restricta promotes alcohol-induced liver injury. Hepatol. Commun.7, e0029 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sánchez-Botet, A. et al. Atypical cyclin P regulates cancer cell stemness through activation of the WNT pathway. Cell Oncol.44, 1273–1286 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao, M. et al. KHDRBS3 accelerates glycolysis and promotes malignancy of hepatocellular carcinoma via upregulating 14-3-3ζ. Cancer Cell Int.23, 244 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alsaif, H. S. et al. Homozygous loss-of-function mutations in AP1B1, encoding beta-1 subunit of adaptor-related protein complex 1, cause MEDNIK-like syndrome. Am. J. Hum. Genet.105, 1016–1022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, Y. et al. Expression profile of hydroxysteroid dehydrogenase-like 2 in polychaete Perinereis aibuhitensis in response to BPA. Life.13, 10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaziri, N. D. et al. Amelioration of nephropathy with apoA-1 mimetic peptide in apoE-deficient mice. Nephrol. Dial. Transplant.25, 3525–3534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong, J. et al. KCNIP3 silence promotes proliferation and epithelial-mesenchymal transition of papillary thyroid carcinoma through activating Wnt/β-catenin pathway. Tissue Cell.75, 101739 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Kianpour, M. et al. Aptamer/doxorubicin-conjugated nanoparticles target membranous CEMIP2 in colorectal cancer. Int. J. Biol. Macromol.245, 125510 (2023). [DOI] [PubMed] [Google Scholar]
- 46.Swystun, L. L., Michels, A. & Lillicrap, D. The contribution of the sinusoidal endothelial cell receptors CLEC4M, stabilin-2, and SCARA5 to VWF-FVIII clearance in thrombosis and hemostasis. J. Thromb. Haemost.21, 2007–2019 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun, C. C. et al. Progress on the study of Popeye domain-containing 3 (POPDC3) in malignancies and striated muscle function and homeostasis. Clin. Genet.103, 617–624 (2023). [DOI] [PubMed] [Google Scholar]
- 48.Gobbi, G. CCNP innovations in neuropsychopharmacology award: The psychopharmacology of psychedelics: Where the brain meets spirituality. J. Psychiatry Neurosci.49, E301–E318 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaufmann, W. K. Cell cycle checkpoints and DNA repair preserve the stability of the human genome. Cancer Metastasis Rev.14, 31–41 (1995). [DOI] [PubMed] [Google Scholar]
- 50.Cheng, T. et al. Inhibiting centrosome clustering reduces cystogenesis and improves kidney function in autosomal dominant polycystic kidney disease. JCI Insight.9, e172047 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cormier, S. A. & Kappen, C. Identification of a chondrocyte-specific enhancer in the Hoxc8 gene. J. Dev. Biol.12, 5 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gong, M. et al. Enhanced expression of CNTD2/CCNP predicts poor prognosis in bladder cancer based on the GSE13507. Front. Genet.12, 579900 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li, Q. et al. Impaired lipophagy-induced microglial lipid droplet accumulation contributes to the buildup of TREM1 in diabetes-associated cognitive impairment. Autophagy19, 2639–2656 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lobov, I. B. et al. The Dll4/Notch pathway controls postangiogenic blood vessel remodeling and regression by modulating vasoconstriction and blood flow. Blood117, 6728–6737 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Box, J. M., Higgins, M. E. & Stuart, R. A. Importance of conserved hydrophobic pocket region in yeast mitoribosomal mL44 protein for mitotranslation and transcript preference. J. Biol. Chem.300, 107519 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin, T.-Y. & Hsu, Y.-H. IL-20 in acute kidney injury: Role in pathogenesis and potential as a therapeutic target. Int. J. Mol. Sci.21, 1009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stenvinkel, P. et al. chronic inflammation in chronic kidney disease progression: Role of Nrf2. Kidney Int. Rep.6, 1775–1787 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gröger, N. et al. SLC4A11 prevents osmotic imbalance leading to corneal endothelial dystrophy, deafness, and polyuria. J. Biol. Chem.285, 14467–14474 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lan, Q. et al. Mechanistic complement of autosomal dominant polycystic kidney disease: The role of aquaporins. J. Mol. Med.102, 773–785 (2024). [DOI] [PubMed] [Google Scholar]
- 60.Yeo, J. H. et al. A role for the mitochondrial protein MRPL44 in maintaining OXPHOS capacity. PLoS ONE10, e0134326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergmann, C. Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front. Pediatr.5, 221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan, X. et al. Selective inhibition of hepatic stellate cell and fibroblast-derived LOXL1 attenuates BDL- and Mdr2(-/-)-induced cholestatic liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol.325, G608–G621 (2023). [DOI] [PubMed] [Google Scholar]
- 63.Chen, H., Li, J., Cao, D. & Tang, H. Construction of a prognostic model for hepatocellular carcinoma based on macrophage polarization-related genes. J. Hepatocell. Carcinoma11, 857–878 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su, K., Yu, T. & Wu, H. Accurate feature selection improves single-cell RNA-seq cell clustering. Brief. Bioinform.22, bbab034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng, X., Zhu, X., Wang, J. & Li, R. Comparison of gene selection methods for clustering single-cell RNA-seq data. Curr. Bioinform.17, 1–14 (2022). [Google Scholar]
- 66.Han, H. A novel feature selection for RNA-seq analysis. bioRxiv 209841 (2017). [DOI] [PubMed]
- 67.Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B57, 289–300 (1995). [Google Scholar]
- 68.Barabási, A.-L. Network Science (Cambridge University Press, 2016).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The transcriptomic data used in this study were obtained from the publicly available NCBI Gene Expression Omnibus (GEO) repository under accession number GSE242476 (Goggolidou et al., 2024). This dataset includes samples from ARPKD patients and age-matched healthy controls.
The code used in this study is available at: https://github.com/Okuinobuo/ARPKD2024/.