

Abstract
Chalcones are indeed a versatile scaffold in medicinal chemistry. Their structure, featuring an α,β-unsaturated carbonyl group, makes them highly reactive and capable of interacting with various biological targets. This reactivity is a key reason why chalcones and their derivatives are of such interest in drug discovery. The continued exploration of chalcone derivatives in medicinal chemistry will likely yield new insights and therapeutic candidates, given their broad spectrum of biological activities and the flexibility in modifying their structures. As chalcone derivatives, pyranochalcones and chromanchalcones are members of a subclass of flavonoids that are widely distributed. Several scientific databases were investigated to compile articles that illustrated the biological functions of chromenochalcones and their derivatives. Preclinical research on chromenochalcones and their derivatives is well covered in this review, highlighting the compounds with enormous significance as antimalarial, anti-inflammatory, antileishmanial, cytotoxic, antibacterial, antifungal, and antioxidant agents. In addition, the article briefly discusses the synthetic pathways employed for the total synthesis of selected pyranochalcones, including mallaophilippens C and E, citrunobin, and lesperol. Consequently, this overview may help research and design novel, potent therapeutic medications based on previously developed methodologies. This review is intended to provide a thorough, authoritative, and critical assessment of the chromenochalcone template for the chemistry community.
Chalcones are indeed a versatile scaffold in medicinal chemistry.
1. Introduction
Chalcone, also known as ‘(E)-1,3-diphenyl-2-propene-1-one’, is a crucial precursor to flavonoids and isoflavonoids. It serves as the open-chain intermediate in the synthesis of flavones. Chalcones refer to the α,β-unsaturated ketones formed when an aryl methyl ketone and an aromatic aldehyde are combined in the presence of a base.1,2 They typically exist in various conjugated forms featuring a benzylideneacetophenone scaffold, where two aromatic rings are linked by an α,β-unsaturated carbonyl bridge. Kostanecki originally conceived the concept and created a range of natural products with chromophoric properties. They undergo a variety of chemical reactions and are discovered to synthesize pyrazoline, isoxazole, and different heterocyclic compounds. Chalcone chemistry has intrigued researchers in the 21st century due to its straightforward chemistry, simple synthesis process, abundance of hydrogen atoms that can be replaced to create a wide range of derivatives, and its potential for various beneficial biological effects.3 Chromenochalcones and pyranochalcones, depicted in Fig. 1, are derivatives of chalcone and belong to a plentiful subclass of flavonoids found extensively in nature. Several pyranochalcones, also known as chromenochalcones, have been documented to display antimalarial, antileishmanial, antimicrobial, antiallergic, chemopreventive, and anti-inflammatory properties as depicted in Fig. 2.4 Certain botanical species that possess these chemical compounds have been utilized in traditional medicinal practices in China as well as in Europe to treat a range of ailments.5,6 This discussion provides a concise overview of naturally occurring and synthetic chromenochalcones (pyranochalcones), focusing on their biological activities and pharmacological properties. Additionally, it offers information on the synthetic routes of certain naturally occurring chromenochalcones (pyranochalcones). This review is supplementary to previous research and encompasses more up-to-date findings on the antimalarial, antileishmanial, cytotoxic, and anti-inflammatory properties of these compounds.
Fig. 1. General structure of the chalcone and chromeno/chromenochalcones.

Fig. 2. Bioactive chromenochalcones with therapeutic potential.4.

2. Antimalarial activity
Plasmodium falciparum and P. vivax are the two primary human malaria parasites. The former, P. falciparum, is the primary cause of the majority of fatalities, and it has acquired resistance to almost all currently accessible medications.7 The increasing prevalence of Plasmodium falciparum resistance to antimalarials has prompted the exploration of alternative treatments. Any resources that can lessen the impact of lethal malaria in this context are valuable to investigate, including the hunt for novel biologically active components derived from plants.8 Among these, both synthetic and naturally occurring chalcones have shown encouraging potential.
Ngameni et al. isolated chromenochalcones, i.e., 4-hydroxylonchocarpin (1) and kanzonol B (2), from the twigs of Dorstenia barteri var. subtriangularis (Moraceae) utilizing chromatographic methods.9 The compounds (1 and 2) were assessed in vitro against the W2 strain of Plasmodium falciparum. The results indicated that both compounds exhibited significant antiplasmodial activity, demonstrating potency with relatively low IC50 values (3.36 μM and 9.61 μM, respectively).9 This confirmed the chromenochalcones as potential leads for the development of antimalarials.

α-Pyranochalcones and pyrazoline analogs were synthesized by Wanare et al. to discover chemically diverse antimalarial leads. The antimalarial activity of the compounds was evaluated by assessing the growth of malaria causing parasites in culture using the microtiter plate-based SYBR-Green-I assay. Among tested compounds, (E)-3-(3-(2,3,4-trimethoxyphenyl)-acryloyl)-2H-chromene-2-one (3) emerged as the most potent analog in the series. It exhibited an IC50 value of 3.1 μg mL−1 against the chloroquine-sensitive Plasmodium falciparum strain 3D7, and an IC50 value of 1.1 μg mL−1 against the chloroquine-resistant field isolate RKL9.10

Frölich et al. conducted an in vitro evaluation of the antiplasmodial properties of xanthohumol, a prominent chalcone derived from hops, alongside seven additional natural or semi-synthetic derivatives. The study evaluated their efficacy against two distinct strains of Plasmodium falciparum and their interactions with glutathione (GSH)-dependent haemin degradation pathways. The antiplasmodial activity was measured using a [3H] hypoxanthine incorporation assay against both a chloroquine-sensitive strain (poW) and a multidrug-resistant clone (Dd2). Out of eight compounds tested, four exhibited activities with IC50 values below 25 μM against at least one of the strains. Notably, two pyrano-derivatives (compounds 4 and 5) in which the prenyl moiety undergoes cyclization to form an additional ring system also exhibit antiplasmodial activity with IC50 values of 16.4 μM and 23.7 μM against poW, respectively, and antiplasmodial activity with IC50 values of 10.7 and 35.0 μM against Dd2 strains of P. falciparum, respectively. The presence of a 2,3-pyrano ring system in compound 4, rather than a free hydroxyl group, clearly impaired its ability to interact with hemin, highlighting the critical role of this structural feature in modulating the observed bioactivity.11

Chromenodihydrochalcones, which were separated from C. ramosissima, were stated as antimalarial agents by our group. The in vitro antimalarial properties of all compounds were assessed at concentrations of 50, 10, and 2 μg mL−1 against the Plasmodium falciparum strain NF-54. The chromenodihydrochalcones (compounds 6–8) exhibited lower levels of activity. Compound 6, involved in chromene formation, featuring a 4′-hydroxyl group and lacking an α–β unsaturated double bond, achieved 100 percent inhibition at a concentration of 10 μg mL−1. Its 4-O-methyl derivative, compound 7, demonstrated the same level of inhibition at a concentration of 50 μg mL−1, whereas compound 8 was inactive at 50 μg mL−1.12

Based on the lead molecule found in natural products, we recently synthesized a library of 88 chalcones with a variety of structural characteristics, including prenylated chalcones, chromanochalcones, chromenochalcones, and chromenodihydrochalcones. Our structure–activity relationship (SAR) studies revealed that the presence of the benzopyran core, along with hydroxyl and aldehyde substituents on ring A, is critical for activity. Additionally, the inclusion of an alkenyl (prenyl or C-5 unit) substituent on the benzopyran core, as seen in compounds 9 to 13, and an aminoalkyl substituent on the hydroxyl groups, as in compounds 14 and 15, as well as a heteroatom at the 3-position on ring A, is significant for enhancing the antimalarial efficacy (16–18) of chalcones. Several compounds from this synthetic series exhibited promising in vitro activity. The chromenochalcones 9, 10, 11, 12, and 13 have an IC50 of 347.37, 125.34, 73.22, 94.71, and 87.59 nM, respectively, against the CQS strain (3D7), and an IC50 of 932, 1093.34, 366.26, 84.20, and >1196.18 nM respectively against the CQR strain (K1). Additionally, certain compounds inhibited the growth of parasitemia and prolonged the lifespan of Swiss mice that were vitiated with the chloroquine-resistant P. yoelii N-67 strain. Compound 9 demonstrated 98.06% parasitemia suppression on day four, whereas compounds 10, 11, 12, and 13 achieved parasitemia suppression rates of 89%, 71%, 88%, and 87%, respectively. Additionally, compounds 9, 10, and 13 were assessed against the cysteine proteases falcipain-2 (FP-2) and falcipain-3 (FP-3) for their inhibitory activity. Compounds 9 and 13 exhibited no inhibitory effects on FP-3. However, at higher concentrations, they stimulated the activity of the enzyme. Compound 10 demonstrated marginal inhibition of FP-3 at lower concentrations; however, it exhibited an intriguing effect of enhancing the enzyme's activity at higher concentrations. Notably, compounds 9, 10, and 13 repressed FP-2 with an IC50 of 13.53, 5.53, and 4.64 μM, respectively, aligning with their observed in vitro antimalarial activity (IC50 of 347.2 nm, 125.3 nM, and 87.5 nM).13

3. Antileishmanial activity
Leishmaniasis is an illness caused by protozoan parasites of the Leishmania donovani genus. The parasites exist in two distinct developmental stages i.e., the ‘extracellular promastigote’, which is transmitted through the sting of the sandfly vector, and the ‘intracellular amastigote’, which is a parasite that exclusively resides within macrophages.14 There are very few treatment options available for leishmaniasis. The current primary medications were first introduced more than five decades ago, and all drug regimens have notable disadvantages. The initial therapy utilizing pentavalent antimony (meglumine antimonate and sodium stibogluconate) is associated with significant and undesirable adverse reactions. In India, the level of resistance to these compounds has reached a point where they are no longer viable for use in numerous regions.15,16
Furthermore, the utilization of pentamidine and amphotericin B, which are second-line medications, is restricted due to their toxicity. Liposomal amphotericin B is a highly efficacious choice; nevertheless, the high cost of these drug formulations restricts their utilization in most endemic areas. Miltefosine, a recently approved medication in India, is the initial oral treatment option for visceral leishmaniasis. Leishmaniasis has been classified by the WHO as a “neglected and emerging disease”, and there is a recognized need for new drugs to combat the parasites responsible for it. Given the population situation in developing countries at risk from the disease, it is necessary to develop new drugs that are selective, non-toxic, affordable, and can be taken orally. Hence, it is crucial to prioritize the quest for groundbreaking medicines that are developed using distinct molecular frameworks and targeted towards unexplored biological systems.17,18 Recently, researchers have discovered that a range of synthetic and naturally occurring compounds called chalcone derivatives show promise as potential treatments for leishmaniasis. These compounds have been tested in laboratory and animal studies, both in vitro and in vivo assays. Licochalcone-A is one of the few naturally occurring chalcones that is currently being studied, despite the testing of numerous synthetic compounds.
We have isolated chromenodihydrochalcones (19–22) and synthesized some chromenochalcones (1 and 23), containing a 2′,2′-dimethyl benzopyran system and tested against intracellular amastigotes residing within murine macrophages and extracellular promastigotes of L. donovani. Our chromenochalcones exhibited inhibition rates ranging from 30% to 89% in promastigote viability and from 22% to 74% in amastigote multiplication at a concentration of 50 μg mL−1. Among these, the most potent compound, chromenochalcone 1, which possesses a hydroxyl group at the 4-position on ring A, demonstrated 84% inhibition against promastigotes and 74% inhibition against amastigotes at a 50 μg mL−1 concentration. Our results showed that the chalcones, which contain a 2′,2′-dimethyl benzopyran system, are more potent antileishmanial agents than the simple chalcones.19 Continuing our research, we created several new chromenochalcones and tested their effectiveness against intracellular amastigotes of L. donovani and in vitro extracellular promastigotes. The most potent compound in this series, compound 17, featuring a pyridine ring at position A, exhibited 99% inhibition of promastigotes at a concentration of 10 μg mL−1, 82% inhibition at 0.25 μg mL−1, and 96% inhibition of amastigotes at 10 μg mL−1.20 Furthermore, Shivahare et al. evaluated the efficacy of compound 17 in a Leishmania donovani–hamster model, administering a dose of 100 mg kg−1 body weight per day for five consecutive days. The treatment resulted in over 84% parasite inhibition by day 7 post-treatment, with sustained activity observed until day 28. Molecular and immunological analyses further revealed the dual mechanism of action of compound 17, functioning both as a direct antiparasitic agent and as a host immunostimulant. Additionally, the study suggested its potential applicability in the treatment of non-healing forms of leishmaniasis.21
Gupta et al. reported the synthesis of various synthetic chalcone derivatives; among them an alkylated amine-containing chromenochalcone (compound 24) exhibited potent antileishmanial activity. The study indicated that the incorporation of alkylated amino substituents on rings A and B significantly enhanced the compound's bioavailability, leading to improved activity compared to its parent analogues (IC50 = 2.04 μM, SI = 162.5) against amastigotes. Given its promising in vitro efficacy against both extracellular promastigotes and intracellular amastigotes, the investigation was extended to an in vivo model using the L. donovani MHOM/IN/80/Dd8 strain in a hamster model. The findings demonstrated that when compound 24, administered intraperitoneally at doses of 50 or 100 mg kg−1 per day for 5 to 10 consecutive days, resulted in 48.54 ± 10.55% parasite inhibition at 50 mg kg−1 per day on day 7 post-treatment.22

Foroumadi et al. synthesized two types of novel chromenochalcones, 25–29 (type A) and 30–34 (type B), to study their antileishmanial activity against the promastigote form of Leishmania major (Table 1). In the primary screening assay, the Leishmania parasite was exposed to a 10 μM concentration of the synthesized compounds 25–29 and 30–34 for 3 consecutive days. The growth inhibitory effect of these compounds was monitored on day 1, day 2, and day 3, with results presented in Table 1. Compounds 25–28 (type A chalcones) demonstrated excellent antileishmanial activity, achieving 100% inhibition at 10 μM. Compound 25 initially showed no inhibition on day 1 but after three days, the compound demonstrated significant inhibitory activity, achieving 93% inhibition. In contrast, the other compounds (type B chalcones) exhibited only limited to moderate inhibition at the same concentration.23
Table 1. In vitro activities of chromene-based chalcones 25–29 and 30–34 against the promastigote form of L. major.

| |||||
|---|---|---|---|---|---|
| Compound | R | % inhibition at the concentration of 10a μM | |||
| Day 1 | Day 2 | Day 3 | IC50b (μM) | ||
| 25 | H | 0.0 | 18.8 | 93 | 30 ± 0.06 |
| 26 | 2-Cl | 100 | 100 | 100 | 5 ± 0.14 |
| 27 | 3-Cl | 100 | 100 | 100 | 0.8 ± 0.26 |
| 28 | 4-Cl | 100 | 100 | 100 | 0.7 ± 0.3 |
| 29 | 2,4-Cl2 | 98.1 | 100 | 100 | 0.75± 0.23 |
| 30 | H | 0.0 | 12.8 | 62.8 | 45 ± 0.15 |
| 31 | 2-Cl | 0.0 | 0.0 | 1.0 | >50 |
| 32 | 3-Cl | 0.0 | 0.0 | 75.2 | 45 ± 0.22 |
| 33 | 4-Cl | 0.0 | 0.0 | 5.4 | >50 |
| 34 | 2,4-Cl2 | 4.3 | 4.3 | 8.4 | >50 |
| Glucantime | 81.97 ± 3.85c | ||||
The growth inhibitory effect of compounds on Leishmania parasite for three consecutive days.
The values represent mean ± SD,
IC50 in mM.
The test compounds' IC50 values against L. major indicate that most compounds possessed good leishmanicidal activity (IC50 ≤ 50 μM) concerning reference drugs (Table 1). The most potent compounds against L. major's promastigote form were chloro substituted type A chalcones (27–29) with IC50 values less than 1.0 μM.
Nazarian et al. synthesized a collection of new chalconoids that incorporate a 6-chloro-2H-chromene-3-yl group. They then assessed the effectiveness of these compounds against the promastigote form of Leishmania major, a parasite responsible for causing leishmaniasis. All the assessed compounds have shown high in vitro antileishmanial activity at concentrations of <3.0 μM. Among these compounds, 35 and 36 (containing 2-chlorophenyl and 2-bromo phenyl, respectively) were more potent with IC50 values of 1.22 ± 0.31 and 1.33 ± 0.52 μM, respectively.24

4. Anti-inflammatory activity
Mast cells, neutrophils, and macrophages are likely to be crucial participants in inflammatory disorders. The activation of microglial cells is also a critical factor in diseases related to inflammation of the central nervous system. Hence, the suppression of the activation of these inflammatory cells emerges as a crucial objective for the design of small molecule drugs to address inflammatory disorders. Nitric oxide (NO) is a key factor in the cytotoxicity caused by macrophages, and the presence of NO may contribute to the development of septic shock.25 Excessive production of nitric oxide (NO) can also lead to the destruction of healthy tissues during both acute and chronic inflammation. This phenomenon is mechanistically closely linked to carcinogenesis.26 Therefore, substances that hinder the production of nitric oxide in macrophages have the potential to be used for anti-inflammatory and cancer chemopreventive drugs.
Three novel chromenochalcones, mallotophilippens C (37), D (38), and E (39), were isolated from the Mallotus philippinensis fruits.27 These compounds effectively inhibited nitric oxide production and suppressed the expression of the inducible nitric oxide synthase (iNOS) gene in a murine macrophage-like cell line (RAW 264.7) activated by lipopolysaccharide (LPS) and recombinant mouse interferon-γ (IFN-γ). Additionally, they down regulated the expression of genes encoding cyclooxygenase-2 (COX-2), interleukin-6 (IL-6), and interleukin-1β (IL-1β). Xanthohumol B 40 isolated from hops (H. lupulus)28 inhibited the production of NO-induced by LPS and INF-γ in the murine macrophage-like cell line, RAW 264.7.

From the methanol extracts of Psoralea corylifolia, Lee et al. through activity guided purification, two inhibitors of NO production from lipopolysaccharide (LPS)-activated microglia were successfully isolated, alongside two compounds that exhibited no inhibitory activity. The active compounds were recognized as chromenoflavanone [7,8-dihydro-8-(4-hydroxyphenyl)-2,2-dimethyl-2H,6H-benzo-(1,2-b:5,4-b′) dipyran-6-one] (compound 41) and 4-hydroxylonchocarpin (compound 1). Notably, compound 1 was isolated from this plant for the first time. Both compounds 41 and 1 inhibited NO production in LPS-activated microglia in a dose-dependent manner, with IC50 values of 11.4 mM and 10.2 mM, respectively. Additionally, they suppressed the expression of iNOS protein and mRNA in LPS-activated microglial cells at a concentration of 10 μM, as evidenced by western blot analysis and RT-PCR experiments. Additionally, these compounds were found to inhibit the degradation of I-κB-α in activated microglia. These findings suggest that compounds 41 and 1 could serve as lead compounds for the development of neuroprotective drugs that inhibit NO overproduction in activated microglial cells.29

Han et al. isolated a novel prenylated chalcone, 3′′,3′′-dimethylpyrano[3′,4′]2,4,2′-trihydroxychalcone (42), from the heartwood of Artocarpus communis. Additionally, two new flavonoids and eight known flavonoids were identified from this plant for the first time. Compound 42 exhibited potent inhibitory activity on nitric oxide (NO) production in LPS-activated RAW 264.7 mouse macrophage cells, with an IC50 value of 18.8 μM.30
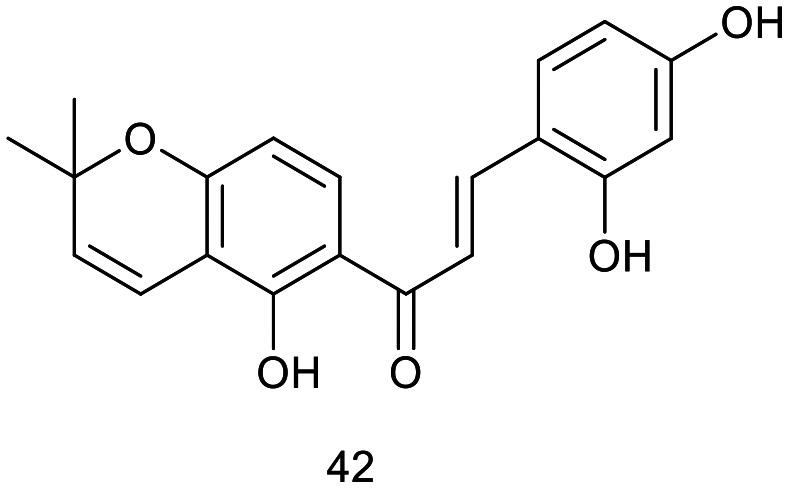
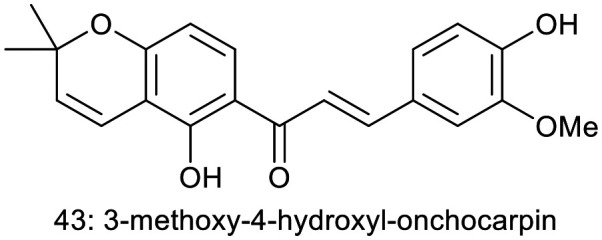
J. H. Jeon et al. synthesized and evaluated the anti-inflammatory activity of a natural chalcone, 3-methoxy-4-hydroxyl-onchocarpin (43), which was originally isolated from the roots of Lonchocarpus utilis. This chalcone demonstrated significant suppression of NO production at a concentration of 10 μM. Furthermore, cell viability assays indicated that the synthetic chalcone did not exhibit cytotoxicity at this concentration. The IC50 value of synthetic chalcone (43) was 3.35 μM.31
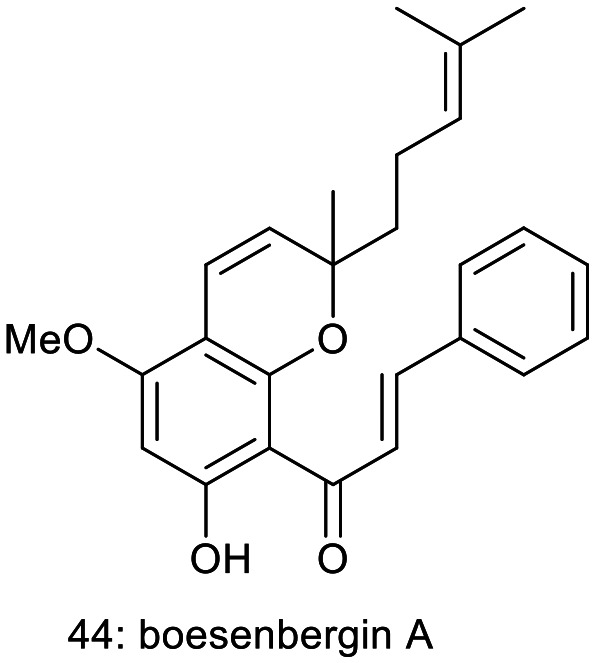
Isa et al. isolated boesenbergin A (44), a chalcone derivative of known structure from Boesenbergia rotunda. They studied the anti-inflammatory, cytotoxic and antioxidant activities of (44). Compound 44 exhibited significant anti-inflammatory activity at concentrations ranging from 12.5 to 50 μg mL−1, without showing considerable cytotoxicity in the murine macrophage cell line RAW 264.7 at 50 μg mL−1. The biological activity observations in this study suggest that compound 44 may be one of the active agents responsible for the reported biological effects of the B. rotunda crude extract.32
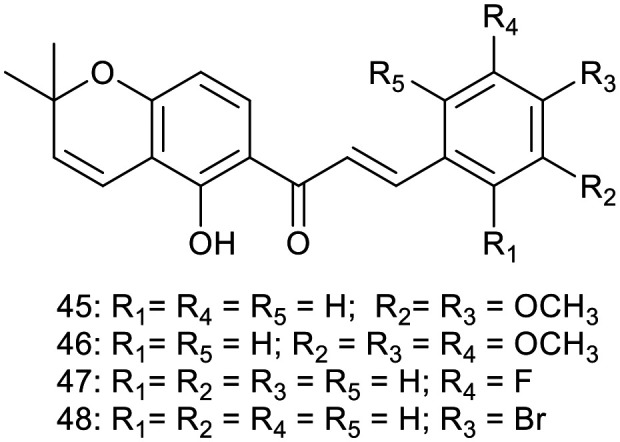
Searching for new anti-inflammatory compounds, Peng et al. designed and synthesized a series of pyranochalcone derivatives. Their inhibitory effects on nitric oxide (NO) production in lipopolysaccharide (LPS)-induced RAW 264.7 cells, as well as their impact on inducible nitric oxide synthase activity, were investigated.33 Among these derivatives, four compounds 45 (it is a naturally occurring chalcone isolated from Lonchocarpus subglaucescensroots34), 46 (it is also a naturally occurring chalcone isolated from Crotalaria ramosissima35), 47, and 48 displayed more potent inhibitory activity towards NO production compared to the positive control indomethacin at a concentration of 10 mM, with inhibition rates of 78.4%, 81.3%, 80.4%, and 79.9%, respectively, versus 59.2% for indomethacin. Furthermore, 45 significantly suppressed inflammation progression in both experimental models of adjuvant-induced arthritis (AIA) and carrageenan-induced hind paw edema. Compound 45 was also identified as a potent iNOS inhibitor, with an IC50 of 7.9 mM, and its binding to the active site of murine iNOS was confirmed through docking studies.
5. Cytotoxic activity
Cancer is a prominent contributor to mortality in contemporary society. Considerable effort has been dedicated to treating different types of cancer for many years, and in recent times, there has been a notable focus on chemoprevention of cancer. Within the realm of small molecule drugs designed to prevent or treat cancer, the presence of an alkenone moiety as a fundamental component of the molecule holds significant potential.36 Alkenones are a class of organic molecules that have a ketene functionality and an unsaturated group (double bond). They can be either acyclic or cyclic. The naturally occurring or synthetic anticancer agents exhibit a conjugated arrangement of these two functional groups. Chalcones, also known as 1,3-diphenyl-2-propen-1-ones, have a chemical structure that consists of two aromatic rings connected by a three-carbon α,β-unsaturated carbonyl system. Chalcones and their derivatives are a class of compounds that have been found to demonstrate significant potential in fighting cancer.37
Dehydrocycloxanthohumol hydrate (40: xanthohumol B) and dehydrocycloxanthohumol (49: xanthohumol C) from hops (Humulus lupulus)38 were assessed for their antiproliferative activity in human breast cancer (MCF-7), colon cancer (HT-29), and ovarian cancer (A-2780) cells in vitro by Miranda et al.5 Compound 49 induced a dose-dependent decrease in the growth of all cancer cell lines tested, with concentrations ranging from 0.1 to 100 μM. After two days of treatment, the IC50 value for compound (49) against MCF-7 cells was 15.7 μM. Following four days of treatment, the IC50 value decreased to 6.87 μM. HT-29 and A-2780 cells displayed greater resistance to compound 48 compared to MCF-7 cells.

Vogel et al. synthesized the minor hop (H. lupulus) chalcones xanthohumol C (49) and 1′′,2′′-dihydroxanthohumol C (50) and tested their cytotoxicity against a HeLa cell line using the MTT cell proliferation assay. These compounds had IC50 values of 12.5 ± 1.7, and 15.4 ± 1.4 μM, respectively.39

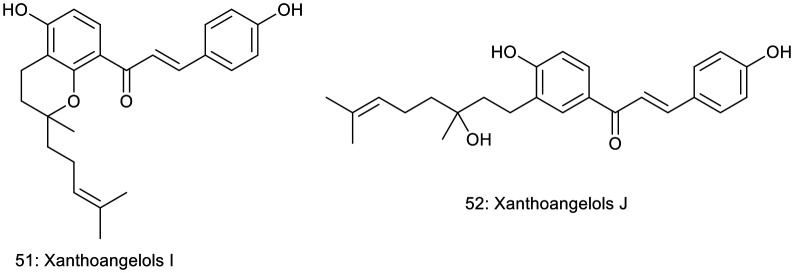
Akihisa et al. obtained xanthoangelols I (51) and J (52) from EtOAc-soluble fractions of Angelica keiskei (Apiaceae) stem exudates. Both compounds feature modified geranyl substituents and demonstrated inhibition of Epstein–Barr virus early antigen induction by 12-O-tetradecanoylphorbol-13-acetate (TPA) in Raji cells, a human Burkitt's lymphoma cell line. The compounds maintained high cell viability (60–70%) at a concentration of 32 nmol (compound to TPA mol ratio of 1000 : 1), indicating very low cytotoxicity at this concentration. Compounds 51 and 52 exhibited potent inhibitory effects, with IC50 values of 273 and 264 mol ratio/32 pmol TPA, respectively. These values indicate greater potency than the reference compound, retinoic acid, which had an IC50 value of 482 mol ratio/32 pmol TPA.40
Cyclokuraridin (53), obtained from MeOH extracts of the roots of Sophora flavescens (Leguminosae), demonstrated medium cytotoxicity (IC50 = 15.1 μg mL−1) in the KB tumor cell line.41

Isa et al. isolated boesenbergin A (compound 44) from Boesenbergia rotunda. The cytotoxicity of compound 44 was evaluated against several cell lines: human hepatocellular carcinoma (HepG2), colon adenocarcinoma (HT-29), non-small cell lung cancer (A549), prostate adenocarcinoma (PC3), and normal hepatic cells (WRL-68). The IC50 values, representing the concentration at which 50% inhibition of cell viability occurred, were 20.22 ± 3.15 μg mL−1 for A549, 10.69 ± 2.64 μg mL−1 for PC3, 20.31 ± 1.34 μg mL−1 for HepG2, 94.10 ± 1.19 μg mL−1 for HT-29, and 9.324 ± 0.24 μg mL−1 for WRL-68.32
Ye et al. isolated fourteen compounds, including one novel chalcone millepachine (54) as active principles from Chinese herbal medicine M. pachycarpa.42 All isolates were assessed for their cytotoxicity against a panel of cancer cell lines, including HepG2, C26, LL2, and B16, with cisplatin serving as a positive control. Furthermore, their apoptosis-inducing potential was evaluated in HeLa-C3 cells, with taxol used as a positive control. Compound 53 demonstrated significant cytotoxicity, with IC50 values of 1.29, 5.14, 0.60, and 2.03 μM against the HepG2, C26, LL2, and B16 cell lines, respectively. In the apoptosis assay, compound 54 exhibited substantial apoptosis-inducing effects, achieving significant results at a concentration of 2 μM within 36 hours. It was identified as the most potent apoptotic inducer among the compounds isolated from M. pachycarpa Benth.

In the ongoing pursuit of novel anticancer agents, Wang et al. designed, synthesized, and evaluated a series of millepachine derivatives for their antitumor activity across six human cancer cell lines. The incorporation of electron-withdrawing substituents on the right phenyl ring was observed to have a detrimental impact on the anti-tumor activity of the compounds. In contrast, the introduction of dialkylamine groups at the para position of the right phenyl ring significantly enhanced antiproliferative activity. Moreover, the presence of electron-donating substituents at the same position was found to be more favorable for biological activity. However, the inclusion of methoxy (–OCH3) and hydroxyl (–OH) functional groups did not result in a notable improvement in anti-tumor efficacy. Furthermore, replacing the right phenyl ring with various heterocyclic frameworks led to a pronounced decline in cytotoxic potential. Nevertheless, the substitution with a naphthalen-2-yl moiety exhibited a positive influence, enhancing antiproliferative activity. Among these derivatives, compound 55 demonstrated significantly greater antiproliferative activity compared to millepachine (compound 54), with a mean IC50 value of 0.64 μM versus 2.86 μM against HepG2, K562, SK-OV-3, HCT116, HT29, and SW620 cells. Additionally, compound 55 promisingly inhibited tubulin polymerization and induced G2/M phase cell cycle arrest in HepG2 cells. In vivo studies revealed that compound 55 achieved an 84.1% reduction in tumor growth, surpassing the effects of both millepachine (54) and the positive control anticancer drug cisplatin, which reduced tumor growth by 69.4% and 62.3%, respectively.43

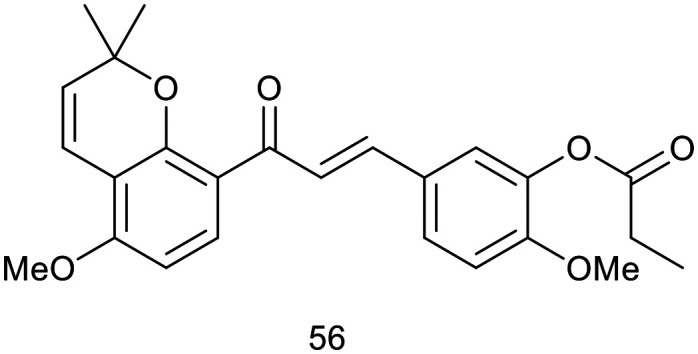
Tubulin, a protein crucial for cell proliferation and mitotic spindle formation,44,45 has been recognised as a significant molecular target for anticancer therapeutics.46 Anticancer drugs exert their effects on microtubules by disrupting the dynamics of tubulin polymerization or depolymerization, thereby inducing mitotic arrest. Two primary classes of anticancer agents function through this mechanism:47 microtubule-destabilizing agents (such as combretastatin vinca alkaloids, and colchicine) and microtubule-stabilizing agents (such as taxol, epothilone, and discodermolide). Recently, D. Cao et al.48 recently synthesized a series of millepachine-based pyranochalcones and found that esterification products exhibited significantly greater potency compared to their ether counterparts. Aliphatic derivatives demonstrated strong antiproliferative activity; however, the introduction of a benzene ring within the substituent group led to a marked reduction in biological efficacy. Also he evaluated their cytotoxicity across a range of human tumor cell lines, including those with a multidrug-resistant phenotype. To elucidate their precise mechanism of action, the effects of selected compounds on tubulin polymerization and tumor cell cycle distribution were analysed. The most promising candidate underwent further evaluation for antitumor efficacy and toxicity in vivo. Notably, compound 56 demonstrated exceptional potency, with IC50 values ranging from 0.09 to 1.30 μM across 21 tumor cell lines, including those with multidrug resistance. Moreover, compound 56 markedly induced G2/M phase cell cycle arrest at a concentration of 0.2 μM, promoted tubulin polymerization into microtubules, and led to microtubule stabilization. The in vivo antitumor efficacy of compound 56 was evaluated in subcutaneous xenograft models using human HepG2 liver tumor cell lines. Over a 20 day treatment period, compound 56 was administered at doses of 2.5, 5, and 10 mg kg−1, resulting in a dose-dependent inhibition of tumor growth by day 20, with T/C values of 34.9%, 32.4%, and 33.9%, respectively. For comparison, taxol at 5 mg kg−1 achieved a T/C value of 32.4%.48
In continuation of the anticancer drug research program, these authors synthesized a series of novel tubulin polymerization inhibitors and evaluated their in vitro and in vivo biological activities. Among the compounds studied, compound 57 exhibited robust antiproliferative activity against various tumor cell lines, with IC50 values ranging from 0.15 to 0.62 μM.49 Compound 57 was also found to induce G2/M phase cell cycle arrest and inhibit tubulin polymerization. The molecular docking studies indicated that 57 binds to the colchicine-binding site of tubulin. In xenograft experiments using BALB/c nude mice with H460 human lung carcinoma, compound 57 demonstrated a more potent anticancer effect than the standard anticancer drug taxol. These results highlight 57 as a promising antimitotic agent with potential for cancer treatment.50

Murthy et al. recently designed and synthesized a series of twelve novel chromanochalcones, which were subsequently assessed for their cytotoxic potential against seven different human cancer cell lines (PC-3, MCF-7, HL-60, MIA PaCa-2, AsPC, MDA-MB-435, and Caco-2). Most of these compounds exhibited significant in vitro antiproliferative activity at micromolar (μM) concentrations. Of particular interest, three compounds 58, 59, and 60 demonstrated the most potent effects against the human leukemia HL-60 cell line, with IC50 values ranging from 6 to 15 μM. In terms of the proposed mechanism of action, cell cycle analysis in the HL-60 cell line revealed a marked elevation in the G0/G1 and sub-G0/G1 populations, suggesting the induction of cell cycle arrest and apoptosis.50 Further studies on DNA fragmentation, mitochondrial depolarization, and nuclear morphology confirmed that these compounds exert their anticancer effects through apoptotic pathways. As a result, compounds 58, 59, and 60 have been identified as promising scaffolds for future research in cancer chemotherapy.

6. Antidiabetic activity
6.1. Diacylglycerol acyltransferase (DGAT) inhibitors
Diabetes mellitus encompasses a group of metabolic disorders characterized by persistent hyperglycemia due to impaired insulin secretion or dysfunctional insulin action on target tissues, leading to disruptions in carbohydrate, lipid, and protein metabolism.51 Diacylglycerol acyltransferase (DGAT), formally known as acyl-CoA: 1,2-diacyl-sn-glycerol O-acyltransferase, plays a pivotal role by catalyzing the transfer of an acyl group from acyl-CoA to diacylglycerol, resulting in the formation of triacylglycerol.52,53 Excessive accumulation of triacylglycerol in certain organs and tissues significantly heightens the risk of developing fatty liver, obesity, and hypertriglyceridemia, which in turn can lead to severe atherosclerosis, diabetes, various metabolic disorders, and impaired function of affected organs.54 Since DGAT is the key enzyme responsible for triacylglycerol synthesis, it is considered a critical target for inhibition in the treatment and prevention of these conditions.
Tabata et al. evaluated a methanol extract of Humulus lupulus (L.) hops for its inhibitory effects on rat liver diacylglycerol acyltransferase (DGAT). Their studies revealed that the extract exhibited DGAT inhibitory activity. Through activity-guided fractionation, they isolated two chalcones: xanthohumol (61) and a novel compound, xanthohumol B (40). Both xanthohumol (61) and xanthohumol B (40) demonstrated DGAT inhibition with IC50 values of 50.3 μM and 194 μM, respectively, in rat liver microsomes. Additionally, these compounds preferentially inhibited triacylglycerol formation in intact Raji cells, suggesting that their inhibitory effects on DGAT are more pronounced in living cells.38

6.2. PPAR-γ ligand-binding activity
PPARs (peroxisome proliferator-activated receptors) are ligand-activated transcription factors that belong to the nuclear receptor superfamily and are integral in regulating the expression of genes involved in glucose and lipid metabolism. PPARs are classified into three distinct subtypes: PPAR-α, PPAR-γ, and PPAR-δ.55,56 PPAR-α is predominantly expressed in the kidney, liver and skeleton muscle, while PPAR-δ is ubiquitously expressed.57 PPAR-γ serves as the primary molecular target for insulin-sensitizing thiazolidinedione drugs, such as troglitazone, pioglitazone, and rosiglitazone.58,59 These thiazolidinedione derivatives exert their effects by activating PPAR-γ, thereby enhancing insulin sensitivity through the promotion of small, functional adipocyte differentiation from pre-adipocytes and the induction of apoptosis in large adipocytes. These large adipocytes are known to excessively produce and secrete adipocytokines such as leptin, TNFα, and free fatty acids.60 Additionally, among natural PPAR-γ ligands, several flavonoids,61,62 isoprenoids,63 and triterpene acids64 have been identified as activators of PPAR-γ.
Kuroda et al. tested the EtOH extract of G. glabra roots against PPAR-γ ligand-binding activity. The extract demonstrated significant PPAR-γ ligand-binding activity, leading to the bioassay-guided isolation of a diverse array of compounds, including nine chalcones (such as compound 62), two 3-arylcoumarins, ten isoflavans, an isoflaven, three pterocarpans, a flavone, a flavanol, three isoflavones, an isoflavane, four flavanones, a 2-arylbenzofuran, and two chromones, among which ten were newly identified compounds. All isolated compounds exhibited notable PPAR-γ ligand-binding activity, with their activity at a concentration of 10 μg mL−1 being three times more potent than that of 0.5 μM troglitazone.65

Recently, Venkateshwarlu Korthikunta et al. conducted a synthesis and pharmacological evaluation of a series of benzofuran-based chromenochalcones to assess their antihyperglycemic and antidyslipidemic properties. Among the synthesized compounds, (E)-3-(2-benzoylbenzofuran-5-yl)-1-(7-hydroxy-2,2-dimethyl-2H-chromen-8-yl)prop-2-en-1-one (63) was identified as particularly efficacious. This compound demonstrated significant improvement in postprandial and fasting blood glucose levels, enhanced oral glucose tolerance, and positively modulated serum lipid profiles, serum insulin levels, and the HOMA-index in db/db mice following a 15 day consecutive treatment regimen.66

7. Antibacterial activity
The increasing prevalence of antibiotic-resistant bacterial strains poses a significant challenge in the field of drug design and discovery. Antimicrobial resistance arises from three main tactics: enzymatic deactivation of the drug, alteration of target sites, and expulsion through efflux.67–69 Bacteria commonly employ the active efflux of toxic compounds as a mechanism to safeguard themselves towards the detrimental effects of poisonous molecules they come across in the suroundings.70 The DOT-T1E strain is distinguished by its remarkable resistance to toxic organic solvents, a characteristic largely attributed to three RND efflux pumps: TtgABC, TtgDEF, and TtgGHI. Notably, TtgABC has been identified as a principal factor in the intrinsic tolerance of Pseudomonas putida DOT-T1E to organic solvents.70
Pongachalcone I (64), a naturally occurring pyranochalcone isolated from Tephrosia deflexa, has demonstrated antibacterial activity against Pseudomonas putida.71 Paul et al. conducted in silico molecular docking studies on pongachalcone I (64) targeting the transcriptional regulator TtgR (PDB code: 2UXI), which acts as a major repressor of the TtgABC efflux pump operon. Despite the bacterium's ability to expel antibiotics such as chloramphenicol, tetracycline, and naringenin, compound 64 exhibits significant antibacterial efficacy against it. The study reveals the significant functions of the residues ASN 110 and CYS 137 in the active site of the enzyme (Fig. 3). This finding is valuable for understanding the molecular mechanism of drug recognition and designing drugs to combat antibiotic-resistant bacteria.72
Fig. 3. The interaction of the ligand with the protein.


Building on their previous work, in silico docking studies on Pseudomonas putida, which is resistant to toxic substances or antibiotics, revealed that the pyranochalcone (64) exhibited an inhibitory effect on the strain. Keeping that in mind, these authors selected several natural pyranochalcones reported in various works of literature that are considered as ligands73 (Fig. 4). The selected ligands were glabrachalcone (65), glabrachromene II (66) (both isolated from Pongamia glabra,74 and Millettia pachycarpa75), glaychalcones A (67) and B (68), (isolated from Glycosmis citrifolia),76 licoagrochalcone B (69) (isolated from Patrinia villosa77 and Glycyrrhiza glabra78), anthyllisone (70) (isolated from Anthyllis hermaniae L),79 citrunobin (71) (isolated from Citrus sinensi,),80 boesenbergins A (44) and B (72) (isolated from Boesenbergia pandurata)81,82 and some other pyranochalcones (73, 74, and 75).
Fig. 4. Naturally occurring pyranochalcones used in molecular docking study.

Molecular docking studies were conducted with naturally occurring pyranochalcones targeting the transcriptional regulator enzyme TtgR of the antibiotic-resistant Pseudomonas putida strain. The pyranochalcones boesenbergin A (44) and B (72), and lonchocarpin (73), along with pongachalcone I (64), were predicted to exhibit significant activity against the multidrug-resistant bacterial strain. In contrast, anthyllisone (70) was identified as the least active compound in this series. The influence of the number and position of methoxy groups in the pyranochalcones on their binding affinity was also analyzed.73 The findings from this study will contribute to the design of a new series of drugs aimed at combating antibiotic-resistant bacteria.
8. Chagas disease
Chagas disease, caused by the protozoan Trypanosoma cruzi, is believed to impact approximately 16–18 million individuals, primarily in South and Central America. In these regions, around 25% of the entire population is susceptible to the disease (World Health Organization).83,84 The implementation of measures to control the insect vector (Triotoma infestans) in areas where it is prevalent has effectively eradicated the transmission of insect bites. Therefore, the primary factors contributing to the transmission of the disease are blood transfusion and congenital transmission. The parasite T. cruzi in its bloodstream form lacks a functional tricarboxylic acid cycle and relies heavily on glycolysis to produce ATP. This significant reliance on glycolysis as a source of energy renders the glycolytic enzymes attractive targets for trypanocidal drug design. Consequently, the 3D structure of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has been elucidated.85 GAPDH facilitates the oxidative phosphorylation of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate. Notably, glycosomal GAPDH presents potential target sites with marked structural differences compared to its human counterpart. Inhibitors targeting these sites have been designed, synthesized, sourced from natural compounds, and subsequently tested for efficacy.
To block agents, Tomazela et al. isolated chalcones 76, 77 and flavones from N. magnifica and evaluated by interaction with the enzyme GAPDH obtained from T. cruzi. The mixture of 76 and 77 showed a GAPDH inhibitory activity of 45% at 105 mg ml−1 concentration. These chalcones act as weak inhibitors compared to the isolated flavanones (99% at 100 mg ml−1).86

9. Antifungal activity
Dermatophytes, a specialized group of fungi, primarily target “keratinized” areas of the body, and dermatomycoses are notoriously difficult to treat effectively. Interestingly, chalcone derivatives have demonstrated selective antifungal activity against dermatophytes, while exhibiting no significant effects on other fungal species. This selective efficacy underscores the potential of chalcone derivatives as targeted therapeutic agents for dermatophytic infections. Crotmadine 78, extracted from the leaves as well as stems of Crotalaria madurensis, demonstrated antifungal properties towards Trichophyton mentagrophytes with an effective concentration of 62.51 μg mL−1.87

10. Antioxidant activity
An antioxidant is a substance that inhibits the oxidative processes occurring in other molecules. Oxidation involves the transfer of electrons or hydrogen atoms from a given molecule to an oxidizing agent. Oxidation reactions could generate free radicals. Consequently, these radicals have the ability to initiate chain reactions. Cellular chain reactions can result in cellular damage or cell death. Antioxidants halt these series of reactions by eliminating free radical intermediates and impeding other oxidation reactions. Antioxidants function by undergoing oxidation themselves, making them effective as reducing agents, such as thiols and ascorbic acid.88
Lespeol (79) was isolated from Lespedeza cyrtobotrya,89Artocarpus communis,90 and from the Artocarpus nobilis fruits, which are distributed in Sri Lanka.91 The compound demonstrated significant inhibitory effects on melanin synthesis in normal human epidermal melanocytes,90 and exhibited robust antioxidant activity against DPPH radicals.91

Boesenbergin A (44), a chalcone derivative of a known structure isolated from Boesenbergia rotunda, was tested for its antioxidant properties. The antioxidant capacity of compound 44 was evaluated using the ORAC assay, with its efficacy compared to the positive control, quercetin. At a concentration of 20 μg mL−1, compound 44 exhibited substantial antioxidant activity, comparable to 11.91 ± 0.23 μM Trolox.32
Vogel et al. synthesized the minor hop (H. lupulus) chalcones xanthohumol C (49) and 1′′,2′′-dihydroxanthohumol C (50). They tested their antioxidative activity. The ORAC-fluorescein assay unfolded the potent antioxidative activity for compounds 49 and 50, with 1.8 ± 0.1 and 1.7 ± 0.2 Trolox equivalents, respectively.39
11. Some more naturally occurring chromenochalcones with miscellaneous activities
Purpurenone (80) and its derivative 81, featuring a β-hydroxypyranochalcone moiety, were isolated from Lonchocarpus subglaucescens, a species native to southeastern Brazil.34 Additionally, purpurenone (80) was identified in L. montanus, an ornamental tree commonly referred to as “cabelouro” or “carrancudo” in Brazil.92 Praecansone B (82) was isolated from Tephrosia aequilata, a plant widely found across tropical regions worldwide.93 Traditionally, the roots of this species are employed in the treatment of venereal diseases, while its leaves are used to alleviate abdominal pain.94,95 Pongapinone A (83) was isolated from the Pongamia pinnata bark, an Indonesian medical plant known as “Warrt”. Pongapinone A (83) showed inhibitory activity towards interleukin-1 production with IC50 2.5 μg mL−1.96

Xanthohumol E (84) was also isolated from Humulus lupulus (hop plant), cultivated in virtually all temperate zones globally.97 Sericone (85), a regioisomer of xanthohumol E (83), was isolated from another plant, Mundulea sericea.98 Glabrachromene (86) was isolated from Pongamia glabra.99 Lawson et al. isolated two retrochromenochalcones 87 and 88 from Lonchocarpus nicou roots' lipophile extract.100 Passador et al. isolated a chromenochalcone (89) from the hexane fraction of the stems of N. magnifica var. magnifica101 T. Fukai et al. isolated a pyranochalcone called glyinflanin G (90) from Glycyrrhiza inflate roots (Leguminosae).102

12. Synthetic routes to biologically interesting chromenochalcone natural products
12.1. Total synthesis of mallotophilippens C and E
Mallotophilippens C (37) and E (39) have exhibited significant inhibition of nitric oxide (NO) production in RAW 264.7 cells activated by interferon-γ (IFN-γ) and lipopolysaccharide (LPS). Furthermore, these compounds effectively suppressed the expression of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), interleukin-6 (IL-6), and interleukin-1β (IL-1β) mRNA.27 These findings indicate their potential utility in the development of new therapeutic agents for treating rheumatoid arthritis, a condition characterized by excessive NO production.
Lee et al. reported a novel and coherent synthetic method for producing biologically significant natural products, “mallotophilippens C” (37) and “E” (39). The synthesis leveraged key strategies, including ethylenediamine diacetate-catalyzed benzopyran formation and base-catalyzed aldol reactions, to achieve the desired compounds.103
The synthesis of mallotophilippen C (37) was carried out as per the sequence shown in Scheme 1. The reflux reaction of 2,4,6-trihydroxy acetophenone (91) with geranyl bromide in the presence of anhydrous potassium carbonate in dry acetone for 24 hours resulted in the formation of compound 92 with a yield of 74%. Subsequently, the reaction of compound 92 with 3-methyl-2-butenal, catalyzed by 20 mol% ethylenediamine diacetate in methylene chloride over a 5 hour period, produced benzopyran (93) with a 66% yield. The observed regioselectivity in this cyclization reaction is likely attributable to the influence of the bulky geranyl group. Subsequently, compound 93 was subjected to protection using 1.1 equivalents of methoxymethyl chloride, resulting in the formation of compound 94 with a yield of 94%. The condensation of compound 94 with aldehyde 95, which was protected with an SEM group, was performed in an ethanolic KOH solution at ambient temperature for 48 hours, yielding the pyranochalcone 96 with a 76% yield. Deprotection of compound 96 using 3 N HCl in methanol at 25 °C for 1 hour produced mallotophilippen C (37) with a 65% yield.
Scheme 1. Synthesis of mallotophillippen C.103.

The total synthesis of mallotophilippen E (39) commenced with 2,4,6-trihydroxy acetophenone (91), as depicted in Scheme 2.103 Initially, compound 91 was reacted with prenyl bromide in the presence of DBU in THF, yielding compound 97 with a 40.2% yield. Following this, compound 97 was treated with citral in the presence of 20 mol% ethylenediamine diacetate in methylene chloride at 25 °C for 8 hours, producing compounds 98 and 99 with yields of 78% and 17%, respectively. Both compounds were efficiently separated via column chromatography and subsequently characterized through spectral analysis. Compound 98 was protected with 1.1 equivalents of SEMCl leading to the formation of compound 100 with a 93% yield. This intermediate was subsequently reacted with aldehyde 101 in an ethanolic KOH solution at room temperature for 48 hours, resulting in the synthesis of pyranochalcone 102 with a 72% yield. The SEM protecting groups were subsequently eliminated by treating the compound with HCl in methanol at 25 °C for 1 hour, ultimately affording compound 39 with a 56% yield.
Scheme 2. Synthesis of mallotophilippen E.103.

12.2. Total synthesis of citrunobin, boesenbergin A, boesenbergin B, xanthohumol C, and glabrachromene
Lee et al. developed an efficient and streamlined synthetic pathway for producing biologically relevant pyranochalcones. This methodology facilitates the synthesis of notable natural products, including citrunobin (71), glabrachromene (86), xanthohumol C (49), and boesenbergins A (44) and B (72). Central to this approach are benzopyran formation reactions catalyzed by ethylenediamine diacetate and base-catalyzed aldol condensations.104
The total synthesis of citrunobin (71) and boesenbergin A (44) was achieved via construction of benzopyrans followed by an aldol reaction, beginning with 2,6-dihydroxy-4-methoxyacetophenone (103), as outlined in Scheme 3.104 The reaction of compound 103 with 3-methyl-2-butenal, utilizing 10 mol% ethylenediamine diacetate in refluxing xylene for 10 hours, resulted in the formation of alloevodionol (103) with a yield of 95%.
Scheme 3. Synthesis of citrunobin and boesenbergin A.104.

To finalize the total synthesis of the natural product citrunobin (71), an aldol reaction was performed. In a model study, the reaction of compound 104 with benzaldehyde (106) in the presence of hydroxide in ethanol at 25 °C for 48 hours resulted in the formation of the potassium compound 108 with a 96.1% yield. Efforts to condense compound 104 with 4-hydroxybenzaldehyde using potassium hydroxide in ethanol were unsuccessful. Subsequently, compound 104 was treated with protected benzaldehyde 107, which afforded the compound 109 with a yield of 90%, followed by deprotection of the MOM ether using 3 N HCl in ethanol at 50 °C for 1 hour resulting in the formation of compound 71 with an 86% yield across two steps. Additionally, the reaction of compound 103 with citral, catalyzed by 10 mol% ethylenediamine diacetate in refluxing xylene for 10 hours, produced the adduct 105 in an 89% yield. Subsequent treatment of compound 105 with benzaldehyde (106) in an ethanolic KOH solution afforded boesenbergin A (44) with a 96% yield.
The synthesis of xanthohumol C (49), boesenbergin B (72), and glabrachromene (86) commenced with 2,4-dihydroxy-6-methoxyacetophenone (110), as detailed in Scheme 4.104 The reaction of compound 110 with citral, catalyzed by 10 mol% ethylenediamine diacetate in refluxing xylene for 10 hours, yielded compound 111 with a 90.2% yield. This adduct, 111, was subsequently treated with benzaldehyde (106) in an ethanolic KOH solution at 25 °C for 48 hours, resulting in the formation of boesenbergin B (72) with a 91% yield.
Scheme 4. Synthesis of boesenbergin B, xanthohumol C and glabrachromene.104.

The reaction of compound 110 with 3-methyl-2-butenal, facilitated by 10 mol% ethylenediamine diacetate in refluxing xylene for 10 hours, yielded isoevodionol (112). This intermediate was then subjected to reaction with MOM ether-protected benzaldehyde (107) in an ethanolic KOH solution at 25 °C for 48 hours, resulting in the formation of compound 113 with an 80% yield. Subsequent cleavage of the MOM ether with 3 N hydrochloric acid in methanol afforded xanthohumol C (49) with an 86.04% yield. Alternatively, the reaction of 112 with piperonal in an ethanolic KOH solution at 25 °C for 48 hours led to the formation of glabrachromene (86) with an 87% yield.
12.3. Total synthesis of pyranochalcones (pongachalcone I, glabrachromene II, glabrachalcone, glaychalcone A, and glaychalcone B)
Lee et al. developed an efficient and highly precise total synthesis method for naturally occurring pyranochalcones. Pongachalcone I (64), glabrachalcone (65), glabrachromene II (66), glaychalcone A (67), glaychalcone B (68) were achieved from readily accessible precursors, 2,4-dihydroxyacetophenone and 2,4-dihydroxy-6-methoxyacetophenone. The pivotal steps in the synthetic strategy involved ethylenediamine diacetate-catalyzed benzopyran formation and aldol reactions.105
The synthesis of benzopyran 115 commenced with 2,4-dihydroxyacetophenone (114), as detailed in Scheme 5.105 The reaction of 114 with 3-methyl-2-butenal, using 10 mol% ethylenediamine diacetate as the catalyst in refluxing toluene for 12 hours, yielded desmethyl isoencecalin (115) with a 52% productivity. Subsequent steps involved the implementation of aldol reactions to complete the synthesis of the targeted natural products. Compound 115 was reacted with the protected benzopyran 116 using KOH in ethanol at ambient temperature for 48 hours, yielding compound 121 with a 71% efficiency. This product was subsequently deprotected with tetrabutylammonium fluoride (TBAF) in hexamethylphosphoramide (HMPA) under refluxing tetrahydrofuran (THF) for 5 hours, resulting in compound 122 with an 89% yield. Additionally, the reaction of 115 with compound 117, using KOH in ethanol at 25 °C for 48 hours, afforded glabrachromene II (66) with a 60.02% yield. In contrast, the treatment of 115 with 2,4,5-trimethoxybenzaldehyde (118) produced glabrachalcone (65) with a 73.24% yield.
Scheme 5. Synthesis of glabrachromene II and glabrachalcone.105.

The total synthesis of pongachalcone I (64) and glychalcones A (67) and B (68) was initiated with 2,4-dihydroxy-6-methoxyacetophenone (110), as outlined in Scheme 6.105 The reaction of 110 with 3-methyl-2-butenal, catalyzed by 10 mol% ethylenediamine diacetate in refluxing toluene for 12 hours, resulted in the formation of isoevodionol (112) with a high yield of 95%. The reaction of isoevodionol (112) with benzaldehyde (106) in the presence of KOH in ethanol at room temperature for 48 hours yielded pongachalcone I (64) with an 87% yield. Similarly, the reaction of 112 with 4-methoxybenzaldehyde (120) under identical conditions produced glychalcone A (67) with an 85% yield. Additionally, the reaction of 112 with 3,4-dimethoxybenzaldehyde (121) resulted in glychalcone B (68) with an 82% yield.
Scheme 6. Synthesis of pongachalcone I, glychalcone A and glychalcone B.105.

12.4. Total synthesis of lespeol
Lespeol (79), a compound obtained from Lespedeza cyrtobotrya89 and Artocarpus communis,90 has been found to effectively inhibit melanin production in normal human epidermal melanocytes89 and exhibit notable antioxidant activity against DPPH radicals.91 Jung et al.106 developed a new, straightforward synthetic methodology for producing lespeol (79) and its derivatives (127–129), which are of considerable biological interest.
The total synthesis of lespeol (79) and its derivatives (127–129) was accomplished starting from compound 123, as detailed in Scheme 7.106 The reaction of 123 with DDQ (4,5-dichloro-3,6-dioxocyclohexa-1,4-diene-1,2-dicarbonitrile) in refluxing toluene for 4 hours yielded the cyclized intermediate 124 with a 60% yield. Subsequent condensation of 124 with various benzaldehydes in an ethanolic KOH solution at 25 °C for 48 hours produced the intermediates 125 and 126 in 80% and 73% yields, respectively. The final step involved treating 125 and 126 with concentrated hydrochloric acid in methanol at room temperature for 10 hours, resulting in the formation of lespeol (79) and its unnatural derivative 127 with yields of 90% and 73%, respectively. Additionally, reactions of 124 with 3,4-dimethoxybenzaldehyde and piperonal (1,3-benzodioxole-5-carboxaldehyde) provided the target compounds 128 and 129 in yields of 72% and 70%, respectively.
Scheme 7. Synthesis of lespeol (79) and derivatives 127–129 (ref. 106).

Conclusions and future perspectives
In medicinal chemistry, using privileged structures represents a strategic approach to drug development. These foundational molecular frameworks provide a basis for generating relevant ligands across targets through targeted structural optimization. Researchers can rapidly develop structurally novel chemotypes by modifying the core structure and introducing side chains to existing active compounds. Chalcone and its derivatives, such as chromenochalcones, are prevalent scaffolds found in numerous naturally occurring compounds, particularly those derived from plants. Additionally, several chalcone derivatives have been developed due to their straightforward synthesis. Building on the traditional ethnopharmacological applications of chromenochalcones, this study emphasizes their most significant biological activities, both in vitro and in vivo, including antimalarial, anti-inflammatory, antileishmanial, cytotoxic, antibacterial, antifungal, and antioxidant agents. Bawazir et al. (2024)107 recently isolated flemingin A, a chromenochalcone derivative from Flemingia grahamiana, which exhibited significant antitubercular activity against Mycobacterium tuberculosis in the low oxygen recovery assay (LORA) while demonstrating minimal cytotoxicity. Furthermore, Korthikunta et al. (2023) synthesized a series of benzofuran-based chromenochalcones and assessed their pharmacological potential for antihyperglycemic and antidyslipidemic effects. Among the synthesized derivatives, one compound demonstrated notable efficacy in improving postprandial and fasting blood glucose levels, enhancing oral glucose tolerance, and regulating the serum lipid profile, serum insulin levels, and the HOMA-index in db/db mice.66 For future advancements, researchers investigating the field of chromochalcones can employ various computational approaches to elucidate critical interactions with M. tuberculosis targets, as well as metabolic pathways associated with antihyperglycemic and antidyslipidemic effects. Despite exhibiting numerous promising biological effects and potential for extensive preclinical studies, the precise mechanisms of action of chromenochalcones remain incompletely understood. Although chalcones are relatively easy to synthesize, developing novel synthetic methods is necessary for future research. This will facilitate the exploration of new biological properties, the investigation of their molecular mechanisms of action, and, most importantly, the identification of their specific biological targets. It is anticipated that novel chromenochalcone-based drugs will be identified through modern drug discovery strategies and new chemical sources. Further research and clinical trials are needed to explore their pharmacological actions, interactions with other compounds or medications, and potential toxicity. Further studies on the chromenochalcone scaffold in areas beyond this review are also expected.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Author contributions
Dr. Rohit Singh: conception and design of the study and acquisition of data. Dr. Vedpal: design and acquisition of data. Ms. Archita Katrolia: revising the manuscript critically for important intellectual content.
Conflicts of interest
The authors have declared that there is no conflict of interest.
Acknowledgments
The authors are thankful to the pro-chancellor and vice-chancellor at the College of Pharmacy, JSS University, Noida, for the constant encouragement of the program on drug discovery from natural products of biological importance. Dr. Narender Tadigoppula is responsible for the overall supervision of the work.
Biographies
Biography
Rohit Singh.

Dr. Rohit Singh is a distinguished researcher specializing in medicinal and natural product chemistry. He earned his Ph.D. from the CSIR-Central Drug Research Institute, Lucknow. His research primarily focuses on the custom synthesis of cephalosporin and the isolation of protodioscin. Additionally, he is dedicated to developing innovative methodologies for natural product isolation and the structural modification of bioactive molecules, including amino acids, terpenoids, and sulfur-containing compounds. His work aims to advance therapeutic solutions for conditions such as cancer, diabetes, malaria, and leishmaniasis. His expertise also extends to synthetic methodology development, emphasizing the construction of pharmaceutically relevant heterocyclic frameworks and employing structure–activity relationship (SAR) and molecular diversity-driven approaches.
Biography
Archita Katrolia.

Ms Archita Katrolia is an experienced academician specializing in the targeted delivery of bioactive natural compounds, with a strong focus on developing and optimizing delivery systems to enhance therapeutic efficacy, stability, and bioavailability.
Biography
Ved Pal.

Dr. Ved Pal has completed his M. Pharm and Ph. D. from JSS College of Pharmacy, Ooty. His research work focused on the extraction, isolation, and characterization of Desmodium species (Fabaceae family). He worked on the inhibition of the 5-lipoxygenase enzyme (5-LOX) and the molecular-level studies to inhibit pro-inflammatory cytokines (IL-4, IL-13, and TNF α) with the help of isolated compounds from Desmodium species for in vitro and in vivo activity and molecular mechanics like docking of their targets to establish the activity on targets.
References
- Mazumder R. Ichudaule A. Ghosh S. D. Ghosh R. Top. Curr. Chem. 2024;382:22. doi: 10.1007/s41061-024-00468-7. [DOI] [PubMed] [Google Scholar]
- Zhuang C. Zhang W. Sheng C. Zhang W. Xing C. Miao Z. Chem. Rev. 2017;117:7762–7810. doi: 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammohan A. Reddy J. S. Sravya G. Rao C. N. Zyryanov G. V. Environ. Chem. Lett. 2020;18:433–458. [Google Scholar]
- Rajendran G. Bhanu D. Aruchamy B. Ramani P. Pandurangan N. Bobba K. N. Oh E. J. Chung H. Y. Gangadaran P. Ahn B.-C. Chalcone: A Promising Bioactive Scaffold in Medicinal Chemistry. Pharmaceuticals. 2022;15:1250. doi: 10.3390/ph15101250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda C. L. Stevens J. F. Helmrich A. Henderson M. C. Rodriguez R. J. Yang Y. H. Deinzer M. L. Barnes D. W. Buhler D. R. Food Chem. Toxicol. 1999;37:271–285. doi: 10.1016/s0278-6915(99)00019-8. [DOI] [PubMed] [Google Scholar]
- Demizu S. Kajiyama K. Hiraga Y. Kinoshita K. Koyama K. Takahashi K. Tamura Y. Okada K. Kinoshita T. Chem. Pharm. Bull. 1992;40:392–395. [Google Scholar]
- Umumararungu T. Nkuranga J. B. Habarurema G. Nyandwi J. B. Mukazayire M. J. Mukiza J. Muganga R. Hahirwa I. Mpenda M. Katembezi A. N. Olawode E. O. Kayitare E. Kayumba P. C. Bioorg. Med. Chem. 2023;88-89:117339. doi: 10.1016/j.bmc.2023.117339. [DOI] [PubMed] [Google Scholar]
- Armstrong J. F. Campo B. Alexander S. P. H. Arendse L. B. Cheng X. Davenport A. P. Faccenda E. Fidock D. A. Godinez-Macias K. P. Harding S. D. Kato N. Lee M. C. S. Luth M. R. Mazitschek R. Mittal N. Niles J. C. Okombo J. Ottilie S. Pasaje C. F. A. Probst A. S. Rawat M. Rocamora F. Sakata-Kato T. Southan C. Spedding M. Tye M. A. Yang T. Zhao N. Davies J. A. Br. J. Pharmacol. 2023;180:1899–1929. doi: 10.1111/bph.16144. [DOI] [PubMed] [Google Scholar]
- Ngameni B. Watchueng J. Boyom F. F. Keumedjio F. Ngadjui B. T. Gut J. Abegaz B. M. Rosenthal P. J. ARKIVOC. 2007;2007:116–123. [Google Scholar]
- Wanare G. Aher R. Kawathekar N. Ranjan R. Kaushik N. K. Sahal D. Bioorg. Med. Chem. Lett. 2010;20:4675–4678. doi: 10.1016/j.bmcl.2010.05.069. [DOI] [PubMed] [Google Scholar]
- Frölich S. Schubert C. Bienzle U. Jenett-Siems K. J. Antimicrob. Chemother. 2005;55:883–887. doi: 10.1093/jac/dki099. [DOI] [PubMed] [Google Scholar]
- Narender T. Shweta K. Tanvir M. S. Rao K. S. Puri S. K. Bioorg. Med. Chem. Lett. 2005;15:2453–2455. doi: 10.1016/j.bmcl.2005.03.081. [DOI] [PubMed] [Google Scholar]
- Tadigoppula N. Korthikunta V. Gupta S. Kancharla P. Khaliq T. Soni A. Srivastava R. K. Srivastava K. Puri S. K. Raju K. S. R. Wahajuddin P. S. Sijwali V. K. Mohammad I. S. J. Med. Chem. 2013;56:31–45. doi: 10.1021/jm300588j. [DOI] [PubMed] [Google Scholar]
- Desjeux P. Comp. Immunol., Microbiol. Infect. Dis. 2004;27:305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- EBioMedicine, 2023, 87, 104440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S. Frasca K. Scherrer S. Henao-Martínez A. F. Newman S. Ramanan P. Suarez J. A. Curr. Trop. Med. Rep. 2021;8:121–132. doi: 10.1007/s40475-021-00232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra J. Saxena A. Singh S. Curr. Med. Chem. 2007;14:1153–1169. doi: 10.2174/092986707780362862. [DOI] [PubMed] [Google Scholar]
- Le Pape P. J. Enzyme Inhib. Med. Chem. 2008;23:708–718. doi: 10.1080/14756360802208137. [DOI] [PubMed] [Google Scholar]
- Narender T. Shweta Gupta S. Bioorg. Med. Chem. Lett. 2004;14:3913–3916. doi: 10.1016/j.bmcl.2004.05.071. [DOI] [PubMed] [Google Scholar]
- Narender T. Khaliq T. Shweta N. Goyal N. Gupta S. Bioorg. Med. Chem. 2005;13:6543–6550. doi: 10.1016/j.bmc.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Shivahare R. Korthikunta V. Chandasana H. Suthar M. K. Agnihotri P. Vishwakarma P. Chaitanya T. K. Kancharla P. Khaliq T. Gupta S. Bhatta R. S. Pratap J. V. Saxena J. K. Gupta S. Tadigoppula N. J. Med. Chem. 2014;57:3342–3357. doi: 10.1021/jm401893j. [DOI] [PubMed] [Google Scholar]
- Gupta S. Shivahare R. Korthikunta V. Singh R. Gupta S. Tadigoppula N. Eur. J. Med. Chem. 2014;81:359–366. doi: 10.1016/j.ejmech.2014.05.034. [DOI] [PubMed] [Google Scholar]
- Foroumadi A. Emami S. Sorkhi M. Nakhjiri M. Nazarian Z. Heydari S. Ardestani S. K. Poorrajab F. Shafiee A. Chem. Biol. Drug Des. 2010;75:590–596. doi: 10.1111/j.1747-0285.2010.00959.x. [DOI] [PubMed] [Google Scholar]
- Nazarian Z. Emami S. Heydari S. Ardestani S. K. Nakhjiri M. Poorrajab F. Shafiee A. Foroumadi A. Eur. J. Med. Chem. 2010;45:1424–1429. doi: 10.1016/j.ejmech.2009.12.046. [DOI] [PubMed] [Google Scholar]
- Thiemermann C. Vane J. Eur. J. Pharmacol. 1990;182:591–595. doi: 10.1016/0014-2999(90)90062-b. [DOI] [PubMed] [Google Scholar]
- Honda T. Gribble G. W. Suh N. Finlay H. J. Rounds B. V. Bore L. Favaloro F. G. Wang Y. Sporn M. B. J. Med. Chem. 2000;43:1866–1877. doi: 10.1021/jm000008j. [DOI] [PubMed] [Google Scholar]
- Daikonya A. Katsuki S. Kitanaka S. Chem. Pharm. Bull. 2004;52:1326–1329. doi: 10.1248/cpb.52.1326. [DOI] [PubMed] [Google Scholar]
- Zhao F. Nozawa H. Daikonnya A. Kondo K. Kitanaka S. Biol. Pharm. Bull. 2003;26:61–65. doi: 10.1248/bpb.26.61. [DOI] [PubMed] [Google Scholar]
- Lee M. H. Kim J. Y. Ryu J.-H. Biol. Pharm. Bull. 2005;28:2253–2257. doi: 10.1248/bpb.28.2253. [DOI] [PubMed] [Google Scholar]
- Han A.-R. Kang Y.-J. Windono T. Lee S. K. Seo E.-K. J. Nat. Prod. 2006;69:719–721. doi: 10.1021/np0600346. [DOI] [PubMed] [Google Scholar]
- Jeon J.-H. Kim S.-J. Kim C. G. Kim J.-K. Jun J.-G. Bull. Korean Chem. Soc. 2012;33:953–957. [Google Scholar]
- Isa N. M. Abdelwahab S. I. Mohan S. Abdul A. B. Sukari M. A. Taha M. M. Syam S. Narrima P. Cheah S. Ahmad S. Mustafa M. R. Braz. J. Med. Biol. Res. 2012;45:524–530. doi: 10.1590/S0100-879X2012007500022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng F. Wang G. Li X. Cao D. Yang Z. Ma L. Ye H. Liang X. Ran Y. Chen J. Qiu J. Xie C. Deng C. Xiang M. Peng A. Wei Y. Chen L. Eur. J. Med. Chem. 2012;54:272–280. doi: 10.1016/j.ejmech.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Magalhães A. F. Azevedo Tozzi A. M. G. Noronha Sales B. H. L. Magalhães E. G. Phytochemistry. 1996;42:1459–1471. [Google Scholar]
- Rao M. S. Narukulla R. Fitoterapia. 2007;78:446–447. doi: 10.1016/j.fitote.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Hussain S. Singh A. Nazir S. U. Tulsyan S. Khan A. Kumar R. Bashir N. Tanwar P. Mehrotra R. J. Cell. Biochem. 2019;120:14213–14225. doi: 10.1002/jcb.28782. [DOI] [PubMed] [Google Scholar]
- Constantinescu T. Lungu C. N. Int. J. Mol. Sci. 2021;22:11306. doi: 10.3390/ijms222111306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata N. Ito M. Tomoda H. Ōmura S. Phytochemistry. 1997;46:683–687. doi: 10.1016/s0031-9422(97)00157-x. [DOI] [PubMed] [Google Scholar]
- Vogel S. Heilmann J. J. Nat. Prod. 2008;71:1237–1241. doi: 10.1021/np800188b. [DOI] [PubMed] [Google Scholar]
- Akihisa T. Tokuda H. Hasegawa D. Ukiya M. Kimura Y. Enjo F. Suzuki T. Nishino H. J. Nat. Prod. 2006;69:38–42. doi: 10.1021/np058080d. [DOI] [PubMed] [Google Scholar]
- Shen C.-C. Lin T.-W. Huang Y.-L. Wan S.-T. Shien B.-J. Chen C.-C. J. Nat. Prod. 2006;69:1237–1240. doi: 10.1021/np060189d. [DOI] [PubMed] [Google Scholar]
- Ye H. Fu A. Wu W. Li Y. Wang G. Tang M. Li S. He S. Zhong S. Lai H. Yang J. Xiang M. Peng A. Chen L. Fitoterapia. 2012;83:1402–1408. doi: 10.1016/j.fitote.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Wang G. Wu W. Peng F. Cao D. Yang Z. Ma L. Qiu N. Ye H. Han X. Chen J. Qiu J. Sang Y. Liang X. Ran Y. Peng A. Wei Y. Chen L. Eur. J. Med. Chem. 2012;54:793–803. doi: 10.1016/j.ejmech.2012.06.034. [DOI] [PubMed] [Google Scholar]
- Jordan M. A. Wilson L. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Heald R. Nogales E. J. Cell Sci. 2002;115:3–4. doi: 10.1242/jcs.115.1.3. [DOI] [PubMed] [Google Scholar]
- Carr M. Greene L. M. Knox A. J. S. Lloyd D. G. Zisterer D. M. Meegan M. J. Eur. J. Med. Chem. 2010;45:5752–5766. doi: 10.1016/j.ejmech.2010.09.033. [DOI] [PubMed] [Google Scholar]
- Hadfield J. A. Ducki S. Hirst N. McGown A. T. Prog. Cell Cycle Res. 2003;5:309–325. [PubMed] [Google Scholar]
- Cao D. Han X. Wang G. Yang Z. Peng F. Ma L. Zhang R. Ye H. Tang M. Wu W. Lei K. Wen J. Chen J. Qiu J. Liang X. Ran Y. Sang Y. Xiang M. Peng A. Chen L. Eur. J. Med. Chem. 2013;62:579–589. doi: 10.1016/j.ejmech.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Wang G. Peng F. Cao D. Yang Z. Han X. Liu J. Wu W. He L. Ma L. Chen J. Sang Y. Xiang M. Peng A. Wei Y. Chen L. Bioorg. Med. Chem. 2013;21:6844–6854. doi: 10.1016/j.bmc.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Murthy Y. L. N. Suhasini K. P. Pathania A. S. Bhushan S. Nagendra Sastry Y. Eur. J. Med. Chem. 2013;62:545–555. doi: 10.1016/j.ejmech.2013.01.027. [DOI] [PubMed] [Google Scholar]
- O'Brien R. M. Granner D. K. Physiol. Rev. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- Bell R. M. and Coleman R. A., in The Enzymes, ed. P. D. Boyer, Academic Press, 1983, vol. 16, pp. 87–111 [Google Scholar]
- Mayorek N. Bar-Tana J. J. Biol. Chem. 1985;260:6528–6532. [PubMed] [Google Scholar]
- Alves-Bezerra M. and Cohen D. E., in Comprehensive Physiology, 2017, pp. 1–22, 10.1002/cphy.c170012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson T. M. Brown P. J. Sternbach D. D. Henke B. R. J. Med. Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- Kersten S. Desvergne B. Wahli W. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Braissant O. Foufelle F. Scotto C. Dauça M. Wahli W. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- Kaplan F. Al-Majali K. Betteridge D. J. P. J Cardiovasc Risk. 2001;8:211–217. doi: 10.1177/174182670100800405. [DOI] [PubMed] [Google Scholar]
- Moller D. E. Nature. 2001;414:821–827. doi: 10.1038/414821a. [DOI] [PubMed] [Google Scholar]
- Okuno A. Tamemoto H. Tobe K. Ueki K. Mori Y. Iwamoto K. Umesono K. Akanuma Y. Fujiwara T. Horikoshi H. Yazaki Y. Kadowaki T. J. Clin. Invest. 1998;101:1354–1361. doi: 10.1172/JCI1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon A. W. Harp J. B. Am J Physiol Cell Physiol. 2001;280:C807–C813. doi: 10.1152/ajpcell.2001.280.4.C807. [DOI] [PubMed] [Google Scholar]
- Liang Y.-C. Tsai S.-H. Tsai D.-C. Lin-Shiau S.-Y. Lin J.-K. FEBS Lett. 2001;496:12–18. doi: 10.1016/s0014-5793(01)02393-6. [DOI] [PubMed] [Google Scholar]
- Takahashi N. Kawada T. Goto T. Yamamoto T. Taimatsu A. Matsui N. Kimura K. Saito M. Hosokawa M. Miyashita K. Fushiki T. FEBS Lett. 2002;514:315–322. doi: 10.1016/s0014-5793(02)02390-6. [DOI] [PubMed] [Google Scholar]
- Sato M. Tai T. Nunoura Y. Yajima Y. Kawashima S. Tanaka K. Biol. Pharm. Bull. 2002;25:81–86. doi: 10.1248/bpb.25.81. [DOI] [PubMed] [Google Scholar]
- Kuroda M. Mimaki Y. Honda S. Tanaka H. Yokota S. Mae T. Bioorg. Med. Chem. 2010;18:962–970. doi: 10.1016/j.bmc.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Korthikunta V. Singh R. Srivastava R. Pandey J. Srivastava A. Chaturvedi U. Mishra A. Srivastava A. K. Tamrakar A. K. Tadigoppula N. RSC Med. Chem. 2023;14:470–481. doi: 10.1039/d2md00341d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. Science. 1994;264:375–382. doi: 10.1126/science.8153624. [DOI] [PubMed] [Google Scholar]
- Spratt B. G. Science. 1994;264:388–393. doi: 10.1126/science.8153626. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Science. 1994;264:382–388. doi: 10.1126/science.8153625. [DOI] [PubMed] [Google Scholar]
- Terán W. Krell T. Ramos J. L. Gallegos M.-T. J. Biol. Chem. 2006;281:7102–7109. doi: 10.1074/jbc.M511095200. [DOI] [PubMed] [Google Scholar]
- Kare M. Kone M. E. K. Boulanger A. Niassy B. Lenouen D. Muckensturm B. Nongonierma A. J. Soc. Ouest-Afr. Chim. 2006;22:41. [Google Scholar]
- Paul S. B. Choudhury S. Bioinformation. 2010;4:473–477. doi: 10.6026/97320630004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S. B. Choudhury S. Bioinformatics. 2010;2:61–66. [Google Scholar]
- Wen R. Lv H.-N. Jiang Y. Tu P.-F. Phytochemistry. 2018;149:56–63. doi: 10.1016/j.phytochem.2018.02.005. [DOI] [PubMed] [Google Scholar]
- Singhai Nabin C. Barua Ram P. Sharma Jogendra N. Baruah A. K. Phytochemistry. 1983;22:1005–1006. [Google Scholar]
- Harborne J. B. Williams C. A. Nat. Prod. Rep. 1998;15:631–652. [Google Scholar]
- Peng J. Fan G. Wu Y. J. Chromatogr. A. 2006;1115:103–111. doi: 10.1016/j.chroma.2006.02.079. [DOI] [PubMed] [Google Scholar]
- Asada Y. Li W. Yoshikawa T. Phytochemistry. 1998;47:389–392. [Google Scholar]
- Pistelli L. Spera K. Flamini G. Mele S. Morelli I. Phytochemistry. 1996;42:1455–1458. [Google Scholar]
- Tian-Shung W. Phytochemistry. 1989;28:3558–3560. [Google Scholar]
- Jaipetch T. Kanghae S. Pancharoen O. Patrick V. Reutrakul V. Tuntiwachwuttikul P. White A. Aust. J. Chem. 1982;35:351–361. [Google Scholar]
- Mahidol C. Tuntiwachwuttikul P. Reutrakul V. Taylor W. C. Aust. J. Chem. 1984;37:1739–1745. [Google Scholar]
- Lidani K. C. F. Andrade F. A. Bavia L. Damasceno F. S. Beltrame M. H. Messias-Reason I. J. Sandri T. L. Front Public Health. 2019;7:166. doi: 10.3389/fpubh.2019.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa A. S. Vermeij D. Ramos, Jr. A. N. Luquetti A. O. Lancet. 2024;403:203–218. doi: 10.1016/S0140-6736(23)01787-7. [DOI] [PubMed] [Google Scholar]
- Rohmer M., in Comprehensive Natural Products Chemistry, ed. S. D. Barton, K. Nakanishi and O. Meth-Cohn, Pergamon, Oxford, 1999, pp. 45–67, 10.1016/B978-0-08-091283-7.00036-9 [DOI] [Google Scholar]
- Tomazela D. M. Pupo M. T. Passador E. A. P. da Silva M. F. d. G. F. Vieira P. C. Fernandes J. B. Fo E. Rodrigues. Oliva G. Pirani J. R. Phytochemistry. 2000;55:643–651. doi: 10.1016/s0031-9422(00)00248-x. [DOI] [PubMed] [Google Scholar]
- Bhakuni D. S. Chaturvedi R. J. Nat. Prod. 1984;47:585–591. doi: 10.1021/np50034a003. [DOI] [PubMed] [Google Scholar]
- Danila A., in Renewable Dyes and Pigments, ed. S. Ul Islam, Elsevier, 2024, pp. 253–269, 10.1016/B978-0-443-15213-9.00006-5 [DOI] [Google Scholar]
- Mori-Hongo M. Yamaguchi H. Warashina T. Miyase T. J. Nat. Prod. 2009;72:63–71. doi: 10.1021/np800535g. [DOI] [PubMed] [Google Scholar]
- Fang S.-C. Hsu C.-L. Yu Y.-S. Yen G.-C. J. Agric. Food Chem. 2008;56:8859–8868. doi: 10.1021/jf8017436. [DOI] [PubMed] [Google Scholar]
- Jayasinghe L. Rupasinghe G. K. Hara N. Fujimoto Y. Phytochemistry. 2006;67:1353–1358. doi: 10.1016/j.phytochem.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Magalhães A. F., Tozzi A. M. G., Magalhães E. G., Sannomiya M., Soriano M. d. P. C. and Perez M. A. J. A. d. A. B. d. C., 2007, 79, 351–367 [DOI] [PubMed]
- Tarus P. K. Machocho A. K. Lang'at-Thoruwa C. C. Chhabra S. C. Phytochemistry. 2002;60:375–379. doi: 10.1016/s0031-9422(02)00078-x. [DOI] [PubMed] [Google Scholar]
- Kokwaro J. O., Medicinal plants of east Africa, East African Literature Bureau, 1976 [Google Scholar]
- Kokwaro J. O., Flora of Tropical East Africa-Anacardiaceae (1986), CRC Press, 1986 [Google Scholar]
- Kitagawa I. Zhang R. Hori K. Tsuchiya K. Shibuya H. Chem. Pharm. Bull. 1992;40:2041–2043. doi: 10.1248/cpb.40.2041. [DOI] [PubMed] [Google Scholar]
- Sugii M. Ohkita M. Taniguchi M. Baba K. Kawai Y. Tahara C. Takaoka M. Matsumura Y. Biol. Pharm. Bull. 2005;28:607–610. doi: 10.1248/bpb.28.607. [DOI] [PubMed] [Google Scholar]
- Lee Y. R. Li X. Lee S. W. Yong C. S. Hwang M. R. Lyoo W. S. Bull. Korean Chem. Soc. 2008;29:1205–1210. [Google Scholar]
- Mahey S. Sharma P. Seshadri T. R. Mukerjee S. K. Indian J. Chem. 1972;10:585–588. [Google Scholar]
- Lawson M. A. Kaouadji M. Chulia A. J. Tetrahedron Lett. 2010;51:6116–6119. [Google Scholar]
- Passador E. A. P. da Silva M. F. das G. F. Fo E. R. Fernandes J. B. Vieira P. C. Pirani J. R. Phytochemistry. 1997;45:1533–1537. [Google Scholar]
- Fukai T. Nomura T. Phytochemistry. 1995;38:759–765. [Google Scholar]
- Lee Y. R. Li X. Kim J. H. J. Org. Chem. 2008;73:4313–4316. doi: 10.1021/jo800367r. [DOI] [PubMed] [Google Scholar]
- Lee Y. R. Xia L. Synthesis. 2007;20:3240–3246. [Google Scholar]
- Lee Y. R. Wang X. Xia L. Molecules. 2007;12:1420–1429. doi: 10.3390/12071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D. H. Lee Y. R. Kim S. H. Helv. Chim. Acta. 2010;93:635–647. [Google Scholar]
- Bawazir A. S. Imam S. S. Yahya B. A. Abdulqawi L. N. Discover Plants. 2024;1:3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.