

Abstract
As one of the nine subtypes of adrenergic receptors (ARs) in the brain, α2C-ARs play essential roles in emotion and memory, and are implicated in neuropsychiatric disorders, including depression, Alzheimer’s disease, substance use disorder, and schizophrenia. A recently developed α2C-AR specific positron emission tomography (PET) radiotracer, [11C]ORM-13070, showed promise for imaging α2C-ARs in the brain. Herein, we prepared highly potent C-11 labelled benzo-1,4-dioxane derivatives and compared them with [11C]ORM-13070, aiming to improve the specific binding signal, as evaluated by in vivo rodent brain PET imaging.
Keywords: Adrenergic receptors, Alpha-2C, Brain imaging, Small-molecule radiotracer, PET imaging
Graphical Abstract

1. Introduction
Adrenergic receptors (ARs) mediate the physiological and psychological effects of norepinephrine (NE) and epinephrine in peripheral organs and in the central nervous system (CNS)[1–3]. There are three subclasses of ARs (α1, α2, and β), each of which is divided into three homologous subtypes that exhibit distinct pharmacological properties and tissue distribution[4–6]. The inhibitory activities of α2-ARs in the presynaptic and postsynaptic brain regions make them very attractive therapeutic and diagnostic targets. Presynaptic α2-ARs work as autoreceptors to control the release of NE and as heteroreceptors to regulate the levels of other neurotransmitters such as dopamine (DA), serotonin, acetylcholine, and γ-aminobutyric acid[7]. Through drug- and behavior-based studies, one of the subtypes of α2-ARs, α2C, has been linked to brain functions such as emotion and cognition, likely due to their dense localization in the striatum and hippocampus[8]. Previously, α2C agonists and antagonists have demonstrated their potential as drugs by restoring the noradrenergic system after disruption by pathological conditions, implying α2C-ARs might be an important controller in many CNS diseases[9]. For example, in the forced swim test, both α2C-knockout (KO) mice[10] and control mice after administration of an α2C-specific antagonist demonstrated decreased immobility time, indicating reduced depressive symptoms[11]. Moreover, due to its high concentration in the basal ganglia and hippocampus of the human brain[8,12–16], the α2C receptor has emerged as an attractive therapeutic target for brain disorders such as depression, Alzheimer’s disease, substance use disorder, and schizophrenia.
Over the years, positron emission tomography (PET) has been used to investigate the role of α2-ARs in vivo with non-selective radioligands such as [11C]R107474[17], [11C]yohimbine[18,19], [11C]mirtazapine[20,21], and [11C]MPTQ[22] (Figure 1A). However, α2C-subtype selective radioligands are necessary to conduct mechanistic studies, increasing our understanding of the method by which adrenergic receptors are related to neuronal diseases. For example, the inhibition of α2C-ARs selectively reduces the levels of DA in the striatum without affecting the levels of DA in the prefrontal cortex, indicating α2C-AR antagonists can be used to treat schizophrenia, whose hyperdopaminergic activities were observed in the mesolimbic pathway[23]. Additionally, drug occupancy studies can be conducted to advance drug development by measuring target engagement. One challenge in developing subtype-specific PET radioligands for α2C-ARs is their low level of expression in the brain (5–10 fmol/mg of protein in rodents)[16], requiring ligands with extremely high affinity and selectivity to perform in vivo quantification. The first subtype-selective radiotracer for α2C-AR, [11C]ORM-13070 (Figure 1A; α2C Ki = 3.8 nM, α2A Ki = 109 nM)[24] was introduced by Arponen et al. in 2014, who reported its potential as an α2C-AR PET radiotracer in rodents[25]. In 2014, Luoto et al. evaluated its metabolic stability, pharmacokinetics, and whole-body radiation dosimetry in humans [12] and in 2015, Lehto et al. studied its test-retest variability of in the human brain and observed <10% striatal variability, supporting its viability as a PET radiotracer for quantification of brain α2C-ARs in the human brain [24].
Figure 1:

A. Some of the reported PET tracers for α2-ARs. B. Potential PET tracers for α2C-ARs.
Since then, it has been utilized to measure extracellular NE in the CNS by Lehto et al. in 2015 [24] and to validate receptor engagement of α2C-AR drug antagonists by Shahid et al. in 2020 [26]. However, [11C]ORM-13070 shows a very fast washout from the brain, little specific binding signal in rodents, and moderate signal in humans. Additionally, two unidentified radio-metabolites have been observed, one of which is known to cross the blood-brain barrier (BBB) and could influence estimations of α2C-AR concentration and occupancy. Herein, we set out to develop radioligands (Figure 1B) with greater affinity for α2C-ARs than ORM-13070 to improve the specific binding signal. We also evaluate the relationship between the rapid washout of these radioligands and brain efflux pumps using efflux pump triple KO mice.
2. Results and Discussion
2.1. Chemistry
Previously, a list of benzo-1,4-dioxane derivatives was reported as potential α2C-AR drugs, including ORM-13070 (Figure 1)[27–29]. Based on the published functional assay data (Kb, Table 2), we selected the most potent antagonists, as they were likely to demonstrate great binding affinity. When compared to known BBB permeable radiotracers (Figure 1A, SUV 0.3 – 3.1), 1-4 displayed desirable in silico properties, including modeled BBB permeability (log[brain]:[blood]). All properties fall within the allowable ranges set by the Lipinski rule of five for BBB permeability (Table 1) [30].
Table 2:
Results of an in-house cell-based assay of 1-4 and ORM-13070. Results are reported as Ki ± standard deviation or as literature Kb values* [28,29].
| Compound | α2A (Ki, nM) | α2C (Ki, nM) | Subtype Selectivity (α2A/α2C) | α2A (nM) | α2C (nM) |
|---|---|---|---|---|---|
| 1 | 1.06±0.52 | 0.25±0.01 | 4.24 | 88* | < 0.1* |
| 2 | 12.3±1.2 | 0.74±0.10 | 16.6 | 306* | 1.62* |
| 3 | 458±26 | 0.00061±0.08 | 751,000 | > 449* | < 0.001* |
| 4 | 2.77±0.58 | 0.40±0.07 | 6.93 | 82* | 0.29* |
| ORM-13070 | 2.8±0.2 | 0.25±0.09 | 11.2 | 109† | 3.8† |
| RS79948 | 0.16±0.12 | 0.29±0.08 | 0.55 | 0.60‡ | 0.77‡ |
Table 1.
Relevant properties such as molecular weight (MW) and topological polar surface area (TPSA) are used to evaluate BBB permeability and potential as a CNS radiotracer of known and proposed radiotracers [16,17,19,21,22,27,29]. Values generated by Stardrop.
| Name | MW (g/mol) | TPSA (Å2) | Log P | Log D | BBB Permeability (log[brain]:[blood]) | Intravenous CNS Score |
|---|---|---|---|---|---|---|
| R107474 | 359.4 | 46.84 | 3.269 | 3.269 | -0.1981 | 0.06429 |
| Yohimbine | 352.5 | 37.3 | 5.382 | 5.382 | 0.03603 | 0.08848 |
| Mirtazapine | 265.4 | 19.37 | 2.134 | 2.548 | 0.5594 | 0.4427 |
| MPTQ | 388.5 | 31.31 | 2.967 | 2.783 | 0.3868 | 0.2609 |
| ORM-13070 | 355.4 | 47.06 | 1.843 | 2.48 | 0.09724 | 0.2062 |
| 1 | 361.5 | 47.06 | 2.226 | 2.361 | −0.1518 | 0.9184 |
| 2 | 372.5 | 51.99 | 2.365 | 1.879 | 0.08818 | 0.1973 |
| 3 | 420.5 | 51.24 | 3.27 | 2.409 | −0.1784 | 0.1561 |
| 4 | 421.5 | 64.88 | 2.433 | 2.771 | −0.07067 | 0.1446 |
The standard compounds ORM-13070, 1, 2, 3, and 4 and their corresponding precursors 1m, 2c, 3c, 4c, and 5b were prepared by modified literature procedures as shown in Scheme 1 [27–29]. In brief, 1,2-dihydroxybenzene (1a) and glycidyl tosylate (1b) were coupled to produce a 1,4-benzodioxane derivative (1c), which was then converted into its tosylate derivative (1d). The intermediate piperazine (1f) was obtained in high yield by heating 1d with tert-butyl piperazine-1-carboxylate followed by -Boc removal under acidic conditions. In parallel, the intermediates 4-bromo-5-(methoxymethyl)-1,3-thiazole (1j) and 4-bromo-5-(((4-methoxybenzyl)oxy)methyl)-1,3-thiazole (1k) were also prepared from 2,4-dibromothiazole-5-carbaldehyde (1g) over 3 steps. Then 1f was subjected to Buchwald-Hartwig amination conditions with 1j to obtain the reference standard (1) and with 1k to obtain p-methoxybenzyl (PMB) protected precursor 1l, which was subsequently deprotected under acidic conditions to obtain the precursor (1m). The Buchwald-Hartwig amination conditions were not successful with TBS derivatives of 1k. To prepare 2 and its precursor (2c), the pyrazole (2a) was synthesized from 1f over 3 steps, wherein 1f was treated with di(1H-imidazol-1-yl)methanethione to obtain a thione intermediate, which was then reacted with hydrazine to get a carbothiohydrazide. Lastly, the thiohydrazide intermediate and ethyl 2-chloroacetoacetate in the presence of conc. hydrochloric acid (HCl) provided 2a in decent yield. The N-H of pyrazole 2a was methylated to form 2b in the presence of sodium tert-butoxide and methyl tosylate. The ethyl ester of 2b was reduced using lithium aluminum hydride (LAH) to make the radiolabeling precursor 2c. Finally, precursor 2c was methylated to prepare the reference standard 2. Similarly, the synthesis of 3 was initiated by heating 1d and tert-butyl (S)-piperidin-3-ylcarbamate followed by -Boc deprotection to obtain intermediate 3a. In parallel, another intermediate 3b was obtained over two steps from 1-(2-methoxyphenyl)ethan-1-one and trimethyl phosphonoacetate. The coupling of 3a and 3b in refluxing toluene provided 3 in good yield, which was subsequently demethylated using boron tribromide (BBr3) to obtain radiolabeling precursor 3c. For the synthesis of 4, the key pyrazole intermediate 4a was prepared from 2-hydrazinyl-6-methoxypyridine and 3-aminocrotononitrile. It was then treated with N-benzyl-2-chloro-N-(2-chloroethyl)ethan-1-amine to obtain the benzyl protected piperazine derivative, which was deprotected to prepare 4b. The reaction of 4b and 1d under basic conditions provided 4, which was subsequently demethylated to obtain radiolabeling precursor 4c. ORM-13070 was prepared from 1f in 3 steps, wherein 1f was treated with ethyl 2-chloronicotinate to obtain 5a, which was further reduced to the precursor 5b. The standard ORM-13070 was obtained upon methylation of 5b.
Scheme 1:

Synthesis schemes of nonradioactive standards (1–4 and ORM-13070), and their precursors 1m, 2c, 3c, 4c, and 5b.
Reagents and Conditions: A. K2CO3, DMF, 60°C, 24 h, 78%; B. TsCl, TEA, CH2Cl2, 0°C - rt, 5 h, 87%; C. K2CO3, CH3CN, reflux, 24 h, 90%; D. TFA:CH2Cl2 = 1:1, 0°C, 1 h, 98%; E. NaBH4, MeOH, 0°C – rt, 24 h, 93%; F. 60 psi H2, 10% Pd/C, Na2CO3, MeOH, rt, 2 d, 87%; G. NaOH, MeI, DMSO, rt, 3 h, 82%; H. 60% NaH, PMBCl, THF, 0°C–60°C, 28 h, 68%; I. Pd(OAc)2, BINAP, Cs2CO3, dioxane, 90°C, 15 h; J. 20% TFA/CH2Cl2, 0°C, 1.5 h, 50%; K. DIPEA, DMF, 90°C, 15 h, 54%; L. 2M LAH, THF, 0°C–rt, 4 h, 82%; M. 60% NaH, THF, CH3I, 0°C–rt, 4 h, 74%; N. i. di(1H-imidazol-1-yl)methanethione, THF, rt, 4 h, ii. hydrazine hydrate, EtOH, rt, 4 h, iii. Ethyl 2-chloroacetoacetate, conc. HCl, rt, 60 h, 74%; O. MeOTs, tBuONa, THF, 0°C-rt, 4 h, 64%; P. 2.0 M LAH in THF, 0°C-rt, 15 h Q. 60% NaH, CH3I, DMF, 0°C-rt, 6 h, 65%; R. i. tert-butyl (S)-piperidin-3-ylcarbamate, K2CO3, DMF, rt–80°C, 20 h, 90%, ii. conc. HCl, dioxane, rt, 20 h, 96%; S. trimethyl phosphonoacetate, 60% NaH, THF, 0°C–rt, 16 h, 86% (cis: trans = 0.9:1); T. NBS, cat. AIBN, CCl4, reflux, 14 h, 71%; U. TEA, toluene, reflux, 4 h, 60%; V. 1M BBr3, DCM, −78°C–rt, 16 h, 67%; W. 3-aminocrotononitrile, AcOH, EtOH, reflux, 4 h, 54%; X. 60% NaH, 0°C–rt, 1 h, BnN(CH2CH2Cl)2, 65°C, 3 h, 49%, ii. 10% Pd/C, HCOONH4, HCOOH, EtOH, 70°C, 1 h, 84%; Y. K2CO3, CH3CN, reflux, 24 h, 41%; Z. LiCl, p-TsOH•H2O, DMF, 110°C, 3 h, 14%.
2.2. Cell-binding assay
The α2A-AR and α2C-AR binding affinity of each compound was measured under an in-house cell-based assay to compare them in parallel (Table 2). All four piperazinyl benzodioxanes (ORM-13070, 1, 2, and 4) possessed hundreds of picomolar to nanomolar affinities for the α2C-AR, while the piperidinyl benzodioxane (3) displayed not only superior affinity (0.6±0.08 pM) but also enhanced subtype selectivity, completely abolishing binding toward α2A-ARs (Table 2).
At this point, we were motivated to compare binding mode of these comopunds in silico docking study using the known protein structure (PDB ID, 6KUW) obtained from PDB (www.rcsb.org). Our preliminary docking study revealed the presence of a hydrophobic pocket where the 3-methoxyphenyl group of compound 3 could interact with edge-to-face π–π interaction with Y402 (Figure 2). There is also close hydrophobic contact between L204 and a α, β-unsaturated γ-lactam ring. These additional interactions may explain exceptionally tight binding of compound 3 to this target. Further optimization of the structure-activity relationship for the piperidine ring template would be valuable.
Figure 2:
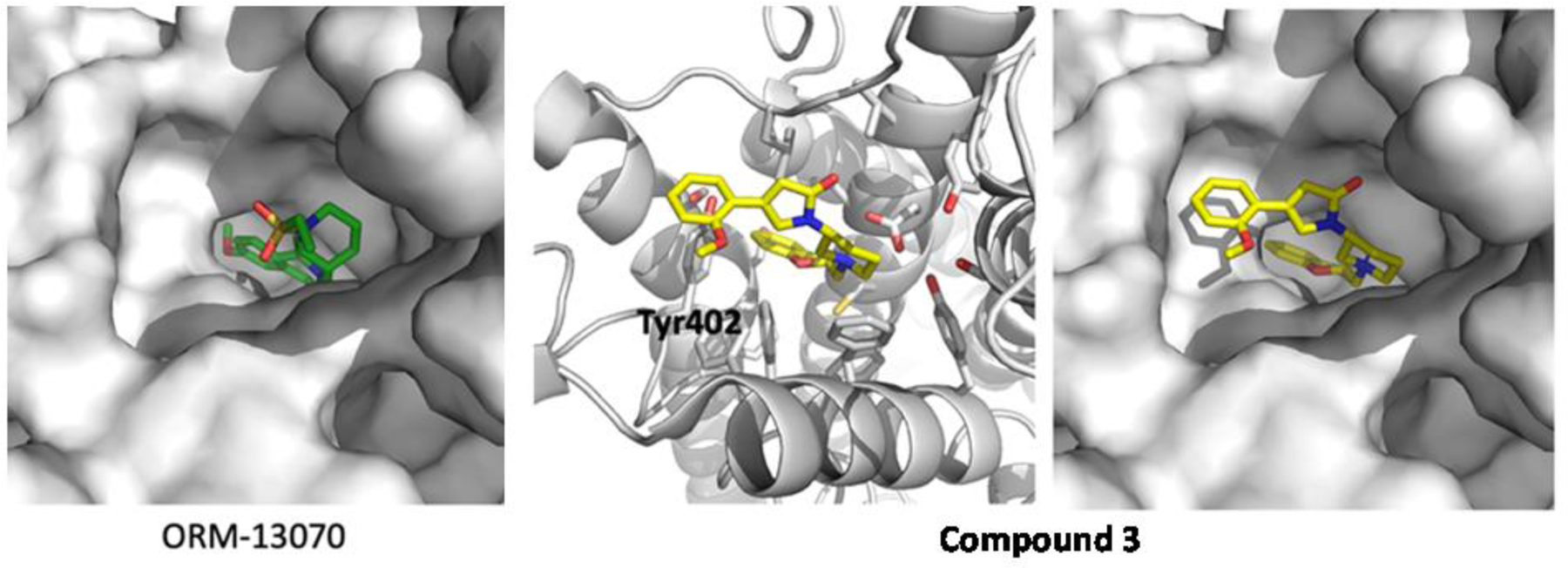
Docking study of ORM-13070 and 3 with human α2C-AR. Two ligands were tested in the binding site of RS79948 on human α2C-AR (PDB, 6KUW)
2.3. Radiochemistry
Three highly potent compounds (1-3) along with ORM-13070 were radiolabeled (Scheme 2) using a similar approach to the published method [25] via O-methylation using [11C]methyl triflate ([11C]MeOTf) with moderate decay-corrected radiochemical yield (RCY) ([11C]ORM-13070, 28.1%±17.6%, n=4; [11C]1, 7.94%±3.1%, n=4; [11C]3, 38.3%, n=1) and high molar activities ([11C]ORM-13070, 1017.5±969 GBq/μmol, n=4; [11C]1, 843±732 GBq/μmol, n=4, Table S1).
Scheme 2.

Radiochemical synthesis of [11C]ORM-13070, [11C]1, [11C]2, and [11C]3.
Reagents and conditions: A. [11C]CH3OTf, TBAOH, MeCN, 80°C, 3 min.
After purification by a semi-preparative HPLC (Figure S1), the radiotracers were formulated with sterile water to lower ethanol (EtOH) concentration below 10% for rodent injection. Radiochemical purity exceeded 99%, and radiosynthesis time lasted 30 min from the end of the bombardment (EOB). Radiochemical stability was demonstrated over 1 hour (Figure S3) and radiolysis showed negligible effect on the radiochemical purity.
2.4. PET imaging studies
2.4.1. Baseline comparison and BBB permeability study
In these preliminary dynamic baseline PET studies, the whole rat brain uptake of [11C]1, [11C]2, and [11C]3 were compared (Figure 3) to that of [11C]ORM-13070. Unfortunately, the most potent radioligand, [11C]3, demonstrated low BBB penetration (n=2), presumably due to its relatively high MW and TPSA. Due to the structural similarity of these compounds, we built a brain uptake model for this scaffold using multiparameter optimization, despite the small sample size (Table S1). We used this model in combination with the initial in vivo results to create cut-off values for BBB permeability, and using these values, compound 4 was excluded from further evaluation due to its high MW and TPSA. Due to both its lower brain uptake (Figure 3) and inferior binding affinity (Table 2), [11C]2 was not examined further, and thus, the pharmacokinetic profile of compound [11C]1 was closely compared with that of [11C]ORM-13070. To begin, both compounds were injected into the same rat on the same day to evaluate their differences without inter-subject variability. Both [11C]ORM-13070 and [11C]1 reached their peaks quickly ([11C]ORM-13070, 1.83 min, n=5; [11C]1, 1.5 min, n=5), had high whole-brain standard uptake value (SUV) ([11C]ORM-13070, 3.38; [11C]1, 3.11), and rapid washout (time to half peak SUV, [11C]ORM-13070, 5.5 min; [11C]1, 4.5 min) (Figure 4). Biodistribution in the brain was also similar, showing high uptake in the striatum (ST) and low in the cerebellum (CB), consistent with previous studies. The standard uptake value ratio (SUVr, SUVST/SUVCB) for [11C]ORM-13070 contained 12.7% greater area under the curve (AUC) than the SUVr of [11C]1 (Figure 4).
Figure 3.

Representative time-averaged images from the first 15 min of PET image acquisition. A. Baseline scans for [11C]ORM-13070, [11C]1, [11C]2, and [11C]3. The upper row shows the trans-axial brain slice PET images, and the bottom row shows the brain slice fused with an MR template. B. Averaged whole-brain uptake of [11C]ORM-13070, [11C]1, [11C]2, and [11C]3 over the whole scan duration.
Figure 4.

Comparison between the SUV of the ST and CB in the same rat (R446) for A. [11C]ORM-13070 and B. [11C]1. C. Comparison of the SUVr (SUVST/SUVCB) of [11C]ORM-13070 and [11C]1. D. PET images of baseline and blocking studies of [11C]ORM-13070 and [11C]1.
2.4.2. Blocking study
The binding specificity of [11C]1 was compared to that of [11C]ORM-13070 by conducting blocking studies with both radiotracers using ORM-13070 as the blocking agent (50 μg/kg in DMSO, injected subcutaneously (s.c.) 10 mins prior to radiotracer injection). The difference in time-activity curves (TACs) between the baseline and blocking scans of [11C]1 was negligible (change in SUVr AUC, 1.02%). In contrast [11C]ORM-13070 showed a significant decrease after blocking (change in SUVr AUC, 11.4%) (Figure 5). Although the in vitro binding properties of both compounds were similar, the in vivo data indicated [11C]ORM-13070 had superior specific binding, confirming that the observed difference in the baseline comparison study was caused by a decrease in specific binding. Both compounds showed little non-specific binding, quick uptake into the CB (time to SUV peak, [11C]ORM-13070, 1.5 min; [11C]1, 1.5 min), and rapid washout from the reference region (time to half peak SUV, [11C]ORM-13070, 3.7 min; [11C]1, 3.3 min) (Figure S4). As binding affinity and BBB permeability are optimized, this property must be preserved.
Figure 5.

Average SUVr of baseline and blocking data for A. [11C]ORM-13070 and B. [11C]1.
2.4.3. Ex vivo brain mice study
Brain uptake of the two selected radioligands, [11C]ORM-13070 and [11C]1, was evaluated in control and KO mice that lack three common efflux transporters: P-glycoprotein (Pgp)-1, Pgp-3, and breast cancer resistance protein (BCRP). We theorized that those active efflux pumps might be responsible for the rapid clearance of these radiotracers from the brain. For both radiotracers, the difference in whole-brain SUV between KO and control mice was not statistically significant (t-test p>0.05, n=4), indicating that the tested compounds are not a substrate of the efflux proteins (Figure 6). It is conceivable that the fast brain washout is due to the low density of α2C-ARs in the brain. Thus, further increasing the ligand’s receptor affinity may slow washout and improve the signal-to-noise (s/n) ratio.
Figure 6.

Comparison between the brain SUV for KO and control mice in A. [11C]ORM-13070 and B. [11C]1.
3. Conclusion
The compounds investigated, [11C]ORM-13070, [11C]1, [11C]2, and [11C]3, were synthesized in moderate radiochemical yields, high molar activities, and desirable radiochemical purities. Unfortunately, the highly potent [11C]3 did not penetrate the BBB, likely due to high MW and TPSA. The selected compound, 1, was highly potent with significant subtype selectivity towards α2C-ARs. Comparisons showed that [11C]ORM-13070 had slower washout from the brain and higher specific binding than [11C]1. Using KO mice, it was determined that the rapid washout from the brain was not due to Pgp-1, Pgp-3, or BCRP. Thus, the fast brain washout is likely due to the low receptor density of α2C-ARs in the rodent brain [16]. Moving forward, a radiotracer with greater affinity for the α2C receptors may be necessary to optimize the s/n ratio.
4. Experimental Section
4.1. General information
4.1.1. Organic chemistry
All reactions were carried out in oven-dried glassware under an atmosphere of nitrogen or argon. Unless specified, all commercial chemicals were purchased from Millipore Sigma (St. Louis, MO, USA) and used without further purification. All solvents such as hexane, ethyl acetate, dichloromethane (DCM), and methanol (MeOH) used for flash chromatography and purification were purchased from VWR (Radnor, PA, USA). Anhydrous solvents such as tetrahydrofuran (THF), dioxane, DCM, and dimethylformamide (DMF) were purchased from Acros Organics (ThermoFisher, Waltham, MA, USA). The chemical structures of the obtained compounds were analyzed by NMR spectroscopy using a Bruker Avance III 500 MHz spectrometer (1H (500 MHz), 13C (125 MHz), Billerica, MA, USA), and low-resolution electrospray ionization (ESI) mass spectrometry (MS) using a Thermo LCQ Deca XP ion trap instrument. For high resolution-mass spectrometry, 10 μL of the sample were dissolved in MeOH or THF, and then diluted in MeOH to acceptable concentrations for MS. They were injected by the EASY-nLC system, and data were acquired by FTMS positive ion mode on an LTQ Orbitrap XL with a mass range of 120–1000 m/z. The chemical shifts of NMR spectra were expressed as parts per million (ppm) downfield or upfield from the reference standard (tetramethylsilane) and the coupling constant (J) was expressed as hertz (Hz). Peak splitting patterns were reported using the following: s = singlet, d = doublet, t = triplet, q = quartet, br = broad, and m = multiplet). Flash chromatography was also performed to purify the products using a Biotage Isolera One Flash Chromatography Instruments (Biotage, Uppsala, Sweden) equipped with a Biotage disposable silica cartridge and disposable glass culture tube. Melting point (mp) of solid compounds were measured using a Fisher-Johns Melting Point Apparatus.
4.1.2. Radiochemistry and PET imaging
[11C]Carbon dioxide (11CO2) was produced from the in-house cyclotron (GE Healthcare, Chicago, IL, USA) using the 14N(p,α)11C reaction, bombarding the nitrogen target containing 1% oxygen. The GE FX-MeI and GE FX-M systems were used for [11C]MeOTf production from 11CO2 and to perform O-methylation of the precursors. The radioactivity of each radiolabeled product was measured by a dose calibrator (Capintec 55tR-PET, Florham Park, NJ, USA). For quality control, analytical HPLC analysis was conducted on an Agilent 1100 series (Santa Clara, CA, USA) monitoring at 254 nm, equipped with a CsI radiodetector (Carrol & Ramsey Instruments, Fort Collins, CO, USA) at a flow rate of 1 mL/min on a Chromolith Speedrod column (50×4.6 mm; Phenomenex, Torrance, CA, USA) using different HPLC solvent systems for each compound (Figure S2). To evaluate radiochemical stability in vitro (Figure S3), the product was measured in HPLC immediately after synthesis and 1 hour later.
All PET scans were conducted on a micro-PET Focus220 scanner (Siemens, Knoxville, TN, USA). Radiotracer injections were performed using a programmable pump (Harvard Apparatus, Holliston, MA, USA). Rat vitals (heart rate, respiratory rate, spO2, and temperature) were monitored during the PET scan using a PhysioSuite module (Kent Scientific, Litchfield, CT, USA). Catheter tubing (BTPE-10) and surgical equipment were purchased from Instetch Laboratories (Plymouth Meeting, PA, USA). Gamma counting was performed on a PerkinElmer Wallac Wizard 1480 (Waltham, MA, USA).
4.2. Organic syntheses
4.2.1. (S)-(2,3-Dihydrobenzo[b][1,4,]dioxin-2-yl)methanol (1c)
To a suspension of 1,2-dihydroxybenzene (1a, 43.81 mmol, 4.824 g) and potassium carbonate (K2CO3, 65.71 mmol, 9.082 g) in anhydrous DMF (45 mL) was added (2R)-(−)-glycidyl tosylate (1b, 43.81 mmol, 10.00 g). The mixture was stirred for 24 hours at 60°C. The reaction aliquot was quenched with sat. ammonium chloride (NH4Cl) solution and extracted three times with ethyl acetate. The residual DMF was further removed in vacuo at 40ºC. The residue was purified by flash chromatography using a gradient of 5% to 30% ethyl acetate in hexane over 40 min to give 1c as a white powder (5.673 g, 78%). mp: 68–69°C, 1H NMR (500 MHz, CDCl3): δ ppm 6.86–6.93 (m, 4H), 4.32 (dd, J = 11 Hz, 2.5 Hz, 1H), 4.27–4.31 (m, 1H), 4.14 (dd, J = 11 Hz, 7.0 Hz, 1H), 3.84–3.96 (m, 2H), 1.97 (d, J = 6.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ ppm 143.26, 143.14, 121.79, 121.72, 117.44, 117.35, 73.53, 65.28, 61.97. LR-MS (ESI): m/z calcd. for C9H10O3 (M+), 166.06; found (M+Na+), 189.27. HR-MS calcd. (M+) 166.06299, found. (M+Na+) 189.05151
4.2.2. (R)-(2,3-Dihydrobenzo[b][1,4]dioxin-2-yl)methyl 4-methylbenzenesulfonate (1d)
To a mixture of 1c (1.824 mmol, 0.303 g) and triethylamine (TEA, 3.648 mmol, 0.51 mL) in anhydrous DCM (3.6 mL) at 0°C was added tosyl chloride (2.189 mmol, 0.417 g) slowly. The mixture was warmed to room temperature and stirred for 5 hours. The reaction aliquot was quenched with saturated NH4Cl solution and extracted three times with DCM. The residue was purified by flash chromatography using a gradient of 5% to 25% ethyl acetate in hexane over 35 min to give 1d as a white powder (0.508 g, 87%). mp: 53–54°C, 1H NMR (500 MHz, CDCl3): δ ppm 7.80 (dd, J = 6.5 Hz, 2.0 Hz, 2H), 7.35 (dd J = 7.5 Hz, 2.0 Hz, 2H), 6.81–6.85 (m, 3H), 6.76–6.79 (m, 1H), 4.39 (qd, J = 2.5 Hz, 1.0 Hz, 1H), 4.18–4.27 (m, 3H) 4.04 (dd, J = 12 Hz, 6.0 Hz, 1H), 2.46 (s, 3H). 13C NMR (125 MHz, CDCl3): δ ppm 145.40, 142.90, 142.36, 132.54, 130.13 (2C), 128.18 (2C), 122.04, 121.88, 117.47, 117.41, 70.43, 67.26, 64.49, 21.82. LR-MS (ESI): m/z calcd. for C9H10O3 (M+), 320.07; found (M+Na+), 343.26. HR-MS: calcd. (M+) 320.07184, found. (M+H+) 321.07839, (M+Na+) 343.05984
4.2.3. tert-Butyl (S)-4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazine-1-carboxylate (1e)
To a mixture of 1d (3.121 mmol, 1.000 g) and 1-Boc-piperazine (9.364 mmol, 1.744 g) in anhydrous THF (13 mL) was added K2CO3 (9.364 mmol, 1.294 g) and the solution was refluxed for 24 hours. The resulting solution was cooled to room temperature and diluted with sodium carbonate (Na2CO3) solution (1:1 mixture of saturated Na2CO3 solution and water). The solution was extracted two times with ethyl acetate. The organic parts were dried over anhydrous sodium sulfate (Na2SO4), filtered, and evaporated. The residue was purified by flash chromatography using a gradient of 5% to 30% ethyl acetate in hexane over 30 min to give 1e as a white crystalline powder after recrystallization in an ethyl acetate and hexane mixture (0.9363 g, 90%). Rf- 0.5, 50% ethyl acetate in hexane. mp: 101°C, 1H NMR (500 MHz, CDCl3): δ ppm 6.82–6.88 (m, 4H), 4.29–4.31 (m, 2H), 4.00 (dd, J = 12 Hz, 7.5 Hz), 3.44 (t, J = 5.0 Hz, 4H) 2.69 (dd, J = 13.5 Hz, 5.5 Hz, 1H), 2.60 (dd, J = 13.5 Hz, 6.0 Hz, 1H), 2.53–2.57 (m, 2H), 2.44–2.49 (m, 2H) 1.46 (s, 9H). 13C NMR (125 MHz, CDCl3): δ ppm 154.86, 143.39, 143.16, 121.68, 121.53, 117.53, 117.24, 79.84, 71.34, 66.94, 58.66, 53.87 (2C), 43–45 (broad s, 2C), 28.56 (3C). LR-MS (ESI): m/z calcd. For C18H26N2O4 (M+), 334.19; found (M+H+), 335.10. HR-MS: calcd. (M+) 334.18926, found (M+H+) 335.19566
4.2.4. (S)-1-((2,3-Dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazine (1f)
A solution of 1e (2.123 mmol, 0.710 g) in a co-solvent of DCM:trifluoroacetic acid (TFA) = 7.1 mL:7.1 mL was stirred at 0°C for 1 hour. The resulting solution was basified with sat. K2CO3 solution:water = 1:1 solution to adjust its pH to be greater than 8. The solution was extracted three times with ethyl acetate, dried over anhydrous Na2SO4, filtered, and evaporated using a rotary evaporator. The residue was further purified with flash chromatography using a gradient of 0% to 25% DCM in MeOH for 20 min to give 1f as a white crystalline powder after recrystallization in ethyl acetate-hexane co-solvent (0.490 g, 98%). mp: 126–127°C, 1H NMR (500 MHz, CDCl3): δ ppm 6.83–6.88 (m, 4H), 4.27–4.32 (m, 1H), 4.25 (dd, J = 11 Hz, 2.0 Hz), 4.00 (dd, J = 11 Hz, 8.0 Hz), 3.21 (t, J = 5.0 Hz, 4H) 2.83–2.88 (m, 4H), 2.77 (dd, J = 13.5 Hz, 6.0 Hz, 1H), 2.67 (dd, J = 13.5 Hz, 5.0 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ ppm 143.20, 142.83, 121.86, 121.75, 117.52, 117.31, 71.36, 66.50, 58.07, 50.57 (2C), 43.69 (2C). LR-MS (ESI): m/z calcd. for C13H18N2O2 (M+), 234.14; found (M+H+), 235.34.
4.2.5. (2,4-Dibromo-1,3-thiazol-5-yl)methanol (1h)
To a solution of 2,4-dibromo-1,3-thiazole-5-carboxaldehyde (1g, 22.15 mmol, 6.000 g, Combi-Blocks, San Diego, CA. USA) in MeOH (44 mL) at 0°C was added sodium borohydride (22.15 mmol, 0.838 g) and the mixture was slowly raised to room temperature and stirred for 24 hours. The resulting solution was quenched with sat. NH4Cl solution and basified to pH=10 with 1 M sodium hydroxide (NaOH) solution. The solution was extracted two times with ethyl acetate, dried over anhydrous Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography using a gradient of 5% to 25% ethyl acetate in hexane over 30 min to give 1h as a yellow powder after recrystallization in hexane (5.626 g, 93%). mp: 86°C, 1H NMR (500 MHz, CDCl3): δ ppm 4.79 (d, J = 5.0 Hz, 2H), 2.16 (t, J = 5.0 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ ppm 137.87, 135.96, 121.77, 58.22. LR-MS (ESI): m/z calcd. for C4H3Br2NOS (M+), 270.83; found (M+H+), 272.34, 274.43, 276.43. Three M+ peaks corresponded with two bromines.
4.2.6. (4-Bromo-1,3-thiazol-5-yl)methanol (1i)
To a solution of 1h (5.496 mmol, 1.500 g) in MeOH (60 mL) at room temperature was added 10% palladium on activated carbon (Pd/C, 0.240 g) and Na2CO3 (12.09 mmol, 1.281 g). The whole mixture was shaken under 60 psi of hydrogen for 2 days at room temperature. The reaction mixture was filtered, and its solvent was removed. The residue was purified by flash chromatography using a gradient of 10% to 40% ethyl acetate in hexane over 30 min to give 1i as a yellow crystal after recrystallization in ethyl acetate-hexane co-solvent (0.930 g, 87%). mp: 63–64°C, 1H NMR (500 MHz, CDCl3): δ ppm 8.72 (s, 1H), 4.86 (d, J = 5.0 Hz, 2H), 2.23 (t, J = 5.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ ppm 153.34, 133.80, 124.85, 58.22. LR-MS (ESI): m/z calcd. for C4H4BrNOS (M+), 192.92; found (M+H+), 194.02, 196.02. Two M+ peaks corresponded with one bromine.
4.2.7. 4-Bromo-5-methoxymethyl-1,3-thiazole (1j)
To a suspension solution of 1i (0.5153 mmol, 0.100 g) and NaOH (0.7328 mmol, 29.3 mg) in 1.1 mL of dimethyl sulfoxide (DMSO) at room temperature was added iodomethane (MeI, 0.7328 mmol, 45.6 μL) and the solution was stirred for 3 hours. The reaction mixture was quenched by sat. NH4Cl solution and extracted three times with ethyl ether. The organic parts were collected, dried over Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography using a gradient of 0% to 10% ethyl acetate in hexane over 20 min to give 1j as a yellow oil after 30 min of vacuum drying with a rotary evaporator (88.4 mg, 82%). 1H NMR (500 MHz, CDCl3): δ ppm 8.73 (s, 1H), 4.61 (s, 2H), 3.41 (s, 3H). 13C NMR (125 MHz, CDCl3): δ ppm 153.48, 131.09, 125.52, 67.02, 58.59. LR-MS (ESI): m/z calcd. for C5H6BrNOS (M+), 206.94; found no M+ peak.
4.2.8. 4-Bromo-5-(4-methoxybenzyloxy)methyl-1,3-thiazole (1k)
To a mixture of 1i (0.5153 mmol, 0.100 g) and sodium hydride (NaH, 60% in mineral oil, 0.5675 mmol, 23 mg) in 1.0 mL of THF at 0°C was added p-methoxybenzyl chloride (0.7730 mmol, 0.11 mL), heated to 60°C, and stirred for 28 hours. Additional NaOH (0.325 mmol, 13 mg) was added to the reaction mixture 20 hours after heating. The solution was diluted with sat. NH4Cl solution and partitioned three times with ethyl acetate. The organic parts were combined, dried over Na2SO4, filtered, and evaporated. The residue was separated by flash chromatography using a gradient of 5% to 30% ethyl acetate in hexane for 25 min to give 1k as a yellowish solid (0.110 g, 68%). mp: 30°C, 1H NMR (500 MHz, CDCl3): δ ppm 8.73 (s, 1H), 7.28 (d, J = 7.0 Hz, 2H) 6.90 (d, J = 7.0 Hz, 2H), 4.67 (s, 2H), 4.53 (s, 2H), 3.81 (s, 3H). 13C NMR (125 MHz, CDCl3): δ ppm 159.65, 153.45, 131.58, 129.69 (2C), 129.44, 125.45, 114.08 (2C), 72.45, 64.32, 55.44. LR-MS (ESI): m/z calcd. for C12H12BrNO2S (M+), 312.98; found (M+H+), 314.12, 316.12. Two M+ peaks corresponded with one bromine.
4.2.9. (S)-4-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-5-(((4-methoxybenzyl)oxy)methyl)-1,3-thiazole (1l)
To a solution of 1f (0.8899 mmol, 0.209 g) and 1k (0.9789 mmol, 0.308 g) in degassed dioxane (4.5 mL) at room temperature was added cesium carbonate (Cs2CO3, 5.339 mmol, 1.740 g), palladium(II) acetate (89.0 μmol, 20.0 mg) and (±)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene (BINAP, 0.178 mmol, 0.111 g). Then, the whole solution was further degassed for 20 min and stirred at 90°C for 16 hours. The reaction mixture was diluted with sat. Na2CO3 solution and extracted three times with ethyl acetate. The organic parts were collected, dried over anhydrous Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography using a gradient from 10% to 70% ethyl acetate in hexane for 30 min to afford 1l as a yellow oil (0.215 g, 52%). 1H NMR (500 MHz, CDCl3): δ ppm 8.55 (s, 1H), 7.28 (d, J = 7.0 Hz, 2H), 6.87–6.90 (m, 4H), 6.83–6.85 (m, 2H), 4.60 (s, 2H), 4.50 (s, 2H), 4.33 (dt, J = 9.5 Hz, 2.0 Hz, 2H), 4.02 (dd, J = 9.5 Hz, 7.0 Hz, 1H), 3.80 (s, 3H), 3.21–3.23 (m, 4H), 2.70–2.74 (m, 3H), 2.64–2.69 (m, 3H) 13C NMR (125 MHz, CDCl3): δ ppm 159.90, 159.56, 149.90, 143.48, 143.29, 129.85, 129.83 (2C), 121.64, 121.48, 117.54, 117.24, 114.79, 114.08 (2C), 71.97, 71.38, 67.08, 62.65, 58.76, 55.45, 54.28 (2C), 51.56 (2C). LR-MS (ESI): m/z calcd. for C25H29N3O4S (M+), 467.19; found (M+H+), 468.20; (M+Na+), 490.23.
4.2.10. (S)-4-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-5-hydroxymethyl1,3-thiazole (1m)
To a solution of 1l (0.3428 mmol, 0.160 g) in DCM (1.6 mL) at 0°C was added TFA (0.4 mL) and stirred for 1.5 hours. The reaction mixture was quenched with sat. Na2CO3 solution:water = 1:1 solution and extracted three times with ethyl acetate. The organic parts were combined, dried over anhydrous Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography using a gradient of 50% to 100% ethyl acetate in hexane over 25 min to give 1m as a yellow solid after recrystallization in ethyl ether-hexane co-solvent (59.2 mg, 50%). 1H NMR (500 MHz, CDCl3): δ ppm 8.54 (s, 1H), 6.83–6.90 (m, 4H), 4.84 (s, 2H), 4.33 (dd, J = 11.5 Hz, 2.0 Hz, 2H), 4.03 (dd, J = 12.0 Hz, 7.0 Hz, 1H), 3.70–3.77 (broad s, 1H), 3.22 (t, J = 4.5 Hz, 4H), 2.74–2.77 (m, 3H), 2.71–2.76 (m, 3H). 13C NMR (125 MHz, CDCl3): δ ppm 158.72, 149.58, 143.42, 143.19, 121.67, 121.51, 119.88, 117.54, 117.25, 71.34, 67.00, 58.61, 57.16, 54.24 (2C), 51.74 (2C). LR-MS (ESI): m/z calcd. for C17H21N3O3S (M+), 347.13; found (M+H+), 348.22; (M+Na+), 370.14.
4.2.11. (S)-4-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-5-(methoxymethyl)thiazole (1)
To a solution of 1f (0.6240 mmol, 0.146 g) and 1j (0.6863 mmol, 0.143 g) in degassed dioxane (6.2 mL) at room temperature was added Cs2CO3 (3.743 mmol, 1.220 g), palladium(II) acetate (62.4 μmol, 14 mg) and BINAP (0.1248 mmol, 78 mg), and stirred at 90°C for 15 hours. The reaction mixture was diluted with sat. Na2CO3 solution and extracted three times with ethyl acetate. The organic parts were collected, dried over anhydrous Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography using a gradient of 10% to 70% ethyl acetate in hexane for 30 min to afford 1 as a yellow oil (0.192 g, 85%). 1H NMR (500 MHz, CDCl3): δ ppm 8.54 (s, 1H), 6.86–6.89 (m, 2H), 6.82–6.83 (m, 2H), 4.54 (s, 2H), 4.32–4.34 (m 2H), 4.02 (dd, J = 9.5 Hz, 6.0 Hz, 1H), 3.38 (s, 3H), 3.24–3.26 (m, 4H), 2.72–2.76 (m, 3H), 2.64–2.67 (m, 3H). 13C NMR (125 MHz, CDCl3): δ ppm 159.75, 149.80, 143.39, 121.55, 121.40, 117.46, 117.17, 114.68, 71.30, 66.97, 65.59, 58.66, 58.07, 54.23 (2C), 51.54 (2C). LR-MS (ESI): m/z calcd. for C18H23N3O3S (M+), 361.15; found (M+H+), 362.50; (M+Na+), 384.57. HR-MS calcd. (M+) 361.14601, found (M+H+) 362.15199
4.2.12. Ethyl (S)-3-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-5-methyl-1H-pyrazole-4-carboxylate (2a)
To a solution of 1f (2.071 mmol, 0.485 g) in THF (3 mL) was added thiocarbonyldiimidazole (2.071 mmol, 0.369 g, Oakwood Chemicals) dropwise and stirred for 4 hours under N2. At this time, TLC has shown complete consumption of the starting material. THF was evaporated, and the crude was diluted with water (30 mL) and extracted with DCM (3×25 mL). Organic layers were dried over anhydrous Na2SO4, concentrated, and the crude was used as such in the next step. The product was a thick red oil. Yield: Crude 0.701 g, 2.069 mmol, 98.6%. TLC-10% MeOH in DCM, Rf 0.65.
To the above intermediate in abs. EtOH (10 mL) was added hydrazine monohydrate slowly (4.358 mmol, 0.360 mL) dissolved in EtOH (2 mL). A white precipitate formation was noticed soon after the addition of hydrazine. The suspension was stirred for 4 more hours at room temperature. After this, volatile components were evaporated to obtain the thiocarbohydrazide intermediate as a white solid and used as such in the next step. TLC- 5% MeOH in DCM, Rf 0.6.
To the thiocarbohydrazide (2.272 mmol, 0.700 g) in abs. EtOH (6 mL) at 0°C was added conc. HCl (6.818 mmol, 0.568 mL) dissolved in abs. EtOH (2 mL), resulting in a clear solution. This is followed by the addition of ethyl 2-chloroacetoacetate (4.544 mmol, 0.628 mL) dissolved in abs. EtOH (1.5 mL). The reaction was slowly warmed to room temperature and stirred for 60 hours. Following this time, the solvent was evaporated, and to the crude was added 0.5 M HCl (30 mL) and water (30 mL). The insoluble solid was filtered and washed with water (10 mL). The water layer was washed with ethyl acetate (10 mL). The water layer was then basified using aq. sodium carbonate (Na2CO3, 30 mL). The product was extracted using ethyl acetate (3×30 mL), and the combined organic layers were dried over Na2SO4 and evaporated to obtain 2a, which was used as such in the next step (0.625 g, 74.3% over 3 steps). 1H NMR (500 MHz, CDCl3) δ 6.79–6.92 (m, 4H), 4.14–4.68 (m, 4H), 4.01 (dd, J = 11.2 Hz, 7.3 Hz, 1H), 3.29 (d, J = 5.5 Hz, 4H), 2.62–2.83 (m, 6H), 2.46 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 163.69, 159.86, 146.47, 143.26, 143.07, 121.49, 121.31, 117.41, 117.05, 100.77, 71.13, 66.90, 59.77, 58.61, 53.72 (2C), 50.01 (2C), 14.41, 13.30. LR-MS (ESI): m/z calcd. for (M+), C20H26N4O4, 386.20; found (M+H+), 387.18
4.2.13. Ethyl (S)-5-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-1,3-dimethyl-1H-pyrazole-4-carboxylate (2b)
To the solution of 2a (2.253 mmol, 0.870 g) in anhydrous THF (6 mL) at 0°C was added methyl tosylate (2.703 mmol, 0.409 mL) dissolved in THF (2 mL). Then sodium tert-butoxide (2.929 mmol, 0.281 g) was added in 2 portions and the reaction was slowly warmed to room temperature and stirred for 4 hours. After, the reaction was quenched with aq. NH4Cl (15 mL) and water (30 mL), then extracted with ethyl acetate (3×25 mL). The organic layers were dried over Na2SO4 and purified using column chromatography using a gradient of 0% to 5% MeOH in DCM to give 2b as a colorless solid (0.580 g, 64.3%) Rf-0.6 in 5% MeOH-DCM. 1H NMR (500 MHz, CDCl3) δ 6.91 – 6.79 (m, 4H), 4.38 – 4.22 (m, 4H), 4.00 (dd, J = 11.6 Hz, 7.6 Hz, 1H), 3.66 (s, 3H), 3.25 (d, J = 6.1 Hz, 4H), 2.84 – 2.56 (m, 6H), 2.45 (s, 3H), 1.35 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 163.65, 158.79, 144.79, 143.28, 143.13, 121.42, 121.24, 117.38, 117.01, 101.03, 71.14, 66.94, 59.62, 58.60, 53.80 (2C), 50.32 (2C), 35.91, 14.42, 11.89. LC-MS (ESI): m/z calcd. for (M+), C21H28N4O4, 400.21; found (M+H+), 401.27
4.2.14. (S)-(5-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-1,3-dimethyl-1H-pyrazol-4-yl)-methanol (2c)
To 2b (1.050 mmol, 0.420 g) dissolved in THF (4 mL) at 0°C was added 2.0 M LAH (2.100 mmol, 1.05 mL) dropwise. The reaction was warmed to room temperature and stirred for 15 hours. Following this, the reaction was quenched with water (85 μL), 15% NaOH (85 μL), and water (2 mL) sequentially, and then ethyl acetate (25 mL) was added and stirred for 1 hour. The solid was filtered and washed with ethyl acetate (2×210 mL). The filtrate was dried to obtain 2c as a white solid (0.340 g, 90.4%). mp: 140–141°C, 1H NMR (500 MHz, CDCl3) δ 6.70–6.99 (m, 4H), 4.51 (s, 2H), 4.32–4.39 (m, 2H), 3.98–4.09 (m, 1H), 3.67 (d, J = 2.3 Hz, 3H), 3.22 (ddd, J = 5.8 Hz, 3.8 Hz, 1.5 Hz, 4H), 2.59–2.91 (m, 6H), 2.21 (d, J = 9.7 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 156.65, 143.33, 143.16, 138.14, 121.49, 121.31, 117.41, 117.08, 107.35, 71.26, 66.96, 58.58, 55.41, 54.08 (2C), 50.56 (2C), 35.62, 9.57. LR-MS (ESI): m/z calcd. for (M+), C19H26N4O3, 358.20; found (M+H+), 359.22. HR-MS calcd. (M+) 358.20049, found (M+H+) 359.20688
4.2.15. (S)-1-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)-4-(4-(methoxymethyl)-1,3-dimethyl-1H-pyrazol-5-yl)-piperazine (2)
To a solution of 2c (0.215 mmol, 0.080 g) in DMF (1 mL) at 0°C was added NaH (60% in mineral oil, 0.323 mmol, 0.012 g) and warmed to room temperature and stirred for 15 min. Following this, MeI (0.323 mmol, 25 μL) dissolved in DMF (0.3 mL) was slowly added and the reaction was stirred for 6 hours. Then the reaction was quenched with aq. NH4Cl (2 mL) and water (10 mL). The water layers were extracted with ethyl acetate (3×10 mL). The combined organic layers were washed with brine (10 mL) and evaporated, and the crude was purified using column chromatography with a gradient of 0% to 5% MeOH in DCM to afford 2 as a colorless oil (0.028 g, 34.1%). 1H NMR (500 MHz, DMSO) δ 6.90 – 6.77 (m, 4H), 4.42 – 4.26 (m, 2H), 4.09 (s, 2H), 3.97 (ddd, J = 11.4, 9.4, 7.2 Hz, 1H), 3.56 (s, 3H), 3.18 (s, 3H), 3.01 (dt, J = 17.0, 4.9 Hz, 4H), 2.61 (ddd, J = 14.0, 6.0, 3.3 Hz, 3H), 2.55 – 2.47 (m, 3H), 2.14 (s, 3H). 13C NMR (126 MHz, DMSO) δ 156.87, 143.50, 143.33, 139.70, 121.76, 121.54, 117.57, 117.30, 104.01, 71.25, 66.72, 63.94, 58.40, 56.98, 53.88 (2C), 50.60 (2C), 35.88, 9.69. HR-MS (ESI): m/z calcd. for (M+), C20H28N4O3, 372.21614; found (M+H+), 373.22236, (M+Na+) 395.20423
4.2.16. (S)-1-(((S)-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperidin-3-amine. Hydrogen chloride (3a)
A mixture of tosylate 1d (5.810 g, 18.156 mmol), tert-butyl (S)-piperidin-3-ylcarbamate (3.631 g, 18.156 mmol) and K2CO3 (4.384 g, 31.773 mmol) in DMF (30 mL) was heated to 80°C for 24 hours, after which the reaction was quenched with water (70 mL), brine (40 mL) and extracted with 50% ethyl acetate in hexane (3×45 mL). The organic layers were combined, dried, and purified using column chromatography with a gradient of 0% to 35% ethyl acetate in hexane to elute the product. Product is a colorless thick oil2. Yield: 4.934 g, 14.178 mmol, 78.3%. 1H NMR (600 MHz, CDCl3) δ 6.90 – 6.82 (m, 4H), 4.41 (tq, J = 13.6, 5.0 Hz, 1H), 4.34 – 4.27 (m, 2H), 4.14 – 4.03 (m, 1H), 4.03 – 3.96 (m, 1H), 3.82 – 3.18 (m, 2H), 2.76 – 2.64 (m, 1H), 2.62 – 2.54 (m, 2H), 2.49 – 2.34 (m, 2H), 1.94 – 1.52 (m, 2H), 1.47 (t, J = 3.2 Hz, 9H). 13C NMR (126 MHz, CDCl3) δ 155.12, 154.95, 143.28, 143.08, 121.45, 121.28, 117.33, 117.05, 79.10, 71.05, 66.81, 65.17, 59.40, 58.43, 54.50, 53.40, 48.96, 46.38, 44.16, 29.48, 28.42, 28.34, 22.41. HR-MS (ESI): m/z calcd. for (M+), C19H28N2O4, 348.20491; found (M+H+), 349.21119
The above product (2.015 g, 14.396 mmol) was dissolved in DCM (5 mL) and dioxane (5 mL). To the solution at room temperature was added conc. HCl (2.27 mL, 23.168 mmol) dissolved in dioxane (5 mL) dropwise. CO2 bubbles were noticed during the addition. The reaction was stirred at room temperature for 20 hours after which MeOH (20 mL) was added and evaporated. Evaporation was repeated with MeOH (2×15 mL) until the excess HCl was gone. The HCl salt of the product was obtained as a white solid. Yield: 1.398 g, 5.637 mmol, 97%. 1H NMR (500 MHz, CDCl3) δ 7.07 – 5.98 (m, 4H), 2.90 (ddt, J = 17.8, 9.3, 4.1 Hz, 2H), 2.73 – 2.61 (m, 2H), 2.62 – 2.42 (m, 1H), 2.30 – 2.21 (m, 1H), 2.03 – 1.89 (m, 1H), 1.87 – 1.78 (m, 1H), 1.77 – 1.68 (m, 1H), 1.66 – 1.44 (m, 1H), 1.38 – 1.20 (m, 3H), 1.13 (dtt, J = 13.3, 10.1, 4.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 143.31, 143.16, 121.41, 121.23, 117.35, 117.03, 71.28, 71.21, 67.06, 66.95, 65.21, 63.59, 63.23, 58.65, 54.61, 47.89, 44.12, 33.78, 23.60. HR-MS (ESI): m/z calcd. for (M+), C14H20N2O2, 248.15248; found (M+H+), 249.15904
4.2.17. Methyl (Z)-4-bromo-3-(2-methoxyphenyl)but-2-enoate (3b)
To the ice-cold solution of NaH (60% in mineral oil, 1.000 g, 25.833 mmol) in 10 mL THF at 0°C was added trimethyl phosphonoacetate (4.701 mL, 25.833 mmol) dissolved in THF (15 mL) dropwise over 5 min, and then the reaction was slowly warmed to room temperature over 30 min, after which was added dropwise 2-methoxyacetophenone (3.191 g, 15.499 mmol) dissolved in THF (10 mL). Then the reaction was stirred at room temperature for 16 hours. Following this time, a TLC showed two new spots. The reaction was worked up using water (30 mL), brine (30 mL), and ethyl acetate (3×30 mL). The organic layers were combined, dried, and purified using flash chromatography with a gradient from 0% to 9% ethyl acetate in hexane to isolate cis (Z)-isomer (1.810 g) as colorless oil and from 9% to 20% ethyl acetate in hexane to isolate trans (E)-isomer (~2 g) as colorless oil. Subsequent bromination was carried out with cis isomer. Yield: 3.810 g, 18.495 mmol, 71.4%. cis (Z): 1H NMR (600 MHz, CDCl3) δ 7.32 (ddd, J = 8.3, 7.4, 1.8 Hz, 1H), 7.16 (dd, J = 7.5, 1.8 Hz, 1H), 6.99 – 6.90 (m, 2H), 5.94 (q, J = 1.4 Hz, 1H), 3.86 – 3.83 (m, 3H), 3.76 (s, 3H), 2.53 (d, J = 1.6 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 167.18, 156.94, 156.35, 133.01, 129.53, 128.78, 120.57, 118.80, 111.03, 55.45, 50.99, 19.90. trans (E): 1H NMR (600 MHz, CDCl3) δ 7.34 – 7.27 (m, 1H), 7.05 (dd, J = 7.4, 1.8 Hz, 1H), 6.98 (dd, J = 7.4, 1.1 Hz, 1H), 6.94 (dd, J = 8.3, 1.0 Hz, 1H), 6.00 (q, J = 1.5 Hz, 1H), 3.82 (s, 3H), 3.56 (s, 3H), 2.18 (d, J = 1.6 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 166.00, 155.34, 153.91, 133.65, 130.38, 130.17, 128.83, 128.02, 120.38, 118.33, 110.81, 55.54, 50.86, 26.17. HR-MS (ESI): m/z calcd. for (M+), C14H20N2O2, 248.15248; found (M+H+), 249.15904. HR-MS (ESI): m/z calcd. for (M+), C12H14O3, 206.09429; found (M+Na+), 229.08265
The above cis product methyl-(Z)-3-(2-methoxyphenyl)but-2-enoate (1.810 g, 8.738 mmol) was dissolved in carbon tetrachloride (CCl4, 30 mL) to which at room temperature was added N-bromosuccinimide (NBS, 1.710 g, 39.612mmol) in one portion after which azobisisobutyronitrile (AIBN, 0.057 g, 0.350 mmol) was added and the reaction was refluxed at 70°C for 14 hours. Following this time, a TLC showed a new spot just below the starting compound and disappearance of the starting compound. The reaction was worked up with water (30 mL), brine (20 mL), and DCM (3×25 mL). The organic layers were combined, dried, and purified using flash chromatography with a gradient from 0% to 10% ethyl acetate in hexane to elute product 3b (1.786 g, 6.289 mmol) as white solid. Yield: 71%. mp: 53–54°C, 1H NMR (300 MHz, CDCl3) δ 7.39 (ddd, J = 8.3, 7.4, 1.8 Hz, 1H), 7.31 – 7.17 (m, 1H), 7.08 – 6.89 (m, 2H), 5.98 (s, 1H), 5.05 (s, 2H), 3.85 (s, 3H), 3.81 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 165.86, 156.37, 154.52, 130.71, 130.45, 128.49, 121.52, 120.73, 110.87, 55.49, 51.53, 28.56. HR-MS (ESI): m/z calcd. for (M+), C12H13BrO3, 284.00481, and 286.00276, found (M+H)+ 285.01106 and 287.00903
4.2.18. 1-((S)-1-(((S)-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperidin-3-yl)-4-(2-methoxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (3)
To the solution of amine 3a (1.218 g, 4.912 mmol) and bromide 3b (1.406 g, 4.912 mmol) in toluene (30 mL) at room temperature was added TEA (0.747 mL, 5.403 mmol). The reaction was then heated at 100°C for 4 hours. Following this time, the reaction was quenched with water (40 mL) and extracted with DCM (3×30 mL). The organic layers were combined, dried, evaporated, and purified using flash chromatography with a gradient from 3% to 4% MeOH in DCM to elute the product 3 (1.226 g, 2.919 mmol) as white solid. Yield: 60.3%. mp: 57–58°C. 1H NMR (600 MHz, CDCl3) δ 7.44 – 7.35 (m, 2H), 7.05 – 6.97 (m, 2H), 6.92 – 6.79 (m, 4H), 6.69 (t, J = 1.5 Hz, 1H), 4.51 – 4.40 (m, 2H), 4.39 – 4.28 (m, 4H), 4.06 (dd, J = 11.6, 7.2 Hz, 1H), 3.93 (s, 3H), 3.07 – 3.01 (m, 1H), 2.84 (dt, J = 11.4, 4.0 Hz, 1H), 2.76 – 2.65 (m, 2H), 2.39 – 2.24 (m, 2H), 1.91 (dq, J = 12.5, 4.2 Hz, 1H), 1.83 – 1.70 (m, 2H), 1.63 – 1.53 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 171.86, 157.97, 151.07, 143.36, 143.12, 131.04, 127.87, 123.69, 121.44, 121.26, 120.98, 120.77, 117.32, 117.13, 111.51, 71.17, 66.69, 58.40, 57.75, 55.41, 54.70, 51.39, 48.02, 28.97, 24.47. HR-MS (ESI): m/z calcd. for (M+), C25H28N2O4, 420.20491; found (M+H+), 421.21070
4.2.19. 1-((S)-1-(((S)-2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperidin-3-yl)-4-(2-hydroxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (3c)
To the solution of 3 (0.401 g, 0.952 mmol) in DCM (14 mL) at −78°C was slowly added 1.0 M DCM solution of boron tribromide (BBr3, 2.597 mL, 3.808 mmol) over 5 min and stirred at the same temperature for 1 hour and at room temperature for 16 hours. Most of the starting compound disappeared and TLC showed a new spot just below the starting compound (5% MeOH in DCM Rf 0.4). The reaction was quenched slowly with aq. sodium bicarbonate (NaHCO3, 25 mL) and worked up with brine (20 mL) and DCM (3×15 mL). The organic layers were combined, dried, evaporated, and purified using flash chromatography with a gradient from 0% to 6% MeOH in DCM to elute the product as white solid. Yield: 0.260 g, 0.641 mmol, 67.3%. mp: 93–94°C, 1H NMR (600 MHz, CDCl3) δ 10.12 (s, 1H), 7.32 (dd, J = 7.8, 1.7 Hz, 1H), 7.23 (ddd, J = 8.6, 7.2, 1.6 Hz, 1H), 7.08 (dd, J = 8.2, 1.2 Hz, 1H), 6.93 (t, J = 1.4 Hz, 1H), 6.91 – 6.78 (m, 5H), 4.60 – 4.49 (m, 2H), 4.39 – 4.24 (m, 3H), 4.04 – 3.93 (m, 1H), 3.06 (dd, J = 10.8, 4.0 Hz, 1H), 2.84 (dt, J = 11.5, 3.9 Hz, 1H), 2.73 – 2.61 (m, 2H), 2.33 (t, J = 10.3 Hz, 1H), 2.26 (td, J = 11.2, 3.1 Hz, 1H), 1.91 (dq, J = 12.6, 4.2 Hz, 1H), 1.83 – 1.68 (m, 1H), 1.60 (qd, J = 11.5, 4.5 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 173.44, 156.64, 152.89, 143.29, 143.05, 131.27, 127.49, 121.71, 121.49, 121.30, 119.62, 118.71, 117.37, 117.22, 117.10, 71.08, 66.67, 60.46, 58.36, 57.63, 54.48, 51.78, 48.47, 28.94, 24.36, 21.08. HR-MS (ESI): m/z calcd. for (M+), C24H26N2O4, 406.18926; found (M+H+), 407.19506
4.2.20. 1-(6-methoxypyridin-2-yl)-3-methyl-1H-pyrazol-5-amine (4a)
The 2-hydrazinyl-6-methoxypyridine (2.010 g, 14.565 mmol) was suspended in EtOH (6 mL), to which was added 3-aminocrotononitrile (1.267 g, 14.565 mmol) and acetic acid (AcOH, 1.750 mL, 29.130 mmol). The reaction was refluxed for 4 hours. Following this time, TLC showed complete consumption of the starting material. The reaction was quenched with water (30 mL) and 15% NaOH (13 mL). The water phase was then extracted with ethyl acetate (3×35 mL). The organic layers were combined, washed with brine (15 mL), and concentrated. The crude was purified using column chromatography with a gradient from 0% to 40% ethyl acetate in hexane to obtain 4a as a white solid. Yield: 1.560 g, 7.647 mmol, 53.4%. mp: 82–84°C. 1H NMR (500 MHz, CDCl3) δ 7.66 (t, J = 8.0 Hz, 1H), 7.46 (dd, J = 7.9, 0.7 Hz, 1H), 6.52 (dd, J = 8.1, 0.7 Hz, 1H), 5.72 (s, 2H), 5.35 (s, 1H), 3.91 (s, 3H), 2.22 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.28, 152.27, 150.95, 148.58, 141.01, 105.24, 105.04, 90.16, 53.59, 14.09. HR-MS (ESI): m/z calcd. for (M+), C10H12N4O, 204.10111; found (M+H+), 205.10751
4.2.21. 1-(1-(6-methoxypyridin-2-yl)-3-methyl-1H-pyrazol-5-yl)piperazine (4b)
To the mixture of 1-(6-methoxypyridin-2-yl)-3-methyl-1H-pyrazol-5-amine 4a (0.500 g, 2.45 mmol) and N-benzyl-2-chloro-N-(2-chloroethyl)ethan-1-amine (0.573 g, 2.450 mmol) in DMF at 0°C was added NaH (60% in mineral oil, 0.196 g, 4.900 mmol) in 2 portions. The reaction was slowly warmed to room temperature and stirred at 65°C for 3 hours. Following this time, the reaction was cooled and quenched with cold water (20 mL) and extracted with ethyl acetate (3×20 mL). The organic layers were combined, washed with brine (15 mL), evaporated, and purified using flash chromatography with a gradient from 0% to 50% ethyl acetate in hexane to isolate the product as yellow oil. Yield: 0.504 g, 1.389 mmol, 49.4%. 1H NMR (500 MHz, CDCl3) δ 7.69 – 7.62 (m, 1H), 7.43 – 7.12 (m, 6H), 6.66 (dd, J = 8.2, 1.1 Hz, 1H), 5.70 – 5.66 (s, 1H), 4.04 – 3.99 (s, 3H), 3.57 – 3.53 (s, 2H), 3.02 – 2.97 (bt, 4H), 2.55 – 2.49 (bt, 4H), 2.33 – 2.27 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 163.47, 153.22, 150.29, 149.90, 140.11, 137.82, 129.13, 128.26, 127.15, 109.80, 108.39, 94.56, 63.05, 53.79, 52.68, 51.50, 14.27.
To the mixture of the above product (0.490 g, 1.349 mmol), ammonium formate (0.750 g, 13.490 mmol), and formic acid (0.550 mL, 13.490 mmol) in EtOH (6 mL) was added 10% Pd/C (0.140 g, 0.1349 mmol) under nitrogen and the solution was stirred at 70°C for an hour. Following this time, 10% Pd/C was filtered off and washed with MeOH (10 mL). The filtrate was evaporated and dissolved in aq. Na2CO3 (30 mL) and ethyl acetate (20 mL). The aqueous layer was further extracted with ethyl acetate (2×20 mL). The organic layers were combined, washed with brine (15 mL), dried over Na2SO4, and concentrated to get 4b colorless oil which was used as such in the next step. TLC 6% MeOH in DCM. Yield: 0.310 g, 84.5%, 0.854 mmol. 1H NMR (600 MHz, CDCl3) δ 7.83 – 7.43 (m, 1H), 7.25 (dd, J = 7.7, 0.8 Hz, 1H), 6.62 (dd, J = 8.2, 0.8 Hz, 1H), 5.65 (s, 1H), 3.96 (s, 3H), 3.29 (s, 1H), 2.97 – 2.89 (s, 8H), 2.24 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 163.47, 153.19, 150.27, 149.85, 140.21, 109.88, 108.27, 94.78, 53.80, 52.21, 45.29, 14.23. HR-MS (ESI): m/z calcd. for (M+), C14H19N5O, 273.15896; found (M+H+), 274.16539
4.2.22. (S)-1-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)-4-(1-(6-methoxypyridin-2-yl)-3-methyl-1H-pyrazol-5-yl)piperazine (4)
To the solution of amine 4b (0.310 g, 0.854 mmol) in acetonitrile (CH3CN, 8 mL) at room temperature was added 1d (0.290 g, 0.939 mmol) followed by K2CO3 (0.390 g, 2.135 mmol) and the reaction was refluxed for 24 hours. Following this time, the reaction was quenched with water (20 mL) and extracted with ethyl acetate (3×20 mL). The organic layers were combined, washed with brine (10 mL), concentrated, and purified using flash chromatography with a gradient from 0% to 50% ethyl acetate in hexane to isolate 4 as colorless solid. Yield: 0.196 g, 41%, 0.736 mmol. Amine (0.105 g) was recovered using 10% 2% ammonia solution/MeOH in DCM. 1H NMR (600 MHz, CDCl3) δ 7.66 (t, J = 7.9 Hz, 1H), 7.29 (dd, J = 7.6, 0.7 Hz, 1H), 6.91 – 6.81 (m, 4H), 6.67 (dd, J = 8.2, 0.7 Hz, 1H), 5.69 (s, 1H), 4.34 – 4.27 (m, 2H), 4.01 (s, 3H), 3.00 (ddd, J = 6.1, 3.8, 1.8 Hz, 4H), 2.75 – 2.55 (m, 6H), 2.29 (s, 3H). 13C NMR (151 MHz, CD Cl3) δ 163.51, 153.06, 150.32, 149.90, 143.26, 143.03, 140.18, 121.54, 121.39, 117.37, 117.11, 109.87, 108.44, 94.73, 71.26, 66.79, 58.47, 53.82, 53.55, 51.54, 14.28. HR-MS (ESI): m/z calcd. for (M+), C23H27N5O3, 421.21139; found (M+H+), 422.21734
4.2.23. 6-(5-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)-3-methyl-1H-pyrazol-1-yl)pyridin-2-ol (4c)
To the solution of 4 (0.150 g, 0.356 mmol) in DMF (2 mL) was added lithium chloride (LiCl, 0.105 g, 2.495 mmol) followed by p-toluenesulfonic acid (p-TsOH•H2O, 0.947 g, 4.964 mmol) and the contents were refluxed (120°C) for 3 hours. A new TLC spot was noticed along with the starting material. Following this time, the reaction was quenched with aq. Na2CO3 (15 mL) and extracted with ethyl acetate (3×15 mL). The organic layers were combined, dried, and the crude was purified with column chromatography using a gradient from 0% to 60% ethyl acetate in hexane for the starting material and using 3% MeOH in DCM to elute the product. TLC 5% MeOH-DCM. Yield: 0.020 g, 0.049 mmol, 14%. 1H NMR (600 MHz, CDCl3) δ 7.46 (dd, J = 9.1, 7.4 Hz, 1H), 6.94 – 6.83 (m, 4H), 6.77 (dd, J = 7.4, 0.9 Hz, 1H), 6.37 (dd, J = 9.2, 0.9 Hz, 1H), 5.85 (s, 1H), 4.38 – 4.29 (m, 2H), 4.11 – 4.00 (m, 1H), 3.03 (t, J = 4.9 Hz, 4H), 2.88 – 2.60 (m, 6H), 2.26 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 162.07, 152.98, 151.27, 143.21, 142.97, 141.82, 141.48, 121.62, 121.47, 117.43, 117.13, 115.85, 98.13, 93.70, 71.31, 66.73, 58.25, 53.34, 52.30, 14.18. HR-MS (ESI): m/z calcd. for (M+), C22H25N5O3, 407.19574; found (M+H+), 408.20034
4.2.24. Ethyl (S)-2-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)nicotinate (5a)
To a stirred solution of 1f (0.346 g, 1.476 mmol) and ethyl 2-chloronicotinate (0.273 g, 1.476 mmol) in DMF (4 mL) at room temperature was added diisopropylethylamine (DIPEA, 0.63 mL, 3.690 mmol) and heated at 90°C for 15 hours. To work up the reaction, it was cooled and quenched with water (20 mL), ethyl acetate (10 mL) and aq. NaHCO3 (30 mL). The aqueous layer was extracted with ethyl acetate (3×30 mL) and the combined organic layers were washed with brine and dried over Na2SO4. The crude was concentrated and purified by column chromatography using a gradient of 20% to 50% ethyl acetate in hexane to elute 5a as a yellow oil (0.300 g, 54%). 1H NMR (500 MHz, CDCl3) δ 8.29 (dd, J = 4.7, 2.0 Hz, 1H), 8.00 (dd, J = 7.6, 1.9 Hz, 1H), 6.95 – 6.82 (m, 4H), 6.76 (dd, J = 7.6, 4.7 Hz, 1H), 4.43 – 4.31 (m, 4H), 4.05 (dd, J = 11.5, 7.6 Hz, 1H), 3.48 (ddd, J = 5.8, 3.9, 1.6 Hz, 4H), 2.80 – 2.68 (m, 3H), 2.68 (m, 3H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 167.43, 159.10, 150.48, 143.31, 143.12, 140.73, 121.52, 121.36, 117.43, 117.10, 114.08, 114.05, 71.21, 66.93, 61.13, 58.62, 53.91, 49.10, 14.33. ESI [M+H]+C21H25N3O4, found 383.18
4.2.25. (S)-(2-(4-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)piperazin-1-yl)pyridin-3-yl)methanol (5b)
To 5a (0.280 g, 0.730 mmol) dissolved in THF (2.5 mL) at 0°C was added 2.0 M THF solution of LAH (0.887 mL, 1.480 mmol) dropwise, producing hydrogen bubbles. The reaction was warmed to room temperature and stirred for 4 hours. Following this time, the reaction was quenched sequentially with water (85 μL), 15% NaOH (85 μL), and water (2 mL), and then ethyl acetate (25 mL) was added, and the mixture was stirred for 1 hour. The solid was filtered and washed with ethyl acetate (2×10mL). The filtrate was dried to obtain 5b as a white solid (0.206 g, 0.604 mmol, 83%). mp: 115–116°C. 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 4.8 Hz, 1H), 7.62 (d, J = 7.4 Hz, 1H), 7.00 (dd, J = 7.5, 4.9 Hz, 1H), 6.86 (ddd, J = 9.9, 7.3, 4.4 Hz, 4H), 4.72 (s, 2H), 4.33 (dd, J = 10.0, 3.4 Hz, 2H), 4.02 (dd, J = 11.6, 7.5 Hz, 2H), 3.18 (t, J = 4.9 Hz, 4H), 2.72 (dtd, J = 25.6, 14.3, 13.5, 7.9 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 160.31, 146.91, 143.26, 143.05, 137.08, 128.26, 121.54, 121.38, 119.07, 117.40, 117.11, 71.19, 66.84, 62.14, 58.43, 54.15, 50.12. ESI [M+H]+C19H23N3O3, found 341.17
4.2.26. (S)-1-((2,3-dihydrobenzo[b][1,4]dioxin-2-yl)methyl)-4-(3-(methoxymethyl)pyridin-2-yl)piperazine (ORM-13070)
To the solution of 5b (0.090 g, 0.263 mmol) in THF (2.5 mL) at 0°C was added NaH (60% in mineral oil, 0.018 g, 0.421 mmol) and warmed to room temperature and stirred for 45 min. Following this time, MeI (23 μL, 0.342 mmol) dissolved in THF (0.3 mL) was slowly added and the reaction was stirred for 4 hours. The reaction was quenched with sat. NH4Cl solution (2 mL) and water (10 mL). The water layers were extracted with ethyl acetate (3×10 mL). The organic layers were combined, washed with brine (10 mL), evaporated, and purified using flash chromatography with a gradient from 0% to 2.5% MeOH in DCM to obtain ORM-13070 as a colorless liquid (0.069 g, 0.194 mmol, 74%). 1H NMR (600 MHz, CDCl3) δ 8.26 (dd, J = 4.9, 1.9 Hz, 1H), 7.71 (dd, J = 7.5, 2.0 Hz, 1H), 6.95 (dd, J = 7.5, 4.8 Hz, 1H), 6.93 (m, 2H), 6.88 – 6.83 (m, 2H), 4.44 (s, 2H), 4.41 – 4.32 (m, 2H), 4.05 (dd, J = 11.5, 7.5 Hz, 1H), 3.44 (s, 3H), 3.24 (t, J = 5.4 Hz, 4H), 2.79 (dt, J = 13.5, 5.2 Hz, 3H), 2.70 (h, J = 6.4, 5.5 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 161.02, 147.01, 143.36, 143.16, 138.07, 124.72, 121.50, 121.35, 117.84, 117.41, 117.13, 71.26, 70.47, 66.95, 58.61, 58.51, 54.19, 50.40. ESI [M+H]+C20H25N3O3, found 355.19
4.3. Radiosynthesis
To a reaction vessel containing the alcohol precursor (~1 mg, 1m, 2c, 3c, 4c, 5b) and tetrabutylammonium hydroxide (2.5 μL) in acetonitrile (200 μL) was trapped [11C]MeOTf in a stream of helium at room temperature. After radioactivity plateaued, the solution was heated for 3 min at 80°C. After cooling the solution to 40°C, the product was diluted with 10% ethanolic phosphate buffer (1 mL, pH=7.2–7.4) and injected onto a semi-preparative HPLC for purification (Figure SI2). After product collection, the radioligand was formulated with sterile water to reduce EtOH concentration below 10%.
4.4. Methods for small animal PET studies
All animal handling and experiment protocols (NIAAA 19–01, MIB-03, MIB-04) were approved by the Animal Care & Use Committee at the NIH, which complies with the guidelines of the U.S. Department of Agriculture’s animal welfare act and the U.S. Public Health Service’s policy on humane care and use of laboratory animals. Male rats (Charles River Laboratories (CRL), Frederick, MD. USA) weighing between 250–350g were housed 1–2 per cage. Male and female Swiss control mice (CRL) and mice with a genetic knockout of three common brain efflux proteins (bred in-house), weighing between 25–45g, were housed 1–6 per cage without regard for animal sex. All animals were housed at the NIH animal handling facility with a 12-hour light/dark cycle and allowed to access food and water as needed. For each study, initial anesthesia was induced under 5.0% isoflurane (Baxter, Deerfield, IL. USA) in oxygen at 1.5 L/min and maintained between 1.0–2.5% isoflurane in oxygen for surgeries and experiments.
Before the PET scan, a 10-minute transmission scan was conducted using a Co-57 point source for attenuation correction. To perform the PET study, data acquisition was started five seconds before radiotracer injection and lasted for 80 min. Radiotracer injections, averaging 17.6 ± 2.6 MBq (n=20), were performed using a pump with a one-minute injection through a penile vein catheter, followed immediately by a 200 μL flush with heparinized saline. All the animal studies conducted are summarized in Table S1. Briefly, for the baseline comparison studies, [11C]ORM-13070 and [11C]1 were injected into one rat across two PET scans, conducted on the same day, to allow for a direct comparison of the tracers. Each compound was used in a blocking study by performing one baseline scan and one blocking scan on the same day with the same animal. For both radiotracers, ORM-13070 was used as the blocking agent (50 μg/kg in DMSO, s.c.).
To conduct mouse ex vivo biodistribution studies, mice were injected by hand through a tail vein catheter with 100 μL of the radiotracer, averaging 1.6 ± 0.59 MBq (n=9), followed immediately by a 50 μL flush with heparinized saline. Control and KO mice were injected with [11C]ORM-13070 and [11C]1. 15 min after injection, the animals were euthanized and whole brain tissue was collected, and the radioactivity was measured by gamma counting. Tissue mass was measured and combined with subject weight and injected dose for SUV calculation.
4.6. Data analysis
Raw emission data obtained in list mode were reconstructed into 22 frames (6×20 sec, 5×60 sec, 4×120 sec, 3×300 sec, 3×600 sec, 1×1200 sec) using 2D filtered back projection to generate a sinogram. PET data were analyzed using PMOD v3.807 (PMOD Technologies, Zurich, Switzerland). To generate TACs, all PET images were co-registered to an MR template and brain uptake was normalized using body weight and injected dose. The regions of interest examined were the ST, due to its relatively high concentration of α2C-ARs, and the CB used as a reference region because of its lack of α2C-ARs. To analyze the ex vivo biodistribution studies, the SUV of the brain was calculated using the counted activity, rat weight, and injected dose.
4.7. Cell-binding assay
4.7.1. Molecular cloning and transfections
Human embryonic kidney (HEK) 293 cells (ATCC, Manassas, VA. USA) were grown at 37°C in Dulbecco’s modified eagle medium (Corning Inc., Corning, NY. USA) supplemented with 10% fetal bovine serum (Corning) and 1% penicillin/streptomycin/amphotericin B (Corning). Complementary DNA (cDNA) constructs containing ADRA2A (GenBank accession #NM_000681) or ADRA2C (GenBank accession #NM_000683) were purchased from GenScript (Piscataway, NJ. USA). Prior to transfection, HEK293 cells were seeded at the appropriate density to ensure 75% cell confluence within 24 hours. Transfection of the cDNA constructs was performed after 24 hours using X-tremeGENE Roche Transfection Reagent (Millipore Sigma) according to the manufacturer’s protocol using a 3:1 transfection reagent to cDNA ratio as previously described [32]. Following 48 hours of transfection, the media was changed to complete media containing the selection agent G418 (400 µg/mL, Corning). Cells were maintained in G418 containing media to generate stable cell lines used in subsequent experiments.
4.7.2. Membrane preparation
Membranes were prepared from HEK293 cells expressing α2A-ARs and α2C-ARs as previously described [6,31]. In brief, cells were lysed in ice-cold lysis buffer (25 mM tris, pH=7.4, 5 mM ethylenediaminetetraacetic acid (EDTA), 1 µg/mL aprotinin, 1 µg/mL leupeptin) and centrifuged at 1,000 g for 5 min at 4°C. The supernatant was centrifuged at 30,000 g for 5 min, and the crude membrane pellet was resuspended in lysis buffer containing 10% glycerol and stored at −80°C until use.
4.7.3. Radioligand binding
Radioligand binding was performed using crude HEK293 membranes overexpressing α2A-ARs and α2C-ARs as previously described [6]. Competitive radioligand binding was performed using a single concentration (1 nM) of [3H]RX821002 (PerkinElmer) with increasing concentrations of α2-AR nonselective antagonist RS79948 (Tocris, Minneapolis, MN. USA) and α2C-AR subtype-selective antagonists 1, 2, 3, 4, and ORM-13070 in binding buffer (250 µL, 75 mM tris pH=7.4, 2 mM EDTA, 12.5 mM magnesium chloride (MgCl2), 1 µg/mL aprotinin, 1 µg/mL leupeptin). Specific radioactive counts plotted as a function of the competitive receptor antagonist concentration, and nonlinear regression analysis was used to determine the concentration of the receptor antagonist that reduced specific [3H]RX821002 binding by 50% (IC50). Equilibrium dissociation constants (Ki) of each competitive antagonist for specific [3H]RX821002 binding sites were calculated using the method of Cheng and Prusoff as described [6].
Supplementary Material
Highlights.
Synthesis and C-11 radiolabeling of benzo-1,4-dioxane-based α2C-AR ligands.
In vitro binding potency and in vivo evaluation of BBB permeability in rat brain.
Investigate the influence of brain efflux pumps on BBB permeability.
Acknowledgments
This research was performed with support from the intramural research program of the National Institute on Alcohol Abuse and Alcoholism (Y1AA-3009, Volkow) and the University of Missouri tier 2 research and creative works strategic investment program project (Kil). The authors would like to thank Kelly O’Connor, Andrew Kelleher, Torben Pearson, Jinpyo Seo, and Gus Bakhoda for their assistance in conducting radiosynthesis and small animal PET imaging studies. The authors are grateful to the NIH Clinical Center PET department (Dr. Peter Herscovitch and Mr. Kris Kim) and the NIMH Molecular Imaging Branch (Drs. Robert Innis, Victor Pike, and Jeih-San Liow) for their PET imaging infrastructure support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Associated content
The supplemental data for this work can be found attached to this article:
Declaration of competing interest
The authors declare no competing financial interests or personal relationships that could have appeared to influence the work presented in this report.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
5. References
- [1].Strosberg AD, Structure, function, and regulation of adrenergic receptors, Protein Sci 2 (1993) 1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Eisenhofer G, Kopin IJ, Goldstein DS, Catecholamine metabolism: A contemporary view with implications for physiology and medicine, Pharmacol. Rev 56 (2004) 331–349. [DOI] [PubMed] [Google Scholar]
- [3].Ahlquist RP, A study of the adrenotropic receptors, Am. J. Physiol 153 (1948) 586–600. [DOI] [PubMed] [Google Scholar]
- [4].Philipp M, Hein L, Adrenergic receptor knockout mice: Distinct functions of 9 receptor subtypes, Pharmacol. Ther 101 (2004) 65–74. [DOI] [PubMed] [Google Scholar]
- [5].Paul J, Bylund B, Lefkowitz J, Eikenburg C, Ruffolo R, Langer Z, Minneman P, International union of pharmacology X. Recommendation for nomenclature of adrenoreceptors: consensus update, Am. Soc. Pharmacol. Exp. Ther 47 (1995) 267–270. [PubMed] [Google Scholar]
- [6].Sastre M, García‐Sevilla JA, α2‐Adrenoceptor subtypes identified by [3H]RX821002 binding in the human brain: The agonist guanoxabenz does not discriminate different forms of the predominant α2A subtype, J. Neurochem 63 (1994) 1077–1085. [DOI] [PubMed] [Google Scholar]
- [7].Uys MM, Shahid M, Harvey BH, Therapeutic potential of selectively targeting the α2C -adrenoceptor in cognition, depression, and schizophrenia—New developments and future perspective, Front. Psychiatry. 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bücheler MM, Hadamek K, Hein L, Two α2-adrenergic receptor subtypes, α2A and α2C, inhibit transmitter release in the brain of gene-targeted mice, Neuroscience. 109 (2002) 819–826. [DOI] [PubMed] [Google Scholar]
- [9].Ordway GA, Effect of noradrenergic lesions on subtypes of α2‐adrenoceptors in rat brain, J. Neurochem 64 (1995) 1118–1126. [DOI] [PubMed] [Google Scholar]
- [10].Sallinen J, Haapalinna A, MacDonald E, Viitamaa T, Lähdesmäki J, Rybnikova E, Pelto-Huikko M, Kobilka BK, Scheinin M, Genetic alteration of the α2-adrenoceptor subtype c in mice affects the development of behavioral despair and stress-induced increases in plasma corticosterone levels, Mol. Psychiatry. 4 (1999) 443–452. [DOI] [PubMed] [Google Scholar]
- [11].Sallinen J, Holappa J, Koivisto A, Kuokkanen K, Chapman H, Lehtimäki J, Piepponen P, Mijatovic J, Tanila H, Virtanen R, Sirviö J, Haapalinna A, Pharmacological characterisation of a structurally novel α2C-adrenoceptor antagonist ORM-10921 and its effects in neuropsychiatric models, Basic Clin. Pharmacol. Toxicol 113 (2013) 239–249. [DOI] [PubMed] [Google Scholar]
- [12].Luoto P, Suilamo S, Oikonen V, Arponen E, Helin S, Herttuainen J, Hietamäki J, Holopainen A, Kailajärvi M, Peltonen JM, Rouru J, Sallinen J, Scheinin M, Virta J, Virtanen K, Volanen I, Roivainen A, Rinne JO, 11C-ORM-13070, a novel PET ligand for brain α2C-adrenoceptors: radiometabolism, plasma pharmacokinetics, whole-body distribution and radiation dosimetry in healthy men, Eur. J. Nucl. Med. Mol. Imaging. 41 (2014) 1947–1956. [DOI] [PubMed] [Google Scholar]
- [13].Haass-Koffler CL, Swift RM, Leggio L, Noradrenergic targets for the treatment of alcohol use disorder, Psychopharmacology (Berl). 235 (2018) 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gilsbach R, Hein L, Are the pharmacology and physiology of α2 adrenoceptors determined by α2-heteroreceptors and autoreceptors respectively?, Br. J. Pharmacol 165 (2012) 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sallinen J, Höglund I, Engström M, Lehtimäki J, Virtanen R, Sirviö J, Wurster S, Savola JM, Haapalinna A, Pharmacological characterization and CNS effects of a novel highly selective α2C-adrenoceptor antagonist JP-1302, Br. J. Pharmacol 150 (2007) 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Alluri SR, Kim SW, Volkow ND, Kil K, PET radiotracers for CNS-adrenergic receptors: Developments and perspectives, Molecule. 25 (2020) 4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Van der Mey M, Windhorst AD, Klok RP, Herscheid JDM, Kennis LE, Bischoff F, Bakker M, Langlois X, Heylen L, Jurzak M, Leysen JE, Synthesis and biodistribution of [11C]R107474, a new radiolabeled α2-adrenoceptor antagonist, Bioorganic Med. Chem 14 (2006) 4526–4534. [DOI] [PubMed] [Google Scholar]
- [18].Nahimi A, Jakobsen S, Munk OL, Vang K, Phan JA, Rodell A, Gjedde A, Mapping α2 adrenoceptors of the human brain with 11C-yohimbine, J. Nucl. Med 56 (2015) 392–398. [DOI] [PubMed] [Google Scholar]
- [19].Jakobsen S, Pedersen K, Smith DF, Jensen SB, Munk OL, Cumming P, Detection of α2 -adrenergic receptors in brain of living pig with 11C-yohimbine, J. Nucl. Med 47 (2006) 2008–2015. [PubMed] [Google Scholar]
- [20].Smith DF, Stork BS, Wegener G, Ashkanian M, Jakobsen S, Bender D, Audrain H, Vase KH, Hansen SB, Videbech P, Rosenberg R, [11C]mirtazapine binding in depressed antidepressant nonresponders studied by PET neuroimaging, Psychopharmacology (Berl). 206 (2009) 133–140. [DOI] [PubMed] [Google Scholar]
- [21].Marthi K, Bender D, Gjedde A, Smith DF, [11C]Mirtazapine for PET neuroimaging: Radiosynthesis and initial evaluation in the living porcine brain, Eur. Neuropsychopharmacol. 12 (2002) 427–432. [DOI] [PubMed] [Google Scholar]
- [22].Prabhakaran J, Majo VJ, Milak MS, Mali P, Savenkova L, Mann JJ, Parsey RV, Kumar JSD, Synthesis and in vivo evaluation of [11C]MPTQ: A potential PET tracer for alpha2A-adrenergic receptors, Bioorganic Med. Chem. Lett 20 (2010) 3654–3657. [DOI] [PubMed] [Google Scholar]
- [23].Uys M, Shahid M, Sallinen J, Dreyer W, Cockeran M, Harvey BH, The α2C-adrenoceptor antagonist, ORM-10921, has antipsychotic-like effects in social isolation reared rats and bolsters the response to haloperidol, Prog. Neuro-Psychopharmacology Biol. Psychiatry. 71 (2016) 108–116. [DOI] [PubMed] [Google Scholar]
- [24].Lehto J, Hirvonen MM, Johansson J, Kemppainen J, Luoto P, Naukkarinen T, Oikonen V, Arponen E, Rouru J, Sallinen J, Scheinin H, Vuorilehto L, Finnema SJ, Halldin C, Rinne JO, Scheinin M, Validation of [11C]ORM-13070 as a PET tracer for alpha2c-adrenoceptors in the human brain, Synapse. 69 (2015) 172– 181. [DOI] [PubMed] [Google Scholar]
- [25].Arponen E, Helin S, Marjamäki P, Grönroos T, Holm P, Löyttyniemi E, Någren K, Scheinin M, Haaparanta-Solin M, Sallinen J, Solin O, A PET tracer for brain α2C adrenoceptors, 11C-ORM-13070: Radiosynthesis and preclinical evaluation in rats and knockout mice, J. Nucl. Med 55 (2014) 1171–1177. [DOI] [PubMed] [Google Scholar]
- [26].Shahid M, Rinne JO, Scheinin M, Virta J, Marjamäki P, Solin O, Arponen E, Sallinen J, Kuokkanen K, Rouru J, Application of the PET ligand [11C]ORM-13070 to examine receptor occupancy by the α2C-adrenoceptor antagonist ORM-12741: translational validation of target engagement in rat and human brain, EJNMMI Res 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Din Belle D, Holm P, Tolvanen A, Wohlfahrt G, Aryl pieprazine and their use as alpha2C antagonists, WO 2010/058060 A1, (2010). [Google Scholar]
- [28].Wang S, Kumpulainen E, Pystynen J, Pohjakallio A, Haikarainen A, Preparation of 2-(1-heteroarylpiperazin-4-yl)methyl-1,4-benzodioxane derivatives useful as α2C antagonists, WO 2016/193551 Al, (2016). [Google Scholar]
- [29].Haikarainen A, Kumpulainen E, Pohjakallio A, Pystynen J, Wang S, Benzodioxane derivatives and Their pharmaceutical use, WO 2018/002437 A1, (2018). [Google Scholar]
- [30].Pollastri MP, Overview on the rule of five, Curr. Protoc. Pharmacol (2010) 1–8. [DOI] [PubMed] [Google Scholar]
- [31].Uhlen AS, Dambrova M, Nasman J, Schioth HB, Gu Y, Wikberg-Matsson A, Wikberg JES, [3H]RS79948–197 binding to human, rat, guinea pig and pig α2A-, α2B- and and α2C-adrenoceptors. Comparison with MK912, RX821002, rauwolscine and yohimbine, Eur. J. Pharmacol 343 (1998) 93–101. [DOI] [PubMed] [Google Scholar]
- [32].Tanner MA, Thomas TP, Grisanti LA, Death receptor 5 contributes to cardiomyocyte hypertrophy through epidermal growth factor receptor transactivation, J. Mol. Cell. Cardiol 136 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.