

Abstract
A major barrier to the impact of genomic diagnosis in patients with congenital malformations is the lack of understanding regarding how sequence variants contribute to disease pathogenesis and whether this information could be used to generate patient-specific therapies. Congenital diaphragmatic hernia (CDH) is among the most common and severe of all structural malformations; however, its underlying mechanisms are unclear. We identified loss-of-function sequence variants in the epigenomic regulator gene SIN3A in two patients with complex CDH. Tissue-specific deletion of Sin3a in mice resulted in defects in diaphragm development, lung hypoplasia, and pulmonary hypertension, the cardinal features of CDH and major causes of CDH-associated mortality. Loss of SIN3A in the lung mesenchyme resulted in reduced cellular differentiation, impaired cell proliferation, and increased DNA damage. Treatment of embryonic Sin3a mutant mice with anacardic acid, an inhibitor of histone acetyltransferase, reduced DNA damage, increased cell proliferation and differentiation, improved lung and pulmonary vascular development, and reduced pulmonary hypertension. These findings demonstrate that restoring the balance of histone acetylation can improve lung development in the Sin3a mouse model of CDH.
INTRODUCTION
Congenital malformations remain the leading cause of infant mortality in the United States and are a major source of newborn morbidity (1). One of the most common and severe anomalies is congenital diaphragmatic hernia (CDH), which occurs in 1 of every 3000 to 3500 live births with a mortality rate of 10 to 50% (2–7). Although abnormal diaphragm development is the hallmark of the disease, the high rate of morbidity and mortality in patients with CDH is due to defects in lung and pulmonary vascular development causing lung hypoplasia and pulmonary hypertension (8, 9). Despite the frequency and severity of CDH, the underlying developmental mechanisms are unclear. Genetic studies in patients and families have identified a growing list of CDH candidate genes implicated in development of the diaphragm (10–13). Loss-of-function studies of these genes in animal models have demonstrated impaired diaphragm development and a direct role for genetic mutations in the mechanisms responsible for lung hypoplasia and pulmonary hypertension (14–19).
Epigenetic regulation of gene expression plays an important role throughout development and in physiological adaptation to birth (20–24). Defects in epigenetic regulation of gene expression have been implicated in the mechanisms responsible for human disease including neurodevelopmental disorders and structural malformations such as congenital heart disease and neural tube defects (22, 25–30). Global disruption of DNA methylation or histone acetylation, two of the most well-studied epigenetic mechanisms of gene regulation, results in early embryonic lethality (31–34). In the lungs, regulation of histone acetylation has been shown to be important for development and in the mechanisms responsible for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, lung cancer, and pulmonary hypertension (35–43). The balance of gene transcription and repression maintained by histone acetylation is controlled by the complementary activity of histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes (43–45). The tightly coordinated activity of these enzymes is necessary for rapid changes in gene expression that occur during development and in disease (46–49).
To direct the timing-, cell-, and genome region–specific action of histone deacetylases, HDAC1 and HDAC2 form multiprotein complexes (50–52). The Switch-insensitive 3 transcription regulator-histone deacetylase (SIN3)–HDAC complex is one of several such complexes that regulate histone acetylation (53–58). The SIN3-HDAC complex is most well-known for its role in transcriptional repression by HDAC and regulation of cell cycling during development (54–60). In addition to its central role in the SIN3-HDAC complex, SIN3 transcription regulator family member A (SIN3A) also activates transcription and has become recognized as a transcriptional coregulator whose function depends on interactions with a wide range of DNA binding cofactors (59, 61–67). During development, SIN3A plays multiple roles in organogenesis and cell lineage specification, whereas loss of SIN3A causes defects in energy metabolism and impaired cell cycling (68–74). Relevant to CDH, SIN3A was demonstrated to play a role in skeletal muscle cell development, maintenance, and function (75). In the developing lungs, loss of SIN3A in foregut endoderm–derived epithelium resulted in failure of branching morphogenesis and cell cycle arrest (76). In adult mice, conditional deletion of Sin3a in type 2 alveolar epithelial cells resulted in p53-dependent senescence and pulmonary fibrosis (77). Haploinsufficiency or sequence variants in the SIN3A gene have been reported in human patients with Witteveen-Kolk syndrome (OMIM 613406), leading to characteristic neurocognitive impairment, growth and feeding difficulties, and distinctive facial features (78–83). Despite the importance of SIN3A during development, to our knowledge, there are no previous reports of SIN3A sequence variants in patients with congenital malformations.
To understand the genetic and developmental mechanisms responsible for CDH, we conducted whole-genome sequencing in patients and family members with the disease. Small deletions that resulted in heterozygous loss of function of SIN3A were identified in two patients with complex CDH. To determine the role that SIN3A plays in the development of the diaphragm and lung mesenchyme, we conducted tissue-specific deletion of Sin3a in mice. We found that SIN3A is required in the skeletal muscle and mesothelium for diaphragm formation. Furthermore, in the lung mesenchyme, SIN3A is required for cellular differentiation, cell cycling, and regulation of DNA damage. Although SIN3A controls gene expression through multiple mechanisms, we found that loss of SIN3A resulted in an imbalance of histone acetylation and deacetylation, which was restored by embryonic inhibition of HAT.
RESULTS
SIN3A loss-of-function sequence variants were identified in patients with CDH
To identify genetic mechanisms responsible for CDH, whole-genome or -exome sequencing was conducted on 827 patient and parent trios enrolled in the Diaphragmatic Hernia Research and Exploration; Advancing Molecular Science (DHREAMS) study (13, 84). Similar to the epidemiological data reported previously for patients with CDH (2–4), 59% of this cohort was male, and 33.5% had complex CDH with additional anomalies including congenital heart disease, neurodevelopmental disorders, skeletal anomalies, genitourinary anomalies, and gastrointestinal anomalies (13). Among the 1153 protein-coding variants identified were 418 gene damaging variants found in 318 patients with CDH (38.4%) (13) including de novo, two– and seven–base pair frameshift deletions in the SIN3A gene in two patients with complex CDH (Fig. 1, A and B). The first patient had a two–base pair deletion and frameshift variant in exon 18 and died during the newborn period with a severe, multisystem phenotype including right-sided CDH, respiratory failure, coarctation of the aorta, imperforate anus, and limb abnormalities (Fig. 1B and table S1). The second patient had a seven–base pair deletion and frameshift variant in exon 11 and also exhibited a multisystem phenotype with left-sided CDH, unilateral pelvic kidney, and a palate defect but less severe lung and pulmonary vascular disease and developed schizoaffective disorder (Fig. 1B and table S1). Although growth and neurodevelopmental disorders have been described in patients with SIN3A gene variants and Witteveen-Kolk syndrome (80, 83), CDH has not previously been identified in patients with damaging SIN3A sequence variants.
Fig. 1. SIN3A sequence variants were present in patients with CDH, and SIN3A is expressed in the developing lungs and diaphragm.

(A) Sanger sequencing of patient and parent trios identified de novo sequence variants in two patients with complex CDH. (B) Characteristics of the patients are provided. (C to E) Immunofluorescence images of SIN3A (magenta) in the lungs and diaphragms of embryonic mice. (D) and (E) represent higher-magnification images of (C). (F to H) Immunofluorescence images of SIN3A in embryonic mouse lungs at E14 (F) with E-cadherin (G, ECAD, green) in the epithelium and vimentin (H, VIMENTIN, green) in the mesenchyme (white indicates overlapping immunofluorescence staining). Scale bars, 100 μm.
To determine whether disruption of SIN3A function might lead to CDH phenotypes, including diaphragm malformation, pulmonary hypertension, or lung hypoplasia, expression of Sin3a was characterized in mice. Sin3a expression was identified in the developing diaphragm and throughout the lungs at embryonic day 12 (E12, Fig. 1, C to E). In addition to its reported expression in the endoderm-derived epithelium (Fig. 1G) (76), Sin3a was expressed throughout the developing lung mesenchyme during embryonic and early postnatal stages (Fig. 1, F to H, and fig. S1, A to E) including in Platelet-derived growth factor receptor alpha (Pdgfrα)–expressing myofibroblasts, Perilipin 2 (Plin2)–expressing lipofibroblasts, Transgelin (Sm22α)–expressing airway and vascular smooth muscle cells, and ETS transcription factor (Erg)–expressing lung endothelial cells (fig. S1, F to I).
SIN3A is required in the skeletal muscle and mesothelium for diaphragm development
To determine whether SIN3A is required for diaphragm formation, conditional deletion of Sin3a was conducted using cell-specific Cre-recombination in cell populations that compose the diaphragm. To investigate the role of SIN3A in the developing diaphragm fibroblasts, Prx1-Cre was used to induce recombination in lateral plate mesoderm–derived fibroblast cells that express Sin3a (Fig. 2, A and B). Compared with controls (Fig. 2C and fig. S2, A to E), Prx1-Cre; Sin3a conditional knockout (CKO) embryos exhibited abnormal limb formation (fig. S2F) but normal diaphragm development with Transcription factor 4 (Tcf4)–expressing fibroblast cells, Myogenic differentiation 1 (Myod1)–expressing skeletal muscle cells, and WT1 transcription factor (Wt1)–expressing mesothelial cells (Fig. 2D and fig. S2, G to J).
Fig. 2. Conditional deletion of Sin3a in the developing mouse diaphragm.
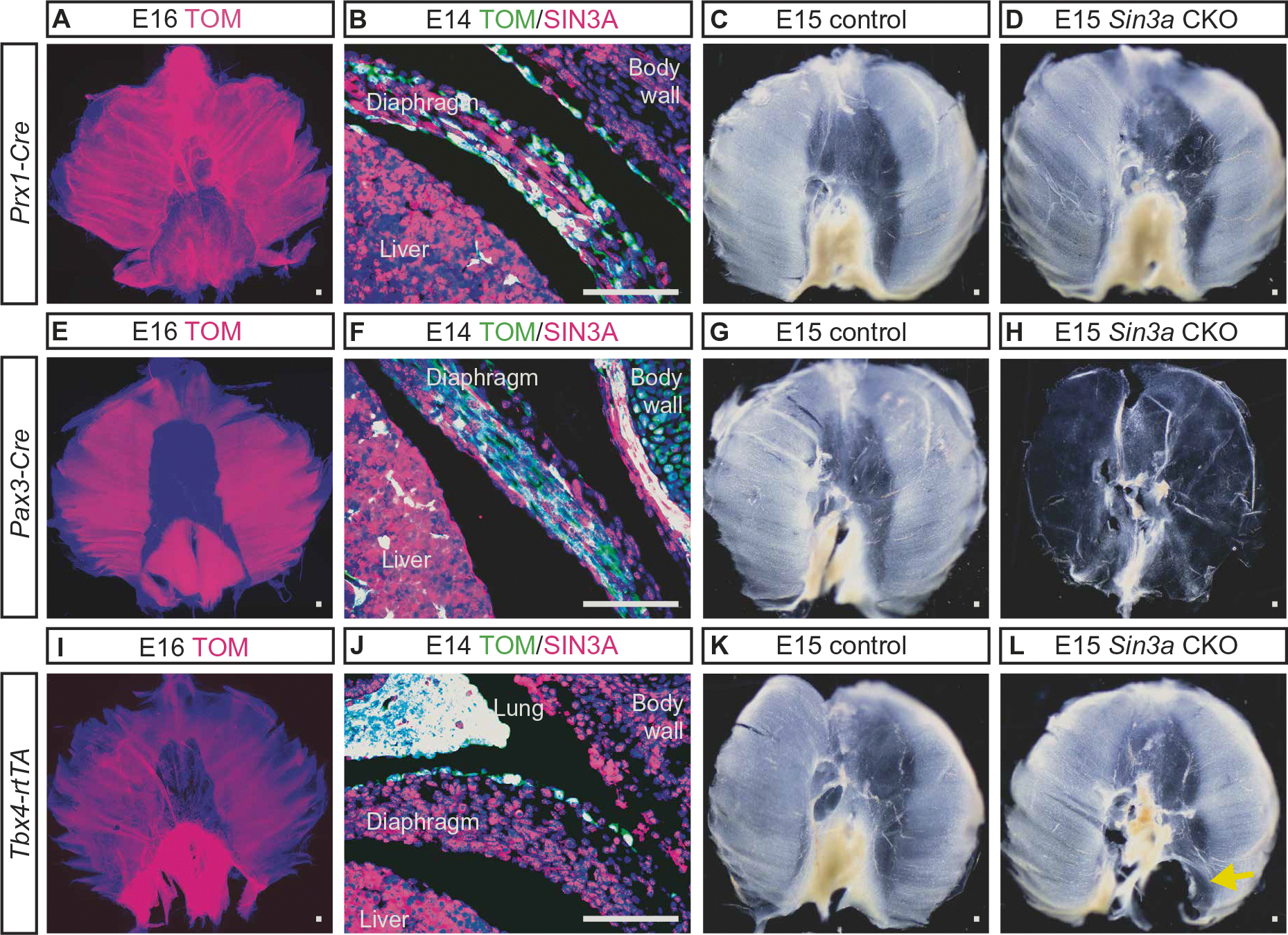
(A to D) Prx1-Cre was used to induce recombination in diaphragm fibroblast cells that express Sin3a. (A) Whole-mount image of E16 Rosa Tomato fluorescence reporter mouse (Rosa Tomato) diaphragm with Prx1-Cre–induced recombination shows recombined Tomato-positive (TOM, magenta) cells throughout the diaphragm. (B) Immunofluorescence image of E14 Rosa Tomato mouse with Prx1-Cre–induced recombination shows TOM (magenta) and SIN3A (green; white indicates overlapping immunofluorescence). (C and D) Whole-mount diaphragm images of E15 control (C) and Prx1-Cre Sin3a CKO deletion embryos (D). (E to H) Pax3-Cre was used to induce recombination in diaphragm skeletal muscle cells that express Sin3a. (E) Whole-mount image of E16 Rosa Tomato mouse diaphragm with Pax3-Cre–induced recombination shows recombined TOM-positive cells (magenta). (F) IF image of E14 Rosa tomato with Pax3-Cre–induced recombination shows TOM (magenta) and SIN3A (green; white indicates overlapping immunofluorescence). (G and H) Whole-mount diaphragm images of E15 control (G) and Pax3-Cre Sin3a CKO embryos (H). (I to L) Tbx4-rtTA; Tet-o-Cre was used to induce recombination in mesothelial cells that expresses Sin3a. (I) Whole-mount image of E16 Rosa Tomato mouse diaphragm with Tbx4-rtTA; Tet-o–induced recombination shows recombined TOM-positive cells (magenta). (J) IF image of E14 Rosa tomato with Pax3-Cre–induced recombination showing TOM (magenta) and SIN3A (green; white indicates overlapping immunofluorescence). (K and L) Whole-mount diaphragm images of E15 control (K) and Tbx4-rtTA; Tet-o-Cre Sin3a CKO embryos (L) with defect in the left posterior lateral portion of the diaphragm (L, yellow arrow). Scale bars, 100 μm.
To determine whether SIN3A is required in the skeletal muscle during diaphragm development, Pax3-Cre was used to induce recombination in somatic mesoderm-derived skeletal muscle that expresses Sin3a (Fig. 2, E and F). Compared with controls (Fig. 2G and fig. S2, A to E), Pax3-Cre; Sin3a CKO embryos had a thin, membranous, and muscle-less diaphragm in addition to other structural defects including anencephaly (Fig. 2H and fig. S2, K to O). The diaphragms of Pax3-Cre; Sin3a CKO embryos did not contain Tcf4-expressing fibroblast or Myod1-expressing skeletal muscle cells and were composed exclusively of Wt1-expressing mesothelial cells (fig. S2, M to O).
To determine whether SIN3A is required in diaphragm mesothelial cells, Tbx4-rtTA; Tet-o-Cre was used to induce recombination in the developing mesothelium that expresses Sin3a beginning at E6 (Fig. 2, I and J). Compared with controls (Fig. 2K and fig. S2, A to E), deletion of Sin3a using this approach resulted in a left-sided, posterior-lateral defect of the diaphragm with herniation of the stomach, intestine, and liver into the left thorax (Fig. 2L and fig. S2, P and Q). This left posterior-lateral diaphragm defect is seen in most patients with CDH. The remaining diaphragms of Tbx4-rtTA; Tet-o-Cre; Sin3a CKO embryos contained Tcf4-expressing fibroblast, Myod1-expressing skeletal muscle, and Wt1-expressing mesothelial cells (Fig. 2, R to T). At the site where the diaphragm failed to close, there was decreased expression of Tcf4, normal expression of Myod1, and increased and disorganized expression of Wt1 (fig. S2, U to W). Together, these data suggested that SIN3A is required in both the somatic mesoderm-derived skeletal muscle and the mesothelium during diaphragm development.
SIN3A is required for development of the lung mesenchyme
To investigate the role of SIN3A during lung development, Tbx4-rtTA; Tet-o-Cre was used to induce deletion of Sin3a beginning at E6 (fig. S3A), resulting in recombination throughout the lung mesenchyme and in the diaphragm (fig. S3B). Tbx4-rtTA; Tet-o-Cre; Sin3a CKO mice were born at the expected Mendelian ratio but died after birth (fig. S3C). Compared with controls (fig. S3D), deletion of Sin3a using this approach resulted in left CDH with liver herniation into the thorax (fig. S3E) and lung hypoplasia (fig. S3F). Histological analysis of the embryonic thorax revealed that, compared with controls (fig. S3G), Tbx4-rtTA; Tet-o-Cre; Sin3a CKO mice had left-sided CDH, rightward deviation of the heart, liver herniation, and lung hypoplasia (fig. S3H). To determine whether lung hypoplasia in Tbx4-rtTA; Tet-o-Cre; Sin3a CKO mice occurred independent of mechanical compression by herniated abdominal organs, mutant lungs were compared to controls at E12, before organ herniation (fig. S3I). Despite the loss of SIN3A, E12 Tbx4-rtTA; Tet-o-Cre; Sin3a CKO lungs were similar to controls (fig. S3I).
To investigate the role of SIN3A during later stages of lung mesenchymal development without mechanical compression caused by herniated abdominal organs, doxycycline was used to induce recombination throughout the lung mesenchyme beginning at E12 (Fig. 3, A and B). Using this approach, compared with controls that had Sin3a expression throughout the lungs (Fig. 3C), Tbx4-rtTA; Tet-o-Cre; Sin3a CKO mice (referred to hereafter as Sin3a CKO) lacked expression of Sin3a in the mesenchyme but retained expression in the epithelium (Fig. 3D). Despite appearing normal at birth and surviving into adulthood, Sin3a CKO mice did not gain weight as rapidly as controls (Fig. 3, E and F). Histological analysis at postnatal day 28 (P28) revealed that, compared with controls (Fig. 3G), Sin3a CKO mice had emphysematous dilation of the distal airspaces (Fig. 3H). Like newborn patients with CDH, Sin3a CKO mice had defects in pulmonary vascular development (Fig. 3, I to L) including decreased lung vessel number and increased vascular smooth muscle wall thickness (Fig. 3, K and L) as well as pulmonary hypertension with right ventricular hypertrophy, increased pulmonary vascular resistance, decreased right ventricular function, and increased right ventricular peak systolic pressure (Fig. 3, M to P). Despite having normal lung size at birth (Fig. 3Q), the first histological evidence of abnormal lung structure was present in Sin3a CKO mice at P0 with thickened interstitium and simplified air spaces (Fig. 3, R to T). Pulmonary vascular defects were also present at P0 in Sin3a CKO mice with decreased lung vessel number but not vascular smooth muscle hypertrophy (Fig. 3, U to X).
Fig. 3. SIN3A is required in the lung mesenchyme.
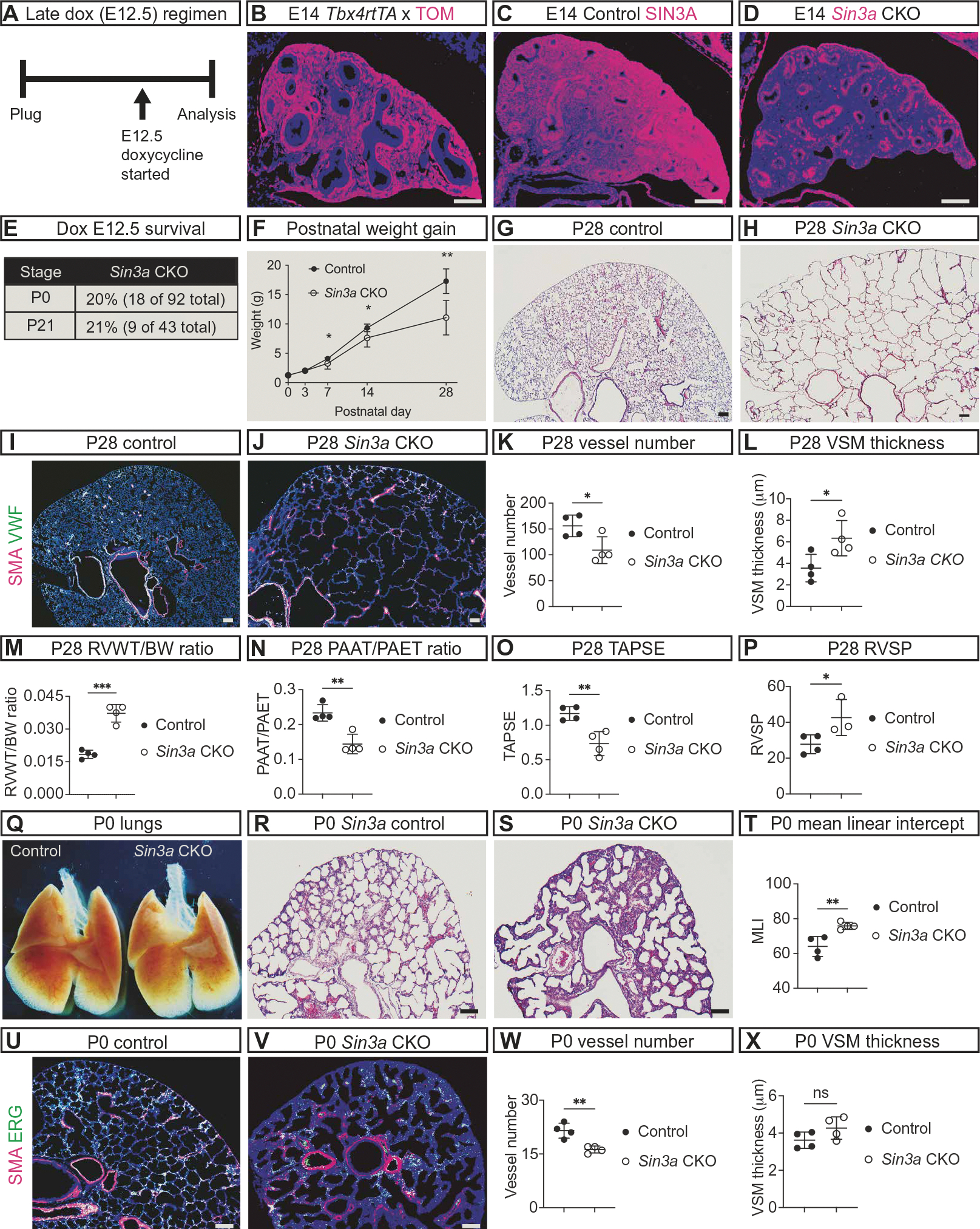
(A) Schematic showing late embryonic (E12) induction of Tbx4-rtTA; Tet-o-Cre with doxycycline (Dox). (B) Immunofluorescence image of E14 Rosa Tomato mouse lung with late (E12) dox induction of Tbx4-rtTA; Tet-o-Cre–mediated recombination shows recombined Tomato-positive cells (TOM, magenta) throughout the lung. (C and D) Images of SIN3A immunofluorescence (magenta) showing Sin3a expression at E14 in control (C) and Sin3a CKO lungs (D). (E) Percentages of Sin3a CKO that survived to P0 and P21. (F) Graph depicting weight gain of control and Sin3a CKO mice from P0 to P28 (P7 *P = 0.03, P14 *P = 0.04, P28 **P = 0.002). (G and H) Histological sections of P28 control (G) and Sin3a CKO (H) lungs. (I to L) Immunofluorescence images of von Willebrand factor (VWF, green) for endothelial cells and smooth muscle alpha actin (SMA, magenta) for smooth muscle cells at P28 in control (I) and Sin3a CKO (J) lungs with graphs comparing the number of vessels (K, *P = 0.03) and width of vascular smooth muscle (L, VSM, *P = 0.04). (M to P) Graphs comparing pulmonary vascular physiology data in P28 control and Sin3a CKO mice including echocardiographic measurement of right ventricular hypertrophy (M, RVWT/BW ***P = 0.0008), pulmonary vascular resistance (N, PAAT/PAET ratio **P = 0.003), and right ventricular function (O, TAPSE **P = 0.008), with peak systolic right ventricular pressure measured by catheterization of the right ventricular (P, RVSP *P = 0.04). (Q to T) Whole-mount image of P0 lungs from control and Sin3a CKO embryos (Q) with histological sections from control (R) and Sin3a CKO (S) lungs and graph showing comparison of airspace simplification (T, MLI **P = 0.008). (U to X) Immunofluorescence images of ETS transcription factor (ERG, green) for endothelial cells and smooth muscle (SMA, magenta) from P0 control (U) and Sin3a CKO (V) lungs and graphs comparing the number of vessels (W, **P = 0.004) and width of smooth muscle (X, P = 0.13). RVWT/BW, right ventricle wall thickness to body weight ratio; PAAT/PAET, pulmonary artery acceleration time to pulmonary artery ejection time ratio; TAPSE, tricuspid valve annular plane excursion during systole; RVSP, right ventricle peak systolic pressure; MLI, mean linear intercept. Statistical comparisons of control and Sin3a CKO mice were made using unpaired t test with statistical significance determined if the P value was <0.05. Scale bars, 100 μm.
SIN3A is required in the lung mesenchyme for myofibroblast, extracellular matrix, endothelial, and alveolar epithelial cell development
To determine the impact of SIN3A loss of function during lung mesenchymal development, the number and type of lung mesenchymal cells were analyzed in control and Sin3a CKO lungs. Compared with controls (Fig. 4A), Sin3a CKO lungs had fewer mesenchymal cells (Fig. 4, B and C). Loss of SIN3A resulted in decreased Pdgfrα–green fluorescent protein (GFP)–labeled myofibroblast precursor cells, extracellular matrix, and Elastin gene expression compared with controls (Fig. 4, D to I). In contrast, lipofibroblast cells were not affected by the loss of SIN3A (Fig. 4, J to L). These data support a primary role for SIN3A in lung mesenchymal cell development in addition to its role in diaphragm formation. Supporting the idea that the mesenchyme plays a central role in directing embryonic lung development, mesenchyme-specific deletion of Sin3a resulted in decreased Erg-expressing alveolar endothelial cells, Surfactant protein C (Sftpc)–expressing type-2 alveolar epithelial cells, and Hop homeobox (Hopx)–expressing type-1 alveolar epithelial cells compared with controls (fig. S4, A to I).
Fig. 4. SIN3A is required in the lung mesenchyme for mesenchymal cell and extracellular matrix development.
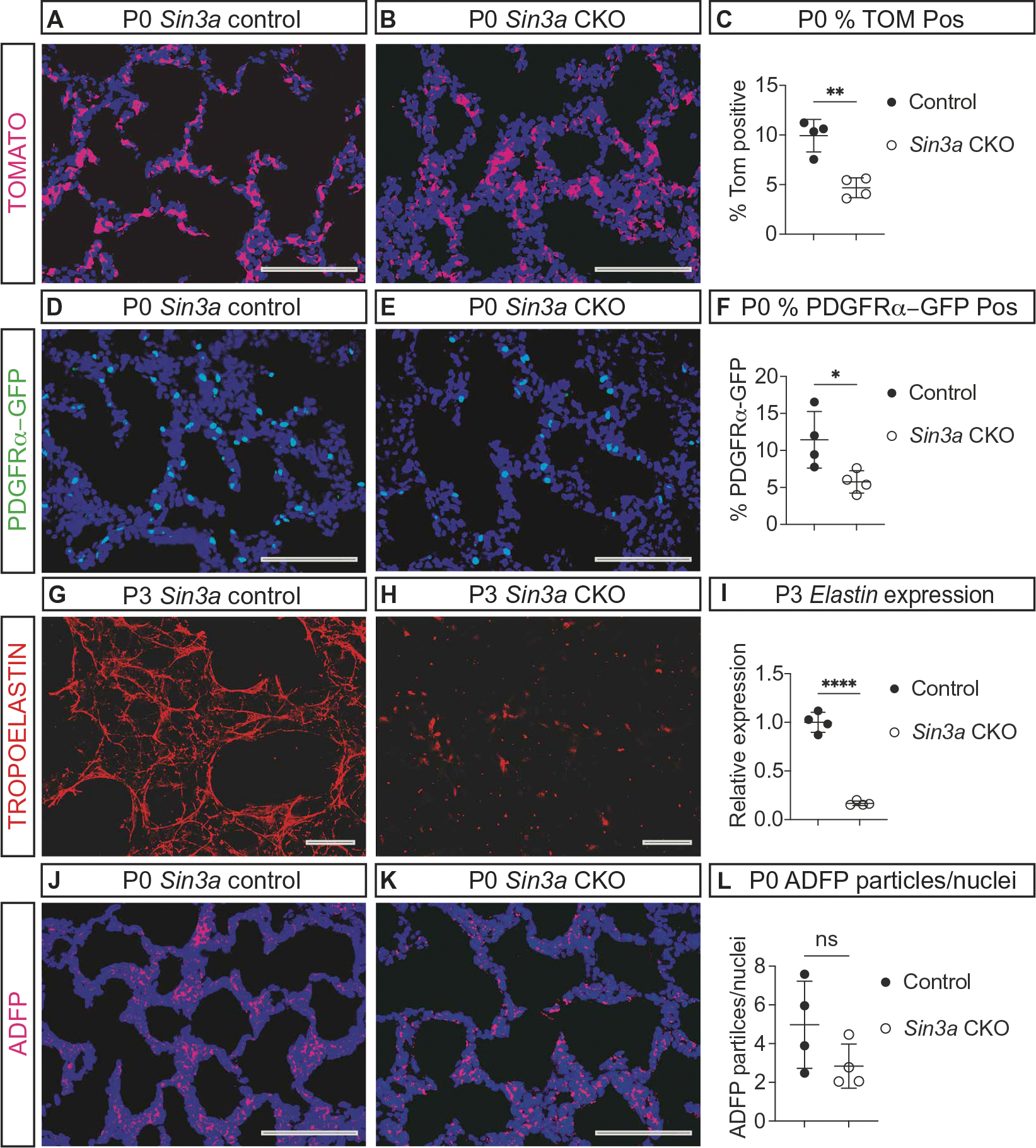
(A and B) Images of P0 Rosa Tomato (TOM, magenta) immunofluorescence with (E12) dox induction of Tbx4-rtTA; Tet-o-Cre–mediated recombination shows recombined TOM-positive cells (magenta) in P0 control (A) and Sin3a CKO (B) mice. (C) Graph comparing the percentage of TOM-positive recombined cells in P0 control and Sin3a CKO mice (**P = 0.003). (D and E) Images of Pdgfrα-GFP (green) immunofluorescence showing myofibroblast precursor cells at P0 in control (D) and Sin3a CKO (E) lungs. (F) Graph comparing the percentage of Pdgfrα-GFP–positive cells (*P = 0.03). (G and H) Images of TROPOELASTIN (red) immunofluorescence in P3 control (G) and Sin3a CKO (H) lungs. (I) Graph comparing expression of Elastin in whole-lung RNA extracts from P3 control and Sin3a CKO lungs (****P < 0.0001). (J and K) Images of adipose differentiation–related protein (ADFP, magenta) immunofluorescence in P0 control (J) and Sin3a CKO (K) lungs. (L) Graph comparing the ratio of ADFP-positive particles per nuclei in P0 control and Sin3a CKO lungs (P = 0.1). Statistical comparisons between control and Sin3a CKO mice were performed using unpaired t test with statistical significance determined if the P value was <0.05. Scale bars, 100 μm.
To explore the role of SIN3A in vascular development during lung formation, Cdh5-Cre was used to delete Sin3a in developing endothelial cells (fig. S5A). SIN3A was previously implicated in the transcriptional response of endothelial cells to hypoxia (85); however, the role of SIN3A during lung endothelial cell or vascular development has not been demonstrated previously. Compared with controls, Cdh5-Cre Sin3a CKO embryos initiated lung development and appeared healthy at E11 (fig. S5, B to E) but developed diffuse hemorrhage between E11 and E12 (fig. S5F), failed to undergo branching morphogenesis (fig. S5, G and H), and experienced disorganized vascular development in the lungs (fig. S5, I to L). These data demonstrated an essential role for SIN3A in endothelial cells for embryonic vascular development.
SIN3A is required for regulation of cell cycling and DNA damage
SIN3A has been demonstrated to direct multiple mechanisms necessary for development, including regulation of cell cycling and cellular differentiation. To determine whether SIN3A plays a similar role in the developing lung mesenchyme, cell proliferation, cell death, and DNA damage were investigated in Sin3a CKO mice. Compared to controls, Sin3a CKO lungs had a similar number of phosphohistone H3 (PPH3)–positive dividing cells (Fig. 5, A to C) but fewer 5-ethynyl-2 deoxyuridine (EDU)–positive cells undergoing G1 phase–to–S phase transition (Fig. 5, D to F). In addition, compared with controls, Sin3a CKO mice had more cleaved caspase 3 (CC3)–positive cells undergoing apoptosis (Fig. 5, G to I). Last, because loss of SIN3A has been shown to result in increased DNA damage (86), immunofluorescence staining for phosphorylated histone variant H2AX at Ser-139 (γH2AX) was used to quantify the number of cells with evidence of DNA damage. Compared with controls, Sin3a CKO lungs showed an increase in the number of γH2AX-positive cells (Fig. 5, J to L). These data suggested that loss of SIN3A in the lung mesenchyme resulted in decreased G1 phase–to–S phase transition, increased programmed cell death, and increased DNA damage.
Fig. 5. SIN3A is required for regulation of cell cycling, apoptosis, and DNA damage.

(A and B) Images of phosphohistone H3 (PPH3, magenta) immunofluorescence in E16 control (A) and Sin3a CKO (B) lungs. (C) Graph comparing the percentage of PPH3-positive cells in E16 control and Sin3a CKO lungs (P = 0.99). (D and E) Images of EDU (magenta) immunofluorescence in E16 control (D) and Sin3a CKO (E) lungs. (F) Graph comparing the percentage of EDU-positive cells in E16 control and Sin3a CKO lungs (**P = 0.004). (G and H) Images of cleaved caspase-3 (CC3, magenta) immunofluorescence in E16 control (G) and Sin3a CKO (H) lungs. (I) Graph comparing the percentage of CC3-positive cells in E16 control and Sin3a CKO lungs (*P = 0.04). (J and K) Images of phosphorylated histone H2AX (γH2AX, magenta) immunofluorescence in E16 control (J) and Sin3a CKO (K) lungs. (L) Graph comparing the percentage of γH2AX-positive cells in E16 control and Sin3a CKO lungs (**P = 0.007). Statistical comparison of control and Sin3a CKO mice was performed using an unpaired t test with statistical significance determined if the P value was <0.05. Scale bars, 100 μm.
Loss of SIN3A transcriptional regulation results in impaired lung mesenchymal cell differentiation
To identify the genetic mechanisms responsible for abnormal lung development due to the loss of SIN3A, RNA was collected from the lungs of four control and four Sin3a CKO mice at three stages (fig. S6A) including E16, when defects in cell cycling and DNA damage were first evident (Fig. 5, D to F and J to L) and when expression of Sin3a was highest (fig. S1A); P0, when the histological phenotype was first evident (Fig. 3, R to T) and when there were changes in the number of mesenchymal, endothelial, and alveolar epithelial cells (Fig. 4, A to F, and fig. S4, A to I); and P3, when there was a reduction in the extracellular matrix (Fig. 4, G to I). Expression of more than 200 genes was altered in Sin3a CKO mice at each stage (tables S2 to S4), and 37 genes were misregulated by the loss of SIN3A at all stages (fig. S6A). Consistent with the known role of SIN3A as a transcriptional repressor, deletion of Sin3a resulted in increased expression of most of these genes (fig. S6B). Among these 37 genes were those expressed in proliferative lung mesenchymal progenitor cells and that encode regulators of cell cycle progression or G1 phase–to–S phase transition, including Ring finger protein 26 (Rnf26), Cytochrome P450 family 26, subfamily A member 1 (Cy-p46a1), Cyclin E1 (Ccne1), HECT domain E3 ubiquitin protein ligase 3 (Hectd3), Ribosomal RNA processing 12 homolog (Rrp12), WD repeat domain 55 (Wdr55), and Transmembrane protein 231 (Tmem231) (fig. S6B).
To investigate the changes in gene expression that occurred specifically in lung mesenchymal cells, flow cytometry was used to isolate mesenchymal cells that underwent recombination indicated by the presence of a CRE-responsive red-fluorescence reporter protein from four control and four Sin3a CKO mice at E16. Compared with controls, Sin3a CKO lung mesenchymal cells had differential expression of more than 4000 genes (Fig. 6A). In contrast to the wholelung gene expression analysis (fig. S6, A and B), the number of genes with increased expression (2155) was similar to the number with decreased expression (2010) in recombined Sin3a CKO mesenchymal cells (Fig. 6A). Among the top 100 misregulated genes were 9 identified in the shared whole-lung gene expression experiment [Glucosylceramidase beta 1 (Gba), Intraflagellar transport 22 (Ift22), Quinoid dihydropteridine reductase (Qdpr), Ribonuclease P 40 subunit (Rpp40), Cyp46a1, Ecotropic viral integration site 5 like (Evi5l), RNA terminal phosphate cyclase-like 1 (Rcl1), Wdr55, and KLF transcription factor 16 (Klf16)] as well as genes expressed in myofibroblasts [Desmin (Des)], lipofibroblasts [Bromodomain adjacent to zinc finger domain 1A (Baz1a)], and extracellular matrix [Glutathione S-transferase omega 1 (Gsto1), ADAM metallopeptidase with thrombospondin type 1 motif 4 (Adamts4), Cartilage acidic protein 1 (Crtac1), Collagen type IX alpha 2 chain (Col9a2), Scavenger receptor class A member 3 (Scara3), and Collagen type XVI alpha 1 chain (Co-l16a1)] (Fig. 6A). Also among the top 100 misregulated genes were those encoding members of the apoptosis pathway [Superoxide dismutase 2 (Sod2), Inhibitor of growth family member 2 (Ing2), Apoptosis enhancing nuclease (Aen), and MAX network transcriptional repressor (Mnt)], involved in DNA repair [Centrosomal protein 164 (Cep164), Centromere protein X (Cenpx), Sirtuin 7 (Sirt7), and Chromatin assembly factor 1 subunit B (Chaf1b)], and SIN3-associated proteins [SAPs: Ing2, Inhibitor of growth family member 1 (Ing1), and Sin3 associated polypeptide (Sap30); Fig. 6A].
Fig. 6. SIN3A regulates a transcriptional program necessary for mesenchymal cell differentiation.

(A) Volcano plot of differentially regulated gene expression in recombined lung mesenchymal cells from E16 Sin3a CKO compared with controls determined by bulk RNA sequencing [genes with −log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301 (dotted lines) with increased expression are colored red, and those with decreased expression are colored blue]. (B) Heatmaps of significantly misregulated individual myofibroblast, matrix fibroblast, lipofibroblast, and mesenchymal proliferative progenitor cell gene expression (*genes with −log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301) in E16 control and Sin3a CKO recombined lung cells determined by bulk RNA sequencing and bar graphs of qRT-PCR gene expression performed using whole-lung RNA extracts from E16 control and Sin3a CKO embryos showing genes expressed in myofibroblasts (Myh11, *P = 0.03; Igfbp5, *P = 0.02; Des, *P = 0.02; and Pdgfrα, *P = 0.02), matrix fibroblasts (Ltbp4, *P = 0.04; Fn1, *P = 0.02; Col4a1, *P = 0.02; and Eln, ***P = 0.0005), lipofibroblasts (Lpl, P = 0.63; Fgf10, *P = 0.03; Tgm2, **P = 0.001; and Adfp, *P = 0.03), and proliferating mesenchymal progenitor cells (Uhrf1, *P = 0.03; Cks2, *P = 0.02; Dek, *P = 0.01; and Cks1b, *P = 0.01). Statistical comparisons of gene expression analyzed by qRT-PCR between control and Sin3a CKO whole-lung RNA were performed using unpaired t test. (C) Uniform Manifold Approximation and Projection (UMAP) of clustered recombined E16 lung mesenchymal cells from control and Sin3a CKO mice analyzed by scRNA-seq categorized into three groups: proliferative progenitor cells, undifferentiated transitional fibroblasts, and differentiating mesenchymal precursor cells. (D and E) Comparison of E16 control and Sin3a CKO UMAPs (D) and bar graphs comparing the percentage of cells within each cluster in the control and Sin3a CKO recombined lung mesenchymal cells (PMP, ***P = 2.67 × 10−26; IF1 and IF2, P = 0.59; Matrix and Lipo, *P = 0.02; Matrix and IF2, P = 0.15; Matrix, Myo, Lipo, and IF, ***P = 6.36 × 10−9; Myo, ***P = 7.63 × 10−39; Matrix, ***P = 7.43 × 10−10; PMP and IF2, P = 0.20; Ebf1, ***P = 7.86 × 10−26; PMP and IF1, P = 0.58; PMP, IF1, and IF2, ***P = 2.18 × 10−10; PMP and Myo, ***P = 6.66 × 10−5; Meso, ***P = 3.67 × 10−9; Immune, ***P = 4.92 × 10−6). Statistical comparison of the distribution of cells within each cluster between control and Sin3a CKO recombined lung mesenchymal cells was conducted using χ2 test. (F and G) UMAPs showing RNA velocity analysis of scRNA-seq data from E16 control and Sin3a CKO recombined lung mesenchymal cells (F) and heatmap of RNA velocity lengths (G). (H) Box and whisker plot comparing RNA velocity length in E16 control and Sin3a CKO recombined lung mesenchymal cell clusters (***P = 4.72 × 10−281), proliferative mesenchymal progenitor cells (PMP, ***P = 8.02 × 10−20 and PMP/myofibroblast, ***P = 2.20 × 10−9), and differentiating mesenchymal precursor cells (H, myofibroblast, ***P = 2.30 × 10−32; matrix fibroblast, ***P = 2.09 × 10−15; and Ebf1 fibroblast cells, ***P = 3.51 × 10−46). Statistical comparisons of RNA velocity between control and Sin3a CKO recombined lung mesenchymal cells analyzed by scRNA-seq were performed using Z test.
Because impaired mesenchymal cell development was a predominant phenotype observed in the Sin3a CKO lungs, we compared the differentially expressed genes with a recently published reference list of genes expressed in mesenchymal cell subpopulations and their precursors (87). Compared with controls, Sin3a CKO mesenchymal cells had decreased expression of myofibroblast, matrix fibroblast, and lipofibroblast genes (Fig. 6B). By contrast, expression of proliferative progenitor genes was increased in Sin3a CKO mesenchymal cells (Fig. 6B). These data were confirmed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) conducted using RNA from whole lungs of E16 Sin3a CKO and littermate control mice (Fig. 6B) and suggested that loss of SIN3A resulted in impaired lung mesenchymal cell differentiation.
To investigate the impact of Sin3a deletion on mesenchymal cell differentiation, recombined lung mesenchymal cells collected from two control and two Sin3a CKO embryos were analyzed using single-cell RNA sequencing (scRNA-seq) at E16 (fig. S7). Using this approach, recombined mesenchymal cells were organized into 14 clusters and broadly categorized as proliferative mesenchymal progenitor cells (PMPs), undifferentiated transitional mesenchymal cells, or differentiating mesenchymal precursor cells (Fig. 6C and fig. S8). Comparison of the relative number of cells within each cluster demonstrated that Sin3a CKO lung mesenchymal cells had an increase in proliferative progenitor cells and a decrease in differentiating myofibroblast, matrix, and Early B cell factor 1 (Ebf1)–expressing lung fibroblast precursor cells (Fig. 6 D and E; fig. S9; and table S5). The reduction in differentiating mesenchymal precursor cells was most notable in the myofibroblast cells (Fig. 6E, ***P = 7.63 × 10−39, and table S5). These results were similar to the histological data (Fig. 4, D to I) and sorted lung mesenchymal gene expression analysis (Fig. 6B) and suggested that defects in lung development in Sin3a CKO mice were due to failure of cell differentiation.
SIN3A is required for lung myofibroblast and matrix fibroblast differentiation
Analysis of spliced and unspliced transcripts using RNA velocity in the scRNA-seq data demonstrated the pattern of differentiation from proliferative mesenchymal cells to transitional undifferentiated mesenchymal cells and then to differentiating mesenchymal precursor cells in control and Sin3a CKO lung mesenchymal cells (Fig. 6F). To determine how Sin3a deletion affected the potential for cell differentiation, we analyzed the rate of differentiation indicated by RNA velocity lengths (88) that were reduced in mesenchymal cells collected from Sin3a CKO mice compared with controls (Fig. 6, G and H). The decrease in RNA velocity lengths was observed in PMPs, transitional undifferentiated mesenchymal cells, and differentiating mesenchymal precursor cells (Fig. 6H). These data suggest that loss of SIN3A resulted in the decreased differentiation potential of mesenchymal cells at all stages of differentiation.
To determine the genetic mechanisms responsible for reduced mesenchymal cell differentiation, Ingenuity Pathway Analysis (IPA) was used to identify pathways misregulated by the loss of SIN3A (Fig. 7A). Among the most misregulated canonical pathways and biological processes were pathways relevant for lung development and the phenotype observed in Sin3a CKO lungs, including idiopathic pulmonary fibrosis, lung formation, vascular development, entry into S phase, fibroblast cell death, and senescence, where SI-N3A has been shown to play a role in lung epithelial cells (Fig. 7A) (77). The misregulated upstream regulators included several that play an important role in mesenchyme and lung myofibroblast development and differentiation (Fig. 7, A to C), including decreased expression of myofibroblast and matrix fibroblast genes [Actin alpha 2, smooth muscle, aorta (Acta2); Myosin, heavy polypeptide 11, smooth muscle (Myh11); Tagln, Transforming growth factor–beta induced (TgAi); Des, Insulin-like growth factor binding protein 5 (IgAp5); Eln, Insulin-like growth factor binding protein 4 (IgAp4); Jun D proto-oncogene (Jund); and Matrix metallopeptidase 2 (Mmp2); Fig. 7B] and decreased expression of transforming growth factor–β (TGFB) and WNT pathway genes [FBJ osteosarcoma oncogene (Fos); Jun proto-oncogene (Jun); Collagen, type I, alpha 1 (Col1a1); Collagen, type III, alpha 1 (Col3a1); Collagen, type IV, alpha 1 (Col4a1); Collagen, type VI, alpha 3 (Col6a3); and Frizzled class receptor 2 (Fzd2); Fig. 7C]. These findings were confirmed using the RNA-seq analysis conducted on sorted lung mesenchymal cells (Fig. 6A) and gene expression analysis conducted on whole-lung RNA extracts at E16, which demonstrated a similar reduction in TGFB and wingless (WNT) pathway gene expression in Sin3a CKO lungs (Fig. 7C). Together, these data suggested that loss of SIN3A resulted in altered gene expression in pathways required for myofibroblast and matrix fibroblast differentiation.
Fig. 7. SIN3A is required for regulation of mesenchymal cell differentiation, cell cycling, DNA damage, and senescence.

(A) IPA of differential gene expression conducted using scRNA-seq data performed on recombined lung mesenchymal cells from E16 control and Sin3a CKO embryos. Shown are data demonstrating misregulation of pathways, processes, and upstream regulators of gene expression and representative differentially regulated genes. (B) Violin plots comparing the expression of genes that regulate fibroblast differentiation in E16 control and Sin3a CKO recombined lung mesenchymal cells analyzed by scRNA-seq in PMP, PMP and myofibroblast (PMP and Myo), Myo, matrix, and Ebf1-expressing fibroblast (Ebf1) cell clusters. (C) Violin plots comparing the expression of TGFB/SMAD and WNT pathway genes in E16 control and Sin3a CKO recombined lung mesenchymal cells analyzed by scRNA-seq in PMP, PMP and Myo, Myo, Matrix, and Ebf1 cell clusters as well as heatmaps comparing the TGFB/SMAD and WNT pathway genes with significantly different expression (*genes with −log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301) in recombined lung mesenchymal cells from E16 control and Sin3a CKO lungs analyzed by bulk RNA sequencing and bar graphs comparing the relative expression of Tgbi (*P = 0.01) and Wnt5b (*P = 0.01) determined by qRT-PCR analysis of whole-lung RNA extracts from E16 control and Sin3a CKO embryos. (D) Violin plots comparing the expression of genes that promote cell cycling (H19, Id3, and Ccnd2) and genes that promote cell death and senescence (Ccne1, Bax, Sod2, and Ing1) in E16 control and Sin3a CKO recombined lung mesenchymal cells analyzed by scRNA-seq and IPA of PMP, PMP and Myo, Myo, Matrix, and Ebf1 cell clusters. (E) Heatmaps comparing the expression of significantly misregulated genes (*genes with − log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301) that promote cell cycle progression and cell cycle arrest in recombined lung mesenchymal cells from E16 control and Sin3a CKO cells analyzed by bulk RNA-seq and bar graphs comparing the relative expression of genes that promote cell cycle progression (Osr1, P = 0.08; Prrx2, P = 0.78; Fgfr1, P = 0.52; Tgf-br2, P = 0.58) or cell cycle arrest (Cdkn2d, **P = 0.002; Cdc14a, ***P = 0.0007; Trp53inp1, *P = 0.04; Ddit **P = 0.001) conducted on whole-lung RNA extracts from E16 control and Sin3a CKO lungs. (F) Heatmaps comparing the expression of significantly misregulated DNA damage checkpoint genes or genes that are expressed during DNA repair (*genes with −log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301) in recombined lung mesenchymal cells from E16 control and Sin3a CKO cells analyzed by bulk RNA-seq and bar graphs comparing the relative expression of DNA damage checkpoint genes (Pidd1, *P = 0.03; Chek2, *P = 0.03; Rps27I, ***P = 0.0003; Usp28, *P = 0.04) or genes expressed during DNA repair (Cenpx, **P = 0.004; Sirt7, **P = 0.001; Aplf, *P = 0.02; Char1b, *P = 0.01) analyzed by qRT-PCR conducted on whole-lung RNA extracts from E16 control and Sin3a CKO lungs. (G) Heatmaps comparing the expression of genes that encode histone methyltransferase, histone demethylase, DNA methyltransferase, and DNA demethylase factors (*genes with –log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301) in recombined lung mesenchymal cells from E16 control and Sin3a CKO cells analyzed by bulk RNA-seq and bar graphs comparing the relative expression of histone methyltransferase (Kmt5b, *P = 0.03), histone demethylase, DNA methyltransferase, and DNA demethylase genes analyzed by qRT-PCR conducted on whole-lung RNA extracts from E16 control and Sin3a CKO lungs. (H) Heatmaps comparing the expression of genes that encode SIN3-HDAC complex core component factors or SAPS (*genes with −log10 false discovery rate >2 and log10 fold change >0.301 or <−0.301) in recombined lung mesenchymal cells from E16 control and Sin3a CKO cells analyzed by bulk RNA-seq and bar graphs comparing the relative expression of SIN3-HDAC core complex (Sap30, *P = 0.01; Sap18, *P = 0.02; and Suds3, **P = 0.004) or SAP genes (Brms1, *P = 0.02, Sap130, **P = 0.001; Arid4b, *P = 0.03; Ing1, **P = 0.005; Ing2, **P = 0.007; and Mxi1, *P = 0.02) analyzed by qRT-PCR conducted on whole-lung RNA extracts from E16 control and Sin3a CKO lungs. Statistical comparisons of gene expression analyzed by qRT-PCR in control and Sin3a CKO whole-lung RNA were performed using unpaired t test. Statistical significance was determined if the P value was <0.05.
SIN3A deletion altered gene expression, leading to impaired cell cycling, DNA damage, and senescence
In addition to the changes in mesenchymal cell differentiation, pathway and differential gene expression analysis of Sin3a CKO mesenchymal cells demonstrated decreased entry into S phase (Fig. 7A) with decreased expression of genes that promote cell cycle progression [H19 imprinted maternally expressed transcript (H19), Inhibitor of DNA binding 3 (Id3), and Cyclin D2 (Ccnd2)]; increased expression of genes associated with cell cycle arrest, cell death, or senescence [Ccne1, BCL2-associated X protein (Bax), Sod2, and Ing1]; and increased expression of Ing2, one of the SAPs (Fig. 7D). These findings were confirmed using the sorted lung mesenchymal RNA-seq analysis (Fig. 6A) and gene expression analysis conducted on RNA extracted from E16 control and Sin3a CKO lungs (Fig. 7, E to H). Loss of SIN3A resulted in reduced expression of genes that promote mesenchymal cell proliferation and increased expression of genes that promote cell cycle arrest (Fig. 7E). Sin3a CKO mice also had increased expression of genes expressed in response to DNA damage and altered expression of genes associated with DNA repair (Fig. 7F). These data supported the conclusion that SIN3A is required during lung development to promote mesenchymal cell differentiation and that loss of SIN3A resulted in impaired cell cycling and increased DNA damage.
Pathway and single-cell gene expression analysis demonstrated that loss of SIN3A resulted in increased expression of genes involved in senescence (Fig. 7, A and D). SIN3A has been demonstrated to regulate genes implicated in senescence, and loss of SIN3A in the lung epithelium was associated with senescence and pulmonary fibrosis (76, 77). Compared with controls, Sin3a CKO mesenchymal cells had differential expression of multiple genes associated with senescence (fig. S10, A to C). This expression analysis did not indicate an increase or decrease in senescence activation because genes that promote or repress senescence were both positively and negatively affected by the loss of SIN3A (fig. S10, A to C). These data suggested that loss of SIN3A in the mesenchyme resulted in an accumulation of progenitor cells, decreased cell cycling, increased DNA damage, and senescence.
SIN3A is required for controlling the balance of histone acetylation
SIN3A regulates gene expression during development through multiple mechanisms including histone and DNA methylation and demethylation (59, 89) and was recently demonstrated to play a role in pulmonary hypertension by regulating methylation of the bone morphogenetic protein receptor type 2 (BMPR2) promoter (90). Gene expression analysis of sorted lung mesenchymal cells from Sin3a CKO and control lungs at E16 demonstrated increased expression of genes that promote both histone methylation and demethylation, whereas genes that promote DNA methylation or demethylation were decreased (Fig. 7G). Gene expression analysis conducted on whole-lung RNA extracts from Sin3a CKO and control mice at E16 did not demonstrate a consistent trend in either histone or DNA methylation (Fig. 7G). Because more of the genes that direct histone methylation or demethylation were misregulated in Sin3a CKO recombined mesenchymal cells, immunofluorescence staining for transcriptional repressing methylation of histone 3 lysine 9 (H3K9Me2 and H3K9Me3) and lysine 27 (H3K27Me3) was conducted at E16 (fig. S11, A to I). These data demonstrated that no significant change in histone methylation was detectable despite the loss of SIN3A during lung mesenchymal development (fig. S11, A to I; P = 0.46 for H3K9Me2, P = 0.64 for H3K9Me3, and P = 0.57 for H3K27Me3).
SIN3A is known to act as a transcriptional repressor through its cooperative interaction with HDACs. The activity of the SIN3-HDAC complex is modulated by multiple cofactors (53, 91, 92). In recombined Sin3a CKO lung mesenchymal cells at E16 (Fig. 6A), expression of the SIN3-HDAC core complex and SAP genes was up-regulated, including Sap30, Sin3-associated polypeptide 18 (Sap18), Suppressor of defective silencing 3 homolog (Suds3), AT-rich interaction domain 4B (Arid4b), RB transcriptional corepressor 1 (Rb1), Breast cancer metastasis-suppressor 1 (Brms1), Sin3A-associated protein 130 (Sap130), Sin3-associated polypeptide (Sap25), Ing1, Ing2, Transcriptional regulator, SIN3B (Sin3b), and MAX interactor 1 dimerization protein (Mxi1) (Fig. 7H). These data were confirmed by gene expression analysis conducted on whole-lung RNA extracts from Sin3a CKO and control embryos at E16 demonstrating misregulated expression of both core SIN3-HDAC and SAP cofactors (Fig. 7H). By contrast, expression of HAT genes CREB-binding protein (Crebbp) and E1A-binding protein p300 (Ep300) was not changed in lung mesenchymal cells from Sin3a CKO embryos (fig. S12).
To determine whether loss of SIN3A in the lung mesenchyme had a direct impact on the balance of histone acetylation, immunofluorescence staining for transcriptional activating acetylation of H3K9 and H3K27 was conducted on Sin3a CKO and control lungs (fig. S13, A to F). These data demonstrated that loss of SIN3A resulted in increased histone acetylation in Sin3a CKO lungs (fig. S13, C and F). The imbalance of histone acetylation and deacetylation observed in Sin3a CKO lungs might be responsible for the observed changes in gene expression and the decrease in mesenchymal cell differentiation, decrease in cell cycling, and increase in DNA damage.
To investigate this hypothesis and to determine whether embryonic inhibition of HDAC recapitulated SIN3A loss of function, untreated control and Sin3a CKO mice (Fig. 8, A to F) were compared to those treated with dimethyl sulfoxide (DMSO) vehicle control (fig. S14, A to F) and to those treated with HDAC inhibitor trichostatin A (TSA) beginning when doxycycline-induced recombination was initiated (Fig. 8G and fig. S14, G to L) (93, 94). Using this approach, there was no difference in the number of EDU-positive cells in untreated Sin3a CKO lungs (Fig. 8B) compared to those treated with DMSO vehicle control (fig. S14, B and C) or those treated with TSA (fig. S14, H and I). In contrast, compared with untreated control lungs (Fig. 8A) or those treated with DMSO (fig. S14A), there was a reduction in the number of EDU-positive cells in control lungs treated with TSA (fig. S14, G and I). The number of EDU-positive cells in control lungs treated with TSA was similar to the number of EDU-positive cells in untreated Sin3a CKO lungs (Fig. 8B and fig. S14, G and I). These data suggested that embryonic HDAC inhibition with TSA resulted in decreased G1 phase–to–S phase transition that was similar to loss of SIN3A function. HDAC inhibition with TSA did not alter the number of cells with evidence of DNA damage indicated by γH2AX staining in either Sin3a CKO or control lungs (Fig. 8, D to F, and fig. S14, D to F and J to L), suggesting that DNA damage observed in Sin3a CKO mice might not be due to decreased HDAC function alone.
Fig. 8. Embryonic inhibition of HAT rescued lung mesenchyme and pulmonary vascular defects in Sin3a CKO mice.

(A and B) Images of EDU (magenta) immunofluorescence in untreated E16 control (A) and Sin3a CKO (B) lungs. (C) Graph comparing the percentage of EDU-positive cells in untreated E16 control and Sin3a CKO lungs (**P = 0.004). (D and E) Images of γH2AX (magenta) immunofluorescence in untreated E16 control (D) and Sin3a CKO (E) lungs. (F) Graph comparing the percentage of γH2AX-positive cells in untreated E16 control and Sin3a CKO lungs (**P = 0.007). (G) Schematic of embryonic treatment regimen used in subsequent experiments starting when doxycycline is given to induce recombination at E12. (H and I) Images of EDU immunofluorescence in anacardic acid (AA)–treated E16 control (H) and Sin3a CKO (I) lungs. (J) Graph comparing the percentage of EDU-positive cells in untreated and AA-treated E16 control and Sin3a CKO lungs (untreated control versus AA-treated control, P = 0.87; untreated Sin3a CKO versus AA-treated Sin3a CKO, ***P = 0.0007; untreated control versus AA-treated Sin3a CKO, P >0.99). (K and L) Images of γH2AX immunofluorescence in AA-treated E16 control (K) and Sin3a CKO (L) lungs. (M) Graph comparing the percentage of γH2AX-positive cells in untreated and AA-treated E16 control and Sin3a CKO lungs (untreated control versus AA-treated control, P = 0.99; untreated Sin3a CKO versus AA-treated Sin3a CKO, *P = 0.01; untreated control versus AA-treated Sin3a CKO, P = 0.48). (N to Q) Images of Pdgfrα-GFP immunofluorescence (GFP, green) in E16 untreated control (N) and Sin3a CKO (O) and AA-treated control (P) and Sin3a CKO (Q) lungs. (R) Graph comparing the percentage of GFP-positive cells in untreated and AA-treated E16 control and Sin3a CKO lungs (untreated control versus AA-treated control, P = 0.94; untreated Sin3a CKO versus AA-treated Sin3a CKO, *P = 0.04; untreated control versus AA-treated Sin3a CKO, P = 0.99). (S to V) Histological sections of E16 untreated control (S) and Sin3a CKO (T) lungs and AA-treated control (U) and Sin3a CKO (V) lungs. (W) Graph comparing the MLI of untreated and AA-treated E16 control and Sin3a CKO lungs (untreated control versus AA-treated control, P = 0.98; untreated Sin3a CKO versus AA-treated Sin3a CKO, P = 0.47; untreated control versus AA-treated Sin3a CKO, P = 0.47). (X to A′) Images of ERG (green) and SMA (magenta) immunofluorescence in E16 untreated control (X) and Sin3a CKO (Y) and AA-treated control (Z) and Sin3a CKO (A′) lungs. (B′) Graph comparing the number of vessels in untreated and AA-treated E16 control and AA-treated Sin3a CKO lungs (untreated control versus AA-treated control, P >0.99; untreated Sin3a CKO versus AA-treated Sin3a CKO, *P = 0.04; untreated control versus AA-treated Sin3a CKO, P = 0.95). Statistical comparisons between untreated control and Sin3a CKO mice (C and F) were performed using unpaired t test. Statistical comparisons between untreated and treated control and Sin3a CKO mice (J, M, R, W, and B′) were made using one-way ANOVA with Tukey’s test for multiple comparisons. Statistical significance was determined when the P value was <0.05. Scale bars, 100 μm.
Inhibition of HAT restores the balance of histone acetylation in Sin3a CKO mice
To determine whether the balance of histone acetylation and deacetylation could be restored despite the absence of SIN3A, control and Sin3a CKO embryos were treated with A-485, a potent and specific inhibitor of HAT p300/CPB (95), beginning at E12 (fig. S14, M to R). Treatment with A-485 resulted in decreased G1 phase–to–S phase transition in control lungs (fig. S14, M and O) and did not change G1 phase–to–S phase transition in Sin3a CKO lungs (fig. S14, N and O) compared to those that were untreated (Fig. 8, A to C). In contrast, compared to untreated control and mutant lungs (Fig. 8, D to F), DNA damage was unchanged in control lungs and decreased in Sin3a CKO lungs treated with A-485 (fig. S14, P to R). These data suggested that potent inhibition of p300/CBP HAT by A-485 could make DNA resistant to damage but not accessible for replication during lung development and further supported the idea that SIN3A plays an important role in regulating the balance of histone acetylation and deacetylation.
We hypothesized that partial inhibition of acetyltransferase might restore the balance of histone acetylation necessary for DNA replication and cell proliferation and reduce DNA damage. To investigate this hypothesis, anacardic acid (AA), a naturally occurring HAT inhibitor that is 1000 times less potent than A-485 (93, 95, 96), was used to treat control and Sin3a CKO embryos following the regimen conducted with TSA and A-485 (Fig. 8, H to M). Compared to the amount of histone acetylation that was observed in untreated control and Sin3a CKO lungs (fig. S13, A to F), embryonic treatment with AA did not change histone acetylation in control lungs and reduced histone acetylation in Sin3a CKO lungs (fig. S15, A to F). Using the same embryonic treatment with AA, there was no difference in the number of EDU-positive cells or in the number of cells with evidence of DNA damage in control lungs treated with AA (Fig. 8, H, J, K, and M). In contrast, the number of EDU-positive cells was increased, whereas the number of cells with evidence of DNA damage was decreased in Sin3a CKO lungs treated with AA (Fig. 8, I, J, L and M).
To determine whether the increased cell proliferation and decreased DNA damage observed in Sin3a CKO embryos after AA treatment were due to inhibition of p300-CBP specifically, we treated control and Sin3a CKO embryos beginning at E12 with diluted A-485 to mimic the potency of AA HAT inhibition. We found that both 1:100 and 1:1000 dilutions of A-485 were sufficient to increase cell proliferation and decrease DNA damage in Sin3a CKO embryos (fig. S16, A to L). These data suggested that partial inhibition of HAT with either AA or A485 was sufficient to restore the balance of histone acetylation despite the loss of SIN3A. This conclusion was further supported by the finding that AA treatment resulted in an increased number of Pdgfrα-GFP–positive myofibroblast precursor cells in Sin3a CKO lungs compared with controls and untreated Sin3a CKO lungs (Fig. 8, N to R), reduced apoptosis (fig. S17, A to F), improved saccular development of the Sin3a CKO lungs (Fig. 8, S to W), and increased lung vessel number (Fig. 8, X to B′). In addition, although there was no change in body weight after treatment with AA (fig. S18A), pulmonary vascular physiological function was improved in Sin3a CKO mice treated with AA with reduced right ventricular hypertrophy (Fulton’s index, fig. S18B) and reduced pulmonary vascular resistance (RVSP, fig. S18C).
These data support the idea that epigenetic regulation of gene expression is controlled by a balance of histone acetylation and deacetylation during embryonic development (fig. S19). Loss of this balance either by genetic deletion of a critical cofactor, such as SI-N3A, or by pharmacological inhibition of HAT or HDAC may result in impaired cell cycling, DNA damage, and impaired cellular differentiation that can be restored through partial inhibition of HAT activity.
DISCUSSION
Regulation of histone acetylation is required to direct the rapid changes in gene expression and DNA replication necessary during development (97–102). The complementary activity of HAT and HDAC enzymes maintains the balance of histone acetylation and therefore controls transcriptional activation or repression as well as DNA replication (99, 102). The balance of HAT and HDAC activity itself is necessary for development (48, 103, 104); however, how failure to maintain this balance contributes to congenital malformations is not clear. Here, we showed that loss of SIN3A function in the developing diaphragm and lungs caused failure of diaphragm formation and lung defects similar to those observed in patients with CDH. In the developing lungs, SIN3A was required to maintain the balance of histone acetylation, whereas loss of SIN3A resulted in defects in lung development and pulmonary hypertension, the two major contributors to mortality in patients with CDH.
The SIN3-HDAC complex is one of many chromatin modifiers that regulate gene expression and DNA replication by controlling the open or compact state of chromatin and thus access of transcription or replication factors to DNA (53, 105). During development, chromatin modifications themselves are affected by changes in the environment, including exposures to toxins and changes in the mother’s health, and translate these changes to the developing embryo (106). These interactions are especially important in human structural malformations where there is an overlapping impact of pathogenic gene variants and environmental mechanisms that contribute to the underlying defects in development (107–110).
Despite research demonstrating that epigenetic regulation of gene expression is essential for mammalian development (20, 21, 23), how loss of epigenetic regulation in humans contributes to structural malformations remains unclear. Mutations in genes that encode chromatin modifiers have been reported in patients with neurodevelopmental diseases and in diseases that occur later in life (25, 26, 111–115). Furthermore, defects in epigenetic gene regulation have been implicated in congenital heart disease and neural tube defects (22, 27–30); however, mutations in genes that encode chromatin modifiers are uncommon among the genes identified in whole-exome or whole-genome sequencing in patients with structural malformations (116). This may be because epigenetic regulation is ubiquitously necessary during development, and disruption of these mechanisms, including loss of a critical co-factor, does not permit embryonic development.
The genetic or epigenetic mechanisms responsible for the difference in phenotypes between patients with Witteveen-Kolk syndrome and the patients in this study are unclear. Identification of Lon peptidase 1, mitochondrial (LONP1) sequence variants in patients with CDH and in patients with cerebral, ocular, dental, auricular, and skeletal syndrome demonstrates that subtle differences in the variants themselves can have a profound impact on the patient phenotype (13, 117). The generation of modeling approaches that incorporate sequence and chromosomal structural variation and the potential functional consequences of these variants will be essential to accurately predict disease phenotype and penetrance as well as to understand more subtle changes that affect disease severity.
In the developing lung epithelium, SIN3A was shown to be required for cell cycling and branching morphogenesis (76). Abnormalities in lung branching have been reported in patients with CDH (118–120); however, the more common phenotypes associated with CDH include pulmonary hypertension and lung hypoplasia (119, 121–123). SIN3A has been associated with regulation of cell cycling, and loss of SIN3A has been demonstrated to result in activation of p53, making senescence a likely subsequent loss-of-function phenotype (59, 124). In adult type II alveolar epithelial cells, loss of SIN3A resulted in p53-dependent activation of senescence and fibrosis (77). Although senescence is a likely downstream phenotype after the loss of SIN3A, our data suggest that the underlying mechanism responsible for this phenotype is loss of SIN3-HDAC regulation of gene expression.
Identifying fetal interventions that improve the outcome of patients with structural malformations is a major objective, especially for CDH (125–129). Although genomic analysis, including fetal exome or genome sequencing, could be a useful method to identify patients likely to respond to fetal interventions, CDH-associated sequence and copy number variants continue to be identified, and therefore, at this time, exome and whole-genome sequencing are more likely to yield a genetic diagnosis than a CDH-specific gene panel. Furthermore, although gene damaging variants have been reported more frequently in patients with complex CDH (11, 130), patients with isolated CDH also have a significant burden of likely pathogenic genomic variants (13, 131). Our hope is that with an increasing number of patients with CDH undergoing genomic analysis, we will be able to establish a pattern of gene variants and associated molecular pathway defects that are common in patients with both complex and isolated forms of the disease.
Making a genetic diagnosis in patients with CDH and determining which genetic variants only affect diaphragm development would help to identify patients more likely to respond to fetal surgical interventions because the lung and pulmonary vascular defects in these patients are more likely to be due to mechanical compression alone. In contrast, determining which genetic variants play a direct role in lung or pulmonary vascular development, as demonstrated in the case of SIN3A, could help to identify patients who may be less likely to respond to a treatment focused on mechanical compression alone. These patients are more likely to have primary defects in lung and pulmonary vascular development and are more likely to have lung hypoplasia and pulmonary hypertension that is not responsive to conventional treatment.
In addition to its role in embryonic lung development, SIN3A was recently implicated in the molecular mechanisms responsible for pulmonary hypertension by epigenetic regulation of BMPR2 expression (90). In this case, intratracheal SIN3A protein administration in rats with experimentally induced pulmonary hypertension restored Bmpr2 expression and decreased pulmonary artery pressure (90). This work demonstrates the role of SIN3A in adult patients and experimental models of pulmonary hypertension and complements our findings that loss of SIN3A resulted in developmental defects in lung and pulmonary vascular development as well as pulmonary hypertension, the major cause of mortality in patients with CDH.
We acknowledge important limitations of our study, including the fact that CDH is a genetically heterogeneous disease with multiple gene variants playing a role and that the mechanisms of abnormal lung and pulmonary vascular development were investigated in a mouse model of CDH. To address these limitations and build on the results related to SIN3A loss of function, we propose future studies to investigate the role of other genomic variants identified in patients with CDH during diaphragm, lung, and pulmonary vascular development. Our hypothesis is that patients with genomic variants that play a direct role in lung and pulmonary vascular development have more severe lung hypoplasia and pulmonary hypertension than patients with genomic variants that only affect diaphragm development. Furthermore, we propose that although CDH is a genetically and phenotypically heterogeneous disease, there may be common pathways that are differentially affected by the genome variants in patients with CDH. Identifying these common molecular pathways will help to subcategorize patients and determine which groups of patients are more or less likely to respond to high-risk fetal interventions including fetal endoluminal tracheal occlusion (FETO), fetal drug and small-molecule inhibitor treatments, and stem cell–based strategies. Last, developing approaches that alter the balance of histone acetylation and deacetylation during embryonic development may be effective for patients with SIN3A sequence variants and other genomic variants that affect the activity of HAT and HDAC enzymes. It will be important to demonstrate abnormal histone acetylation and associated changes in gene expression in lung cells from patients to determine whether loss of SIN3A causes increased histone acetylation as we observed in our mouse models of lung and pulmonary vascular defects associated with CDH. It is also essential to consider that the balance of HAT and HDAC is likely to be different in different cell populations and during different stages of development. Global inhibition of HAT enzymes might be effective at restoring normal development in one cell population while causing a pathological imbalance in neighboring populations. Therefore, developing cell-specific targeting strategies and approaches that target a narrow time during development might be necessary for this strategy to be effective and safe. Despite these limitations, we demonstrated that making a genetic diagnosis in patients with CDH enabled us to determine the underlying developmental mechanisms of abnormal diaphragm and lung formation and to identify an approach that improved lung development.
MATERIALS AND METHODS
Study design
The objective of this study was to determine the role of Sin3a during diaphragm and lung mesenchyme development. Our work included genomic evaluation of patients with CDH, tissue and cell lineage–specific deletion of Sin3a in mice during diaphragm and lung development, gross and histological analysis of Sin3a conditional deletion (Sin3a CKO) diaphragm and lung tissue, transcriptomic analysis of whole lung and recombined lung mesenchymal cells, and pulmonary vascular physiological analysis using transthoracic echocardiography and right heart catheterization in mice. Sin3a CKO mice and littermate controls were treated with HDAC and HAT inhibitors. Mice were selected for these experiments on the basis of their Sin3a CKO or control genotypes. For histological experiments, four control and four Sin3a CKO lung samples were used, and each histological experiment was performed at least two times. Quantification of histological images and physiological studies was conducted by blinded observers. For transcriptomic analysis of whole lungs and sorted recombined lung mesenchymal cells, four control and four Sin3a CKO lungs were analyzed. For sorted recombined mesenchymal single-cell transcriptomic analysis, two control and two Sin3a CKO lungs were used. Outliers were recorded and included.
Human genomic analysis
Blood and saliva samples from 827 patient and parent trios enrolled in the DHREAMS study were analyzed by whole-genome and -exome sequencing as described previously (13, 84). Signed, informed consent was obtained from participants, and all studies were approved by the institutional review board at each participating institution and the Columbia University Irving Medical Center Institutional Review Board. Clinical data from medical records were prospectively collected and entered in a central Research Electronic Data Capture (REDCap) database. A full description of the methods used for patient and control sequencing analysis, sequence variant confirmation, and the impact of sequence variants on gene transcript or protein structure or function was published previously (13, 84).
Animal handling and genetics
Mice were housed and handled according to an animal care committee–approved protocol in a temperature-controlled environment with regular light and dark cycling, free access to food and water, and no more than five mice per cage. All experimental procedures were performed in an American Association for Accreditation of Laboratory Animal Care–accredited laboratory animal facility. All mice were bred on a mixed genetic background, and age-matched littermates were used as controls for comparison. Both male and female mice were used in all experiments.
Recombination of the Sin3a flox allele (59) was achieved by crossing with the previously described Pax3-Cre [the Jackson Laboratory, 005549 (132)], Prx1-Cre[the Jackson Laboratory, 005584 (133)], Cdh5-Cre [the Jackson Laboratory, 006137 (134)], or Tbx4-rtTA; Tet-o-Cre alleles (135). Tbx4-rtTA was induced by feeding pregnant mice doxycycline (Teklad) starting at E6.5 or E12.5. Cre-positive, Sin3a-heterogenous littermates were used as controls.
Statistical analysis
To determine statistical significance, unpaired, two-sided t tests were performed for comparison between two groups using Welch’s correction, and one-way analysis of variance (ANOVA) was used for comparison between multiple groups for histology and physiology-based experiments. Statistical significance was determined by Z test for RNA velocity experiments and χ2 test of distribution of single-cell clusters. Statistical significance for differential gene expression was adjusted with a Benjamin-Hochberg FDR correction at the 5% level.
Supplementary Material
Acknowledgments:
We thank G. David (New York University Grossman School of Medicine) and W. Shi (Keck School of Medicine, University of Southern California) for sharing mouse strains; the University of Wisconsin-Madison Gene Expression Center and Bioinformatics Resource Center for help conducting and analyzing the RNA sequencing experiments; the University of Wisconsin Carbone Cancer Center FLOW Cytometry Laboratory, where lung mesenchymal cell sorting was conducted; and the University of Wisconsin-Madison Optical Imaging Core and the University of California, San Diego School of Medicine Microscopy Core, where the confocal imaging was conducted. We also thank A. Ikeda and the members of the Ikeda laboratory (University of Wisconsin-Madison) and X. Sun and members of the Sun laboratory (University of California, San Diego) for discussion and review of this manuscript. We also thank the patients and family members who have participated in the DHREAMs study.
Funding:
This work was supported by the following: Translational Science Career Development Award KL2TR000428 (D.J.M.), a subaward from the National Center for Advancing Translational Sciences (NCATS) grant to the University of Wisconsin-Madison Institute for Clinical and Translational Research UL1TR000427; National Institutes of Health grants NHLBI R01HL146859 (D.J.M.), NHLBI R01HL132366, R01HL136998, X01HL140543 (Y.S. and W.K.C.), and NICHD P01HD068250 (Y.S., W.K.C., and D.J.M.); National Key Research and Development Program of China 2019YFE0119400 (H.T.); and Natural Science Foundation of China 81770059, 81970052, and 82170057 (H.T.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All raw sequencing data and processed files have been deposited in GEO under the series reference GSE246195. Mice carrying the Sin3a flox allele (59) were shared by G. David under a material agreement with New York University Grossman School of Medicine. Mice carrying the Tbx4-rtTA; Tet-o-Cre alleles (135) were shared by W. Shi under a material agreement with Keck School of Medicine, University of Southern California.
REFERENCES AND NOTES
- 1.Mortality in the United States, 2019; 2020. https://cdc.gov/nchs/data/databriefs/db395-H.pdf.
- 2.Balayla J, Abenhaim HA, Incidence, predictors and outcomes of congenital diaphragmatic hernia: A population-based study of 32 million births in the United States. J. Matern. Fetal Neonatal Med. 27, 1438–1444 (2014). [DOI] [PubMed] [Google Scholar]
- 3.McGivern MR, Best KE, Rankin J, Wellesley D, Greenlees R, Addor MC, Arriola L, de Walle H, Barisic I, Beres J, Bianchi F, Calzolari E, Doray B, Draper ES, Garne E, Gatt M, Haeusler M, Khoshnood B, Klungsoyr K, Latos-Bielenska A, O’Mahony M, Braz P, Donnell BM, Mullaney C, Nelen V, Queisser-Luft A, Randrianaivo H, Rissmann A, Rounding C, Sipek A, Thompson R, Tucker D, Wertelecki W, Martos C, Epidemiology of congenital diaphragmatic hernia in Europe: A register-based study. Arch. Dis. Child. Fetal Neonatal Ed. 100, F137–F144 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Paoletti M, Raffler G, Gaffi MS, Antounians L, Lauriti G, Zani A, Prevalence and risk factors for congenital diaphragmatic hernia: A global view. J. Pediatr. Surg. 55, 2297–2307 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Aly H, Bianco-Batlles D, Mohamed MA, Hammad TA, Mortality in infants with congenital diaphragmatic hernia: A study of the United States National Database. J. Perinatol. 30, 553–557 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Politis MD, Bermejo-Sánchez E, Canfield MA, Contiero P, Cragan JD, Dastgiri S, de Walle HEK, Feldkamp ML, Nance A, Groisman B, Gatt M, Benavides-Lara A, Hurtado-Villa P, Kallén K, Landau D, Lelong N, Lopez-Camelo J, Martinez L, Morgan M, Mutchinick OM, Pierini A, Rissmann A, Šípek A, Szabova E, Wertelecki W, Zarante I, Bakker MK, Kancherla V, Mastroiacovo P, Nembhard WN, International Clearinghouse of Birth Defects Surveillance and Research, Prevalence and mortality in children with congenital diaphragmatic hernia: A multicountry study. Ann. Epidemiol. 56, 61–69.e3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramakrishnan R, Salemi JL, Stuart AL, Chen H, O'Rourke K, Obican S, Kirby RS, Trends, correlates, and survival of infants with congenital diaphragmatic hernia and its subtypes. Birth Defects Res. 110, 1107–1117 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Wynn J, Krishnan U, Aspelund G, Zhang Y, Duong J, Stolar CJH, Hahn E, Pietsch J, Chung D, Moore D, Austin E, Mychaliska G, Gajarski R, Foong Y-L, Michelfelder E, Potolka D, Bucher B, Warner B, Grady M, Azarow K, Fletcher SE, Kutty S, Delaney J, Crombleholme T, Rosenzweig E, Chung W, Arkovitz MS, Outcomes of congenital diaphragmatic hernia in the modern era of management. J. Pediatr. 163, 114–119.e1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seabrook RB, Grover TR, Rintoul N, Weems M, Keene S, Brozanski B, DiGeronimo R, Haberman B, Hedrick H, Gien J, Ali N, Chapman R, Daniel J, Harrison HA, Johnson Y, Porta NFM, Uhing M, Zaniletti I, Murthy K, Children’s Hospital Neonatal Consortium Congenital Diaphragmatic Hernia Focus Group, Treatment of pulmonary hypertension during initial hospitalization in a multicenter cohort of infants with congenital diaphragmatic hernia (CDH). J. Perinatol. 41, 803–813 (2021). [DOI] [PubMed] [Google Scholar]
- 10.Bogenschutz EL, Fox ZD, Farrell A, Wynn J, Moore B, Yu L, Aspelund G, Marth G, Yandell M, Shen Y, Chung WK, Kardon G, Deep whole-genome sequencing of multiple proband tissues and parental blood reveals the complex genetic etiology of congenital diaphragmatic hernias. HGG Adv. 1, 100008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longoni M, High FA, Qi H, Joy MP, Hila R, Coletti CM, Wynn J, Loscertales M, Shan L, Bult CJ, Wilson JM, Shen Y, Chung WK, Donahoe PK, Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum. Genet. 136, 679–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, Zhu N, Kitaygorodsky A, Hernan R, Aspelund G, Lim F-Y, Crombleholme T, Cusick R, Azarow K, Danko ME, Chung D, Warner BW, Mychaliska GB, Potoka D, Wagner AJ, Fiky ME, Wilson JM, Nickerson D, Bamshad M, High FA, Longoni M, Donahoe PK, Chung WK, Shen Y, De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLOS Genet. 14, e1007822 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiao L, Xu L, Yu L, Wynn J, Hernan R, Zhou X, Farkouh-Karoleski C, Krishnan US, Khlevner J, De A, Zygmunt A, Crombleholme T, Lim FY, Needelman H, Cusick RA, Mychaliska GB, Warner BW, Wagner AJ, Danko ME, Chung D, Potoka D, Kosiński P, McCulley DJ, Elfiky M, Azarow K, Fialkowski E, Schindel D, Soffer SZ, Lyon JB, Zalieckas JM, Vardarajan BN, Aspelund G, Duron VP, High FA, Sun X, Donahoe PK, Shen Y, Chung WK, Rare and de novo variants in 827 congenital diaphragmatic hernia probands implicate LONP1 as candidate risk gene. Am. J. Hum. Genet. 108, 1964–1980 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merrell AJ, Ellis BJ, Fox ZD, Lawson JA, Weiss JA, Kardon G, Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat. Genet. 47, 496–504 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ackerman KG, Wang J, Luo L, Fujiwara Y, Orkin SH, Beier DR, Gata4 is necessary for normal pulmonary lobar development. Am. J. Respir. Cell Mol. Biol. 36, 391–397 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR, Fog2 is required for normal diaphragm and lung development in mice and humans. PLOS Genet. 1, 58–65 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell MK, Longoni M, Wells J, Maalouf FI, Tracy AA, Loscertales M, Ackerman KG, Pober BR, Lage K, Bult CJ, Donahoe PK, Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 109, 2978–2983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carmona R, Cañete A, Cano E, Ariza L, Rojas A, Muñoz-Chápuli R, Conditional deletion of WT1 in the septum transversum mesenchyme causes congenital diaphragmatic hernia in mice. eLife 5, e16009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCulley DJ, Wienhold MD, Hines EA, Hacker TA, Rogers A, Pewowaruk RJ, Zewdu R, Chesler NC, Selleri L, Sun X, PBX transcription factors drive pulmonary vascular adaptation to birth. J. Clin. Invest. 128, 655–667 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S, Zhang N, Duan F, Ming J, Zhang X, Niu W, Song W, Jin H, Guo Y, Dai S, Hu L, Fang L, Wang Q, Li Y, Li W, Na J, Xie W, Sun Y, Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557, 256–260 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Santos F, Hendrich B, Reik W, Dean W, Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 241, 172–182 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dreßen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L, Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 9, 391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Yuan P, Yan Z, Yang M, Huo Y, Nie Y, Zhu X, Qiao J, Yan L, Single-cell multiomics sequencing reveals the functional regulatory landscape of early embryos. Nat. Commun. 12, 1247 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U, Sartori C, Fetal programming of pulmonary vascular dysfunction in mice: Role of epigenetic mechanisms. Am. J. Physiol. Heart Circ. Physiol. 301, H247–H252 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY, Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Shirohzu H, Kubota T, Kumazawa A, Sado T, Chijiwa T, Inagaki K, Suetake I, Tajima S, Wakui K, Miki Y, Hayashi M, Fukushima Y, Sasaki H, Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 112, 31–37 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Greene NDE, Stanier P, Moore GE, The emerging role of epigenetic mechanisms in the etiology of neural tube defects. Epigenetics 6, 875–883 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, Kim R, Kline JK, Lee T, Leipzig J, Lopez A, Mane SM, Mitchell LE, Newburger JW, Parfenov M, Pe’er I, Porter G, Roberts AE, Sachidanandam R, Sanders SJ, Seiden HS, State MW, Subramanian S, Tikhonova IR, Wang W, Warburton D, White PS, Williams IA, Zhao H, Seidman JG, Brueckner M, Chung WK, Gelb BD, Goldmuntz E, Seidman CE, Lifton RP, De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao J, Wu Q, Huang Y, Wang L, Su Z, Ye H, The role of DNA methylation in syndromic and non-syndromic congenital heart disease. Clin. Epigenetics 13, 93 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chowdhury S, Erickson SW, MacLeod SL, Cleves MA, Hu P, Karim MA, Hobbs CA, Maternal genome-wide DNA methylation patterns and congenital heart defects. PLOS ONE 6, e16506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li E, Bestor TH, Jaenisch R, Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Okano M, Bell DW, Haber DA, Li E, DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C, Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 21, 2672–2681 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN, Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Tian Y, Morley MP, Lu MM, Demayo FJ, Olson EN, Morrisey EE, Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2-Bmp4/Rb1 regulatory pathway. Dev. Cell 24, 345–358 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Frank DB, Morley MP, Zhou S, Wang X, Lu MM, Lazar MA, Morrisey EE, HDAC3-dependent epigenetic pathway controls lung alveolar epithelial cell remodeling and spreading via miR-17–92 and TGF-β signaling regulation. Dev. Cell 36, 303–315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ, Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 352, 1967–1976 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Ito K, Caramori G, Lim S, Oates T, Chung KF, Barnes PJ, Adcock IM, Expression and activity of histone deacetylases in human asthmatic airways. Am. J. Respir. Crit. Care Med. 166, 392–396 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Cosío BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF, Adcock IM, Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am. J. Respir. Crit. Care Med. 170, 141–147 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Li M, Zheng Y, Yuan H, Liu Y, Wen X, Effects of dynamic changes in histone acetylation and deacetylase activity on pulmonary fibrosis. Int. Immunopharmacol. 52, 272–280 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Robinson CM, Neary R, Levendale A, Watson CJ, Baugh JA, Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res. 13, 74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gan Y, Shen YH, Wang J, Wang X, Utama B, Wang XL, Role of histone deacetylation in cell-specific expression of endothelial nitric-oxide synthase. J. Biol. Chem. 280, 16467–16475 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bogaard HJ, Mizuno S, Al Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF, Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 183, 1402–1410 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Ruiz-Carrillo A, Wangh LJ, Allfrey VG, Processing of newly synthesized histone molecules. Science 190, 117–128 (1975). [DOI] [PubMed] [Google Scholar]
- 45.Annunziato AT, Seale RL, Histone deacetylation is required for the maturation of newly replicated chromatin. J. Biol. Chem. 258, 12675–12684 (1983). [PubMed] [Google Scholar]
- 46.Kuo MH, Allis CD, Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20, 615–626 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Lee J-H, Hart SRL, Skalnik DG, Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis 38, 32–38 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Haberland M, Montgomery RL, Olson EN, The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Schölz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, Brickman JM, Kesicki EA, Lai A, Bromberg KD, Cole PA, Choudhary C, Time-resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 174, 231–244.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly RDW, Cowley SM, The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 41, 741–749 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dümpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G, Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D, Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams GE, Chandru A, Cowley SM, Co-repressor, co-activator and general transcription factor: The many faces of the Sin3 histone deacetylase (HDAC) complex. Biochem. J. 475, 3921–3932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE, Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 89, 341–347 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Laherty CD, Yang W-M, Sun J-M, Davie JR, Seto E, Eisenman RN, Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 89, 349–356 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM, Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 89, 373–380 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Kadosh D, Struhl K, Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89, 365–371 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D, Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 89, 357–364 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Dannenberg J-H, David G, Zhong S, van der Torre J, Wong WH, Depinho RA, mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 19, 1581–1595 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pile LA, Schlag EM, Wassarman DA, The SIN3/RPD3 deacetylase complex is essential for G2 phase cell cycle progression and regulation of SMRTER corepressor levels. Mol. Cell. Biol. 22, 4965–4976 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshimoto H, Ohmae M, Yamashita I, The Saccharomyces cerevisiae GAM2/SIN3 protein plays a role in both activation and repression of transcription. Mol. Gen. Genet. 233, 327–330 (1992). [DOI] [PubMed] [Google Scholar]
- 62.Vidal M, Strich R, Esposito RE, Gaber RF, RPD1 (SIN3/UME4) is required for maximal activation and repression of diverse yeast genes. Mol. Cell. Biol. 11, 6306–6316 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das TK, Sangodkar J, Negre N, Narla G, Cagan RL, Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 32, 3184–3197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baltus GA, Kowalski MP, Tutter AV, Kadam S, A positive regulatory role for the mSin3A-HDAC complex in pluripotency through Nanog and Sox2. J. Biol. Chem. 284, 6998–7006 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, Vercauteren K, Saelens X, Meuleman P, Leroux-Roels G, De Bosscher K, Boutros M, Tavernier J, The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl. Acad. Sci. U.S.A. 109, 12058–12063 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernstein BE, Tong JK, Schreiber SL, Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. U.S.A. 97, 13708–13713 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Banks CAS, Zhang Y, Miah S, Hao Y, Adams MK, Wen Z, Thornton JL, Florens L, Washburn MP, Integrative modeling of a Sin3/HDAC complex sub-structure. Cell Rep. 31, 107516 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neufeld TP, Tang AH, Rubin GM, A genetic screen to identify components of the sina signaling pathway in Drosophila eye development. Genetics 148, 277–286 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pile LA, Wassarman DA, Chromosomal localization links the SIN3-RPD3 complex to the regulation of chromatin condensation, histone acetylation and gene expression. EMBO J. 19, 6131–6140 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma V, Swaminathan A, Bao R, Pile LA, Drosophila SIN3 is required at multiple stages of development. Dev. Dyn. 237, 3040–3050 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Pennetta G, Pauli D, The Drosophila Sin3 gene encodes a widely distributed transcription factor essential for embryonic viability. Dev. Genes Evol. 208, 531–536 (1998). [DOI] [PubMed] [Google Scholar]
- 72.Swaminathan A, Pile LA, Regulation of cell proliferation and wing development by Drosophila SIN3 and String. Mech. Dev. 127, 96–106 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Cowley SM, Iritani BM, Mendrysa SM, Xu T, Cheng PF, Yada J, Liggitt HD, Eisenman RN, The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol. Cell. Biol. 25, 6990–7004 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.David G, Grandinetti KB, Finnerty PM, Simpson N, Chu GC, Depinho RA, Specific requirement of the chromatin modifier mSin3B in cell cycle exit and cellular differentiation. Proc. Natl. Acad. Sci. U.S.A. 105, 4168–4172 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Oevelen C, Bowman C, Pellegrino J, Asp P, Cheng J, Parisi F, Micsinai M, Kluger Y, Chu A, Blais A, David G, Dynlacht BD, The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol. Cell. Biol. 30, 5686–5697 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yao C, Carraro G, Konda B, Guan X, Mizuno T, Chiba N, Kostelny M, Kurkciyan A, David G, McQualter JL, Stripp BR, Sin3a regulates epithelial progenitor cell fate during lung development. Development 144, 2618–2628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR, Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 203, 707–717 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mefford HC, Rosenfeld JA, Shur N, Slavotinek AM, Cox VA, Hennekam RC, Firth HV, Willatt L, Wheeler P, Morrow EM, Cook J, Sullivan R, Oh A, McDonald MT, Zonana J, Keller K, Hannibal MC, Ball S, Kussmann J, Gorski J, Zelewski S, Banks V, Smith W, Smith R, Paull L, Rosenbaum KN, Amor DJ, Silva J, Lamb A, Eichler EE, Further clinical and molecular delineation of the 15q24 microdeletion syndrome. J. Med. Genet. 49, 110–118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Narumi-Kishimoto Y, Araki N, Migita O, Kawai T, Okamura K, Nakabayashi K, Kaname T, Ozawa Y, Ozawa H, Takada F, Hata K, Novel SIN3A mutation identified in a Japanese patient with Witteveen-Kolk syndrome. Eur. J. Med. Genet. 62, 103547 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Witteveen JS, Willemsen MH, Dombroski TCD, van Bakel NHM, Nillesen WM, van Hulten JA, Jansen EJR, Verkaik D, Veenstra-Knol HE, van Ravenswaaij-Arts CMA, Wassink-Ruiter JSK, Vincent M, David A, Le Caignec C, Schieving J, Gilissen C, Foulds N, Rump P, Strom T, Cremer K, Zink AM, Engels H, de Munnik SA, Visser JE, Brunner HG, Martens GJM, Pfundt R, Kleefstra T, Kolk SM, Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat. Genet. 48, 877–887 (2016). [DOI] [PubMed] [Google Scholar]
- 81.van Dongen LCM, Wingbermühle E, Dingemans AJM, Bos-Roubos AG, Vermeulen K, Pop-Purceleanu M, Kleefstra T, Egger JIM, Behavior and cognitive functioning in Witteveen-Kolk syndrome. Am. J. Med. Genet. A 182, 2384–2390 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Latypova X, Vincent M, Mollé A, Adebambo OA, Fourgeux C, Khan TN, Caro A, Rosello M, Orellana C, Niyazov D, Lederer D, Deprez M, Capri Y, Kannu P, Tabet AC, Levy J, Aten E, den Hollander N, Splitt M, Walia J, Immken LL, Stankiewicz P, Walter KM, Suchy S, Louie RJ, Bell S, Stevenson RE, Rousseau J, Willem C, Retiere C, Yang X-J, Campeau PM, Martinez F, Rosenfeld JA, Caignec CL, Küry S, Mercier S, Moradkhani K, Conrad S, Besnard T, Cogné B, Katsanis N, Bézieau S, Poschmann J, Davis EE, Isidor B, Haploinsufficiency of the Sin3/HDAC corepressor complex member SIN3B causes a syndromic intellectual disability/autism spectrum disorder. Am. J. Hum. Genet. 108, 929–941 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Balasubramanian M, Dingemans AJM, Albaba S, Richardson R, Yates TM, Cox H, Douzgou S, Armstrong R, Sansbury FH, Burke KB, Fry AE, Ragge N, Sharif S, Foster A, De Sandre-Giovannoli A, Elouej S, Vasudevan P, Mansour S, Wilson K, Stewart H, Heide S, Nava C, Keren B, Demirdas S, Brooks AS, Vincent M, Isidor B, Küry S, Schouten M, Leenders E, Chung WK, van Haeringen A, Scheffner T, Debray F-G, White SM, Valenzuela Palafoll MI, Pfundt R, Newbury-Ecob R, Kleefstra T, Comprehensive study of 28 individuals with SIN3A-related disorder underscoring the associated mild cognitive and distinctive facial phenotype. Eur. J. Hum. Genet. 29, 625–636 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu L, Wynn J, Ma L, Guha S, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Bucher B, LeDuc CA, Costa K, Stolar C, Aspelund G, Arkovitz MS, Chung WK, De novo copy number variants are associated with congenital diaphragmatic hernia. J. Med. Genet. 49, 650–659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tiana M, Acosta-Iborra B, Puente-Santamaría L, Hernansanz-Agustin P, Worsley-Hunt R, Masson N, García-Rio F, Mole D, Ratcliffe P, Wasserman WW, Jimenez B, Del Peso L, The SIN3A histone deacetylase complex is required for a complete transcriptional response to hypoxia. Nucleic Acids Res. 46, 120–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McDonel P, Demmers J, Tan DWM, Watt F, Hendrich BD, Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev. Biol. 363, 62–73 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu X, Rowan SC, Liang J, Yao C, Huang G, Deng N, Xie T, Wu D, Wang Y, Burman A, Parimon T, Borok Z, Chen P, Parks WC, Hogaboam CM, Weigt SS, Belperio J, Stripp BR, Noble PW, Jiang D, Categorization of lung mesenchymal cells in development and fibrosis. iScience 24, 102551 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ, Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 89.Coenen-van der Spek J, Relator R, Kerkhof J, McConkey H, Levy MA, Tedder ML, Louie RJ, Fletcher RS, Moore HW, Childers A, Farrelly ER, Champaigne NL, Lyons MJ, Everman DB, Rogers RC, Skinner SA, Renck A, Matalon DR, Dills SK, Monteleone B, Demirdas S, Dingemans AJM, Kaat LD, Kolk SM, Pfundt R, Rump P, Sadikovic B, Kleefstra T, Butler KM, DNA methylation episignature for Witteveen-Kolk syndrome due to SIN3A haploinsufficiency. Genet. Med. 25, 63–75 (2023). [DOI] [PubMed] [Google Scholar]
- 90.Bisserier M, Mathiyalagan P, Zhang S, Elmastour F, Dorfmüller P, Humbert M, David G, Tarzami S, Weber T, Perros F, Sassi Y, Sahoo S, Hadri L, Regulation of the methylation and expression levels of the BMPR2 gene by SIN3a as a novel therapeutic mechanism in pulmonary arterial hypertension. Circulation 144, 52–73 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kadamb R, Mittal S, Bansal N, Batra H, Saluja D, Sin3: Insight into its transcription regulatory functions. Eur. J. Cell Biol. 92, 237–246 (2013). [DOI] [PubMed] [Google Scholar]
- 92.Chaubal A, Pile LA, Same agent, different messages: Insight into transcriptional regulation by SIN3 isoforms. Epigenetics Chromatin 11, 17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dekker FJ, van den Bosch T, Martin NI, Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov. Today 19, 654–660 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Menegola E, Di Renzo F, Broccia ML, Prudenziati M, Minucci S, Massa V, Giavini E, Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Res. B Dev. Reprod. Toxicol. 74, 392–398 (2005). [DOI] [PubMed] [Google Scholar]
- 95.Zhou F, Liu Q, Zhang L, Zhu Q, Wang S, Zhu K, Deng R, Liu Y, Yuan G, Wang X, Zhou L, Selective inhibition of CBP/p300 HAT by A-485 results in suppression of lipogenesis and hepatic gluconeogenesis. Cell Death Dis. 11, 745 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peng C, Zhu J, Sun HC, Huang XP, Zhao WA, Zheng M, Liu LJ, Tian J, Inhibition of histone H3K9 acetylation by anacardic acid can correct the over-expression of Gata4 in the hearts of fetal mice exposed to alcohol during pregnancy. PLOS ONE 9, e104135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Verdone L, Agricola E, Caserta M, Di Mauro E, Histone acetylation in gene regulation. Brief. Funct. Genomic. Proteomic. 5, 209–221 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Verdone L, Caserta M, Di Mauro E, Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 83, 344–353 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Struhl K, Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12, 599–606 (1998). [DOI] [PubMed] [Google Scholar]
- 100.McCool KW, Xu X, Singer DB, Murdoch FE, Fritsch MK, The role of histone acetylation in regulating early gene expression patterns during early embryonic stem cell differentiation. J. Biol. Chem. 282, 6696–6706 (2007). [DOI] [PubMed] [Google Scholar]
- 101.Bar-Ziv R, Voichek Y, Barkai N, Chromatin dynamics during DNA replication. Genome Res. 26, 1245–1256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Unnikrishnan A, Gafken PR, Tsukiyama T, Dynamic changes in histone acetylation regulate origins of DNA replication. Nat. Struct. Mol. Biol. 17, 430–437 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Peserico A, Simone C, Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 371832 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haery L, Thompson RC, Gilmore TD, Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 6, 184–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gelato KA, Fischle W, Role of histone modifications in defining chromatin structure and function. Biol. Chem. 389, 353–363 (2008). [DOI] [PubMed] [Google Scholar]
- 106.Legoff L, D'Cruz SC, Tevosian S, Primig M, Smagulova F, Transgenerational inheritance of environmentally induced epigenetic alterations during mammalian development. Cell 8, 1559 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ramakrishnan R, Stuart AL, Salemi JL, Chen H, O'Rourke K, Kirby RS, Maternal exposure to ambient cadmium levels, maternal smoking during pregnancy, and congenital diaphragmatic hernia. Birth Defects Res. 111, 1399–1407 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Caspers KM, Oltean C, Romitti PA, Sun L, Pober BR, Rasmussen SA, Yang W, Druschel C, National Birth Defects Prevention Study, Maternal periconceptional exposure to cigarette smoking and alcohol consumption and congenital diaphragmatic hernia. Birth Defects Res. A Clin. Mol. Teratol. 88, 1040–1049 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Finnell RH, Caiaffa CD, Kim S-E, Lei Y, Steele J, Cao X, Tukeman G, Lin YL, Cabrera RM, Wlodarczyk BJ, Gene environment interactions in the etiology of neural tube defects. Front. Genet. 12, 659612 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Workalemahu T, Grantz KL, Grewal J, Zhang C, Louis GMB, Tekola-Ayele F, Genetic and environmental influences on fetal growth vary during sensitive periods in pregnancy. Sci. Rep. 8, 7274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Santen GWE, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EAJ, Hilhorst-Hofstee Y, Wessels MW, den Hollander NS, Ruivenkamp CAL, van Ommen G-JB, Breuning MH, den Dunnen JT, van Haeringen A, Kriek M, Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380 (2012). [DOI] [PubMed] [Google Scholar]
- 112.Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A, Histone acetylation and disease. Cell. Mol. Life Sci. 58, 728–736 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F, Paz MF, Herranz M, Palacios J, Arango D, Orntoft TF, Aaltonen LA, Schwartz S Jr., Esteller M, A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 38, 566–569 (2006). [DOI] [PubMed] [Google Scholar]
- 114.Cho KS, Elizondo LI, Boerkoel CF, Advances in chromatin remodeling and human disease. Curr. Opin. Genet. Dev. 14, 308–315 (2004). [DOI] [PubMed] [Google Scholar]
- 115.Brookes E, Shi Y, Diverse epigenetic mechanisms of human disease. Annu. Rev. Genet. 48, 237–268 (2014). [DOI] [PubMed] [Google Scholar]
- 116.Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, Spiegel E, Brennan K, Stong N, Jobanputra V, Ren Z, Zhu X, Mebane C, Nahum O, Wang Q, Kamalakaran S, Malone C, Anyane-Yeboa K, Miller R, Levy B, Goldstein DB, Wapner RJ, Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 393, 758–767 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Strauss KA, Jinks RN, Puffenberger EG, Venkatesh S, Singh K, Cheng I, Mikita N, Thilagavathi J, Lee J, Sarafianos S, Benkert A, Koehler A, Zhu A, Trovillion V, McGlincy M, Morlet T, Deardorff M, Innes AM, Prasad C, Chudley AE, Lee INW, Suzuki CK, CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 96, 121–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nose K, Kamata S, Sawai T, Tazuke Y, Usui N, Kawahara H, Okada A, Airway anomalies in patients with congenital diaphragmatic hernia. J. Pediatr. Surg. 35, 1562–1565 (2000). [DOI] [PubMed] [Google Scholar]
- 119.Ameis D, Khoshgoo N, Keijzer R, Abnormal lung development in congenital diaphragmatic hernia. Semin. Pediatr. Surg. 26, 123–128 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Ryan CA, Finer NN, Etches PC, Tierney AJ, Peliowski A, Congenital diaphragmatic hernia: Associated malformations—Cystic adenomatoid malformation, extralobular sequestration, and laryngotracheoesophageal cleft: Two case reports. J. Pediatr. Surg. 30, 883–885 (1995). [DOI] [PubMed] [Google Scholar]
- 121.Kluth D, Tenbrinck R, von Ekesparre M, Kangah R, Reich P, Brandsma A, Tibboel D, Lambrecht W, The natural history of congenital diaphragmatic hernia and pulmonary hypoplasia in the embryo. J. Pediatr. Surg. 28, 456–463 (1993). [DOI] [PubMed] [Google Scholar]
- 122.Mohseni-Bod H, Bohn D, Pulmonary hypertension in congenital diaphragmatic hernia. Semin. Pediatr. Surg. 16, 126–133 (2007). [DOI] [PubMed] [Google Scholar]
- 123.Thébaud B, Mercier JC, Dinh-Xuan AT, Congenital diaphragmatic hernia. A cause of persistent pulmonary hypertension of the newborn which lacks an effective therapy. Biol. Neonate 74, 323–336 (1998). [DOI] [PubMed] [Google Scholar]
- 124.Wilkinson DS, Tsai W-W, Schumacher MA, Barton MC, Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming growth factor β-mediated transcription repression. Mol. Cell. Biol. 28, 1988–1998 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Antounians L, Catania VD, Montalva L, Liu BD, Hou H, Chan C, Matei AC, Tzanetakis A, Li B, Figueira RL, da Costa KM, Wong AP, Mitchell R, David AL, Patel K, De Coppi P, Sbragia L, Wilson MD, Rossant J, Zani A, Fetal lung underdevelopment is rescued by administration of amniotic fluid stem cell extracellular vesicles in rodents. Sci. Transl. Med. 13, eaax5941 (2021). [DOI] [PubMed] [Google Scholar]
- 126.Okolo F, Zhang G, Rhodes J, Gittes GK, Potoka DA, Intra-amniotic sildenafil treatment promotes lung growth and attenuates vascular remodeling in an experimental model of congenital diaphragmatic hernia. Fetal Diagn. Ther. 47, 787–799 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Mous DS, Kool HM, Burgisser PE, Buscop-van Kempen MJ, Nagata K, Boerema-de Munck A, van Rosmalen J, Dzyubachyk O, Wijnen RMH, Tibboel D, Rottier RJ, Treatment of rat congenital diaphragmatic hernia with sildenafil and NS-304, selexipag's active compound, at the pseudoglandular stage improves lung vasculature. Am. J. Physiol. Lung Cell. Mol. Physiol. 315, L276–L285 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Jeanty C, Kunisaki SM, MacKenzie TC, Novel non-surgical prenatal approaches to treating congenital diaphragmatic hernia. Semin. Fetal Neonatal Med. 19, 349–356 (2014). [DOI] [PubMed] [Google Scholar]
- 129.Verla MA, Style CC, Olutoye OO, Prenatal intervention for the management of congenital diaphragmatic hernia. Pediatr. Surg. Int. 34, 579–587 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Yu L, Sawle AD, Wynn J, Aspelund G, Stolar CJ, Arkovitz MS, Potoka D, Azarow KS, Mychaliska GB, Shen Y, Chung WK, Increased burden of de novo predicted deleterious variants in complex congenital diaphragmatic hernia. Hum. Mol. Genet. 24, 4764–4773 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brosens E, Peters NCJ, van Weelden KS, Bendixen C, Brouwer RWW, Sleutels F, Bruggenwirth HT, van Ijcken WFJ, Veenma DCM, Cochius-Den Otter SCM, Wijnen RMH, Eggink AJ, van Dooren MF, Reutter HM, Rottier RJ, Schnater JM, Tibboel D, de Klein A, Unraveling the genetics of congenital diaphragmatic hernia: An ongoing challenge. Front. Pediatr. 9, 800915 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA, Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev. Biol. 280, 396–406 (2005). [DOI] [PubMed] [Google Scholar]
- 133.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ, Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002). [DOI] [PubMed] [Google Scholar]
- 134.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML, VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767 (2006). [DOI] [PubMed] [Google Scholar]
- 135.Zhang W, Menke DB, Jiang M, Chen H, Warburton D, Turcatel G, Lu CH, Xu W, Luo Y, Shi W, Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 11, 111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All raw sequencing data and processed files have been deposited in GEO under the series reference GSE246195. Mice carrying the Sin3a flox allele (59) were shared by G. David under a material agreement with New York University Grossman School of Medicine. Mice carrying the Tbx4-rtTA; Tet-o-Cre alleles (135) were shared by W. Shi under a material agreement with Keck School of Medicine, University of Southern California.