

Abstract
Background
Pineal parenchymal tumor of intermediate differentiation (PPTID) is a rare tumor. This study aims to evaluate patient outcomes and propose a treatment algorithm based on existing literature and our case series.
Methods
This prospective observational study includes seven patients diagnosed with PPTID through histopathology. We analyzed their clinical presentation, magnetic resonance imaging findings, surgical approaches, histopathological and immunohistochemical analysis, adjuvant treatments, and outcomes. We conducted univariate and multivariate statistical analyses.
Results
The mean patient age was 40 years, with a male predominance. All patients presented with hydrocephalus, four of which required cerebrospinal fluid diversion procedures. The average tumor size was 3.13cm, with 85.7% showing brain invasion. Surgical outcomes included one gross total resection, two near total resections, and four subtotal resections. The supracerebellar infratentorial (Krause) approach was used in 71.4% of cases. About 85.7% were diagnosed with grade 3 PPTID. Five patients received adjuvant radiotherapy. The analysis showed each additional mitosis unit decreased survival by 0.17 units, equating to roughly 2 months (P < 0.016). Follow-up ranged from 6 to 120 months, with a 5-year survival rate of 57.1%. Factors influencing survival included the extent of tumor resection, brain invasion, tumor grade, and adjuvant treatment, although it was not statistically significant due to the small sample size.
Conclusion
We propose a treatment algorithm for PPTID and highlight the importance of further research to understand its biological characteristics. Safe maximal resection appears beneficial for higher-grade PPTID, but the role of adjuvant treatment after complete resection of lower-grade tumors remains uncertain.
Keywords: Hydrocephalus, Pineal gland, Pineal parenchymal tumor of intermediate differentiation, Pineal parenchymal tumor, Radiotherapy, Safe maximal resection

INTRODUCTION
The pineal gland, traditionally addressed as Descartes’ seat of the soul, is a pine-cone-shaped structure located in the superior aspect of the posterior third ventricular recess.[47] The gland has been theorized to be a center of “rational thought.” It secretes an indoleamine, “melatonin,” which is responsible not only for orchestrating the circadian rhythm in vertebrates but also for recently discovered antineoplastic properties.[16] About 95% of the pineal gland comprises pinealocytes with dendritic projections and 5% glial cells.[15] Unfortunately, both of the above cell lines are harbingers of neoplasia.[7,44,68] The most common pineal region tumors are germ cell tumors, and they account for 50% of intracranial germ cell tumors.[49] On the contrary, parenchymal tumors are relatively rare compared to germ cell tumors and comprise <1% of CNS tumors.[12,67] In 2021, the World Health Organization (WHO) classified pineal parenchymal tumors as pineocytoma, pineoblastoma, papillary pineal tumors, pineal parenchymal tumors of intermediate differentiation (PPTID), and desmoplastic myxoid tumor of the pineal region – SMARCB1-mutant.[38] Pineal parenchymal tumors are neoplasms arising from pineocytes. Pineocytomas are benign tumors and have an indolent course. The other end of the spectrum comprises pineoblastomas, which are aggressive tumors with a 5-year survival rate of <60%.[35,36]
PPTIDs represent an even rarer subset within the pineal parenchymal tumors (10–50%).[20,40] PPTID was first described by Schild et al. in 1993 and was included as a part of the WHO classification in 2000.[57] Verily, these entities are regarded as neoplasms with histology blending with both pineocytoma and pineoblastoma.[40] The optimal treatment strategy for this type of tumor has not been devised due to its rarity. The question of whether such tumors can be effectively treated with surgery, a combination of adjuvant chemotherapy and radiation, or radiotherapy alone remains a topic of ongoing debate.[1]
Therefore, we bring to light a series of seven cases of PPTID from a tertiary care neurosurgical unit in South Asia, which can add to understanding the progress and outcome of this ailment.
MATERIALS AND METHODS
The Institutional Ethics Board review clearance was obtained (IRB:48/2023). Informed consent was obtained from all patients and their next of kin.
Patients
Seven patients with histologically proven PPTID treated in the Department of Neurosurgery, Kasturba Medical College and Hospital, Manipal, Karnataka, India, between January 2014 and January 2024 were a part of this cohort. This study analyzed the clinical records, clinical presentation, preoperative neurological deficits, radiological features, surgical strategy executed, histopathological features, immuno-histochemistry markers, postoperative complications/neurological deficits, adjuvant treatment, recurrence of tumor, 5-year outcomes based on modified Rankin scale (mRS) dichotomized into good as 0–3 and poor as 4–6, and overall survival. The data of the above seven patients are shown in Table 1. Moreover, we conducted an exhaustive search on PUBMED and MEDLINE, employing MeSH terms and specific keywords such as “pineal parenchymal tumors” and “pineal parenchymal tumors of intermediate differentiation.” Since 2008, we have gathered and reviewed all accessible articles on PPTID. When the available data were deemed sufficient to infer the extent of resection, adjuvant treatment, and outcomes, we compiled this information into Table 2. The purpose of this literature review was to understand how, over time and with increased awareness among clinicians, the trends in diagnosis, treatment, and outcomes have evolved.
Table 1:
Summary of the seven patients in our case series.

Table 2:
Summary of articles from 2008 on PPTID.

Caveats in managing PPTID
Broadly, the following issues were factored into consideration for optimizing and executing a holistic treatment strategy individualized to each of our patients:
Treatment for acute hydrocephalus – Endoscopic Third Ventriculostomy (ETV), Ventriculoperitoneal Shunt (VP shunt), or External Ventricular Drain (EVD).
Tissue diagnosis – Endoscopic third ventriculostomy/ stereotactic biopsy versus open surgical biopsy?
Pre-operative imaging – Plane of tumor on magnetic resonance imaging (MRI) with adjacent vital structures, that is, brainstem, thalamus, and deep venous system.
Approach to be undertaken for the definitive surgery – Supracerebellar infratentorial (Krause), Infraoccipital transtentorial (Poppen), or Interhemispheric transcallosal?[48]
The extent of resection – based on the plane of tumor preoperatively and intraoperatively.
Adjuvant radiation is based on histopathological examination, the extent of resection, and recurrence.
We defined the extent of resection as
Gross-total resection
Complete microscopic resection and no evidence of residual tumor on postoperative imaging
Near-total resection
Complete microscopic resection with some radiological evidence of residual tumor on postoperative imaging.
Sub-total resection
50–70% tumor decompression performed. A significant amount of the tumor was not removed due to adhesion/ loss of plane with adjacent vital structures.
Statistical analysis
Due to the non-parametric nature of our data set and the small sample size due to disease rarity, we applied the Spearman correlation coefficient and Fischer’s exact test to identify any possible association. Univariate and multivariate statistical analyses, including log-rank test and Cox regression analysis for assessing all variables against survival outcomes, were conducted using IBM Statistical Package for the Social Sciences software for Mac OS.
RESULTS
Demographics and clinical symptomatology
All our patients were adults with a mean age of 40.43 ± 10.03 (years). Five (71.4%) of the group were male, and 2 (28.6%) were female. All seven patients had either clinical symptoms or radiological evidence of hydrocephalus. Apart from hydrocephalus, the clinical presentation was predominantly that of headache (71.4%), papilledema (71.4%), projectile vomiting (57.1%), diplopia (42.9%), and others, which are shown in Figure 1. None of the tumors were detected incidentally.
Figure 1:

Graph depicting the clinical symptomatology in our patient cohort.
Imaging characteristics
On MRI of the brain with intravenous Gadolinium contrast, most patients had a bulky, solid tumor with heterogeneous contrast enhancement (85.7 %). The mean tumor size was 3.13 ± 1.34 cm. Four patients had calcifications (57.1%). In 85.7 % of cases, there was adjacent brain invasion in the form of loss of plane superiorly between the thalamus, tectal plate, 4th ventricular floor, or complete brainstem involvement inferiorly. Preoperative complete neuraxial screening was not performed. All patients had negative tumor markers such as alpha-fetoprotein, human chorionic gonadotropin, and carcinoembryonic antigen.
Cerebrospinal fluid (CSF) diversion
Four out of seven patients required CSF diversion (57.1%). Two patients underwent ETV (28.57%). In patients where ETV was undertaken to treat the hydrocephalus, tumor biopsy was precluded by the inter-thalamic adhesions obstructing the path.
Definitive surgery
Our strategy for the surgical corridor was planned based on the displacement of the internal cerebral veins, the vein of Galen, and the lateral/anterior extent of the tumor. If the veins were displaced cranially, Krause’s approach (71.4%) was favored over Poppen’s (14.3 %). In one patient, due to the anterior projection of the tumor into the 3rd ventricle and vein of Galen draping the tumor posteriorly, an interhemispheric trans-callosal, trans-choroidal approach was utilized.
Grade of resection and complications
The extent of tumor resection and the need for adjuvant treatment was tailored to individual patients based on preoperative MRI and intraoperative tumor plane with an adjacent normal eloquent brain. In only 1 patient (14.3 %), gross total resection was undertaken. The other six patients underwent either a subtotal (57.1%) or a near-total resection (28.6%) due to poorly defined arachnoid planes. Two patients developed immediate postoperative complications. One patient developed a tectal infarct and became vegetative, following which his mRS did not improve. The other patient developed an operative site hematoma, which was managed conservatively with anti-edema measures, after which the patient improved. We postulate that the complications were due to a lack of clear arachnoid planes, distorted anatomy, and significant vascularity of the tumor, which is a challenge in itself to secure hemostasis in a deep, narrow surgical corridor besieged by vital structures.
Histopathological examination and immunohistochemistry
Six out of seven patients in our study group had grade 3 PPTID (85.71%). The presence of high mitoses diagnosed a higher grade of the tumor [Figure 2a] and the Ki-67 index [Figure 2b]. The mean (standard deviation [SD]) of Mitoses was 10.43 (8.12). The median (interquartile range [IQR]) of Mitoses was 6.00 (5–13). The Mitoses ranged from 5 to 26. The mean (SD) of Ki-67 was 19.14 (19.97). The median (IQR) of Ki-67 was 12.00 (7-26). The Ki-67 ranged from 1 to 55. Six patients (85.7%) were positive for synaptophysin [Figure 2c].
Figure 2:
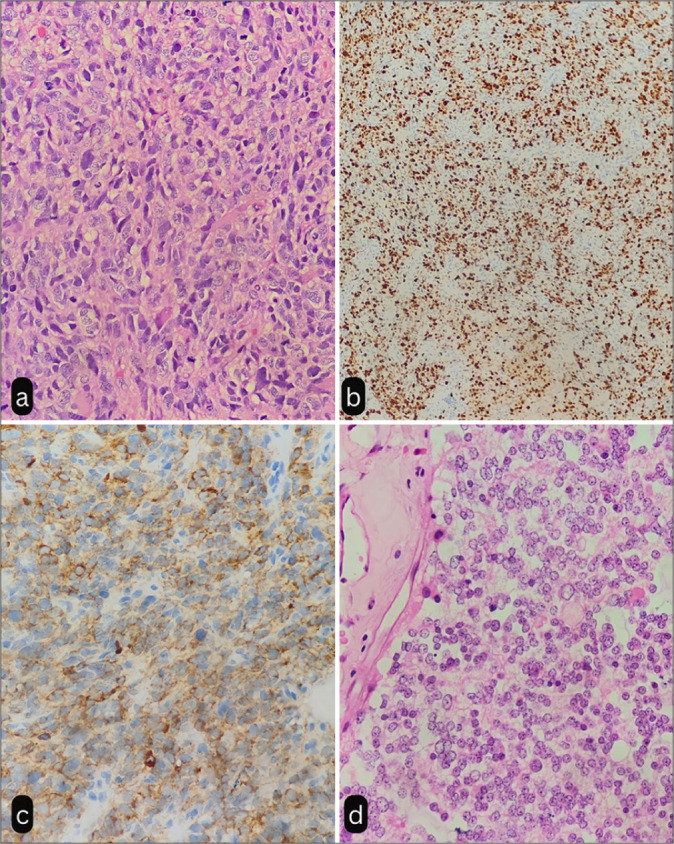
(a) Tumor composed of sheets of neoplastic tumor cells showing pleomorphism and increased mitosis suggestive of pineal parenchymal tumor of intermediate differentiation (H&E, ×40). (b) Tumor cells showing high Ki67 index (×10). (c) Tumor cells showing immunoreactivity to Synaptophysin (×40). (d) Photomicrograph showing uniform cells with round nuclei, moderate amount of eosinophilic cytoplasm, and absence of mitosis (H&E, ×40). H & E: Hematoxylin and Eosin Stain
Tumor grade change
One patient demonstrated a grade change from pineocytoma [Figure 2d] to PPTID after a 5-year disease-free interval.
Following the second surgery, she developed hydrocephalus, for which she underwent a VP shunt. During the second surgery, only a subtotal decompression of the tumor was plausible due to adhesions with adjacent structures.
Mitoses of tumor and survival
Non-parametric tests (Spearman Correlation) were used to explore the correlation between the two variables, as at least one of the variables was not normally distributed. Mitoses and survival (Years) had a strong negative correlation, which was statistically significant (rho = −0.85, P = 0.016). For every 1 unit increase in mitoses, the survival (Years) decreases by 0.17 units, that is, around 2 months [Figure 3].
Figure 3:

Co-relation between mitoses and survival based on Spearman’s Co-relation coefficient.
Adjuvant treatment
All patients with Karnofsky Performance Status (KPS) of more than 70 were subjected to adjuvant whole-brain radiation therapy (RT) with a boost to the pineal region. The typical radiation dosage administered was 55 Gy delivered in 25–30 fractions. In two patients (28.5%), the residual tumor was stable and showed a good response to RT. In one patient, the tumor demonstrated a remarkable response, and there was complete radiological remission of the disease on follow-up. Two patients (28.5%) developed a recurrence of the tumor despite RT. One patient (14.2%) did not receive RT since the biopsy was suggestive of pineocytoma, and she had undergone a gross total resection. After 60 months, she developed a recurrence. After the second surgery, she is receiving RT and is on follow-up. Two patients (28.5%) did not receive RT due to aggressive disease progression and poor KPS.
Recurrence and leptomeningeal spread
Three patients (42.8%) developed recurrence after 6 months, 18 months, and 60 months, respectively, after the first surgery. One patient succumbed to the aggressive recurrent disease, while the other two patients are on follow-up treatment. Two patients (28.5%) developed leptomeningeal seeding of the tumor on follow-up imaging.
Outcome and survival
Four out of 7 patients (57.1%) are on follow-up with good outcomes (mRS <3). One patient had an aggressive tumor occupying the whole of the fourth ventricle and was adherent to the floor at the time of diagnosis. One patient developed an extensive recurrence with leptomeningeal spread, following which the patient and family opted for palliative care. The mean survival rate in our patient data set was 35 months ± 2 months (51.7%) over a follow-up of 120 months. All patients were on regular, compliant follow-up. The 5-year progression-free rate was 71.43 % in this cohort of patients. However, due to the small sample size, we could not demonstrate a statistically significant association between the tumor grade and progression-free survival/overall survival.
Illustrative cases
Case 2
A 50-year-old male presented with a homogenous T1 isointense non-contrast-enhancing tumor on MRI [Figure 4a]. Planning the approach to a posterior third ventricular tumor is essential in attaining good tumor decompression without causing undue complications. On a more detailed analysis of the MRI, we noticed that he had a very steep tentorial angle with the vein of the Galen complex draping the tumor posteriorly. Hence, a transcallosal, subchoroidal approach was advocated for this case. Near-total tumor resection was achieved in this case [Figure 4b].
Figure 4:

(a) Case 2: magnetic resonance imaging (MRI) – brain, mid-sagittal section T1 contrast sequence: Non-enhancing pineal region tumor, with vein of Galen Complex draping the tumor posteriorly. (b) Case 2: Postoperative computed tomography (CT) brain: Craniotomy defect centered around the coronal suture, Interhemispheric trans callosal sub-choroidal approach was advocated. Near-total resection (NTR) was achieved. (c) Case 3: Before radiotherapy (RT) – MRI brain axial T1 contrast sequence demonstrating the extensive nature of the lesion extending rostrally to involve the thalamus (d) Case 3: After RT–MRI brain axial T1 contrast, depicting good response to RT. (e) Case 4: Preoperative CT brain showing homogenous hyperdensity suggesting that pineal parenchymal tumor of intermediate differentiation can not only have speckled or peripheral calcifications. (f) Case 4: Postoperative MRI demonstrating NTR. (g) Case 3: Before RT–MRI brain T1 contrast mid-sagittal section showing a diffuse heterogeneous contrast-enhancing tumor involving the thalamus and callosum. (h) Case 3: After RT– MRI brain T1 post contrast sequence mid- sagittal section. Complete radiological remission of the lesion is noted. The neurovascular structures, including the vein of Galen complex, can be well delineated. (i) Case 7: Homogenous contrast-enhancing lesion on T1 MRI, with visibly well-defined planes. HPE was suggestive of pineocytoma. (j) Case 7: Postoperative CT brain. Gross-total resection of the tumor was achieved. (k) Case 7: MRI brain after 5 years of disease-free interval depicting an aggressive recurrence with poor planes and supra-tentorial extension. (l) Case 6: Neuraxis screening for this patient revealed diffuse leptomeningeal contrast-enhancing lesions suggestive of drop metastasis.
Case 4
A 48-year-old male presented with a homogenously calcified lesion in the pineal recess [Figure 4c]. He underwent a near-total resection. The tumor was dissected from the midbrain. Part of the tumor adherent to the vein of the Galen complex was not removed [Figure 4d].
Case 5
A 25-year-old female presented with signs and symptoms suggestive of raised intracranial pressure (ICP). MRI brain with contrast revealed a large tumor in the pineal recess extending rostrally to involve the thalamus [Figure 4e and f]. She underwent tumor decompression and biopsy. Post-surgery, she was subjected to radiotherapy. She received 55Gy over 30 fractions with a pineal region boost. This patient was diagnosed with high-grade PPTID and underwent a subtotal decompression. However, she demonstrated an excellent response to radiotherapy, and there was complete radiological remission on follow-up imaging [Figure 4g and h].
Case 7
A 48-year-old lady presented with symptoms suggestive of raised ICP. MRI brain evaluation revealed homogenous enhancing posterior third ventricular tumor [Figure 4i]. She underwent gross total resection as demonstrated on the postoperative computed tomography brain [Figure 4j]. The biopsy was suggestive of pineocytoma. Five years later, she presented with similar complaints, and the MRI showed an aggressive recurrence of the tumor with a significant component extending above the tentorium [Figure 4k]. This time, due to the adherence of the tumor to the deep venous system, only a subtotal decompression was achieved. Biopsy revealed grade 3 PPTID.
DISCUSSION
Since the incorporation of PPTID into the WHO classification system in 2000, more discoveries and advancements have been made in diagnosing and treating these tumors.[1,61,68,75] Nevertheless, to this day, there remains a lack of consensus on the optimal approach and guidelines for directing treatment options for this tumor. What exacerbates the situation is that the incorrect grouping of these tumors may underestimate the likelihood of CSF seeding (as illustrated in Figure 4l, which depicts a patient from our cohort exhibiting leptomeningeal dissemination) and prognosis.[57]
PPTID can manifest across all age demographics, predominantly in adults.[25,45,57] All our patients were adults with a mean age of 40 years. Contrary to the available literature, most of our patients were male (71.4%).[15,39,42] Some series highlight equal sex distribution.[11] Our patients exhibited symptoms consistent with hydrocephalus, aligning with findings from most series. In addition, we observed that Parinaud syndrome is a rare presentation, even when radiological evidence indicates the involvement of the midbrain tectum.
Many authors emphasize that gross total resection of tumors translates into better overall survival. It is crucial for the skilled neurosurgeon to carefully weigh the pros and cons of pursuing an aggressive gross total resection, considering the potential neurological morbidity involved. We opt for a safe maximal resection followed by adjuvant radiation akin to that utilized for pineoblastoma or a high-grade glioma to mitigate the risk of neurological deficits.[5,41,60,63] It is imperative to remain cognizant that injury to critical structures such as the vein of the Galen complex, posterior thalamus, or midbrain tectum can result in significant morbidity. When the tumor exhibits well-defined arachnoid planes, advocating for a gross total resection is feasible. In our series, most patients (85.7%) underwent either a near-total or subtotal resection followed by adjuvant radiation. Our outcomes align with those reported in other series. Endoscopic surgery or endoscope-assisted microsurgery has gained popularity over the past few years, and studies have demonstrated better gross total resection and reduced rates of hydrocephalus.[72]
The histopathological diagnosis is the most crucial yet perplexing aspect of PPTID. These tumors exhibit significant histological heterogeneity, and their behavior often defies prediction.[26] While some authors propose an aggressive treatment approach regardless of the tumor grade, others advocate for tumor decompression followed by adjuvant radiation therapy.[11,13] Many have utilized adjuvant temozolomide chemotherapy, whereas others have used procarbazine, vincristine, lomustine, nimustine, carboplatin, and interferon B in different combinations.[69,74] It was traditionally considered that chemotherapeutic agents have poor blood-brain barrier penetration.[24] However, post-irradiation, there is a possibility that due to capillary bed disruption, the blood-brain barrier is more permissible to chemotherapeutic agents, which could be the rationale.[6] Nonetheless, some authors even propose that we might be overtreating PPTID, suggesting that it can be managed effectively with radiation therapy alone.[13] We opted not to administer chemotherapy to any of our patients. Among the cohort, three out of seven patients exhibited stable disease and are alive, while one patient demonstrated an excellent response to radiotherapy. This suggests a potential radiosensitivity of the tumor. However, Chalif et al., in a recent study, concluded that radiotherapy does not affect the outcomes of grade 2 PPTID.[8]
In general, PPTIDs demonstrate moderately high cellularity, mild-to-moderate nuclear atypia, and moderate mitotic activity, displaying diffuse or lobulated growth patterns.[37] Further prognostic stratification is facilitated by assessing the extent of necrosis, the mitotic index, and the presence of neurofilament protein.[4,14,25] The 2007 WHO Classification of Central Nervous System Tumors considers 2 variables – proliferative activity and immunoreactivity for neurofilament protein – as outcome predictors.[11,56] These tumors can generally present a positive expression for neurofilament, synaptophysin, renal S antigen, and chromogranin A.[15] Neurofilament protein expression has a very debatable role in predicting outcomes. Jouvet et al. suggested that neurofilament is expressed only in low-grade PPTID (grade 2) and correlates with better outcomes.[25] However, various studies over the last decade have questioned its utility.[9,75] Due to the variability of neurofilament expression, other immunohistochemistry markers, such as the MIB-1 labeling index (MIB-LI) and Ki-67, have been considered more reliable for diagnostic purposes.[15,27,53] MIB-LI and mitoses have a significant implication in the prognostication of the disease and in determining recurrence.[2,19,21,27,64] More recently, it has been discovered that an in-frame mutation in the KBTBD4 gene is positive in approximately 80% of cases and is specific to PPTID. Consequently, this discovery can aid in clearly distinguishing PPTID from pineocytoma, allowing for a more definitive diagnosis beyond solely relying on pineocytomatous pseudo-rosettes.[65]
Numerous authors have made efforts to classify the tumor based on histopathological characteristics. Yu et al. classified tumors into high-risk and low-risk groups. The high-risk group comprised a mitotic count ≥3/10 high-power field (HPF) or Ki67 LI ≥5, whereas the low-risk group was defined as mitotic count <3/10 HPF and Ki67 LI <5.[75] The former category is associated with dismal outcomes. Chatterjee et al. have proposed a classification for PPTIDs with mitosis <4/10 HPF and Ki-67 <5% as grade II and cases with mitosis >4/10 HPF and/or Ki-67 >5% as grade III PPTID.[9] While these classification systems can assist in forecasting outcomes and planning adjuvant treatment for the high-risk category, they do not assist in devising holistic tumor management strategies. In our series, all patients had mitoses > 5/10 HPF, and 6 out of 7 patients (85.7%) had Ki-67 >5%. Only an increase in mitotic count demonstrated a correlation with survival and achieved statistical significance. We did not find a significant statistical association between Ki-67 and outcomes. In one patient, the tumor exhibited a grade change, initially presenting with absent mitoses, which subsequently increased to 15–20/10 HPFs and Ki-67 of 3 % upon diagnosis as PPTID grade 3. Apart from our patient, various other reports have documented grade changes in pineal tumors.[3,28,31,52] From these instances, one may deduce that PPTID is an intermediary pathological condition capable of originating or transitioning from a pineocytoma, displaying the propensity to advance into or imitate the characteristics of a pineoblastoma. This may suggest that it is unlikely that PPTID arises de novo.
In 2011, the guidelines set forth by the British Neurooncology Society for rare tumors offer a delineated algorithm elucidating the procedural steps to be undertaken in managing a tumor located within the pineal region. The British Neuro-oncology Society recommends surgical resection as the primary intervention for PPTID while indicating that the efficacy of radiotherapy remains uncertain.[1,58] Indeed, the current landscape reveals a notable absence of consensus regarding diagnostic methodologies and therapeutic strategies. However, similar to the above guidelines and review by Mallick et al., we have devised a simple pragmatic algorithm that can help clinicians tailor their treatment for PPTID [Figure 5].[42] Due to a lack of strong evidence, we have not incorporated adjuvant chemotherapy into the algorithm. However, the algorithm serves as a foundational framework for managing patients and a stepping stone for future research.
Figure 5:

A practical management algorithm for pineal parenchymal tumor of intermediate differentiation. PPTID: Pineal parenchymal tumor of intermediate differentiation, ETV: Endoscopic third ventriculostomy, GTR: Gross total resection, STR: Sub total resection, NTR: Near total resection, MRI: Magnetic resonance imaging, RT: Radiotherapy, CSF: Cerebrospinal fluid
Limitations
Due to the rarity of the disease, data can only be derived from a small sample size case series. This indicates the need for systematic reviews. We humbly acknowledge the criticism for not incorporating neuraxial screening at the presentation. We strongly emphasize that MRI screening of the neuraxis is essential in PPTID. However, we remain uncertain about the necessity of routine CSF sampling. Given the diversity in the existing literature and our small sample size, conducting a quantitative meta-analysis was not feasible. This is primarily due to the limited availability of literature, which consists mainly of case reports and a few small case series.
CONCLUSION
Drawing on insights and observations from various eminent authors over the past two decades, a tentative consensus may be reached suggesting that low-grade PPTID could potentially undergo gross total resection safely, given the preservation of the tumor-arachnoid interface. Conversely, in cases of high-grade PPTID, surgical precision is paramount to avoid grave neurological deficits/morbidity. Following near-total or subtotal resection, adjuvant chemo/radiotherapy becomes imperative for comprehensive management and better overall survival.
Definitive lacunae revolve around these pivotal questions: -
In the scenario where a high-grade lesion has undergone gross total resection, is adjuvant therapy mandatory, or is there an acceptable level of risk in opting for vigilant follow-up with regular imaging?
Should patients with low-grade PPTID, despite gross total resection, require rigorous follow-up imaging or adjuvant therapy, considering its potential for progression to a higher-grade recurrence?
The above questions are still devoid of answers and will require larger multicenter observational studies to address.
Acknowledgement
The authors would like to thank Ms. Shubavati, Chief Secretary, Department of Neurosurgery, for her constant assistance and support in executing this study.
Footnotes
How to cite this article: Srinivasan S, Hegde A, Nair R, Jampani RT, Ashraf M, Chigurupati D, et al. Pineal parenchymal tumor of intermediate differentiation: Case series and literature review: Is it time for a consensus? Surg Neurol Int. 2025;16:138. doi: 10.25259/SNI_1068_2024
Contributor Information
Siddharth Srinivasan, Email: siddharth.srinivasan93@gmail.com.
Ajay Hegde, Email: dr.ajayhegde@gmail.com.
Rajesh Nair, Email: rajesh.nair@manipal.edu.
Ravi Teja Jampani, Email: jcravitejachowdary@gmail.com.
Mohammad Ashraf, Email: mohammad.ashraf6@nhs.scot.
Dhanwanth Chigurupati, Email: dhanwanth.chigurupati@nhs.scot.
Bharat Kumar Raju, Email: bharatkmr@gmail.com.
Susanth Subramanian, Email: susanth.s@manipal.edu.
Udgam Baxi, Email: udgambaxi@gmail.com.
Yasaswi Kanneganti, Email: yasaswi2u@gmail.com.
Sarah Johnson, Email: johnson.sarah27@mayo.edu.
Bhavna Nayal, Email: bhavna.nayal@manipal.edu.
Geeta Vasudevan, Email: geeta.v@manipal.edu.
Deepak Nayak, Email: deepak.nayak@manipal.edu.
Girish Menon, Email: girish.menon@manipal.edu.
Ethical Approval
The research/study approved by the Institutional Review Board at Manipal Academy of Higher Education, approval number: IRB/48/2023 dated June 05 2023
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial Support and Sponsorship
Nil.
Conflicts of Interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
REFERENCES
- 1.Amato-Watkins AC, Lammie A, Hayhurst C, Leach P. Pineal parenchymal tumours of intermediate differentiation-An evidence-based review of a new pathological entity. Br J Neurosurg. 2016;30:11–5. doi: 10.3109/02688697.2015.1096912. [DOI] [PubMed] [Google Scholar]
- 2.Arivazhagan A, Anandh B, Santosh V, Chandramouli BA. Pineal parenchymal tumors--utility of immunohistochemical markers in prognostication. Clin Neuropathol. 2008;27:325–33. doi: 10.5414/npp27325. [DOI] [PubMed] [Google Scholar]
- 3.Bando T, Ueno Y, Shinoda N, Imai Y, Ichikawa K, Kuramoto Y, et al. Therapeutic strategy for pineal parenchymal tumor of intermediate differentiation (PPTID): Case report of PPTID with malignant transformation to pineocytoma with leptomeningeal dissemination 6 years after surgery. J Neurosurg. 2018;130:2009–15. doi: 10.3171/2018.2.JNS171876. [DOI] [PubMed] [Google Scholar]
- 4.Bielle F, Navarro S, Bertrand A, Cornu P, Mazeron JJ, Jouvet A, et al. Late dural relapse of a resected and irradiated pineal parenchymal tumor of intermediate differentiation. Clin Neuropathol. 2014;33:424–7. doi: 10.5414/NP300764. [DOI] [PubMed] [Google Scholar]
- 5.Bonosi L, Marrone S, Benigno UE, Buscemi F, Musso S, Porzio M, et al. Maximal safe resection in glioblastoma surgery: A systematic review of advanced intraoperative image-guided techniques. Brain Sci. 2023;13:216. doi: 10.3390/brainsci13020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown WR, Blair RM, Moody DM, Thore CR, Ahmed S, Robbins ME, et al. Capillary loss precedes the cognitive impairment induced by fractionated whole-brain irradiation: A potential rat model of vascular dementia. J Neurol Sci. 2007;257:67–71. doi: 10.1016/j.jns.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Carr C, O’Neill BE, Hochhalter CB, Strong MJ, Ware ML. Biomarkers of pineal region tumors: A review. Ochsner J. 2019;19:26–31. doi: 10.31486/toj.18.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalif EJ, Murray RD, Mozaffari K, Chillakuru YR, Shim T, Monfared A, et al. Malignant pineal parenchymal tumors in adults: A national cancer database analysis. Neurosurgery. 2022;90:807. doi: 10.1227/neu.0000000000001915. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee D, Lath K, Singla N, Kumar N, Radotra BD. Pathologic prognostic factors of pineal parenchymal tumor of intermediate differentiation. Appl Immunohistochem Mol Morphol. 2019;27:210–5. doi: 10.1097/PAI.0000000000000565. [DOI] [PubMed] [Google Scholar]
- 10.Chen YL, Tai LH, Lieu AS. Recurrent pineal parenchymal tumor of intermediate differentiation with intratumoral hemorrhage: A case report and review of the literature. Rare Tumors. 2023;15:20363613231177537. doi: 10.1177/20363613231177537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choque-Velasquez J, Resendiz-Nieves JC, Jahromi BR, Colasanti R, Raj R, Tynninen O, et al. Pineal parenchymal tumors of intermediate differentiation: A long-term follow-up study in Helsinki neurosurgery. World Neurosurg. 2019;122:e729–39. doi: 10.1016/j.wneu.2018.10.128. [DOI] [PubMed] [Google Scholar]
- 12.Cuccia F, Mortellaro G, Cespuglio D, Valenti V, DE Gregorio G, Quartuccio E, et al. A Case report of adult pineoblastoma occurring in a pregnant woman. Anticancer Res. 2019;39:2627–31. doi: 10.21873/anticanres.13386. [DOI] [PubMed] [Google Scholar]
- 13.Das P, Mckinstry S, Devadass A, Herron B, Conkey DS. Are we over treating Pineal Parenchymal tumour with intermediate differentiation? Assessing the role of localised radiation therapy and literature review. SpringerPlus. 2016;5:26. doi: 10.1186/s40064-015-1502-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fauchon F, Jouvet A, Paquis P, Saint-Pierre G, Mottolese C, Ben Hassel M, et al. Parenchymal pineal tumors: A clinicopathological study of 76 cases. Int J Radiat Oncol Biol Phys. 2000;46:959–68. doi: 10.1016/s0360-3016(99)00389-2. [DOI] [PubMed] [Google Scholar]
- 15.Favero G, Bonomini F, Rezzani R. Pineal gland tumors: A review. Cancers. 2021;13:1547. doi: 10.3390/cancers13071547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Favero G, Moretti E, Bonomini F, Reiter RJ, Rodella LF, Rezzani R. Promising antineoplastic actions of melatonin. Front Pharmacol. 2018;9:1086. doi: 10.3389/fphar.2018.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fèvre-Montange M, Vasiljevic A, Frappaz D, Champier J, Szathmari A, Aubriot Lorton MH, et al. Utility of Ki67 immunostaining in the grading of pineal parenchymal tumours: A multicentre study. Neuropathol Appl Neurobiol. 2012;38:87–94. doi: 10.1111/j.1365-2990.2011.01202.x. [DOI] [PubMed] [Google Scholar]
- 18.Fomchenko EI, Erson-Omay EZ, Kundishora AJ, Hong CS, Daniel AA, Allocco A, et al. Genomic alterations underlying spinal metastases in pediatric H3K27M-mutant pineal parenchymal tumor of intermediate differentiation: Case report. J Neurosurg Pediatr. 2019;25:121–30. doi: 10.3171/2019.8.PEDS18664. [DOI] [PubMed] [Google Scholar]
- 19.Fukuoka K, Sasaki A, Yanagisawa T, Suzuki T, Wakiya K, Adachi J, et al. Pineal parenchymal tumor of intermediate differentiation with marked elevation of MIB-1 labeling index. Brain Tumor Pathol. 2012;29:229–34. doi: 10.1007/s10014-012-0089-x. [DOI] [PubMed] [Google Scholar]
- 20.Han SJ, Clark AJ, Ivan ME, Parsa AT, Perry A. Pathology of pineal parenchymal tumors. Neurosurg Clin N Am. 2011;22:335–40. vii. doi: 10.1016/j.nec.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Heim S, Beschorner R, Mittelbronn M, Keyvani K, Riemenschneider MJ, Vajtai I, et al. Increased mitotic and proliferative activity are associated with worse prognosis in papillary tumors of the pineal region. Am J Surg Pathol. 2014;38:106–10. doi: 10.1097/PAS.0b013e31829e492d. [DOI] [PubMed] [Google Scholar]
- 22.Ito K, Aihara Y, Chiba K, Oda Y, Kawamata T. A case of a pineal parenchymal tumor of intermediate differentiation with bifocal lesions differentiated by negative placental alkaline phosphatase in the spinal fluid. Childs Nerv Syst. 2024;40:2935–9. doi: 10.1007/s00381-024-06429-1. [DOI] [PubMed] [Google Scholar]
- 23.Ito T, Kanno H, Sato K, Oikawa M, Ozaki Y, Nakamura H, et al. Clinicopathologic study of pineal parenchymal tumors of intermediate differentiation. World Neurosurg. 2014;81:783–9. doi: 10.1016/j.wneu.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Jacus MO, Daryani VM, Harstead KE, Patel YT, Throm SL, Stewart CF. Pharmacokinetic properties of anticancer agents for the treatment of CNS tumors: Update of the literature. Clin Pharmacokinet. 2016;55:297–311. doi: 10.1007/s40262-015-0319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jouvet A, Saint-Pierre G, Fauchon F, Privat K, Bouffet E, Ruchoux MM, et al. Pineal parenchymal tumors: A correlation of histological features with prognosis in 66 cases. Brain Pathol Zurich Switz. 2000;10:49–60. doi: 10.1111/j.1750-3639.2000.tb00242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang YJ, Bi WL, Dubuc AM, Martineau L, Ligon AH, Berkowitz AL, et al. Integrated genomic characterization of a pineal parenchymal tumor of intermediate differentiation. World Neurosurg. 2016;85:96–105. doi: 10.1016/j.wneu.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 27.Kanno H, Nishihara H, Oikawa M, Ozaki Y, Murata J, Sawamura Y, et al. Expression of O6-methylguanine DNA methyltransferase (MGMT) and immunohistochemical analysis of 12 pineal parenchymal tumors. Neuropathology. 2012;32:647–53. doi: 10.1111/j.1440-1789.2012.01315.x. [DOI] [PubMed] [Google Scholar]
- 28.Kato H, Tanei T, Nishimura Y, Nagashima Y, Ishii M, Nishii T, et al. Pineal parenchymal tumor of intermediate differentiation with late spinal dissemination 13 years after initial surgery: Illustrative case. J Neurosurg Case Lessons. 2023;5:CASE22475. doi: 10.3171/CASE22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kerezoudis P, Yolcu YU, Laack NN, Ruff MW, Khatua S, Daniels DJ, et al. Survival and associated predictors for patients with pineoblastoma or pineal parenchymal tumors of intermediate differentiation older than 3 years: Insights from the National Cancer Database. Neurooncol Adv. 2022;4:vdac057. doi: 10.1093/noajnl/vdac057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khodayari B, Lien WW. Clinical outcomes for primary parenchymal tumor of intermediate differentiation. Int J Radiat Oncol Biol Phys. 2018;102:e265. [Google Scholar]
- 31.Kim BS, Kim DK, Park SH. Pineal parenchymal tumor of intermediate differentiation showing malignant progression at relapse. Neuropathology. 2009;29:602–8. doi: 10.1111/j.1440-1789.2008.00994.x. [DOI] [PubMed] [Google Scholar]
- 32.Komakula S, Warmuth-Metz M, Hildenbrand P, Loevner L, Hewlett R, Salzman K, et al. Pineal parenchymal tumor of intermediate differentiation: Imaging spectrum of an unusual tumor in 11 cases. Neuroradiology. 2011;53:577–84. doi: 10.1007/s00234-010-0794-2. [DOI] [PubMed] [Google Scholar]
- 33.Kumar R, Dayal S, Krishna M. Pineal parenchymal tumor with intermediate differentiation-a case report and review of literature from rural India. Indian J Neurosurg. 2020;9:55–7. [Google Scholar]
- 34.Kunigelis KE, Kleinschmidt-DeMasters BK, Youssef AS, Lillehei KO, Ormond DR. Clinical features of pineal parenchymal tumors of intermediate differentiation (PPTID): A single-institution series. World Neurosurg. 2021;155:e229–35. doi: 10.1016/j.wneu.2021.08.056. [DOI] [PubMed] [Google Scholar]
- 35.Liu AP, Gudenas B, Lin T, Orr BA, Klimo P, Kumar R, et al. Risk-adapted therapy and biological heterogeneity in pineoblastoma: Integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials. Acta Neuropathol (Berl) 2020;139:259–71. doi: 10.1007/s00401-019-02106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu AP, Priesterbach-Ackley LP, Orr BA, Li BK, Gudenas B, Reddingius RE, et al. WNT-activated embryonal tumors of the pineal region: Ectopic medulloblastomas or a novel pineoblastoma subgroup? Acta Neuropathol (Berl) 2020;140:595–7. doi: 10.1007/s00401-020-02208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis DN, Perry A, Reifenberger G, von Deimling A, FigarellaBranger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol (Berl) 2016;131:803–20. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 38.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, FigarellaBranger D, et al. The 2021 WHO Classification of tumors of the central nervous system: A summary. Neurooncology. 2021;23:1231–51. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Low JT, Kirkpatrick JP, Peters KB. Pineal parenchymal tumors of intermediate differentiation treated with ventricular radiation and temozolomide. Adv Radiat Oncol. 2021;7:100814. doi: 10.1016/j.adro.2021.100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu VM, Luther EM, Eichberg DG, Morell AA, Shah AH, Komotar RJ, et al. Prognosticating survival of pineal parenchymal tumors of intermediate differentiation (PPTID) by grade. J Neurooncol. 2021;155:165–72. doi: 10.1007/s11060-021-03863-y. [DOI] [PubMed] [Google Scholar]
- 41.Lutterbach J, Fauchon F, Schild SE, Chang SM, Pagenstecher A, Volk B, et al. Malignant pineal parenchymal tumors in adult patients: Patterns of care and prognostic factors. Neurosurgery. 2002;51:44–55. doi: 10.1097/00006123-200207000-00006. discussion 55-6. [DOI] [PubMed] [Google Scholar]
- 42.Mallick S, Benson R, Rath GK. Patterns of care and survival outcomes in patients with pineal parenchymal tumor of intermediate differentiation: An individual patient data analysis. Radiother Oncol J Eur Soc Ther Radiol Oncol. 2016;121:204–8. doi: 10.1016/j.radonc.2016.10.025. [DOI] [PubMed] [Google Scholar]
- 43.Martínez H, Nagurney M, Wang ZX, Eberhart CG, Heaphy CM, Curtis MT, et al. ATRX Mutations in pineal parenchymal tumors of intermediate differentiation. J Neuropathol Exp Neurol. 2019;78:703–8. doi: 10.1093/jnen/nlz050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayol Del Valle M, De Jesus O. StatPearls. Treasure Island, FL: StatPearls Publishing; 2024. Pineal gland cancer. Available from: http://www.ncbi.nlm.nih.gov/books/NBK560567 [Last accessed on 2024 Mar 21] [PubMed] [Google Scholar]
- 45.Mena H, Rushing EJ, Ribas JL, Delahunt B, McCarthy WF. Tumors of pineal parenchymal cells: A correlation of histological features, including nucleolar organizer regions, with survival in 35 cases. Hum Pathol. 1995;26:20–30. doi: 10.1016/0046-8177(95)90110-8. [DOI] [PubMed] [Google Scholar]
- 46.Miyazaki A, Makino K, Shinojima N, Yamashita S, Mikami Y, Mukasa A. Spinal dissemination of pineal parenchymal tumors of intermediate differentiation over 10 years after initial treatment: A case report. Cureus. 2024;16:e57147. doi: 10.7759/cureus.57147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montaser AS, Cho EY, Catalino MP, Hanna J, Smith TR, Laws ER. A Surgical perspective on the association between cystic lesions of the pineal gland (descartes’ seat of the soul) and the pituitary (the master gland) J Neurol Surg Part B Skull Base. 2022;83(S2):e598–602. doi: 10.1055/s-0041-1735635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagao S, Kuyama H, Murota T, Suga M, Tanimoto T, Kawauchi M, et al. Surgical approaches to pineal tumors: Complications and outcome. Neurol Med Chir (Tokyo) 1988;28:779–85. doi: 10.2176/nmc.28.779. [DOI] [PubMed] [Google Scholar]
- 49.Nagasawa DT, Lagman C, Sun M, Yew A, Chung LK, Lee SJ, et al. Pineal germ cell tumors: Two cases with review of histopathologies and biomarkers. J Clin Neurosci. 2017;38:23–31. doi: 10.1016/j.jocn.2016.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nam JY, Gilbert A, Cachia D, Mandel J, Fuller GN, Penas-Prado M, et al. Pineal parenchymal tumor of intermediate differentiation: A single-institution experience. Neurooncol Pract. 2020;7:613–9. doi: 10.1093/nop/npaa024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park JH, Kim JH, Kwon DH, Kim CJ, Khang SK, Cho YH. Upfront stereotactic radiosurgery for pineal parenchymal tumors in adults. J Korean Neurosurg Soc. 2015;58:334–40. doi: 10.3340/jkns.2015.58.4.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park TH, Kim SK, Phi JH, Park CK, Kim YH, Paek SH, et al. Survival and malignant transformation of pineal parenchymal tumors: A 30-year retrospective analysis in a single-institution. Brain Tumor Res Treat. 2023;11:254–65. doi: 10.14791/btrt.2023.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pusztaszeri M, Pica A, Janzer R. Pineal parenchymal tumors of intermediate differentiation in adults: Case report and literature review. Neuropathology. 2006;26:153–7. doi: 10.1111/j.1440-1789.2006.00657.x. [DOI] [PubMed] [Google Scholar]
- 54.Rahmanzade R, Pfaff E, Banan R, Sievers P, Suwala AK, Hinz F, et al. Genetical and epigenetical profiling identifies two subgroups of pineal parenchymal tumors of intermediate differentiation (PPTID) with distinct molecular, histological and clinical characteristics. Acta Neuropathol (Berl) 2023;146:853–6. doi: 10.1007/s00401-023-02638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Samkari AM, Alshehri FD, AlMehdar AS, Matar MY. Pineal parenchymal tumors of intermediate differentiation: A case report and literature review. Cureus. 2023;15:e50139. doi: 10.7759/cureus.50139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scheithauer BW, Fuller GN, VandenBerg SR. The 2007 WHO Classification of tumors of the nervous system: Controversies in surgical neuropathology. Brain Pathol. 2008;18:307–16. doi: 10.1111/j.1750-3639.2008.00179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schild SE, Scheithauer BW, Schomberg PJ, Hook CC, Kelly PJ, Frick L, et al. Pineal parenchymal tumors: Clinical, pathologic, and therapeutic aspects. Cancer. 1993;72:870–80. doi: 10.1002/1097-0142(19930801)72:3<870::aid-cncr2820720336>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 58.Senft C, Raabe A, Hattingen E, Sommerlad D, Seifert V, Franz K. Pineal parenchymal tumor of intermediate differentiation: Diagnostic pitfalls and discussion of treatment options of a rare tumor entity. Neurosurg Rev. 2008;31:231–6. doi: 10.1007/s10143-008-0126-8. [DOI] [PubMed] [Google Scholar]
- 59.Shrateh ON, Jobran AW, Owienah H, Sweileh T, Abulihya M, Shahin N, et al. Large pineal parenchymal tumor of intermediate differentiation causing compression with resultant obstructive hydrocephalus: A case report. Ann Med Surg. 2023;85:480. doi: 10.1097/MS9.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stoiber EM, Schaible B, Herfarth K, Schulz-Ertner D, Huber PE, Debus J, et al. Long term outcome of adolescent and adult patients with pineal parenchymal tumors treated with fractionated radiotherapy between 1982 and 2003--a single institution’s experience. Radiat Oncol Lond Engl. 2010;5:122. doi: 10.1186/1748-717X-5-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takase H, Tanoshima R, Singla N, Nakamura Y, Yamamoto T. Pineal parenchymal tumor of intermediate differentiation: A systematic review and contemporary management of 389 cases reported during the last two decades. Neurosurg Rev. 2022;45:1135–55. doi: 10.1007/s10143-021-01674-3. [DOI] [PubMed] [Google Scholar]
- 62.Tandean S, Siahaan AM, Loe ML, Indharty RS, Julijamnasi, Sitorus MS, et al. Case report: Implantation metastasis following stereotactic biopsy of pineal parenchymal tumor of intermediate differentiation in an adult patient: An exceptionally rare complication. Front Neurol. 2022;13:1019955. doi: 10.3389/fneur.2022.1019955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tate MC, Rutkowski MJ, Parsa AT. Contemporary management of pineoblastoma. Neurosurg Clin N Am. 2011;22:409–12. doi: 10.1016/j.nec.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Tsumanuma I, Tanaka R, Washiyama K. Clinicopathological study of pineal parenchymal tumors: Correlation between histopathological features, proliferative potential, and prognosis. Brain Tumor Pathol. 1999;16:61–8. doi: 10.1007/BF02478904. [DOI] [PubMed] [Google Scholar]
- 65.Vasiljevic A. Pineal parenchymal tumors of intermediate differentiation: in need of a stringent definition to avoid confusion. Scientific commentary on ‘Genetical and epigenetical profiling identifies two subgroups of pineal parenchymal tumors of intermediate differentiation (PPTID) with distinct molecular, histological and clinical characteristics.’. Acta Neuropathol (Berl) 2024;147:34. doi: 10.1007/s00401-024-02684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verma A, Epari S, Bakiratharajan D, Sahay A, Goel N, Chinnaswamy G, et al. Primary pineal tumors-Unraveling histological challenges and certain clinical myths. Neurol India. 2019;67:491. doi: 10.4103/0028-3886.258045. [DOI] [PubMed] [Google Scholar]
- 67.Villano JL, Propp JM, Porter KR, Stewart AK, Valyi-Nagy T, Li X, et al. Malignant pineal germ-cell tumors: An analysis of cases from three tumor registries. Neuro-Oncology. 2008;10:121–30. doi: 10.1215/15228517-2007-054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang KY, Chen MM, Malayil Lincoln CM. Adult primary brain neoplasm, including 2016 World Health Organization classification. Radiol Clin North Am. 2019;57:1147–62. doi: 10.1016/j.rcl.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 69.Watanabe T, Mizowaki T, Arakawa Y, Iizuka Y, Ogura K, Sakanaka K, et al. Pineal parenchymal tumor of intermediate differentiation: Treatment outcomes of five cases. Mol Clin Oncol. 2014;2:197–202. doi: 10.3892/mco.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Webb M, Johnson DR, Mahajan A, Brown P, Neth B, Kizilbash SH, et al. Clinical experience and outcomes in patients with pineal parenchymal tumor of intermediate differentiation (PPTID): A single-institution analysis. J Neurooncol. 2022;160:527–34. doi: 10.1007/s11060-022-04174-6. [DOI] [PubMed] [Google Scholar]
- 71.Wu X, Wang W, Lai X, Zhou Y, Zhou X, Li J, et al. CD24 and PRAME are novel grading and prognostic indicators for pineal parenchymal tumors of intermediate differentiation. Am J Surg Pathol. 2020;44:11. doi: 10.1097/PAS.0000000000001350. [DOI] [PubMed] [Google Scholar]
- 72.Xin C, Xiong Z, Yan X, Zolfaghari S, Cai Y, Ma Z, et al. Endoscopic-assisted surgery versus microsurgery for pineal region tumors: A single-center retrospective study. Neurosurg Rev. 2021;44:1017–22. doi: 10.1007/s10143-020-01283-6. [DOI] [PubMed] [Google Scholar]
- 73.Yamashita S, Takeshima H, Hata N, Uchida H, Shinojima N, Yokogami K, et al. Clinicopathologic analysis of pineal parenchymal tumors of intermediate differentiation: A multi-institutional cohort study by the Kyushu Neuro-Oncology Study Group. J Neurooncol. 2023;162:425–33. doi: 10.1007/s11060-023-04310-w. [DOI] [PubMed] [Google Scholar]
- 74.Yi J, Kim H, Choi Y, Seol Y, Kahng D, Choi Y, et al. Successful treatment by chemotherapy of pineal parenchymal tumor with intermediate differentiation: A case report. Cancer Res Treat. 2013;45:244–9. doi: 10.4143/crt.2013.45.3.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu T, Sun X, Wang J, Ren X, Lin N, Lin S. Twenty-seven cases of pineal parenchymal tumours of intermediate differentiation: mitotic count, Ki-67 labelling index and extent of resection predict prognosis. J Neurol Neurosurg Psychiatry. 2016;87:386–95. doi: 10.1136/jnnp-2014-309805. [DOI] [PubMed] [Google Scholar]