

Abstract

This report presents a method for the synthesis of 2-aroyl cyclobutanones via the reaction of in situ-generated cyclopropanones with acyl sulfonium ylides representing a formal carbene insertion into cyclopropanones. The reaction is highly stereoselective in the case of 2-substituted cyclopropanones, and the cyclobutanones thus obtained are well suited to α-alkylation, offering versatile synthetic applications.
Introduction
Cyclobutanones are highly valuable intermediates in organic synthesis.1 Additionally, cyclobutanes serve as crucial building blocks in synthetic chemistry due to their high ring strain. Moreover, these structures are frequently encountered as key motifs in bioactive natural products and pharmaceuticals.2,3 Traditionally, cyclobutanones have been prepared via [2 + 2]-cycloaddition reactions of ketenes and olefins or enol ethers.4,5 Over the years, significant advancements have been made using different metal catalysts to enhance efficiency and selectivity.6 For instance, gold,7 ruthenium,8 and palladium9 catalysts have been widely used for cyclobutanone synthesis through semipinacol-type ring-expansion reactions of cyclopropanols.10 In 2012, Hashmi and co-workers demonstrated a gold-catalyzed oxidative rearrangement of propargyl alcohols to 1,3-diketones (Scheme 1a).11 Later, in 2020 Zhang and co-workers reported the C–H insertion of an oxidatively generated gold carbene with tert-butyl alkynyl ketones, leading to the formation of strained cyclobutanones (Scheme 1b).12
Scheme 1. Previous Work and Our Design.

Notably, Lindsay and co-workers established the viability of enantioenriched 1-sulfonylcyclopropanols (SCPs) for synthesizing chiral β-lactams through a stereospecific [1,2]-shift involving a hydroxylamine hemiaminal intermediate (Scheme 1c).13 Recently, our group reported a straightforward and efficient one-pot procedure for synthesizing various 2-aryl-2-vinyl-substituted cyclobutanones. This approach makes use of readily available 1-tosylcyclopropanol as a cyclopropanone precursor and cinnamyl sulfonium salts as starting materials (Scheme 1d). The process involves the nucleophilic attack of an in situ-formed sulfonium ylide on the electrophilic carbonyl of the cyclopropanone, which ultimately, after a series of proton shifts, leads to the formation of a leaving group and triggers the subsequent ring-expansion.14 Building on this strategy, we hypothesized that a similar approach could allow a one-pot synthesis of 2-aroylcyclobutanones from SCPs and acyl sulfonium salts as starting materials (Scheme 1e).
Results and Discussion
We began our investigations employing the literature-known benzoyl sulfonium bromide salt resulting from the reaction of bromoacetophenone and thiolane as the precursor to sulfonium ylide 2.15 However, when we reacted SCP 1a with this sulfonium salt in the presence of different bases, the reaction primarily led to an undesired product resulting from the nucleophilic substitution of thiolane by the sulfinate anion generated in the cyclopropanone formation. To overcome this issue, we modified the reaction by directly utilizing sulfonium ylide 2a which was obtained by reacting the benzoyl sulfonium salt with aqueous NaOH. Notably, the stability of the obtained sulfonium ylides strongly depends on the substituent at the arene core. Therefore, the sulfonium ylides were directly submitted to the cyclobutanone synthesis without further purification. This refined approach enabled the subsequent reaction with SCP 1a, promoting the formation of the 2-benzoyl cyclobutanone 3a.
Following a thorough optimization of the reaction conditions (see Supporting Information for detailed information), we observed that strong bases such as LiHMDS or KOH (Table 1, entries 1 and 2) were not suitable to facilitate the desired reaction. Instead, K3PO4 proved to be more effective in promoting product formation (entry 3). Further investigations into the reaction parameters revealed that both the choice of solvent (entries 3 to 5) and the reaction temperature (entries 5 to 9) played critical roles in achieving a successful transformation. Toluene emerged as the optimal solvent, while low concentration of SCP and elevated temperatures were essential for maximizing efficiency. Based on these findings, we conducted the reaction using K3PO4 as the base and toluene as the solvent at 100 °C. Notably, within 30 min complete conversion of 1a was confirmed by GC/MS analysis, leading to 46% isolated yield of 3a. The resulting cyclobutanones were found to be moisture-sensitive and exhibited instability under humid conditions at room temperature, leading to ring-opening into the corresponding keto carboxylic acid over time (see SI). Storage under an atmosphere of argon at −20 °C significantly helped to increase stability. With optimized reaction conditions in hand, we examined the substrate scope of the transformation (Scheme 2). When reacting unsubstituted SCP 1a with various 4-aryl-substituted sulfonium ylides 2a–g, 2-aroylcyclobutanones 3a–g were obtained in moderate yields. The reaction demonstrated tolerance for both electron-withdrawing and electron-donating groups.
Table 1. Optimization Table of the Reaction Conditionsa.

| entryb | solvent (c/M)c | base (eq.) | T [°C] | yield [%]d,e |
|---|---|---|---|---|
| 1 | CH2Cl2 (0.05) | LiHMDS (1.0) | –78 to r.t. | <5 |
| 2 | CH2Cl2 (0.05) | KOH (1.0) | –78 to r.t. | – |
| 3 | CH2Cl2 (0.05) | K3PO4 (1.0) | –78 to r.t. | 13 |
| 4 | DME (0.05) | K3PO4 (1.5) | –78 to r.t. | 17 |
| 5 | PhMe (0.05) | K3PO4 (1.5) | –78 to r.t. | 23 |
| 6 | PhMe (0.01) | K3PO4 (1.5) | 0 to r.t. | 24 |
| 7 | PhMe (0.01) | K3PO4 (1.5) | r.t. | 19 |
| 8 | PhMe (0.03) | K3PO4 (1.5) | 100 | 52 (46) |
| 9 | PhMe (0.01) | K3PO4 (1.5) | 50 | 22 |
See Supporting Information (SI) for detailed information.
Reactions were carried out on a 0.1 mmol scale with respect to the SCP 1a.
Concentration with respect to SCP 1a.
Yields refer to 1H NMR yield against a 1,3,5-trimethoxybenzene standard.
Yields in parentheses refer to isolated and purified products on a 0.3 mmol scale.
Scheme 2. Cyclobutanone Substrate Scope.

Isolated yields on a 0.3 mmol scale if not stated otherwise.
Reaction performed on a 1 mmol scale.
Next, we turned our attention to preparing and utilizing several substituted SCPs. Reacting the geminal dimethyl-substituted SCP 1b with sulfonium ylides 2a and 2b afforded the cyclobutanones 3h and 3i in 31 and 26% yield, respectively. When phenyl-substituted SCP 1c was reacted with sulfonium ylide 2a 2,3-disubstituted cyclobutanone 3j was isolated in 25% yield as the sole cyclobutanone product of the reaction. When methyl-substituted SCP 1d was reacted with sulfonium ylides 2a and 2b 2,3-disubstituted cyclobutanones 3k and 3l were isolated in 50 and 57% yields, respectively. Careful NMR analyses of our products, as well as X-ray diffraction analysis of cyclobutanone 3o confirmed that the trans-substituted cyclobutanones were obtained. Furthermore, when enantiomerically pure SCP 1d (>99% ee) was used in the reaction with sulfonium ylide 2a, cyclobutanone 3k was obtained (92% ee) with only slight loss of enantiopurity confirming the stereospecificity of the reaction (see Supporting Information for further information). Gratifyingly, the fused SCP 1e demonstrated good reactivity with sulfonium ylides 2a, 2b and 2d producing 3m, 3n and 3o in 54, 41 and 30% yield, respectively. In addition, our method shows scalability. Reacting the fused SCP 1e and sulfonium ylide 2a on a 1 mmol scale yielded cyclobutanone 3m in 60% yield.
Based on our observations, the following mechanistic scenario is proposed (Scheme 3). Upon formation of cyclopropanone I from the SCP 1 under basic conditions, ylide 2 preferentially attacks the carbonyl of cyclopropanone I from the opposite face with respect to residue R, forming cyclopropoxide intermediate II. Subsequently, elimination of thiolane III facilitates ring-expansion via a stereospecific [1,2]-shift to afford the trans-substituted cyclobutanone 3.
Scheme 3. Plausible Mechanism.

Notably, no other regioisomeric cyclobutanones were observed as the carbon center with higher electron density migrates similar to Baeyer–Villiger rearrangements, i.e., in our case the higher substituted bond.13a,13d,13e This is easily understandable in terms of MO theory since the positive inductive effect of the substituents raises the energy level of the respective filled σ orbital.
When measuring the NMR spectra of these compounds we were surprised that no enol forms were observed as it is most commonly the case in 1,3-dicarbonyl compounds. Theoretically, two enol forms might be possible (Figure 1). Both of them would involve a further sp2-hybridized carbon in the four-membered ring. The formation of such structures would increase the strain energy of the four-membered rings tremendously. Simple density functional theory (DFT) studies (M062X/def2-QZVP, D3, CPCM (chloroform)) we performed showed that (b) and (c) are +2.3 and +5.9 kcal/mol higher in energy then the 1,3-diketone (a). Consequently, the equilibrium between the ketone and the enol is strongly in favor of the ketone. More precisely, only 2% enol formation is expected.
Figure 1.

Different enol forms and their energies (M062X/def2-QZVP, D3, CPCM(chloroform)).
Cyclobutanone 3m has emerged as a valuable substrate for alkylation, facilitating the formation of all-carbon quaternary centers.16 Thus, we performed several transformations on the cyclobutanones obtained in this study (Scheme 4). Alkylation of 3m in the presence of K2CO3 and ethyl iodide diastereoselectively gave ethyl-substituted cyclobutanone 4 in 31% yield. Attack took place from the convex less hindered side of the molecule as it was proven by NOE investigations. Reaction of 3m with methylamine led to ring-opening, producing cyclohexane 5 in 50% yield. Allylation of 3m with the cinnamyl sulfonium salt 6(14) diastereoselectively afforded cyclobutanone 7 in 68% yield. The configuration of the quaternary stereogenic center was confirmed again by NOE correlation of the allylic protons with the bridgehead protons of the fused ring system.
Scheme 4. Follow-up Reactions.

Conclusions
An effective method for the stereoselective synthesis of trans-2,3-disubstituted aroyl cyclobutanones is reported via the insertion of sulfonium ylides into cyclopropanones which were generated in situ from stable SCP precursors. This protocol is characterized by mild reaction conditions and moderate functional group tolerance. Additionally, alkyl-substituted cyclobutanones were synthesized with ease and efficiency.
Experimental Section
General Procedures (GP1) for the Synthesis of Sulfonium Ylides 2a–2g
Sulfonium ylides were synthesized following the procedure reported by Zefirov and co-workers.15 A solution of tetrahydrothiophene (132 mg, 1.50 mmol, 0.13 mL, 1.00 equiv) and the respective acyl bromide (1.50 mmol, 1.00 equiv) in anhydrous acetonitrile (0.35 M, 4 mL) under inert atmosphere (argon) was stirred at r.t. for 48 h. The resulting reaction mixture was then filtered and the solid residue was washed with diethyl ether and dried under vacuum affording the sulfonium salts as white solids. Subsequently, an aqueous solution of sodium hydroxide (1 M, 1.65 mmol, 1.65 mL, 1.10 equiv) was added dropwise to a stirred suspension of the crude sulfonium salt in H2O (5 mL) at 0 °C. The reaction mixture was allowed to stir at r.t. for 12 h and then extracted with CHCl3 (2 × 15 mL). The combined organic phases were dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to yield the respective sulfonium ylides 2a–2g.
1-Phenyl-2-(tetrahydro-1λ4-thien-1-ylidene)ethan-1-one (2a)
Prepared according to GP1 from
2-bromo-1-phenylethan-1-one (299 mg) afforded sulfonium ylide 2a (220 mg, 1.07 mmol, 71%) as a pale-yellow solid. m.p.:
108–109 °C. FTIR (ATR):  [cm–1] = 2944, 1664,
1590, 1559, 1509, 1478, 1446, 1394, 1330, 1308, 1274, 1214, 1175,
1096, 1068, 1020, 1001. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.81–7.74 (m, 2H), 7.37–7.30 (m,
3H), 4.33 (s, 1H), 3.68–3.52 (m, 2H), 3.19–2.98 (m,
2H), 2.84–2.59 (m, 2H), 2.06–1.91 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) =
181.7, 140.8, 129.3, 127.7, 126.2, 51.5, 43.1, 28.5. HRMS (ESI, Orbitrap):
C12H15OS, calcd.: 207.0838, found: 207.0835
[M + H]+.
[cm–1] = 2944, 1664,
1590, 1559, 1509, 1478, 1446, 1394, 1330, 1308, 1274, 1214, 1175,
1096, 1068, 1020, 1001. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.81–7.74 (m, 2H), 7.37–7.30 (m,
3H), 4.33 (s, 1H), 3.68–3.52 (m, 2H), 3.19–2.98 (m,
2H), 2.84–2.59 (m, 2H), 2.06–1.91 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) =
181.7, 140.8, 129.3, 127.7, 126.2, 51.5, 43.1, 28.5. HRMS (ESI, Orbitrap):
C12H15OS, calcd.: 207.0838, found: 207.0835
[M + H]+.
1-(4-Fluorophenyl)-2-(tetrahydro-1λ4-thien-1-ylidene)ethan-1-one (2b)
Prepared according to GP1 from
2-bromo-1-(4-fluorophenyl)ethanone (326 mg) afforded sulfonium ylide 2b (202 mg, 0.90 mmol, 60%) as a yellow solid. m.p.: 76–77
°C. FTIR (ATR):  [cm–1] = 2937, 1669,
1597, 1513, 1497, 1385, 1210, 1153, 1083, 1012. 1H NMR
(500 MHz, CDCl3): δ (ppm) = 7.82–7.70 (m,
2H), 7.04–6.95 (m, 2H), 4.26 (s, 1H), 3.66–3.54 (m,
2H), 3.09 (dddd, J = 11.3, 6.2, 5.1, 1.3 Hz, 2H),
2.82–2.66 (m, 2H), 2.06–1.92 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 180.4,
163.6 (d, J = 248.3 Hz), 137.0 (d, J = 3.1 Hz), 128.2 (d, J = 8.4 Hz), 114.5 (d, J = 21.3 Hz), 51.5, 43.1, 28.6. 19F NMR (471
MHz, CDCl3): δ (ppm) = – 112.3 (m). HRMS (ESI,
Orbitrap): C12H14OFS, calcd.: 225.0744, found:
225.0740 [M + H]+.
[cm–1] = 2937, 1669,
1597, 1513, 1497, 1385, 1210, 1153, 1083, 1012. 1H NMR
(500 MHz, CDCl3): δ (ppm) = 7.82–7.70 (m,
2H), 7.04–6.95 (m, 2H), 4.26 (s, 1H), 3.66–3.54 (m,
2H), 3.09 (dddd, J = 11.3, 6.2, 5.1, 1.3 Hz, 2H),
2.82–2.66 (m, 2H), 2.06–1.92 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 180.4,
163.6 (d, J = 248.3 Hz), 137.0 (d, J = 3.1 Hz), 128.2 (d, J = 8.4 Hz), 114.5 (d, J = 21.3 Hz), 51.5, 43.1, 28.6. 19F NMR (471
MHz, CDCl3): δ (ppm) = – 112.3 (m). HRMS (ESI,
Orbitrap): C12H14OFS, calcd.: 225.0744, found:
225.0740 [M + H]+.
1-(4-Chlorophenyl)-2-(tetrahydro-1λ4-thien-1-ylidene)ethan-1-one (2c)
Prepared according to GP1 from
2-bromo-1-(4-chlorophenyl)ethanone (350 mg) afforded sulfonium ylide 2c (223 mg, 0.93 mmol, 62%) as a red oil. FTIR (ATR):  [cm–1] = 2941, 1915,
1670, 1577, 1506, 1483, 1399, 1379, 1308, 1274, 1202, 1172, 1133,
1086, 1011. 1H NMR (400 MHz, CDCl3): δ
(ppm) = 7.74–7.68 (m, 2H), 7.32–7.27 (m, 2H), 4.32 (s,
1H), 3.76–3.46 (m, 2H), 3.16–2.98 (m, 2H), 2.87–2.67
(m, 2H), 2.05–1.93 (m, 2H). 13C{1H} NMR
(101 MHz, CDCl3): δ (ppm) = 180.9, 139.8, 135.8,
128.5, 128.3, 52.7, 43.7, 29.2. HRMS (ESI, Orbitrap): C12H14OClS, calcd.: 241.0459, found: 241.0456 [M + H]+.
[cm–1] = 2941, 1915,
1670, 1577, 1506, 1483, 1399, 1379, 1308, 1274, 1202, 1172, 1133,
1086, 1011. 1H NMR (400 MHz, CDCl3): δ
(ppm) = 7.74–7.68 (m, 2H), 7.32–7.27 (m, 2H), 4.32 (s,
1H), 3.76–3.46 (m, 2H), 3.16–2.98 (m, 2H), 2.87–2.67
(m, 2H), 2.05–1.93 (m, 2H). 13C{1H} NMR
(101 MHz, CDCl3): δ (ppm) = 180.9, 139.8, 135.8,
128.5, 128.3, 52.7, 43.7, 29.2. HRMS (ESI, Orbitrap): C12H14OClS, calcd.: 241.0459, found: 241.0456 [M + H]+.
1-(4-Bromophenyl)-2-(tetrahydro-1λ4-thien-1-ylidene)ethan-1-one (2d)
Prepared according to GP1 from
2-bromo-1-(4-bromophenyl)ethanone (417 mg) afforded sulfonium ylide 2d (297 mg, 1.05 mmol, 70%) as a red oil. FTIR (ATR):  [cm–1] = 2942, 1916,
1671, 1576, 1511, 1480, 1396, 1375, 1308, 1294, 1272, 1201, 1173,
1133, 1083, 1067, 1007. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.66–7.62 (m, 2H), 7.48–7.44 (m,
2H), 4.30 (s, 1H), 3.71–3.53 (m, 2H), 3.17–3.04 (m,
2H), 2.82–2.68 (m, 2H), 2.06–1.95 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) =
181.0, 140.3, 131.5, 128.6, 124.2, 52.7, 43.7, 29.2. HRMS (ESI, Orbitrap):
C12H14OBrS, calcd.: 284.9949, found: 284.9945
[M + H]+.
[cm–1] = 2942, 1916,
1671, 1576, 1511, 1480, 1396, 1375, 1308, 1294, 1272, 1201, 1173,
1133, 1083, 1067, 1007. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.66–7.62 (m, 2H), 7.48–7.44 (m,
2H), 4.30 (s, 1H), 3.71–3.53 (m, 2H), 3.17–3.04 (m,
2H), 2.82–2.68 (m, 2H), 2.06–1.95 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) =
181.0, 140.3, 131.5, 128.6, 124.2, 52.7, 43.7, 29.2. HRMS (ESI, Orbitrap):
C12H14OBrS, calcd.: 284.9949, found: 284.9945
[M + H]+.
2-(Tetrahydro-1λ4-thien-1-ylidene)-1-(p-tolyl)ethan-1-one (2e)
Prepared
according to GP1 from 2-bromo-1-(4-methylphenyl)ethanone
(320 mg) afforded sulfonium ylide 2e (284 mg, 1.29 mmol,
86%) as a red oil. FTIR (ATR):  [cm–1] = 2943, 1664,
1589, 1558, 1509, 1478, 1393, 1329, 1307, 1273, 1174, 1096, 1020,
1001. 1H NMR (500 MHz, CDCl3): δ (ppm)
= 7.68 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 4.30 (s, 1H), 3.60 (dtd, J = 11.6,
6.4, 3.0 Hz, 2H), 3.14 – 3.02 (m, 2H), 2.80 – 2.69 (m,
2H), 2.34 (s, 3H), 1.98 (m, 2H). 13C{1H} NMR
(126 MHz, CDCl3): δ (ppm) = 181.9, 139.6, 138.2,
128.7, 126.5, 51.5, 43.4, 28.8, 21.4. HRMS (ESI, Orbitrap): C13H16ONaS, calcd.: 243.0814, found: 243.0818 [M
+ Na]+.
[cm–1] = 2943, 1664,
1589, 1558, 1509, 1478, 1393, 1329, 1307, 1273, 1174, 1096, 1020,
1001. 1H NMR (500 MHz, CDCl3): δ (ppm)
= 7.68 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 4.30 (s, 1H), 3.60 (dtd, J = 11.6,
6.4, 3.0 Hz, 2H), 3.14 – 3.02 (m, 2H), 2.80 – 2.69 (m,
2H), 2.34 (s, 3H), 1.98 (m, 2H). 13C{1H} NMR
(126 MHz, CDCl3): δ (ppm) = 181.9, 139.6, 138.2,
128.7, 126.5, 51.5, 43.4, 28.8, 21.4. HRMS (ESI, Orbitrap): C13H16ONaS, calcd.: 243.0814, found: 243.0818 [M
+ Na]+.
1-(4-Methoxyphenyl)-2-(tetrahydro-1λ4-thien-1-ylidene)ethan-1-one (2f)
Prepared according to GP1 from
2-bromo-1-(4-methoxyphenyl)ethanone (344 mg) afforded sulfonium ylide 2f (235 mg, 0.99 mmol, 66%) as a red oil. FTIR (ATR):  [cm–1] = 2933, 1664,
1589, 1543, 1509, 1478, 1393, 1330, 1307, 1270, 1174, 1094, 1020,
1007. 1H NMR (300 MHz, CDCl3): δ (ppm)
= 7.80–7.69 (m, 2H), 6.91–6.78 (m, 2H), 4.27 (s, 1H),
3.82 (s, 3H), 3.62 (dt, J = 12.9, 7.3 Hz, 2H), 3.08
(dt, J = 11.9, 6.4 Hz, 2H), 2.85–2.67 (m,
2H), 2.07–1.87 (m, 2H). 13C{1H} NMR:
Sulfonium ylide 2f shows low stability in solution and
undergoes decomposition. Therefore, it was not possible to obtain
a clean 13C NMR spectrum. HRMS (ESI, Orbitrap): C13H17O2S calcd.: 237.0949, found: 237.0945 [M
+ H]+.
[cm–1] = 2933, 1664,
1589, 1543, 1509, 1478, 1393, 1330, 1307, 1270, 1174, 1094, 1020,
1007. 1H NMR (300 MHz, CDCl3): δ (ppm)
= 7.80–7.69 (m, 2H), 6.91–6.78 (m, 2H), 4.27 (s, 1H),
3.82 (s, 3H), 3.62 (dt, J = 12.9, 7.3 Hz, 2H), 3.08
(dt, J = 11.9, 6.4 Hz, 2H), 2.85–2.67 (m,
2H), 2.07–1.87 (m, 2H). 13C{1H} NMR:
Sulfonium ylide 2f shows low stability in solution and
undergoes decomposition. Therefore, it was not possible to obtain
a clean 13C NMR spectrum. HRMS (ESI, Orbitrap): C13H17O2S calcd.: 237.0949, found: 237.0945 [M
+ H]+.
2-(Tetrahydro-1λ4-thien-1-ylidene)-1-(4-(trifluoromethyl)phenyl)ethan-1-one (2g)
Prepared according to GP1 from
2-bromo-1-(4-trifluoromethyl phenyl)ethanone (400 mg) afforded sulfonium
ylide 2g (218 mg, 0.79 mmol, 53%) as a pale-yellow solid.
m.p.: 72–73 °C. FTIR (ATR):  [cm–1] = 2936, 1614,
1587, 1519, 1505, 1443, 1393, 1325, 1273, 1206, 1151, 1107, 1014. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.89–7.83
(m, 2H), 7.62–7.56 (m, 2H), 4.36 (s, 1H), 3.71–3.55
(m, 2H), 3.20–3.05 (m, 2H), 2.84–2.69 (m, 2H), 2.03
(dqd, J = 11.4, 7.2, 2.2 Hz, 2H). 13C{1H} NMR: Sulfonium ylide 2g shows low stability
in solution and undergoes decomposition. Therefore, it was not possible
to obtain a clean 13C NMR spectrum. 19F NMR
(471 MHz, CDCl3): δ (ppm) = – 62.5 (m). HRMS
(ESI, Orbitrap): C13H14OF3S calcd.:
275.0712, found: 275.0714 [M + H]+.
[cm–1] = 2936, 1614,
1587, 1519, 1505, 1443, 1393, 1325, 1273, 1206, 1151, 1107, 1014. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.89–7.83
(m, 2H), 7.62–7.56 (m, 2H), 4.36 (s, 1H), 3.71–3.55
(m, 2H), 3.20–3.05 (m, 2H), 2.84–2.69 (m, 2H), 2.03
(dqd, J = 11.4, 7.2, 2.2 Hz, 2H). 13C{1H} NMR: Sulfonium ylide 2g shows low stability
in solution and undergoes decomposition. Therefore, it was not possible
to obtain a clean 13C NMR spectrum. 19F NMR
(471 MHz, CDCl3): δ (ppm) = – 62.5 (m). HRMS
(ESI, Orbitrap): C13H14OF3S calcd.:
275.0712, found: 275.0714 [M + H]+.
General Procedure (GP2) for the Synthesis of Cyclobutanones 3a–3o
A flame-dried (2×) 25 mL Schlenk flask was charged with SCP 1a–1h (0.30 mmol, 1.00 equiv), K3PO4 (0.45 mmol, 1.50 equiv) and the corresponding sulfonium ylide 2a–2g (0.45 mmol, 1.50 equiv). The flask was evacuated and backfilled with argon (2×) before anhydrous Toluene (12 mL) was added. The reaction mixture was degassed by freezing pump thaw (3×), placed into a preheated oil bath at 100 °C and stirred for the indicated time. Afterward the reaction mixture was cooled to r.t., solids were removed by filtration through filter paper and the filtrate was concentrated under reduced pressure. Purification by flash column chromatography (SiO2, n-pentane/EtOAc) yielded the respective cyclobutanones 3a–3o.
2-Benzoylcyclobutan-1-one (3a)
Prepared
according to GP2 from SCP 1a (64 mg) and
sulfonium ylide 2a (93 mg) for 30 min. Purification by
flash column chromatography (SiO2, n-pentane/EtOAc
19:1) afforded the cyclobutanone 3a (24 mg, 0.14 mmol,
46%) as a yellow oil. Rf: 0.44 (n-pentane/EtOAc 9:1). FTIR (ATR):  [cm–1] = 2965, 2943,
2699, 1691, 1673, 1596, 1580, 1449, 1410, 1377, 1320, 1308, 1285,
1231, 1190, 1155, 1071. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.10–8.01 (m, 2H), 7.65–7.40 (m,
3H), 5.25–5.14 (m, 1H), 3.25–3.16 (m, 2H), 2.88 (dddd, J = 11.7, 9.4, 8.4, 6.4 Hz, 1H), 2.26–2.13 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ
(ppm) = 200.9, 190.4, 136.3, 134.0, 129.8, 129.1, 71.0, 46.9, 12.0.
HRMS (GC-APCI, QTOF): C11H11O2, calcd.:
175.0754, found: 175.0751 [M + H]+.
[cm–1] = 2965, 2943,
2699, 1691, 1673, 1596, 1580, 1449, 1410, 1377, 1320, 1308, 1285,
1231, 1190, 1155, 1071. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.10–8.01 (m, 2H), 7.65–7.40 (m,
3H), 5.25–5.14 (m, 1H), 3.25–3.16 (m, 2H), 2.88 (dddd, J = 11.7, 9.4, 8.4, 6.4 Hz, 1H), 2.26–2.13 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ
(ppm) = 200.9, 190.4, 136.3, 134.0, 129.8, 129.1, 71.0, 46.9, 12.0.
HRMS (GC-APCI, QTOF): C11H11O2, calcd.:
175.0754, found: 175.0751 [M + H]+.
2-(4-Fluorobenzoyl)cyclobutan-1-one (3b)
Prepared according to GP2 from SCP 1a (64
mg) and sulfonium ylide 2b (101 mg) for 30 min. Purification
by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3b (26
mg, 0.14 mmol, 45%) as a yellow oil. Rf: 0.33 (n-pentane/EtOAc 9:1). FTIR (ATR):  [cm–1] = 2970, 2936,
1777, 1668, 1592, 1505, 1410, 1308, 1288, 1212, 1195, 1160, 1144,
1074, 1055, 1033. 1H NMR (500 MHz, CDCl3): δ
(ppm) = 8.16–8.02 (m, 2H), 7.22–7.10 (m, 2H), 5.20–5.07
(m, 1H), 3.27–3.11 (m, 2H), 2.93–2.81 (m, 1H), 2.27–2.12
(m, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 200.8, 188.7, 166.5 (d, J = 255.7
Hz), 132.8 (d, J = 2.9 Hz), 132.5 (d, J = 9.6 Hz), 116.2 (d, J = 22.2 Hz), 71.0, 46.8,
11.9. 19F NMR (471 MHz, CDCl3): δ (ppm)
= – 104.28 (m). HRMS (GC-APCI, QTOF): C11H10FO2, calcd.: 193.0659, found: 193.0656 [M + H]+.
[cm–1] = 2970, 2936,
1777, 1668, 1592, 1505, 1410, 1308, 1288, 1212, 1195, 1160, 1144,
1074, 1055, 1033. 1H NMR (500 MHz, CDCl3): δ
(ppm) = 8.16–8.02 (m, 2H), 7.22–7.10 (m, 2H), 5.20–5.07
(m, 1H), 3.27–3.11 (m, 2H), 2.93–2.81 (m, 1H), 2.27–2.12
(m, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 200.8, 188.7, 166.5 (d, J = 255.7
Hz), 132.8 (d, J = 2.9 Hz), 132.5 (d, J = 9.6 Hz), 116.2 (d, J = 22.2 Hz), 71.0, 46.8,
11.9. 19F NMR (471 MHz, CDCl3): δ (ppm)
= – 104.28 (m). HRMS (GC-APCI, QTOF): C11H10FO2, calcd.: 193.0659, found: 193.0656 [M + H]+.
2-(4-Chlorobenzoyl)cyclobutan-1-one (3c)
Prepared according to GP2 from SCP 1a (64
mg) and sulfonium ylide 2c (108 mg) for 30 min. Purification
by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3c (23
mg, 0.11 mmol, 37%) as a yellow oil. Rf: 0.26 (n-pentane:EtOAc 9:1). FTIR (ATR):  [cm–1] = 2970, 2918,
1777, 1708, 1663, 1587, 1572, 1487, 1401, 1299, 1239, 1215, 1197,
1178, 1145, 1088, 1040, 1008. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.07–7.91 (m, 2H), 7.53–7.39
(m, 2H), 5.23–5.05 (m, 1H), 3.35–3.15 (m, 2H), 2.89–2.64
(m, 1H), 2.31–2.07 (m, 1H). 13C{1H} NMR
(126 MHz, CDCl3): δ (ppm) = 200.1, 188.6, 140.0,
134.1, 130.6, 128.9, 70.5, 46.3, 11.4. HRMS (GC-APCI, QTOF): C11H10ClO2, calcd.: 209.0364, found: 209.0360
[M + H]+.
[cm–1] = 2970, 2918,
1777, 1708, 1663, 1587, 1572, 1487, 1401, 1299, 1239, 1215, 1197,
1178, 1145, 1088, 1040, 1008. 1H NMR (500 MHz, CDCl3): δ (ppm) = 8.07–7.91 (m, 2H), 7.53–7.39
(m, 2H), 5.23–5.05 (m, 1H), 3.35–3.15 (m, 2H), 2.89–2.64
(m, 1H), 2.31–2.07 (m, 1H). 13C{1H} NMR
(126 MHz, CDCl3): δ (ppm) = 200.1, 188.6, 140.0,
134.1, 130.6, 128.9, 70.5, 46.3, 11.4. HRMS (GC-APCI, QTOF): C11H10ClO2, calcd.: 209.0364, found: 209.0360
[M + H]+.
2-(4-Bromobenzoyl)cyclobutan-1-one (3d)
Prepared according to GP2 from SCP 1a (64
mg) and sulfonium ylide 2d (127 mg) for 30 min. Purification
by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3d (28
mg, 0.11 mmol, 37%) as a yellow oil. Rf: 0.27 (n-pentane:EtOAc 9:1). FTIR (ATR):  [cm–1] = 2802, 1695,
1581, 1486, 1450, 1433, 1401, 1372, 1301, 1279, 1225, 1192, 1115,
1071, 1012. 1H NMR (500 MHz, CDCl3): δ
(ppm) = 7.98–7.87 (m, 2H), 7.70–7.56 (m, 2H), 5.20–5.06
(m, 1H), 3.28–3.14 (m, 2H), 2.95–2.80 (m, 1H), 2.20
(dtd, J = 11.7, 9.2, 7.9 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 200.6,
189.4, 135.1, 132.4, 131.2, 129.4, 71.0, 46.9, 11.9. HRMS (GC-APCI,
QTOF): C11H10BrO2, calcd.: 252.9859,
found: 252.9854 [M + H]+.
[cm–1] = 2802, 1695,
1581, 1486, 1450, 1433, 1401, 1372, 1301, 1279, 1225, 1192, 1115,
1071, 1012. 1H NMR (500 MHz, CDCl3): δ
(ppm) = 7.98–7.87 (m, 2H), 7.70–7.56 (m, 2H), 5.20–5.06
(m, 1H), 3.28–3.14 (m, 2H), 2.95–2.80 (m, 1H), 2.20
(dtd, J = 11.7, 9.2, 7.9 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 200.6,
189.4, 135.1, 132.4, 131.2, 129.4, 71.0, 46.9, 11.9. HRMS (GC-APCI,
QTOF): C11H10BrO2, calcd.: 252.9859,
found: 252.9854 [M + H]+.
2-(4-Methylbenzoyl)cyclobutan-1-one (3e)
Prepared according to GP2 from SCP 1a (64
mg) and sulfonium ylide 2e (99 mg) for 30 min. Purification
by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3e (19
mg, 0.10 mmol, 34%) as a colorless solid. m.p.: 110–111 °C. Rf: 0.45 (n-pentane:EtOAc 9:1).
FTIR (ATR):  [cm–1] = 2965, 1768,
1667, 1604, 1315, 1297, 1223, 1204, 1180, 1142, 1055, 1009. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.02–7.88
(m, 2H), 7.32–7.25 (m, 2H), 5.22–5.10 (m, 1H), 3.25–3.15
(m, 2H), 2.94–2.79 (m, 1H), 2.42 (s, 3H), 2.25–2.09
(m, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) = 201.2, 190.0, 145.0, 134.0, 129.9, 129.8, 71.0,
46.9, 22.2, 12.1. HRMS (GC-APCI, QTOF): C12H12O2, calcd.: 188.0832, found: 188.0830 [M]+.
[cm–1] = 2965, 1768,
1667, 1604, 1315, 1297, 1223, 1204, 1180, 1142, 1055, 1009. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.02–7.88
(m, 2H), 7.32–7.25 (m, 2H), 5.22–5.10 (m, 1H), 3.25–3.15
(m, 2H), 2.94–2.79 (m, 1H), 2.42 (s, 3H), 2.25–2.09
(m, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ (ppm) = 201.2, 190.0, 145.0, 134.0, 129.9, 129.8, 71.0,
46.9, 22.2, 12.1. HRMS (GC-APCI, QTOF): C12H12O2, calcd.: 188.0832, found: 188.0830 [M]+.
2-(4-Methoxybenzoyl)cyclobutan-1-one (3f)
Prepared according to GP2 from SCP 1a (64
mg) and sulfonium ylide 2f (106 mg) for 30 min. Purification
by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3f (20
mg, 0.10 mmol, 32%) as a colorless solid. m.p.: 118–119 °C. Rf: 0.23 (n-pentane:EtOAc 9:1).
FTIR (ATR):  [cm–1] = 2836, 1706,
1670, 1596, 1573, 1510, 1461, 1415, 1373, 1312, 1289, 1252, 1233,
1195, 1177, 1115, 1077, 1021. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.10–7.98 (m, 2H), 6.99–6.92
(m, 2H), 5.21–5.07 (m, 1H), 3.87 (s, 3H), 3.23–3.10
(m, 2H), 2.92–2.79 (m, 1H), 2.25–2.09 (m, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ
(ppm) = 201.5, 188.9, 164.4, 132.2, 129.5, 114.4, 70.7, 56.0, 46.8,
12.1. HRMS (GC-APCI, QTOF): C12H13O3, calcd.: 205.0859, found: 205.0853 [M + H]+.
[cm–1] = 2836, 1706,
1670, 1596, 1573, 1510, 1461, 1415, 1373, 1312, 1289, 1252, 1233,
1195, 1177, 1115, 1077, 1021. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.10–7.98 (m, 2H), 6.99–6.92
(m, 2H), 5.21–5.07 (m, 1H), 3.87 (s, 3H), 3.23–3.10
(m, 2H), 2.92–2.79 (m, 1H), 2.25–2.09 (m, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ
(ppm) = 201.5, 188.9, 164.4, 132.2, 129.5, 114.4, 70.7, 56.0, 46.8,
12.1. HRMS (GC-APCI, QTOF): C12H13O3, calcd.: 205.0859, found: 205.0853 [M + H]+.
2-(4-(Trifluoromethyl)benzoyl)cyclobutan-1-one (3g)
Prepared according to GP2 from SCP 1a (64 mg) and sulfonium ylide 2g (123 mg) for 30 min.
Purification by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3g (28 mg, 0.12 mmol, 38%) as a yellow solid. m.p.: 88–89 °C. Rf: 0.25 (n-pentane:EtOAc 9:1).
FTIR (ATR):  [cm–1] = 2925, 1784,
1685, 1581, 1513, 1457, 1409, 1319, 1283, 1223, 1167, 1130, 1110,
1063, 1015. 1H NMR (700 MHz, CDCl3): δ
(ppm) = 8.22–8.11 (m, 2H), 7.82–7.70 m, 2H), 5.28–5.12
(m, 1H), 3.31–3.16 (m, 2H), 2.98–2.85 (m, 1H), 2.29–2.16
(m, 1H). 13C{1H} NMR (176 MHz, CDCl3): δ (ppm) = 199.9, 189.2, 138.7 (d, J = 1.3
Hz), 134.9 (d, J = 32.6 Hz), 129.8, 125.9 (d, J = 3.8 Hz), 123.7 (d, J = 272.8 Hz), 71.1,
46.7, 11.6. 19F NMR (471 MHz, CDCl3): δ
(ppm) = – 63.21 (m). HRMS (GC-APCI, QTOF): C12H10F3O2, calcd.: 243.0633, found: 243.0632
[M + H]+.
[cm–1] = 2925, 1784,
1685, 1581, 1513, 1457, 1409, 1319, 1283, 1223, 1167, 1130, 1110,
1063, 1015. 1H NMR (700 MHz, CDCl3): δ
(ppm) = 8.22–8.11 (m, 2H), 7.82–7.70 m, 2H), 5.28–5.12
(m, 1H), 3.31–3.16 (m, 2H), 2.98–2.85 (m, 1H), 2.29–2.16
(m, 1H). 13C{1H} NMR (176 MHz, CDCl3): δ (ppm) = 199.9, 189.2, 138.7 (d, J = 1.3
Hz), 134.9 (d, J = 32.6 Hz), 129.8, 125.9 (d, J = 3.8 Hz), 123.7 (d, J = 272.8 Hz), 71.1,
46.7, 11.6. 19F NMR (471 MHz, CDCl3): δ
(ppm) = – 63.21 (m). HRMS (GC-APCI, QTOF): C12H10F3O2, calcd.: 243.0633, found: 243.0632
[M + H]+.
(R)-2-Benzoyl-3,3-dimethylcyclobutan-1-one (3h)
Prepared according to GP2 from
SCP 1b (68 mg) and sulfonium ylide 2a (93
mg) for 30 min. Purification by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3h (19 mg, 0.09 mmol, 31%) as a yellow oil. Rf: 0.39 (n-pentane:EtOAc 19:1). FTIR
(ATR):  [cm–1] = 2954, 1690,
1597, 1580, 1448, 1362, 1324, 1230, 1177, 1116, 1009. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.88–7.84
(m, 2H), 7.61–7.56 (m, 1H), 7.51–7.45 (m, 2H), 4.69
(dd, J = 2.1, 0.7 Hz, 1H), 2.98–2.95 (m, 2H),
1.60 (s, 3H), 1.28 (s, 3H). 13C{1H} NMR (101
MHz, CDCl3): δ (ppm) = 201.7, 194.1, 136.8, 133.4,
128.6, 128.2, 74.2, 59.0, 32.3, 29.5, 23.8. HRMS (GC-APCI, QTOF):
C13H15O2, calcd.: 203.1067, found:
203.1065 [M + H]+.
[cm–1] = 2954, 1690,
1597, 1580, 1448, 1362, 1324, 1230, 1177, 1116, 1009. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.88–7.84
(m, 2H), 7.61–7.56 (m, 1H), 7.51–7.45 (m, 2H), 4.69
(dd, J = 2.1, 0.7 Hz, 1H), 2.98–2.95 (m, 2H),
1.60 (s, 3H), 1.28 (s, 3H). 13C{1H} NMR (101
MHz, CDCl3): δ (ppm) = 201.7, 194.1, 136.8, 133.4,
128.6, 128.2, 74.2, 59.0, 32.3, 29.5, 23.8. HRMS (GC-APCI, QTOF):
C13H15O2, calcd.: 203.1067, found:
203.1065 [M + H]+.
(R)-2-(4-Fluorobenzoyl)-3,3-dimethylcyclobutan-1-one (3i)
Prepared according to GP2 from SCP 1b (68 mg) and sulfonium ylide 2b (101 mg) for 30 min.
Purification by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3i (17 mg, 0.08 mmol, 26%) as a yellow oil. Rf: 0.31 (n-pentane:EtOAc 19:1). FTIR (ATR):  [cm–1] = 2957, 1690,
1575, 1580, 1447, 1362, 1324, 1232, 1177, 1116, 1009. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.93–7.87
(m, 2H), 7.18–7.13 (m, 2H), 4.63 (d, J = 1.10
Hz, 1H), 2.97 (d, 2H), 1.60 (s, 3H), 1.29 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 201.9,
192.9, 166.5 (d, J = 256.0 Hz), 133.7 (d, J = 3.0 Hz), 131.5 (d, J = 9.7 Hz), 116.4
(d, J = 22.0 Hz), 74.6, 59.5, 32.9, 30.1, 24.4. 19F NMR (471 MHz, CDCl3): δ (ppm) = –
104.16 (m). HRMS (GC-APCI, QTOF): C13H14FO2, calcd.: 221.0975, found: 221.0973 [M + H]+.
[cm–1] = 2957, 1690,
1575, 1580, 1447, 1362, 1324, 1232, 1177, 1116, 1009. 1H NMR (500 MHz, CDCl3): δ (ppm) = 7.93–7.87
(m, 2H), 7.18–7.13 (m, 2H), 4.63 (d, J = 1.10
Hz, 1H), 2.97 (d, 2H), 1.60 (s, 3H), 1.29 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 201.9,
192.9, 166.5 (d, J = 256.0 Hz), 133.7 (d, J = 3.0 Hz), 131.5 (d, J = 9.7 Hz), 116.4
(d, J = 22.0 Hz), 74.6, 59.5, 32.9, 30.1, 24.4. 19F NMR (471 MHz, CDCl3): δ (ppm) = –
104.16 (m). HRMS (GC-APCI, QTOF): C13H14FO2, calcd.: 221.0975, found: 221.0973 [M + H]+.
(2R,3S)-2-Benzoyl-3-phenylcyclobutan-1-one (3j)
Prepared according to GP2 from
SCP 1c (82 mg) and sulfonium ylide 2a (93
mg) for 30 min. Purification by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3j (19 mg, 0.08 mmol, 25%) as a colorless solid. m.p.: 123–124
°C. Rf: 0.49 (n-pentane:EtOAc
9:1). FTIR (ATR): 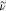 [cm–1] = 1784, 1669,
1596, 1449, 1322, 1297, 1270, 1229, 1139, 1080. 1H NMR
(500 MHz, CDCl3): δ (ppm) = 8.01–7.89 (m,
2H), 7.53–7.46 (m, 1H), 7.43–7.38 (m, 2H), 7.29–7.26
(m, 2H), 7.25 (t, J = 1.9 Hz, 1H), 7.21–7.15
(m, 2H), 5.08 (dt, J = 7.5, 2.5 Hz, 1H), 4.42 (dt, J = 9.9, 7.8 Hz, 1H), 3.47 (ddd, J = 17.9,
9.8, 2.3 Hz, 1H), 3.35 (ddd, J = 18.0, 8.1, 2.6 Hz,
1H). 13C{1H} NMR (126 MHz, CDCl3):
δ (ppm) = 199.2, 190.1, 142.4, 136.2, 134.2, 129.9, 129.3, 129.1,
127.4, 127.1, 78.0, 51.9, 30.3. HRMS (GC-APCI, QTOF): C17H15O2, calcd.: 251.1067, found: 251.1066 [M
+ H]+.
[cm–1] = 1784, 1669,
1596, 1449, 1322, 1297, 1270, 1229, 1139, 1080. 1H NMR
(500 MHz, CDCl3): δ (ppm) = 8.01–7.89 (m,
2H), 7.53–7.46 (m, 1H), 7.43–7.38 (m, 2H), 7.29–7.26
(m, 2H), 7.25 (t, J = 1.9 Hz, 1H), 7.21–7.15
(m, 2H), 5.08 (dt, J = 7.5, 2.5 Hz, 1H), 4.42 (dt, J = 9.9, 7.8 Hz, 1H), 3.47 (ddd, J = 17.9,
9.8, 2.3 Hz, 1H), 3.35 (ddd, J = 18.0, 8.1, 2.6 Hz,
1H). 13C{1H} NMR (126 MHz, CDCl3):
δ (ppm) = 199.2, 190.1, 142.4, 136.2, 134.2, 129.9, 129.3, 129.1,
127.4, 127.1, 78.0, 51.9, 30.3. HRMS (GC-APCI, QTOF): C17H15O2, calcd.: 251.1067, found: 251.1066 [M
+ H]+.
(2R,3S)-2-Benzoyl-3-methylcyclobutan-1-one (3k)
Prepared according to GP2 from
SCP 1d (64 mg) and sulfonium ylide 2a (93
mg) for 30 min. Purification by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3k (28 mg, 0.15 mmol, 50%) as a yellow oil. Rf: 0.27 (n-pentane:EtOAc 19:1). FTIR
(ATR): 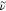 [cm–1] = 2965, 1679,
1597, 1447, 1411, 1371, 1292, 1260, 1221, 1179, 1160, 1002. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.07–7.99
(m, 2H), 7.64–7.45 (m, 3H), 4.89–4.57 (m, 1H), 3.42–3.20
(m, 2H), 2.93–2.65 (m, 1H), 1.47–1.33 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ
(ppm) = 200.4, 190.7, 136.5, 133.9, 129.7, 129.0, 77.2, 52.9, 21.0,
20.8. HRMS (GC-APCI, QTOF): C12H13O2, calcd.: 189.0910, found: 189.0908 [M + H]+.
[cm–1] = 2965, 1679,
1597, 1447, 1411, 1371, 1292, 1260, 1221, 1179, 1160, 1002. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.07–7.99
(m, 2H), 7.64–7.45 (m, 3H), 4.89–4.57 (m, 1H), 3.42–3.20
(m, 2H), 2.93–2.65 (m, 1H), 1.47–1.33 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ
(ppm) = 200.4, 190.7, 136.5, 133.9, 129.7, 129.0, 77.2, 52.9, 21.0,
20.8. HRMS (GC-APCI, QTOF): C12H13O2, calcd.: 189.0910, found: 189.0908 [M + H]+.
(2R,3S)-2-(4-Fluorobenzoyl)-3-methylcyclobutan-1-one (3l)
Prepared according to GP2 from
SCP 1d (64 mg) and sulfonium ylide 2b (101
mg) for 30 min. Purification by flash column chromatography (SiO2, n-pentane/EtOAc 19:1) afforded the cyclobutanone 3l (35 mg, 0.17 mmol, 57%) as a yellow oil. Rf: 0.23 (n-pentane:EtOAc 19:1). FTIR
(ATR): 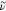 [cm–1] = 2976, 2765,
1777, 1670, 1596, 1506, 1446, 1411, 1371, 1285, 1234, 1209, 1156. 1H NMR (700 MHz, CDCl3): δ (ppm) = 8.09–8.02
(m, 2H), 7.20–7.13 (m, 2H), 4.76–4.55 (m, 1H), 3.38–3.19
(m, 2H), 2.90–2.68 (m, 1H), 1.47–1.30 (m, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ
(ppm) = 199.8, 188.6, 166.0 (d, J = 255.9 Hz), 132.5
(d, J = 3.0 Hz), 132.0 (d, J = 9.5
Hz), 115.8 (d, J = 22.0 Hz), 76.7, 52.4, 20.5, 20.4. 19F NMR (659 MHz, CDCl3): δ (ppm) = –
104.33 (m). HRMS (GC-APCI, QTOF): C12H12FO2, calcd.: 207.0816, found: 207.0814 [M + H]+.
[cm–1] = 2976, 2765,
1777, 1670, 1596, 1506, 1446, 1411, 1371, 1285, 1234, 1209, 1156. 1H NMR (700 MHz, CDCl3): δ (ppm) = 8.09–8.02
(m, 2H), 7.20–7.13 (m, 2H), 4.76–4.55 (m, 1H), 3.38–3.19
(m, 2H), 2.90–2.68 (m, 1H), 1.47–1.30 (m, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ
(ppm) = 199.8, 188.6, 166.0 (d, J = 255.9 Hz), 132.5
(d, J = 3.0 Hz), 132.0 (d, J = 9.5
Hz), 115.8 (d, J = 22.0 Hz), 76.7, 52.4, 20.5, 20.4. 19F NMR (659 MHz, CDCl3): δ (ppm) = –
104.33 (m). HRMS (GC-APCI, QTOF): C12H12FO2, calcd.: 207.0816, found: 207.0814 [M + H]+.
(1S,6R,8R)-8-Benzoylbicyclo[4.2.0]octan-7-one (3m)
Prepared
according to GP2 from SCP 1e (76 mg) and
sulfonium ylide 2a (93 mg) for 30 min. Purification by
flash column chromatography (SiO2, n-pentane/EtOAc
49:1) afforded cyclobutanone 3m (37 mg, 0.16 mmol, 54%)
as a pale-yellow solid. On a 1 mmol scale: Prepared according to GP2. A flame-dried (2×) 100 mL Schlenk flask was charged
with SCP 1e (252 mg), K3PO4 (318
mg) and corresponding sulfonium ylide 2a (310 mg). The
flask was evacuated and backfilled with argon (2×) before anhydrous
Toluene (40 mL) was added. The reaction mixture was degassed by freezing
pump thaw (1×), placed into a preheated oil bath at 100 °C
and stirred for the 30 min. Afterward the reaction mixture was cooled
to r.t., solids were removed by filtration through filter paper and
the filtrate was concentrated under reduced pressure. Purification
by flash column chromatography (SiO2, n-pentane/EtOAc 49:1) yielded respective cyclobutanone 3m (136 mg, 0.60 mmol, 60%) as a pale-yellow solid. m.p.: 62–63
°C. Rf: 0.29 (n-pentane:EtOAc
19:1). FTIR (ATR): 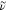 [cm–1] = 2929, 2854,
1761, 1663, 1596, 1581, 1448, 1340, 1326, 1309, 1285, 1264, 1221,
1192, 1159, 1137, 1034, 1024. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.21–7.97 (m, 2H), 7.68–7.39
(m, 3H), 4.69 (dd, J = 3.6, 1.7 Hz, 1H), 3.54 (dd, J = 10.2, 7.9 Hz, 1H), 3.28–3.00 (m, 1H), 2.29–2.09
(m, 1H), 2.03–1.83 (m, 1H), 1.68–1.51 (m, 2H), 1.51–1.46
(m, 1H), 1.41–1.11 (m, 3H). 13C{1H} NMR
(126 MHz, CDCl3): δ (ppm) = 200.5, 190.8, 136.2,
133.3, 128.9, 128.6, 76.7, 56.9, 27.6, 24.9, 22.3, 22.2, 21.1. HRMS
(GC-APCI, QTOF): C15H17O2, calcd.:
229.1223, found: 229.1225 [M + H]+.
[cm–1] = 2929, 2854,
1761, 1663, 1596, 1581, 1448, 1340, 1326, 1309, 1285, 1264, 1221,
1192, 1159, 1137, 1034, 1024. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.21–7.97 (m, 2H), 7.68–7.39
(m, 3H), 4.69 (dd, J = 3.6, 1.7 Hz, 1H), 3.54 (dd, J = 10.2, 7.9 Hz, 1H), 3.28–3.00 (m, 1H), 2.29–2.09
(m, 1H), 2.03–1.83 (m, 1H), 1.68–1.51 (m, 2H), 1.51–1.46
(m, 1H), 1.41–1.11 (m, 3H). 13C{1H} NMR
(126 MHz, CDCl3): δ (ppm) = 200.5, 190.8, 136.2,
133.3, 128.9, 128.6, 76.7, 56.9, 27.6, 24.9, 22.3, 22.2, 21.1. HRMS
(GC-APCI, QTOF): C15H17O2, calcd.:
229.1223, found: 229.1225 [M + H]+.
(1S,6R,8R)-8-(4-Fluorobenzoyl)bicyclo[4.2.0]octan-7-one (3n)
Prepared according to GP2 from SCP 1e (76 mg) and sulfonium ylide 2b (101 mg) for 30 min.
Purification by flash column chromatography (SiO2, n-pentane/EtOAc 49:1) afforded the cyclobutanone 3n (30 mg, 0.12 mmol, 41%) as a colorless solid. m.p.: 50–51
°C. Rf: 0.30 (n-pentane:EtOAc
19:1). FTIR (ATR):  [cm–1] = 2983, 2929,
2855, 1763, 1675, 1595, 1504, 1447, 1409, 1351, 1318, 1279, 1262,
1215, 1190, 1154, 1101, 1059, 1033, 1010. 1H NMR (500 MHz,
CDCl3): δ (ppm) = 8.16–8.04 (m, 2H), 7.19–7.13
(m, 2H), 4.64 (dd, J = 3.7, 1.8 Hz, 1H), 3.51 (ddt, J = 10.7, 7.4, 2.2 Hz, 1H), 3.15 (dddd, J = 11.0, 8.6, 7.5, 3.6 Hz, 1H), 2.23–2.11 (m, 1H), 2.00–1.88
(m, 1H), 1.67–1.51 (m, 2H), 1.51–1.47 (m, 1H), 1.36–1.24
(m, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 200.8, 189.4, 166.1 (d, J = 255.6
Hz), 132.8 (d, J = 2.8 Hz), 131.9 (d, J = 9.3 Hz), 116.0 (d, J = 22.2 Hz), 76.8, 57.1,
27.8, 25.0, 22.5, 22.5, 21.4. 19F NMR (471 MHz, CDCl3): δ (ppm) = – 104.46 (m). HRMS (GC-APCI, QTOF):
C15H16FO2, calcd.: 247.1129, found:
247.1126 [M + H]+.
[cm–1] = 2983, 2929,
2855, 1763, 1675, 1595, 1504, 1447, 1409, 1351, 1318, 1279, 1262,
1215, 1190, 1154, 1101, 1059, 1033, 1010. 1H NMR (500 MHz,
CDCl3): δ (ppm) = 8.16–8.04 (m, 2H), 7.19–7.13
(m, 2H), 4.64 (dd, J = 3.7, 1.8 Hz, 1H), 3.51 (ddt, J = 10.7, 7.4, 2.2 Hz, 1H), 3.15 (dddd, J = 11.0, 8.6, 7.5, 3.6 Hz, 1H), 2.23–2.11 (m, 1H), 2.00–1.88
(m, 1H), 1.67–1.51 (m, 2H), 1.51–1.47 (m, 1H), 1.36–1.24
(m, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 200.8, 189.4, 166.1 (d, J = 255.6
Hz), 132.8 (d, J = 2.8 Hz), 131.9 (d, J = 9.3 Hz), 116.0 (d, J = 22.2 Hz), 76.8, 57.1,
27.8, 25.0, 22.5, 22.5, 21.4. 19F NMR (471 MHz, CDCl3): δ (ppm) = – 104.46 (m). HRMS (GC-APCI, QTOF):
C15H16FO2, calcd.: 247.1129, found:
247.1126 [M + H]+.
(1S,6R,8R)-8-(4-Bromobenzoyl)bicyclo[4.2.0]octan-7-one (3o)
Prepared according to GP2 from SCP 1e (76 mg) and sulfonium ylide 2d (128 mg) for 30 min.
Purification by flash column chromatography (SiO2, n-pentane/EtOAc 49:1) afforded cyclobutanone 3o (28 mg, 0.09 mmol, 30%) as a colorless solid. m.p.: 87–88
°C. Rf: 0.39 (n-pentane:EtOAc
19:1). FTIR (ATR):  [cm–1] = 2937, 2849,
1771, 1666, 1577, 1395, 1322, 1281, 1217, 1192, 1066, 1036, 1002. 1H NMR (700 MHz, CDCl3): δ (ppm) = 7.97–7.90
(m, 2H), 7.67–7.56 (m, 2H), 4.63 (dd, J =
3.7, 1.8 Hz, 1H), 3.58–3.45 (m, 1H), 3.20–3.09 (m, 1H),
2.24– 2.12 (m, 1H), 2.01–1.91 (m, 1H), 1.64–1.59
(m, 1H), 1.55 (dddd, J = 13.1, 10.6, 9.2, 6.3 Hz,
2H), 1.36–1.24 (m, 3H). 13C{1H} NMR (176
MHz, CDCl3): δ (ppm) = 200.5, 190.0, 135.1, 132.2,
130.7, 129.0, 76.9, 57.2, 27.8, 25.0, 22.5, 22.5, 21.4. HRMS (GC-APCI,
QTOF): C15H16BrO2, calcd.: 307.0328,
found: 307.0328 [M + H]+.
[cm–1] = 2937, 2849,
1771, 1666, 1577, 1395, 1322, 1281, 1217, 1192, 1066, 1036, 1002. 1H NMR (700 MHz, CDCl3): δ (ppm) = 7.97–7.90
(m, 2H), 7.67–7.56 (m, 2H), 4.63 (dd, J =
3.7, 1.8 Hz, 1H), 3.58–3.45 (m, 1H), 3.20–3.09 (m, 1H),
2.24– 2.12 (m, 1H), 2.01–1.91 (m, 1H), 1.64–1.59
(m, 1H), 1.55 (dddd, J = 13.1, 10.6, 9.2, 6.3 Hz,
2H), 1.36–1.24 (m, 3H). 13C{1H} NMR (176
MHz, CDCl3): δ (ppm) = 200.5, 190.0, 135.1, 132.2,
130.7, 129.0, 76.9, 57.2, 27.8, 25.0, 22.5, 22.5, 21.4. HRMS (GC-APCI,
QTOF): C15H16BrO2, calcd.: 307.0328,
found: 307.0328 [M + H]+.
(1S,6R,8S)-8-Benzoyl-8-ethylbicyclo[4.2.0]octan-7-one (4)
A flame-dried (2×) microwave vial was charged with cyclobutanone 3m (23 mg, 0.10 mmol, 1.00 equiv) and K2CO3 (22 mg, 0.16 mmol, 1.60 equiv). The vial was evacuated and
backfilled with argon (2×) before anhydrous acetone (1.4 mL)
was added. The reaction mixture was placed into preheated oil bath
at 50 °C, then EtI (78 mg, 0.50 mmol, 0.04 mL, 5.00 equiv) was
added dropwise to the reaction mixture and stirred for 24 h. Upon
completion, the reaction mixture was cooled to r.t., the solids were
removed by filtration through filter paper and the mixture was concentrated
in vacuo. Purification by flash column chromatography
(SiO2, n-pentane/EtOAc 49:1) yielded ethyl-substituted
cyclobutanone 4 (8 mg, 0.03 mmol, 31%) as a colorless
oil. Rf: 0.38 (n-pentane:EtOAc
19:1). FTIR (ATR):  [cm–1] = 2932, 2857,
1777, 1667, 1595, 1449, 1412, 1238. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.23–7.73 (m, 2H), 7.70–7.35
(m, 3H), 3.58 (t, J = 8.7 Hz, 1H), 2.72 (td, J = 9.9, 7.2 Hz, 1H), 2.45–2.24 (m, 1H), 2.19–2.08
(m, 2H), 2.07–1.93 (m, 1H), 1.51–1.38 (m, 3H), 1.28–0.98
(m, 3H), 0.91 (t, J = 7.5 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 207.0,
196.3, 135.6, 132.9, 129.3, 128.3, 80.6, 52.1, 34.9, 31.2, 27.3, 22.4,
21.9, 20.4, 10.3. HRMS (GC-APCI, QTOF): C17H21O2, calcd.: 257.1547, found: 257.1543 [M + H]+.
[cm–1] = 2932, 2857,
1777, 1667, 1595, 1449, 1412, 1238. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.23–7.73 (m, 2H), 7.70–7.35
(m, 3H), 3.58 (t, J = 8.7 Hz, 1H), 2.72 (td, J = 9.9, 7.2 Hz, 1H), 2.45–2.24 (m, 1H), 2.19–2.08
(m, 2H), 2.07–1.93 (m, 1H), 1.51–1.38 (m, 3H), 1.28–0.98
(m, 3H), 0.91 (t, J = 7.5 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 207.0,
196.3, 135.6, 132.9, 129.3, 128.3, 80.6, 52.1, 34.9, 31.2, 27.3, 22.4,
21.9, 20.4, 10.3. HRMS (GC-APCI, QTOF): C17H21O2, calcd.: 257.1547, found: 257.1543 [M + H]+.
N-Methyl-2-(2-oxo-2-phenylethyl)cyclohexane-1-carboxamide (5)
A microwave vial was charged with cyclobutanone 3m (46 mg, 0.20 mmol, 1.00 equiv). Methylamine (40% in H2O, 0.80 mmol, 0.06 mL, 4.00 equiv) was added and the reaction
mixture was stirred for 4 h at r.t. The mixture was extracted twice
with CH2Cl2. The combined organic phases were
dried over MgSO4, filtered and concentrated under vacuum.
Purification by flash column chromatography (SiO2, n-pentane/EtOAc 1:1) yielded cyclohexane 5.
(26 mg, 0.10 mmol, 50%) as a pale-yellow solid. m.p.: 108–109
°C. Rf: 0.30 (n-pentane:EtOAc
1:1). FTIR (ATR):  [cm–1] = 3292, 2939,
2859, 1687, 1632, 1597, 1559, 1448, 1326, 1217, 1008. 1H NMR (700 MHz, CDCl3): δ (ppm) = 7.98–7.93
(m, 2H), 7.58–7.41 (m, 3H), 5.61 (s, 1H), 3.04 (dd, J = 6.8, 1.4 Hz, 2H), 2.74 (d, J = 4.8
Hz, 3H), 2.55 (dtd, J = 11.1, 7.0, 4.3 Hz, 1H), 2.46
(dt, J = 8.4, 4.4 Hz, 1H), 1.87–1.57 (m, 5H),
1.51–1.34 (m, 3H). 13C{1H} NMR (176 MHz,
CDCl3): δ (ppm) = 200.5, 175.3, 137.4, 133.2, 128.7,
128.3, 45.7, 39.5, 34.0, 29.5, 26.5, 26.2, 23.8, 23.0. HRMS (ESI,
Orbitrap): C16H22NO2, calcd.: 260.1645,
found: 260.1641 [M + H]+.
[cm–1] = 3292, 2939,
2859, 1687, 1632, 1597, 1559, 1448, 1326, 1217, 1008. 1H NMR (700 MHz, CDCl3): δ (ppm) = 7.98–7.93
(m, 2H), 7.58–7.41 (m, 3H), 5.61 (s, 1H), 3.04 (dd, J = 6.8, 1.4 Hz, 2H), 2.74 (d, J = 4.8
Hz, 3H), 2.55 (dtd, J = 11.1, 7.0, 4.3 Hz, 1H), 2.46
(dt, J = 8.4, 4.4 Hz, 1H), 1.87–1.57 (m, 5H),
1.51–1.34 (m, 3H). 13C{1H} NMR (176 MHz,
CDCl3): δ (ppm) = 200.5, 175.3, 137.4, 133.2, 128.7,
128.3, 45.7, 39.5, 34.0, 29.5, 26.5, 26.2, 23.8, 23.0. HRMS (ESI,
Orbitrap): C16H22NO2, calcd.: 260.1645,
found: 260.1641 [M + H]+.
(1S,6R,8S)-8-Benzoyl-8-cinnamylbicyclo[4.2.0]octan-7-one (7)
In a flame-dried (2×) 25 mL Schlenk flask, cyclobutanone 3m (68 mg, 0.30 mmol, 1.00 equiv), K2CO3 (66 mg, 0.48 mmol, 1.60 equiv) and cinnamyl sulfonium salt 614 (171 mg, 0.60 mmol, 2.00 equiv) were added.
The flask was evacuated and backfilled with argon (2×) before
anhydrous acetone (14 mL) was added. The reaction mixture was placed
into preheated oil bath at 45 °C and the reaction mixture was
stirred until full consumption of the starting material was indicated
by TLC analysis. Upon completion, the reaction mixture was cooled
to r.t., the solids were removed by filtration through filter paper
and the mixture was concentrated under vacuum. Purification by flash
column chromatography (SiO2, n-pentane/EtOAc
49:1) yielded cyclobutanone 7 (70 mg, 0.20 mmol, 68%)
as a colorless solid. m.p.: 88–87 °C. Rf: 0.32 (n-pentane:EtOAc 19:1). FTIR
(ATR):  [cm–1] = 2927, 2852,
1767, 1660, 1596, 1579, 1493, 1445, 1299, 1259, 1212, 1180, 1156,
1118, 1075, 1027. 1H NMR (300 MHz, CDCl3): δ
(ppm) = 8.04–7.99 (m, 2H), 7.60–7.43 (m, 3H), 7.39–7.26
(m, 3H), 7.26–7.13 (m, 2H), 6.29 (dt, J =
15.7, 1.2 Hz, 1H), 6.08 (ddd, J = 15.7, 7.9, 6.9
Hz, 1H), 3.64–3.44 (m, 1H), 3.17–2.92 (m, 2H), 2.79
(td, J = 9.8, 7.3 Hz, 1H), 2.21–1.99 (m, 2H),
1.50 (td, J = 12.7, 6.3 Hz, 3H), 1.24–0.96
(m, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 206.5, 196.7, 136.8, 136.1, 134.1, 133.1, 129.8,
128.6, 128.5, 127.7, 126.5, 123.7, 80.2, 52.4, 41.2, 34.3, 27.5, 22.6,
22.1, 20.6. HRMS (ESI, Orbitrap): C24H24O2Na, calcd.: 367.1669, found: 367.1667 [M + Na]+.
[cm–1] = 2927, 2852,
1767, 1660, 1596, 1579, 1493, 1445, 1299, 1259, 1212, 1180, 1156,
1118, 1075, 1027. 1H NMR (300 MHz, CDCl3): δ
(ppm) = 8.04–7.99 (m, 2H), 7.60–7.43 (m, 3H), 7.39–7.26
(m, 3H), 7.26–7.13 (m, 2H), 6.29 (dt, J =
15.7, 1.2 Hz, 1H), 6.08 (ddd, J = 15.7, 7.9, 6.9
Hz, 1H), 3.64–3.44 (m, 1H), 3.17–2.92 (m, 2H), 2.79
(td, J = 9.8, 7.3 Hz, 1H), 2.21–1.99 (m, 2H),
1.50 (td, J = 12.7, 6.3 Hz, 3H), 1.24–0.96
(m, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ (ppm) = 206.5, 196.7, 136.8, 136.1, 134.1, 133.1, 129.8,
128.6, 128.5, 127.7, 126.5, 123.7, 80.2, 52.4, 41.2, 34.3, 27.5, 22.6,
22.1, 20.6. HRMS (ESI, Orbitrap): C24H24O2Na, calcd.: 367.1669, found: 367.1667 [M + Na]+.
Acknowledgments
This research was supported by DAAD (Ph.D. Fellowship to I.A.). Stefan Braukmüller is gratefully acknowledged for measuring, evaluation, and discussion of the NMR experiments. We thank Burkhard Butschke for the X-ray diffraction experiments. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no. INST 40/575-1 FUGG (JUSTUS2 cluster).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.5c00167.
Detailed experimental procedures and analytical data for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Devi P.; Rutledge P. J. Cyclobutanone Analogues of β-Lactam Antibiotics: β-Lactamase Inhibitors with Untapped Potential?. ChemBioChem 2017, 18, 338–351. 10.1002/cbic.201600529. [DOI] [PubMed] [Google Scholar]; b Belluš D.; Ernst B. Cyclobutanones and Cyclobutenones in Nature and in Synthesis. Angew. Chem., Int. Ed. 1988, 27, 797–827. 10.1002/anie.198807971. [DOI] [Google Scholar]; c Namyslo J. C.; Kaufmann D. E. The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev. 2003, 103, 1485–1538. 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]; d Leemans E.; D’hooghe M.; De Kimpe N. Ring Expansion of Cyclobutylmethylcarbenium Ions to Cyclopentane or Cyclopentene Derivatives and Metal-Promoted Analogous Rearrangements. Chem. Rev. 2011, 111, 3268–3333. 10.1021/cr100295j. [DOI] [PubMed] [Google Scholar]
- a Lee-Ruff E.; Mladenova G. Enantiomerically pure cyclobutane derivatives and their use in organic synthesis. Chem. Rev. 2003, 103, 1449–1483. 10.1021/cr010013a. [DOI] [PubMed] [Google Scholar]; b Van Der Kolk M. R.; Janssen M. A. C. H.; Rutjes F. P. J. T.; Blanco-Ania D. Cyclobutanes in Small-Molecule Drug Candidates. ChemMedChem 2022, 17, e202200020 10.1002/cmdc.202200020. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hui C.; Liu Y.; Jiang M.; Wu P. Cyclobutane-Containing Scaffolds in Bioactive Small Molecules. Trends Chem. 2022, 4, 677–681. 10.1016/j.trechm.2022.04.006. [DOI] [Google Scholar]
- a Dembitsky V. M. Bioactive Cyclobutane-Containing Alkaloids. J. Nat. Med. 2007, 62, 1–33. 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]; b Hong Y. J.; Tantillo D. J. How Cyclobutanes Are Assembled in Nature—Insights from Quantum Chemistry. Chem. Soc. Rev. 2014, 43, 5042–5050. 10.1039/c3cs60452g. [DOI] [PubMed] [Google Scholar]; c Li J.; Gao K.; Bian M.; Ding H. Recent Advances in the Total Synthesis of Cyclobutane-Containing Natural Products. Org. Chem. Front. 2020, 7, 136–154. 10.1039/C9QO01178A. [DOI] [Google Scholar]
- a Otto P.; Huisgen R. Cycloadditionen Des Diphenylketens an 1,3-Diene. Tetrahedron Lett. 1968, 9, 4491–4495. 10.1016/S0040-4039(01)99168-3. [DOI] [Google Scholar]; b Binsch G.; Feiler L. A.; Huisgen R. Sterischer Ablauf Und Mechanismus Der Cycloadditionen Der Ketene an CC-Doppelbindungen. Tetrahedron Lett. 1968, 9, 4497–4501. 10.1016/S0040-4039(01)99169-5. [DOI] [Google Scholar]; c Huisgen R.; Feiler L. A.; Binsch G. Cycloadditionen der Ketene, V. Stereospezifische Cyclobutanon-Bildung aus Ketenen mit cis-trans-isomeren Vinyläthern. Chem. Ber. 1969, 102, 3460–3474. 10.1002/cber.19691021025. [DOI] [Google Scholar]; d Huisgen R.; Mayr H. Kinetics of 2 + 2 Cycloadditions of Diphenylketene to Enol Ethers; The Structure of the Orientation Complexes. Tetrahedron Lett. 1975, 16, 2965–2968. 10.1016/S0040-4039(00)75045-3. [DOI] [Google Scholar]
- For reviews, see:; a Snider B. B. Intramolecular Cycloaddition Reactions of Ketenes and Keteniminium Salts with Alkenes. Chem. Rev. 1988, 88, 793–811. 10.1021/cr00087a005. [DOI] [Google Scholar]; b Hyatt J. A.; Raynolds P. W. Ketene Cycloadditions. Org. React. 1994, 45, 159–237. 10.1002/0471264180.or045.02. [DOI] [Google Scholar]
- Sweis R. F.; Schramm M. P.; Kozmin S. A. Silver-Catalyzed [2 + 2] Cycloadditions of Siloxy Alkynes. J. Am. Chem. Soc. 2004, 126, 7442–7443. 10.1021/ja048251l. [DOI] [PubMed] [Google Scholar]
- Zanini M.; Cataffo A.; Echavarren A. M. Synthesis of Cyclobutanones by Gold(I)-Catalyzed [2 + 2] Cycloaddition of Ynol Ethers with Alkenes. Org. Lett. 2021, 23, 8989–8993. 10.1021/acs.orglett.1c03499. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Xie J.; Maulide N. Stereoselective, Dual-Mode Ruthenium-Catalyzed Ring Expansion of Alkynylcyclopropanols. J. Am. Chem. Soc. 2008, 130, 17258–17259. 10.1021/ja807894t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Yasukata T. A Catalytic Asymmetric Wagner–Meerwein Shift. J. Am. Chem. Soc. 2001, 123, 7162–7163. 10.1021/ja010504c. [DOI] [PubMed] [Google Scholar]
- For a recent review, see:; Biletskyi B.; Colonna P.; Masson K.; Parrain J.-L.; Commeiras L.; Chouraqui G. Small Rings in the Bigger Picture: Ring Expansion of Three- and Four-Membered Rings to Access Larger All-Carbon Cyclic Systems. Chem. Soc. Rev. 2021, 50, 7513–7538. 10.1039/D0CS01396J. [DOI] [PubMed] [Google Scholar]
- Rudolph M.; Wang T.; Shi S.; Hashmi S. K. Regioselectivity Switch: Gold(I)-Catalyzed Oxidative Rearrangement of Propargyl Alcohols to 1,3-Diketones. J. Org. Chem. 2012, 77, 7761–7767. 10.1021/jo301381z. [DOI] [PubMed] [Google Scholar]
- Zheng Z.; Wang Y.; Ma X.; Li Y.; Zhang L. Non-Diazo C–H Insertion Approach to Cyclobutanones through Oxidative Gold Catalysis. Angew. Chem., Int. Ed. 2020, 59, 17398–17402. 10.1002/anie.202003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Poteat C. M.; Jang Y.; Jung M.; Johnson J. D.; Williams R. G.; Lindsay V. N. G. Enantioselective Synthesis of Cyclopropanone Equivalents and Application to the Formation of Chiral β-Lactams. Angew. Chem., Int. Ed. 2020, 59, 18655–18661. 10.1002/anie.202006786. [DOI] [PubMed] [Google Scholar]; b Rivera R. M.; Jang Y.; Poteat C. M.; Lindsay V. N. G. General Synthesis of Cyclopropanols via Organometallic Addition to 1-Sulfonylcyclopropanols as Cyclopropanone Precursors. Org. Lett. 2020, 22, 6510–6515. 10.1021/acs.orglett.0c02303. [DOI] [PubMed] [Google Scholar]; c Jang Y.; Lindsay V. N. G. Synthesis of Cyclopentenones with Reverse Pauson–Khand Regiocontrol via Ni-Catalyzed C–C Activation of Cyclopropanone. Org. Lett. 2020, 22, 8872–8876. 10.1021/acs.orglett.0c03246. [DOI] [PubMed] [Google Scholar]; d Poteat C. M.; Lindsay V. N. G. Stereospecific Synthesis of Enantioenriched Alkylidenecyclobutanones via Formal Vinylidene Insertion into Cyclopropanone Equivalents. Org. Lett. 2021, 23, 6482–6487. 10.1021/acs.orglett.1c02303. [DOI] [PubMed] [Google Scholar]; e Jung M.; Muir J. E.; Lindsay V. N. G. Expedient Synthesis of Spiro[3.3]Heptan-1-Ones via Strain-Relocating Semipinacol Rearrangements. Tetrahedron 2023, 134, 133296 10.1016/j.tet.2023.133296. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Jang Y.; Deng W.; Sprague I. S.; Lindsay V. N. G. Divergent Synthesis of β-Fluoroamides via Silver-Catalyzed Oxidative Deconstruction of Cyclopropanone Hemiaminals. Org. Lett. 2023, 25, 5389–5394. 10.1021/acs.orglett.3c01992. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Rivera R. M.; Ferrin Z. R.; Lindsay V. N. G. Iron-Catalyzed Oxidative Rearrangement of Cyclopropanone Hemiaminals: General Access to Pyrroloindolones from Indoles. Org. Lett. 2024, 26, 4738–4743. 10.1021/acs.orglett.4c01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange M.; Werz D. B. Ring-Enlargement of in Situ Generated Cyclopropanones by the Reaction with Sulfonium Ylides: One-Pot Synthesis of Cyclobutanones. Org. Lett. 2024, 26, 9871–9876. 10.1021/acs.orglett.4c03661. [DOI] [PubMed] [Google Scholar]
- Podrugina T. A.; Alferova V. A.; Mironov A. V.; Matveeva E. D.; Gleiter R.; Zefirov N. S. Mixed arsonium–iodonium and sulfonium–iodonium ylides: synthesis and characteristics. Tetrahedron 2016, 72, 6955–6962. 10.1016/j.tet.2016.09.027. [DOI] [Google Scholar]
- Reeves C. M.; Eidamshaus C.; Kim J.; Stoltz B. M. Enantioselective construction of α-quaternary cyclobutanones by catalytic asymmetric allylic alkylation. Angew. Chem., Int. Ed. 2013, 52, 6718–6721. 10.1002/anie.201301815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.