

Abstract
Facile generation of specific mutations enables the study of functional variations in natural populations and advances genetic engineering applications. Here, we present a new approach, Mutagenesis by Template-guided Amplicon Assembly (MEGAA), for the rapid construction of kilobase-sized DNA variants. With this method, many mutations can be made at a time at over 90% efficiency per target in a predictable manner. We devised a robust and iterative protocol for an open-source lab automation robot that allows desktop production and long-read sequencing validation of variants. Using this system, we demonstrated the construction of 31 natural SARS-CoV2 spike gene variants and 10 recoded E. coli genome fragments, with each 4 kb region containing up to 150 mutations. Furthermore, 125 defined combinatorial AAV2 cap gene variants were easily built using the system that exhibited viral packaging enhancements of up to 10-fold compared to wild-type. The MEGAA platform enables generation of multi-site sequence variants quickly, cheaply, and in a scalable manner for diverse applications in biotechnology.
INTRODUCTION
Construction and manipulation of kilobase-sized DNA building blocks is foundational to synthetic biology and synthetic genomics1, 2. At the gene and pathway level, synthetic or engineered sequences can be applied to a design-build-test-learn (DBTL) framework to optimize for a desired function3, 4. At the genome scale, de novo synthesis and genome assembly can be used to explore synthetic genome designs5–9. However, despite significant advances in DNA synthesis over the last two decades10, current methods still suffer from size limits, synthesis fidelity, and long lead times. Building synthetic DNA remains expensive, labor intensive, and impractical for gigabase genomes such as those of animals and plants11. Breakthroughs in deep learning and computational design are now able to generate thousands to millions of synthetic variants12, 13, but most protein-sized sequences cannot be synthesized and experimentally tested at scale, which underscores an important unmet need in the field.
From a conceptual perspective, de novo synthesis is fundamentally ill-suited for making gene variants where specific mutations are dispersed across a “wild-type” core sequence. This is because significant time and resources are wasted in making a common core sequence from scratch. Beyond de novo synthesis, current alternatives have numerous drawbacks. Strategies using cellular machineries such as double-stranded DNA recombineering14, base editing15, or prime editing16 require assembling complicated constructs and are not readily multiplexable (i.e., target >2 distinct sites at a time). Other published or commercial mutagenesis protocols using primers can make only one or a couple of mutations at a time at best and often require an existing cloned vector17–20. More multiplexable oligonucleotide-mediated allelic replacement methods21, 22 rely on DNA transformation into cells and screening of many colonies that add significant time, labor, and cost burdens. To address these shortcomings, we present here a new in vitro variant synthesis platform called Mutagenesis by Template-guided Amplicon Assembly (MEGAA) that can produce kilobase-long sequence variants rapidly, in a scalable manner, and at high fidelity.
RESULTS
Overview of template-mediated variant synthesis
MEGAA uses a seed DNA material to generate an initial template for subsequent annealing, extension, and ligation of oligo pools that carry mutations of interest (Fig. 1a). The generated variant is then specifically amplified against the initial template to yield the final high-fidelity product. In the first step, the input seed DNA is amplified by PCR using a Q5U® hot start high-fidelity DNA polymerase where dTTPs are substituted with dUTPs. This results in MEGAA templates where all thymine (T) bases are replaced by uracil (U) bases. In the second step, the U-containing template is combined with Taq DNA ligase, Q5U® hot start high-fidelity DNA polymerase, dNTP, and the desired mutagenic pool of oligos and a forward extension primer at 500 to 1,000-fold molar excess of the template as a single-pot reaction in a compatible buffer (see Methods). Then oligo annealing, extension, and ligation reactions proceed. Since the Q5U® polymerase does not exhibit strand displacement activity nor 5’ to 3’ exonuclease activity, once mutagenic oligos are annealed to the template, the polymerase will only gap fill between oligos and allow subsequent ligation by Taq DNA ligase. Rapid oligo annealing is performed from 95°C down to 4°C at a rate of 3°C/sec. Fast annealing with excess oligos is crucial for avoiding renaturation of the U-template DNA. Furthermore, this prevents Taq ligase from unwarranted ligation before the single-stranded variant allele is fully gap-filled. The assembled single-stranded variant allele, which has incorporated the mutagenic oligos, are then amplified by PCR using a Q5® hot-start high-fidelity DNA polymerase that cannot extend off of U-containing templates. Archaeal polymerases such as Q5 bind tightly to uracil nucleotides, which stall DNA polymerization23. Q5U® is a modified Q5 DNA polymerase that contains a mutation in the uracil-binding pocket to enable amplification of templates containing uracil and inosine bases24. This enables specific amplification of the variant amplicon from the MEGAA reaction for direct downstream applications (e.g., cloning, sequencing, or transformation).
Figure 1: The MEGAA method for DNA variant synthesis.

(a) Overview of the MEGAA protocol. (b) Testing rsgA templates of different lengths and target positions (left panel; the numbers on the right side of the bar show the sizes of the fragments, and the span of the bar are the starting and ending positions of the fragments) with their corresponding MEGAA reaction products (middle panel; asterisks indicate U-containing templates), and variant generation efficiency (right panel; the numbers in purple on the right side of the bar show the percentages of fully complete variants). (c) Efficiency of MEGAA per target site across different rsgA templates. (d) Correlation between mean MEGAA efficiency across 9 targets versus rsgA template size.
To accurately and rapidly analyze the full-length MEGAA products in parallel, we developed a low cost long-read sequencing pipeline using the Oxford Nanopore MinION platform with a PCR barcoding scheme that allowed multiplexing of up to 96 samples per run (Supplementary Fig. 1). A custom variant calling pipeline was used to assess MEGAA efficiency across target sites (see Methods). With this setup, MEGAA products can be analyzed cost-effectively (~$2.90 per sample), with sufficient accuracy for unique variant identification, and with a reduced turnaround time (from 2 days by Sanger sequencing to only ~2 hours).
We first piloted the ability of MEGAA to generate variants using oligo pools containing 1, 3, 6 or 9 oligos for a 1,192 bp DNA template (rsgA gene from E. coli K-12), with each oligo (20–39 nt) containing a 2–5 base substitution of the template sequence (Supplementary Table 1). The efficiency and completeness of each variant synthesis by MEGAA was assessed by nanopore sequencing. Variants from oligo pools containing 1 oligo were generated at >90% efficiency, while >70% were generated completely in a 3-pool reaction, 35% were generated completely in a 6-pool reaction, and 2.5% were generated completely in a 9-pool reaction with ~25% of variants having 8 or 9 mutations. (Supplementary Fig. 2a). In larger oligo pools (e.g., 9-pool), we noted that targets near the 5’ region were generally more efficiently converted than those at the 3’ region (Supplementary Fig. 2b). We included a head-to-head experiment to compare MEGAA efficiency with other commercial directed mutagenesis kits. For single-site mutagenesis, MEGAA efficiency was higher than commercial kits, (i.e., 93.5% for MEGAA versus 85.2%, 87.6%, and 80.7% for Q5® Site-Directed Mutagenesis Kit, QuikChange II Site-Directed Mutagenesis Kit, and QuikChange Lightning Multi Site-Directed Mutagenesis Kit, respectively) (Supplementary Fig. 2a). Importantly, MEGAA was substantially more efficient than the commercial kits in multiplex target reactions. While only 3.6% of sequences were completely mutated for a 6-oligo pool reaction in the QuikChange Lighting reaction, 35.4% of sequences in MEGAA were completely mutated.
Next, we tested the capacity of MEGAA to work on templates of different sizes ranging from 1 kb to 13 kb. We generated 16 U-templates of different sizes (rsgA1- rsgA16) by amplifying the rsgA gene region of the E. coli K-12 genome. Then, we performed separate MEGAA reactions using the same 9-oligo pool designed against a shared 1 kb region across the different sized U-templates (Fig. 1b). In general, MEGAA products had robust amplicon bands at the expected sizes. Templates larger than 10 kb (e.g., rsgA16) did not produce a detectable amplicon. From nanopore sequencing of MEGAA products, we observed varying levels of completeness in MEGAA product yield, with more than 40% of all products having at least 5 of 9 targets converted for almost all templates (Fig. 1b). The mean MEGAA efficiency across all target sites reached as high as 75% for the rsgA6 template (1,192 bp) and as low as 29% for the rsgA15 template (9,156 bp) (Fig. 1c). We observed target-specific differences in MEGAA efficiency that were consistent across all template sizes. In general, 5’ targets were more efficiently generated than 3’ targets, suggesting global factors governing oligo assembly (Fig. 1c). Targets s5 and s7 were less efficiently generated than expected, which implies that local oligo annealing factors are also at play. Overall, the size of the template correlated with MEGAA efficiency, with shorter templates more efficiently converted (Fig. 1d). To verify that these results hold true for another template, we repeated the experiment on 16 additional templates (pheS1-pheS16) derived from the E. coli K-12 genome near the pheS gene using a 12-oligo pool. The same trends were observed—most target sites were made at high efficiency with some variation in some targets (Supplementary Fig. 3). Together, these findings indicated that MEGAA is efficient and multiplexable across different templates of up to 10 kb in length and can be amenable for further improvements.
Optimization of variant synthesis and iterative cycling
We hypothesized that the reduced MEGAA efficiency near the 3’ region of templates was due to extension of the template without having the oligos annealed in their proper place. Therefore, a strategy was devised where oligos were designed to have a gradation of melting temperatures (Tm), with 5’ oligos having the lowest Tm (47 °C) and 3’ oligos having the highest Tm (64 °C) (Supplementary Fig. 4). This strategy should support a more ordered assembly process whereby 3’ oligos first anneal to the U-template before 5’ oligos, which would increase the likelihood of generating a more fully converted variant. We performed a head-to-head comparison of this new oligo design (Design-2) with the prior oligo design (Design-1) where all oligos had the same Tm. With Design-2 oligos, the resulting MEGAA variants had a notably improved mean MEGAA efficiency of 86% per target (versus 75% for Design-1), with nearly all target positions performing better, specially s7 (Supplementary Fig. 4). We also tested the opposite oligo design (Design-3) with 5’ oligos having the highest Tm and 3’ oligos having the lowest Tm, which yielded even lower oligo incorporation at the 3’ region and thus further confirmed our oligo design principle (Supplementary Fig. 5). Using the Design-2 strategy, we further showed that MEGAA could operate on templates with GC contents ranging from 29% to 63%, yielding mean conversion rates per target of 91.7% and 81.0% respectively (Supplementary Fig. 6). To characterize off-target mutations in MEGAA products, we further cloned products into shuttle vectors, transformed them into cells, and isolated selected colonies for Sanger sequencing, which did not reveal any additional mutations outside of MEGAA target sites.
Conceptually, MEGAA could be repeatedly cycled such that the output from one round is used as the direct input of the next round, which could further enhance MEGAA product conversion towards the target genotype (Fig. 2a). Using the rsgA6 template and the Design-1 or Design-2 for 9-oligo pools, we developed and tested a protocol whereby the MEGAA product from the prior round is reamplified into U-containing templates for the next round of MEGAA reactions without any laborious cell transformation nor clonal purification steps. For Design-1 oligos, as more MEGAA rounds are performed, the fraction of fully converted variants increased, reaching near completion after the 5th round (Fig. 2b, Supplementary Fig. 7). For Design-2 oligos, the desired variant was almost completely generated after just 2 or 3 rounds, highlighting the substantially improved performance using the more optimized oligo design. Importantly, the conversion state of the variant product over multiple MEGAA cycles can be modeled using a simple binominal distribution (see Methods). For Design-2 oligos, our experimental data matches the model prediction of an overall MEGAA efficiency per site between 80–90% per cycle, while Design-1 oligos gave a more varying MEGAA efficiency between 50–70% (Fig. 2c). Therefore, MEGAA can be computationally modeled and experimentally tuned to generate variants of different levels of mutational saturation across a population.
Figure 2: MEGAA cycling, optimization, modeling, and automation.

(a) Schematic of MEGAA cycling to regenerate inputs for additional rounds. (b) Variants generated across increasing number of MEGAA rounds, with random oligo annealing design (Design-1) and ordered oligo annealing design (Design-2). (c) Modeling MEGAA cycling efficiency using a binomial process to assess the fraction of all target sites converted at a given mean conversion rate (μ). Solid lines are population distributions at different conversion rates (0.1–0.95) predicted by the model. Dotted line is Design-1 data and Design-2 data over multiple MEGAA cycles. (d) The MEGAAtron platform to automate the design and synthesis of variants and their validation by nanopore sequencing.
Automated desktop construction of DNA sequence variants
To generalize and standardize our variant synthesis platform, we used a low-cost open-source liquid handling and nucleic acid amplification workstation (Opentrons OT-2) to execute MEGAA reactions in an automated end-to-end pipeline dubbed MEGAAtron (Fig. 2d; Supplementary Table 2). First, a MEGAA design tool (MEGAA-dt) was developed to generate sequences of mutagenesis oligos based on input templates and desired changes by automatically optimizing for high MEGAA efficiency and low resource requirements (Supplementary Fig. 8, Methods). MEGAA oligos are ordered from commercial vendors individually or as premixed pools. Reagents, templates, and oligos are loaded onto the MEGAAtron robotic system, which can produce 24 different variants in a single run, including all steps of the protocol through a MEGAA round (or multiple rounds) from PCR amplification to product purification (Fig. 2d). The resulting MEGAA products are assessed by nanopore sequencing for quality control and efficiency characterization. The overall turnaround time of the pipeline once all inputs are ready (e.g., oligo pool, initial template) is less than 6 hours with a cost ranging from ~$20 per variant (depending on variant type) including oligos, consumables, and sequencing, which is 10 times cheaper than commercialized gene synthesis (Supplementary Fig. 9). To obtain truly 100% clonal variants, an additional cloning step can be performed, and a minimal amount of colony sequencing is needed to identify the desired variant based on nanopore sequencing analysis (e.g., 3 out of 4 colonies picked are expected to contain the perfect variant from nanopore reads).
Gene and genome-scale templated variant synthesis
Using the MEGAAtron system, we sought to showcase gene to genome-scale uses of MEGAA for fast and cheap templated variant synthesis. We first explored a new capacity to generate viral variants that would otherwise require total gene synthesis. Fast variant production of key viral components can facilitate the testing of neutralizing antibodies and therapies against variants25, 26 and help establish zoonotic transmission paths for better pandemic preparedness27. We chose the 3,822 bp S gene that encode the Spike protein from the SARS-CoV-2 virus, which has been extensively characterized by surveillance sequencing during the ongoing global pandemic since late 2019. We assimilated a set of 31 representative natural S gene variants from different SARS-CoV2 lineages from around the world, encompassing major variants of interest (VOI) and variants of concern (VOC) as of fall 2021 (Fig. 3a). Across the 31 S gene variants, 66 unique mutations are present, with some variants containing up to 13 substitutions and deletions. Oligos were commercially synthesized for each target site and separately pooled to produce their respective variants on the MEGAAtron system. Within 12 hours and two round of MEGAA reactions, we successfully generated all S gene variants to a high degree of saturation as assessed by full-length nanopore sequencing (Fig. 3a, Supplementary Table 3). In 27 of 31 variants, we observed the correct complete variant sequence in >50% of single-molecule reads from nanopore sequencing. This means that 1 of every 2 molecules in each MEGAA product had the perfect sequence, which we were able to confirm by Sanger sequencing of select cloned variants. Furthermore, for 14 variants with residue substitutions, 89.3 ± 8.6% of the nanopore reads were fully mutated. Of the 17 deletion-containing variants, 65.5 ± 16.6% showed fully mutated nanopore reads. Notably, variant ID31, which contains a 21bp deletion along with 6 separate residue substitutions, exhibits complete variant generation in 70% of the reads, thus demonstrating the versatility of our method in making different mutation types. Beyond defined variants, we further explored the generation of complex variant populations by MEGAA using oligos with degenerate bases to target multiple sites. Using a 6-pool or a 9-pool oligo set with NNS base degeneracies on the B.1.617.2 (Delta) S gene template, we produced complex yet even variant pools estimated to contain >1E9 and >3.5E13 unique sequences for the 6-pool and 9-pool MEGAA reactions, respectively (Supplementary Fig. 10).
Figure 3: Generation of SARS-CoV2 spike gene variants and E. coli codon compressed recoded fragment using MEGAA.

(a) 31 natural spike gene variant sequences are individually made with MEGAA. MEGAA yield after 2 cycles measured by nanopore sequencing is shown. Asterisks indicate additional mutations in spike gene variants according to WHO. (b) Generation of recoded genomes by systematically removing codons in E. coli genome. (c) MEGAA reaction results on 10 fragments showing fraction of recoded target sites in each fragment. MEGAA yield after 1 cycle measured by nanopore sequencing is shown. The numbers in black on the top of the bar show the percentages of fully complete variants and the numbers in purple show the percentages of >90% complete variants for each fragment (N.A. indicate the value is less than 0.1%) (d) Recoding efficiency across all target sites in 3 representative fragments.
Next, we applied MEGAAtron in a synthetic biology application involving genome-scale codon replacement (Fig. 3b). Several recent studies explored the generation of synthetic genomes with recoded and reduced codon assignments that could provide biocontainment to viral infections and expansion of the genetic code with non-natural amino acids9, 28, 29. Thus far, these efforts required either multiplex oligo-recombineering21 or de novo synthesis of kilobase-sized fragments and subsequent hierarchical assemblies into full genomes6, which are highly resource intensive approaches. We sought to showcase MEGAA as a new “templated synthetic genome synthesis” framework that is facile, less expensive, and more scalable. A codon replacement scheme (TTA→CTC, TTG→CTA, AGA→AGA, AGG→CGA, TCG→AGC, TCA→AGT) was adopted for the E. coli K-12 genome based on prior recoding strategies5, 6 (Fig. 3b). MEGAAtron was used in a proof-of-concept study to generate 10 recoded fragments each at ~3.6 kb in length using E. coli K12 genomic DNA as the seed sequence (Supplementary Table 4). A total of 428 codon changes were made across this 36 kb genomic region. The resulting MEGAA products were pooled and analyzed by nanopore sequencing. Impressively, many fragments with >50 codon changes (e.g., Frag-9 and Frag-10) had >70% of products with >75% of targets recoded from a single cycle of MEGAA (Fig. 3c). In general, most sites were efficiently targeted (78.8% efficiency) although some outliers were observed (Fig. 3d, Supplementary Fig. 11), which may require further oligo design optimizations. Importantly, these fragments were generated in less than 3 days at 20 times lower cost than by commercial de novo gene synthesis. Once generated, these 3.6 kb fragments could then be combined into larger blocks by established genome assembly methods6. We anticipate that this approach will be useful for recoding bacterial and eukaryotic genomes11.
Generating gene therapy carrier variants using MEGAA
Adeno-associated viruses (AAVs) have emerged as a safe and promising viral vector for DNA-based gene therapy, with over 149 past or ongoing clinical trials30. The AAV capsid consists of 60 molecules of viral proteins encoded by the cap gene in the 4.8 kb single-stranded DNA genome of AAV. Mutations in the cap gene can lead to a variety of altered viral properties including changes in tissue tropism, packaging efficiency, thermal stability, and neutralization escape by canonical antibodies31. An elegant recent study32 generated a comprehensive single-residue saturation mutagenesis library of the cap gene in AAV2 and found many variable regions of the cap gene that individually modified AAV properties. However, the combinatorial effects of multiple distant mutations were not explored. We used MEGAAtron to build AAV variants each containing up to 6 insertions at selected sites along the capsid protein that individually showed enhanced packaging efficiency based on saturation insertion data32 (Fig. 4a, Supplementary Fig. 12). In particular, we chose negatively charged residues of aspartic acid (D) or glutamic acid (E) to insert into the capsid protein at residue positions 37/38, 139/140, 190/191, 447/448, 501/502, or 591/592, which in general are surface-facing on the capsid. Altering the AAV surface charge can impact various viral particle properties and enhance purification by ion exchange during manufacturing33, 34. Each variant also contained a unique 24 bp barcode that enable rapid identification and quantification by short-read Illumina sequencing. Variants were produced using MEGAAtron in an arrayed format with defined 1 to 12 mutant oligo combinations and cloned into pAAV-CMV plasmid. Isolates were verified by nanopore sequencing (Supplementary Table 5, Supplementary Fig. 13). In total, 192 barcoded clones were verified corresponding to 125 unique variants, including 24 wild-type barcoded variants (Supplementary Table 5). Plasmids carrying each variant were equally pooled and transfected together into HEK293T cells to assess viral packaging efficiency by Illumina barcodes sequencing (Methods). Packaging efficiency was quantified as the abundance of variants in the virus pool relative to the plasmid pool.
Figure 4: AAV2 cap gene engineering using MEGAAtron.
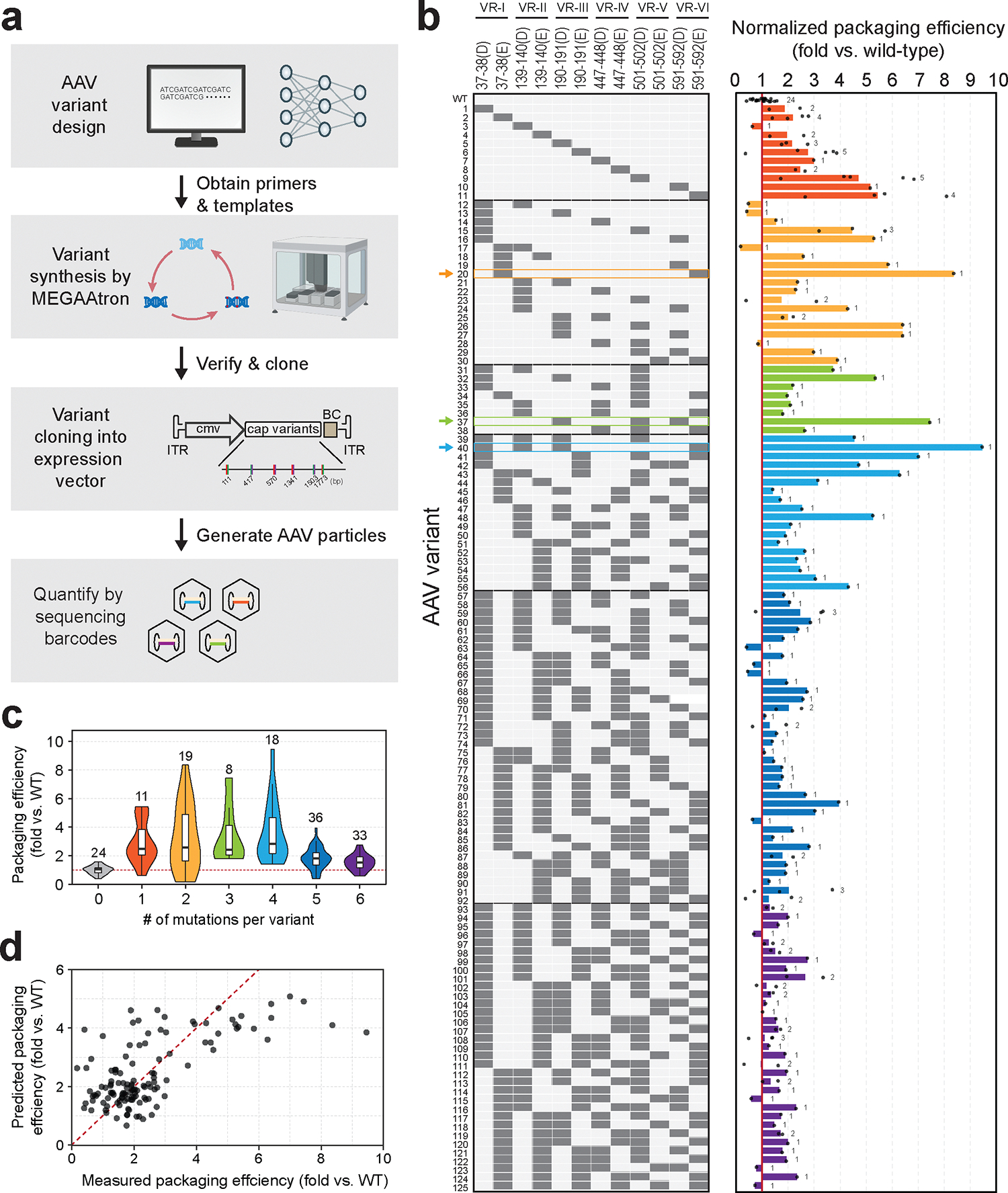
(a) Overview of AAV2 variant generation workflow. (b) Package efficiency of 125 AAV2 variants normalized to wild-type levels. Six mutation sites VR-I to VR-VI are noted on the left. Dots in the plot represent individual barcoded replicates, and numbers next to the bars represent numbers of barcoded replicates for AAV variants used in this study. (c) Violin plot showing packaging efficiency of variants based on number of mutations per variant. Numbers above the violin plot represent number of barcoded replicates (for WT) or number of variants (for AAV2 variants) shown in the plot. Definition of box-plot elements: center line: median; box limits: upper and lower 25th quartiles; whiskers: 1.5x interquartile range. (d) Plot showing measured versus predicted packaging efficiency versus using a linear regression model.
We first confirmed that in general single residue D or E insertions at each of the 6 chosen sites showed improvements in AAV2 packaging efficiency compared to the wild-type, which correlated well with previous data32 and thus verified our quantitative assay (Supplementary Fig. 14). Next, we explored the combinatorial variants in the library and identified several that exhibited substantially improved packaging efficiency (Fig. 4b). Notably, Var20 (37/38E, 591/592E), Var37 (190/191D, 501/502D, 591/592D) and Var40 (37/38D, 139/140D, 190/191D, 591/592E) showed 8.4-fold, 7.4-fold and 9.5-fold improvement over wild-type, respectively. Interestingly, we observed that variants containing 5 or 6 insertions had much poorer packaging efficiencies overall (mean of 1.9-fold and 1.5-fold, respectively) than variants with 1 to 4 mutations (mean of 2.9 to 3.7-fold, respectively) (Fig. 4c). These results suggest that an excess in negative surface charge residues substantially reduced improvements in AAV packaging and indicate an upper limit to guiding combinatorial optimizations of this set of variant designs. Nevertheless, even with a limited combinatorial survey of 4-site or fewer variants, high-performing mutants were identified within our AAV2 library.
Finally, we applied our library data to a linear regression model to investigate mutation determinants of AAV2 packaging (Methods). In general, the linear model was able to predict improved packaging efficiency to a reasonable level (adjusted R2 of 0.383, p-value of 5.7e-10) (Fig. 4d, Supplementary Fig. 15). Interesting, we found that in general 591/592(E) and 190/191(E) mutations had statistically significant positive coefficients in the linear model (p<0.05). On the other hand, 447/448(D/E) mutations had large negative coefficients and 139/140(D/E) had small negative coefficients that were both statistically significant (p<0.05). These results highlight that a linear model can capture meaningful information of multi-site D/E mutational effects on AAV2 packaging efficiency, while also suggesting non-linear combinatorial effects that will warrant more sophisticated models35, 36.
DISCUSSION
Genetic variants are key for understanding biological function and evolution. The capacity to build variants quickly and cheaply from an existing template can accelerate new biological discoveries and biotechnology. MEGAA offers an unprecedented capability to generate 10–100s of multi-site variations across kilobases of DNA at high efficiency in a matter of hours with automation. We showed that MEGAA can be cycled to drive the mutagenesis reaction to near completion or be used to generate degenerate variations in DNA of up to 10 kb in length. Although iterative PCRs during MEGAA cycling may potentially accumulate amplification errors, we can assess the DNA sequence fidelity as a function of PCR cycles, which shows that PCR-associated errors is minimal for over 5 MEGAA cycles (e.g., >89% of a 2 kb template maintains the perfect sequence over 5 cycles) (Supplementary Fig. 16). We further showed that the distribution of variants can be reliably modeled to offer increased control of the in vitro mutagenesis reaction process. This variant synthesis platform can be more economic than de novo gene synthesis for long sequences. In this study, our 125-member AAV2 variant library costs ~$33,000 to build through a commercial de novo gene synthesis vendor (2.2 kb at $0.12/bp) and can take up to 3 weeks to obtain, compared to ~$2,900 (at $0.01/bp, reagents cost) using the MEGAAtron platform done in a few days by a single person. Finally, we provided a systematic comparison of the advantages of the MEGAA platform compared to other commercial kits and published methods20 (Supplementary Table 6, Supplementary Fig. 17).
Clonal generation of a perfect sequence is a costly and time-consuming step in gene synthesis that also applies to MEGAA, but it could be side-stepped if MEGAA efficiency is sufficiently high. The platform could be further improved by exploring other DNA polymerases to increase amplicon length and combined with pathway-scale DNA assembly methods to yield 100-kb-sized fragments37, 38. Use of more sophisticated DNA folding and annealing models39 could help provide sequence-based MEGAA efficiency predictions and oligo design. Desktop DNA synthesizers can further improve MEGAA turnaround time, while better long-read sequencing technologies will further enhance accurate analysis and validation of MEGAA products. Implementation of droplet-based DNA synthesis strategies40 could further increase the throughput of MEGAA. We envision that template-mediated synthesis to rapidly iterate many genetic designs will be a crucial part of the synthetic biology arsenal to tackle pressing challenges facing the world.
Methods
Chemical, oligonucleotide, and enzyme reagents
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. The CloneJET PCR cloning system was purchased from Thermo Fisher Scientific. Mutagenic oligos and sequencing primers were purchased from Integrated DNA Technologies. All enzymes were purchased from New England BioLabs.
Strains, viruses, and culture conditions
Genomic DNA from Escherichia coli strain K-12 MG1655 was used as the U-containing DNA template for MEGAA studies and codon replacement experiments. Plasmid pcDNA3.1 SARS-CoV-2 S D614G was obtained from Addgene to produce the SARS-CoV2 S gene variants. Plasmids pAAV-CMV vector, pRC2-mi342 vector and pHelper vector were purchased from Takara Bio Inc. (Cat. #6230) to produce the AAV2 capsid variants and for AAV packaging. NEB® Turbo Competent Escherichia coli was used for cloning reactions using standard protocols.
In silico design of MEGAA oligos
MEGAA oligos were designed to target a template sequence using a custom python script (MEGAA-dt) available at https://github.com/hym0405/MEGAAdt. MEGAA-dt script takes reference template sequences and desired mutations information as input and generates designs of mutagenesis oligos and sequences of final variants. Briefly, desired mutations of each variant are evaluated based on their proximity to determine potential oligo regions and mutations too close to each other will be covered by the same oligo. Next, the numbers of perfectly matched bases at 5’ end and 3’ end of oligos are determined sequentially based on their melting temperatures to make oligos assemble in order (lower melting temperatures for upstream oligos). Mutagenesis oligos are then evaluated for their length and distances to adjacent oligo and sequences of final oligo designs and variants are generated.
MEGAA protocol and MEGAAtron automation system
In the first step, a MEGAA template is generated using an input DNA source (e.g., wild-type genomic DNA) by PCR amplification with a Q5U® hot start high-fidelity DNA polymerase (New England Biolabs, Cat. #M0515L) with a buffer mix where dTTPs are replaced with dUTPs. Q5U® hot start high-fidelity DNA polymerase is able to use dUTPs at the same fidelity as dTTPs, and as a result the MEGAA template contains uracil (U) bases instead of thymine (T) bases. In the second step, a mix of the MEGAA template, Q5U® hot start high-fidelity DNA polymerase, Taq DNA ligase (New England Biolabs, Cat. #M0208L), and dNTP is made. Phosphorylated mutagenic oligos (~30–40 nucleotides) containing the desired mutations (i.e., substitutions, insertion and deletions) and a forward extension primer are also added to the mix at 500 to 1,000-fold excess of the template. Oligo annealing, extension, and ligation reactions then proceed in the single-pot reaction. Rapid oligo annealing (95°C→4°C at a rate of 3°C/sec) is performed on a standard thermal cycler. To increase MEGAA reactions throughput, a liquid handling robot (OT-2; Opentrons, Brooklyn, NY) equipped with magnetic, temperature, and PCR modules was used to automate the MEGAA reaction. A detailed step-by-step protocol for the method is included in Supplementary Methods and the detailed description and setup breakdown of MEGAAtron system can be found in Supplementary Table 2.
MEGAA characterization experiments
A U-containing DNA template (rsgA6, 1,192 bp) was generated by PCR using a Q5U® hot start high-fidelity DNA polymerase with primers rsgA-F0/R0. In the meantime, 15 U-templates of different sizes (1.2–13 kb) were by amplifying the rsgA gene region of the E. coli genome with primers from rsgA-F1 to rsgA-F5 with rsgA-R0, rsgA-R1 to rsgA-R5 with rsgA-F0, rsgA-F1 to rsgA-F5 with rsgA-R1 to rsgA-R5 respectively. Another set of 16 U-templates (1.8–12 kb) were by amplifying the pheS gene region with a similar approach (Supplementary Table 1). For rsgA6 fragment, 4 individual MEGAA reactions are performed with 1, 3, 6 and 9 phosphorylated mutantic oligos. For each rsgA/pheS gene region mutagenesis, 9-target and 12-target phosphorylated mutantic oligo pool were added to MEGAA reactions. 3 rsgA6 variants (1, 3, and 6-target), rsgA1-rsgA15 and pheS1-pheS15 amplicons were prepared for nanopore sequencing.
MEGAA optimization experiments
Oligo pools OP1 (OP1.1- OP1.9) and OP2 (OP2.1- OP2.9) were designed to target 9 sites in rsgA gene. Oligos in OP1 were designed with a similar melting temperature. However, Oligos in OP2 were designed to have a gradation of melting temperature from 47°C to 64°C. 1,192 bp DNA U-containing template was by amplifying the rsgA gene region of the E. coli genome with primers rsgA-F0 and rsgA-R0. After the separate MEGAA reactions were performed, PCR amplicons (rsgA6) were cleaned up and performed via iteratively cycled MEGAA. 5- and 3-cycle MEGAA reactions were performed with OP1 and OP2 respectively. After each MEGAA reaction, rsgA6r1-r5 and rsgA6r1-r3 PCR amplicons from MEGAA OP1 and OP2 were prepared for nanopore sequence respectively.
Comparison of the mutagenesis efficiency of MEGAA and commercial kit experiments
MEGAA does not require the template to already be cloned into a circular plasmid DNA. Since circular plasmids are required for all these other methods as input, we firstly generated target plasmids (pJET1.2-rsgA6) by cloning the linear DNA fragments (rsgA6 template in Fig. 1b) into pJET1.2/blunt (Thermo Scientific #K1231). Mutagenesis was then performed according to the manufacturer’s instructions. Subsequently, the mutagenesis efficiency of different methods was assessed by Nanopore sequencing. In brief, transformation was performed for mutated products and all colonies were scraped from plates and pooled together. On average, more than 500 colonies were obtained for commercial kits. Targeted 1.2 kb fragments were then amplified from the pooled colonies using uniquely barcoded primers targeting plasmid backbone region to avoid amplifying endogenous rsgA gene in E. coli genome. Finally, amplified products were subjected to gel examination and desired bands were excised for Nanopore sequencing.
SARS-CoV-2 S mutagenesis experiment
U-containing S gene templates were PCR amplified using pcDNA3.1 SARS-CoV-2 S D614G plasmid1 as the DNA template with primers SARS-CoV-2 S_tempF and SARS-CoV-2 S_tempR. Mutagenesis of the SARS-CoV-2 S was performed via a MEGAA reaction with the modification that primer lengths were adjusted to ensure ordered oligo annealing. We designed mutagenic oligos containing target codons to generate all representative variants from alpha to lambda variants. Meanwhile, oligos containing degenerate bases (NNS) were designed to generate all combinations based on B.1.617.2 and AY.2 variants. Finally, 33 MEGAA reactions were carried out with 64 defined oligos and 10 degenerated oligos (Supplementary Table 1). All variants were prepared for Nanopore sequencing after cleaning up with SPRI beads.
Genome recoding mutagenesis experiment
Approximately 36 kb DNA chunks in the E. coli K-12 genome were randomly chosen to be recoded with synonymous mutation DNA by compressed redundant codons (TTA→CTC, TTG→CTA, AGA→AGA, AGG→CGA, TCG→AGC, TCA→AGT). The DNA chucks were split into 10 fragments with 17–54bp overlaps. 10 paired primers 36K-F1/R1 to 36K-F10/R10 were designed and applied to amplify 10 U-containing DNA templates respectively. Meanwhile, 289 mutagenic oligos were designed with an ordered oligo annealing strategy to cover 1,015 mutated bases in the 36 kb DNA. Then 10 oligo pools, which contained 14 to 40 mutagenic oligos per pool rather than individual oligos were synthesized for MEGAA reactions. Following the MEGAA reaction steps, nanopore sequencing was applied to verify the recoding products.
AAV cap mutagenesis experiment
U-containing DNA of the wildtype barcoded AAV2 cap gene were generated by two round PCR amplifications of the pRC2-mi342 vector (Takara Bio Inc, Cat. #6230) with primers AAV2-tempF/ AAV2-tempR1 and AAV2-tempF/ AAV2-tempR2. (Supplementary Fig. 13) AAV2-tempR1 and AAV2-tempR2 included 12bp random barcoding DNA. 12 oligos were designed to carry D or E insertions in the 6 variable regions (VR), VR-I to VR-VI, which positions were 35–40, 132–152, 188–192, 445–460, 490–500, 576–596 in capsid protein respectively. After oligo phosphorylation, MEGAA reactions were carried out with the oligo pool covering 6 variable regions. All cap variants were assessed on nanopore sequencing. The pAAV-CMV vector was linearized using EcoRI and BamHI restriction enzymes. Unique barcoded wildtype cap gene and MEGAA variants were digested using EcoRI and BamHI. The two products were purified using SPRI beads, ligated using T4 DNA ligase (New England Biolabs, Cat. #M0202M), and incubated at 16°C overnight. A pooled plasmid was transformed into competent NEB Turbo cells and grown for 10 hours at 37 °C. Individual colonies were picked for colony PCR with Oxford Nanopore sequencing barcoded primers. Indexed amplicons were pooled equimolar and sequenced on Oxford Nanopore platform to identify mutations sites as well as barcode sequences.
Virus was produced using AAVpro® Helper Free System (Takara Bio Inc, Cat. #6230), with minor adjustments. Briefly, a 150-mm cell culture dish (Thermo Scientific™ 150468) was inoculated with 6.0 × 106 293T cells in DMEM culture medium supplemented with 1× GlutaMAX™, 1× Pen/Strep antibiotic, and 5% FBS according to standard cell culture protocols. The 293T cells were split into 10× 150-mm cell culture dishes for the experiment when cells were approximately 90% confluent. Two days after splitting the cells, PEI transfection was performed with PEI:DNA mass ratio of 3:1 with 36 μg pR2-mi342, 70 μg pHelper, and 0.25 μg pool variants, which included 125 unique barcoded pAAV-CMV-aav2cap variants along with 24 barcoded cap wildtype plasmids. The culture medium was completely replaced with fresh DMEM containing 1× GlutaMAX™, 1× Pen/Strep antibiotic, and 5% FBS at 12 hours after transfection. 50% media volume (200 mL) was added after 72 hours. After 5 days, isolation of AAV2 particles from AAV-producing cells was performed according to the AAVpro® Helper Free System instructions. Nuclease treatment was performed by adding 1/100 volume of 1 M MgCl2 solution to the supernatant mixture obtained from AAVpro® Helper Free System and along with TURBO™ DNase (Thermo Scientific™ Cat. AM1907) to a final concentration of 0.4 U/μl. The supernatant was collected after 5,000×g centrifuge for 10 minutes at 4 °C and run purification of desalting and concentration of the AAV2 particles based on the protocol of AAVpro® Purification Kit (Takara Bio Inc, Cat. #6232).
To evaluate packaging efficiency of different variants, barcode regions of variants for input plasmids and virus particles were quantitatively amplified and sequenced on the NextSeq platform. Briefly, 1μL purified AAV2 particles (1×108 GC/μL) and input plasmids pool were first subjected to a 12-cycle PCR amplification using AAV_bcRead primer pairs and SPRI beads cleanup to generate amplicon of barcodes regions. Next, a quantitative PCR reaction was performed to add indexed Illumina TruSeq adapters to the amplicon and advanced to the final extension step during exponential amplification. Yielding libraries were then purified by gel electrophoresis and sequenced on the Illumina NextSeq platform (2×75 paired-end mode; Control Software v4.0) with 20% PhiX spike-in (Illumina FC-110–3001) according to manufacturer’s instruction. Sequences of primers used for library preparation for barcode sequencing are provided in Supplementary Table 7.
Raw sequencing reads of variants barcode amplicon were analyzed by in-house script to calculate the variants packaging efficiency. Briefly, barcode sequences of the variants were extracted from reads and matched to variant identity based on references from Nanopore sequencing. Reads mapped to each variant were then counted and the relative abundances were calculated as: RA (variant-X) = reads count mapped to variant-X / total mapped reads count. Next, variant relative abundances between input plasmid and yielding virus particles were compared to quantify packaging efficiency: efficiency (variant-X) = RA (variant-X) in virus pool / RA (variant-X) in plasmids pool. The efficiency was then normalized by WT variants to generate final variant packaging efficiency used in downstream analysis: normalized efficiency(variant-X) = efficiency (variant-X) / average of efficiency (WT variants). To explore the determinants of AAV2 packaging efficiency, a linear regression model was constructed in R v4.1.2 to predict packaging efficiency based on the binarized mutation profile of all 125 variants, and predicted efficiency as well as coefficients of mutation sites were extracted from the linear model to evaluate the overall performance and combinatorial effect of each site.
Nanopore sequencing and data analysis
To determine overall variant generation efficiency, we implemented a barcoded Oxford Nanopore strategy to sequence the full length of generated variants. Briefly, unique dual 12-bp barcodes were added to both ends of the variants by PCR amplification, and yielded barcoded variants were pooled together and purified by gel electrophoresis. Approximately 300 fmol pooled variants after cleanup were subjected to Oxford Nanopore library preparation and sequencing following the manufacturer’s instructions. Variants underwent Nanopore sequencing using the protocol ‘Amplicons by Ligation (SQK-LSK110)’ (Oxford Nanopore Technologies). Both MinION Flow Cell R9.4.1(FLO-MIN106D) and R10.4 (FLO-MIN112) were used for sequencing on a MinION with the MinKNOW v21.11.8 (ONT). Base-calling was performed with Guppy v3.6.0 (ONT) in GPU mode. Full-length reads were first demultiplexed based on barcodes of both ends using an in-house Python script and subjected to quality filtering to only keep high-quality reads (no more than 3-bp mismatches and 1-bp gap in 20-bp region of both 5’ and 3’ ends). Demultiplexed reads were then aligned to reference sequence by MUSCLE2 v3.8.31 using default setting. Variant generation efficiency was then calculated based on reads alignment using an in-house Python script. In-house scripts used for Oxford Nanopore sequencing data analysis can be accessed at https://github.com/hym0405/MEGAAdt.
Analytical model of MEGAA cycling process
Using a binomial distribution, we can predict the completeness of MEGAA reactions () at MEGAA cycle with the average oligo incorporation efficiency per locus () through the formula . The completeness metric indicates the fraction (or % completeness) of all target sites mutated in the end-product mix with meaning 100% of products have 100% mutations at all target sites. In this model, a of could mean that either 50% of products have all sites mutated or 100% of products have half of their sites mutated. Mapping our experimental data to this simple model gives us an estimated average oligo incorporation efficiency of 0.8 to 0.9 for Design-2 oligos (i.e., 80–90% mutagenesis efficiency per site per MEGAA round), compared to 0.5 to 0.7 for Design-1 oligos (50–70% mutagenesis efficiency).
Statistics & Reproducibility
The gel electrophoresis in Fig. 1b and Supplementary Fig. 3b were repeated more than three times independently and similar results were obtained. No statistical method was used to predetermine sample size as the number analyzed depends on the yield from experiments. Data exclusion was based on sequencing coverage or amplicons quality to remove technical artifacts. The experiments were not randomized, and the researchers were blinded to samples as different designs were processed together in the experiment. Blinding in analysis was not possible and all analyses were performed with the same parameters.
Supplementary Material
Acknowledgements
We thank Guillaume Urtecho, Junming Qian, and Lei Huang for technical support and Kristin Beiswenger and other members of the Wang laboratory for advice and comments on the manuscript. H.H.W. acknowledges funding support from the NSF (MCB-2032259), DoE (47879/SCW1710), NIH (1R01AI132403, 1R01DK118044, 1R01EB031935, #75N93021C00014), ONR (N00014–17-1–2353), Burroughs Wellcome Fund (1016691), Irma T. Hirschl Trust, and Schaefer Research Award.
Footnotes
Competing Interests Statement
H.H.W. is a scientific advisor of SNIPR Biome, Kingdom Supercultures, Fitbiomics, Arranta Bio, VecX Biomedicines, Genus PLC, and a scientific co-founder of Aclid, all of whom are not involved in the study. A patent application on methods described in this paper has been filed by Columbia University. The remaining authors declare no additional competing interests.
Code Availability
Scripts used for Oxford Nanopore sequencing data analysis can be accessed at https://github.com/hym0405/MEGAAdt.
Data Availability
Processed packaging efficiency data in previous study was obtained from GitHub (https://github.com/churchlab/AAV_fitness_landscape) to identify insertion sites with potential enhanced packaging efficiency for AAV variants design and correlate with packaging efficiency obtained in this study. The sequencing data generated in this study have been submitted to the NCBI BioProject database under accession number PRJNA834093.
References
- 1.Bartley BA, Beal J, Karr JR & Strychalski EA Organizing genome engineering for the gigabase scale. Nat Commun 11, 689 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esvelt KM & Wang HH Genome-scale engineering for systems and synthetic biology. Mol Syst Biol 9, 641 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brophy JA & Voigt CA Principles of genetic circuit design. Nat Methods 11, 508–520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Blasi R, Zouein A, Ellis T & Ceroni F Genetic Toolkits to Design and Build Mammalian Synthetic Systems. Trends Biotechnol 39, 1004–1018 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Ostrov N et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Fredens J et al. Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell LA et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 355 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Hutchison CA 3rd et al. Design and synthesis of a minimal bacterial genome. Science 351, aad6253 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Lajoie MJ et al. Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoose A, Vellacott R, Storch M, Freemont PS & Ryadnov MG DNA synthesis technologies to close the gene writing gap. Nature Reviews Chemistry (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boeke JD et al. The Genome Project-Write. Science 353, 126–127 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blazejewski T, Ho HI & Wang HH Synthetic sequence entanglement augments stability and containment of genetic information in cells. Science 365, 595–598 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Sharan SK, Thomason LC, Kuznetsov SG & Court DL Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc 4, 206–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–+ (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H & Naismith JH An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8, 91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tseng WC, Lin JW, Hung XG & Fang TY Simultaneous mutations up to six distal sites using a phosphorylation-free and ligase-free polymerase chain reaction-based mutagenesis. Anal Biochem 401, 315–317 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Kitzman JO, Starita LM, Lo RS, Fields S & Shendure J Massively parallel single-amino-acid mutagenesis. Nat Methods 12, 203–206, 204 p following 206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cozens C & Pinheiro VB Darwin Assembly: fast, efficient, multi-site bespoke mutagenesis. Nucleic Acids Res 46, e51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang HH et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiCarlo JE et al. Yeast oligo-mediated genome engineering (YOGE). ACS Synth Biol 2, 741–749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasken RS, Schuster DM & Rashtchian A Archaebacterial DNA polymerases tightly bind uracil-containing DNA. J Biol Chem 271, 17692–17696 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Abellan-Schneyder I, Schusser AJ & Neuhaus K ddPCR allows 16S rRNA gene amplicon sequencing of very small DNA amounts from low-biomass samples. BMC Microbiol 21, 349 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 602, 676–681 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Iketani S et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 604, 553–556 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harvey WT et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19, 409–424 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robertson WE et al. Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 372, 1057–1062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rovner AJ et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 518, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuzmin DA et al. The clinical landscape for AAV gene therapies. Nat Rev Drug Discov 20, 173–174 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Bartel MA, Weinstein JR & Schaffer DV Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther 19, 694–700 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Ogden PJ, Kelsic ED, Sinai S & Church GM Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 366, 1139–1143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu G et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J Virol Methods 140, 183–192 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Hsu HL et al. Structural characterization of a novel human adeno-associated virus capsid with neurotropic properties. Nat Commun 11, 3279 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bryant DH et al. Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol 39, 691–696 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Zhu D et al. Machine learning-based library design improves packaging and diversity of adeno-associated virus (AAV) libraries. 2021.2011.2002.467003 (2021). [Google Scholar]
- 37.Jia H, Guo Y, Zhao W & Wang K Long-range PCR in next-generation sequencing: comparison of six enzymes and evaluation on the MiSeq sequencer. Sci Rep 4, 5737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis T, Adie T & Baldwin GS DNA assembly for synthetic biology: from parts to pathways and beyond. Integr Biol (Camb) 3, 109–118 (2011). [DOI] [PubMed] [Google Scholar]
- 39.McDevitt S, Rusanov T, Kent T, Chandramouly G & Pomerantz RT How RNA transcripts coordinate DNA recombination and repair. Nature Communications 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plesa C, Sidore AM, Lubock NB, Zhang D & Kosuri S Multiplexed gene synthesis in emulsions for exploring protein functional landscapes. Science 359, 343–347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 1.Yurkovetskiy L et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 183, 739–751 e738 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed packaging efficiency data in previous study was obtained from GitHub (https://github.com/churchlab/AAV_fitness_landscape) to identify insertion sites with potential enhanced packaging efficiency for AAV variants design and correlate with packaging efficiency obtained in this study. The sequencing data generated in this study have been submitted to the NCBI BioProject database under accession number PRJNA834093.