

Abstract
Structural, functional, and molecular‐level changes in the aging heart are influenced by a dynamic interplay between immune signaling and cellular metabolism that is referred to as immunometabolism. This review explores the crosstalk between cellular metabolic pathways including glycolysis, oxidative phosphorylation, fatty acid metabolism, and the immune processes that govern cardiac aging. With a rapidly aging population that coincides with increased cardiovascular risk and cancer incidence rates, understanding the immunometabolic underpinnings of cardiac aging provides a foundation for identifying therapeutic targets to mitigate cardiac dysfunction. Aging alters the immune environment of the heart by concomitantly driving the changes in immune cell metabolism, mitochondrial dysfunction, and redox signaling. Shifts in these metabolic pathways exacerbate inflammation and impair tissue repair, creating a vicious cycle that accelerates cardiac functional decline. Treatment with cancer therapy further complicates this landscape, as aging‐associated immunometabolic disruptions augment the susceptibility to cardiotoxicity. The current review highlights therapeutic strategies that target the immunometabolic axis to alleviate cardiac aging pathologies. Interventions include modulating metabolic intermediates, improving mitochondrial function, and leveraging immune signaling pathways to restore cardiac health. Advances in immunometabolism thus hold significant potential for translating preclinical findings into therapies that improve the quality of life for the aging population and underscore the need for approaches that address the immunometabolic mechanisms of cardiac aging, providing a framework for future research.
Keywords: cardiac aging, immunometabolism, inflammaging, oxidative phosphorylation
Subject Categories: Basic Science Research, Metabolism, Oxidant Stress, Aging
Nonstandard Abbreviations and Acronyms
- AS
aortic stenosis
- CANTOS
Canakinumab Anti‐inflammatory Thrombosis Outcome Study
- CoQ10
coenzyme Q10
- ECM
extracellular matrix
- EMBRACE
Evaluation of Myocardial Effects of MTP‐131 for Reducing Reperfusion Injury in Patients With Acute Coronary Events
- HSC
hematopoietic tem cell
- IMICA
Interleukin‐6 Inhibition for Modulating Inflammation After Cardiac Arrest
- MMP
matrix metalloproteinase
- MMPOWER
Safety, Tolerability, and Efficacy of MTP‐131 for the Treatment of Mitochondrial Myopathy
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- OXPHOS
oxidative phosphorylation
- PANDA
Protective Effect of CoQ10 Against Negative Inflammatory Response and Organ Dysfunction in Cardiovascular Surgery
- ROS
reactive oxygen species
- TGF‐β
transforming growth factor‐β
- Tregs
regulatory T cells
The term immunometabolism centers around the interplay between the disciplines of immunology and cellular metabolism. Although immunometabolism was described as an emerging frontier in 2011 by Mathis and Shoelson, 1 the foundation of the interaction goes back to the early 1900s. It is based on the preclinical and clinical observations that indicated the influence of systemic metabolism on the activation, function, and differentiation of the immune responses and vice versa. The scope of immunometabolism relies on the role of metabolic intermediates and regulators of pathways including glycolysis, oxidative phosphorylation (OXPHOS), amino acid and fatty acid metabolism, that is, ATP, nicotinamide adenine dinucleotide, and reactive oxygen species (ROS), that interact with immune signaling and function. 2 , 3 , 4 In the context of aging, endogenous alterations in metabolic pathways lead to a consequential impact on the immune system, a concept known as “inflammaging,” wherein chronic low‐grade inflammation due to biological aging and multiple comorbidities has metabolic implications. 5 , 6
The US population is rapidly aging, and the number of individuals aged ≥65 years will double between 2010 and 2030. 7 Coinciding with an aging population, US cancer incidence rates in these individuals are predicted to increase by 67% in the same time frame. 8 Among the ≈15.5 million cancer survivors in the United States in 2022, 67% of cancer survivors were aged ≥65 years. 9
As cancer survival rates improve, there is an increasing need to address chronic adverse sequelae affecting the quality of life of cancer survivors. Cardiotoxicity is defined as the occurrence of cardiac electrophysiology dysfunction or muscle damage encompassing numerous heterogeneous side effects including arrhythmias, changes in blood pressure, myocardial ischemia, thrombosis, and impairment in systolic or diastolic function. 10 As a well‐recognized side effect of cancer therapy, cardiotoxicity affects the structural, functional, and molecular aspects of the heart. The risk of cardiotoxicity due to conventional cancer therapies increases exponentially in older populations due to endogenous physiological and cellular changes including endothelial dysfunction, structural changes, mitochondrial dysfunction, immunosenescence, and inflammation as well as associated comorbidities. 11 Immunometabolic changes have been shown to negatively impact cardiac aging by contributing to pathologies including arrhythmias, heart failure, and atherosclerosis. 12 , 13 Targeting the metabolic pathways that regulate the remodeling of the aging heart thus presents a favorable avenue that can be strategically exploited to design therapeutic interventions that modulate the immune responses in various pathologies like cancer and chronic normal tissue damage due to cancer therapies. This review will focus on the various aspects of immunometabolism in the context of aging and its cardiac implications. Furthermore, it will provide recent insights into preclinical and therapeutic interventions currently utilized in immunometabolism.
Structural and Functional Landscape of the Aging Heart
Biological aging encompasses a progressive decline in the structural and functional aspects of cellular living systems and organs. A global decline in the aging heart often manifests as reduced diastolic function, increased left ventricular (LV) hypertrophy, atrial fibrillation, and increased susceptibility to infections due to immune dysfunction. While some of these changes are related to pathology or disease, many arise naturally as part of the endogenous aging process. These aging‐induced structural, functional, and molecular changes in the heart tissues can predispose older individuals to cancer therapy–induced normal tissue complications (Figure 1). This predisposition has been proposed to occur through aging‐related increases in mitochondrial dysfunction and DNA damage, 14 cellular senescence, 15 and endothelial dysfunction. 16 These immunometabolic alterations not only contribute to cardiac functional decline but also drive structural changes that compound diastolic dysfunction, leading to heart failure.
Figure 1. Landscape of the aging heart.
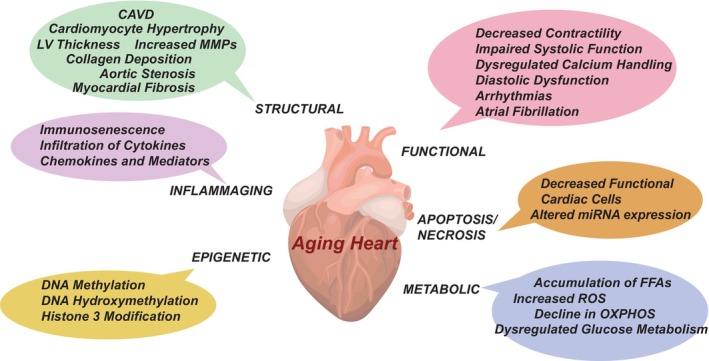
Various facets of cardiac aging include structural, functional, inflammaging, apoptosis/necrosis, epigenetic modification, and metabolic changes. CAVD indicates calcified aortic valve disease; FFA, free fatty acid; LV, left ventricular; OXPHOS, oxidative phosphorylation; and ROS, reactive oxygen species.
Morphological and structural changes in cardiac tissues include myocardial fibrosis, characterized by increased collagen deposition leading to increased myocardial stiffness and reduced conformity, which impairs diastolic function and contributes to heart failure. 17 , 18 Age‐associated cardiac hypertrophy is characterized by shifts in the immune cell subsets, particularly in the proinflammatory M1 macrophage subset in the ischemic heart tissue that consequently contributes to a chronic inflammatory state exacerbating cardiac dysfunction. 19 Age‐associated extracellular matrix (ECM), myocardial remodeling, and cardiac hypertrophy are structural hallmarks that serve as a segway for immunometabolism and are associated with increased collagen deposition and increased degradation capacity of the ECM because of increased expression of matrix metalloproteinases (MMPs) in cardiac fibroblasts, vascular smooth muscle cells, and leukocytes. 20 , 21 Several studies have indicated that ECM controls and guides the movement and positioning of immune cells. For example, T cells prefer sites containing thin ECM fibers compared with densely cross‐linked collagen matrices. 22 Studies have also shown that MMP‐9 triggers the activation of cytokines and chemokines, including interleukin‐1β, interleukin‐6, interleukin‐8, tumor necrosis factor‐α (TNF‐α), and transforming growth factor‐β (TGF‐β) by cleaving their inactive precursors into active forms or by binding to specific receptors, thereby regulating the fibrotic and inflammatory cascade during cardiac remodeling. 23 , 24 , 25 , 26 For example, MMP‐9 cleaves pro–interleukin‐1β to activate the inflammatory cytokine interleukin‐1β, enhancing the inflammatory response. 27 , 28 , 29 Likewise, MMP‐9–mediated cleavage has been shown to activate TGF‐β, contributing to increased collagen synthesis and tissue remodeling. 30 , 31 In addition to the ECM components, tissue remodeling also influences cellular responses in cardiac tissues.
Progressive cardiomyocyte hypertrophy is another hallmark of cardiac aging, resulting in the enlargement of myocytes as a response to the loss of myocytes and increased functional demands, reducing the compensatory capacity of the heart tissue. 32 ANP (atrial natriuretic peptide) and BNP (brain‐type natriuretic peptide) have been identified as markers of aging‐associated hypertrophy. 33 , 34 In addition to cardiomyocyte enlargement, increased LV thickness without any changes in LV mass due to aging can reduce the internal dimension and volume of diastole, altering the filling capacity. 35 Valvular degeneration, often indicated by calcification of the heart valves, especially the aortic valve, can lead to aortic stenosis (AS) and impaired valve function. 36 The Tromsø study demonstrated that the prevalence of AS increases with aging, with the average values being 0.2% (50–59 years), 1.3% (60–69 years), 3.9% (70–79 years) and 9.8% (80–89 years). 37 Calcified aortic valve disease due to aging is a bidirectional inflammation‐associated disease characterized by ossified aortic valves containing macrophages, T lymphocytes, and foam cells. 37 , 38 Calcified aortic valve disease progression thus contributes to the overall cardiac health affecting the valve structure and the functional capacity of the heart.
The age‐associated cardiac functional landscape presents with decreased contractility impairing systolic function and β‐adrenergic desensitization, thereby diminishing the heart's ability to increase heart rate and contractility during stress and exercise. 39 Furthermore, studies have shown that impaired relaxation due to increased myocardial stiffness and calcium handling dysregulation in cardiomyocytes from older individuals has been associated with diastolic dysfunction. 40 , 41 Electrophysiological conduction abnormalities as a result of aging have been associated with a degenerated sinoatrial and atrioventricular node, and conduction pathways can lead to arrhythmias with an increased risk for bradycardia and atrial fibrillation. 42 , 43 , 44 The pathophysiology of arrhythmias is associated with apoptosis of cardiac myocytes that contribute to progressive pump failure thereby exacerbating cardiac dysfunction. Additionally, studies have shown arrhythmogenicity of fibro‐fatty infiltrations wherein the size and percentage of adipose infiltrates influenced wave propagation and the onset of arrhythmias. 45
Increased levels of apoptosis and necrosis, concomitant with the limited regenerative capacity of cardiomyocytes due to aging, leads to a decrease in the number of functional cardiac cells. 46 , 47 Furthermore, microRNAs have also recently emerged as a regulator of physiological pathways, including aging of the cardiovascular system. 48 Age‐associated increased expression of microRNA 34a and decreased level of microRNA 146a has been observed in endothelial cells that induce apoptosis and reduce oxidative stress via downregulation of nicotinamide adenine dinucleotide phosphate oxidase (NOX) 4 expression, a catalytic subunit of NOX. 49 , 50 , 51 Increased steady‐state levels of reactive oxygen species (ROS) due to 1‐ and 2‐electron reductions of molecular oxygen in the mitochondrial electron transport chain can contribute to higher levels of mitochondrial oxidative damage to DNA and proteins of OXPHOS, causing a decline in ATP production. 52
Age‐associated epigenetic dysregulation also affects the transcription of genes involved in cardiac function. 53 Methylation at 5‐cytosine (5‐methylcytosine) is a key epigenetic modification associated with the onset of pathology. 53 Interestingly, DNA methylome profiling in failing cardiomyocytes of mice returned to a fetal methylation pattern implicating that failing cardiomyocytes may undergo cellular reprogramming that affects their function, regenerative potential, and stress response resembling the plasticity seen in fetal cardiomyocytes. 54 This epigenetic reprogramming presents avenues for therapeutic interventions that promote a stable methylation pattern that potentially enhances overall cardiac function and promotes better outcomes in heart failure patients. Dysregulated DNA hydroxymethylation patterns including alterations in 5‐hydroxymethylcytosine could lead to maladaptive repair programs promoting cardiac fibrosis hypertrophy and apoptosis, all of which directly affect cardiac function. 55 Other posttranslational modifications including linker and core histone modifications are also involved in cardiovascular aging and pathologies. For example, H3K27ac, which functions as an active promoter and enhancer, has been implicated as a promoter of age‐related genes in murine models. 56 Other histone modifications of posttranslational modifications on histone H3 including H3K79me2, 57 H3K9me2, 58 and H4K20me3 59 have also been associated with pathological cardiac hypertrophy and aging. Although increased ROS promotes autophagy, repression of autophagy is observed in cardiac aging, possibly due to the exhaustion of the autophagic machinery due to the persistent and progressive increase in ROS with age. 60
Cardiac aging is often associated with changes in the metabolic landscape including bioenergetics and metabolism whereby aging hearts demonstrate decreased oxidation of fatty acids with accumulation of free fatty acids and lipid droplets in heart tissue and increased glucose utilization a common feature in hypertrophic or dilated cardiomyopathy associated with cardiac aging. 61 , 62 This observation has been exploited using dietary interventions, including long‐term calorie restriction, and has been shown to significantly alleviate age‐associated mitochondrial dysfunction and offer cardiac protection. 63 Transcriptional profile data of genes coding for the mitochondrial proteins from the ventricles of young (6 months) and old (24 months) mice showed a 95% downregulation of mitochondrial electron transport chain genes. 64 Interestingly, the transcriptional downregulation of the mitochondrial electron transport chain genes in the ventricles of old mice contribute to increased metabolic stress that potentially imbalances the pro‐ and anti‐inflammatory responses associated with age‐associated heart disease.
Morphological and functional manifestations in the aging heart thus contribute to a deterioration in cardiac performance. An insight into the crosstalk between immunometabolism and these age‐related changes can prove crucial for developing effective therapeutic strategies aimed at alleviating the risks of chronic implications, particularly in older patients.
Metabolism in the Aging Heart
Age‐related cardiac degeneration due to impairments in structural, metabolic, and functional flexibilities contributes to an overall decline in cardiac efficiency, leading to the development of various cardiac pathologies (Figure 2). Endogenous aging is often associated with a progressive decline in mitochondrial efficiency due to aberrant mitochondrial function, increased ROS production, decreased ATP production, and metabolic reprogramming, all of which can cause changes in immune cell metabolism. 62 , 65 Decreased oxidative capacity of the mitochondria, concomitant with increased oxidative damage, results in damage to essential macromolecules, including DNA, mitochondrial DNA, lipids, and proteins. 66 , 67 Cardiac output and myocardial efficiency serve as an index for the cardiac ATP supply. 65 Perturbations in ATP production can lead to impairment in the contractile function of cardiomyocytes lowering the cardiac efficiency. Studies in a murine model of metabolic heart disease suggested that impairments in ATP production contribute to diastolic dysfunction. 68 Age‐related impairments in ATP production and increased oxidative damage can thus result in metabolic and functional inflexibility in cardiomyocytes, impairing contractility and increasing susceptibility to cardiac pathologies. Modulation of mitochondrial metabolism and ATP generation may offer therapeutic avenues to mitigate diastolic dysfunction and improve overall cardiac health in older patients.
Figure 2. Structural, metabolic, and functional impairments in the aging heart.
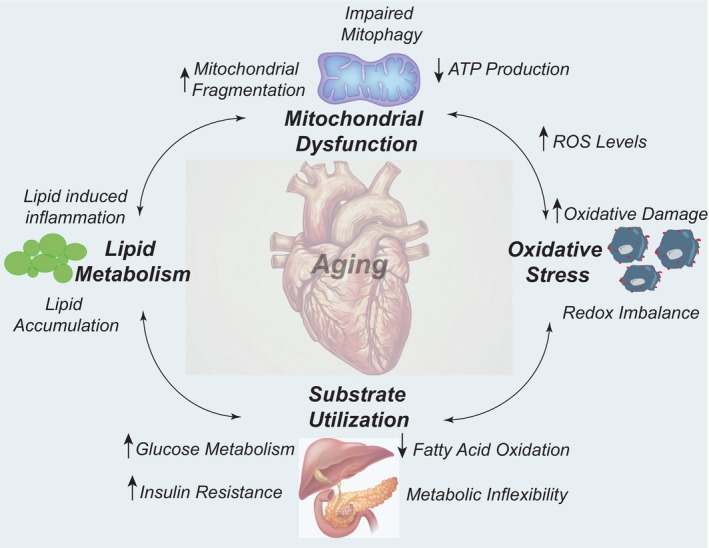
Mitochondrial dysfunction manifests collectively as impairments in mitophagy, mitochondrial dynamics, and bioenergetics (ATP production) concomitantly with increased ROS levels, contributing to oxidative damage. Thus, the redox imbalance causes alterations in substrate utilization, including decreased fatty acid oxidation, increased glucose metabolism, and insulin resistance leading to metabolic inflexibility, lipid accumulation, and inflammation. Increased mitochondrial ROS also causes cardiac lipid accumulation109,110 leading to cardiomyopathy in the aging heart. ROS indicates reactive oxygen species.
Age‐associated increased production of superoxide from the mitochondrial OXPHOS and other cellular sources contributes to oxidative damage that exacerbates mitochondrial electron transport chain dysfunction, creating a vicious cycle of oxidative stress.35 OXPHOS is fueled by fatty acid β‐oxidation by providing reducing equivalents, namely, nicotinamide adenine dinucleotide and reduced flavin adenine dinucleotide that enter OXPHOS through complexes I and III. 69 Aging leads to decreased fatty acid β‐oxidation in the mitochondrial matrix of cardiomyocytes, resulting in the generation of nicotinamide adenine dinucleotide and flavin adenine dinucleotide, bolstering glucose metabolism and an increased production of cytokines including TNF‐α, interleukin‐1β, and interleukin‐6 creating a state of metabolic/substrate inflexibility. 70 , 71 , 72 , 73 , 74 , 75 Decreased fatty acid β‐oxidation along with increased glycolysis in cardiomyocytes, is a hallmark of metabolic remodeling that occurs in cardiac hypertrophy. 76 Additionally, increased levels of glycolysis and glucose metabolism at the expense of fatty acid β‐oxidation and increased insulin secretion have been shown to correlate with immune activation. 77 Age‐associated increase in the generation of mitochondrial ROS also contributes to increased cardiac lipid accumulation, leading to cardiomyopathy associated with the downregulation of peroxisome proliferator‐activated receptor that regulates cardiac fatty acid metabolism. 78 , 79 Peroxisome proliferator‐activated receptor is ubiquitous in almost every immune cell type and is considered anti‐inflammatory with increased expression associated with low levels of proinflammatory cytokines including TNF, interleukin‐1, and interleukin‐6. 80
In addition to OXPHOS and cellular energy production, the selective degradation of dysfunctional mitochondria occurs through mitophagy, which aids in mitochondrial quality control. 81 Accumulation of damaged mitochondria due to impairments in mitophagy can contribute to age‐related implications including neurodegeneration, cancer, diabetes, and inflammation disorders. 82 Age‐associated decreased levels of Parkin and PTEN‐induced kinase 1, regulators of mitophagy can lead to accumulation of dysfunctional mitochondria, increasing oxidative stress levels thereby contributing to impaired cardiac contractility, myocardial stiffness, and development of fibrosis. 83 , 84 Studies have shown that age‐associated impairments in mitophagy can also lead to inflammation. For example, impaired mitophagy can result in cytosolic leakage of mitochondrial DNA that can activate the cyclic GMP‐AMP synthase–stimulator of interferon genes pathway of macrophage inflammatory response. 85 Furthermore, age‐associated impaired mitophagy results in dysfunctional mitochondria, activating the NOD‐like receptor protein 3 inflammasome, augmenting the inflammatory response, and contributing to systemic inflammation. 86
The overall metabolic shift in the aging heart coupled with the functional decline of mitochondrial oxidative metabolism thus contributes to oxidative stress, lipid accumulation, and inflammation driving toward the pathology of cardiomyopathy and impaired cardiac function. The cardiac milieu in the aging heart is thus a collective dysregulation of various pivotal aspects of metabolism that are highly interlinked and can be attributed causatively to age‐associated cardiac pathologies.
Immune Landscape and the Aging Heart
Cardiac aging is driven by degenerative structural, functional, and metabolic changes that result in a visceral increased inflammatory status due to impairments in the various aspects of the innate immune response caused by endogenous aging and exposure to stress. The accurate functional crosstalk between metabolism and the immune landscape during aging is thus deemed crucial for maintaining cardiac homeostasis to prevent the exacerbated development of age‐associated cardiac implications.
Chronic low‐grade inflammation, coined as “inflammaging” is an evolutionary perspective on the concept of “immunosenescence” that centers around the idea that aging is associated with increased infiltration of immune cells as a result of provoked antigenic load and stress (physical, chemical, and biological), which exacerbates tissue damage and overall fibrosis. 6 The adaptive immune response is capable of retaining specific memory related to stress as we age; activated T cells following stress will shrink and retain a subset of memory T cells that are prone to senescence with age and thus produce increased amounts of proinflammatory cytokines, mediators, and chemokines. 87 , 88 , 89
Systemic and tissue‐specific (cardiac) low‐grade inflammation, commonly referred to as “inflammaging” is often associated with physiological changes such as aging and obesity and closely resembles immunosenescence. 90 It is typically characterized by high levels of proinflammatory markers, TGF‐β, TNF‐α, and C‐reactive protein. 6 , 91 The levels of several immune mediators were found to be increased with age including interferon‐γ, interleukin‐2, interleukin‐6, and interleukin‐8. 92 , 93 Increased levels of proinflammatory markers and cytokines cause a persistent inflammatory environment with changes in subsets of the myeloid cell population. This in conjunction with impaired repair, fibroblast activation, and altered mechanical properties, consequently leads to an overall functional decline and contributes to the development of myocardial fibrosis. 94 , 95
Age‐associated pathologies are often associated with changes in the different subsets of myeloid cell populations and immunosenescence. Macrophages represent the primary source of MMPs and tissue inhibitors of metalloproteinases and, along with myofibroblasts, have been shown to play a crucial role in the secretion of ECM components and their remodeling. 94 , 96 Macrophages are phagocytic cells with high plasticity and can change their functional phenotypic states from proinflammatory, antitumorigenic to anti‐inflammatory, protumorigenic, or wound healing macrophages in response to stress within the tissue microenvironment including infections, tissue injury, and cancer. 97 , 98 Activated to a proinflammatory phenotype (M1) in the early stages of the inflammation process, macrophages secrete proinflammatory cytokines and chemokines, including TNF‐α, interleukin‐6, interleukin‐12, and chemokine (C‐X‐C motif) ligands (CXCL9 and CXCL10) that subsequently recruit helper cluster of differentiation (CD) 4+ T cells and effector CD8+ T cells. 97 , 99 , 100 Additionally, M1 macrophages release factors responsible for fibroblast activation, stimulating the production of ECMs, leading to excessive collagen deposition and fibrosis, increased stiffness, and diastolic dysfunction. 101 Aging has been associated with increased polarization of macrophages to the M1 phenotype resulting in an endogenously heightened inflammatory state. 97 Moreover, age‐associated decline in the regenerative capacity of the heart along with the inflammatory input from the M1 macrophages can impede repair mechanisms leading to poor recovery in older patients. 97 In contrast, reparative macrophages polarize to a fibrotic/wound healing (M2) phenotype following phagocytosis of apoptotic neutrophils characterized by decreased expression of interleukin‐6 and TNF‐α and an increase in TGF‐β, galectin‐3, and interleukin‐10. 102 , 103 , 104 While M2 macrophages are typically associated with repair and anti‐inflammatory responses, excessive activation can lead to the overproduction of ECM, increased production of profibrotic factors, and fibrosis. 105 Interestingly, increased expression of anti‐inflammatory macrophages was observed in the lung 106 and muscle 107 of C57BL/6J mice with aging. Age‐related ROS accumulation resulting from altered mitochondrial metabolism leads to the accumulation of monocyte‐derived C‐C chemokine receptor type 2+ macrophages to replace cardiac resident macrophages and induced vascular endothelial cells to express high levels of vascular cell adhesion molecule 1, which furthers the monocyte infiltration into cardiac tissues contributing to inflammation and tissue damage. 108 , 109 Following infiltration into the cardiac tissues, the recruited monocytes can differentiate into macrophages, releasing inflammatory mediators, thereby contributing to tissue damage and remodeling under pathologies like myocardial infarction. 108 Furthermore, increased accumulation of cardiac‐specific macrophages can induce the secretion of MMP9 that supports the progression of macrophages to the inflammatory phenotype (M1), which contributes to a chronic inflammation state in the aging heart, thereby activating collagen accumulation to promote interstitial fibrosis. 110 , 111
T‐cell dysregulation impacts the aging heart due to profound changes induced due to immunosenescence. Studies have shown a progressive decrease in the population of naïve T cells and an increased population of memory and effector T cells. 6 , 112 , 113 , 114 The skewing of T‐cell subsets to a memory phenotype occurs concurrently with the downregulation of costimulatory factors, including CD27 and CD28, which are aspects of human T‐cell senescence and increase the susceptibility to aging‐related pathologies like cardiovascular complications and cancer. 115 CD27 and CD28 provide a costimulatory signal and promote the assembly of the T‐cell complex to amplify the T‐cell receptor signals and support T‐cell expansion. 116 CD28 has been shown to promote cell cycle entry and activity as well as promote the survival of activated T cells through the upregulation of the anti‐apoptotic regulator of the Bcl‐2 family, Bcl‐xL. 117 Age‐related decrease in helper T cell populations has been linked to reduced myocardial inflammation and preserved cardiac function relative to age‐matched controls. 118 Furthermore, a unique population of memory CD4+ T cells referred to as senescence‐associated T cells expressing programmed cell death 1, CD153, a TNF family protein, and osteopontin have been identified to increase progressively in aging mice. 119 , 120 Senescence‐associated T cells classically express programmed cell death 1, CD153, and osteopontin, which are markers attributed to immunosenescense and chronic low‐grade inflammation. 121 Osteopontin is involved in the regulation of cardiac fibrosis, with elevated levels of osteopontin observed in both the myocardium and the plasma of patients with both dilated and hypertrophic cardiomyopathy, as a result of fibroblast activation. 122 Recently, a growing body of research has identified a subpopulation of age‐associated granzyme K–expressing CD8+ T cells that are distinct from T‐effector memory cells that express markers of tissue homing and exhaustion 123 and are suggested to be an intermediate memory‐like preterminally differentiated population of nonnaïve cells. 124 These clonally expanded memory CD8+ T cells have been shown to accumulate in atherosclerotic plaques and are identified as proatherogenic in aged mice. 125 Additionally, the age‐associated decline in telomere length and the consequent DNA damage during replicative senescence is driven by the homeostatic proliferation of T cells wherein T cells divide in the absence of a cognate antigen to reconstitute the lymphoid populations. 126 , 127 , 128 In the aging heart, dysregulation of T cells can present in several ways: (1) as senescent CD4+ T cells leading to inflammation and age‐related cardiac dysfunction, 121 (2) changes in the makeup of cardiac‐resident leukocyte population with age, 35 (3) secretion of proinflammatory cytokines that promote vascular pathology and increase the risk of cardiovascular disease, 129 and (4) a decline in the rate of thymic T cells with age. 130 Inflammaging has been shown to tip the scales toward memory T cells over the naïve cell population with CD4+ T cells polarizing toward a T‐helper 17 phenotype, consequently leading to the production of proinflammatory cytokines (interleukin‐17 and interferon‐γ), a concept often associated with atherosclerotic plaques within arteries, promoting the progression of the disease. 131 , 132 , 133 This T‐helper 17 phenotype is often induced by a metabolic switch, resulting in the upregulation of glycolysis, facilitating sustained inflammation. Thus, the implications of aging‐associated immune senescence depend not only on the balance between the different T‐cell subsets and their functional decline but also influence the overall immune response including the activity/frequency of other immune cells like neutrophils.
Neutrophils respond to stress or injury by migrating to injured tissue sites to phagocytose pathogens, producing ROS that alleviates the sterile inflammation, which typically occurs in the absence of infection to aid in the clearance of damaged tissue debris. 134 The differentiation of hematopoietic stem cells (HSCs) into mature neutrophils also known as granulopoiesis, is the de novo production of neutrophils in the hematopoietic cords of the bone marrow. 135 , 136 The stemness of HSCs decreases as a function of age toward a fate of myeloid lineage due to inflammation, dysfunctional mitochondrial metabolism, replicative stress, and reduced telomere length. 137 Accumulation of gene mutations, particularly in stem cells, increases the proliferative potential of mutant HSCs, predisposing tissues to hematopoietic malignancies and cardiovascular disease. Sano et al demonstrated that Tet‐2 deficiency in HSCs exacerbates cardiac dysfunction in a murine heart failure model, primarily through the activation of interleukin‐1β–NOD‐like receptor protein 3 inflammasome signaling. 138 Although aged HSCs shift toward a myeloid lineage, studies suggest that this may have minimal effect on neutrophil populations in healthy individuals. 139 , 140 However, murine studies consistently show an age‐associated increase in neutrophils within lymphoid organs. 139 , 141 , 142 An age‐associated increase in CD16high/CD62Llow immunosuppressive neutrophils was observed in conjunction with a low phagocytic index and reduced ROS production that increases the susceptibility to infections in older patients. 143 Interestingly, age‐associated impairment in the myocardium correlates with a shift in the cardiac‐specific leukocytes and an increased population of neutrophils and monocytes. 118 These studies are suggestive of the concept that inflammaging in the heart renders mice susceptible to cardiac dysfunction.
Over the past few decades, substantial evidence has indicated immunosenescence as a risk factor for cardiovascular disease with an impact on myocardial physiology and function, thereby affecting the susceptibility to cardiovascular pathologies. 91 , 118 , 144 With the significance of different subsets of leukocyte populations, including neutrophils, T cells, B cells, and macrophages/monocytes in cardiac repair and remodeling, the global landscape of immune response can thus impact the development and progression of cardiovascular diseases.
Clinical Interventions in the Aging Heart Pathologies
Modulation of immune tolerance by suppressing age‐associated inflammation to augment cardiac remodeling and metabolism has gained research precedence in the field of aging research. The age‐associated functional and metabolic decline in cardiac tissues has warranted the need for easily implementable, nontoxic therapeutic interventions that bolster the endogenous cardiovascular regenerative and reparative capacity. Several metabolic pathways and their metabolites have emerged as promising avenues to target immunometabolism in aging heart tissues, including mitochondria‐targeted biologics, anti‐inflammatory therapies, metabolic modulators, and lifestyle interventions.
With the detrimental role of oxidative stress and mitochondrial impairment in age‐associated cardiac functional decline and the associated pathologies, there is a rising interest in using conventional antioxidant therapies to mitigate the endogenous effects of aging on cardiac function. Some commonly used nonspecific antioxidants include NOX inhibitors and N‐acetylcysteine. Studies using pharmacological NOX inhibitors, apocynin, and VAS2870 have demonstrated improved β‐adrenergic stimulated contractility and intracellular calcium handling in aged rat hearts. 145 Additionally, treatment with GLX481304, a pharmacological inhibitor of Nox 2 and 4, has been shown to inhibit ROS production in isolated mouse cardiomyocytes and improve contractility and function following ischemia–reperfusion injury in mice via direct effects on the cardiomyocytes by inhibiting a ROS‐induced modification of the contractile system. 146 Moreover, NOX4 has been shown to regulate cardiac intermediatory metabolism via H2O2‐specific signaling. 147 , 148 Although different isoforms of NOXs have been favorably used to modulate ROS and thereby modify the redox environment, the functional diversity of NOX enzymes in the cardiovascular system has prevented using specific NOX inhibitors to alleviate age‐associated cardiac pathologies. N‐acetylcysteine is known for its antioxidant and anti‐inflammatory properties and acts by upregulating the rate of glutathione synthesis, suppressing nuclear factor κ B, and decreasing the production of proinflammatory cytokines, i.e., interleukin‐1, interleukin‐6, and TNF. 149 Several studies have focused on the clinical implications of N‐acetylcysteine, which include protection against coronary artery disease, myocardial infarction, atrial fibrillation, and cardiomyopathy. 150 , 151 , 152
Mitochondrially targeted antioxidants that specifically reduce mitochondrial ROS have also been clinically used to target the implications on the aging heart. Mitoquinone, a ubiquinol antioxidant with high bioavailability, attached to a triphenylphosphonium moiety, enables it to cross cell membranes and accumulate in the mitochondrial matrix to reduce mitochondrial ROS and improve mitochondrial function. 153 , 154 Preclinical models have shown that treatment with 200‐μM mitoquinone in drinking water for up to 7 weeks decreased MitoSOX oxidation in bone marrow–derived neutrophils in both male and female mice along with reduced neutrophil ROS and neutrophil extracellular traps formation. 155 A clinical trial in 2018 showed that mitoquinone was well tolerated in healthy older adults and led to a 42% increase in brachial artery flow‐mediated dilation following treatment with 20 mg/day mitoquinone for 6 weeks, along with alleviation in mitochondrial ROS‐related suppression of endothelial function and lower aortic stiffness (Mitochondrial‐Targeted Antioxidant Supplementation for Improving Age‐Related Vascular Dysfunction in Humans, NCT04851288). 156 Furthermore, the plasma levels of oxidized low‐density lipoprotein were also found to be lower in the mitoquinone‐treated groups relative to placebo, indicating that targeting mitochondria ROS may be of significance in treating age‐associated vascular dysfunction. 156 Furthermore, in a double‐blinded, randomized clinical trial, mitoquinone and endurance training, individually and in combination, effectively enhanced cardiovascular health by reducing inflammation and increasing antioxidant defense. 157 The study suggested that the combination of mitoquinone and endurance training significantly decreased systolic blood pressure, improved cardiac function indices including LV mass and LV end‐systolic diameter, and reduced interleukin‐6 levels via modulation of microRNA‐222. 157 Additionally, an ongoing clinical trial at the University of Connecticut is assessing the effects of mitoquinone supplementation on vasodilation, mobility, and cognitive performance in frail older adults (The Mito‐Frail Trial: Effects of MitoQ on Vasodilation, Mobility and Cognitive Performance in Frail Older Adults, NCT06027554).
Another mitochondria‐targeted synthetic tetrapeptide, elamipretide, also known as bendavia, has emerged as a mitochondria‐specific therapeutic that has been shown to restore mitochondrial bioenergetics by targeting cardiolipin and accumulating in the inner mitochondrial membrane. 158 , 159 , 160 , 161 Elamipretide has been shown to reduce the generation of mitochondrial ROS and optimize ATP synthesis and mitochondrial electron transport thereby preventing cardiac pathologies. 158 , 159 , 160 , 161 Additionally, elamipretide has also been in several clinical trials for acute and chronic cardiac events (EMBRACE [Evaluation of Myocardial Effects of MTP‐131 for Reducing Reperfusion Injury in Patients With Acute Coronary Events], NCT01572909; A Study to Evaluate the Effects of 4 Weeks Treatment With Subcutaneous Elamipretide on Left Ventricular Function in Subjects With Stable Heart Failure With Preserved Ejection Fraction, NCT02814097; Effect of Elamipretide on Left Ventricular Function in Subjects With Stable Heart Failure With Reduced Ejection Fraction, NCT02788747; A Phase 2 Study to Evaluate the Cardiac and Renal Effects of Short Term Treatment With Elamipretide in Patients Hospitalized With Congestion Due to Heart Failure, NCT02914665; A Study Investigating the Safety, Tolerability, and Pharmacokinetics of MTP‐131 in Subjects With Congestive Heart Failure, NCT02388464) and mitochondrial disorders (MMPOWER [Safety, Tolerability, and Efficacy of MTP‐131 for the Treatment of Mitochondrial Myopathy], NCT02367014). The highly lipophilic quinone, coenzyme Q10 (CoQ10), or ubiquinone, is a potent antioxidant that has been shown to facilitate mitochondrial‐associated ATP production. 162 In addition to serving as an electron carrier in the mitochondria, the anti‐inflammatory role of CoQ10 is attributed to its ability to repress inflammatory gene expression, thereby improving immune function. Preclinical data demonstrate a pathophysiological role for CoQ10 in heart failure and other cardiovascular events with improved outcomes following CoQ10 supplementation. 163 CoQ10 has been in clinical trials for >30 years as an intervention for cardiovascular diseases, including heart failure (PANDA [Protective Effect of CoQ10 Against Negative Inflammatory Response and Organ Dysfunction in Cardiovascular Surgery], NCT04444349) and coronary artery disease (Coenzyme Q10 in Relation of the Lipid Peroxidation, Antioxidant Enzyme Activities in Coronary Artery Disease Patients, NCT01163500).
Several anti‐inflammatory therapies have also been employed to maintain homeostasis in the endogenous inflammation‐prone environment of the aging heart. For example, increased levels of interleukin‐6 in older patients have demonstrated an increased risk for cardiovascular death by 69%, thereby increasing the risk of “all‐cause” death. 164 Cytokine inhibitors that target proinflammatory cytokines like interleukin‐6 can aid in decreasing age‐associated chronic inflammation in the heart. More specifically, tocilizumab, an antagonist for the interleukin‐6 receptor, has shown potential in dampening systemic inflammation, thereby improving age‐associated cardiac outcomes to mitigate organ injury in patients. 165 In a clinical trial for modulation of systemic inflammatory response after out‐of‐hospital cardiac arrest, treatment with tocilizumab was shown to profoundly decrease the levels of C‐reactive protein (−84%) and leukocytes (−34%) (IMICA [Interleukin‐6 Inhibition for Modulating Inflammation After Cardiac Arrest], NCT03863015). 165 Increased circulating levels of TNF have been associated with increased prevalence of atherosclerosis in older patients. 166 As such, TNF antagonists that have the potential to reduce inflammation have been investigated for their potential benefits in age‐associated cardiovascular pathologies, including rheumatoid arthritis, and atherosclerosis, and have been associated with a decreased risk of myocardial infarction 167 and acute coronary syndrome. 168 Upregulation in macrophage‐associated inflammatory cytokines, including TNF, interleukin‐1β, and interleukin‐6, has been seen in rheumatoid arthritis, with interleukin‐6 as a crucial factor in plaque destabilization and atherothrombosis. 169 Etanercept (Enbrel) is a soluble, TNF receptor–IgG1 fusion protein that binds and inhibits TNF, thereby preventing TNF‐mediated inflammatory responses. Etanercept has been approved by the Food and Drug Administration for rheumatoid arthritis. 170 Although the pathological role of TNF has been assessed using population‐based studies in cardiac pathologies with elevated serum levels of the proinflammatory cytokine, the complex nature of cytokine‐based therapies presents with disease‐associated benefits and side effects. As such, anti‐TNF therapies have been criticized for their poor cardiac clinical outcomes including increased deaths, increasing hospitalizations, and worsening congestive heart failure. 171 , 172 The CANTOS (Canakinumab Anti‐inflammatory Thrombosis Outcome Study, NCT01327846) trial demonstrated that inhibition of interleukin‐1β (a highly pleiotropic, proinflammatory cytokine) in patients with previous myocardial infarction and a high C‐reactive protein lowered the rate of recurrent cardiovascular events compared with placebo. 173 Notably, interleukin‐1β inhibition has been shown to reduce the T‐helper 17/regulatory T cell (Treg) imbalance and promote the differentiation of naïve CD4+ T cells into T‐helper 17 cells. 174 Additionally, anakinra (kineret), interleukin‐1 receptor antagonist, has been employed to treat a wide spectrum of cardiovascular diseases. 175 , 176 Other immunomodulatory therapies include infliximab, a TNF inhibitor that has been shown to enhance Treg function by increasing their proliferation and capacity to suppress proinflammatory cytokine production. 177 It needs to be noted that although effective in modulating the proinflammatory response, cytokine‐targeting therapies have been shown to increase the risk of infection particularly due to immune suppression. In CANTOS, canakinumab was associated with increased rates of neutropenia, cellulitis, pseudomembranous colitis, fatal infection, or sepsis, limiting its use in nonautoimmune conditions including atherosclerosis. 173 Additionally, the TNF inhibitor infliximab significantly increases the risk of developing bacterial infections due to its immunosuppressive effects as well as latent tuberculosis infections due to the risk of reactivation of latent tuberculosis. 178 The efficacy of repurposed therapeutic drugs thus needs to be evaluated to outweigh the risk of adverse side effects in cardiovascular and metabolic disease conditions.
Therapies that modulate specific immune cell subsets have also been employed to alleviate the cardiac inflammatory phenotype. Tregs are the immunosuppressive subset of CD4+ T cells that alternate between glycolysis and OXPHOS for proliferation. 179 Tregs play a key role in the modulation of the inflammatory cascade and maintain immune homeostasis. 179 Characterized by the expression of CD4, CD25, and the transcription factor forkhead box P3, Tregs are derived from the thymus or peripheral naïve T cells. 180 Tregs secrete anti‐inflammatory cytokines including interleukin‐10 and TGF‐β that aid in immune activation and mitigate tissue damage. 180 Naïve T cells undergo differentiation into 4 different subsets including T‐helper T cells, T‐helper 2 cells, T‐helper 17 cells, and Tregs. 180 , 181 Among these, T‐helper 17 has emerged as a marker in the context of cardiovascular diseases. T‐helper 17 cells generate increased levels of the proinflammatory cytokine interleukin‐17 in patients with acute coronary syndrome 182 and heart failure. 180 , 183 Increased T‐helper 17 cells relative to Tregs create a functional imbalance in the T‐helper 17/Treg ratio thereby promoting an inflammatory phenotype and disease progression. Recent studies have shown that elderly patients with atrial fibrillation enhance the Treg population in the spleen and blood upon oral administration of Bacteroides fragilis, an anaerobic, gram‐negative bacterium commonly found in the microbiota of the human colon. 184 Ex vivo expansion or adoptive transfer therapies of Tregs have emerged in recent years as a clinical approach in cardiovascular pathologies. The global burden of an aging population and the increasing number of “cancer survivors” populace is associated with an enhanced risk of cardiovascular diseases due to endogenous aging implications or as a chronic impression of cancer therapies. Although the field of immunometabolic interventions has been of much research interest, there is still a lack of rigorous clinical evidence/data from randomized trials that tests the effects of targeted immune interventions on metabolism and vice versa. In terms of improving aging heart pathologies, targeting immunometabolic pathways and their modulation has been effective in alleviating chronic inflammatory changes and metabolic impairments that are characteristic of the aging heart to enhance cardiac function and repair, reduce the incidence of age‐related cardiovascular diseases, and ultimately improve the quality of life in the elderly population.
Sources of Funding
This work was supported by National Institutes of Health grants UO1 AI184289 and R21 CA270742‐01.
Disclosures
None.
Acknowledgments
Author contributions: Conceptualization: Drs Mapuskar and Allen; validation: Drs Allen, London, Mapuskar, and Houtman; investigation and resources: Drs Mapuskar and Houtman; writing—original draft preparation: Dr Mapuskar; writing—review and editing: Drs Allen, London, Houtman, and Zacharias. All authors have read and agreed to the published version of the manuscript.
This manuscript was sent to Nadia R. Sutton, MD, MPH, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 10.
References
- 1. Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol. 2011;11:81–83. doi: 10.1038/nri2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iwasaki Y, Takeshima Y, Fujio K. Basic mechanism of immune system activation by mitochondria. Immunol Med. 2020;43:142–147. doi: 10.1080/25785826.2020.1756609 [DOI] [PubMed] [Google Scholar]
- 3. Pajak B, Zielinski R, Priebe W. The impact of glycolysis and its inhibitors on the immune response to inflammation and autoimmunity. Molecules. 2024;29:19. doi: 10.3390/molecules29061298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang S, Lv K, Liu Z, Zhao R, Li F. Fatty acid metabolism of immune cells: a new target of tumour immunotherapy. Cell Death Dis. 2024;10:39. doi: 10.1038/s41420-024-01807-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teissier T, Boulanger E, Cox LS. Interconnections between Inflammageing and Immunosenescence during ageing. Cells. 2022;11:48. doi: 10.3390/cells11030359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm‐aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x [DOI] [PubMed] [Google Scholar]
- 7. SEER Cancer Statistics Review (CSR) 1975–2014. https://seer.cancer.gov/archive/csr/1975_2014/.
- 8. American Community Survey Reports. https://www.census.gov/content/dam/Census/library/publications/2018/acs/ACS‐38.pdf.
- 9. American Cancer Society ‐ Cancer Treatment & Survivorship ‐ Facts & Figures. 2022–2024. https://www.cancer.org/content/dam/cancer‐org/research/cancer‐facts‐and‐statistics/cancer‐treatment‐and‐survivorship‐facts‐and‐figures/2022‐cancer‐treatment‐and‐survivorship‐fandf‐acs.pdf.
- 10. Raschi E, Vasina V, Ursino MG, Boriani G, Martoni A, De Ponti F. Anticancer drugs and cardiotoxicity: insights and perspectives in the era of targeted therapy. Pharmacol Ther. 2010;125:196–218. doi: 10.1016/j.pharmthera.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 11. Bocchi EA, Avila MS, Ayub‐Ferreira SM. Aging, cardiotoxicity, and chemotherapy. Aging (Albany NY). 2019;11:295–296. doi: 10.18632/aging.101776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeBerge M, Chaudhary R, Schroth S, Thorp EB. Immunometabolism at the heart of cardiovascular disease. JACC Basic Transl Sci. 2023;8:884–904. doi: 10.1016/j.jacbts.2022.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marelli‐Berg FM, Aksentijevic D. Immunometabolic cross‐talk in the inflamed heart. Cell Stress. 2019;3:240–266. doi: 10.15698/cst2019.08.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patel J, Baptiste BA, Kim E, Hussain M, Croteau DL, Bohr VA. DNA damage and mitochondria in cancer and aging. Carcinogenesis. 2020;41:1625–1634. doi: 10.1093/carcin/bgaa114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen W, He J, Hou T, Si J, Chen S. Common Pathogenetic mechanisms underlying aging and tumor and means of interventions. Aging Dis. 2022;13:1063–1091. doi: 10.14336/AD.2021.1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bloom SI, Islam MT, Lesniewski LA, Donato AJ. Mechanisms and consequences of endothelial cell senescence. Nat Rev Cardiol. 2023;20:38–51. doi: 10.1038/s41569-022-00739-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- 18. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–574. doi: 10.1007/s00018-013-1349-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Esfahani NS, Wu Q, Kumar N, Ganesan LP, Lafuse WP, Rajaram MVS. Aging influences the cardiac macrophage phenotype and function during steady state and during inflammation. Aging Cell. 2021;20:e13438. doi: 10.1111/acel.13438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, Khalil RA. Matrix Metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol. 2018;81:241–330. doi: 10.1016/bs.apha.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML. The impact of aging on cardiac extracellular matrix. Geroscience. 2017;39:7–18. doi: 10.1007/s11357-017-9959-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sutherland TE, Dyer DP, Allen JE. The extracellular matrix and the immune system: a mutually dependent relationship. Science. 2023;379:eabp8964. doi: 10.1126/science.abp8964 [DOI] [PubMed] [Google Scholar]
- 23. Baran P, Hansen S, Waetzig GH, Akbarzadeh M, Lamertz L, Huber HJ, Ahmadian MR, Moll JM, Scheller J. The balance of interleukin (IL)‐6, IL‐6.Soluble IL‐6 receptor (sIL‐6R), and IL‐6.sIL‐6R.sgp130 complexes allows simultaneous classic and trans‐signaling. J Biol Chem. 2018;293:6762–6775. doi: 10.1074/jbc.RA117.001163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, Lee SR, Yang SH. The role of tumor necrosis factor alpha (TNF‐alpha) in autoimmune disease and current TNF‐alpha inhibitors in therapeutics. Int J Mol Sci. 2021;22:16. doi: 10.3390/ijms22052719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramana KV. Tumor necrosis factor‐alpha converting enzyme: implications for ocular inflammatory diseases. Int J Biochem Cell Biol. 2010;42:1076–1079. doi: 10.1016/j.biocel.2010.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindsey ML, Iyer RP, Jung M, DeLeon‐Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post‐myocardial infarction remodeling. J Mol Cell Cardiol. 2016;91:134–140. doi: 10.1016/j.yjmcc.2015.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schonbeck U, Mach F, Libby P. Generation of biologically active IL‐1 beta by matrix metalloproteinases: a novel caspase‐1‐independent pathway of IL‐1 beta processing. J Immunol. 1998;161:3340–3346. doi: 10.4049/jimmunol.161.7.3340 [DOI] [PubMed] [Google Scholar]
- 28. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Opdenakker G, Van den Steen PE, Dubois B, Nelissen I, Van Coillie E, Masure S, Proost P, Van Damme J. Gelatinase B functions as regulator and effector in leukocyte biology. J Leukoc Biol. 2001;69:851–859. doi: 10.1189/jlb.69.6.851 [DOI] [PubMed] [Google Scholar]
- 30. Kobayashi T, Kim H, Liu X, Sugiura H, Kohyama T, Fang Q, Wen FQ, Abe S, Wang X, Atkinson JJ, et al. Matrix metalloproteinase‐9 activates TGF‐beta and stimulates fibroblast contraction of collagen gels. Am J Physiol Lung Cell Mol Physiol. 2014;306:L1006–L1015. doi: 10.1152/ajplung.00015.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)‐9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139:32–40. doi: 10.1016/j.pharmthera.2013.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–1568. doi: 10.1161/01.res.68.6.1560 [DOI] [PubMed] [Google Scholar]
- 33. Sheydina A, Riordon DR, Boheler KR. Molecular mechanisms of cardiomyocyte aging. Clin Sci (Lond). 2011;121:315–329. doi: 10.1042/CS20110115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Younes A, Boluyt MO, O'Neill L, Meredith AL, Crow MT, Lakatta EG. Age‐associated increase in rat ventricular ANP gene expression correlates with cardiac hypertrophy. Am J Phys. 1995;269:H1003–H1008. doi: 10.1152/ajpheart.1995.269.3.H1003 [DOI] [PubMed] [Google Scholar]
- 35. Akasheva DU, Plokhova EV, Tkacheva ON, Strazhesko ID, Dudinskaya EN, Kruglikova AS, Pykhtina VS, Brailova NV, Pokshubina IA, Sharashkina NV, et al. Age‐related left ventricular changes and their association with leukocyte telomere length in healthy people. PLoS One. 2015;10:e0135883. doi: 10.1371/journal.pone.0135883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhatia N, Basra SS, Skolnick AH, Wenger NK. Aortic valve disease in the older adult. J Geriatr Cardiol. 2016;13:941–944. doi: 10.11909/j.issn.1671-5411.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eveborn GW, Schirmer H, Heggelund G, et al. The evolving epidemiology of valvular aortic stenosis. The Tromsø study. Heart. 2013;99:5–400. doi: 10.1136/heartjnl-2012-302265 [DOI] [PubMed] [Google Scholar]
- 38. Ohukainen P, Ruskoaho H, Rysa J. Cellular mechanisms of Valvular thickening in early and intermediate calcific aortic valve disease. Curr Cardiol Rev. 2018;14:264–271. doi: 10.2174/1573403X14666180820151325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ferrara N, Komici K, Corbi G, Pagano G, Furgi G, Rengo C, Femminella GD, Leosco D, Bonaduce D. Beta‐adrenergic receptor responsiveness in aging heart and clinical implications. Front Physiol. 2014;4:396. doi: 10.3389/fphys.2013.00396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dai X, Hummel SL, Salazar JB, Taffet GE, Zieman S, Schwartz JB. Cardiovascular physiology in the older adults. J Geriatr Cardiol. 2015;12:196–201. doi: 10.11909/j.issn.1671-5411.2015.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vakka A, Warren JS, Drosatos K. Cardiovascular aging: from cellular and molecular changes to therapeutic interventions. J Cardiovasc Aging. 2023;3:3. doi: 10.20517/jca.2023.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alghamdi AM, Boyett MR, Hancox JC, Zhang H. Cardiac pacemaker dysfunction arising from different studies of Ion Channel remodeling in the aging rat heart. Front Physiol. 2020;11:546508. doi: 10.3389/fphys.2020.546508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goyal P, Rich MW. Electrophysiology and heart rhythm disorders in older adults. J Geriatr Cardiol. 2016;13:645–651. doi: 10.11909/j.issn.1671-5411.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mirza M, Strunets A, Shen WK, Jahangir A. Mechanisms of arrhythmias and conduction disorders in older adults. Clin Geriatr Med. 2012;28:555–573. doi: 10.1016/j.cger.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Coster T, Claus P, Kazbanov IV, Haemers P, Willems R, Sipido KR, Panfilov AV. Arrhythmogenicity of fibro‐fatty infiltrations. Sci Rep. 2018;8:2050. doi: 10.1038/s41598-018-20450-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goldspink DF, Burniston JG, Tan LB. Cardiomyocyte death and the ageing and failing heart. Exp Physiol. 2003;88:447–458. doi: 10.1113/eph8802549 [DOI] [PubMed] [Google Scholar]
- 47. Mallat Z, Fornes P, Costagliola R, Esposito B, Belmin J, Lecomte D, Tedgui A. Age and gender effects on cardiomyocyte apoptosis in the normal human heart. J Gerontol A Biol Sci Med Sci. 2001;56:M719–M723. doi: 10.1093/gerona/56.11.m719 [DOI] [PubMed] [Google Scholar]
- 48. Seeger T, Boon RA. MicroRNAs in cardiovascular ageing. J Physiol. 2016;594:2085–2094. doi: 10.1113/JP270557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lazzeroni D, Villatore A, Souryal G, Pili G, Peretto G. The aging heart: a molecular and clinical challenge. Int J Mol Sci. 2022;23:23. doi: 10.3390/ijms232416033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vasa‐Nicotera M, Chen H, Tucci P, Yang AL, Saintigny G, Menghini R, Mahe C, Agostini M, Knight RA, Melino G, et al. miR‐146a is modulated in human endothelial cell with aging. Atherosclerosis. 2011;217:326–330. doi: 10.1016/j.atherosclerosis.2011.03.034 [DOI] [PubMed] [Google Scholar]
- 51. Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF, Lu MH, Tang Y, Yu HY, Sun SH. Repression of the miR‐17‐92 cluster by p53 has an important function in hypoxia‐induced apoptosis. EMBO J. 2009;28:2719–2732. doi: 10.1038/emboj.2009.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mapuskar KA, Flippo KH, Schoenfeld JD, Riley DP, Strack S, Hejleh TA, Furqan M, Monga V, Domann FE, Buatti JM, et al. Mitochondrial superoxide increases age‐associated susceptibility of human dermal fibroblasts to radiation and chemotherapy. Cancer Res. 2017;77:5054–5067. doi: 10.1158/0008-5472.CAN-17-0106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Herman AB, Occean JR, Sen P. Epigenetic dysregulation in cardiovascular aging and disease. J Cardiovasc Aging. 2021;1:1. doi: 10.20517/jca.2021.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, Wurch A, Bonisch U, Gunther S, Backofen R, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014;5:5288. doi: 10.1038/ncomms6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang W, Song M, Qu J, Liu GH. Epigenetic modifications in cardiovascular aging and diseases. Circ Res. 2018;123:773–786. doi: 10.1161/CIRCRESAHA.118.312497 [DOI] [PubMed] [Google Scholar]
- 56. Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez‐Gonzalez A, Delhomme N, Ghavi‐Helm Y, Wilczynski B, Riddell A, Furlong EE. Tissue‐specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44:148–156. doi: 10.1038/ng.1064 [DOI] [PubMed] [Google Scholar]
- 57. Schubeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O'Neill LP, Turner BM, Delrow J, et al. The histone modification pattern of active genes revealed through genome‐wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18:1263–1271. doi: 10.1101/gad.1198204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rhodes B, Kroger J. Parental bonding and separation‐individuation difficulties among late adolescent eating disordered women. Child Psychiatry Hum Dev. 1992;22:249–263. doi: 10.1007/BF00707667 [DOI] [PubMed] [Google Scholar]
- 59. Dambacher S, Hahn M, Schotta G. The compact view on heterochromatin. Cell Cycle. 2013;12:2925–2926. doi: 10.4161/cc.26179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stadtman ER. Protein oxidation in aging and age‐related diseases. Ann N Y Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x [DOI] [PubMed] [Google Scholar]
- 61. Borcherding N, Jia W, Giwa R, Field RL, Moley JR, Kopecky BJ, Chan MM, Yang BQ, Sabio JM, Walker EC, et al. Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte‐derived mitochondria into the blood. Cell Metab. 2022;34:1499–1513.e8. doi: 10.1016/j.cmet.2022.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xie S, Xu SC, Deng W, Tang Q. Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications. Signal Transduct Target Ther. 2023;8:114. doi: 10.1038/s41392-023-01378-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li SJ, Lin YH, Chiang CH, Wang PY, Chen CY. Early‐onset dietary restriction maintains mitochondrial health, autophagy and ER function in the left ventricle during aging. J Nutr Biochem. 2022;101:108944. doi: 10.1016/j.jnutbio.2022.108944 [DOI] [PubMed] [Google Scholar]
- 64. Preston CC, Oberlin AS, Holmuhamedov EL, Gupta A, Sagar S, Syed RH, Siddiqui SA, Raghavakaimal S, Terzic A, Jahangir A. Aging‐induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech Ageing Dev. 2008;129:304–312. doi: 10.1016/j.mad.2008.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yaniv Y, Juhaszova M, Sollott SJ. Age‐related changes of myocardial ATP supply and demand mechanisms. Trends Endocrinol Metab. 2013;24:495–505. doi: 10.1016/j.tem.2013.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Srivastava S. The mitochondrial basis of aging and age‐related disorders. Genes (Basel). 2017;8:8. doi: 10.3390/genes8120398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–322. doi: 10.1023/B:CANC.0000031769.14728.bc [DOI] [PubMed] [Google Scholar]
- 68. Luptak I, Sverdlov AL, Panagia M, Qin F, Pimentel DR, Croteau D, Siwik DA, Ingwall JS, Bachschmid MM, Balschi JA, et al. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J Mol Cell Cardiol. 2018;116:106–114. doi: 10.1016/j.yjmcc.2018.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem. 2010;285:29834–29841. doi: 10.1074/jbc.M110.139493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Quinn KM, Palchaudhuri R, Palmer CS, La Gruta NL. The clock is ticking: the impact of ageing on T cell metabolism. Clin Transl Immunology. 2019;8:e01091. doi: 10.1002/cti2.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, Durovic B, Jauch A, Hess C. Rapid effector function of memory CD8+ T cells requires an immediate‐early glycolytic switch. Nat Immunol. 2013;14:1064–1072. doi: 10.1038/ni.2687 [DOI] [PubMed] [Google Scholar]
- 72. van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, Chang CH, Smith AM, Ai T, Faubert B, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hyyti OM, Ledee D, Ning XH, Ge M, Portman MA. Aging impairs myocardial fatty acid and ketone oxidation and modifies cardiac functional and metabolic responses to insulin in mice. Am J Physiol Heart Circ Physiol. 2010;299:H868–H875. doi: 10.1152/ajpheart.00931.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chung KW. Advances in understanding of the role of lipid metabolism in aging. Cells. 2021;10:10. doi: 10.3390/cells10040880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Russo L, Lumeng CN. Properties and functions of adipose tissue macrophages in obesity. Immunology. 2018;155:407–417. doi: 10.1111/imm.13002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kolwicz SC Jr, Olson DP, Marney LC, Garcia‐Menendez L, Synovec RE, Tian R. Cardiac‐specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure‐overload hypertrophy. Circ Res. 2012;111:728–738. doi: 10.1161/CIRCRESAHA.112.268128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fischer HJ, Sie C, Schumann E, Witte AK, Dressel R, van den Brandt J, Reichardt HM. The insulin receptor plays a critical role in T cell function and adaptive immunity. J Immunol. 2017;198:1910–1920. doi: 10.4049/jimmunol.1601011 [DOI] [PubMed] [Google Scholar]
- 78. Drosatos K. Fatty old hearts: role of cardiac lipotoxicity in age‐related cardiomyopathy. Pathobiol Aging Age Relat Dis. 2016;6:32221. doi: 10.3402/pba.v6.32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pol CJ, Lieu M, Drosatos K. PPARs: protectors or opponents of myocardial function? PPAR Res. 2015;2015:1–19. doi: 10.1155/2015/835985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mansouri RM, Bauge E, Staels B, Gervois P. Systemic and distal repercussions of liver‐specific peroxisome proliferator‐activated receptor‐alpha control of the acute‐phase response. Endocrinology. 2008;149:3215–3223. doi: 10.1210/en.2007-1339 [DOI] [PubMed] [Google Scholar]
- 81. Ding Q, Qi Y, Tsang SY. Mitochondrial biogenesis, mitochondrial dynamics, and Mitophagy in the maturation of Cardiomyocytes. Cells. 2021;10:10. doi: 10.3390/cells10092463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Babbar M, Basu S, Yang B, Croteau DL, Bohr VA. Mitophagy and DNA damage signaling in human aging. Mech Ageing Dev. 2020;186:111207. doi: 10.1016/j.mad.2020.111207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Quinn PMJ, Moreira PI, Ambrosio AF, Alves CH. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun. 2020;8:189. doi: 10.1186/s40478-020-01062-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu M, Lv J, Pan Z, Wang D, Zhao L, Guo X. Mitochondrial dysfunction in heart failure and its therapeutic implications. Front Cardiovasc Med. 2022;9:945142. doi: 10.3389/fcvm.2022.945142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Choudhuri S, Chowdhury IH, Garg NJ. Mitochondrial regulation of macrophage response against pathogens. Front Immunol. 2020;11:622602. doi: 10.3389/fimmu.2020.622602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Litwiniuk A, Baranowska‐Bik A, Domanska A, Kalisz M, Bik W. Contribution of mitochondrial dysfunction combined with NLRP3 Inflammasome activation in selected neurodegenerative diseases. Pharmaceuticals (Basel). 2021;14:14. doi: 10.3390/ph14121221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, Henson SM. Human CD8(+) EMRA T cells display a senescence‐associated secretory phenotype regulated by p38 MAPK. Aging Cell. 2018;17:17. doi: 10.1111/acel.12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chou JP, Effros RB. T cell replicative senescence in human aging. Curr Pharm Des. 2013;19:1680–1698. doi: 10.2174/138161213805219711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fulop T, Larbi A, Pawelec G, Khalil A, Cohen AA, Hirokawa K, Witkowski JM, Franceschi C. Immunology of aging: the birth of Inflammaging. Clin Rev Allergy Immunol. 2023;64:109–122. doi: 10.1007/s12016-021-08899-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Golonka RM, Xiao X, Abokor AA, Joe B, Vijay‐Kumar M. Altered nutrient status reprograms host inflammation and metabolic health via gut microbiota. J Nutr Biochem. 2020;80:108360. doi: 10.1016/j.jnutbio.2020.108360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–522. doi: 10.1038/s41569-018-0064-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Koelman L, Pivovarova‐Ramich O, Pfeiffer AFH, Grune T, Aleksandrova K. Cytokines for evaluation of chronic inflammatory status in ageing research: reliability and phenotypic characterisation. Immun Ageing. 2019;16:11. doi: 10.1186/s12979-019-0151-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Boeing H, Korfmann A, Bergmann MM. Recruitment procedures of EPIC‐Germany. European investigation into cancer and nutrition. Ann Nutr Metab. 1999;43:205–215. doi: 10.1159/000012787 [DOI] [PubMed] [Google Scholar]
- 94. Suthahar N, Meijers WC, Sillje HHW, de Boer RA. From inflammation to fibrosis‐molecular and cellular mechanisms of myocardial tissue Remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep. 2017;14:235–250. doi: 10.1007/s11897-017-0343-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117:1450–1488. doi: 10.1093/cvr/cvaa324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–2114. doi: 10.1161/ATVBAHA.108.173898 [DOI] [PubMed] [Google Scholar]
- 97. Duong L, Radley HG, Lee B, Dye DE, Pixley FJ, Grounds MD, Nelson DJ, Jackaman C. Macrophage function in the elderly and impact on injury repair and cancer. Immun Ageing. 2021;18:4. doi: 10.1186/s12979-021-00215-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–147. doi: 10.1146/annurev-pathmechdis-012418-012718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Medzhitov R, Janeway C Jr. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x [DOI] [PubMed] [Google Scholar]
- 100. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015 [DOI] [PubMed] [Google Scholar]
- 101. Zaidi Y, Aguilar EG, Troncoso M, Ilatovskaya DV, DeLeon‐Pennell KY. Immune regulation of cardiac fibrosis post myocardial infarction. Cell Signal. 2021;77:109837. doi: 10.1016/j.cellsig.2020.109837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF‐beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T. Regulation of alternative macrophage activation by galectin‐3. J Immunol. 2008;180:2650–2658. doi: 10.4049/jimmunol.180.4.2650 [DOI] [PubMed] [Google Scholar]
- 104. Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012;13:31. doi: 10.1186/1471-2172-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Long H, Lichtnekert J, Andrassy J, Schraml BU, Romagnani P, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes account for organ injury, regeneration or atrophy. Front Immunol. 2023;14:1194988. doi: 10.3389/fimmu.2023.1194988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gomez CR, Hirano S, Cutro BT, Birjandi S, Baila H, Nomellini V, Kovacs EJ. Advanced age exacerbates the pulmonary inflammatory response after lipopolysaccharide exposure. Crit Care Med. 2007;35:246–251. doi: 10.1097/01.CCM.0000251639.05135.E0 [DOI] [PubMed] [Google Scholar]
- 107. Wang Y, Wehling‐Henricks M, Samengo G, Tidball JG. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle‐derived nitric oxide. Aging Cell. 2015;14:678–688. doi: 10.1111/acel.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen R, Zhang H, Tang B, Luo Y, Yang Y, Zhong X, Chen S, Xu X, Huang S, Liu C. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. 2024;9:130. doi: 10.1038/s41392-024-01840-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Orlandi A, Francesconi A, Marcellini M, Ferlosio A, Spagnoli LG. Role of ageing and coronary atherosclerosis in the development of cardiac fibrosis in the rabbit. Cardiovasc Res. 2004;64:544–552. doi: 10.1016/j.cardiores.2004.07.024 [DOI] [PubMed] [Google Scholar]
- 110. Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol. 2022;19:250–264. doi: 10.1038/s41569-021-00624-2 [DOI] [PubMed] [Google Scholar]
- 111. Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD. Secreted protein acidic and rich in cysteine facilitates age‐related cardiac inflammation and macrophage M1 polarization. Am J Physiol Cell Physiol. 2015;308:C972–C982. doi: 10.1152/ajpcell.00402.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zanni F, Vescovini R, Biasini C, Fagnoni F, Zanlari L, Telera A, Di Pede P, Passeri G, Pedrazzoni M, Passeri M, et al. Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: a contribution to understand the relationship between inflammation and immunosenescence. Exp Gerontol. 2003;38:981–987. doi: 10.1016/s0531-5565(03)00160-8 [DOI] [PubMed] [Google Scholar]
- 113. Sansoni P, Vescovini R, Fagnoni F, Biasini C, Zanni F, Zanlari L, Telera A, Lucchini G, Passeri G, Monti D, et al. The immune system in extreme longevity. Exp Gerontol. 2008;43:61–65. doi: 10.1016/j.exger.2007.06.008 [DOI] [PubMed] [Google Scholar]
- 114. Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory‐phenotype cells. Nat Immunol. 2011;12:478–484. doi: 10.1038/ni.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li M, Yao D, Zeng X, Kasakovski D, Zhang Y, Chen S, Zha X, Li Y, Xu L. Age related human T cell subset evolution and senescence. Immun Ageing. 2019;16:24. doi: 10.1186/s12979-019-0165-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003;198:1369–1380. doi: 10.1084/jem.20030916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Noel PJ, Boise LH, Green JM, Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. 1996;157:636–642. doi: 10.4049/jimmunol.157.2.636 [DOI] [PubMed] [Google Scholar]
- 118. Ramos GC, van den Berg A, Nunes‐Silva V, Weirather J, Peters L, Burkard M, Friedrich M, Pinnecker J, Abesser M, Heinze KG, et al. Myocardial aging as a T‐cell‐mediated phenomenon. Proc Natl Acad Sci USA. 2017;114:E2420–E2429. doi: 10.1073/pnas.1621047114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Minato N, Hattori M, Hamazaki Y. Physiology and pathology of T‐cell aging. Int Immunol. 2020;32:223–231. doi: 10.1093/intimm/dxaa006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tahir S, Fukushima Y, Sakamoto K, Sato K, Fujita H, Inoue J, Uede T, Hamazaki Y, Hattori M, Minato N. A CD153+CD4+ T follicular cell population with cell‐senescence features plays a crucial role in lupus pathogenesis via osteopontin production. J Immunol. 2015;194:5725–5735. doi: 10.4049/jimmunol.1500319 [DOI] [PubMed] [Google Scholar]
- 121. Suda M, Paul KH, Minamino T, Miller JD, Lerman A, Ellison‐Hughes GM, Tchkonia T, Kirkland JL. Senescent cells: a therapeutic target in cardiovascular diseases. Cells. 2023;12:12. doi: 10.3390/cells12091296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res. 2008;102:319–327. doi: 10.1161/CIRCRESAHA.107.160408 [DOI] [PubMed] [Google Scholar]
- 123. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, Brioschi S, Shchukina I, Kerndl M, Bambouskova M, et al. Comprehensive profiling of an aging immune system reveals clonal GZMK(+) CD8(+) T cells as conserved Hallmark of Inflammaging. Immunity. 2021;54:99–115.e12. doi: 10.1016/j.immuni.2020.11.005 [DOI] [PubMed] [Google Scholar]
- 124. Duquette D, Harmon C, Zaborowski A, Michelet X, O'Farrelly C, Winter D, Koay HF, Lynch L. Human Granzyme K is a feature of innate T cells in blood, tissues, and tumors, responding to cytokines rather than TCR stimulation. J Immunol. 2023;211:633–647. doi: 10.4049/jimmunol.2300083 [DOI] [PubMed] [Google Scholar]
- 125. Tyrrell DJ, Wragg KM, Chen J, Wang H, Song J, Blin MG, Bolding C, Vardaman D 3rd, Giles K, Tidwell H, et al. Clonally expanded memory CD8(+) T cells accumulate in atherosclerotic plaques and are pro‐atherogenic in aged mice. Nat Aging. 2023;3:1576–1590. doi: 10.1038/s43587-023-00515-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rufer N, Brummendorf TH, Kolvraa S, Bischoff C, Christensen K, Wadsworth L, Schulzer M, Lansdorp PM. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J Exp Med. 1999;190:157–167. doi: 10.1084/jem.190.2.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hickman SP, Turka LA. Homeostatic T cell proliferation as a barrier to T cell tolerance. Philos Trans R Soc Lond Ser B Biol Sci. 2005;360:1713–1721. doi: 10.1098/rstb.2005.1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Shirakawa K, Sano M. T cell Immunosenescence in aging, obesity, and cardiovascular disease. Cells. 2021;10:10. doi: 10.3390/cells10092435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Henein MY, Vancheri S, Longo G, Vancheri F. The role of inflammation in cardiovascular disease. Int J Mol Sci. 2022;23:23. doi: 10.3390/ijms232112906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Palmer DB. The effect of age on thymic function. Front Immunol. 2013;4:316. doi: 10.3389/fimmu.2013.00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Moro‐Garcia MA, Alonso‐Arias R, Lopez‐Larrea C. When aging reaches CD4+ T‐cells: phenotypic and functional changes. Front Immunol. 2013;4:107. doi: 10.3389/fimmu.2013.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, Esposito B, Perez N, Yasukawa H, Van Snick J, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin‐17 in atherosclerosis. J Exp Med. 2009;206:2067–2077. doi: 10.1084/jem.20090545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387–401. doi: 10.1038/s41569-020-0352-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Peiseler M, Kubes P. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Invest. 2019;129:2629–2639. doi: 10.1172/JCI124616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553:418–426. doi: 10.1038/nature25022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sreejit G, Johnson J, Jaggers RM, Dahdah A, Murphy AJ, Hanssen NMJ, Nagareddy PR. Neutrophils in cardiovascular disease: warmongers, peacemakers, or both? Cardiovasc Res. 2022;118:2596–2609. doi: 10.1093/cvr/cvab302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. 2019;20:303–320. doi: 10.1038/s41580-019-0103-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. Tet2‐mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL‐1beta/NLRP3 Inflammasome. J Am Coll Cardiol. 2018;71:875–886. doi: 10.1016/j.jacc.2017.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Van Avondt K, Strecker JK, Tulotta C, Minnerup J, Schulz C, Soehnlein O. Neutrophils in aging and aging‐related pathologies. Immunol Rev. 2023;314:357–375. doi: 10.1111/imr.13153 [DOI] [PubMed] [Google Scholar]
- 140. Zinovkin RA, Zamyatnin AA. Mitochondria‐targeted drugs. Curr Mol Pharmacol. 2019;12:202–214. doi: 10.2174/1874467212666181127151059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady‐state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. doi: 10.1016/j.trsl.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tomay F, Wells K, Duong L, Tsu JW, Dye DE, Radley‐Crabb HG, Grounds MD, Shavlakadze T, Metharom P, Nelson DJ, et al. Aged neutrophils accumulate in lymphoid tissues from healthy elderly mice and infiltrate T‐ and B‐cell zones. Immunol Cell Biol. 2018;96:831–840. doi: 10.1111/imcb.12046 [DOI] [PubMed] [Google Scholar]
- 143. Sauce D, Dong Y, Campillo‐Gimenez L, Casulli S, Bayard C, Autran B, Boddaert J, Appay V, Elbim C. Reduced oxidative burst by primed neutrophils in the elderly individuals is associated with increased levels of the CD16bright/CD62Ldim immunosuppressive subset. J Gerontol A Biol Sci Med Sci. 2017;72:163–172. doi: 10.1093/gerona/glw062 [DOI] [PubMed] [Google Scholar]
- 144. Marc Appel SF. Gustavo Campos Ramos. Myocardial inflammation comes of age. Curr Opin Physio. 2020;19:8–54. doi: 10.1016/j.cophys.2020.09.006 [DOI] [Google Scholar]
- 145. Valdes A, Treuer AV, Barrios G, Ponce N, Fuentealba R, Dulce RA, Gonzalez DR. NOX inhibition improves beta‐adrenergic stimulated contractility and intracellular calcium handling in the aged rat heart. Int J Mol Sci. 2018;19:19. doi: 10.3390/ijms19082404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Szekeres FLM, Walum E, Wikstrom P, Arner A. A small molecule inhibitor of Nox2 and Nox4 improves contractile function after ischemia‐reperfusion in the mouse heart. Sci Rep. 2021;11:11970. doi: 10.1038/s41598-021-91575-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Nabeebaccus AA, Reumiller CM, Shen J, Zoccarato A, Santos CXC, Shah AM. The regulation of cardiac intermediary metabolism by NADPH oxidases. Cardiovasc Res. 2023;118:3305–3319. doi: 10.1093/cvr/cvac030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Schurmann C, Rezende F, Kruse C, Yasar Y, Lowe O, Fork C, van de Sluis B, Bremer R, Weissmann N, Shah AM, et al. The NADPH oxidase Nox4 has anti‐atherosclerotic functions. Eur Heart J. 2015;36:3447–3456. doi: 10.1093/eurheartj/ehv460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Pawlas N, Nierwinska K, Stolecka A, Gabryel B, Pudelko A, Malecki A. Effects of N‐acetylcysteine and ebselen on arachidonic acid release from astrocytes and neurons cultured in normoxic or simulated ischemic conditions. Pharmacol Rep. 2009;61:941–946. doi: 10.1016/s1734-1140(09)70153-7 [DOI] [PubMed] [Google Scholar]
- 150. Phaelante A, Rohde LE, Lopes A, Olsen V, Tobar SA, Cohen C, Martinelli N, Biolo A, Dal‐Pizzol F, Clausell N, et al. N‐acetylcysteine plus Deferoxamine improves cardiac function in Wistar rats after non‐reperfused acute myocardial infarction. J Cardiovasc Transl Res. 2015;8:328–337. doi: 10.1007/s12265-015-9633-5 [DOI] [PubMed] [Google Scholar]
- 151. Raghu G, Berk M, Campochiaro PA, Jaeschke H, Marenzi G, Richeldi L, Wen FQ, Nicoletti F, Calverley PMA. The multifaceted therapeutic role of N‐Acetylcysteine (NAC) in disorders characterized by oxidative stress. Curr Neuropharmacol. 2021;19:1202–1224. doi: 10.2174/1570159X19666201230144109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Shafiei E, Bahtoei M, Raj P, Ostovar A, Iranpour D, Akbarzadeh S, Shahryari H, Anvaripour A, Tahmasebi R, Netticadan T, et al. Effects of N‐acetyl cysteine and melatonin on early reperfusion injury in patients undergoing coronary artery bypass grafting: a randomized, open‐labeled, placebo‐controlled trial. Medicine (Baltimore). 2018;97:e11383. doi: 10.1097/MD.0000000000011383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Oskuye ZZ, Mehri K, Mokhtari B, Bafadam S, Nemati S, Badalzadeh R. Cardioprotective effect of antioxidant combination therapy: a highlight on MitoQ plus alpha‐lipoic acid beneficial impact on myocardial ischemia‐reperfusion injury in aged rats. Heliyon. 2024;10:e28158. doi: 10.1016/j.heliyon.2024.e28158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Young ML, Franklin JL. The mitochondria‐targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg‐AD mice. Mol Cell Neurosci. 2019;101:103409. doi: 10.1016/j.mcn.2019.103409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Fortner KA, Blanco LP, Buskiewicz I, Huang N, Gibson PC, Cook DL, Pedersen HL, Yuen PST, Murphy MP, Perl A, et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus‐prone MRL‐lpr mice. Lupus Sci Med. 2020;7:7. doi: 10.1136/lupus-2020-000387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Rossman MJ, Santos‐Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M, Gioscia‐Ryan RA, Murphy MP, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71:1056–1063. doi: 10.1161/HYPERTENSIONAHA.117.10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Masoumi‐Ardakani Y, Najafipour H, Nasri HR, Aminizadeh S, Jafari S, Safi Z. Moderate endurance training and MitoQ improve cardiovascular function, oxidative stress, and inflammation in hypertensive individuals: the role of miR‐21 and miR‐222: a randomized, double‐blind, clinical trial. Cell J. 2022;24:577–585. doi: 10.22074/cellj.2022.8089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171:2017–2028. doi: 10.1111/bph.12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chavez JD, Tang X, Campbell MD, Reyes G, Kramer PA, Stuppard R, Keller A, Zhang H, Rabinovitch PS, Marcinek DJ, et al. Mitochondrial protein interaction landscape of SS‐31. Proc Natl Acad Sci USA. 2020;117:15363–15373. doi: 10.1073/pnas.2002250117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Szeto HH. First‐in‐class cardiolipin‐protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–2050. doi: 10.1111/bph.12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014;96:672–683. doi: 10.1038/clpt.2014.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7(Suppl):S41–S50. doi: 10.1016/j.mito.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 163. Tsai KL, Huang YH, Kao CL, Yang DM, Lee HC, Chou HY, Chen YC, Chiou GY, Chen LH, Yang YP, et al. A novel mechanism of coenzyme Q10 protects against human endothelial cells from oxidative stress‐induced injury by modulating NO‐related pathways. J Nutr Biochem. 2012;23:458–468. doi: 10.1016/j.jnutbio.2011.01.011 [DOI] [PubMed] [Google Scholar]
- 164. Li H, Liu W, Xie J. Circulating interleukin‐6 levels and cardiovascular and all‐cause mortality in the elderly population: a meta‐analysis. Arch Gerontol Geriatr. 2017;73:257–262. doi: 10.1016/j.archger.2017.08.007 [DOI] [PubMed] [Google Scholar]
- 165. Meyer MAS, Wiberg S, Grand J, Meyer ASP, Obling LER, Frydland M, Thomsen JH, Josiassen J, Moller JE, Kjaergaard J, et al. Treatment effects of Interleukin‐6 receptor antibodies for modulating the systemic inflammatory response after out‐of‐hospital cardiac arrest (the IMICA trial): a double‐blinded, placebo‐controlled, single‐center, randomized, clinical trial. Circulation. 2021;143:1841–1851. doi: 10.1161/CIRCULATIONAHA.120.053318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bruunsgaard H, Skinhoj P, Pedersen AN, Schroll M, Pedersen BK. Ageing, tumour necrosis factor‐alpha (TNF‐alpha) and atherosclerosis. Clin Exp Immunol. 2000;121:255–260. doi: 10.1046/j.1365-2249.2000.01281.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Low AS, Symmons DP, Lunt M, Mercer LK, Gale CP, Watson KD, Dixon WG, Hyrich KL; British Society for Rheumatology Biologics Register for Rheumatoid A, the BCCC . Relationship between exposure to tumour necrosis factor inhibitor therapy and incidence and severity of myocardial infarction in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:654–660. doi: 10.1136/annrheumdis-2016-209784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ljung L, Askling J, Rantapaa‐Dahlqvist S, Jacobsson L; Group AS . The risk of acute coronary syndrome in rheumatoid arthritis in relation to tumour necrosis factor inhibitors and the risk in the general population: a national cohort study. Arthritis Res Ther. 2014;16:R127. doi: 10.1186/ar4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Interleukin‐6 Receptor Mendelian Randomisation Analysis C , Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, Guo Y, Chung C, Peasey A, et al. The interleukin‐6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379:1214–1224. doi: 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Weinblatt ME, Bathon JM, Kremer JM, Fleischmann RM, Schiff MH, Martin RW, Baumgartner SW, Park GS, Mancini EL, Genovese MC. Safety and efficacy of etanercept beyond 10 years of therapy in north American patients with early and longstanding rheumatoid arthritis. Arthritis Care Res. 2011;63:373–382. doi: 10.1002/acr.20372 [DOI] [PubMed] [Google Scholar]
- 171. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti TNFTACHFI. Randomized, double‐blind, placebo‐controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor‐alpha, in patients with moderate‐to‐severe heart failure: results of the anti‐TNF therapy against congestive heart failure (ATTACH) trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2 [DOI] [PubMed] [Google Scholar]
- 172. Hussain A, Tarahomi T, Singh L, Bollampally M, Heydari‐Kamjani M, Kesselman MM. Cardiovascular risk associated with TNF alpha inhibitor use in patients with rheumatoid arthritis. Cureus. 2021;13:e17938. doi: 10.7759/cureus.17938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 174. Liu X, Chen J, Yue S, Zhang C, Song J, Liang H, Liang C, Chen X. NLRP3‐mediated IL‐1beta in regulating the imbalance between Th17 and Treg in experimental autoimmune prostatitis. Sci Rep. 2024;14:18829. doi: 10.1038/s41598-024-69512-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin‐1 in heart disease. Circulation. 2013;128:1910–1923. doi: 10.1161/CIRCULATIONAHA.113.003199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Buckley LF, Abbate A. Interleukin‐1 blockade in cardiovascular diseases: a clinical update. Eur Heart J. 2018;39:2063–2069. doi: 10.1093/eurheartj/ehy128 [DOI] [PubMed] [Google Scholar]
- 177. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti‐TNFalpha therapy. J Exp Med. 2004;200:277–285. doi: 10.1084/jem.20040165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Andersen NN, Jess T. Risk of infections associated with biological treatment in inflammatory bowel disease. World J Gastroenterol. 2014;20:16014–16019. doi: 10.3748/wjg.v20.i43.16014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, Carbone F, Fontana S, Horvath TL, La Cava A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–941. doi: 10.1016/j.immuni.2010.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wang X, Zhou H, Liu Q, Cheng P, Zhao T, Yang T, Zhao Y, Sha W, Zhao Y, Qu H. Targeting regulatory T cells for cardiovascular diseases. Front Immunol. 2023;14:1126761. doi: 10.3389/fimmu.2023.1126761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Miossec P, Kolls JK. Targeting IL‐17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794 [DOI] [PubMed] [Google Scholar]
- 182. Cheng X, Yu X, Ding YJ, Fu QQ, Xie JJ, Tang TT, Yao R, Chen Y, Liao YH. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol. 2008;127:89–97. doi: 10.1016/j.clim.2008.01.009 [DOI] [PubMed] [Google Scholar]
- 183. Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta. 2010;411:1963–1968. doi: 10.1016/j.cca.2010.08.013 [DOI] [PubMed] [Google Scholar]
- 184. Zhang Y, Sun D, Zhao X, Luo Y, Yu H, Zhou Y, Gao Y, Han X, Duan Y, Fang N, et al. Bacteroides fragilis prevents aging‐related atrial fibrillation in rats via regulatory T cells‐mediated regulation of inflammation. Pharmacol Res. 2022;177:106141. doi: 10.1016/j.phrs.2022.106141 [DOI] [PubMed] [Google Scholar]