

Abstract
Mechanobiology is essential for cardiovascular structure and function and regulates the normal physiological and pathological processes of the cardiovascular system. Cells in the cardiovascular system are extremely sensitive to their mechanical environment, and once mechanical stimulation is abnormal, the homeostasis mechanism is damaged or lost, leading to the occurrence of pathological remodeling diseases. In the past 20 years, many articles concerning the mechanobiology of cardiovascular homeostasis and remodeling have been published. To better understand the current development status, research hotspots and future development trends in the field, this paper uses CiteSpace software for bibliometric analysis, quantifies and visualizes the articles published in this field in the past 20 years, and reviews the research hotspots and emerging trends. The regulatory effects of mechanical stimulation on the biological behavior of endothelial cells, smooth muscle cells and the extracellular matrix, as well as the mechanical-related remodeling mechanism in heart failure, have always been research hotspots in this field. This paper reviews the research advances of these research hotspots in detail. This paper also introduces the research status of emerging hotspots, such as those related to cardiac fibrosis, homeostasis, mechanosensitive transcription factors and mechanosensitive ion channels. We hope to provide a systematic framework and new ideas for follow-up research on mechanobiology in the field of cardiovascular homeostasis and remodeling and promote the discovery of more therapeutic targets and novel markers of mechanobiology in the cardiovascular system.
Keywords: Mechanobiology, Cardiovascular homeostasis, Cardiovascular remodeling, Bibliometrics, Research status, Research trends
1. Introduction
Mechanobiology focuses on how mechanical stimulation interacts with chemical signals to regulate cell and tissue functions and their molecular mechanisms. With the stress-growth law proposed by Y.C. Fung in 1990, which mentioned that the remodeling of blood vessels involving the growth or resorption of cells and extracellular materials is linked to mechanical stresses,1 researchers have gradually turned their attention to the mechanism of the mechanical regulation of life processes. With the accelerated development of molecular biology and cell biology, a new interdisciplinary field, mechanobiology, has gradually emerged. Mechanobiology studies not only reveal the biomechanical mechanisms of normal growth, development and aging but also clarify the role of mechanical stimulation in the pathogenesis of diseases.2 Although the vast majority of cells in the body are subjected to mechanical stimulation, most current research on mechanobiology focuses on the cardiovascular system.3, 4, 5 The cardiovascular system, which is centered on the heart, pumps blood out of the heart to the entire body through the heart's mechanical pumping action, achieving energy transfer and material exchange. This process involves many mechanical effects, including the mechanical force generated by the beating of the heart and the mechanical stimulation of blood pressure and flow through blood vessels. Mechanical stimulation is a mechanical signal that is the main regulator of cell structure and function. It causes disturbances in the position and morphology of cells and the vascular microenvironment. Mechanosensitive sensors on cell membranes can sense these mechanical changes and transduce them into chemical signals, activating multiple signaling pathways and responding by remodeling, growth, and repair.6
Cardiovascular homeostasis and remodeling have always been research hotspots in the field of cardiovascular diseases. Cardiovascular homeostasis is a physiological state in which cardiovascular physiological functions as well as internal and external environmental compositions and properties are maintained in a relatively dynamic balance, whereas cardiovascular remodeling is a dynamic pathological process.7 Mechanical stimulation is a key regulator of cardiovascular homeostasis and pathological remodeling.8 Under physiological conditions, the cardiovascular system senses mechanical stimulation and then maintains the relative balance of cardiovascular composition, structure and function through various regulatory mechanisms to achieve cardiovascular homeostasis.9 When mechanical stimulation is abnormal or the cardiovascular mechanosensation or mechanotransduction capacity is impaired, pathological cardiovascular remodeling can occur.9, 10, 11 Different cardiovascular cells and components respond differently to abnormal mechanical stimulation, causing different cardiovascular remodeling events. Pathological remodeling changes in the cardiovascular system are manifested in both structure and function. Structural changes include intimal hyperplasia, thickening or thinning of the media, adventitial fibrosis, and ventricular concentric hypertrophy,10,12 which in turn affect functional changes manifested as changes in vascular compliance and vascular regulation disorders.9,13 The biological processes involved include the synthesis, degradation, and reorganization of the extracellular matrix (ECM), as well as the excessive proliferation, migration, and apoptosis of endothelial cells (ECs) and vascular smooth muscle cells (VSMCs).14, 15, 16 Pathological mechanical stimulation or mechanotransduction induces cardiovascular remodeling leading to a range of cardiovascular diseases, including hypertension, atherosclerosis (AS), heart failure (HF), and restenosis after vein grafting.17, 18, 19 Therefore, in cardiovascular homeostasis and remodeling, a deep understanding of the biological effects of mechanical stimulation and how to produce biological effects is of great theoretical and clinical importance for identifying potential mechanics-related drug targets and new biomarkers and laying a mechanobiological foundation for the prevention and treatment of pathological cardiovascular remodeling diseases.
Currently, with the continuous development of mechanobiology in cardiovascular homeostasis and remodeling, the number of related articles has increased rapidly. Therefore, it is necessary to conduct a retrospective analysis of published articles in this field to better understand the status of past research and provide guidance for future research. Bibliometrics is a branch of informatics. It quantitatively analyzes relevant information in the scientific literature to understand the knowledge structure and research trends in a certain research field. It is an effective tool for objectively studying the current status of a discipline and reflecting its development.20 Recently, many reviews related to bibliometrics have been published.21, 22, 23 By using bibliometric analysis software to construct a knowledge graph, a comprehensive literature review of the proposed research dimension is conducted. It usually takes scientific publications as input, quickly organizes many published articles, generates interactive visualization maps of complex structures, visualizes the structure and dynamics of a certain research field, and then analyzes the results to determine the main research topics and frontiers.24 CiteSpace is a bibliometric software for visual analysis of scientific literature. It presents the structure, laws and distribution of scientific knowledge by drawing a series of visual maps, helping researchers to better understand the development context of the research field.25 This article uses CiteSpace bibliometric analysis software to conduct a bibliometric analysis of the field of mechanobiology in cardiovascular homeostasis and remodeling, comprehensively analyzes the development status and trends of this field, better elucidates the current research hotspots and emerging trends in this field, and promotes the development of this field.
2. Bibliometric methods
2.1. Data collection and retrieval strategy
The Web of Science is the world's largest information resource platform, covering the fields of natural sciences, engineering technology, biomedicine, social sciences, arts and humanities. It also includes the world's highest-level authoritative academic journals, monographs, conferences, abstracts and conference proceedings with powerful citation indexing and academic influence evaluation functions. In this work, the Web of Science Core Collection (WoSCC) is used as the data source, and the data are searched from 2004 to 2024 November as the time period. The search terms used were ALL=((cardiac OR vascular OR artery OR arterial OR vessel OR cardiovascular) AND (remodel∗ OR homeostasis)) AND (mechanobiology OR mechanics). A total of 2872 studies were retrieved, and 2796 studies were analyzed via bibliometrics after the duplicate articles were deleted. Standard bibliometric indicators were used to analyze the number of published articles, countries, institutions, authors, cited references, and keywords in CiteSpace 6.4.R1.
2.2. Bibliometric analysis tool - CiteSpace
This study selected 6.4.R1 from CiteSpace and used 2796 articles retrieved from WoSCC as the data source. This analysis included 2212 articles, 354 reviews, 95 proceedings papers, etc (Fig. 1). We Select January 2004 to November 2024 for time slicing, with each slice set to 1 year, the selection standard Top N set to 50, and the node types selected as author, institution, country, keyword, and reference to visualize the obtained valid literature data. The analysis results include annual publication analysis, national and institutional cooperation analysis, author analysis and citation explosion keywords. The relevant content is visualized in an information graph, and the key nodes and their connections in different graphs are analyzed. The higher the frequency of occurrence in the graph is, the larger the node. The color and thickness of the inner circle of a node represent the frequency of occurrence at different times, whereas the purple outer circle of the node indicates its high betweenness centrality, expressing the intermediary connection role of the node in the collinear network, which can be considered a turning point and plays a key role in the information graph. The connecting lines between nodes are represented as co-occurrence or co-citation relationships, and the thickness of the connecting lines indicates the strength of co-occurrence. The more connections there are and the thicker the connecting lines between nodes are, the closer the relationship is. Finally, the research hotspots are visually displayed through an information graph, and research trends are predicted.
Fig. 1.

Types of publications in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
3. Bibliometric analysis
3.1. Annual publication volume analysis
Since 2004, there have been a total of 2796 research articles in this field, increasing from 30 in 2004 to over 240, showing an overall upward trend. In 2021, more than 200 papers were published, reaching its peak. Within two years, the number of published articles remained at approximately 240. As of the search date, the number of papers published in 2024 was 197. The regression curves predict that the number of publications in this field will be 245 in 2024 (Fig. 2). With increasing social attention given to cardiovascular disease and the continuous development and improvement of strategies for disease modeling and treatment, this field has also received widespread attention. The stable upward trend of published articles indicates that this field has become a hot research topic in recent years and has attracted widespread attention.
Fig. 2.

Number of publications in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
3.2. Collaborative relationship analysis
3.2.1. Country/Regional cooperation network
A total of 82 nodes and 893 connections of the country cooperation network map were obtained after visualized analysis with "country" as the node. Each node represents a country. The larger the node is, the more articles published by that country. The top five countries are the United States, China, Italy, the United Kingdom, and Germany(Fig. 3). The United States has the highest number of publications, with a total of 1449 articles (Fig. 4). Through CiteSpace visualization, it is possible to analyze the cooperative relationships between countries (Fig. 3), where the multiple and dense connections between countries indicate close cooperation between them. The cooperation between countries is as follows. The United States has close ties with China, the United Kingdom, Italy, Germany, Canada, France, and many of the countries shown (Fig. 3); China has close ties with South Africa, Britain, Germany, Canada, France and other European and American countries, as well as Japan, South Korea and other Asian countries; Italy has close ties with France, Greece, Canada, the Netherlands and Australia; Britain has close ties with Switzerland, Singapore, Greece, Spain and India; and Germany has close ties with Belgium, Ireland, South Korea, Japan and South Africa. The top five countries in terms of centrality are the United States (0.35), South Africa (0.11), England (0.07), Egypt (0.07), and Slovenia (0.06). The centrality of America and South Africa is greater than 0.1, and the high centrality in the national cooperation network also indicates that these 2 countries have a certain influence on cardiovascular homeostasis and remodeling in the field of mechanobiology.
Fig. 3.

Network diagram of the effects of national cooperation in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
Fig. 4.
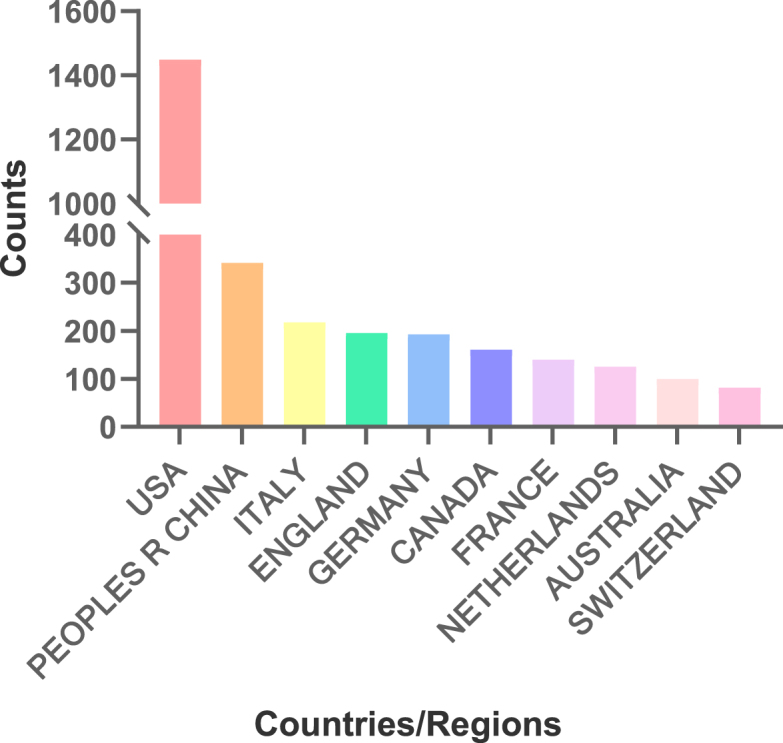
Top 10 countries in the field of mechanobiology in cardiovascular homeostasis and remodeling in terms of publication volume.
3.2.2. Institutional cooperation network
The information graph of articles published by institutions in various countries from 2004 to 2024 includes 441 nodes and 2906 connections. According to the institutional cooperation network (Fig. 5), the top ten institutions with the greatest number of publications can be obtained, among which the top five institutions are the University of California System, Harvard University, Yale University, Pennsylvania Commonwealth System of Higher Education (PCSHE), and University System of Georgia (Table 1). In the network diagram of institutional cooperation (Fig. 5), University of California System and University of California Davis, University of California Berkeley, University of California Irvine, Austral University, University of Illinois System, University of California San Francisco, University of Illinois Chicago Hospital have close ties in the field of mechanobiology in cardiovascular homeostasis and remodeling. Harvard University works closely with Beth Israel Deaconess Medical Center, Harvard Medical School, and the Broad Institute. Yale University and Texas A&M University College Station, Texas A&M University System, Universitat Politecnica de Valencia and Universite de Montpellier have close links in this field. Pennsylvania Commonwealth System of Higher Education (PCSHE) and the University of Pittsburgh, the Mayo Clinic, the University of Pittsburgh Palermo, Boston Children's Hospital, the University of Limerick, Pennsylvania State University, Pennsylvania State University and Duke University cooperate and exchange. There are close ties between the University System of Georgia and the Georgia Institute of Technology, the US Department of Veterans Affairs, the Veterans Health Administration (VHA), Augusta University, the Atlanta VA Medical Center, and the Atlanta VA Health Care System.
Fig. 5.

Network diagram of institutional cooperation in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
Table 1.
Top 9 institutions in terms of publication volume from 2004 to 2024.
| Institution | Counts | Countries |
|---|---|---|
| University of California System | 159 | USA |
| Harvard University | 118 | USA |
| Yale University | 98 | USA |
| Pennsylvania Commonwealth System of Higher Education (PCSHE) | 93 | USA |
| University System of Georgia | 92 | USA |
| University of Texas System | 86 | USA |
| Georgia Institute of Technology | 85 | USA |
| Institut National de la Sante et de la Recherche Medicale (Inserm) | 84 | FRANCE |
| Stanford University | 76 | USA |
The CiteSpace visualization of institutional collaboration network graphs reveals that the nodes of institutions such as University of Maryland Baltimore (0.27), CNRS - National Institute for Biology (INSB) (0.24), Johns Hopkins University (0.22), Brigham & Women's Hospital (0.2), National Institutes of Health (NIH) - USA (0.2), University of Toronto (0.19), University of California System (0.18), and Centre National de la Recherche Scientifique (CNRS) (0.17) all have purple outer circles, indicating that these institutions have high centrality and play important roles in research and development in this field (Fig. 5). Currently, the main research in this field is focused on schools, with few hospitals conducting related research. Approximately ninety percent of the top nine institutions in terms of publication volume are from the United States, which has a certain leading position in research in this field.
3.2.3. Author collaboration network
Through the analysis of the number of articles published by authors in the field of mechanobiology in cardiovascular homeostasis and remodeling, as well as the collaboration network, the collaboration network diagram includes 744 nodes and 1144 connections. The size of the nodes in the collaboration diagram represents the number of articles published by the authors, and the thickness of the connection lines reflects the strength of their collaboration relationship. According to the institutional cooperation network (Fig. 6), the top ten authors with the highest number of publications can be obtained, among which the top five authors are Jay D. Humphrey, and Marijana. Tadic, Dao Wen. Wang, Cesare. Cuspidi, Rudolph L. Gleason. Nearly half of the top 10 authors in the past 20 years are from the United States (Table 2).
Fig. 6.

Author collaboration network in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
Table 2.
Top 10 authors in terms of publication volume from 2004 to 2024.
| Authors | Counts | Affiliations | Countries |
|---|---|---|---|
| Jay D. Humphrey | 61 | Yale University | USA |
| Marijana. Tadic | 25 | Tadic, Marijana | GERMANY |
| Dao Wen. Wang | 24 | Union Hospital, Tongji Medical College, Huazhong University of Science and Technology | PEOPLES R CHINA |
| Cesare. Cuspidi | 21 | Università Milano-Bicocca | ITALY |
| Rudolph L. Gleason | 20 | Georgia Institute of Technology | USA |
| Hai-Chao. Han | 17 | University of Texas at San Antonio | USA |
| Seungik. Baek | 16 | Michigan State University | USA |
| Chen. Chen | 16 | Xidian University | PEOPLES R CHINA |
| David A. Vorp | 15 | University of Pittsburgh Swanson School of Engineering | USA |
| Gerhard A. Holzapfel | 15 | Graz University of Technology | AUSTRIA |
The cooperative network diagram closely related to national and institutional cooperation is different. The cooperative relationships among scholars are relatively scattered, and scholars are closely connected on a small scale (Fig. 6). David A. Vorp and Jay D. Humphrey jointly published information about abdominal aortic aneurysms and biomechanical behavior research.26 Marijana Tadic and Cesare. Cuspidi closely collaborated in the field of vascular research. Together, they have participated in a number of studies in the areas of hypertension, cardiac mechanics, and cardiology. Specifically in patients with hypertension, they collaborated to investigate the relationship between left atrial enlargement (LAE) and changes in heart structure and function, as well as the impact of these changes on prognosis.27 Moreover, the prevalence of LAE and its association with left ventricular hypertrophy are also discussed in untreated and treated hypertension patients.28 These collaborative results not only improve the understanding of vascular disease mechanisms, but also provide valuable insights for clinical practice.
The collaboration between Seungik Vorp. Baek and David A., in the field of vascular research, are reflected mainly in the joint research projects and published academic papers. They have collaborated on a number of occasions, including a special issue of Annals of Biomedical Engineering exploring cardiovascular biomechanics and biofluids, in particular, modeling cardiovascular structures.29 In addition, they collaborated on the publication of research on multiscale modeling of blood vessels via a fluid‒solid growth framework, which involves advances in medical imaging, vascular biology, and biomechanics and is expected to play an increasingly important role in clinical decision-making processes. Their collaboration highlights the importance of applying biomechanics and biofluidics to the progression of vascular disease and shows how to analyze aneurysm enlargement or wall stress through computational models. These collaborative results not only promote an understanding of the biomechanics of vascular diseases but also provide a new perspective and method for clinical treatment. The cooperative work of Chen, Chen and Wang, Dao Wen focused mainly on the mechanical pathogenic mechanism involved in the occurrence and development of HF and revealed a series of mechanically regulated signaling pathways.30, 31, 32 The roles of miR-665, mammalian target protein complex 1 (mTORC1) and phosphodiesterase 5 (PDE5) in heart failure are closely related to the biomechanical properties of the heart. MiR-665 affects cardiac function by affecting phosphatase and tensin cognates, mTORC1 affects vascular tone and blood pressure via Yes-associated proteins, and PDE5 inhibition prevents cardiac fibrosis by affecting Smad signaling, all of which are important components of cardiac biomechanics research. Understanding how these molecules and signaling pathways regulate the mechanical properties of the heart could provide new strategies for the treatment of heart disease.
3.3. Research hotspot analysis
High-frequency keywords often reflect research hotspots in the field. To further study the current research hotspots and trends of mechanobiology in cardiovascular homeostasis and remodeling, a keyword co-occurrence network diagram (Fig. 7) was generated through CiteSpace, which contains 597 nodes and 6016 connections. The top 10 most frequently occurring keywords included "extracellular matrix", "shear stress", "smooth muscle cells", "heart failure", "endothelial cells", "blood pressure", "blood flow", "computational fluid dynamics", "myocardial infarction", and "hypertension", indicating that these are the most studied directions by researchers in the past 20 years. The research hotspots in the field of cardiovascular homeostasis and remodeling in mechanobiology influence each other. The fluid shear stress generated by blood flow mainly acts on ECs, and the mechanical load caused by blood pressure mainly acts on SMCs and cardiomyocytes. These cardiovascular cells regulate the remodeling process of the ECM in a paracrine manner. Cardiovascular cells can also influence each other in a paracrine manner. These findings indicate that cardiovascular remodeling and homeostasis constitute a complex overall process that is inseparable from the division of labor and cooperation among various cardiovascular components. On the basis of the top five most frequent keywords in the keyword co-occurrence network map, the following focuses on the role of the biological behavior of ECs, smooth muscle cells and the ECM under mechanical stimulation in cardiovascular remodeling and homeostasis, as well as the mechanical regulatory mechanism in the occurrence and development of HF.
Fig. 7.

Cooccurrence of key words in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
3.3.1. Mechanobiology of the endothelial cell response to shear stress
ECs, as the cells in direct contact with blood in vessels, are continuously exposed to hemodynamics and play an important role in the regulation of cardiovascular homeostasis and remodeling by mechanical stimulation, which has become the focus of researchers. Shear stress caused by blood flow is the main mechanical force sensed by ECs.17,33 ECs have excellent mechanosensitive properties and mechanotransduction ability and can continuously sense the magnitude, pulsation and direction of shear stress.8,34,35 ECs convert mechanical signals into intracellular biochemical signals through specific mechanosensitive sensors, such as ion channels, integrins, the glycocalyx and adhesion molecules.33,36 This signal transduction process causes changes in the morphology, structure, function and gene expression of ECs and subsequently affects cardiovascular homeostasis or remodeling.37, 38, 39 The hemodynamics of the cardiovascular system is complex, and shear stress manifests itself in many forms. In the vertical part of the cardiovascular system, blood flow is in a single direction, and shear stress manifests itself as laminar shear stress(LSS),40 whereas in arterial bifurcations, bends, or valves, where blood flow is weaker and direction changes are complex, shear stress manifests itself as low oscillatory shear stress(OSS).41,42 Different forms of shear stress stimulate ECs and cause different biological processes. Many studies have reported the protective effects of physiological LSS on endothelial function, including promoting the formation of tight cell junctions in ECs and exerting normal barrier functions.43,44 It also regulates the balance of vasodilator and vasomotor substances secreted by ECs to maintain normal vascular tension.45,46 Many studies have also investigated the role of LSS in maintaining the anti-proliferative, anti-inflammatory and anti-thrombotic phenotypes of ECs.47, 48, 49, 50 Recent studies have also shown that LSS promotes EC polarization and directional migration by regulating the remodeling of adhesion junctions between ECs and plays an important role in repairing and maintaining vascular integrity after vascular injury.51
Sustained stimulation by pathological shear stress induces endothelial dysfunction leading to cardiovascular pathological remodeling diseases, of which atherosclerosis is the most common and studied disease.17,38,52, 53, 54 Shear stress is the main cause of the focal distribution of atherosclerotic lesions.55,56 In AS-susceptible areas, ECs are exposed to OSS and low shear stress, increasing the expression of proinflammatory factors and cell adhesion factors57, 58, 59 and activating the oxidative pathways that cause EC damage and the subsequent formation and development of AS lesions.58,60, 61, 62 In addition, OSS or low shear stress disrupts the barrier function of ECs63 and promotes the deposition of lipids and inflammatory cells under the endothelium, leading to inward remodeling of the vascular wall and causing the occurrence of atherosclerotic stenosis.64,65 Similarly, pathological shear stress is also a major cause of heart valve disease,66, 67, 68 and there is a spatial correlation between the location of the lesion and hemodynamics. Studies have shown that lipid deposition and calcification of the aortic valve are more likely to occur on the aortic side,69, 70, 71 and shear stress manifests as low OSS, which may be related to the fact that low OSS promotes EC damage.72,73 Moreover, abnormal shear stress in heart valve ECs promotes endothelial–mesenchymal transformation (EndMT), causing pathological remodeling of the aortic valve and leading to valvular dysfunction.74 Low OSS plays an important role in the pathological remodeling process of aortic aneurysms.75, 76, 77 Aortic aneurysms are in a disturbed blood flow environment. ECs are chronically stimulated by low OSS, and the pathological phenotype is activated, which manifests as increased oxidative stress, EndMT, and increased EC permeability.78,79 In addition, ECs of the pathological phenotype secrete a variety of key growth factors and matrix-regulating proteins that regulate ECM remodeling and SMC phenotypic transformation,80 and these processes are associated with aortic aneurysm growth and rupture.80,81 Hemodynamic disturbances are also a major cause of poor prognosis in some surgical procedures such as angioplasty and stent implantation, with altered hemodynamics in the postprocedural region manifesting as low OSS, which promotes neoplastic endothelial proliferation by inducing EC injury.82, 83, 84, 85 Abnormal shear stress-induced EC dysfunction is also the primary cause of vascular stenosis after arterial bypass grafting.86 When veins are suddenly exposed to the shear stress in the arterial system, and the magnitude of the shear stress is much greater than the shear stress previously carried by venous ECs, EC inflammation and apoptosis are included, promoting subsequent angiogenesis and intimal hyperplasia.87, 88, 89 In summary, the effects of hemodynamics on EC physiology and pathology have been extensively studied and will continue to be a major focus in the future. Elucidating the role of blood flow patterns in EC dysfunction can provide insights into the pathogenesis of lesions in specific regions of the cardiovascular system and help develop therapeutic strategies based on hemodynamics.
3.3.2. Mechanobiology in the extracellular matrix
The ECM is a three-dimensional structural entity within the cardiovascular system that mainly includes elastin, collagen, glycosaminoglycans (GAGs) and related proteoglycans. It is the main component of the cardiovascular microenvironment,90 providing mechanical support for cells and maintaining the normal structure‒function relationship of the cardiovascular system.91,92 It is also the main conductor of mechanical force within the cardiovascular system, transmitting key signals to vascular cells, cardiomyocytes and interstitial cells.93, 94, 95 Under physiological conditions, the ECM remains relatively stable to maintain normal cardiovascular function.96 In pathological vascular remodeling events caused by abnormal mechanical forces, ECM remodeling is a key link, which manifests as changes in the composition of matrix proteins, matrix metalloproteinase-mediated matrix protein degradation, and changes in the topological characteristics of the ECM.97, 98, 99 When blood vessels are exposed to LSS, the ECM composition is altered to exhibit pro-inflammatory and pro-tissue repair phenotypes, promoting the generation of an intravascular inflammatory microenvironment.100,101 Increased circumferential stress can also trigger changes in the ECM components of the vascular wall, manifested as the breakage of elastic fibers and excessive deposition of collagen,102, 103, 104, 105 while also affecting the rearrangement of matrix proteins and proteoglycans.105,106 The topological structure of the ECM can also change due to pressure overload.105,107 The pathological remodeling of the ECM can lead to changes in vascular structure and function, manifested as thickening of the arterial wall, increased vascular stiffness, decreased compliance, and increased peripheral resistance.90,106,108 A study revealed that the speed of the blood pressure pulse wave also affects the composition of the ECM, indicating that, in hypertensive patients, the relative content of collagen is positively correlated with the speed of the pulse wave, whereas the relative content of elastin is negatively correlated with it.109 The occurrence and development of aortic aneurysms are related to changes in the ECM.110, 111, 112 Pathological remodeling of the ECM in aortic aneurysms can affect the strength of the vascular wall. When the wall stress exceeds the wall strength, aortic dissection or rupture occurs.113 Mechanical stimulation can also affect the ECM remodeling of heart valves. OSS increases the production of collagen and GAG.66,114 Both cyclic stretching and pressure overload lead to remodeling of the ECM of the aortic valve, causing degenerative aortic valve disease.115,116 Changes in the composition and structure of the ECM can also affect the response of cardiovascular cells to mechanical stimulation. Fibronectin can increase the mechanical sensitivity of cells to high shear stress, whereas the presence of collagen types I and IV and laminin can improve the mechanical sensitivity of cells to low shear stress.117 In addition, the topological characteristics of the ECM help to resist the inflammatory and proliferative activation of ECs by turbulent flow.101 In Marfan syndrome mice, the ECM structure of the heart valves is destroyed, and the proper structure-function relationship of the heart cannot be maintained under mechanical stimulation, resulting in pathological remodeling of the heart valves and myxomatous valvular disease.92 In summary, ECM remodeling under mechanical stimulation has been a popular topic that researchers have explored over the past 20 years. The discovery of new mechanisms and perspectives in this field is a continuous improvement in the understanding of the pathogenesis of cardiovascular pathological remodeling diseases.
3.3.3. Mechanobiology in vascular smooth muscle cells
VSMCs are the main cellular components of the tunica media of the vascular wall and are mainly subjected to mechanical stimulation generated by pulsatile blood pressure in the form of cyclical stretch.118 VSMCs are also important participants in vascular homeostasis and remodeling under mechanical stimulation.119, 120, 121 VSMCs have a high degree of plasticity. Under vascular homeostasis, VSMCs exhibit a contractile phenotype to maintain normal vascular tension and function.14,122 However, in a hemodynamic environment that promotes pathological vascular remodeling, VSMCs undergo phenotypic switching and differentiate into a more migratory, secretory, and proliferative synthetic phenotype, promoting the formation of vascular lesions.123, 124, 125 An increasing number of studies have shown that mechanical stimulation regulates VSMC phenotypic switching through various mechanisms.126 In the event of an acute increase in blood pressure, smooth muscle cells contract rapidly to maintain normal wall stress.127 In arterial remodeling induced by chronically high blood pressure, the contribution of vascular smooth muscle is most pronounced, including chronic vascular constriction and luminal narrowing due to dysfunction and aberrant proliferation of SMCs, as well as vascular fibrosis resulting from the dedifferentiation of SMCs into myofibroblasts.126,128,129 In addition, vascular smooth muscle is a key regulator of ECM remodeling after increased circumferential stress. It participates in vascular remodeling by secreting a variety of matrix metalloproteinases to regulate the remodeling of the ECM,130,131 causing pathological remodeling of conduction arteries and resistance arteries.130 The abnormal adaptation of SMCs to increased circumferential stress is also the main cause of pathological remodeling in aortic aneurysms.132,133 In addition, VSMCs respond to mechanical stimulation of increased blood pressure by altering cell adhesion and the expression of ECM molecules.134,135 In a healthy state, VSMCs are not directly exposed to shear stress. When atherosclerotic lesions rupture or invasive techniques such as angioplasty cause damage to ECs, VSMCs are directly exposed to blood flow, and receiving shear stress stimulation, leading to corresponding changes in intracellular molecular signaling and function. Physiological LSS inhibits the proliferation of VSMCs by increasing the expression of nitric oxide synthase and AMP-activated protein kinase phosphorylation.136 However, low shear stress and OSS promote the proliferation and migration of VSMCs to the intima, resulting in an EC phenotype, with a significant increase in the expression of EC markers,137 which contributes to the formation of focal neointima and promotes the formation of atherosclerotic stenosis lesions.138, 139, 140 After endarterectomy, the exposure of VSMCs to high LSS leads to increased apoptosis, which helps reduce restenosis after vascular injury.141 Similarly, in vascular grafts, high shear stress can inhibit smooth muscle cell proliferation and reduce neointima formation.124 In summary, the phenotypic switching of VSMCs under mechanical stimulation has always been a hot area of research for researchers, and its mechanism of action may be a potential therapeutic target in the future.
3.3.4. Mechanobiology in heart failure
HF is a clinical syndrome and the endpoint of remodeling in most cardiovascular diseases.142 Clinical manifestations include impaired cardiac pumping function and the inability of cardiac output to meet the metabolic needs of systemic tissues. Pathological features include ventricular enlargement and relative thinning of the ventricular wall.143 In the early stage of mechanical overload, the heart is able to maintain cardiac output and cardiac systolic and diastolic function to maintain cardiovascular homeostasis through compensatory mechanisms of myocardial hypertrophy. However, as mechanical overload persists, cardiac remodeling becomes decompensated hypertrophy, accompanied by changes in structure, molecules, metabolism, and the ECM, leading to cardiac dysfunction and eventually HF.144 Both pressure and volume overload are important pathogenic factors of HF,145 leading to progressive remodeling of ventricular geometry and myocardial structure. These remodeling processes affect the mechanical properties of the heart, which in turn aggravates the cardiac remodeling process and eventually results in HF. Excessive ECM deposition is observed in HF induced by pressure overload, contributing to ventricular wall thickening,146,147 whereas increased ECM degradation contributes to ventricular wall thinning and ventricular cavity dilation in HF induced by volume overload.148 In the context of myocardial hypertrophy and dysfunction caused by pressure overload, many studies have shown that inflammation is an important pathogenic mechanism that causes cardiac dysfunction by inducing cardiomyocyte death and damage.149, 150, 151, 152 In addition, mitochondrial dysfunction is a key pathogenic factor. Pressure overload promotes the development of HF by downregulating mitochondrial autophagy and causing mitochondrial dysfunction.153 Similarly, the activation of the cardiac immune response mechanism is also involved in adverse cardiac remodeling and HF caused by mechanical overload.154 Studies have shown that under the action of hemodynamic mechanical forces, metabolic remodeling of the heart precedes structural remodeling. Pressure overload reduces the expression of cardiac pyruvate kinase muscle isoenzymes, reduces the utilization of glucose by cardiomyocytes, and subsequently leads to cardiomyocyte death, aggravating myocardial dysfunction and fibrosis and ultimately causing HF.155 Recent studies have shown that hypoxia can protect against the development of cardiac hypertrophy and HF caused by pressure overload by increasing the expression of hypoxia-inducible factor-1α and reducing the expression of genes related to pathological remodeling.156 Many studies have also been conducted on the pathological remodeling mechanisms of HF volume overload. During volume overload, the left ventricle undergoes extensive remodeling, manifested as chamber dilation, increased myocardial wall stress, and progressive myocardial cell contractile dysfunction. The mechanisms involved include myocardial and EC apoptosis, altered cardiac fibroblast function, and extensive ECM remodeling.148 Volume overload also leads to the upregulation of metabolic genes related to myocardial stress and glycolysis in the ventricle.157 Hypertension and adverse ventricular remodeling after myocardial infarction can lead to HF. Hemodynamic changes after acute myocardial infarction cause pathological remodeling of cardiomyocytes, leading to left ventricular systolic and diastolic dysfunction.158 In hypertension, enhanced cyclic stretch induces a hypertrophic response in cardiomyocytes by inducing NF-kB activation and increasing VEGF secretion, promoting pathological myocardial remodeling.159 Research on the mechanobiology of heart failure has focused mainly on the influence of mechanical regulatory mechanisms under volume overload and pressure overload on the biological behavior of cardiac cells and ECM, which will continue to be a direction of interest in the future.
3.4. Emerging trends analysis
Keywords with the strongest citation bursts (Fig. 8) can indicate emerging research hotspots and provide a basis for exploring future research trends and cutting-edge topics. In the research on mechanobiology in cardiovascular homeostasis and remodeling, the keywords whose citation bursts have continued to the present are responses, homeostasis, fibrosis, and mechanisms, indicating that these four keywords may represent future research trends. In recent years, the mechanical regulatory mechanisms of cardiovascular remodeling and homeostasis have been continuously discovered, among which mechanosensitive transcription factors and mechanosensitive ion channel piezo1 have received the most attention. In the past, researchers focused their attention on the effects of mechanical stimulation on cardiovascular remodeling. In recent years, researchers have gradually focused on the role of mechanical regulation in cardiovascular homeostasis. In cardiac remodeling, cardiac fibrosis has recently attracted increasing attention from researchers, and mechanical-related regulatory mechanisms have been continuously discovered. The following is a detailed introduction to the research progress of these emerging research hotspots in the mechanobiology of cardiovascular remodeling and homeostasis.
Fig. 8.

Top 20 keywords with the strongest citation bursts in the field of mechanobiology in cardiovascular homeostasis and remodeling from 2004 to 2024.
3.4.1. Mechanotransduction transcription factors
A core mechanism by which mechanical force affects cell behavior involves changes in gene expression, and various mechanosensitive transcription factors play key roles in this process. Mechanical stimulation regulates gene expression by activating different mechanosensitive transcription factors, thereby affecting the function of cardiovascular cells.160,161 These include Krüppel-like factor 2, Krüppel-like factor 2, NF-kB and Yes associated protein (YAP)/WW domain-containing transcription regulator 1 (TAZ, also known as WWTR1)/TEA-domain transcription factor (TEAD).162, 163, 164, 165 In recent years, researchers have focused on the effect of mechanical stimulation on YAP/TAZ/TEAD and its role in cardiovascular homeostasis and remodeling and have obtained many valuable findings. YAP/TAZ, mechanosensitive transcriptional coactivators that regulate the TEAD family of transcription factors, are the final effector molecules downstream of Hippo signaling.166 It plays an integral role in the development of the cardiovascular system and the maintenance of cardiovascular homeostasis.167, 168, 169 Many studies have shown that it is the most important mechanosensitive transcription factor activated by disturbance,170, 171, 172, 173 and can shuttle between the cytoplasm and the nucleus to respond to mechanotransduction.172 Changes in cell morphology, rigidity and topology of the ECM, shear stress and stretching can affect the distribution of YAP within the cell and the corresponding biological effects.174 The role of YAP in the mechanical conduction of ECs has been continuously studied and has attracted increasing attention. Studies have shown that LSS can inhibit YAP activity in ECs, thereby inhibiting inflammation and playing an antiatherosclerotic role, whereas OSS induces YAP activation and translocation into the nucleus of ECs, leading to increased EC proliferation and monocyte adhesion factor expression, resulting in the occurrence of key pathogenic events in atherosclerosis, such as intimal thickening and foam cell formation.173,175 These studies have laid a theoretical foundation for the use of YAP as a therapeutic target for mechanically stimulated atherosclerotic diseases. In myocardial ischemia‒reperfusion injury, LSS exerts a protective effect on microvascular ECs by activating YAP and alleviating myocardial injury.176 In hypertension, blood pressure overload increases YAP nuclear translocation in vascular ECs, promoting oxidative stress and inhibiting NO bioavailability, leading to endothelial dysfunction.177 In heart valves, YAP activation is also affected by mechanical forces, affecting valve growth and remodeling.67 In addition to ECs, YAP/TAZ also play important roles in the mechanotransduction of smooth muscle cells. Proper YAP/TAZ mechanotransduction in VSMCs is a key factor in maintaining the contractile phenotype and function of vascular smooth muscle and is essential for maintaining vascular homeostasis. In pressure overload, SMC-specific YAP/TAZ loss leads to decreased vasocontractility, impaired myogenic response, and increased compliance.178,179 In addition, YAP/TAZ loss also affects the regulation of ECM remodeling by SMCs under pressure overload, which manifests as reduced assembly of elastic fibers and increased degradation of elastin, leading to the formation of aneurysms.180,181 Pathological mechanical stretch downregulates enhancer of zeste homolog 2 via activation of the YAP-TEAD1 pathway, thereby exacerbating VSMC apoptosis and vascular remodeling.182 These studies suggest that the balance of YAP/TAZ activation represents the balance of vascular cell function and that the deletion or increased nuclear translocation of YAP/TAZ causes the activation of downstream pathogenic signaling pathways, leading to the occurrence of vascular pathological remodeling. At present, the role of YAP/TAZ in cardiovascular homeostasis and remodeling is still in the stage of continuous in-depth study. In the future, many studies are needed to elucidate its specific mechanism of action and explore the regulatory factors affecting its activation/inactivation balance to provide a new perspective for the treatment of cardiovascular remodeling diseases.
3.4.2. Piezo1 in mechanobiology
Piezo1 is a mechanosensitive cationic channel on the cell membrane that responds to different mechanical forces and converts them into intracellular signals, participating in the occurrence and development of cardiovascular remodeling diseases.183, 184, 185 In recent years, its pathogenic mechanism in cardiovascular remodeling induced by mechanical stimulation has been continuously explored and enriched. In the heart, piezo1 is a key mechanical sensor that initiates mechanical-chemical signal transduction to cause cardiac remodeling.186,187 Abnormal mechanical stretch upregulates piezo1 in cardiomyocytes, and the mechanical signal is converted into an intracellular oxidative stress signal.188 Piezo1 is an important regulator of stretch-induced the expression of genes related to cardiac remodeling in cardiac fibroblasts.189 After myocardial infarction, abnormal mechanical stress and mechanical load on the myocardium cause piezo1 upregulation, which induces pathological ventricular remodeling by activating multiple signaling pathways, including those that mediate inflammatory cascades190 and disrupt intracellular calcium cycling dynamics in cardiomyocytes.191 In pulmonary hypertension, enhanced cyclic stretch promotes endothelial–mesenchymal transition and stimulates SMC proliferation by upregulating piezo1,192 whereas high shear stress caused by high flow causes the upregulation of piezo1 in ECs and activates endothelial inflammation.193 The pressure overload-induced activation of piezo1 causes platelet activation and thrombosis, promoting the occurrence of hypertensive complications.194 In addition, under pressure overload, piezo1 upregulation disrupts cardiomyocyte calcium homeostasis causing myocardial hypertrophic remodeling.195 The role of piezo1 in AS has been extensively studied, and many new discoveries concerning the role of piezo1 in hemodynamically related atherosclerosis have recently been reported. The upregulation of piezo1 under OSS causes the activation and nuclear translocation of YAP, thereby mediating endothelial dysfunction and promoting the formation of atherosclerotic lesions.196, 197, 198 In advanced atherosclerotic lesions, the activation of piezo1 is associated with increased plaque stability. The activation of piezo1 promotes foam cell apoptosis, reduces the expression of inflammatory factors, and increases the collagen content.199 In addition, piezo1 has been found to be the target of many traditional Chinese medicines for the treatment of atherosclerosis which delay the progression of atherosclerotic lesions by inhibiting the expression of piezo1.198,200 Activated neutrophils release neutrophil extracellular traps (NETs) to mediate inflammation, which plays an important role in the formation of early atherosclerotic lesions. A recent study revealed that low shear stress downregulates piezo1 to promote the generation of NETs.201 In the response of SMCs to increased ECM rigidity, piezo1 is among the key mechanotransduction molecules whose activation causes an increase in SMC volume.202 The discovery of these new mechanisms has provided some new insights into the mechanotransduction function of piezo1, which is a continuous improvement in the biological function of piezo1 and further enhances its status in cardiovascular diseases related to mechanical regulation.
3.4.3. Cardiovascular homeostasis
In the past, researchers have focused mainly on the effects of mechanical stimulation on cardiovascular remodeling, but in recent years researchers have focused on the role of mechanical regulation in cardiovascular homeostasis. Mechanical homeostasis is the most important aspect of cardiovascular homeostasis and requires a combination of mechanical stimulation and mechanotransduction induced biological responses to maintain it.9,203 The regulation of homeostasis is characterized by negative feedback regulation. In the cardiovascular system, when mechanical stimulation deviates from the optimal value or set value, the corresponding negative feedback regulatiory mechanism is activated to restore the system to the normal range.204,205 Blood pressure and flow homeostasis are essential for the physiological function of the cardiovascular system to ensure normal cardiovascular perfusion to cope with changing metabolic and physiological needs.206 When surgical operations or changes in tissue perfusion cause blood flow to increase or decrease, ECs sense that shear stress deviates from the set point. ECs respond quickly by regulating vasoconstriction or dilation through corresponding mechanisms, thereby changing the diameter of the blood vessels, maintaining blood flow shear stress within the physiological range, and maintaining cardiovascular homeostasis.206 When blood pressure increases acutely, the first to sense it in arterioles are SMCs, which maintain local hemodynamic homeostasis through myogenic contraction.179,207 The homeostasis of the ECM is also important for cardiovascular homeostasis, which is reflected in the balance of quantity, composition, assembly and mechanical properties.107,208 In different layers of the vascular wall, the components of the ECM also differ in terms of site specificity, which is a prerequisite for the normal physiological function of the cardiovascular system in the mechanical environment.209 Most ECM components are not static but rather dynamically changing, constantly produced, degraded and assembled. In this process, the production and degradation of the ECM maintain a relatively balanced state,95 and these relatively balanced states are the prerequisites for maintaining mechanical properties.107 The state of cells is also important in the regulation of cardiovascular homeostasis, and this state is reflected in the phenotype of cells. Changes in the cell phenotype affect the perception of mechanical stimulation, thereby affecting the biological processes caused by subsequent mechanical transduction. ECs are the barrier cells of the cardiovascular system, and maintaining the normal phenotype of ECs is crucial for maintaining cardiovascular homeostasis.210 Studies have shown that a more flexible ECM promotes longer elongation and tighter cell junctions in ECs under LSS, and promotes the production of nitric oxide, indicating that the quality of the ECM is related to the phenotype of ECs. Furthermore, suggests that the homeostasis of the interaction between the ECM and ECs is crucial for the maintenance of vascular homeostasis under hemodynamic conditions.211 Maintaining the contractile phenotype of VSMC is beneficial for resisting the pathological remodeling of the ECM caused by pressure overload and preventing the occurrence of aortic aneurysms.212,213 Mitochondria are the energy centers of cells, and their homeostasis is crucial for the maintaining of the contractility phenotype of SMCs.214 The homeostasis of the interaction between SMCs and the ECM can maintain normal vascular compliance and prevent arteriosclerosis.118 Recent studies have shown that maintaining glutathione homeostasis in SMCs can prevent VSMC dysfunction and is an important means to improve pathological remodeling of the ECM in atherosclerosis and offset arterial stiffness.215 In the future, the mechanical regulatory mechanisms of cardiovascular homeostasis will be revealed by an increasing number of studies, and the focus will gradually be refined from overall cardiovascular homeostasis to the homeostasis of the components of the cardiovascular system as well as the interactions among the components.
3.4.4. Mechanically regulated cardiac fibrosis
Cardiac fibrosis is the pathophysiological basis of HF.151,216 In recent years, many studies have investigated the role of mechanical regulation in cardiac fibrosis. Cardiac fibrosis is significantly related to hemodynamic changes. Pathological hemodynamics drives cardiac fibroblasts to differentiate into myofibroblasts through mechanotransduction,217, 218, 219 causing excessive accumulation of ECM components and secretion of pro-fibrotic factors.220, 221, 222 Myocardial mechanical overload has been confirmed to be the main driver of myocardial fibrosis in living myocardial slices that preserve structure and function.223 Abnormalities in cardiac mechanics can also promote cardiac fibrosis. In patients with mitral valve prolapse, myocardial fibrosis was found to be associated with impaired myocardial longitudinal strain caused by mitral valve prolapse.224 Another study revealed that decreased myocardial mechanical parameters, such as global longitudinal strain, circumferential strain and myocardial strain, can reflect the severity of myocardial fibrosis.225 Activated fibroblasts and myofibroblasts are the main cellular effectors of cardiac fibrosis.226,227 Studies have shown that ECs may also acquire a fibrotic phenotype through endothelial–mesenchymal transition under mechanical overload conditions, thereby promoting cardiac fibrosis.228 In pressure overload mediated cardiac fibrosis, macrophage-mediated inflammation also plays a role in the differentiation of fibroblasts into myofibroblasts.229 Many studies have confirmed that there is two-way communication between fibroblasts and the ECM. The mechanical properties of the ECM affect the behavior of fibroblasts, and the state of fibroblasts will also regulates the remodeling of the ECM. There is also a positive feedback loop between the two.230,231 A decrease in the viscoelasticity of the ECM and an increase in linear elasticity will activate the mechanically sensitive pathway and cause fibroblast activation.232,233 Activated fibroblasts are the main source of ECM proteins, causing increased ECM remodeling.167,234 Cardiac fibroblasts can also sense mechanical stress through mechanosensitive receptors, ion channels, and integrins, activating the intracellular fibrosis cascade and leading to fibrosis caused by pressure overload.235, 236, 237, 238, 239 The transformation of cardiac fibroblasts into myofibroblasts under pressure overload is significantly associated with intracellular metabolic changes, manifested by increased aerobic glycolysis and lactate production.240 A recent study revealed that pressure overload activates reactive oxygen species production pathways to induce mitochondrial oxidative damage and promote myocardial fibrosis.241 Exosomes also play important roles in cardiac fibrosis caused by pressure overload. Enhanced mechanical stretch induces cardiomyocytes to regulate the activation of cardiac fibroblasts through an exosome-mediated paracrine mechanism, thereby promoting cardiac fibrosis.242 Crosstalk between fibroblasts and cardiomyocytes plays an important role in the process of cardiac fibrosis. A recent study revealed that during volume overload-induced cardiac fibrosis, integrinbeta-like1 is upregulated in fibroblasts, promoting cardiomyocyte hypertrophy.243 Mechanical stimulation is an important driving factor of cardiac fibrosis. Although the mechanical-related pathogenic mechanism has received much attention from researchers in recent years, there is still much room for research in the future, including mechanism regulating the crosstalk between cardiomyocytes and fibroblasts under mechanical stimulation, as well as the positive feedback regulation between fibroblast activation and ECM remodeling.
4. Summary and prospects
In this study, we used CiteSpace bibliometric analysis software to analyze the scientific literature concerning the mechanobiology of cardiovascular homeostasis and remodeling over the past 20 years and quantitatively and visually reviewed the research achievements and progress in this field. First, we conducted a quantitative analysis of basic information such as annual publication volume, authors, institutions, and countries. Second, we conducted a co-occurrence analysis of keywords, obtained hot keywords in the field, and summarized the research progress of hot keywords. Finally, we analyzed the citation burst keywords, obtained the emerging research directions in the field, and explained its current research status.
Cardiovascular disease is currently the leading cause of death worldwide, and most cardiovascular diseases are associated with pathological remodeling caused by mechanical stimulation.244,245 Over the past 20 years, owing to the continuous in-depth research on cardiovascular mechanobiology in various countries and the continuous development of technical means such as cell biology, molecular biology, in vitro mechanical stimulation models and mechanical simulation software, the number of published articles in this field has steadily increased. Among them, endothelial dysfunction induced by abnormal shear stress is the most concerning aspect for researchers, and many studies have been conducted on this topic. The focus of the research is this regulatory mechanism of abnormal stress on EC phenotypes. A large number of mechanosensitive signaling cascades have been discovered, and a deep understanding of these mechanosensitive signaling molecules will provide new potential targets for the treatment of cardiovascular pathological remodeling diseases. In the cardiovascular system, biological processes under mechanical stimulation are regulated by interactions between components within the system, including interactions between cardiovascular cells and the interaction between the ECM and cardiovascular cells. In addition, under mechanical stimulation, there are interactions between vascular cells that mediate vascular homeostasis. When blood flow increases and shear stress increases, ECs secrete vasodilator cytokines, inducing rapid relaxation of VSMCs and expansion of the arterial diameter, thereby restoring wall shear stress to the initial level.246 The ECM can also initiate mechanical signaling through matrix–cell interactions to affect the behavior and function of vascular cells, playing a vital role in cardiovascular homeostasis and remodeling.99,118,204 The quality and quantity of the ECM also affect the stiffness of blood vessels.107 ECM–cell mechanotransduction interactions are mediated by focal adhesions and integrins. Focal adhesions connect the ECM and the cytoskeleton, and integrins transmit information about the chemical composition and mechanical state of the ECM to cells.247,248 The most critical aspect of the effects of abnormal mechanical stimulation on cardiovascular cells, the ECM and the interactions between them is the process of cellular mechanotransduction.58,100 Mechanosensitive channels and mechanosensitive transcription factors play important roles in mechanotransduction and act as intermediate hubs for mechanical stimulation to elicit biological effects in cells.161 Cardiovascular remodeling diseases such as hypertension and HF usually occur under abnormal mechanical stimulation through positive feedback. Abnormal mechanical stimulation activates related signaling pathways, such as inflammation, oxidative stress, and immunity, causing pathological remodeling, such as vascular stiffness and myocardial hypertrophy, and this structural and functional remodeling further promotes the activation of pathogenic signaling pathways.249 Among these research hotspots and emerging research directions, mechanobiology has been continuously improved in cardiovascular homeostasis and remodeling, more mechanisms will be discovered in the future, and more potential therapeutic targets are expected to be identified in the clinic.
Although mechanobiology-related research on cardiovascular homeostasis and remodeling has made good progress in the past 20 years, there are still some challenges and difficulties in the future development process, which need to be solved and explored by researchers in the future. (1) More in depth, multiomics, and multidimensional research should be conducted on the mechanobiology of cardiovascular homeostasis and remodeling. Research progress on the role of mechanical stimulation in EC and VSMC dysfunction has revealed that its mechanism of action and effects, including mechanical sensing and mechanical transduction, have been continuously studied in depth. An increasing number of membrane-localized mechanical sensors and intracellular signaling molecule pathways have been discovered.250,251 Conducting more in-depth and comprehensive research will provide a theoretical basis for the development of new drugs and enable these findings to truly enter the clinic, which is highly important for improving the precision treatment of cardiovascular remodeling diseases. (2) The transmission of mechanical force within blood vessels requires physically interconnected structures, such as the ECM, focal adhesions, cytoskeleton and cell nucleus.99,252 The interaction mechanisms involved will require more research in the future, which will help to fully understand the mechanobiology of cardiovascular homeostasis and remodeling. (3) Currently, a complete quantitative system for evaluating the severity of lesions via mechanical stimulation is lacking. Many articles have reported the role of mechanical stimulation, such as pressure, circumferential wall stress, and shear stress, in diseases such as atherosclerosis, hypertension, and HF. However, it is still difficult to obtain accurate values at the lesion site, which limits its clinical application. Currently, most of them are simulated through computational fluid dynamics (CFD).253,254 The accuracy of the numerical values depends largely on the accuracy of CFD modeling, which limits the precise quantification of the relationship between mechanical stimulation and the severity of lesions. In the future, technology can be continuously improved to improve the accuracy of computational fluid dynamics simulations. In combination with medical imaging, advanced computational fluid dynamics and flow field testing technologies, cardiovascular system modeling and quantitative analysis can be carried out to explore new noninvasive detection technologies for cardiovascular diseases and personalized treatment and surgical design to provide mechanobiology solutions for the diagnosis, treatment and early warning of cardiovascular diseases.
CRediT authorship contribution statement
Wei Liao: Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Yuxi Huang: Visualization, Software, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xiangxiu Wang: Visualization, Software, Methodology, Investigation. Ziqiu Hu: Software, Investigation. Chuanrong Zhao: Writing – review & editing, Writing – original draft, Visualization, Resources, Project administration, Methodology, Investigation, Funding acquisition, Conceptualization. Guixue Wang: Writing – review & editing, Writing – original draft, Resources, Project administration, Funding acquisition, Conceptualization.
Ethical approval
This study does not contain any studies with human or animal subjects performed by any of the authors.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by the National Key Technology R & D Program of China (No. 2023YFB3810100), the National Natural Science Foundation of China (No. 12032007, No. 32201071, No. 12302406), the Science and Technology Innovation Project of JinFeng Laboratory, Chongqing, China (No. jfkyjf202203001), and the Fundamental Research Funds for the Central Universities (No. 2024CDJCGJ-016, No. 2023CDJYGRH-ZD03), Chongqing Traditional Chinese Medicine Innovation Team Construction Project (No. [2022]33). The author thanks Professor Chaojun Tang from Soochow University for his enthusiastic assistance in revising the language of this paper and the support from the Public Experiment Center of State Bioindustrial Base (Chongqing), China.
Footnotes
This article is part of a special issue entitled: Cardiovascular Mechano published in Mechanobiology in Medicine.
Contributor Information
Chuanrong Zhao, Email: zhaocr@cqu.edu.cn.
Guixue Wang, Email: wanggx@cqu.edu.cn.
References
- 1.Fung Y-c. Springer Science & Business Media; 2013. Biomechanics: Motion, Flow, Stress, and Growth. [Google Scholar]
- 2.Jiang Z.-L. Mechanobiology research in China. Mechanobiology in Medicine. 2023;1(1) [Google Scholar]
- 3.Wang Y., Shyy J.Y., Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu Rev Biomed Eng. 2008;10:1–38. doi: 10.1146/annurev.bioeng.010308.161731. [DOI] [PubMed] [Google Scholar]
- 4.Zhao C., Wang X., Wang G. Hot topics and emerging trends in mechanobiology research. Sichuan Da Xue Xue Bao Yi Xue Ban. 2024;55(1):1–5. doi: 10.12182/20240160104. (in Chinese) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H.Z.C., Wang G. Advances in vascular biomechanics and mechanobiology. Medical Biomechanics. 2024;39(1):17–23. (in Chinese) [Google Scholar]
- 6.Martinac B., Nikolaev Y.A., Silvani G., et al. Cell membrane mechanics and mechanosensory transduction. Curr Top Membr. 2020;86:83–141. doi: 10.1016/bs.ctm.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Weber K.T., Anversa P., Armstrong P.W., et al. Remodeling and reparation of the cardiovascular system. J Am Coll Cardiol. 1992;20(1):3–16. doi: 10.1016/0735-1097(92)90130-f. [DOI] [PubMed] [Google Scholar]
- 8.Baeyens N., Schwartz M.A. Biomechanics of vascular mechanosensation and remodeling. Mol Biol Cell. 2016;27(1):7–11. doi: 10.1091/mbc.E14-11-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humphrey J.D., Schwartz M.A. Vascular mechanobiology: homeostasis, adaptation, and disease. Annu Rev Biomed Eng. 2021;23:1–27. doi: 10.1146/annurev-bioeng-092419-060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura M., Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 11.Wang X., Khalil R.A. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol. 2018;81:241–330. doi: 10.1016/bs.apha.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein L.W. Atherosclerosis regression, vascular remodeling, and plaque stabilization. J Am Coll Cardiol. 2007;49(2):271–273. doi: 10.1016/j.jacc.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 13.Schiffrin E.L. How structure, mechanics, and function of the vasculature contribute to blood pressure elevation in hypertension. Can J Cardiol. 2020;36(5):648–658. doi: 10.1016/j.cjca.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Chen R., McVey D.G., Shen D., Huang X., Ye S. Phenotypic switching of vascular smooth muscle cells in atherosclerosis. J Am Heart Assoc. 2023;12(20) doi: 10.1161/JAHA.123.031121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohindra R., Agrawal D.K., Thankam F.G. Altered vascular extracellular matrix in the pathogenesis of atherosclerosis. J Cardiovasc Transl Res. 2021;14(4):647–660. doi: 10.1007/s12265-020-10091-8. [DOI] [PubMed] [Google Scholar]
- 16.Souilhol C., Harmsen M.C., Evans P.C., Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res. 2018;114(4):565–577. doi: 10.1093/cvr/cvx253. [DOI] [PubMed] [Google Scholar]
- 17.Chiu J.J., Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91(1):327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia M., Li Q., Guo J., et al. Deletion of BACH1 attenuates atherosclerosis by reducing endothelial inflammation. Circ Res. 2022;130(7):1038–1055. doi: 10.1161/CIRCRESAHA.121.319540. [DOI] [PubMed] [Google Scholar]
- 19.Thompson A.A.R., Lawrie A. Targeting vascular remodeling to treat pulmonary arterial hypertension. Trends Mol Med. 2017;23(1):31–45. doi: 10.1016/j.molmed.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Chen C., Hu Z., Liu S., Tseng H. Emerging trends in regenerative medicine: a scientometric analysis in CiteSpace. Expet Opin Biol Ther. 2012;12(5):593–608. doi: 10.1517/14712598.2012.674507. [DOI] [PubMed] [Google Scholar]
- 21.Liu X., Zhao S., Tan L., et al. Frontier and hot topics in electrochemiluminescence sensing technology based on CiteSpace bibliometric analysis. Biosens Bioelectron. 2022;201 doi: 10.1016/j.bios.2021.113932. [DOI] [PubMed] [Google Scholar]
- 22.Tan L., Wang X., Yuan K., et al. Structural and temporal dynamics analysis on drug-eluting stents: history, research hotspots and emerging trends. Bioact Mater. 2023;23:170–186. doi: 10.1016/j.bioactmat.2022.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan K., Deng C., Tan L., et al. Structural and temporal dynamics analysis of zinc-based biomaterials: history, research hotspots and emerging trends. Bioact Mater. 2024;35:306–329. doi: 10.1016/j.bioactmat.2024.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X., Liu Y. Visualization analysis of high-speed railway research based on CiteSpace. Article. Transp Policy. 2020;85:1–17. [Google Scholar]
- 25.Chen C. Searching for intellectual turning points: progressive knowledge domain visualization. Proc. Natl. Acad. Sci. U.S.A. 2004;101(Suppl 1):5303–5310. doi: 10.1073/pnas.0307513100. Suppl 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krawiec J.T., Vorp D.A. Adult stem cell-based tissue engineered blood vessels: a review. Biomaterials. 2012;33(12):3388–3400. doi: 10.1016/j.biomaterials.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Cuspidi C., Giovannini R., Tadic M. Left atrial stiffness: a novel marker of hypertension-mediated organ damage on the horizon? Hypertens Res. 2021;44(3):365–367. doi: 10.1038/s41440-020-00582-1. [DOI] [PubMed] [Google Scholar]
- 28.Tadic M., Cuspidi C., Grassi G., Ivanovic B. Gender-specific therapeutic approach in arterial hypertension - challenges ahead. Pharmacol Res. 2019;141:181–188. doi: 10.1016/j.phrs.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 29.Finol E.A., Di Martino E.S., Baek S. Cardiovascular biomechanics and biofluids: a special issue with a focus on modeling of cardiovascular structures. Ann Biomed Eng. 2013;41(7):1309–1310. [Google Scholar]
- 30.Fan J., Zhang X., Nie X., et al. Nuclear miR-665 aggravates heart failure via suppressing phosphatase and tensin homolog transcription. Sci China Life Sci. 2020;63(5):724–736. doi: 10.1007/s11427-018-9515-1. [DOI] [PubMed] [Google Scholar]
- 31.Yao L., He J., Li B., et al. Regulation of YAP by mammalian target of rapamycin complex 1 in endothelial cells controls blood pressure through COX-2/mPGES-1/PGE(2) cascade. Hypertension. 2019;74(4):936–946. doi: 10.1161/HYPERTENSIONAHA.119.12834. [DOI] [PubMed] [Google Scholar]
- 32.Gong W., Yan M., Chen J., Chaugai S., Chen C., Wang D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor beta-induced Smad signaling. Front Med. 2014;8(4):445–455. doi: 10.1007/s11684-014-0378-3. [DOI] [PubMed] [Google Scholar]
- 33.Davies P.F. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75(3):519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C., Baker B.M., Chen C.S., Schwartz M.A. Endothelial cell sensing of flow direction. Arteriosclerosis, thrombosis, Vasc Biol. 2013;33(9):2130–2136. doi: 10.1161/ATVBAHA.113.301826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blackman B.R., Garcia-Cardena G., Gimbrone M.A., et al. A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J Biomech Eng. 2002;124(4):397–407. doi: 10.1115/1.1486468. [DOI] [PubMed] [Google Scholar]
- 36.Orr A.W., Helmke B.P., Blackman B.R., Schwartz M.A. Mechanisms of mechanotransduction. Dev Cell. 2006;10(1):11–20. doi: 10.1016/j.devcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 37.Hamrangsekachaee M., Wen K., Bencherif S.A., Ebong E.E. Atherosclerosis and endothelial mechanotransduction: current knowledge and models for future research. Am J Physiol Cell Physiol. 2023;324(2):C488–C504. doi: 10.1152/ajpcell.00449.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X., Shen Y., Shang M., Liu X., Munn L.L. Endothelial mechanobiology in atherosclerosis. Cardiovasc Res. 2023;119(8):1656–1675. doi: 10.1093/cvr/cvad076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chatterjee S. Endothelial mechanotransduction, redox signaling and the regulation of vascular inflammatory pathways. Front Physiol. 2018;9:524. doi: 10.3389/fphys.2018.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6(1):16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karau K.L., Krenz G.S., Dawson C.A. Branching exponent heterogeneity and wall shear stress distribution in vascular trees. Am J Physiol Heart Circ Physiol. 2001;280(3):H1256–H1263. doi: 10.1152/ajpheart.2001.280.3.H1256. [DOI] [PubMed] [Google Scholar]
- 42.Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol. 2017;219(2):382–408. doi: 10.1111/apha.12725. [DOI] [PubMed] [Google Scholar]
- 43.Tarbell J.M. Shear stress and the endothelial transport barrier. Cardiovasc Res. 2010;87(2):320–330. doi: 10.1093/cvr/cvq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang F., Zhang Y., Zhu J., et al. Laminar flow protects vascular endothelial tight junctions and barrier function via maintaining the expression of long non-coding RNA MALAT1. Front Bioeng Biotechnol. 2020;8:647. doi: 10.3389/fbioe.2020.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo F., Li X., Peng J., et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann Biomed Eng. 2014;42(9):1978–1988. doi: 10.1007/s10439-014-1033-5. [DOI] [PubMed] [Google Scholar]
- 46.Pan S. Molecular mechanisms responsible for the atheroprotective effects of laminar shear stress. Antioxidants Redox Signal. 2009;11(7):1669–1682. doi: 10.1089/ars.2009.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang J., Pu Y., Zhang H., et al. KLF2 mediates the suppressive effect of laminar flow on vascular calcification by inhibiting endothelial BMP/SMAD1/5 signaling. Circ Res. 2021;129(4):e87–e100. doi: 10.1161/CIRCRESAHA.120.318690. [DOI] [PubMed] [Google Scholar]
- 48.Psefteli P.M., Kitscha P., Vizcay G., et al. Glycocalyx sialic acids regulate Nrf2-mediated signaling by fluid shear stress in human endothelial cells. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishii T., Warabi E., Mann G.E. Mechanisms underlying unidirectional laminar shear stress-mediated Nrf2 activation in endothelial cells: Amplification of low shear stress signaling by primary cilia. Redox Biol. 2021;46 doi: 10.1016/j.redox.2021.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Sun H.Y., Kumar S., Puerta M.D.M., Jo H., Rezvan A. ZBTB46 is a shear-sensitive transcription factor inhibiting endothelial cell proliferation via gene expression regulation of cell cycle proteins. Lab Invest. 2019;99(3):305–318. doi: 10.1038/s41374-018-0060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bougaran P., Bats M.L., Delobel V., et al. ROR2/PCP a new pathway controlling endothelial cell polarity under flow conditions. Arteriosclerosis, thrombosis, Vasc Biol. 2023;43(7):1199–1218. doi: 10.1161/ATVBAHA.123.319106. [DOI] [PubMed] [Google Scholar]
- 52.Gimbrone M.A., Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y., Qiu J., Luo S., et al. High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis. Regen Biomater. 2016;3(4):257–267. doi: 10.1093/rb/rbw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu J., Lei D., Hu J., et al. Effect of intraplaque angiogenesis to atherosclerotic rupture-prone plaque induced by high shear stress in rabbit model. Regen Biomater. 2017;4(4):215–222. doi: 10.1093/rb/rbx007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.J M., L N., A V., et al. Wall shear stress alteration: a local risk factor of atherosclerosis. Curr Atheroscler Rep. 2022;24(3):143–151. doi: 10.1007/s11883-022-00993-0. [DOI] [PubMed] [Google Scholar]
- 56.Zhou M., Yu Y., Chen R., et al. Wall shear stress and its role in atherosclerosis. Front Cardiovasc Med. 2023;10 doi: 10.3389/fcvm.2023.1083547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim S., Woo C.H. Laminar flow inhibits ER stress-induced endothelial apoptosis through PI3K/Akt-Dependent signaling pathway. Mol Cells. 2018;41(11):964–970. doi: 10.14348/molcells.2018.0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davies P.F., Civelek M., Fang Y., Fleming I. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res. 2013;99(2):315–327. doi: 10.1093/cvr/cvt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Babendreyer A., Molls L., Dreymueller D., Uhlig S., Ludwig A. Shear stress counteracts endothelial CX3CL1 induction and monocytic cell adhesion. Mediat Inflamm. 2017;2017 doi: 10.1155/2017/1515389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin X., Zhang K., Qiu J., et al. Uptake of oxidative stress-mediated extracellular vesicles by vascular endothelial cells under low magnitude shear stress. Bioact Mater. 2022;9:397–410. doi: 10.1016/j.bioactmat.2021.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Z., Liu H., Zhao X., et al. Oridonin attenuates low shear stress-induced endothelial cell dysfunction and oxidative stress by activating the nuclear factor erythroid 2-related factor 2 pathway. BMC Complement Med Ther. 2022;22(1):180. doi: 10.1186/s12906-022-03658-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z., Wang F., Kong X., Gao X., Gu Y., Zhang J. Oscillatory shear stress induces oxidative stress via TLR4 activation in endothelial cells. Mediat Inflamm. 2019;2019 doi: 10.1155/2019/7162976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qu K., Wang C., Huang L., et al. Oscillatory shear stress-induced downregulation of TET1s injures vascular endothelial planar cell polarity by suppression of actin polymerization. APL Bioeng. 2023;7(3) doi: 10.1063/5.0141289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gulino-Debrac D. Mechanotransduction at the basis of endothelial barrier function. Tissue Barriers. 2013;1(2) doi: 10.4161/tisb.24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao R., Sun R., Ye Y., et al. Low shear stress-induced blockage of autophagic flux impairs endothelial barrier and facilitates atherosclerosis in mice. Exp Cell Res. 2024;439(1) doi: 10.1016/j.yexcr.2024.114071. [DOI] [PubMed] [Google Scholar]
- 66.Dayawansa N.H., Baratchi S., Peter K. Uncoupling the vicious cycle of mechanical stress and inflammation in calcific aortic valve disease. Front Cardiovasc Med. 2022;9 doi: 10.3389/fcvm.2022.783543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang M., Lin B.Y., Sun S., Dai C., Long F., Butcher J.T. Shear and hydrostatic stress regulate fetal heart valve remodeling through YAP-mediated mechanotransduction. Elife. 2023;12 doi: 10.7554/eLife.83209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fan L., Yao D., Fan Z., et al. Beyond VICs: shedding light on the overlooked VECs in calcific aortic valve disease. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2024;178 doi: 10.1016/j.biopha.2024.117143. [DOI] [PubMed] [Google Scholar]
- 69.Richards J., El-Hamamsy I., Chen S., et al. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol. 2013;182(5):1922–1931. doi: 10.1016/j.ajpath.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weinberg E.J., Mack P.J., Schoen F.J., Garcia-Cardena G., Kaazempur Mofrad M.R. Hemodynamic environments from opposing sides of human aortic valve leaflets evoke distinct endothelial phenotypes in vitro. Cardiovasc Eng. 2010;10(1):5–11. doi: 10.1007/s10558-009-9089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun L., Rajamannan N.M., Sucosky P. Defining the role of fluid shear stress in the expression of early signaling markers for calcific aortic valve disease. PloS one. 2013;8(12) doi: 10.1371/journal.pone.0084433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pang K.T., Ghim M., Sarathchandra P., et al. Shear-mediated ALK5 expression regulates endothelial activation. Biochem Biophys Res Commun. 2023;642:90–96. doi: 10.1016/j.bbrc.2022.12.058. [DOI] [PubMed] [Google Scholar]
- 73.Parra-Izquierdo I., Sanchez-Bayuela T., Lopez J., et al. Interferons are pro-inflammatory cytokines in sheared-stressed human aortic valve endothelial cells. Int J Mol Sci. 2021;22(19) doi: 10.3390/ijms221910605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faure E., Bertrand E., Gaste A., Plaindoux E., Deplano V., Zaffran S. Side-dependent effect in the response of valve endothelial cells to bidirectional shear stress. Int J Cardiol. 2021;323:220–228. doi: 10.1016/j.ijcard.2020.08.074. [DOI] [PubMed] [Google Scholar]
- 75.Mutlu O., Salman H.E., Al-Thani H., El-Menyar A., Qidwai U.A., Yalcin H.C. How does hemodynamics affect rupture tissue mechanics in abdominal aortic aneurysm: focus on wall shear stress derived parameters, time-averaged wall shear stress, oscillatory shear index, endothelial cell activation potential, and relative residence time. Comput Biol Med. 2023;154 doi: 10.1016/j.compbiomed.2023.106609. [DOI] [PubMed] [Google Scholar]
- 76.Bappoo N., Syed M.B.J., Khinsoe G., et al. Low shear stress at baseline predicts expansion and aneurysm-related events in patients with abdominal aortic aneurysm. Circ Cardiovasc Imaging. 2021;14(12):1112–1121. doi: 10.1161/CIRCIMAGING.121.013160. [DOI] [PubMed] [Google Scholar]
- 77.Li S.T.X., Qu C. The effect of wall shear stress on atherosclerosis and aneurysm. Chin J Arteriosclerosis. 2024;32(5):451–455. (in Chinese) [Google Scholar]
- 78.Hasan M., Al-Thani H., El-Menyar A., Zeidan A., Al-Thani A., Yalcin H.C. Disturbed hemodynamics and oxidative stress interaction in endothelial dysfunction and AAA progression: focus on Nrf2 pathway. Int J Cardiol. 2023;389 doi: 10.1016/j.ijcard.2023.131238. [DOI] [PubMed] [Google Scholar]
- 79.Bjorck H.M., Du L., Pulignani S., et al. Altered DNA methylation indicates an oscillatory flow mediated epithelial-to-mesenchymal transition signature in ascending aorta of patients with bicuspid aortic valve. Sci Rep. 2018;8(1):2777. doi: 10.1038/s41598-018-20642-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van de Pol V., Kurakula K., DeRuiter M.C., Goumans M.J. Thoracic aortic aneurysm development in patients with bicuspid aortic valve: what is the role of endothelial cells? Front Physiol. 2017;8:938. doi: 10.3389/fphys.2017.00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Filiberto A.C., Spinosa M.D., Elder C.T., et al. Endothelial pannexin-1 channels modulate macrophage and smooth muscle cell activation in abdominal aortic aneurysm formation. Nat Commun. 2022;13(1):1521. doi: 10.1038/s41467-022-29233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ward M.R., Tsao P.S., Agrotis A., Dilley R.J., Jennings G.L., Bobik A. Low blood flow after angioplasty augments mechanisms of restenosis: inward vessel remodeling, cell migration, and activity of genes regulating migration. Arteri Throm Vasc Biol. 2001;21(2):208–213. doi: 10.1161/01.atv.21.2.208. [DOI] [PubMed] [Google Scholar]
- 83.Sanmartin M., Goicolea J., Garcia C., et al. Influencia de la tension de cizallamiento en la reestenosis intra-stent: estudio in vivo con reconstruccion 3D y dinamica de fluidos computacional. Rev Esp Cardiol. 2006;59(1):20–27. [PubMed] [Google Scholar]
- 84.Koskinas K.C., Chatzizisis Y.S., Antoniadis A.P., Giannoglou G.D. Role of endothelial shear stress in stent restenosis and thrombosis: pathophysiologic mechanisms and implications for clinical translation. J Am Coll Cardiol. 2012;59(15):1337–1349. doi: 10.1016/j.jacc.2011.10.903. [DOI] [PubMed] [Google Scholar]
- 85.Shishido K., Antoniadis A.P., Takahashi S., et al. Effects of low endothelial shear stress after stent implantation on subsequent neointimal hyperplasia and clinical outcomes in humans. J Am Heart Assoc. 2016;5(9) doi: 10.1161/JAHA.115.002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu C.C.F. High wall shear stress caused by arterio-venous fistula reduces neointimal hyperplasia induced by stent implantation. Chin J Arteriosclerosis. 2021;29(2):129–134. (in Chinese) [Google Scholar]
- 87.Ward A.O., Angelini G.D., Caputo M., et al. NF-kappaB inhibition prevents acute shear stress-induced inflammation in the saphenous vein graft endothelium. Sci Rep. 2020;10(1) doi: 10.1038/s41598-020-71781-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ward A.O., Caputo M., Angelini G.D., George S.J., Zakkar M. Activation and inflammation of the venous endothelium in vein graft disease. Atherosclerosis. 2017;265:266–274. doi: 10.1016/j.atherosclerosis.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 89.Jackson M.L., Bond A.R., Ascione R., Johnson J.L., George S.J. FGL2/FcgammaRIIB signalling mediates arterial shear stress-mediated endothelial cell apoptosis: implications for coronary artery bypass vein graft pathogenesis. Int J Mol Sci. 2024;25(14) doi: 10.3390/ijms25147638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin P.K., Davis G.E. Extracellular matrix remodeling in vascular disease: Defining its regulators and pathological influence. Arteriosclerosis, thrombosis, Vasc Biol. 2023;43(9):1599–1616. doi: 10.1161/ATVBAHA.123.318237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Halper J. Basic components of vascular connective tissue and extracellular matrix. Adv Pharmacol. 2018;81:95–127. doi: 10.1016/bs.apha.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 92.Gonzalez B.A., Harmeyer S.W., Song T., Sadayappan S., Yutzey K.E. Dynamic changes in mitral valve extracellular matrix, tissue mechanics and function in a mouse model of Marfan syndrome. Matrix Biol. 2024;126:1–13. doi: 10.1016/j.matbio.2024.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tschumperlin D.J. Mechanotransduction. Compr Physiol. 2011;1(2):1057–1073. doi: 10.1002/cphy.c100016. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y., Chatterjee E., Li G., Xu J., Xiao J. Force-sensing protein expression in response to cardiovascular mechanotransduction. EBioMedicine. 2024;110 doi: 10.1016/j.ebiom.2024.105412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang D., Brady T., Santhanam L., Gerecht S. The extracellular matrix mechanics in the vasculature. Nat Cardiovasc Res. 2023;2(8):718–732. doi: 10.1038/s44161-023-00311-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mongkoldhumrongkul N., Latif N., Yacoub M.H., Chester A.H. Effect of side-specific valvular shear stress on the content of extracellular matrix in aortic valves. Cardiovasc Eng Technol. 2018;9(2):151–157. doi: 10.1007/s13239-016-0280-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma Z., Mao C., Jia Y., Fu Y., Kong W. Extracellular matrix dynamics in vascular remodeling. Am J Physiol Cell Physiol. 2020;319(3):C481–C499. doi: 10.1152/ajpcell.00147.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yanagisawa H., Yokoyama U. Extracellular matrix-mediated remodeling and mechanotransduction in large vessels during development and disease. Cell Signal. 2021;86 doi: 10.1016/j.cellsig.2021.110104. [DOI] [PubMed] [Google Scholar]
- 99.Yamashiro Y., Thang B.Q., Ramirez K., et al. Matrix mechanotransduction mediated by thrombospondin-1/integrin/YAP in the vascular remodeling. Proc. Natl. Acad. Sci. U.S.A. 2020;117(18):9896–9905. doi: 10.1073/pnas.1919702117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Russo T.A., Banuth A.M.M., Nader H.B., Dreyfuss J.L. Altered shear stress on endothelial cells leads to remodeling of extracellular matrix and induction of angiogenesis. PloS one. 2020;15(11) doi: 10.1371/journal.pone.0241040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakayama K.H., Surya V.N., Gole M., et al. Nanoscale patterning of extracellular matrix alters endothelial function under shear stress. Nano Lett. 2016;16(1):410–419. doi: 10.1021/acs.nanolett.5b04028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wagenseil J.E., Mecham R.P. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res. 2012;5(3):264–273. doi: 10.1007/s12265-012-9349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kohn J.C., Lampi M.C., Reinhart-King C.A. Age-related vascular stiffening: causes and consequences. Front Genet. 2015;6:112. doi: 10.3389/fgene.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu Q., Chakravorty A., Bathgate R.A., Dart A.M., Du X.J. Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension. 2010;55(5):1260–1266. doi: 10.1161/HYPERTENSIONAHA.109.149369. [DOI] [PubMed] [Google Scholar]
- 105.Briones A.M., Arribas S.M., Salaices M. Role of extracellular matrix in vascular remodeling of hypertension. Curr Opin Nephrol Hypertens. 2010;19(2):187–194. doi: 10.1097/MNH.0b013e328335eec9. [DOI] [PubMed] [Google Scholar]
- 106.Laurent S., Boutouyrie P. The structural factor of hypertension: large and small artery alterations. Circ Res. 2015;116(6):1007–1021. doi: 10.1161/CIRCRESAHA.116.303596. [DOI] [PubMed] [Google Scholar]
- 107.Cai Z., Gong Z., Li Z., Li L., Kong W. Vascular extracellular matrix remodeling and hypertension. Antioxidants Redox Signal. 2021;34(10):765–783. doi: 10.1089/ars.2020.8110. [DOI] [PubMed] [Google Scholar]
- 108.Garoffolo G., Pesce M. Vascular dysfunction and pathology: focus on mechanical forces. Vasc Biol. 2021;3(1):R69–R75. doi: 10.1530/VB-21-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.You B.A., Shen L., Li J.F., Chen Y.G., Gu X.H., Gao H.Q. The correlation between carotid-femoral pulse wave velocity and composition of the aortic media in CAD patients with or without hypertension. Swiss Med Wkly. 2012;142 doi: 10.4414/smw.2012.13546. [DOI] [PubMed] [Google Scholar]
- 110.Jiang W.J., Ren W.H., Liu X.J., et al. Disruption of mechanical stress in extracellular matrix is related to Stanford type A aortic dissection through down-regulation of Yes-associated protein. Aging (Albany NY) 2016;8(9):1923–1939. doi: 10.18632/aging.101033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shen Y.H., LeMaire S.A., Webb N.R., Cassis L.A., Daugherty A., Lu H.S. Aortic aneurysms and dissections series. Arteriosclerosis, thrombosis, Vasc Biol. 2020;40(3):e37–e46. doi: 10.1161/ATVBAHA.120.313991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seim B.E., Holt M.F., Ratajska A., et al. Markers of extracellular matrix remodeling and systemic inflammation in patients with heritable thoracic aortic diseases. Front Cardiovasc Med. 2022;9 doi: 10.3389/fcvm.2022.1073069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miller K., Mufty H., Catlin A., et al. Is there a relationship between stress in walls of abdominal aortic aneurysm and symptoms? J Surg Res. 2020;252:37–46. doi: 10.1016/j.jss.2020.01.025. [DOI] [PubMed] [Google Scholar]
- 114.Dahal S., Huang P., Murray B.T., Mahler G.J. Endothelial to mesenchymal transformation is induced by altered extracellular matrix in aortic valve endothelial cells. J Biomed Mater Res, Part A. 2017;105(10):2729–2741. doi: 10.1002/jbm.a.36133. [DOI] [PubMed] [Google Scholar]
- 115.Balachandran K., Sucosky P., Jo H., Yoganathan A.P. Elevated cyclic stretch alters matrix remodeling in aortic valve cusps: implications for degenerative aortic valve disease. Am J Physiol Heart Circ Physiol. 2009;296(3):H756–H764. doi: 10.1152/ajpheart.00900.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Konduri S., Xing Y., Warnock J.N., He Z., Yoganathan A.P. Normal physiological conditions maintain the biological characteristics of porcine aortic heart valves: an ex vivo organ culture study. Ann Biomed Eng. 2005;33(9):1158–1166. doi: 10.1007/s10439-005-5506-4. [DOI] [PubMed] [Google Scholar]
- 117.Lai A., Thurgood P., Cox C.D., et al. Piezo1 response to shear stress is controlled by the components of the extracellular matrix. ACS Appl Mater Interfaces. 2022;14(36):40559–40568. doi: 10.1021/acsami.2c09169. [DOI] [PubMed] [Google Scholar]
- 118.Lacolley P., Regnault V., Segers P., Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev. 2017;97(4):1555–1617. doi: 10.1152/physrev.00003.2017. [DOI] [PubMed] [Google Scholar]
- 119.Liu S., Lin Z. Vascular smooth muscle cells mechanosensitive regulators and vascular remodeling. J Vasc Res. 2022;59(2):90–113. doi: 10.1159/000519845. [DOI] [PubMed] [Google Scholar]
- 120.Elmarasi M., Elmakaty I., Elsayed B., et al. Phenotypic switching of vascular smooth muscle cells in atherosclerosis, hypertension, and aortic dissection. J Cell Physiol. 2024;239(4) doi: 10.1002/jcp.31200. [DOI] [PubMed] [Google Scholar]
- 121.Miano J.M., Fisher E.A., Majesky M.W. Fate and state of vascular smooth muscle cells in atherosclerosis. Circulation. 2021;143(21):2110–2116. doi: 10.1161/CIRCULATIONAHA.120.049922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Frismantiene A., Philippova M., Erne P., Resink T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018;52:48–64. doi: 10.1016/j.cellsig.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 123.Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 124.Qiu J., Zheng Y., Hu J., et al. Biomechanical regulation of vascular smooth muscle cell functions: from in vitro to in vivo understanding. J R Soc Interface. 2014;11(90) doi: 10.1098/rsif.2013.0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shinge S.A.U., Zhang D., Achu Muluh T., Nie Y., Yu F. Mechanosensitive Piezo1 channel evoked-mechanical signals in atherosclerosis. J Inflamm Res. 2021;14:3621–3636. doi: 10.2147/JIR.S319789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Scherer C., Pfisterer L., Wagner A.H., et al. Arterial wall stress controls NFAT5 activity in vascular smooth muscle cells. J Am Heart Assoc. 2014;3(2) doi: 10.1161/JAHA.113.000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Touyz R.M., Alves-Lopes R., Rios F.J., et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114(4):529–539. doi: 10.1093/cvr/cvy023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiao X., Yu H., Du Z., et al. Vascular smooth muscle cells specific deletion of angiopoietin-like protein 8 prevents angiotensin II-promoted hypertension and cardiovascular hypertrophy. Cardiovasc Res. 2023;119(9):1856–1868. doi: 10.1093/cvr/cvad089. [DOI] [PubMed] [Google Scholar]
- 129.Liu G., Hitomi H., Hosomi N., et al. Mechanical stretch augments insulin-induced vascular smooth muscle cell proliferation by insulin-like growth factor-1 receptor. Exp Cell Res. 2011;317(17):2420–2428. doi: 10.1016/j.yexcr.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 130.Brown I.A.M., Diederich L., Good M.E., et al. Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arteriosclerosis, thrombosis, Vasc Biol. 2018;38(9):1969–1985. doi: 10.1161/ATVBAHA.118.311229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McCormick M.L., Gavrila D., Weintraub N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arteriosclerosis, thrombosis, Vasc Biol. 2007;27(3):461–469. doi: 10.1161/01.ATV.0000257552.94483.14. [DOI] [PubMed] [Google Scholar]
- 132.Qian W., Hadi T., Silvestro M., et al. Microskeletal stiffness promotes aortic aneurysm by sustaining pathological vascular smooth muscle cell mechanosensation via Piezo1. Nat Commun. 2022;13(1):512. doi: 10.1038/s41467-021-27874-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pedroza A.J., Shad R., Dalal A.R., et al. Acute induced pressure overload rapidly incites thoracic aortic aneurysmal smooth muscle cell phenotype. Hypertension. 2022;79(4):e86–e89. doi: 10.1161/HYPERTENSIONAHA.121.18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Baker A.B., Ettenson D.S., Jonas M., Nugent M.A., Iozzo R.V., Edelman E.R. Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine TGF-beta signaling pathway. Circ Res. 2008;103(3):289–297. doi: 10.1161/CIRCRESAHA.108.179465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Belo V.A., Guimaraes D.A., Castro M.M. Matrix metalloproteinase 2 as a potential mediator of vascular smooth muscle cell migration and chronic vascular remodeling in hypertension. J Vasc Res. 2015;52(4):221–231. doi: 10.1159/000441621. [DOI] [PubMed] [Google Scholar]
- 136.Kim S.A., Sung J.Y., Woo C.H., Choi H.C. Laminar shear stress suppresses vascular smooth muscle cell proliferation through nitric oxide-AMPK pathway. Biochem Biophys Res Commun. 2017;490(4):1369–1374. doi: 10.1016/j.bbrc.2017.07.033. [DOI] [PubMed] [Google Scholar]
- 137.Wang H., Yan S., Chai H., et al. Shear stress induces endothelial transdifferentiation from mouse smooth muscle cells. Biochem Biophys Res Commun. 2006;346(3):860–865. doi: 10.1016/j.bbrc.2006.05.196. [DOI] [PubMed] [Google Scholar]
- 138.Duivenvoorden R., Vanbavel E., de Groot E., et al. Endothelial shear stress: a critical determinant of arterial remodeling and arterial stiffness in humans--a carotid 3.0-T MRI study. Circ Cardiovasc Imaging. 2010;3(5):578–585. doi: 10.1161/CIRCIMAGING.109.916304. [DOI] [PubMed] [Google Scholar]
- 139.Goldman J., Zhong L., Liu S.Q. Negative regulation of vascular smooth muscle cell migration by blood shear stress. Am J Physiol Heart Circ Physiol. 2007;292(2):H928–H938. doi: 10.1152/ajpheart.00821.2006. [DOI] [PubMed] [Google Scholar]
- 140.Yao G., Li H., Zuo X., et al. Oscillatory shear stress promotes vein graft intimal hyperplasia via NADPH oxidase-related pathways. Front Surg. 2023;10 doi: 10.3389/fsurg.2023.1073557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fitzgerald T.N., Shepherd B.R., Asada H., et al. Laminar shear stress stimulates vascular smooth muscle cell apoptosis via the Akt pathway. J Cell Physiol. 2008;216(2):389–395. doi: 10.1002/jcp.21404. [DOI] [PubMed] [Google Scholar]
- 142.McMurray J.J., Pfeffer M.A. Heart failure. Lancet. 2005;365(9474):1877–1889. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- 143.Boorsma E.M., Ter Maaten J.M., Damman K., et al. Congestion in heart failure: a contemporary look at physiology, diagnosis and treatment. Nat Rev Cardiol. 2020;17(10):641–655. doi: 10.1038/s41569-020-0379-7. [DOI] [PubMed] [Google Scholar]
- 144.Wu J., You J., Wang S., Zhang L., Gong H., Zou Y. Insights into the activation and inhibition of angiotensin II type 1 receptor in the mechanically loaded heart. Circ J. 2014;78(6):1283–1289. doi: 10.1253/circj.cj-14-0470. [DOI] [PubMed] [Google Scholar]
- 145.Uriel N., Sayer G., Annamalai S., Kapur N.K., Burkhoff D. Mechanical unloading in heart failure. J Am Coll Cardiol. 2018;72(5):569–580. doi: 10.1016/j.jacc.2018.05.038. [DOI] [PubMed] [Google Scholar]
- 146.Barallobre-Barreiro J., Radovits T., Fava M., et al. Extracellular matrix in heart failure: role of ADAMTS5 in proteoglycan remodeling. Circulation. 2021;144(25):2021–2034. doi: 10.1161/CIRCULATIONAHA.121.055732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Travers J.G., Wennersten S.A., Pena B., et al. HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation. 2021;143(19):1874–1890. doi: 10.1161/CIRCULATIONAHA.120.046462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hutchinson K.R., Stewart J.A., Jr., Lucchesi P.A. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol. 2010;48(3):564–569. doi: 10.1016/j.yjmcc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Korf-Klingebiel M., Reboll M.R., Polten F., et al. Myeloid-Derived growth factor protects against pressure overload-induced heart failure by preserving Sarco/Endoplasmic reticulum Ca(2+)-ATPase expression in cardiomyocytes. Circulation. 2021;144(15):1227–1240. doi: 10.1161/CIRCULATIONAHA.120.053365. [DOI] [PubMed] [Google Scholar]
- 150.Li X., You J., Dai F., et al. TAK1 activation by NLRP3 deficiency confers cardioprotection against pressure overload-induced cardiomyocyte pyroptosis and hypertrophy. JACC Basic Transl Sci. 2023;8(12):1555–1573. doi: 10.1016/j.jacbts.2023.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bacmeister L., Schwarzl M., Warnke S., et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019;114(3):19. doi: 10.1007/s00395-019-0722-5. [DOI] [PubMed] [Google Scholar]
- 152.Wang G., Kong B., Shuai W., Fu H., Jiang X., Huang H. 3,3-Dimethyl-1-butanol attenuates cardiac remodeling in pressure-overload-induced heart failure mice. J Nutr Biochem. 2020;78 doi: 10.1016/j.jnutbio.2020.108341. [DOI] [PubMed] [Google Scholar]
- 153.Shirakabe A., Zhai P., Ikeda Y., et al. Drp1-Dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133(13):1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang Y., Bauersachs J., Langer H.F. Immune mechanisms in heart failure. Eur J Heart Fail. 2017;19(11):1379–1389. doi: 10.1002/ejhf.942. [DOI] [PubMed] [Google Scholar]
- 155.Li Q., Li C., Elnwasany A., et al. PKM1 exerts critical roles in cardiac remodeling under pressure overload in the heart. Circulation. 2021;144(9):712–727. doi: 10.1161/CIRCULATIONAHA.121.054885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Froese N., Szaroszyk M., Galuppo P., et al. Hypoxia attenuates pressure overload-induced heart failure. J Am Heart Assoc. 2024;13(3) doi: 10.1161/JAHA.123.033553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Havlenova T., Skaroupkova P., Miklovic M., et al. Right versus left ventricular remodeling in heart failure due to chronic volume overload. Sci Rep. 2021;11(1) doi: 10.1038/s41598-021-96618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Demirkiran A., van der Geest R.J., Hopman L., et al. Association of left ventricular flow energetics with remodeling after myocardial infarction: new hemodynamic insights for left ventricular remodeling. Int J Cardiol. 2022;367:105–114. doi: 10.1016/j.ijcard.2022.08.040. [DOI] [PubMed] [Google Scholar]
- 159.Leychenko A., Konorev E., Jijiwa M., Matter M.L. Stretch-induced hypertrophy activates NFkB-mediated VEGF secretion in adult cardiomyocytes. PloS one. 2011;6(12) doi: 10.1371/journal.pone.0029055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.da Silva A.R., Gunawan F., Boezio G.L.M., et al. egr3 is a mechanosensitive transcription factor gene required for cardiac valve morphogenesis. Sci Adv. 2024;10(20) doi: 10.1126/sciadv.adl0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Niu N., Xu S., Xu Y., Little P.J., Jin Z.G. Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol Sci. 2019;40(4):253–266. doi: 10.1016/j.tips.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McSweeney S.R., Warabi E., Siow R.C. Nrf2 as an endothelial mechanosensitive transcription factor: going with the flow. Hypertension. 2016;67(1):20–29. doi: 10.1161/HYPERTENSIONAHA.115.06146. [DOI] [PubMed] [Google Scholar]
- 163.Meli V.S., Veerasubramanian P.K., Downing T.L., Wang W., Liu W.F. Mechanosensation to inflammation: roles for YAP/TAZ in innate immune cells. Sci Signal. 2023;16(783) doi: 10.1126/scisignal.adc9656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zheng Q., Zou Y., Teng P., et al. Mechanosensitive Channel PIEZO1 senses shear force to induce KLF2/4 expression via CaMKII/MEKK3/ERK5 Axis in endothelial cells. Cells. 2022;11(14) doi: 10.3390/cells11142191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Paolini A., Fontana F., Pham V.C., Rodel C.J., Abdelilah-Seyfried S. Mechanosensitive Notch-Dll4 and Klf2-Wnt9 signaling pathways intersect in guiding valvulogenesis in zebrafish. Cell Rep. 2021;37(1) doi: 10.1016/j.celrep.2021.109782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Berecz T., Yiu A., Vittay O., et al. Transcriptional co-activators YAP1-TAZ of Hippo signalling in doxorubicin-induced cardiomyopathy. ESC Heart Fail. 2022;9(1):224–235. doi: 10.1002/ehf2.13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mia M.M., Cibi D.M., Ghani S., et al. Loss of Yap/Taz in cardiac fibroblasts attenuates adverse remodelling and improves cardiac function. Cardiovasc Res. 2022;118(7):1785–1804. doi: 10.1093/cvr/cvab205. [DOI] [PubMed] [Google Scholar]
- 168.Ong Y.T., Andrade J., Armbruster M., et al. A YAP/TAZ-TEAD signalling module links endothelial nutrient acquisition to angiogenic growth. Nat Metab. 2022;4(6):672–682. doi: 10.1038/s42255-022-00584-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li R., Shao J., Jin Y.J., et al. Endothelial FAT1 inhibits angiogenesis by controlling YAP/TAZ protein degradation via E3 ligase MIB2. Nat Commun. 2023;14(1):1980. doi: 10.1038/s41467-023-37671-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mannion A.J., Zhao H., Zhang Y., et al. Regulation of YAP promotor accessibility in endothelial mechanotransduction. Arteriosclerosis, thrombosis, Vasc Biol. 2024;44(3):666–689. doi: 10.1161/ATVBAHA.123.320300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu J., Zhao C., Xiao X., et al. Endothelial discoidin domain receptor 1 senses flow to modulate YAP activation. Nat Commun. 2023;14(1):6457. doi: 10.1038/s41467-023-42341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dupont S., Morsut L., Aragona M., et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 173.Wang L., Luo J.Y., Li B., et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540(7634):579–582. doi: 10.1038/nature20602. [DOI] [PubMed] [Google Scholar]
- 174.Ritsvall O., Albinsson S. Emerging role of YAP/TAZ in vascular mechanotransduction and disease. Microcirculation. 2024;31(4) doi: 10.1111/micc.12838. [DOI] [PubMed] [Google Scholar]
- 175.Wang K.C., Yeh Y.T., Nguyen P., et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 2016;113(41):11525–11530. doi: 10.1073/pnas.1613121113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang Q., Cao Y., Liu Y., et al. Shear stress inhibits cardiac microvascular endothelial cells apoptosis to protect against myocardial ischemia reperfusion injury via YAP/miR-206/PDCD4 signaling pathway. Biochem Pharmacol. 2021;186 doi: 10.1016/j.bcp.2021.114466. [DOI] [PubMed] [Google Scholar]
- 177.Pang Z.D., Sun X., Bai R.Y., et al. YAP-galectin-3 signaling mediates endothelial dysfunction in angiotensin II-induced hypertension in mice. Cell Mol Life Sci. 2023;80(2):38. doi: 10.1007/s00018-022-04623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xu Q., Zhuo K., Zhang X., et al. The role of angiotensin II activation of yes-associated protein/PDZ-binding motif signaling in hypertensive cardiac and vascular remodeling. Eur J Pharmacol. 2024;962 doi: 10.1016/j.ejphar.2023.176252. [DOI] [PubMed] [Google Scholar]
- 179.Daoud F., Arevalo Martinez M., Holmberg J., et al. YAP and TAZ in vascular smooth muscle confer protection against hypertensive Vasculopathy. Arteriosclerosis, thrombosis, Vasc Biol. 2022;42(4):428–443. doi: 10.1161/ATVBAHA.121.317365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Arevalo Martinez M., Ritsvall O., Bastrup J.A., et al. Vascular smooth muscle-specific YAP/TAZ deletion triggers aneurysm development in mouse aorta. JCI Insight. 2023;8(17) doi: 10.1172/jci.insight.170845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liu Y.N., Lv X., Chen X., et al. Specific overexpression of YAP in vascular smooth muscle attenuated abdominal aortic aneurysm formation by activating elastic fiber assembly via LTBP4. J Cardiovasc Transl Res. 2023;16(1):65–76. doi: 10.1007/s12265-022-10278-1. [DOI] [PubMed] [Google Scholar]
- 182.Zhong H.Y., Yuan C., Liu X.L., et al. Mechanical stretch aggravates vascular smooth muscle cell apoptosis and vascular remodeling by downregulating EZH2. Int J Biochem Cell Biol. 2022;151 doi: 10.1016/j.biocel.2022.106278. [DOI] [PubMed] [Google Scholar]
- 183.Douguet D., Patel A., Xu A., Vanhoutte P.M., Honore E. Piezo ion channels in cardiovascular mechanobiology. Trends Pharmacol Sci. 2019;40(12):956–970. doi: 10.1016/j.tips.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 184.Lai A., Cox C.D., Chandra Sekar N., et al. Mechanosensing by Piezo1 and its implications for physiology and various pathologies. Biol Rev Camb Phil Soc. 2022;97(2):604–614. doi: 10.1111/brv.12814. [DOI] [PubMed] [Google Scholar]
- 185.Jin C., Su S., Yu S., et al. Essential roles of PIEZO1 in mammalian cardiovascular system: from development to diseases. Cells. 2024;13(17) doi: 10.3390/cells13171422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yuan W., Zhang X., Fan X. The role of the Piezo1 mechanosensitive channel in heart failure. Curr Issues Mol Biol. 2023;45(7):5830–5848. doi: 10.3390/cimb45070369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang J., Zhang K., Du R., et al. The Janus-faced role of Piezo1 in cardiovascular health under mechanical stimulation. Genes Dis. 2023;10(5):1956–1968. doi: 10.1016/j.gendis.2022.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jiang F., Yin K., Wu K., et al. The mechanosensitive Piezo1 channel mediates heart mechano-chemo transduction. Nat Commun. 2021;12(1):869. doi: 10.1038/s41467-021-21178-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ploeg M.C., Munts C., Prinzen F.W., Turner N.A., van Bilsen M., van Nieuwenhoven F.A. Piezo1 mechanosensitive ion channel mediates stretch-induced Nppb expression in adult rat cardiac fibroblasts. Cells. 2021;10(7) doi: 10.3390/cells10071745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sun M., Mao S., Wu C., et al. Piezo1-Mediated neurogenic inflammatory cascade exacerbates ventricular remodeling after myocardial infarction. Circulation. 2024;149(19):1516–1533. doi: 10.1161/CIRCULATIONAHA.123.065390. [DOI] [PubMed] [Google Scholar]
- 191.Su S.A., Zhang Y., Li W., et al. vol. 6. Research (Wash D C).; 2023. p. 165. (Cardiac Piezo1 Exacerbates Lethal Ventricular Arrhythmogenesis by Linking Mechanical Stress with Ca(2+) Handling after Myocardial Infarction). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang Z., Chen J., Babicheva A., et al. Endothelial upregulation of mechanosensitive channel Piezo1 in pulmonary hypertension. Am J Physiol Cell Physiol. 2021;321(6):C1010–C1027. doi: 10.1152/ajpcell.00147.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen J., Miao J., Zhou D., et al. Upregulation of mechanosensitive channel Piezo1 involved in high shear stress-induced pulmonary hypertension. Thromb Res. 2022;218:52–63. doi: 10.1016/j.thromres.2022.08.006. [DOI] [PubMed] [Google Scholar]
- 194.Zhao W., Wei Z., Xin G., et al. Piezo1 initiates platelet hyperreactivity and accelerates thrombosis in hypertension. J Thromb Haemostasis. 2021;19(12):3113–3125. doi: 10.1111/jth.15504. [DOI] [PubMed] [Google Scholar]
- 195.Zhang Y., Su S.A., Li W., et al. Piezo1-Mediated mechanotransduction promotes cardiac hypertrophy by impairing calcium homeostasis to activate Calpain/Calcineurin signaling. Hypertension. 2021;78(3):647–660. doi: 10.1161/HYPERTENSIONAHA.121.17177. [DOI] [PubMed] [Google Scholar]
- 196.Mao J., Yang R., Yuan P., et al. Different stimuli induce endothelial dysfunction and promote atherosclerosis through the Piezo1/YAP signaling axis. Arch Biochem Biophys. 2023;747 doi: 10.1016/j.abb.2023.109755. [DOI] [PubMed] [Google Scholar]
- 197.Lan Y., Lu J., Zhang S., et al. Piezo1-Mediated mechanotransduction contributes to disturbed flow-induced atherosclerotic endothelial inflammation. J Am Heart Assoc. 2024;13(21) doi: 10.1161/JAHA.123.035558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wang Y.M., Chu T.J., Wan R.T., Niu W.P., Bian Y.F., Li J. Quercetin ameliorates atherosclerosis by inhibiting inflammation of vascular endothelial cells via Piezo1 channels. Phytomedicine. 2024;132 doi: 10.1016/j.phymed.2024.155865. [DOI] [PubMed] [Google Scholar]
- 199.Pourteymour S., Fan J., Majhi R.K., et al. PIEZO1 targeting in macrophages boosts phagocytic activity and foam cell apoptosis in atherosclerosis. Cell Mol Life Sci. 2024;81(1):331. doi: 10.1007/s00018-024-05372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pan X., Xu H., Ding Z., et al. Guizhitongluo Tablet inhibits atherosclerosis and foam cell formation through regulating Piezo1/NLRP3 mediated macrophage pyroptosis. Phytomedicine. 2024;132 doi: 10.1016/j.phymed.2024.155827. [DOI] [PubMed] [Google Scholar]
- 201.Zhu Y., Wang T., Yang Y., et al. Low shear stress exacerbates atherosclerosis by inducing the generation of neutrophil extracellular traps via Piezo 1-mediated mechanosensation. Atherosclerosis. 2024;391 doi: 10.1016/j.atherosclerosis.2024.117473. [DOI] [PubMed] [Google Scholar]
- 202.Johnson R.T., Solanki R., Wostear F., et al. Piezo1-mediated regulation of smooth muscle cell volume in response to enhanced extracellular matrix rigidity. Br J Pharmacol. 2024;181(11):1576–1595. doi: 10.1111/bph.16294. [DOI] [PubMed] [Google Scholar]
- 203.Lv H., Ai D. Hippo/yes-associated protein signaling functions as a mechanotransducer in regulating vascular homeostasis. J Mol Cell Cardiol. 2022;162:158–165. doi: 10.1016/j.yjmcc.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 204.Yamashiro Y., Yanagisawa H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin Sci (Lond) 2020;134(17):2399–2418. doi: 10.1042/CS20190488. [DOI] [PubMed] [Google Scholar]
- 205.Morales C.R., Pedrozo Z., Lavandero S., Hill J.A. Oxidative stress and autophagy in cardiovascular homeostasis. Antioxidants Redox Signal. 2014;20(3):507–518. doi: 10.1089/ars.2013.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Baeyens N., Nicoli S., Coon B.G., et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. Elife. 2015;4 doi: 10.7554/eLife.04645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hill M.A., Meininger G.A. Arteriolar vascular smooth muscle cells: mechanotransducers in a complex environment. Int J Biochem Cell Biol. 2012;44(9):1505–1510. doi: 10.1016/j.biocel.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zhang L., Feng Q., Kong W. ECM microenvironment in vascular homeostasis: new targets for atherosclerosis. Physiology. 2024;39(5) doi: 10.1152/physiol.00028.2023. [DOI] [PubMed] [Google Scholar]
- 209.Fu Y., Zhou Y., Wang K., Li Z., Kong W. Extracellular matrix interactome in modulating vascular homeostasis and remodeling. Circ Res. 2024;134(7):931–949. doi: 10.1161/CIRCRESAHA.123.324055. [DOI] [PubMed] [Google Scholar]
- 210.Pei J., Cai L., Wang F., et al. LPA(2) contributes to vascular endothelium homeostasis and cardiac remodeling after myocardial infarction. Circ Res. 2022;131(5):388–403. doi: 10.1161/CIRCRESAHA.122.321036. [DOI] [PubMed] [Google Scholar]
- 211.Kohn J.C., Zhou D.W., Bordeleau F., et al. Cooperative effects of matrix stiffness and fluid shear stress on endothelial cell behavior. Biophys J. 2015;108(3):471–478. doi: 10.1016/j.bpj.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhao G., Zhao Y., Lu H., et al. BAF60c prevents abdominal aortic aneurysm formation through epigenetic control of vascular smooth muscle cell homeostasis. J Clin Investig. 2022;132(21) doi: 10.1172/JCI158309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Du L.J., Sun J.Y., Zhang W.C., et al. NCOR1 maintains the homeostasis of vascular smooth muscle cells and protects against aortic aneurysm. Cell Death Differ. 2023;30(3):618–631. doi: 10.1038/s41418-022-01065-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Xia Y., Zhang X., An P., Luo J., Luo Y. Mitochondrial homeostasis in VSMCs as a central hub in vascular remodeling. Int J Mol Sci. 2023;24(4) doi: 10.3390/ijms24043483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhang J., Xie S.A., Wang J., et al. Echinatin maintains glutathione homeostasis in vascular smooth muscle cells to protect against matrix remodeling and arterial stiffening. Matrix Biol. 2023;119:1–18. doi: 10.1016/j.matbio.2023.03.007. [DOI] [PubMed] [Google Scholar]
- 216.Kong P., Christia P., Frangogiannis N.G. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549–574. doi: 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gong L., Wang S., Shen L., et al. SLIT3 deficiency attenuates pressure overload-induced cardiac fibrosis and remodeling. JCI Insight. 2020;5(12) doi: 10.1172/jci.insight.136852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Moore-Morris T., Guimaraes-Camboa N., Banerjee I., et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Investig. 2014;124(7):2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Adapala R.K., Katari V., Kanugula A.K., Ohanyan V., Paruchuri S., Thodeti C.K. Deletion of endothelial TRPV4 protects heart from pressure overload-induced hypertrophy. Hypertension. 2023;80(11):2345–2356. doi: 10.1161/HYPERTENSIONAHA.123.21528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Xiang F.L., Fang M., Yutzey K.E. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun. 2017;8(1):712. doi: 10.1038/s41467-017-00840-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Li W., Zhang Z., Li X., et al. CGRP derived from cardiac fibroblasts is an endogenous suppressor of cardiac fibrosis. Cardiovasc Res. 2020;116(7):1335–1348. doi: 10.1093/cvr/cvz234. [DOI] [PubMed] [Google Scholar]
- 222.Frangogiannis N.G. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450–1488. doi: 10.1093/cvr/cvaa324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nunez-Toldra R., Kirwin T., Ferraro E., et al. Mechanosensitive molecular mechanisms of myocardial fibrosis in living myocardial slices. ESC Heart Fail. 2022;9(2):1400–1412. doi: 10.1002/ehf2.13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Nagata Y., Bertrand P.B., Baliyan V., et al. Abnormal mechanics relate to myocardial fibrosis and ventricular arrhythmias in patients with mitral valve prolapse. Circ Cardiovasc Imaging. 2023;16(4) doi: 10.1161/CIRCIMAGING.122.014963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hou X., Xiong X., Li X., et al. Predictive value of cardiac magnetic resonance mechanical parameters for myocardial fibrosis in hypertrophic cardiomyopathy with preserved left ventricular ejection fraction. Front Cardiovasc Med. 2022;9 doi: 10.3389/fcvm.2022.1062258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pesce M., Duda G.N., Forte G., et al. Cardiac fibroblasts and mechanosensation in heart development, health and disease. Nat Rev Cardiol. 2023;20(5):309–324. doi: 10.1038/s41569-022-00799-2. [DOI] [PubMed] [Google Scholar]
- 227.Chalise U., Hale T.M. Fibroblasts under pressure: cardiac fibroblast responses to hypertension and antihypertensive therapies. Am J Physiol Heart Circ Physiol. 2024;326(1):H223–H237. doi: 10.1152/ajpheart.00401.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhang L., Guo Y.N., Liu J., et al. Plantamajoside attenuates cardiac fibrosis via inhibiting AGEs activated-RAGE/autophagy/EndMT pathway. Phytother Res. 2023;37(3):834–847. doi: 10.1002/ptr.7663. [DOI] [PubMed] [Google Scholar]
- 229.Zhang N., Ma Q., You Y., et al. CXCR4-dependent macrophage-to-fibroblast signaling contributes to cardiac diastolic dysfunction in heart failure with preserved ejection fraction. Int J Biol Sci. 2022;18(3):1271–1287. doi: 10.7150/ijbs.65802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Long Y., Niu Y., Liang K., Du Y. Mechanical communication in fibrosis progression. Trends Cell Biol. 2022;32(1):70–90. doi: 10.1016/j.tcb.2021.10.002. [DOI] [PubMed] [Google Scholar]
- 231.Nguyen-Truong M., Wang Z. Biomechanical properties and mechanobiology of cardiac ECM. Adv Exp Med Biol. 2018;1098:1–19. doi: 10.1007/978-3-319-97421-7_1. [DOI] [PubMed] [Google Scholar]
- 232.Niro F., Fernandes S., Cassani M., et al. Fibrotic extracellular matrix impacts cardiomyocyte phenotype and function in an iPSC-derived isogenic model of cardiac fibrosis. Transl Res. 2024;273:58–77. doi: 10.1016/j.trsl.2024.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Emig R., Knodt W., Krussig M.J., et al. Piezo1 channels contribute to the regulation of human atrial fibroblast mechanical properties and matrix stiffness sensing. Cells. 2021;10(3) doi: 10.3390/cells10030663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Garoffolo G., Casaburo M., Amadeo F., et al. Reduction of cardiac fibrosis by Interference with YAP-dependent transactivation. Circ Res. 2022;131(3):239–257. doi: 10.1161/CIRCRESAHA.121.319373. [DOI] [PubMed] [Google Scholar]
- 235.Adapala R.K., Katari V., Teegala L.R., Thodeti S., Paruchuri S., Thodeti C.K. TRPV4 mechanotransduction in fibrosis. Cells. 2021;10(11) doi: 10.3390/cells10113053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bartoli F., Evans E.L., Blythe N.M., et al. Global PIEZO1 Gain-of-function Mutation causes cardiac hypertrophy and fibrosis in mice. Cells. 2022;11(7) doi: 10.3390/cells11071199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zou Y., Pan L., Shen Y., et al. Cardiac Wnt5a and Wnt11 promote fibrosis by the crosstalk of FZD5 and EGFR signaling under pressure overload. Cell Death Dis. 2021;12(10):877. doi: 10.1038/s41419-021-04152-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Li R., Frangogiannis N.G. Integrins in cardiac fibrosis. J Mol Cell Cardiol. 2022;172:1–13. doi: 10.1016/j.yjmcc.2022.07.006. [DOI] [PubMed] [Google Scholar]
- 239.Meagher P.B., Lee X.A., Lee J., Visram A., Friedberg M.K., Connelly K.A. Cardiac fibrosis: key role of integrins in cardiac homeostasis and remodeling. Cells. 2021;10(4) doi: 10.3390/cells10040770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.McCarty M.F. Nutraceutical, Dietary, and Lifestyle Options for prevention and treatment of ventricular hypertrophy and heart failure. Int J Mol Sci. 2021;22(7) doi: 10.3390/ijms22073321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Peng F., Liao M., Jin W., et al. 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal Transduct Targeted Ther. 2024;9(1):133. doi: 10.1038/s41392-024-01816-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tang C., Hou Y.X., Shi P.X., et al. Cardiomyocyte-specific Peli1 contributes to the pressure overload-induced cardiac fibrosis through miR-494-3p-dependent exosomal communication. FASEB J official Publ Fed Am Soc Exp Biol. 2023;37(1) doi: 10.1096/fj.202200597R. [DOI] [PubMed] [Google Scholar]
- 243.Chen X., Li X., Wu X., et al. Integrin beta-like 1 mediates fibroblast-cardiomyocyte crosstalk to promote cardiac fibrosis and hypertrophy. Cardiovasc Res. 2023;119(10):1928–1941. doi: 10.1093/cvr/cvad104. [DOI] [PubMed] [Google Scholar]
- 244.Beech D.J., Kalli A.C. Force sensing by piezo channels in cardiovascular health and disease. Arteriosclerosis, thrombosis, Vasc Biol. 2019;39(11):2228–2239. doi: 10.1161/ATVBAHA.119.313348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cecchi E., Giglioli C., Valente S., et al. Role of hemodynamic shear stress in cardiovascular disease. Atherosclerosis. 2011;214(2):249–256. doi: 10.1016/j.atherosclerosis.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 246.Hahn C., Schwartz M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10(1):53–62. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Humphrey J.D., Dufresne E.R., Schwartz M.A. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15(12):802–812. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sun Z., Guo S.S., Fässler R. Integrin-mediated mechanotransduction. J cell Biol. 2016;215(4):445–456. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lai A., Zhou Y., Thurgood P., et al. Endothelial response to the combined biomechanics of vessel stiffness and shear stress is regulated via Piezo1. ACS Appl Mater Interfaces. 2023;15(51):59103–59116. doi: 10.1021/acsami.3c07756. [DOI] [PubMed] [Google Scholar]
- 250.Li J., Zhu J., Gray O., et al. Mechanosensitive super-enhancers regulate genes linked to atherosclerosis in endothelial cells. J cell Biol. 2024;223(3) doi: 10.1083/jcb.202211125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Xu Y., Xu H., Yan J., Song G. Mechanical force induced activation of adhesion G protein–coupled receptor. Mechanobiology in Medicine. 2024;2(3) [Google Scholar]
- 252.Zhu H., Miao R., Wang J., Lin M. Advances in modeling cellular mechanical perceptions and responses via the membrane-cytoskeleton-nucleus machinery. Mechanobiology in Medicine. 2024;2(1) [Google Scholar]
- 253.Lee S.N., Lin A., Dey D., Berman D.S., Han D. Application of quantitative assessment of coronary atherosclerosis by coronary computed tomographic angiography. Korean J Radiol. 2024;25(6):518–539. doi: 10.3348/kjr.2023.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ma Y.S., Gao J., Xie Y.H., Ren G.S., Zhang Z.K. The important role of wall shear stress in coronary atherosclerosis: finite element study of stenosis coronary artery. Chin J Arteriosclerosis. 2018;26(8):831–835. (in Chinese) [Google Scholar]