

Abstract
A significant 19th century public health problem was that the inhabitants of many houses containing wallpaper decorated with green arsenical pigments experienced illness and death. The problem was caused by certain fungi that grew in the presence of inorganic arsenic to form a toxic, garlic-odored gas. The garlic odor was actually put to use in a very delicate microbiological test for arsenic. In 1933, the gas was shown to be trimethylarsine. It was not until 1971 that arsenic methylation by bacteria was demonstrated. Further research in biomethylation has been facilitated by the development of delicate techniques for the determination of arsenic species. As described in this review, many microorganisms (bacteria, fungi, and yeasts) and animals are now known to biomethylate arsenic, forming both volatile (e.g., methylarsines) and nonvolatile (e.g., methylarsonic acid and dimethylarsinic acid) compounds. The enzymatic mechanisms for this biomethylation are discussed. The microbial conversion of sodium arsenate to trimethylarsine proceeds by alternate reduction and methylation steps, with S-adenosylmethionine as the usual methyl donor. Thiols have important roles in the reductions. In anaerobic bacteria, methylcobalamin may be the donor. The other metalloid elements of the periodic table group 15, antimony and bismuth, also undergo biomethylation to some extent. Trimethylstibine formation by microorganisms is now well established, but this process apparently does not occur in animals. Formation of trimethylbismuth by microorganisms has been reported in a few cases. Microbial methylation plays important roles in the biogeochemical cycling of these metalloid elements and possibly in their detoxification. The wheel has come full circle, and public health considerations are again important.
INTRODUCTION
Biomethylation and Bioalkylation
The synthesis and transfer of methyl groups is an important and widely distributed metabolic process. The following natural products containing one or more methyl groups attached to nitrogen atoms were discovered early in the 19th century: creatine, creatinine, choline, and trimethylamine (113). Another compound, trimethylglycine ([trimethylammonio]acetate), originally named lycine (from Lycium barbarum) was later renamed betaine (from Beta vulgaris) (113). This discovery contributed the term “betaine” to chemical nomenclature to describe zwitterionic types, for instance (CH3)n X+-CH2-COO− (if X = N, n = 3; if X = S, n = 2).
The first observation of a biological methylation (16) came when His, with an interest in the detoxification of aromatic compounds, administered pyridine to a dog (122). N-Methylpyridine was excreted in the urine. Similar examples of the methylation of aromatic compounds were the conversion, xanthine → methylxanthine in rabbits, and nicotinic acid → trigonelline in dogs (2).
Much later, nutritional experiments showing, for example, that rats could substitute homocystine for methionine in the diet if choline was present led to the concept of transmethylation. This was defined initially as the transfer of a methyl group from one compound to form another N-methyl or S-methyl compound (80). A very important role for methionine, a methylated sulfur compound (111), was slowly recognized, and S-adenosylmethionine (SAM) was identified as the product of the enzymatic activation of methionine in transmethylation reactions (206). The role of SAM as the methyl donor in hundreds of methylation reactions is now well established (36, 37). The mechanism for the de novo biosynthesis of methyl groups has also been determined (90).
Methylation of metalloids.
The C, O, N, and S atoms of organic compounds frequently function as methyl group acceptors in primary and secondary metabolic processes (37, 206). This article concerns another phenomenon, the use of metalloids as methyl group acceptors with the major emphasis on the production of volatile compounds by microorganisms. Because of its widespread distribution and many uses in agriculture, industry and medicine, most of the research has concerned arsenic. Like nitrogen, arsenic is a member of group 15 of the periodic table (International Union of Pure and Applied Chemistry recommendation). Work with other members of this group, antimony and bismuth, will also be included. It is of interest that antimony and arsenic share some chemical and toxicological properties (96). Microbial biosynthesis of methylated metalloids is now of considerable academic interest, and the physiological actions of the methylated products are of concern in disciplines such as medicine, toxicology and environmental studies.
The term “biomethylation” describes the formation of both volatile and nonvolatile methylated compounds of metals and metalloids. For arsenic, the major volatile compounds formed by biomethylation have the structure, (CH3)nAsH3−n; for n = 1, 2, and 3, the products are mono-, di-, and trimethylarsine, respectively. The major nonvolatile compounds are methylarsonate and dimethylarsinate. The nomenclature of the arsenic oxyacids is somewhat confusing (Table 1). Potassium arsenite preparations have a variable composition; the commercial article corresponding approximately to KAsO2 · HAsO2. The “meta-acid” HAsO2 (i.e., H3AsO3 minus H2O) and its salts are not known in solution. Acids will generally be referred to as ionized forms (e.g., arsenate for arsenic acid), since these are presumably present under physiological conditions.
TABLE 1.
Nomenclature of arsenic compoundsa
| Group and compound | Formula |
|---|---|
| Pentavalent | |
| Arsenic acid (arsenate) | (HO)2AsO(OH) |
| Methylarsonic acid (methylarsonate) | (CH3)(HO)AsO(OH) |
| Dimethylarsinic acid (dimethylarsinate) | (CH3)2AsO(OH) |
| Trivalent | |
| Arsenous acid (arsenite) | (HO)2As(OH) |
| Methylarsonous acid (methylarsonite) | (CH3)(OH)As(OH) |
| Dimethylarsinous acid (dimethylarsinite) | (CH3)2As(OH) |
The above names are those used in this review. Alternative names and abbreviations are as follows: for arsenic acid, orthoarsenic acid and arsenic(V) acid; for methylarsonic acid, methanearsonic acid, methylarsinic acid, and monomethylarsonic acid (abbreviations, MMAV and MMAA); for dimethylarsinic acid, dimethylarsonic acid, cacodylic acid, and hydroxydimethylarsine oxide (abbreviations, DMA and DMAA); for arsenous acid, arsenious acid and arsonic (III) acid; for methylarsonous acid, monomethylarsonous acid (abbreviation, MMAIII).
The related term “bioalkylation” refers to “processes involving living cells that cause an alkyl group to become directly bonded through a carbon atom to some ‘heavy element’ (defined as any element with atomic number larger than 10)” (60, 236). In view of the extensive occurrence of bioalkylation with a methyl group, i.e., biomethylation, the term “bioalkylation” specifically refers to “processes involving alkyl groups other than methyl.” For the simplest alkyl groups such as ethyl, propyl, etc., this process is rare. However, As(C2H5)(CH3)2 has been recorded in landfill and sewage gas, and As(C2H5)3 has been recorded in landfill gas; they are likely formed by microbial action, thus representing a true bioalkylation (86). Natural gas probably contains As(C2H5)(CH3)2 and As(C2H5)3, and chemical mechanisms for their formation have been discussed (129). Relatively nonvolatile arsenic species, As (C2H5)(CH3)2, As(C2H5)2(CH3), and As(C2H5)3, are present in river and harbor sediments (144) as well as nonvolatile Sb(C2H5)3 derivatives. Triethyllead is a toxic metabolic product of tetraethyllead (112) and volatile lead species with methyl and ethyl groups have been detected in sewage gas (86).
The term “bioalkylation” has sometimes been used for more-complex naturally occurring materials, for instance, trimethylarsonium betaine, (CH3)3As+CH2COO−, discovered originally in Australian rock lobster (Panulirus longipes cygnus); lipoidal substances such as O-phosphatidyl trimethylarsoniumlactic acid (Fig. 1A), found in a marine diatom; and arsenoribofuranosides (Fig. 1B), found in marine algae (110), diatoms, the terrestrial alga Nostoc commune var. flagelliforme (148), and in oyster tissue (244). These structural types are not present in bacteria and fungi. However, methylarsonate and dimethylarsinate were found in several edible mushrooms (collected in Switzerland and Slovenia) and, as well, arsenobetaine (35). These materials occur in a wide range of terrestrial mushrooms and lichens; other compounds include arsenocholine and the tetramethylarsonium ion (140, 145, 146, 150, 220) and small amounts of some arsenoribofuranosides (140). The use of bioalkylation in this context appears incongruous; all of the materials contain methyl groups attached to the arsenic atom so presumably the biosynthetic process involved is biomethylation.
FIG. 1.

Examples of arsenolipids. (A) O-Phosphatidyltrimethylarsonium lactic acid; (B) an arsenoribofuranoside. Several variations on this structure are observed. In each case, R is a fatty acyl residue.
Although biomethylation of arsenic is very widespread, occurring not only in microorganisms but also in algae, plants, animals, and humans, it is not universal. In the animal kingdom, several monkeys (marmoset, squirrel, and tamarind), chimpanzees, and guinea pigs do not methylate arsenic to any significant extent (14). There are reviews of biomethylation in general (14, 43, 84, 94, 204, 236) and the methylation of arsenic in particular (15, 119, 173, 227, 232, 235).
The selected topics discussed here are a small component of a very wide area, the overall global role of metalloid elements. In particular, arsenic compounds are widely distributed and transformed on earth not only in the lithosphere but also in the atmosphere and hydrosphere (91, 168). Determination of arsenic species in the environment and the health effects of arsenic exposure have been reviewed (31, 49, 56, 75, 156, 164, 184, 185, 216, 235). The separation and identification of volatile arsines in gases (e.g., in headspaces above microbial cultures) is relatively straightforward, but the identification of nonvolatile components is more difficult. The nonvolatile metabolites may be converted to volatile arsines by hydride generation, for example, by reaction with sodium borohydride as a reducing agent. The advantages of hydride generation atomic absorption spectrometry (AAS) in analysis come from its much lower detection limits—parts per billion compared to parts per million for normal AAS—and from a lowered requirement upon matching the sample matrix which befalls normal AAS (59). At a pH of <4, arsenite is reduced to arsine itself, AsH3; other conversions of oxyacids can be represented as follows: (CH3)nAsO(OH)3−n → (CH3)nAsH3−n where n = 1 to 3 (68, 69). Hence, methylarsonate and dimethylarsinate yield, respectively, monomethylarsine and dimethylarsine. Trimethylarsine oxide yields trimethylarsine. With such techniques, nonvolatile arsenic compounds can be partially identified as mono-, di-, or trimethyl species. Hydride generation can be coupled to separation instruments such as gas chromatography-mass spectrometry (MS) but most often is used with gas chromatography using atomic absorption spectrometric detection (9). With that said, high-pressure liquid chromatography-inductively coupled plasma-MS (HPLC-ICP-MS) is probably the most powerful tool.
Also becoming more widely used for nonvolatile organometalloid analysis are even more powerful so-called hyphenated instrumental techniques such as ion or HPLC-ICP-MS (110, 141, 147). And finally, the extraction of biological materials and subsequent determination of organoarsenicals has very recently been undertaken using microwave digestion followed by UV irradiation, hydride generation, and fluorescence spectrometry (244). This is similar to microwave-assisted extraction of arsenic in soils (114).
There is a wide difference in the arsenic level of marine and terrestrial organisms. In land creatures, the arsenic level is typically about 1 ppm (dry weight), but for marine organisms the values range from a few parts per million to as much as 100 ppm (5, 149, 164). There are practical consequences. While for the U.S. population the normal adult value for urinary arsenic excretion is <50 μg day−1, the value for Japanese adults is three times as high (179). This level of urinary arsenic probably reflects a greater dietary use of marine organisms. A detailed evaluation of “arsenic eaters” has discussed the possibility of the chronic ingestion of arsenic trioxide by otherwise healthy people in hopes of improving a range of factors, from the beauty of women, prophylaxis against disease, to increased sexual potency (197).
Early history.
Hofmeister observed that subcutaneous administration of 0.06 g of sodium tellurate to a 3-kg dog gave a strong garlic odor in the expired breath of the dog 30 min later (123). Similar odors were obtained when various organ pulps were treated with tellurium compounds. Influenced by the previously described work of His on pyridine methylation, Hofmeister obtained inadequate evidence that the odorous material in the expired dog breath was “tellurmethyl,” i.e., dimethyl telluride, (CH3)2Te. Nevertheless, he has been credited as the first to observe the biomethylation of a metalloid. Moreover, he certainly recognized the overall significance of methyl group transfer and stated that when pyridine or tellurium are administered, methylation of these compounds may occur, while under normal conditions other methylated compounds such as choline and creatine may be formed. However, he did not propose a specific methyl group donor.
Hofmeister was not the first to have noted odor production during metalloid metabolism. The formation of odorous materials from both selenium and tellurium compounds was described in animal experiments before his work; thus, a garlic smell (“Knoblauchgeruch”) was observed in 1824 on dissection of a rabbit poisoned with telluric acid (99). These odors, however, were attributed to hydrogen selenide or hydrogen telluride (for details, see reference 48). Interestingly, a 1951 organic chemistry text states that dimethyl telluride has “the most abominable odour of all organometallics” (50).
ARSENIC FUNGI
Wallpaper: Hazardous to Health
Just before Hofmeister's work, some fungi had been shown to produce a garlic odor when grown in the presence of arsenic. This work, not noted by Hofmeister, marks the beginning of the study of biomethylation of metalloids by microorganisms; surprisingly, the work involved problems with wallpaper.
In the 17th century, a dissertation by Caroli de la Font concerning “The Nature and Causes of the Plague… deducing the Pestilential venom from the Air infected and corrupted chiefly by Arsenical Exhalations” had been reviewed in the Philosophical Transactions of the Royal Society (10). In the 1809 abridgment of this series, the dissertation was omitted and replaced with the statement that this work was “filled with absurd conjectures and reasoning” (127). However, some two centuries later, a type of toxic “arsenical exhalation” did come to prominence. Beginning about 1800, cases of poisoning in Germany were ascribed to wallpapers (and tapestries) printed with arsenical pigments.
Some of the decorative pigments used in printing were bright green arsenical materials such as Scheele's green (copper arsenite, CuHAsO3) and Schweinfurt (or Schweinfurth) green, also termed Paris green, Vienna green, or emerald green (copper acetoarsenite, 3CuO · As2O3 · Cu[OOC · CH3]). The use of these pigments was extensive (20). In 1871, arsenical papers were used “in the majority of dwelling houses, from the palace down to the navvy's hut. It is rare to meet with a house where arsenic is not visible on the walls of at least some of the rooms” (12). Moreover, arsenical pigments were used for coloring purposes in many other ways, even in foodstuffs. Many cases of poisoning were recorded (130), and a leading article in 1860 stated that “In the most emphatic manner, we feel it to be a duty to call the attention of medical practitioners to recent lamentable case of fatal poisoning, occasioned by the atrocious practice of coloring hanging-papers with arsenic pigments” (11).
The toxic effects observed in rooms coated with green pigments might have resulted from inhalation of arsenic-containing particles. However, poisoning was also observed where arsenical paper had been covered by fresh paper lacking arsenic, hence formation of dust particles was unlikely. The distinguished chemist Leopold Gmelin recorded in an 1839 newspaper article that an adverse, mouse-like odor was usually present in rooms where poisoning had occurred (100; a translation was kindly provided as a personal communication to T. G. Chasteen by M. Wiggli). He believed that the smell was caused by arsenic that had volatilized as alkarsine [cacodyl oxide, (CH3)2AsOAs(CH3)2]. Cacodyl itself is denigrated by the Oxford English Dictionary as “of most disgusting odour.” Whether volatilization of arsenic was possible and caused poisoning was a much debated question (40, 157, 207, 208).
Work of Bartolomeo Gosio
Slowly, it was realized that damp and moldy conditions favored the production of a volatile arsenic compound. The Italian physician Bartolomeo Gosio initiated a controlled program to determine how arsenic preparations of use in industry and the domestic economy became a health hazard (101, 102, 104, 105, 107). Although a biography of Gosio (3) lists a “nota preventiva,” dated 1890, on the role of fungi in the “gasification” of arsenic, this may be misdated. Gosio stated that he worked on this subject from May 1891 to April 1892; a preliminary account of his work was given in 1891 (103). He used a simple technique to obtain microorganisms producing a garlic odor when grown on a potato mash containing arsenic trioxide (arsenous oxide). Such a substrate, exposed to the air of a cellar, quickly developed colonies of fungi and bacteria and produced the garlic odor. Since pure cultures of the bacteria were odor-free, the gasification was ascribed to the action of fungi.
Initially, Gosio focused on arsenic volatilization by “mucor mucedo [sic],” presumably the common bread mold. Copper arsenite, the basis for the arsenical colors used in dyeing, was converted to the garlic-odored gas, but sulfide-containing pigments (e.g., orpiment and realgar) were not. Gosio aspirated gas from mixed microbial cultures into a silver nitrate solution; within 3 days there was a marked reduction of the silver salt. The presence of arsenic in the liquid after removal of silver was shown by the chemical Marsh test (102, 104). From 800 g of potato mash containing 0.12 g of arsenic trioxide, Gosio obtained 2.8 mg of metallic arsenic corresponding to the gasification of 4.3 mg of “As2O3 [sic].” This was a small yield to be sure, but it was a clear demonstration of arsenic volatilization by some saprophytes. Arsenical gases were also obtained from “hangings colored with Scheele's and Schweinfurth's greens, through the vegetation of the mucor… hence the danger to those who live in such an atmosphere.” At this time, Gosio mistakenly believed that the gas contained arsine itself.
Another fungus showing a very active production of arsenical gas when grown with arsenic compounds was isolated from a piece of carrot exposed to air (102, 104). It was identical to “penicillium brevicaule [sic]” discovered earlier on rotted paper (“papier putride”). Gosio stated that this organism developed such an intensity of arsenical gas that it was dangerous to approach it (“il est dangereux de s'en approcher”). A rat exposed to the vapor was quickly killed (“Un rat qu'on expose à ces sortes d'émanations tombe rapidement dans des convulsions mortelles”), and a small mouse placed in a vessel with the fungus producing the gas died after a few seconds. When Emmerling reported contradictory results with Mucor mucedo and Aspergillus glaucus, Gosio vigorously defended his work and regretted that Emmerling had not attempted to obtain suitable cultures from him (82, 106). At the Harvard Laboratory, an American investigator, Charles Robert Sanger, had investigated cases “of chronic arsenical poisoning” from wallpapers but had been unable to demonstrate the volatilization of arsenic. Unlike Emmerling, he corresponded with Gosio, received cultures of Penicillium brevicaule, and confirmed Gosio's results (207, 208). Gosio's work was recognized by naming the garlic-odored volatile material as “Gosio gas.” It is, perhaps, the only gas named eponymously.
P. brevicaule is now named formally as Scopulariopsis brevicaulis (Sacc.) Bainier (200). Historically, Penicillium and Scopulariopsis species have often been confused; there is, however, one important difference—Scopulariopsis species are never green. Scopulariopsis species, especially S. brevicaulis, are abundant in nature in material such as soil, stored grain and forage, and slowly decaying semidry vegetation (200). Quite typically, Gosio's organism was isolated from rotting paper and a slice of carrot.
Gosio had also shown volatilization of arsenic to a garlic-odored product with A. glaucus and Aspergillus virens, Mucor ramosus, Cephalothecium roseum, and Sterigmatocystis ochracea. Such organisms, termed “arsenic fungi,” were later shown to be more common than previously supposed (237); thus, active strains were found among other aspergilli (Aspergillus fischeri, Aspergillus sydowi). From soil samples containing enough arsenic to prevent normal growth of certain crops, active gas producers were also obtained; they were two strains of Fusarium, a strain of Paecilomyces, and a sterile brown fungus. The volatile materials were not identified chemically. This work led to the definition of three microbial responses to arsenic: some microorganisms were inhibited by it, some tolerated arsenic but did not produce arsenical gas, and some tolerated it with production of gas. Further work with arsenic fungi is described later.
Microbiological Test for Arsenic
Gosio developed a sensitive microbiological test for arsenic based on the garlic odor (102, 105, 107, 108). A material possibly containing arsenic was extracted with water or dilute acid, and the extract was evaporated. Small quantities of the residue were added to a substrate and incubation with S. brevicaulis followed. After a few hours at 25 to 30°C, the presence of arsenic was indicated by a garlic odor. In an improved method, the suspected material was added to a pregrown culture on a potato slice; a garlic odor developed in about 10 min when arsenic was present. As little as 1 μg of arsenic trioxide in 1 g of material could be detected; the odor threshold for Gosio gas (trimethylarsine) in dilute aqueous solution is now said to be 2 ng kg−1 (75). Czapek's solution agar can also be used for growth of S. brevicaulis (222). Earlier research has been summarized (165). Although qualitatively more delicate than the Marsh test, the Gosio method is not adapted for quantitative use. A modern use of it is described later in connection with the bacterial methylation of arsenic.
WORK OF CHALLENGER AND ASSOCIATES (“LEEDS SCHOOL”)
When Gosio gas was passed into a solution of mercuric chloride in dilute HCl (Biginelli's solution) a crystalline precipitate was formed. A small vial of the Gosio/Biginelli mercurichloride is to this day preserved at The Museum of the History of Medicine (Museo di Storia della Medicina) in Rome, Italy. Under the printed name “Laboratorio Batteriologico della Sanità Pubblica” is handwritten, “arsina penicillare comp. mercurico” (C. Serarcangeli, personal communication). From analysis of this material (25, 26) and of the gas itself (107) it appeared that the gas was diethylarsine, (C2H5)2AsH. More recently, the aspiration of culture headspace gases into Biginelli's solution has been termed “chemofocusing” (65).
Gosio Gas Is Trimethylarsine
When the reaction between Gosio gas and HgCl2 was reexamined by Challenger and his associates, two crystalline mercuric chloride precipitates were obtained, depending on the time of aspiration (40, 46). These compounds were the di- and mono-mercurichloride of trimethylarsine, (CH3)3As · 2HgCl2 and (CH3)3As · HgCl2, respectively. Hence, Gosio gas was trimethylarsine. Ironically, trimethylarsine had been discovered as early as 1854, whereas diethylarsine was not known until after Gosio's work. Trimethylarsine is a colorless, oily liquid with a boiling point of 51 to 53°C (760 mm). It was obtained by reaction of magnesium methyl iodide, prepared in pentyl ether, with arsenic trichloride in xylene (81) among other more modern methods.
Trimethylarsine was obtained from the organic compounds sodium methylarsonate, CH3AsO(ONa)2, and sodium dimethylarsinate, (CH3)2AsOONa, by action of S. brevicaulis. Mixed methylarsines containing other alkyl groups were formed by growth of S. brevicaulis on suitable substrates (40) (Table 2).
TABLE 2.
Formation of mixed alkyl arsines by S. brevicaulis
| Substratea | Formula | Product |
|---|---|---|
| Ethylarsonic acid | (C2H5)AsO(OH)2 | (CH3)2As(C2H5) |
| Diethylarsinic acid | (C2H5)2AsO(OH) | (CH3)As(C2H5)2 |
| Propylarsonic acid | (C3H7)AsO(OH)2 | (CH3)2As(C3H7) |
| Methylpropylarsinic acid | (C3H7)(CH3)AsO(OH) | (CH3)2As(C3H7) |
| Allylarsonic acid | (C3H5)AsO(OH)2 | (CH3)2As(C3H5) |
The substrate names have been modernized from those used by the Challenger group (40).
Challenger Mechanism for Arsenic Biomethylation
The Leeds School suggested a mechanism for trimethylarsine formation (40, 41, 42) that, in its broad details, has stood the test of time. It was proposed before SAM was identified as a nearly universal methyl donor in biological systems and when compounds such as formaldehyde, choline, and betaine were considered to be possible methyl donors in S. brevicaulis. It was assumed that a “positive methyl group,” CH3+, would need to be generated and transferred to the metalloid. For the purpose of the present discussion it will be taken that SAM is, in fact, the methyl donor, and evidence for the assumption is presented later. To proceed from arsenate to trimethylarsine, four two-electron reductions are required. Each reduction produces a compound with an electron lone pair on the arsenic atom; with the exception of the final step, each reduction product is then methylated by SAM. Basic mechanisms for reduction using a hydride ion and methylation using SAM are shown in Fig. 2, as well as a complete and modern version of the individual reaction steps from arsenate to trimethylarsine in Fig. 3. A chemical model for the Challenger mechanism uses (CH3)3S+PF6− as the methyl donor and SO2 as the reducing agent (13a).
FIG. 2.

Typical reactions of the Challenger mechanism. The top line indicates a mechanism for the reduction, As(V) → As(III), resulting in an unshared pair of electrons on As. Structures are as follows: R1 = R2 = OH, arsenate; R1 = CH3, R2 = OH, methylarsonate; R1 = R2 = CH3, dimethylarsinate. For reduction of trimethylarsine oxide to trimethylarsine, the process is a little different. Following proton addition, the structure H-O-As+(CH3)3 reacts with hydride ion leading to elimination of H2O. The bottom line indicates the methylation of an As(III) structure with SAM [shown in abbreviated form as CH3-S+-(C)2]. A proton is released and SAM is converted to S-adenosylhomocysteine [abbreviated form, S-(C)2].
FIG. 3.

Challenger mechanism for the conversion of arsenate to trimethylarsine. (A) Arsenate; (B) arsenite; (C) methylarsonate; (D) methylarsonite; (E) dimethylarsinate; (F) dimethylarsinite; (G) trimethylarsine oxide; (H) trimethylarsine. The top line of structures shows the As(V) intermediates. The vertical arrows indicate the reduction reactions to the As(III) intermediates (bottom line), and the diagonal arrows indicate the methylation steps by SAM (see Fig. 2 for details of the reduction and methylation processes).
Two of the proposed intermediates are poorly defined As(III) compounds—methylarsonous acid, CH3As(OH)2, and dimethylarsinous acid, (CH3)2AsOH. For experimental purposes, use is made of the fact that aqueous solutions of the cyclic methylarsine oxide, (CH3AsO)x, also termed methylarsonous acid anhydride, contain methylarsonous acid: (CH3AsO)x + H2O → CH3As(OH)2. This acid is also present when diiodomethylarsine, CH3AsI2, is dissolved in water. Similarly, dimethylarsinous acid may exist as its anhydride, tetramethylarsinous acid anhydride, (CH3)2As-O-As(CH3)2 (239): [(CH3)2As]2O+ H2O → 2(CH3)2AsOH.
It is of interest that methylarsonous acid has been detected in human urine by using ion-pair chromatographic separation of compounds with hydride generation atomic fluorescence spectrometry detection (153, 154). The individuals who were examined normally drank water with a high arsenic level, and urine samples were analyzed after treatment of some of the subjects with the chelating agent sodium 2,3-dimercapto-1-propanesulfonate.
Understanding the chemical details of the overall scheme has sometimes caused difficulty, in part because of confusion between the term “oxidation number,” which seeks to define electron distribution, and “valency” (or “valence”), which refers to the number of combining entities. The normal valency of arsenic is 3 or 5 (e.g., AsF3 and AsF5), and the most prominent oxidation states are −III, +III, and +V; the latter two states, As(III) and As(V), are of most concern here. The reductive steps are straightforward in concept but not in practice. Thus, arsenate with the oxidation number +V and a positive charge on the As atom is converted by a two-electron reduction to arsenite, with an oxidation number of +III [As(V) → As(III)]. The other reductions are conceptually similar (Fig. 3).
To convert arsenate to trimethylarsine, the reductions are likely to be enzyme catalyzed with reductants providing the necessary 2e−. The reductants are almost certainly thiols, and particular attention has been given to glutathione (GSH) and lipoic acid (6,8-dithiooctanoic acid). There is little experimental evidence from microbial systems, but it is assumed that the reactions resemble those for formation of methylarsonate and dimethylarsinate in mammals (239).
A disulfide redox couple, for instance involving GSH (equation 1), would drive the reduction of As(V) to As(III) in the general process of equation 2 (74).
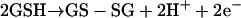 |
(1) |
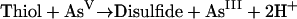 |
(2) |
GSH does reduce arsenate to arsenite in vitro (equation 3), and further reaction eventually forms arsenotriglutathione [tri(γ-glutamylcysteinylglycinyl)-trithioarsenite] (equation 4):
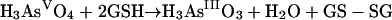 |
(3) |
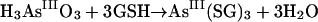 |
(4) |
For the second reductive step, there is evidence that both in vitro and in vivo GSH does function as a reductant (239). Again, the mechanism is complicated by the ability of arsenic compounds to react with thiols. In model experiments, disodium methylarsonate was easily reduced by cysteine, GSH, or mercaptoethanol, to give derivatives of As(III) (74). For the overall reaction of equation 5, individual steps (equations 6, 7, and 8) were postulated:
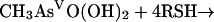 |
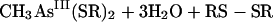 |
(5) |
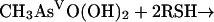 |
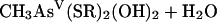 |
(6) |
 |
(7) |
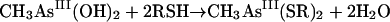 |
(8) |
Similar equations can be written for the subsequent reduction steps; it does seem likely that in vivo compounds with RS groups attached to the arsenic atom are involved. Dithiol derivatives such as lipoic acid could be involved, perhaps as a membrane-bound or enzyme-bound component. Thus, for arsenite itself, the reaction of equations 9 and 10 might be involved with the formation of cyclic disulfide derivatives.
 |
(9) |
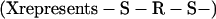 |
 |
(10) |
The final step of the Challenger mechanism, the reduction of trimethylarsine oxide to trimethylarsine, was possible with a variety of thiol reagents (cysteine, dimercaptopropanol, lipoic acid, mercaptoacetic acid, and mercaptoethanol) (73). On the basis of kinetic and other observations, the detailed mechanism of equations 11 to 14 was postulated. Enzyme- or membrane-bound lipoic acid was regarded as a likely in vivo reductant.
 |
(11) |
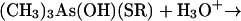 |
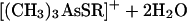 |
(12) |
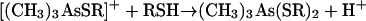 |
(13) |
 |
(14) |
If reductive steps using GSH occur in vivo, the recycling of GS-SG to GSH by GSH reductase is necessary. It is of interest that this enzyme is inhibited by various arsenic compounds, including arsenotriglutathione (230, 231).
The methylation steps are at first sight somewhat confusing, since in all cases an oxidation has occurred, As(III) → As(V). To keep the process in balance there must have been a concomitant reduction. Since no redox cofactor is involved, the process appears at first glance to be a simple methyl transfer process. However, a detailed analysis of the reaction indicates that the necessary reduction is found in the corresponding decrease in oxidation number from −II to −IV for the transferred carbon. Using a formal method for assigning oxidation states to redox-labile elements in organic compounds helps in following the reduction and methylation steps with arsenic as shown in Fig. 4 (117).
FIG. 4.

Changes in oxidation numbers on As and C during methylation according to Hanselmann's method (117). Abbreviations: Ox, oxidation number; E.N., electronegative (than).
Role of SAM
Since the methyl group of [14CH3]methionine was transferred to arsenic in the formation of trimethylarsine by S. brevicaulis (47), the postulated role for SAM was substantiated. Similar work has confirmed methyl transfer processes from [C2H3]methionine to materials such as As2O3, CH3AsO3Na2, and (CH3)2AsO2H by the fungi S. brevicaulis and Gliocladium roseum and the yeast Cryptococcus humiculus (for taxonomy of this yeast, see below); the transfers resulted in the formation of 2H-labeled trimethylarsine (66). The latter organism and S. brevicaulis carried out the previously unknown conversion of disodium butylarsonate, n-C4H9AsO3Na2, to butyldimethylarsine.
Nonvolatile di- and trimethylarsenic species were identified in cultures of C. humiculus, and a dimethylarsenic compound was identified in cultures of the marine alga Polyphysa penicilus (68, 69) by the hydride generation technique (see above). When [C2H3]methionine was used as a methyl donor, the 2H-labeled nonvolatile metabolites produced 2H-labeled arsines. These observations were consistent with the operation of the Challenger mechanism, with methyl groups being derived from methionine by way of SAM. Moreover, trimethylarsine formation from (CH3)2AsO(OH) by these organisms was inhibited by ethionine. Ethionine is an antagonist for methionine; hence, these observations support the role of a methionine-based synthetic path (66, 75).
ARSENIC METHYLATION
Fungi and Yeasts
Challenger et al. observed that Penicillium chrysogenum and strains of Penicillium notatum, Aspergillus niger, and A. fischeri did not methylate arsenous acid in the usual bread crumb cultures (27). A. glaucus and Aspergillus versicolor formed trimethylarsine to a limited extent. However, all of these organisms converted methylarsonate to trimethylarsine. Trimethylarsine was also formed from dimethylarsinate by P. notatum, P. chrysogenum, and two strains of A. niger. The latter organism converted ethylarsonate to dimethylethylarsine and P. chrysogenum converted allyllarsonate to dimethylallylarsine.
A wood-rotting basidiomycete, Lenzites trabea, produced a garlic odor when growing on wood treated with arsenical preservatives. When 65 species of wood-rotting fungi were examined with growth in presence of As2O3, only L. trabea and Lenzites saepiaria produced an unidentified, garlic-odored gas (176). Another wood decaying fungus, Phaeolus schweinitzii, produced volatile methylated products of arsenic when grown with “As2O5” (19). The strong garlic smell aided the recognition of plates containing this organism in isolation work with a selective medium. More recent work has confirmed the volatilization of arsenic by this fungus, but again the product was not specifically identified (189). The pathogenic fungus Trichophyton rubrum produced a “peculiar, nauseating, garlic-like odor” when it was grown in the presence of arsenate or arsenite (262). The volatile product contained an unidentified arsenic component(s).
A historical connection, where the wheel comes full circle, may be noted here. The widely used wood preservative, chromated copper arsenate, is converted to trimethylarsine by the yeast C. humiculus (72). Chromated copper arsenate is quite similar to the arsenical wallpaper pigments that had triggered Gosio's work.
The work of the Leeds School was carried out by tedious chemical operations, usually the formation and analysis of mercuric chloride complexes. Moreover, the useful radioactive isotopes of arsenic were not available; however, as already noted 14C was used in studies of methyl transfer reactions. With the advent of improved analytical techniques such as gas chromatography and gas chromatography-MS, and with the availability of arsenic isotopes, the detection of trimethylarsine and related compounds can be carried out with less labor and greater certainty. A simple gas chromatographic assay was used to monitor production of trimethylarsine by three organisms isolated from sewage and described as fungi (53). Two of these organisms, G. roseum and a Penicillium species, converted methylarsonate and dimethylarsinate to trimethylarsine under a range of culture conditions; however, neither arsenate nor arsenite was volatilized. The third organism, identified tentatively as Candida humicola was said later (69) to have been reclassified as Apiotrichum humicola. It had been deposited with the American Type Culture Collection as ATCC 26699 and in the American Type Culture Collection catalog it is now described as Cryptococcus humiculus (Daszewska) Golubev. The name C. humiculus is used throughout this article; the organism is clearly a yeast. When grown at pH 5.0, it produced small amounts of trimethylarsine from arsenate, arsenite, and methylarsonate, with dimethylarsinate being the best substrate for volatilization.
The variables affecting the methylation of arsenic by C. humiculus have been studied. Only a brief account will be given here since two authoritative reviews are available (52, 75). All of the organic As(V) compounds of the Challenger pathway (methylarsonate, dimethylarsinate, trimethylarsine oxide) have been observed in cell extracts metabolizing arsenate, and all of them are precursors for trimethylarsine biosynthesis (70, 195). Preconditioning of cells with dimethylarsinate improves trimethylarsine formation from arsenate and dimethylarsinate. In later experiments, the nonvolatile components of C. humiculus and S. brevicaulis were identified (67). These experiments used low levels (1 ppm) of the arsenic substrates, and under these conditions there was no detectable formation of trimethylarsine with either organism.
With methylarsonate as substrate, C. humiculus produced low levels of dimethylarsinate and trimethylarsine oxide by the second day, with slow increases of both metabolites over 4 weeks. For S. brevicaulis, the metabolism was much slower, with trace amounts of dimethylarsinate and trimethylarsine oxide not appearing until after 2 weeks of incubation.
This was the first report of trimethylarsine oxide as an end product metabolite for the two organisms; under the experimental conditions, it was the major product rather than trimethylarsine. Apparently the organisms tolerated low levels of this oxide, and further “detoxification” by conversion to trimethylarsine was not necessary. An extended model (Fig. 5) for arsenic methylation involving both cells and medium was proposed (67). The observed reaction rates suggested a possible sequential transfer of 2 methyl groups from SAM to methylarsonate as indicated. These observations emphasized that the methylation of arsenic by microorganisms is a very complex process despite the general simplicity of the Challenger mechanism.
FIG. 5.

Expanded version of the Challenger mechanism. This mechanism, proposed for C. humiculus, indicates roles for components both in the cells themselves and in the culture medium. The double vertical lines indicate cell walls. (A) Phosphate transport system; (B) thiols and/or dithiols; (C) active transport system; (D) active/passive transport; (E) passive diffusion. Abbreviations: MMAV, methylarsonic acid; DMA, dimethylarsinic acid; TMAO, trimethylarsine oxide. This diagram is redrawn from scheme 2 of reference 67.
Phosphate strongly inhibited formation of trimethylarsine in cultures growing with arsenate, arsenite, and monomethylarsonate but had no effect when dimethylarsinate was used (54). The reason for this behavior is not clear (75) but it is of interest that phosphorous and arsenic are both members of group 15 of the periodic table. Sodium salts of selenite and selenate were also strongly inhibitory, and sodium tellurate was a moderate inhibitor. C. humiculus is known to methylate both selenate and selenite to dimethylselenide (52).
C. humiculus produced a garlic odor when grown in the presence of benzenearsonic acid (phenylarsonic acid, C6H5As[OH]2) (65). By use of the chemofocusing technique, the volatile product was shown to be C6H5As(CH3)2. Similarly, methylphenylarsinic acid, (C6H5)(CH3)AsO(OH), was converted to the same dimethylphenylarsine. Arsanilic acid, H2NC6H4AsO(OH)2, was not methylated, but 2-hydroxy-4-aminobenzene-arsonic acid yielded trimethylarsine.
The fungicide methylarsine sulfide, (CH3AsS)x (Rhizoctol), produced a garlic odor when grown with C. humiculus; the volatile material was a mixture of trimethylarsine and methylarsine in the ratio 90:10. The conversion was rapid, with 50% of added arsenic being converted in 3 days; compare arsenate or dimethylarsinate → trimethylarsine, which resulted in 1% conversion in 10 days (71). Methylarsine sulfide was actually more toxic to C. humiculus than either arsenite or the corresponding oxide, ([CH3]2AsO)x, suggesting that at least in this case, methylation was not a detoxification process. Methylarsine oxide was also rapidly transformed by C. humiculus to dimethylarsinate (24 h); small amounts of unidentified volatile arsines were also formed.
The marine yeast Rhodotorula rubra an obligate aerobe, converted arsenate to arsenite, methylarsonate, and dimethylarsinate. Unidentified “volatile arsines” were also produced (243).
Modern analytical techniques for the determination of arsenic species continue to be developed and used (64, 110, 147, 149, 151-154, 190-192, 244). In one study a Penicillium species was isolated from an agricultural evaporation pond in an area with high concentrations of soil arsenic. The organism formed trimethylarsine from methylarsonate; addition of carbohydrate and sugar acids to the minimal medium suppressed volatilization, whereas glutamine, isoleucine, and phenylalanine had a stimulatory effect (128). Various elements known to be toxic to living systems generally inhibited the conversion of methylarsonate to trimethylarsine by this Penicillium sp. (92). The most inhibitory element was V, followed by Ni, Sn, B, Te, Mn, Mo, Cr, and Ag; at low concentrations the following elements were stimulatory: Hg, Fe, Cu, Al, Zn, and Se. In soil contaminated more than 30 years ago by arsenic trioxide from a cattle dipping vat, a low level of in situ volatilization of arsenic was demonstrated by subsurface probes. From this soil, a Fusarium sp. producing unidentified volatile arsenicals was isolated (238). A recent monograph provides more information in this area (92a).
Bacteria
Gosio's pioneering work on arsenic volatilization by fungi was merely the tip of a very large iceberg. However, the bacteria that he isolated from his mixed cultures did not produce a garlic odor in the presence of arsenic. In 1945, Challenger asserted that “the weight of the evidence would indicate that bacteria are unable to produce volatile methyl derivatives of arsenic, selenium and tellurium, the statements to the contrary being based on observations of odor alone” (40); another reviewer stated that there was not a single case recorded in which a methylation was performed by bacteria (16). However, the possibility of bacterial methylation had not been overlooked. People taking dimethylarsinate (cacodylate) by ingestion were known to exhale a garlic odor, and this was believed to be due to bacterial decomposition of the arsenic compound (198). Several strains of fecal bacteria (named as follows [with the genus not specified] mesentericus vulgatus, B. mesentericus ruber, and B. subtilis) produced a garlic odor when grown in the presence of either dimethylarsinate or arsenate. Although all strains examined produced the odor from dimethylarsinate, one strain each of B. mesentericus vulgatus and B. mesentericus ruber and three strains of B. subtilis did not produce Gosio gas from arsenate. Later workers using cultures of these organisms obtained from a culture collection, rather than being isolated from feces, could not repeat these results (45). Similarly, organisms named B. coli communis and B. lactis aerogenes did not yield trimethylarsine in the presence of arsenous oxide (again, the genus was not specified). Despite these apparently negative results, it does seem quite possible that bacterial methylation of arsenic using organisms isolated from feces had been observed. More recent evidence suggests that mammalian, intestinal bacteria are responsible for a portion of arsenic metabolism by a pathway different from that of animals. Escherichia coli strains from rat cecal contents converted dimethylarsinate into two unidentified, nonvolatile compounds and converted trimethylarsine oxide into a further, nonvolatile, unidentified material (147). Kuroda et al. (147) have detected trimethylarsine oxide in cultures of E. coli amended with dimethylarsinate. The fecal culture of rats, from which the bacteria were isolated, reduced arsenate to arsenite. Along with the isolated strains themselves, the urine and feces of those rats produced unidentified arsenic compounds. These unknowns were different from arsenate, arsenite, methylarsonic acid, dimethylarsinic acid, trimethylarsine oxide, tetramethylarsonium ion, arsenocholine, and arsenobetaine.
The metabolism of arsenic compounds widely used in agriculture has been studied in mixed microbial populations from soil. While evidence was obtained for arsenic volatilization, it was usually not clear whether bacteria or fungi were involved. For example, when 14C-labeled dimethylarsinate was added to three soil samples under aerobic conditions, 35% of the added arsenic was converted to a volatile material or materials over a 24-week period, and 41% was converted to 14CO2 and AsO43− (253). Under conditions considered to be anaerobic, the amount of volatilized arsenic increased to 61% and a garlic-like odor was observed. If the conditions (flooding with water) were indeed anaerobic, fungal growth would have been unlikely, and the observed volatilization might have resulted from a mixed bacterial culture. The volatilization of arsenic from a soil treated with [74As]H3AsO4, sodium [14C]methylarsonate, and [14C]dimethylarsinate was most rapid under aerobic conditions, but significant yields of di- and trimethylarsine were obtained when the experimental flasks were swept with nitrogen (252). The formation of methylarsine was not detected.
Air samples collected over sodium arsenite-treated lawn grass for 2-day periods, with sample collections usually at 2- or 3-h intervals, reliably gave small amounts of trimethylarsine after about 20 h (29). Dimethylarsine was observed in small amounts only in the last two 2-h intervals. From methylarsonate, trimethylarsine formation began after the first 2-h interval and continued throughout the experiment with substantial amounts after about 14 h. Specific organisms were not isolated from the mixed cultures, but the formation of dimethylarsine, not usually a fungal product, suggests bacterial action.
When three soil samples were incubated with arsenate, arsenite, methylarsonate, and dimethylarsinate, methyl- and dimethylarsine were only produced from methylarsonate and dimethylarsinate; some arsine production was also observed. Surprisingly, two soil bacteria (a Pseudomonas sp. and an Alcaligenes sp.) produced only arsine when incubated anaerobically with arsenate or arsenite (51). Arsine (AsH3) formation is relatively uncommon but is discussed later in connection with anaerobic organisms.
Five bacterial species, (Corynebacterium sp., E. coli, Flavobacterium sp., Proteus sp., and Pseudomonas sp.) isolated from the environment and acclimatized to growth with sodium arsenate for 6 months, transformed arsenate to arsenite, and all of them produced dimethylarsine. The Pseudomonas sp. formed all three of the methylated arsines (214). Arsenic accumulated by the cells was in a protein fraction (212). Six bacterial species (Achromobacter sp., Aeromonas sp., Alcaligenes sp., Flavobacterium sp., Nocardia sp., and Pseudomonas sp.) produced both mono- and dimethylarsine from methylarsonate; only two of them produced trimethylarsine. A Nocardia sp. was the only organism that produced all of the methylarsines from this substrate (213).
C. humiculus was known to reduce trimethylarsine oxide to trimethylarsine (195), and this oxide was also reduced under both aerobic and anaerobic conditions by several bacteria (194). The most active (142 nmol min−1 g of cells−1 [wet weight] at 37°C) of four anaerobes (Fusobacterium nucleatum, Veillonella alcalescens, and two unidentified skin organisms) was V. alcalescens. However, anaerobically grown Staphylococcus aureus gave the highest rate for any of the organisms tested anaerobically (208 nmol min−1 g of cells−1 [wet weight]). Similar results were obtained using aerobic organisms; a marine pseudomonad showed the highest activity (585 nmol min−1 g of cells−1 [wet weight]) with substantial activity (range, 80 to 170 nmol min−1 g of cells−1 [wet weight]) for B. subtilis, E. coli, S. aureus (grown aerobically), and two unidentified skin aerobes. A further skin aerobe and Streptococcus sanguis were much less active. The same conversion was shown by river water, some sea sediments and sewage sludge (low levels in both cases analyzed by odor alone), and rumen fluid (under both aerobic and anaerobic conditions). The authors noted that “the odor of trimethylarsine becomes immediately apparent if some of the oxide is placed on the skin,” presumably by the action of the mixed microbial flora (194).
Arsenic-resistant Pseudomonas putida, isolated from a contaminated Chlorella culture, contained a nonvolatile trimethylarsenic species and excreted into the medium all three methylated species (170). Similar results were obtained with arsenic-resistant Klebsiella oxytoca and a Xanthomonas sp. (169). None of these organisms formed volatile arsenic compounds.
A sample of soil with a low level of arsenic contamination (As, 1.5 ppm) yielded two arsenic-resistant, nonmethylating bacteria (Bacillus sp. and Pseudomonas fluorescens) and a new, arsenic-resistant organism, assigned to the Flavobacterium-Cytophaga groups, with methylating capacity. It produced only trimethylarsine as the volatile material; arsenite was transformed more rapidly than arsenate. Dimethylarsinate did not yield trimethylarsine (124). Contaminated soil (contaminated with As, Cr, and Cu) from a wood-impregnating plant had methylating activity (production of methylarsonate and dimethylarsinate). There was no mention of arsenic volatilization in this work (241).
A brief report has described the bioleaching of columns of arsenic-contaminated soil percolated with a nutrient medium (240). Arsenic was converted to unidentified volatiles, most efficiently under anaerobic conditions, thus suggesting possible bacterial action. Using garden soil spiked with As2S3, the undefined microbial population converted the insoluble material to “nearly 50% water-soluble form within two months” (240). At the same time, small amounts of unidentified volatile arsenicals were produced. Bacterial leaching of arsenic-contaminated materials was suggested as a possible means for bioremediation. Solubilization of arsenic by oxidation from the mineral arsenopyrite (FeAsS) by mesophilic and moderately thermophilic acidophiles has been described without mention of any formation of volatile products (79, 240).
Role of Anaerobic Microorganisms
Following the landmark discovery of biomethylation of mercury (32, 136, 251), it was shown that Methanobacterium bryantii produced dimethylarsine from several arsenic compounds (175). The initial chance observation of alkylarsine synthesis was made when a strong, nauseating, garlic-like odor was detected in reaction mixtures containing cell extract, hydrogen, ATP, arsenate, and methylcobalamin. In fact, the intense garlic odor was used cautiously as a rapid qualitative assay for the formation of alkylarsines--an interesting revival of Gosio's biological method!
Further work on the anaerobic volatilization of arsenic has focused on anaerobic ecosystems such as sewage sludge and the use of pure cultures of anaerobes. As noted earlier, sewage sludge had yielded arsenic volatilizing organisms in 1973. Soon thereafter, sludge from an anaerobic digester was treated with [74As]Na2HAsO4 and then incubated anaerobically (172, 173). Volatile, radioactive arsenic compounds were produced, as evidenced by the use of rubber traps, but when the sludge was preheated to 90°C for 15 min, volatilization did not occur. Unfortunately, experimental difficulties with the system precluded extensive observations. Some evidence for the assumption that methanogens were producing dimethylarsine was obtained by taking advantage of the fact that the main precursor of methane formation in sewage sludge was carbon atom 2 (the methyl carbon) of acetate. In experiments with [2-14C]acetate, radioactivity was incorporated into the traps only when arsenate was present. Other preliminary experiments indicated that rumen fluid and compost were particularly effective in volatilizing arsenic from [74As]Na2HAsO4.
A more-sophisticated analytical technique (on-line coupling of gas chromatography with ICP-MS) later detected volatile metal and metalloid species in landfill and sewage gases (85, 86, 121). For arsenic, the gases from both sources contained (CH3)2AsH, (CH3)3As, and, interestingly, (CH3)2C2H5As. Only sewage gas formed CH3AsH2, and only landfill gas formed (C2H5)3As. Particularly noteworthy is the formation of the ethylated species. Assuming that the ethyl group transfers are a result of microbial action and thus an instance of bioalkylation, it would be of considerable interest to identify the organisms responsible. Similar work has shown that gases released from anaerobic wastewater treatment plants contained arsine as well as the three trimethylarsines (178).
An interesting feature of the work with the anaerobic ecosystems just described was the formation of arsine itself (86, 178). Although the formation of arsine from arsenate and arsenite by soil bacteria (Pseudomonas sp. and Alcaligenes sp.) was known (51), the microbial formation of this nonmethylated metabolite is uncommon. However, as will be seen, arsine formation by pure cultures of anaerobes has been described.
Cell extracts of Desulfovibrio vulgaris strain 803 produced an unidentified, garlic-odored gas when incubated with sodium arsenate (175), and unpublished results (75) have indicated the production of both di- and trimethylarsine from arsenate by cell extracts of Methanobacterium thermoautotrophicum. When this organism was grown under stable chemostat conditions with H2 and CO2 as growth substrates, arsenate was transformed to volatile products, mainly arsine itself with small amounts of dimethylarsine (17). Lowered phosphate concentrations increased the efficiency of the volatilization by 25%.
Enrichment cultures of anaerobes isolated from an arsenic-contaminated lake in the Canadian sub-Arctic region with five selective media showed extensive formation of methylarsenicals (30). Anaerobic oligotrophs were most active, producing a high proportion of dimethylarsinate. Sulfate-reducing consortia with acetate produced mainly trimethylarsine oxide, with relative concentrations of methylarsonate and dimethylarsinate increasing with time. Sulfate-reducing organisms with lactate produced only trimethylarsine oxide by day 4, with a range of products being produced on extended incubation. Iron- and manganese-reducing organisms grew poorly, producing only low levels of methylated arsenicals. Some of the compounds were probably methylarsenic(III) thiols, and volatile arsines were also produced in some cases. In sulfate-reducing (acetate) cultures, AsH3 was formed with monomethylarsine as the major volatile compound. Sulfate-reducing (lactate) cultures gave AsH3 and trimethylarsine as the major products.
Several representatives of sewage sludge microflora (methanogenic archaea) and sulfate-reducing anaerobes have been examined in pure culture in the presence of various arsenic concentrations (178). The most efficient organism for arsenic volatilization was Methanobacterium formicicum; arsine and all three methylarsines, as well as an unidentified volatile arsenic compound, were produced over the As concentration range of 0.05 to 0.3 mM KH2AsO4. Methanosarcina barkeri produced only arsine over the same range, and Methanobacterium thermoautotrophicum formed arsine at 0.1, 0.3, and 0.5 mM concentrations, especially so at the higher arsenic levels. Three organisms—Clostridium collagenovorans, D. vulgaris, and Desulfovibrio gigas—produced small amounts of trimethylarsine. The facultative marine anaerobe Serratia marinorubra converted arsenate to arsenite and methylarsonate when grown aerobically; volatile arsines were not produced under either aerobic or anaerobic conditions (243).
Methyl Donor in Anaerobes
While SAM is clearly the methyl group donor for many arsenic methylation processes, the situation with respect to bacteria and anaerobic microorganisms is less clear-cut. Dimethylarsine formation by M. bryantii required H2, ATP, and a methyl donor in addition to the substrate [74As]Na2HAsO4 (175). Except for arsenate, the same materials are required for methane biosynthesis. The most efficient methyl donor was methylcobalamin, with carbon dioxide as a poor second. Neither serine nor 5-methyltetrahydrofolate was a methyl donor. The addition of vitamin B12 did not lead to dimethylarsine formation, and ethylcobalamin did not transfer the ethyl group. These results suggested that the reaction with methylcobalamin was enzyme catalyzed; there was, in fact, a linear relationship between the amount of methylcobalamin used and the yield of dimethylarsine. It is clear that the transfer of methyl groups from methylcobalamin to arsenic over and above that of nonenzymatic controls was demonstrated (175). Methylcobalamin has been increasingly implicated in a range of methylation reactions in addition to its well-known role in methionine biosynthesis (94, 143, 226).
Since arsenite inhibited methane biosynthesis, it seemed possible that methyl transfer to arsenite led to the formation of a nonvolatile compound. By the use of [14CH3]methylcobalamin, this compound was identified as methylarsonate (175). An abbreviated, Challenger-like mechanism was postulated for dimethylarsine synthesis, with dimethylarsinate undergoing a 4e− reduction as the final step: arsenate → arsenite → methylarsonate → dimethylarsinate → dimethylarsine. Unfortunately, in the schematic mechanism presented by these authors, there appears to be confusion between oxidation numbers and valency. The methyl groups were represented as derived from methylcobalamin with formation of vitamin B12r (notation representing the demethylated form of B12).
One difficulty is the existence of nonenzymatic methyl transfers from this methylcobalamin, e.g., to mercury (251). The methylation of arsenite by methylcobalamin occurred at a low rate in the presence or absence of rat liver extract (33). In 1973, the nonenzymatic transfer of the methyl group of methylcobalamin to arsenite was shown to require a reducing agent such as dithioerythritol (209, 210). Typically, incubation of methylcobalamin, As2O3, and dithioerythritol in water gave arsine, methylarsine, and methane. The reductant, Zn/NH4Cl, behaved similarly. Using [C2H3]cobalamin, the arsenical product was C2H3AsH2, demonstrating transfer of an intact methyl group. There was no alkyl transfer from ethylcobalamin. Selenite inhibited arsenite methylation and selenite underwent methylation to dimethylselenide in the absence of arsenite.
Arsenite methylation by methylcobalamin in the presence of GSH has also been observed (259). The reaction was studied in capped tubes, one reason for this being “to prevent any as yet unknown volatile intermediates from escaping” (259). After treatment of the reaction mixtures with H2O2 to oxidize arsenical intermediates to the As(V) state, methylarsonate and smaller amounts of dimethylarsinate were identified. In view of this experimental procedure, the initial products of methyl transfer were not identified. In addition to containing arsenite, methylcobalamin, and GSH, the complex incubation mixtures contained vitamin B12 and SAM. The unexplained presence of the latter slightly increased the yield of methylarsonate; in the absence of GSH, methylation was not observed. Sodium 2,3-dimercapto-1-propane-sulfonate and sodium selenite also functioned as reducing agents; in combination these two agents behaved synergistically.
Work with E. coli cultures isolated from rats fed dimethylarsinate for 6 months has shown that cysteine but not GSH was required for dimethylarsinate metabolism. Thus, although GSH contributes to arsenic metabolism in animals, there may be a different metabolic pathway in bacteria (147).
Some workers add vitamin B12 (34) to assays for methyl transfer in animal extracts, but there is little evidence for the requirement. Methylcobalamin, added to incubations of rat liver cytosol, produced no effect (228). However, for arsenic methylation by the microbial contents of mouse intestinal cecum, methylcobalamin addition increased production of methylarsonate from inorganic forms of arsenic but had little effect on the formation of dimethylarsinate. These increases may have resulted from increased production of SAM by the cecal microflora or from the use of methylcobalamin in undefined enzymatically catalyzed methylation reactions (116).
The possible chemistry for nonenzymatic and enzyme-catalyzed methyl transfers from methylcobalamin to arsenic has been discussed in detail (83, 196, 203, 209, 210, 250). There are strongly held opinions, particularly with respect to the methyl donor situation in anaerobic organisms. Wood et al. stated that, taking into account all of the available experimental data, “we believe that the biomethylation of arsenic by methyl-B12 occurs in anaerobic ecosystems” (203). However, Cullen and Reimer claimed that “until strong evidence is advanced, there seems little need to invoke a different mechanism for arsenic biomethylation by bacteria from that discussed above for fungi” (75). Moreover, in a discussion at a 1978 symposium dedicated to Challenger, Cullen expressed himself more forcefully: “We are brain-washed by the idea that methyl B12 is involved in all of these reactions. We firmly believe that there is no B12 involved in arsenic methylation. Professor Wood pointed out that the nature of the methylating species had to be established in nature. Much of our discussion now involves base-on, base-off details, yet the compound may not in fact be the methylating agent” (62). It should be noted that the literature contains confusing statements with respect to oxidation numbers and electron flow details (66, 261).
Clearly, there is no general consensus as to a mechanism for anaerobic dimethylarsine formation. There are two general explanations for the observed results with M. bryantii: either there is a direct transfer from methylcobalamin or, if not, then methylcobalamin transfers methyl to a further carrier molecule. Three mechanisms are available for the direct transfer of methyl from methylcobalamin, represented as CH3-Co(III). The methyl group may be transferred as an anion (equation 15), as a radical (equation 16), or as a cation (equation 17):
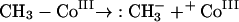 |
(15) |
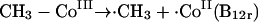 |
(16) |
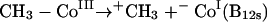 |
(17) |
In terms of the enzymatic situation, the cation transfer (equation 17) would appear to be advantageous; it is analogous to the situation with SAM. This mechanism requires formation of −Co(I), also represented as B12s. However, the compound formed after methyl transfer was B12r (equation 16); it was described as the normal cobamide product formed by demethylation of methylcobalamin (175). If correct, this identification suggests the intervention of a methyl radical, and in fact, consideration of the standard reduction potential (−0.67 V) for As(V) or As(III) is suggestive of a reductive homolytic cleavage (83, 249).
An alternative explanation (209, 210) invoked a role for a thiol, likely GSH, in formation of methane, acetate, and methylarsines by anaerobes. For the first two metabolites, the process is more complex than originally postulated, but the overall concept may be valid for methyl transfer to arsenic. The role of the thiol is that of a reductant; in fact, methylcobalamin can be reductively cleaved nonenzymatically in neutral or weakly acid solutions to form methane. The addition of the thiol to the cobalt atom promoted the heterolytic cleavage of the Co-C bond, leading to formation of a methyl carbanion (or similar species) which may be enzyme bound. Such a species was described as a “kryptocarbanion”; however, “cryptocarbanion” would be more appropriate.
For the enzyme reaction, two thiol units were involved as well as linkage of methylcobalamin as a corrinoid protein (Fig. 6). The methyl carbanion was represented as attacking arsenic in the ionized state and carrying three positive charges (equations 18 to 20). For arsenite the ionization would be represented as As(OH)3 ↔ As3+(OH−)3. Other species would presumably be possible, for instance, those with GSH replacing the OH groups: As(SG)3 ↔ As3+(SG−)3.
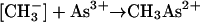 |
(18) |
 |
(19) |
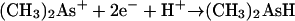 |
(20) |
The ionized species, CH3As2+, would correspond to methylarsonate or a similar structure [CH3As(OH)2 → CH3As2+(OH−)2], and (CH3)2As+ would correspond to dimethylarsinate [(CH3)2AsOH → (CH3)2As+(OH−)] in the usual Challenger mechanism. This proposed mechanism requires production of Co(I), i.e., B12s rather than the observed B12r. Perhaps an interconversion occurred during workup of the enzymatic incubation mixtures. For the nonenzymatic transfer, this mechanism was dismissed (259) as “less likely in the highly reductive conditions of our experiments,” and instead a “nucleophilic attack on the Co-C bond by an arsenite-GSH complex” was proposed. It is not clear why the reaction conditions for this work were characterized as “highly reducing.” The tubes, although capped, contained air, and the incubation components in addition to arsenite and methylcobalamin were the required GSH in Tris-HCl buffer with Mg2+, SAM and vitamin B12. “Highly reducing” seems to be an exaggeration.
FIG. 6.

Possible mechanism for formation of a “cryptocarbanion” from a methyl cobalamin. E indicates an enzyme containing two SH groups (209, 210).
If the transfer is not direct, a possible intermediate would be required; the case of methane biosynthesis may provide a model. It is known that the methyl transfer processes for methane biosynthesis are very complex, with a multitude of cofactors and enzymes being involved; in addition, certain proteins contain bound cobalamin (89, 131, 155, 245). One material that plays an important role in methane biosynthesis is coenzyme M (CoM), HS-CH2-CH2-SO3−; it accepts a methyl from a corrinoid protein forming methyl-CoM, CH3-S-CH2-CH2-SO3−. In fact, amino acid sequences are known for two CoM methyl transferases from M. barkeri and Methanobacterium thermoautotrophicum (89, 155, 163). Methyl-CoM is the last intermediate before methane, being reduced with the involvement of 7-mercaptoheptanoylthreonine phosphate (93).
It has been suggested that for metal alkylation, CoM would be a molecule of interest (173), and it has also been suggested that in methanogens arsenic methylation was coupled to the methane biosynthetic pathway (235), apparently implying a role for CoM. Moreover, it has been asserted that “Methanobacterium methylates arsenate in a side reaction of methane biosynthesis using CoM (2,2′-dithiodiethanesulphonic acid)” (126). The McBride and Wolfe paper (175) cited in reference 126 in support of this statement does not refer to CoM, and another paper by the same authors in the same year (174) describing CoM does not mention arsenic methylation. In 1971, the CoM structure was not known, and in any case, the disulfide form of CoM, CoM-S-S-CoM, noted by Hughes and Poole (126) is not likely to be an acceptor for methyl groups. Another assertion without experimental support is that for M. bryantii the pathway “involves methylation by a low molecular weight cofactor Coenzyme M (CoM)” (234).
Cullen has cited unpublished observations showing that methyl transfer does take place in model systems from methyl CoM to arsenic(III) (61). Apart from this, there is apparently no experimental evidence for a role for CoM in arsenic methylation by microorganisms, and as with so many other parts of the arsenic story, the picture is incomplete. A chemically attractive possibility for a methyl donor to arsenic would be a sulfonium derivative of CoM, (CH3)2-S+-CH2-CH2-SO3−, but methyl transfer to arsenite from this sulfonium compound could not be demonstrated (75). Moreover, S-adenosyl-CoM and methylsulfonium adenosyl-CoM, did not stimulate methane formation in Methanobacterium thermoautotrophicum extracts (63).
A possible intermediate for methyl transfer from methylcobalamin is, of course, SAM. The latter material is clearly synthesized by methanogens and has significant metabolic activity. For instance, methyl-CoM reductase from Methanobacterium thermoautotrophicum contains five modified amino acids, of which four involve methylation: 1-N-methylhistidine, 5-(S)-methylarginine, 2-(S)-methylglutamine, and S-methylcysteine (note that “S” indicates substitution at sulfur and “[S]” indicates a specific configuration). Methionine is taken up from culture medium by this organism and used for protein synthesis and in SAM-dependent methylation reactions (but not in methyl-CoM-dependent methane biosynthesis). The methyl groups of the four modified amino acids derive from methionine, almost certainly by way of SAM (211). Moreover, SAM is involved in methyl transfer to uroporphyrinogen III in Methanobacterium ivanovii (28) and in the biosynthesis of the methylated pterin in methanopterins in M. formicicum and Methanococcus volta (247).
However, apart from the role of methylcobalamin in methionine synthase, there is apparently no simple mechanism by which a methyl group from this corrinoid would become the methyl group of SAM; if methionine synthase were to be involved, methionine adenosyltransferase would also be required. The latter enzyme has, in fact, been purified to homogeneity from Methanococcus jannaschii and has been cloned and expressed in E. coli (109). In other words, for this hypothesis to be viable, the cell extracts of M. bryantii must also have contained both methionine synthase and methionine adenosyltransferase as well as the necessary substrates and cofactors for these enzymes and in addition must have contained methyl transferases for arsenic methylation and their associated redox cofactors. Perhaps this is not impossible, but it seems a tall order.
ENZYMOLOGY OF BIOMETHYLATION
The enzymology of arsenic biomethylation is complicated by the several oxidation states for this element, the previously discussed propensity of arsenic to react with sulfur compounds, and the fact that only very small amounts of the arsenic compounds are present in biological specimens. The chemical intermediates and reactions in the metabolism of arsenate are assumed to be generally the same in microorganisms and animals. However, in microorganisms, the reactions tend to proceed to methylarsines, whereas in mammalian species the major urinary metabolite is generally dimethylarsinate and only a very small amount of it is further reduced. There is considerable variation in the arsenic metabolic pathways among and between various animal species. With the exception of arsenate reductase (see below) most of the enzymatic experiments involve mammalian systems.
Enzymes Catalyzing Reductions
Arsenate reductase.
Although GSH can reduce arsenate to arsenite nonenzymatically, several thiol-requiring arsenate reductases, are involved in connection with bacterial resistance to arsenic. While these enzymes are not known to have a connection with biomethylation they may be usefully described here as possible models for enzymes reducing As(V) compounds in methylation. This is especially so since no microbial reductases involved in biomethylation have been described. Moreover, the arsenate reductases utilize reductants such as thioredoxin and glutaredoxin rather than GSH which is usually postulated to play a role in biomethylation. Two of the sources for arsenate reductases are E. coli and S. aureus. Strains of both of these bacteria are known to convert trimethylarsine oxide to trimethylarsine, and in E. coli, dimethylarsinate is converted to trimethylarsine oxide.
For some bacterial strains that are resistant to heavy metals (including here, arsenic), the resistance determinants are located on plasmids (205, 217-219, 233). In E. coli and S. aureus there are operons induced by arsenate, arsenite, and antimony(III); each of these ions induces all three resistances. The arsenic resistance plasmid, pI258, of S. aureus contains three genes: arsA (which encodes a repressor regulator protein), arsB (which encodes a membrane efflux protein), and arsC (137, 138, 177). The last of these genes, required only for arsenate resistance, codes for the reductase ArsC, or more specifically, ArsCsa to distinguish it from the subsequently discussed ArsCec from E. coli. This reductase is required since arsenite is the species transported from cells.
The ArsCsa enzyme is a monomer of about 14.5 kDa, with 131 amino acid residues (137). This reductase coupled to thioredoxin, thioredoxin reductase, and NADPH. Two enzyme-bound cysteine residues, Cys82 and Cys89, were identified as a redox couple (177). The disulfide form of ArsCsa was only regenerated by thioredoxin (not by GSH), confirming the thioredoxin specificity.
In E. coli the arsenic resistance plasmid is termed R773. The ArsCec reductase, a 16-kDa protein, contained 141 amino acid residues and had an essential thiol component—Cys12 (98, 162). In addition, a histidine residue, His8, was required for catalytic activity by lowering the pKa of the cysteine thiolate (97). Unlike ArsCsa it requires GSH (186) and glutaredoxin 2 (Grx2-SH); thioredoxin is not involved (162, 215). The suggested reaction sequence is as follows where ES− is Cys12 of ArsC:
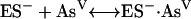 |
 |
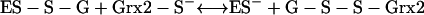 |
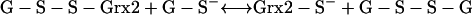 |
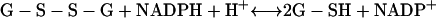 |
The secondary structure and overall protein fold for ArsCec were determined by use of multidimensional, multinuclear magnetic resonance (224). A model fold was structurally homologous to E. coli glutaredoxin, thiol transferases, and GSH S-transferases that are α/β proteins. The ArsCec protein contained large regions of extensive mobility, especially at the active site.
A gene cluster on chromosome XVI of Saccharomyces cerevisiae, confers arsenate resistance, and the arsenate reductase is labeled Acr2p (182). There is a requirement for GSH and glutaredoxin and a mixed disulfide between GSH and Acr2p is probably formed. There is no role for thioredoxin. A homolog of this reductase, Ygr203p, a 148 amino acid residue protein, was overexpressed in E. coli, and the protein was crystallized (180). X-ray diffraction data were obtained, but the crystal structure has not yet been solved.
A few anaerobes use arsenate, selenate, or in one case selenite as terminal electron acceptors, typically with reduction to arsenite or selenite. In view of a recent review (225) only a brief discussion is given here. The respiratory arsenate reductase from Chrysiogenes arsenatis gen. nov., sp. nov., in which acetate is the electron donor consists of two subunits, ArrA (87 kDa) and ArrB (29 kDa) (142). Molybdenum, iron, zinc, and acid-labile sulfur are required cofactors; this enzyme is very different from other arsenate reductases. More recently two further arsenate- and sulfate-reducing bacteria, Desulfomicrobium strains Ben-RA and Ben-RB, were isolated (166). In the Ben-RA strain, which cannot grow with arsenate as an electron acceptor, arsenate is reduced by an enzyme encoded by a chromosomally borne gene that is homologous with the arsC gene of E. coli plasmid R773. The nature of the electron donor for the reductase was not determined. For strain Ben-RB, arsenate was reduced by a membrane-bound enzyme, probably associated with a c-type cytochrome; the major cytochrome in this organism is c551. It thus differs from the arsenate reductase of C. arsenatis.
Human liver arsenate reductase uses (nonnatural) dithiothreitol as the electron donor with GSH, thioredoxin or reduced lipoic acid having much less activity (199). Partially purified enzyme, with an apparent molecular mass of about 72 kDa, required an unidentified, heat-stable cofactor (not a divalent cation).
Methylarsonate reductase.
The only other reduction studied at the enzyme level is that of methylarsonate. Rabbit liver enzyme, purified 118-fold, required GSH. Dithiothreitol alone did not substitute for GSH; however, in the presence of GSH, dithiothreitol almost doubled the formation of methylarsonite. This enzyme was rate-limiting in arsenic biotransformation. The same reductase was also identified in human liver (258).
Enzymes Catalyzing Methyl Group Transfers
The methyltransferases involved in biomethylation of arsenic have interesting chemical properties since they have a “hidden” redox component (Fig. 4). Such activities have been mainly investigated for the mammalian arsenate → dimethylarsinate transformation. Arsenite methyltransferase activity has been studied and partially purified in various animal tissues (14, 118, 258). Methylarsonite transferases have been purified from rabbit liver and Chang human hepatocytes (248, 260). Reviews describing them are available (14, 15, 227). There are apparently no accounts of the characterization of specific arsenic methyltransferases from microorganisms.
ARSENIC BIOMETHYLATION AS DETOXIFICATION?
Whether biomethylation of arsenic is a detoxification mechanism, particularly in animals, has been discussed at length (1, 14, 24, 125, 171, 227, 229). The most poisonous form of arsenic for humans (as measured by experiments that determine a lethal dose for some other test organism) is arsenous acid, As(OH)3, or its anion arsenite, materials commonly present in the environment. In general As(III) derivatives, e.g., arsenite, are more toxic than As(V) compounds, e.g., arsenate (229). Toxicity here, in general, refers to measurements of concentrations that comprise a lethal dose for 50% of the test subjects at some predetermined time limit after exposure. Analogously, selenite (more than selenate) and tellurite (more than tellurate) appear to be the most toxic oxyanionic forms of those metalloids to bacteria (21, 257). In fact, one detoxification mechanism is simply the oxidation of arsenite to arsenate (75). While methylation is often assigned a role in detoxification (242) this assumption may not be universally true. For instance, the As(III) compound, methylarsine oxide [(CH3AsO)n] is more toxic to C. humiculus than arsenite (71). The corresponding sulfide [(CH3AsS)n] is even more toxic than the oxide or arsenite. The order of toxicity for various compounds in Chang human hepatocyte cultures was as follows (from most to least toxic except as indicated: methylarsonous acid, arsenite, arsenate, methylarsonic acid = dimethylarsinic acid (193). There is evidence that organoarsenicals may produce toxic and carcinogenic effects that differ from those of inorganic arsenic (228).
Although arsine and the methylarsines are undoubtedly very toxic in the gas form, a surprising result was obtained when trimethylarsine dissolved in olive oil was administered to mice by mouth (256). Under these conditions the acute toxicity of trimethylarsine “was found to be incomparably low”; the 50% lethal dose was calculated to be 7,870 mg kg of body weight−1. The trimethylarsine was rapidly excreted in urine after oxidation to trimethylarsine oxide. The detoxification effect may in some cases be merely a function of increased volatility of more highly methylated forms. In other words, outgassing may be a significant means of removing metalloids from an organism's environment.
BIOMETHYLATION OF ANTIMONY
Introduction
Like arsenic, antimony is widely distributed on earth, usually at low concentrations, e.g., 4.3 to 7.9 ppm in soils (167). In addition to terrestrial sources, this element can be detected in oceans and freshwater environments in the parts per billion range. The metalloid and its inorganic compounds find a variety of industrial uses; the element is a frequent component of lead alloys. It is used in the ceramic and plastic industries and is widely present in many materials as a fire retardant. A few organic compounds of antimony are used in medicine. Tartar emetic, antimony potassium tartrate, C8H4K2O12Sb2 · 3H2O, is used as an anthelmintic in treatment of parasitic infections and as an animal expectorant. Some other organic antimony derivatives have also been used as anthelmintics, and at one time antimonial materials were used in the treatment of syphilis.
Early information on the determination of antimony species in nature has been summarized (167). Within the last few years, improved methods for the detection of small amounts of antimony and determination of antimony species have been published (58, 59, 78, 139, 144, 159-161).
Antimony and SIDS
Prior to the middle nineteen nineties there was little evidence for microbial methylation of antimony (55). However, this possibility became of interest when a possible cause for sudden infant death syndrome (SIDS) (also known as cot-death) was suggested (202). The hypothesis was that flame retardants in mattresses and covers might undergo methylation to toxic gases by the action of S. brevicaulis or other microorganisms. Since one flame retardant was antimony, trimethylstibine might have been produced. It was claimed that S. brevicaulis had actually been isolated from damp crib mattresses (202). The hypothesis was vociferously supported by some groups, and there were accusations of a cover-up of important facts by “authorities” (223). Later work indicated that the predominant organism isolated from crib mattresses was not S. brevicaulis but a mix of common environmental Bacillus sp. (246). A very detailed reexamination of old evidence and consideration of new information, carried out by an independent advisory committee for the British Department of Health, found no justification for the toxic-gas hypothesis (Lady Limerick, final report to British Government Department of Health, 1998 [www.doh.gov.uk/limerch.htm]).
The nonmicrobial leaching of antimony (as Sb2O3) from polyvinylchloride crib mattresses could account for the high variability associated with antimony levels in livers for both SIDS victims and other infants and for the elevated Sb levels in the hair of some healthy infants (132). The polyurethane inner-foam of crib mattresses might be a site for toxic gas formation of group 15 elements, but determination of the level of Sb, As, P, and Bi in crib mattress foam showed no correlation with the occurrence of SIDS. No volatile forms were detected in the headspace of mixed or monoseptic cultures of anaerobes containing polyurethane foams. There was no evidence for a causal relationship between levels of trimethylantimony or total trimethylantimony forms and SIDS (135).
Microbial Methylation of Antimony
Challenger has described a curious case of chronic antimony poisoning in Vienna, Austria, at the beginning of the 20th century. The problem was in a house decorated with silk curtains mordanted with an antimony compound. Although a volatile antimony compound was a prime suspect, none could be identified (43). Challenger and Ellis investigated growth of S. brevicaulis on media containing tartar emetic (44). The cultures were without odor, and aspiration of the air over them through Biginelli's solution gave no precipitate. Other attempts to volatilize antimony using P. notatum in addition to S. brevicaulis were made by one of Challenger's students (18). It appeared that stibonic acids, RSbO(OH)2, would be useful substrates, and early experiments utilized the more readily available phenylstibonic acid (R = C6H5). Sterile air was slowly aspirated over cultures of both organisms and then into concentrated nitric acid. The latter solution was then carefully analyzed for antimony. Only P. notatum gave a positive result, the amount of antimony being just detectable. Since the expected product, phenyldimethylstibine, might have been oxidized in the air stream, the experiments were repeated with an air-nitrogen mixture containing 8% oxygen. Under these conditions, neither organism produced a volatile antimony compound; however, P. notatum produced small amounts of a volatile antimony compound from growth in presence of potassium antimonate (KSbO3).
A possible mechanism for biomethylation of phenylstibonic acid to phenyldimethylstibine would involve alternate reductions and methylations as in the Challenger mechanism for arsenic biomethylation. The final step would be the reduction of phenyldimethylstibine oxide, C6H5(CH3)2SbO → C6H5(CH3)2Sb. To provide evidence for reductive capabilities, both organisms were grown with trimethylstibine dihydroxide, (CH3)3Sb(OH)2 [i.e., (CH3)3SbO·H2O]. Trimethylstibine, detected both chemically and by odor, was produced in both cases. Thus, it was clear that the reduction Sb(V) → Sb(III) was biogenically possible and that trimethylstibine was sufficiently stable to be detected when formed in fungal cultures. Another potential intermediate in formation of trimethylstibine was dimethylstibonic acid, (CH3)2SbO(OH). This compound was also converted to a volatile material; the best results were obtained with S. brevicaulis rather than with P. notatum. This careful work did establish a very low level of volatilization of antimony by fungi, but the amounts of volatile material were too small for identification. Fungi grown repeatedly in the presence of sodium dimethylstibonate or potassium antimonate showed no increase in antimony volatilization (18).
Despite other inconclusive results on the microbial volatilization of antimony (95, 189) the process is now well established. The reduction of trimethyldibromoantimony to trimethylstibine by P. fluorescens K27, an organism isolated from the selenium-contaminated Kesterson reservoir in central California, was observed but this organism did not biomethylate tartar emetic or potassium hexahydroxyantimonate. However, the headspace of certain soil samples (from another site) amended with either of these compounds and augmented with nitrate-containing growth media contained trimethylstibine. These experiments clearly established the anaerobic microbial biomethylation of antimony; however, the responsible microorganisms were not identified (115).
In similar experiments, mixed anaerobic soil enrichment cultures containing tartar emetic were also shown to produce trimethylantimony (134). Six soil types and three culture media were used; the formation of trimethylantimony was dependent on both the soil sample and the medium and was variable even for replicate analyses.
Other work with complex technology established a low level of antimony methylation under aerobic conditions with S. brevicaulis (7, 133). In typical experiments, the antimony volatilized was about one-millionth of that added. Small amounts of nonvolatile di- and trimethylantimony compounds were detected in some experiments with Sb(III) compounds but not Sb(V) compounds.
S. brevicaulis cultures incorporated labeled methyl groups (13C2H3) from [13C2H3]-l-methionine into nonvolatile tri- and dimethylantimony compounds. Hence, SAM is likely the microbial methylating agent for antimony (6, 8). Dimethylantimony species were shown to be true intermediates in the trimethylstibine biosynthetic pathway. Isotope incorporation into trimethylstibine from [2H3]-d-methionine was much less than that from the l enantiomer. This degree of stereospecificity indicated that the pathway was enzymatic. Since similar differences for l- versus d-methionine occur for arsenic biomethylation it is probable that the mechanism for the methylation of these metalloids is the same. Antimony biomethylation has not been studied at the enzymatic level.
When sewage sludge containing a substantial amount of Sb (1.7 ± 0.3 mg/kg [dry weight]) was incubated anaerobically, trimethylstibine was detected in the headspace gas as a dominant volatile metalloid. With pure cultures of various anaerobes, trimethylstibine was produced by C. collagenovorans and D. vulgaris (but not D. gigas). Methanobacterium thermoautotrophicum, and M. barkeri formed only very small amounts. One organism, M. formicicum, was a particularly active methylator, producing mono- and dimethylstibine as well as the trimethyl derivative and, in addition, stibine, SbH3, itself (178). Trimethylstibine was also present in landfill as well as sewage gases. Concentrations of trimethylstibine in thermophilic sewage digestion gases in the range of 6.7 to 14.7 μg m−3 were proposed to originate from biological sources after this metalloid was supplied from anthropogenic sources (86). Volatile antimony species were detected in gases over hot springs in British Columbia, Canada (120). Analytical methods for antimony determination at trace levels in environmental samples have been reviewed (183, 221).
Small amounts (1 to 12 ng liter−1) of nonvolatile methylstibonic acid, CH3SbO(OH)2, and dimethylstibinic acid, (CH3)2SbO(OH), were detected in rivers and seawaters (4, 5). The compounds were present at levels ranging from 1 to 12 ng liter−1. While these observations were probably the first to identify naturally occurring methylantimony compounds, there was no proof for a biological origin. Moreover, doubt has been cast upon these results, with further evaluation of the hydride generation process suggesting that some chemical species were, in fact, method artifacts (77). Methylated antimony species were detected in geothermal waters in New Zealand (120), but it was not clear whether these compounds were derived by biological (e.g., action of algae) or purely chemical means. Pondweed samples (Potamogetan pectinatus) from Canadian lakes influenced by mine effluents contained substantial levels of antimony (48 to 68 ppm [dry mass]). Antimony species in extracts made with acetic acid were obtained as stibines following reduction; these hydrides were SbH3 produced from the Sb(III) species and the mono-, di-, and trimethylstibines (78). Cultures of the marine diatom Thalassiosira nana converted 125SbCl3 to a stibnolipid. The structure was probably the same as that of 3′-O-(5″-dimethyl arsenoso-5″-deoxyribosyl)-phosphatidylglycerol (Fig. 1B), with Sb replacing As (22). There is little evidence for antimony methylation in mammals (96).
The wood rot fungus P. schweinitzii has produced trimethylarsine and trimethylantimony in solution. While trimethylarsine production from samples with added sodium arsenite was slight, separate samples amended with potassium antimony tartrate yielded conversion of up to 0.7% antimony to the methylated forms. Fungal cultures with added potassium hexahydroxyantimonate produced substantially less methylated antimony (9).
The fate of trimethylstibine in the atmosphere is important because knowledge of its lifetime contributes to an understanding of the involvement of the volatile forms in biogeochemical cycling. Craig et al. (57) have investigated the oxidation products of trimethylstibine in atmospheric oxygen and found the products to be trimethylstibine oxide as “a range of cyclic and linear oligomers” containing stibine oxide units. If these oxidized products can be metabolized by microorganisms, then the remobilization of antimony in biological systems can occur. The kinetics of this oxidation appear to be slower than those proposed by the only other antimony oxidation investigations in the literature (187, 188).
METHYLATION OF BISMUTH
Bismuth, the heaviest of the group 15 periodic table elements, occurs on earth in concentrations somewhat lower than those of antimony, i.e., at a crustal abundance of 0.17 to 0.2 ppm (167). Like antimony and arsenic, it is used in many ways. It is more metallic in character than these elements and forms low-melting-point alloys such as Wood's metal (Bi, 50%; Pb, 25%; Sn, 12.5%; and Cd, 12.5% [melting point, 154°F or 67.8°C]). Bismuth compounds have important applications in cosmetics, and bismuth is a semiconductor dopant.
Bismuth preparations have long been used by physicians, and most of the world's production of bismuth (about 50%) is used in the pharmaceutical industry (76, 201, 254). The 1899 edition of Merck's Manual lists 15 bismuth preparations, compared with only five for antimony (13). These preparations range from “Bismal,” bismuth methylene digallate (to be used as “an intestinal astringent especially in diarrheas not benefited by opiates”) to bismuth valerianate (basic bismuth valerate, to be used as a “sedative and antispasmodic for nervous headaches, cardialgia, chorea, etc.”). Bismuth compounds continue to be used in the treatment of intestinal problems as antacids (bismuth carbonate), as hemostatic agents (bismuth gallate), and in treatment of diarrhea (bismuth salicylate). The well-known, over-the-counter preparation Pepto-Bismol (Procter and Gamble) contains bismuth subsalicylate, HO-C6H4-COOBiO, and is said to provide “soothing relief for indigestion, upset stomach, diarrhea, nausea, and heartburn.” With the recognition of the role of Helicobacter pylori in the ulceration of the gut, much attention has been refocused on the use of bismuth preparations. Eradication of H. pylori infections is generally achieved with a combination therapy of three agents—a bismuth compound, an antibiotic (e.g., amoxicillin or metronidazole), and an anti (H+) secretory agent (255).
Soluble, inorganic bismuth, as bismuth nitrate, was more easily absorbed and then eliminated by rats fed diets augmented with thiols such as l-cysteine, homocysteine, or mercaptopropionic acid (38, 39). Nonthiols such as methionine or alanine did not produce this effect. It is feasible that enteric microbial reduction and methylation were enhanced by these thiol-containing additions and that the metabolic products enhanced bismuth elimination; however, bismuth analogs of arsenical sugars were not investigated.
For detection of small amounts of bismuth a merging-zones flow injection-AAS has been used; volatile alkylbismuth species were generated with sodium tetraethylborate (181). The detection limit for urinary bismuth was 1.6 μg liter−1.
Trimethylbismuth has been identified in landfill and sewage gas (86, 87), and experiments with sediments also showed production of trimethylbismuth under anaerobic conditions. The annual worldwide generation of trimethybismuth was estimated at 30 to 4,980 kg. There was little evidence for nonvolatile methylbismuth compounds in sediments and freshwaters (88). Anaerobic wastewater treatment gases contained trimethylbismuth, and pure cultures of M. formicicum produced significant amounts of trimethylbismuth from Bi(NO3)3. Very small amounts of trimethylbismuth were produced by C. collagenovorans (178).
EPILOGUE
The Italian physician Bartolomeo Gosio, who gave his name to a toxic gas, also discovered the fungal metabolite now known as mycophenolic acid and demonstrated its antibiotic action against anthrax bacillus (23). Most of his career was spent in the Italian Public Health Service, especially dealing with malaria, tuberculosis, and influenza. He died in Rome in 1944 at a time when that city was still occupied by the Nazis. Although brief obituaries were published in Italian, the English-speaking world was preoccupied with World War II, and his death was not noted. Gosio must have known that his eponymous gas had been identified as trimethylarsine, but he could never have imagined the volume of work on biomethylation that was yet to come.
Acknowledgments
Support by a Robert A. Welch departmental grant at Sam Houston State University is gratefully acknowledged by T.G.C.
Much help in obtaining reference materials was kindly provided by D. L. Johnston and A. Rogers, Langley Library, University of Pittsburgh.
REFERENCES
- 1.Abernathy, C. O., Y.-P. Liu, D. Longfellow, H. V. Aposhian, B. Beck, B. Fowler, R. Goyer, R. Menzer, T. Rossman, C. Thompson, and M. Waalkes. 1999. Arsenic: health effects, mechanisms of actions, and research issues. Environ. Health Perspect. 107:593-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerman, D. 1912. Über das Vorkommen von Trigonellin und Nikotinsäure in Harn nach Verfütterung von Nikotinsäure. Z. Biol. 59:17-22. [Google Scholar]
- 3.Agrifoglio, L. 1954. Bartolomeo Gosio. 1865-1944, p. 100-105. In Igienisti Italiani degli Ultimi Cento Anni. U. Hoepli, Milan, Italy.
- 4.Andreae, M. O., J.-F. Asmodé, P. Foster, and L. Van't dack. 1981. Determination of antimony(III), antimony(V), and methylantimony species in natural waters by atomic absorption spectrometry with hydride generation. Anal. Chem. 53:1766-1771. [Google Scholar]
- 5.Andreae, M. O., and P. N. Froelich. 1984. Arsenic, antimony, and germanium biogeochemistry in the Baltic Sea. Tellus 36:101-117. [Google Scholar]
- 6.Andrewes, P., W. R. Cullen, J. Feldmann, I. Koch, and E. Polishchuk. 1999. Methylantimony compound formation in the medium of Scopulariopsis brevicaulis cultures; 13CD3-L-methionine as a source of the methyl group. Appl. Organomet. Chem. 13:681-687. [Google Scholar]
- 7.Andrewes, P., W. R. Cullen, J. Feldmann, I. Koch, E. Polishchuk, and K. J. Reimer. 1998. The production of methylated organoantimony compounds by Scopulariopsis brevicaulis. Appl. Organomet. Chem. 12:827-842. [Google Scholar]
- 8.Andrewes, P., W. R. Cullen, and E. Polishchuk. 2000. Antimony biomethylation by Scopulariopsis brevicaulis: characterization of intermediates and the methyl donor. Chemosphere 41:1717-1725. [DOI] [PubMed] [Google Scholar]
- 9.Andrewes, P., W. R. Cullen, E. Polishchuk, and K. J. Reimer. 2001. Antimony biomethylation by the wood rotting fungus Phaeolus schweinitzii. Appl. Organomet. Chem. 15:473-480. [Google Scholar]
- 10.Anonymous. 1671. Review of Dissertationes duæ Medicæ de Venero Pestilenti: studio Caroli de la Font, M. D. et in Acad. Avenion. Prof. Primar. Amstelodami. Philos. Trans. R. Soc. VI:2210-2211. [Google Scholar]
- 11.Anonymous. 1860. Leading article. Lancet 24 Nov.:518-519. [Google Scholar]
- 12.Anonymous. 1871. Arsenic in wall papers. Br. Med. J. 22 July:101-102. [Google Scholar]
- 13.Anonymous. 1999. Merck's manual of the materia medica, Merck & Co., New York, N.Y. (Facsimile version of 1899 edition.)
- 13a.Antonio, T., A. K. Chopoa, W. R. Cullen, and D. Dolphin. 1979. A model for the biological methylation of arsenic. J. Inorg. Nuclear Chem. 41:1220-1221. [Google Scholar]
- 14.Aposhian, H. V. 1997. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu. Rev. Pharmacol. Toxicol. 37:397-419. [DOI] [PubMed] [Google Scholar]
- 15.Aposhian, H. V., R. Zakharyan, Y. Wu, S. Healy, and M. M. Aposhian. 1997. Enzymatic methylation of arsenic compounds. II. An overview, p. 297-321. In C. O. Abernathy, R. L. Calderon, and W. R. Chappell (ed.), Arsenic: exposure and health effects. Chapman & Hall, London, United Kingdom.
- 16.Bach, S. J. 1945. Biological methylation. Biol. Rev. Camb. Philos. Soc. 20:158-176. [DOI] [PubMed] [Google Scholar]
- 17.Bachofen, R., L. Birch, U. Buchs, P. Ferloni, I. Flynn, G. Jud, H. Tahedl, and T. G. Chasteen. 1995. Volatilization of arsenic compounds by microorganisms, p. 103-108. In R. E. Hinchee, J. L. Means, and D. R. Burris (ed.), Bioremediation of inorganics. Batelle Press, Columbus, Ohio.
- 18.Barnard, D. 1947. Studies on the metabolism of certain Aspergilli and Penicillia. Ph.D. thesis. University of Leeds, Leeds, United Kingdom.
- 19.Barrett, D. K. 1978. An improved selective medium for isolation of Phaeolus schweinitzii. Trans. Br. Mycol. Soc. 71:507-508. [Google Scholar]
- 20.Bartrip, P. W. J. 1994. How green was my valance? Environmental arsenic poisoning and the Victorian domestic ideal. Eng. Hist. Rev. 109:891-913. [Google Scholar]
- 21.Basnayake, R. S. T., J. H. Bius, O. M. Akpolat, and T. G. Chasteen. 2001. Production of dimethyl telluride and elemental tellurium by bacteria amended with tellurite or tellurate. Appl. Organomet. Chem. 15:499-510. [Google Scholar]
- 22.Benson, A. A., and R. A. Cooney. 1988. Antimony metabolites in marine algae, p. 135-137. In P. J. Craig and F. Glockling (ed.), Organometallic compounds in the environment. Principles and reactions. Longmans, Harlow, United Kingdom.
- 23.Bentley, R. 2001. Bartolomeo Gosio, 1863-1944. An appreciation. Adv. Appl. Microbiol. 48:229-250. [PubMed] [Google Scholar]
- 24.Bertolero, F., G. Pozzi, E. Sabbioni, and U. Saffiotti. 1987. Cellular uptake and metabolic reduction of pentavalent to trivalent arsenic as determinants of cytotoxicity and morphological transformation. Carcinogenesis 8:803-808. [DOI] [PubMed] [Google Scholar]
- 25.Biginelli, P. 1900. Composizione e costituzione chimica del gas arsenicale delle tappezzerie. Nota I. Atti Reale Accad. Lincei 9:210-214. [Google Scholar]
- 26.Biginelli, P. 1900. Composizione e costituzione chimica del gas arsenicale delle tappezzerie. Nota II. Atti Reale Accad. Lincei 9:242-249. [Google Scholar]
- 27.Bird, M. L., F. Challenger, P. T. Charlton, and J. O. Smith. 1948. Studies on biological methylation. 11. The action of moulds on inorganic and organic compounds of arsenic. Biochem. J. 43:73-83. [PMC free article] [PubMed] [Google Scholar]
- 28.Blanche, F., C. Robin, M. Couder, D. Faucher, L. Cauchois, B. Cameron, and J. Crouzet. 1991. Purification, characterization, and molecular cloning of S-adenosyl-l-methionine: uroporphyrinogen III methyltransferase from Methanobacterium iranorii. J. Bacteriol. 173:4637-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braman, R. S. 1975. Arsenic in the environment, p. 108-123. In E. A. Woolson (ed.), Arsenical pesticides. American Chemical Society Symposium series 7. American Chemical Society, Washington, D.C.
- 30.Bright, D. A., S. Brock, W. R. Cullen, G. M. Hewitt, J. Jafaar, and K. J. Reimer. 1994. Methylation of arsenic by anaerobic microbial consortia isolated from a lake sediment. Appl. Organomet. Chem. 8:415-422. [Google Scholar]
- 31.Brinckman, F. E., and J. M. Bellama (ed.). 1978. Organometals and organometalloids. Occurrence and fate in the environment. American Chemical Society Symposium series 82. American Chemical Society, Washington, D.C.
- 32.Bryant, M. P., E. A. Wolin, M. J. Wolin, and R. S. Wolfe. 1967. Methanobacillus omelianskii, a symbiotic association of two species of bacteria. Arch. Mikrobiol. 59:20-31. [DOI] [PubMed] [Google Scholar]
- 33.Buchet, J. P., and R. Lauwerys. 1985. Study of inorganic arsenic methylation by rat liver in vitro: relevance for the interpretation of observations in man. Arch. Toxicol. 57:125-129. [DOI] [PubMed] [Google Scholar]
- 34.Buchet, J. P., and R. Lauwerys. 1988. Role of thiols in the in vitro methylation of inorganic arsenic by rat liver cytosol. Biochem. Pharmacol. 37:3149-3153. [DOI] [PubMed] [Google Scholar]
- 35.Byrne, A. R., Z. Slejkovec, T. Stijve, L. Fay, W. Gössler, J. Gailer, and K. J. Irgolic. 1995. Arsenobetaine and other arsenic species in mushrooms. Appl. Organomet. Chem. 9:305-313. [Google Scholar]
- 36.Cantoni, G. L. 1965. S-adenosylmethionine revisited, p. 21-32. In S. K. Shapiro and F. Schlenk (ed.), Transmethylation and methionine biosynthesis. University of Chicago Press, Chicago, Ill.
- 37.Cantoni, G. L., and A. Razin (ed.). 1985. Biochemistry and biology of DNA methylation. A. R. Liss, New York, N.Y.
- 38.Chaleil, D., F. Lefevre, P. Allain, and G. J. Martin. 1981. Enhanced bismuth digestive absorption in rats by some sulfhydryl compounds: NMR study of complexes formed. J. Inorg. Biochem. 15:213-221. [DOI] [PubMed] [Google Scholar]
- 39.Chaleil, D., J. P. Regnault, P. Allain, R. Motta, G. Raynaud, and P. Bouvet. 1988. Action d'une flore microbienne méthanogène d'origine humaine sur l'absorption et la fixation du bismuth chez le rat. Ann. Pharm. Fr. 46:133-137. [PubMed] [Google Scholar]
- 40.Challenger, F. 1945. Biological methylation. Chem. Rev. 36:315-361. [Google Scholar]
- 41.Challenger, F. 1951. Biological methylation. Adv. Enzymol. 12:429-491. [DOI] [PubMed] [Google Scholar]
- 42.Challenger, F. 1955. Biological methylation. Q. Rev. Chem. Soc. (London) 9:255-286. [Google Scholar]
- 43.Challenger, F. 1978. Biosynthesis of organometallic and organometalloid compounds, p. 1-22. In F. E. Brinckman and J. M. Bellama (ed.), Organometals and organometalloids. Occurrence and fate in the environment. American Chemical Society Symposium series 82. American Chemical Society, Washington, D.C.
- 44.Challenger, F., and L. Ellis. 1935. The formation of organo-metalloid compounds by micro-organisms. Part III. Methylated alkyl and dialkyl arsines. J. Chem. Soc. 396-400.
- 45.Challenger, F., and C. Higginbottom. 1935. The production of trimethylarsine by Penicillium brevicaule (Scopulariopsis brevicaulis). Biochem. J. 29:1757-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Challenger, F., C. Higginbottom, and L. Ellis. 1933. The formation of organo-metalloid compounds by microorganisms. Part I. Trimethylarsine and dimethylethylarsine. J. Chem. Soc. 1933:95-101. [Google Scholar]
- 47.Challenger, F., D. B. Lisle, and P. B. Dransfield. 1954. Studies in biological methylation. Part XIV. The formation of trimethylarsine and dimethyl selenide in mould cultures from methyl sources containing 14C. J. Chem. Soc. 1954:1760-1771. [Google Scholar]
- 48.Challenger, F., and H. E. North. 1934. The production of organo-metalloid compounds by micro-organisms. Part II. Dimethyl selenide. J. Chem. Soc. 1934:68-71. [Google Scholar]
- 49.Chappell, W. R., C. O. Abernathy, and R. L. Calderon (ed.). 1999. Arsenic exposure and health effects. Elsevier Science Ltd., Oxford, United Kingdom.
- 50.Chatt, J. 1951. Metal and metalloid compounds of the alkyl radicals, p. 415-458. In E. H. Rodd (ed.), Chemistry of carbon compounds, vol. IA. Elsevier Publishing Co., Amsterdam, The Netherlands. [Google Scholar]
- 51.Cheng. C.-N., and D. D. Focht. 1979. Production of arsine and methylarsines in soil and in culture. Appl. Environ. Microbiol. 38:494-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox, D. P. 1975. Microbiological methylation of arsenic, p. 80-95. In E. A. Woolson (ed.). Arsenical pesticides. American Chemical Society Symposium series 7. American Chemical Society, Washington, D.C.
- 53.Cox, D. P., and M. Alexander. 1973. Production of trimethylarsine gas from various arsenic compounds by three sewage fungi. Bull. Environ. Contam. Toxicol. 9:84-88. [DOI] [PubMed] [Google Scholar]
- 54.Cox, D. P., and M. Alexander. 1973. Effect of phosphate and other anions on trimethylarsine formation by Candida humicola. Appl. Microbiol. 25:408-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Craig, P. J. 1986. Other organometallic compounds in the environment, p. 345-364. In P. J. Craig (ed.), Organometallic compounds in the environment. Principles and reactions. Longmans, Harlow, United Kingdom.
- 56.Craig, P. J. (ed.). 1986. Organometallic compounds in the environment. Principles and reactions. Longmans, Harlow, United Kingdom.
- 57.Craig, P. J., S. A. Forster, R. O. Jenkins, G. Lawson, D. Miller, and N. Ostah. 2001. Use of mass spectroscopic techniques to elucidate the nature of the products of the oxidation of trimethylstibine in air. Appl. Organomet. Chem. 15:527-532. [Google Scholar]
- 58.Craig, P. J., S. N. Forster, R. O. Jenkins, and D. Miller. 1999. An analytical method for the detection of methylantimony species in environmental matrices: methylantimony levels in some UK plant material. Analyst 124:1243-1248. [Google Scholar]
- 59.Craig, P. J., S. N. Forster, R. O. Jenkins, D. P. Miller, N. Ostah, L. M. Smith, and T.-A. Morris. 2000. Occurrence, formation and fate of organoantimony compounds in marine and terrestrial environments, p. 265-280. In A. Gianguzza, E. Pelizzetti, and S. Sammartano (ed.), Chemical processes in the environment. Springer-Verlag, Berlin, Germany.
- 60.Craig, P. J., and F. Glockling (ed.). 1988. The biological alkylation of heavy elements. Special publication no. 66. Royal Society of Chemistry, London, United Kingdom.
- 61.Cullen, W. R. 1978. Discussion of a paper by McBride et al., p. 113. In F. E. Brinckman and J. M. Bellama (ed.), Organometals and organometalloids. Occurrence and fate in the environment. American Chemical Society Symposium series 82. American Chemical Society, Washington, D.C.
- 62.Cullen, W. R. 1978. Discussion of a paper by J. S. Thayer, p. 201. In F. E. Brinckman and J. M. Bellama (ed.), Organometals and organometalloids. Occurrence and fate in the environment. American Chemical Society Symposium series 82. American Chemical Society, Washington, D.C.
- 63.Cullen, W. R., D. Dolphin, D. Hoffman, and E. Trip. 1984. S-adenosylcoenzyme M and (±) S-methyl adenosylcoenzyme M. Preparation and role in methane biosynthesis. Can. J. Chem. 62:2136-2139. [Google Scholar]
- 64.Cullen, W. R., G. K. Eigendorf, and S. A. Pergantis. 1993. Desorption chemical ionization mass spectrometry of arsenic compounds present in the marine and terrestrial environment. Rapid Commun. Mass Spec. 7:33-36. [Google Scholar]
- 65.Cullen, W. R., A. E. Erdman, B. C. McBride, and A. W. Pickett. 1983. The identification of dimethylphenylarsine as a microbial metabolite using a simple method of chemofocusing. J. Microbiol. Methods 1:297-303. [Google Scholar]
- 66.Cullen, W. R., C. L. Froese, A. Lui, B. C. McBride, D. J. Patmore, and M. Reimer. 1977. The aerobic methylation of arsenic by microorganisms in the presence of L-methionine-methyl-d3. J. Organomet. Chem. 139:61-69. [Google Scholar]
- 67.Cullen, W. R., H. Li, G. Hewitt, K. J. Reimer, and N. Zalunardo. 1994. Identification of extracellular arsenical metabolites in the growth medium of the microorganisms Apiotrichum humicola and Scopulariopsis brevicaulis. Appl. Organomet. Chem. 8:303-311. [Google Scholar]
- 68.Cullen, W. R., H. Li, S. A. Pergantis, G. K. Eigendorf, and L. G. Harrison. 1994. The methylation of arsenate by a marine alga Polyphysa peniculus in the presence of L-methionine-methyl-D3. Chemosphere 28:1009-1019. [Google Scholar]
- 69.Cullen, W. R., H. Li, S. A. Pergantis, G. K. Eigendorf, and M. A. Mosi. 1995. Arsenic biomethylation by the microorganism Apiotrichum humicola in the presence of L-methionine-methyl-d3. Appl. Organomet. Chem. 9:507-515. [Google Scholar]
- 70.Cullen, W. R., B. C. McBride, and A. W. Pickett. 1979. The transformation of arsenicals by Candida humicola. Can. J. Microbiol. 25:1201-1205. [DOI] [PubMed] [Google Scholar]
- 71.Cullen, W. R., B. C. McBride, H. Manji, A. W. Pickett, and J. Reglinski. 1989. The metabolism of methylarsine oxide and sulfide. Appl. Organomet. Chem. 3:71-78. [Google Scholar]
- 72.Cullen, W. R., B. C. McBride, A. W. Pickett, and J. Reglinski. 1984. The wood preservative chromated copper arsenate is a substrate for trimethylarsine biosynthesis. Appl. Environ. Microbiol. 47:443-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cullen, W. R., B. C. McBride, and J. Reglinski. 1984. The reduction of trimethylarsine oxide to trimethylarsine by thiols: a mechanistic model for the biological reduction of arsenicals. J. Inorg. Biochem. 21:45-60. [Google Scholar]
- 74.Cullen, W. R., B. C. McBride, and J. Reglinski. 1984. The reaction of methylarsenicals with thiols: some biological implications. J. Inorg. Biochem. 21:179-194. [Google Scholar]
- 75.Cullen, W. R., and K. J. Reimer. 1989. Arsenic speciation in the environment. Chem. Rev. 89:713-764. [Google Scholar]
- 76.Dill, K., and E. L. McGown. 1994. The biochemistry of arsenic, bismuth and antimony compounds, p. 695-713. In S. Patai (ed.), The chemistry of organic arsenic, antimony and bismuth compounds. John Wiley & Sons, Ltd., Chichester, United Kingdom.
- 77.Dodd, M., S. L. Grundy, K. J. Reimer, and W. R. Cullen. 1992. Methylated antimony(V) compounds: synthesis, hydride generation properties and implications for aquatic speciation. Appl. Organomet. Chem. 6:207-211. [Google Scholar]
- 78.Dodd, M., S. A. Pergantis, W. R. Cullen, H. Li, G. K. Eigendorf, and K. J. Reimer. 1996. Antimony speciation in freshwater plant extracts by using hydride generation-gas chromatography-mass spectrometry. Analyst 121:223-228. [Google Scholar]
- 79.Dopson, M., and E. B. Lindström. 1999. Potential role of Thiobacillus caldus in arsenopyrite bioleaching. Appl. Environ. Microbiol. 65:36-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.du Vigneaud, V., and J. R. Rachele. 1965. The concept of transmethylation in mammalian metabolism and its establishment by isotopic labeling through “in vivo” experimentation, p. 1-20. In S. K. Shapiro and F. Schlenk (ed.), Transmethylation and methionine biosynthesis. University of Chicago Press, Chicago, Ill.
- 81.Dyke, W. J. C., and W. J. Jones. 1930. The interaction between alkyl Grignard reagents and arsenic trichloride. J. Chem. Soc. 2426-2430.
- 82.Emmerling, O. 1897. Bemerkung zu vorstehender Entgegnung des Herrn Gosio. Ber. Dtsch. Chem. Ges. 30:1026. [Google Scholar]
- 83.Fanchiang, Y.-T., W. P. Ridley, and J. M. Wood. 1978. Kinetic and mechanistic studies on B12-dependent methyl transfer to certain toxic metal ions, p. 54-64. In F. E. Brinckman and J. M. Bellama (ed.), Organometals and organometalloids. Occurrence and fate in the environment. American Chemical Society Symposium series 82. American Chemical Society, Washington, D.C.
- 84.Fatoki, O. S. 1997. Biomethylation in the natural environment. S. Afr. J. Sci. 93:366-370. [Google Scholar]
- 85.Feldmann, J., R. Grümping, and A. V. Hirner. 1994. Determination of volatile metal and metalloid compounds in gases from domestic waste deposits with GC/ICP-MS. Fresenius J. Anal. Chem. 350:228-234. [Google Scholar]
- 86.Feldmann, J., and A. V. Hirner. 1995. Occurrence of volatile metal and metalloid species in landfill and sewage gases. Int. J. Environ. Anal. Chem. 60:339-359. [Google Scholar]
- 87.Feldman, J., and J. Kleimann. 1997. Flüchtige metallverbindungen im Faulgas. Korrespond. Abwasser 44:99-104. [Google Scholar]
- 88.Feldmann, J., E. M. Krupp, D. Glindemann, A. V. Hirner, and W. R. Cullen. 1999. Methylated bismuth in the environment. Appl. Organomet. Chem. 13:739-748. [Google Scholar]
- 89.Ferguson, D. J., J. A. Krzycki, and D. A. Grahame. 1996. Specific roles of methylcobamide: coenzyme M methyltransferase isozymes in metabolism of methanol and methylamines in Methanosarcina barkeri. J. Biol. Chem. 271:5189-5194. [DOI] [PubMed] [Google Scholar]
- 90.Florkin, M. 1979. Biosynthesis of methionine, cysteine and cystine, p. 83-111. In M. Florkin and E. H. Stotz (ed.), Comprehensive biochemistry, vol. 33B. A history of biochemistry, part V. The unravelling of biosynthetic pathways (continued). Elsevier Scientific Publishing Co., Amsterdam, The Netherlands. [Google Scholar]
- 91.Francesconi, K. A., and J. S. Edmonds. 1994. Biotransformation of arsenic in the marine environment, p. 221-226. In J. O. Nriagu (ed.), Arsenic in the environment. Part I, Cycling and characterization. Wiley and Sons, Inc., New York, N.Y.
- 92.Frankenberger, W. T. 1998. Effects of trace elements on arsenic volatilization. Soil Biol. Biochem. 30:269-274. [Google Scholar]
- 92a.Frankenberger, W. T. 2001. Environmental chemistry of arsenic. Marcel Dekker, Inc., New York, N.Y.
- 93.Friedmann, H. C., A. Klein, and R. K. Thauer. 1990. Structure and function of the nickel porphinoid coenzyme F430, and of its enzyme, methyl coenzyme M reductase. FEMS Microbiol. Rev. 87:339-348. [DOI] [PubMed] [Google Scholar]
- 94.Gadd, G. M. 1993. Microbial formation and transformation of organometallic and organometalloid compounds. FEMS Microbiol. Rev. 11:297-316. [Google Scholar]
- 95.Gates, P. N., H. A. Harrop, J. B. Pridham, and B. Smethhurst. 1997. Can microorganisms convert antimony trioxide or potassium antimonyl tartrate to methylated stibines? Sci. Tot. Environ. 205:215-221. [DOI] [PubMed] [Google Scholar]
- 96.Gebel, T. 1997. Arsenic and antimony: comparative approach on mechanistic toxicology. Chem.-Biol. Interact. 107:131-144. [DOI] [PubMed] [Google Scholar]
- 97.Gladysheva, T., J. Liu, and B. P. Rosen. 1996. His-8 lowers the p Ka of the essential cys-12 residue of the ArsC arsenate reductase of plasmid R773. J. Biol. Chem. 271:33256-33260. [DOI] [PubMed] [Google Scholar]
- 98.Gladysheva, T. B., K. L. Oden, and B. P. Rosen. 1994. Properties of the arsenate reductase of plasmid R773. Biochemistry 33:7288-7293. [DOI] [PubMed] [Google Scholar]
- 99.Gmelin, C. G. 1824. Versuche über die Wirkungen des Baryts, Strontians, Chroms, Molybdäns, Wolframs, Tellurs, Titans, Osmiums, Platins, Iridiums, Rhodiums, Palladiums, Nickels, Kobalts, Urans, Ceriums, Eisens und Mangans auf den thierischen Organismus. H. Laupp'sche Buchhandlung, Tübingen, Germany.
- 100.Gmelin, L. 1839. Warnung vor gewissen grünen Tapeten und Anstrichen. Karlsruher Z. November 24, suppl. 326.
- 101.Gosio, B. 1892. Azione di alcune muffe sui composti fissi d'arsenico. Riv. Ig. Sanità Pubblica 3:201-230. [Google Scholar]
- 102.Gosio, B. 1892. Súl riconoscimento dell'arsenico per mezzo di alcune muffe. Riv. Ig. Sanità Pubblica 3:261-273. [Google Scholar]
- 103.Gosio, B. 1892. Action of microphytes on solid compounds of arsenic: a recapitulation. Science 19:104-106. [DOI] [PubMed] [Google Scholar]
- 104.Gosio, B. 1893. Action de quelques moisissures sur les composés fixes d'arsenic. Arch. Ital. Biol. 18:253-265. [Google Scholar]
- 105.Gosio, B. 1893. Sur le reconnaissance de l'arsenic au moyen de certaines moisissures. Contribution à la toxicologie de l'arsenic. Arch. Ital. Biol. 18:298-305. [Google Scholar]
- 106.Gosio, B. 1897. Zur Frage, wodurch die Giftigkeit arsenhaltiger Tapeten bedingt wird. Ber. Dtsch. Chem. Ges. 30:1024-1027. [Google Scholar]
- 107.Gosio, B. 1901. Recherches ultérieures sur la biologie et sur le chimisme des arsenio-moisissures. Arch. Ital. Biol. 35:201-211. [Google Scholar]
- 108.Gosio, B. 1932. Sulla ricerca dell'arsenico per via biologica. Ist. Sieroterap. Milan Bol. 11:597-602. [Google Scholar]
- 109.Graham, D. E., C. L. Bock, C. Schalk-Hihi, Z. J. Lu, and G. D. Markham. 2000. Identification of a highly diverged class of S-adenosylmethionine synthetases in the Archaea. J. Biol. Chem. 275:4055-4059. [DOI] [PubMed] [Google Scholar]
- 110.Granchinho, S., E. Polishchuk, W. R. Cullen, and K. J. Reimer. 2001. Biomethylation and bioaccumulation of arsenic(V) by marine alga Fucus gardneri. Appl. Organomet. Chem. 15:553-560. [Google Scholar]
- 111.Greenstein, J. P., and M. Winitz. 1961. Chemistry of the amino acids, vol. 3, p. 2125-2155. John Wiley & Sons, New York, N.Y.
- 112.Griffiths, D. E., and J. Usta. 1988. Effects of trialkyllead compounds in mitochondrial membrane potential, p. 205-210. In P. J. Craig and F. Glockling (ed.), The biological alkylation of heavy elements. Special publication 66. Royal Society of Chemistry, London, United Kingdom.
- 113.Guggenheim, M. 1940. Die biogenen Amine (und ihre Bedeutung für die Physiologie und Pathologie des pflanzlichen und tierischen Stoffwechsels). S. Karger, Basel, Switzerland.
- 114.Gürleyük, H., J. F. Tyson, and P. C. Uden. 2000. Determination of extractable arsenic in soils using slurry sampling-on-line microwave extraction-hydride generation-atomic absorption spectrometry. Spectrochim. Acta B 55:935-945. [Google Scholar]
- 115.Gürleyük, H., V. Van Fleet-Stadler, and T. G. Chasteen. 1997. Confirmation of the biomethylation of antimony compounds. Appl. Organomet. Chem. 11:471-483. [Google Scholar]
- 116.Hall, L. L., S. E. George, M. J. Kohan, M. Styblo, and D. J. Thomas. 1997. In vitro methylation of inorganic arsenic in mouse intestinal cecum. Toxicol. Appl. Pharmacol. 147:101-109. [DOI] [PubMed] [Google Scholar]
- 117.Hanselmann, K. W. 1991. Microbial energetics applied to waste repositories. Experientia 47:645-687. [Google Scholar]
- 118.Healy, S. M., E. A. Casarez, F. Ayala-Fierro, and H. V. Aposhian. 1998. Enzymatic methylation of arsenic compounds. V. Arsenite methyltransferase activity in tissues of mice. Toxicol. Appl. Pharmacol. 148:65-70. [DOI] [PubMed] [Google Scholar]
- 119.Healy, S. M., E. Wildfang, R. A. Zakharyan, and H. V. Aposhian. 1999. Diversity of inorganic arsenite biotransformation. Biol. Trace Elem. Res. 68:249-266. [DOI] [PubMed] [Google Scholar]
- 120.Hirner, A. V., J. Feldmann, E. Krupp, R. Grümping, R. Goguel, and W. R. Cullen. 1998. Metal(loid)organic compounds in geothermal gases and waters. Org. Geochem. 29:1765-1778. [Google Scholar]
- 121.Hirner, A. V., E. Krupp, F. Schulz, M. Koziol, and W. Hofmeister. 1998. Organometal(loid) species in geochemical exploration: preliminary qualitative results. J. Geochem. Explor. 64:133-139. [Google Scholar]
- 122.His, W. 1887. Ueber das Stoffwechselproduct des Pyridins. Arch. Exp. Pathol. Pharmakol. 22:253-260. [Google Scholar]
- 123.Hofmeister, F. 1894. Ueber methylirung im Thierkörper. Arch. Exp. Pathol. Pharmakol. 33:198-215. [Google Scholar]
- 124.Honshopp, S., N. Brunken, A. Nehrkorn, and H. J. Breunig. 1996. Isolation and characterization of a new arsenic methylating bacterium from soil. Microbiol. Res. 151:37-41. [DOI] [PubMed] [Google Scholar]
- 125.Hopenhayn-Rich, C., M. L. Biggs, D. A. Kalman, L. E. Moore, and A. H. Smith. 1996. Arsenic methylation patterns before and after changing from high to low concentrations of arsenic in drinking water. Environ. Health Perspect. 104:1200-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hughes, M. N., and R. K. Poole. 1989. Metals and micro-organisms, p. 277. Chapman & Hall, London, United Kingdom.
- 127.Hutton, C., G. Shaw, and R. Pearson. 1809. An account of some books. Book 73, p. 2210. Philos. Trans. R. Soc. I:611-614. [Google Scholar]
- 128.Huysmans, K. D., and W. T. Frankenberger. 1991. Evolution of trimethylarsine by a Penicillium sp. isolated from agricultural evaporation pond water. Sci. Tot. Environ. 105:13-28. [DOI] [PubMed] [Google Scholar]
- 129.Irgolic, K. J., and B. K. Puri. 1990. Organic arsenic compounds in petroleum and natural gas. In J. A. C. Broekaert, S. Gücer, and F. Adams (ed.), Metal speciation in the environment, NATO ASI Series, vol. G 23. Springer-Verlag, Berlin, Germany.
- 130.Jackson, W. A. 1969. To die or not to dye. Poisoning from arsenical pigments in the 19th century. Pharm. Hist. 26:27-31. [PubMed] [Google Scholar]
- 131.Jaun, B. 1993. Methane formation by methanogenic bacteria: redox chemistry of coenzyme F430, p. 287-337. In H. Sigel and A. Sigel (ed.), Metal ions in biological systems, vol. 29. Marcel Dekker, Inc., New York, N.Y. [Google Scholar]
- 132.Jenkins, R. O., P. J. Craig, W. Goessler, and K. J. Irgolic. 1998. Antimony leaching from cot mattresses and sudden infant death syndrome (SIDS). Hum. Exp. Toxicol. 17:138-139. [DOI] [PubMed] [Google Scholar]
- 133.Jenkins, R. O., P. J. Craig, W. Goessler, D. Miller, N. Ostah, and K. J. Irgolic. 1998. Biomethylation of inorganic antimony compounds by an aerobic fungus: Scopulariopsis brevicaulis. Environ. Sci. Technol. 32:882-885. [Google Scholar]
- 134.Jenkins, R. O., P. J. Craig, D. P. Miller, L. C. A. M. Stoop, N. Ostah, and T.-A. Morris. 1998. Antimony biomethylation by mixed cultures of microorganisms under anaerobic conditions. Appl. Organomet. Chem. 12:449-455. [Google Scholar]
- 135.Jenkins, R. O., T.-A. Morris, P. J. Craig, W. Goessler, N. Ostah, and K. M. Wills. 2000. Evaluation of cot mattress inner foam as a potential site for microbial generation of toxic gases. Hum. Exp. Toxicol. 19:693-702. [DOI] [PubMed] [Google Scholar]
- 136.Jensen, S., and Jernelöv, A. 1969. Biological methylation of mercury in aquatic organisms. Nature 223:753-754. [DOI] [PubMed] [Google Scholar]
- 137.Ji, G., E. A. E. Garber, L. G. Armes, C.-M. Chen, J. A. Fuchs, and S. Silver. 1994. Arsenate reductase of Staphylococcus aureus plasmid pI258. Biochemistry 33:7294-7299. [DOI] [PubMed] [Google Scholar]
- 138.Ji, G., and S. Silver. 1992. Reduction of arsenate to arsenite by the ArsC protein of the arsenic resistance operon of Staphylococcus aureus plasmid pI258. Proc. Natl. Acad. Sci. USA 89:9474-9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Koch, I., C. F. Harrington, K. J. Reimer, and W. R. Cullen. 1997. Simplex optimisation of conditions for the determination of antimony in environmental samples by using electrothermal atomic absorption spectrometry. Talanta 44:1241-1251. [DOI] [PubMed] [Google Scholar]
- 140.Koch, I., L. Wang, K. J. Reimer, and W. R. Cullen. 2000. Arsenic species in terrestrial fungi and lichens from Yellowknife, NWT, Canada. Appl. Organomet. Chem. 14:245-252. [Google Scholar]
- 141.Krachler, M., and H. Emons. 2001. Speciation of antimony for the 21st century: promises and pitfalls. Trends Anal. Chem. 20:79-90. [Google Scholar]
- 142.Krafft, T., and J. M. Macy. 1998. Purification and characterization of the respiratory arsenate reductase from Chrysiogenes arsenatis. Eur. J. Biochem. 255:647-653. [DOI] [PubMed] [Google Scholar]
- 143.Kräutler, B. 1990. Chemistry of methylcorrinoids related to their roles in bacterial C1 metabolism. FEMS Microbiol. Rev. 87:349-354. [DOI] [PubMed] [Google Scholar]
- 144.Krupp, E. M., R. Grümping, U. R. R. Furchtbar, and A. V. Hirner. 1996. Speciation of metals and metalloids in sediments with LTGC/ICP-MS. Fresenius J. Anal. Chem. 354:546-549. [DOI] [PubMed] [Google Scholar]
- 145.Kuehnelt, D., W. Goessler, and K. J. Irgolic. 1997. Arsenic compounds in terrestrial organisms I: Collybia maculata, Collybia butyracea and Amanita muscaria from arsenic smelter sites in Austria. Appl. Organomet. Chem. 11:289-296. [Google Scholar]
- 146.Kuehnelt, D., W. Goessler, and K. J. Irgolic. 1997. Arsenic compounds in terrestrial organisms II: arsenocholine in the mushroom Amanita muscaria. Appl. Organomet. Chem. 11:459-470. [Google Scholar]
- 147.Kuroda, K., K. Yoshida, A. Yasukawa, H. Wanibuchi, S. Fukushima, and G. Endo. 2001. Enteric bacteria may play a role in mammalian arsenic metabolism. Appl. Organomet. Chem. 15:548-552. [Google Scholar]
- 148.Lai, V. W.-M., W. R. Cullen, C. F. Harrington, and K. J. Reimer. 1997. The characterization of arsenosugars in commercially available algal products including a Nostoc species of terrestrial origin. Appl. Organomet. Chem. 11:797-803. [Google Scholar]
- 149.Lai, V. W.-M., W. R. Cullen, and S. Ray. 2001. Arsenic speciation in sea scallop gonads. Appl. Organomet. Chem. 15:533-538. [Google Scholar]
- 150.Larsen, E. H., M. Hansen, and W. Gössler. 1998. Speciation and health risk consideration of arsenic in the edible mushroom Laccaria amethystina collected from contaminated and uncontaminated locations. Appl. Organomet. Chem. 12:285-291. [Google Scholar]
- 151.Le, X.-C., W. R. Cullen, and K. J. Reimer. 1993. Determination of urinary arsenic and impact of dietary arsenic intake. Talanta 40:185-193. [DOI] [PubMed] [Google Scholar]
- 152.Le, S. X. C., W. R. Cullen, and K. J. Reimer. 1994. Speciation of arsenic compounds in some marine organisms. Environ. Sci. Technol. 28:1598-1604. [DOI] [PubMed] [Google Scholar]
- 153.Le, X. C., X. Lu, M. Ma, W. R. Cullen, H. V. Aposhian, and B. Zheng. 2000. Speciation of key arsenic metabolic intermediates in human urine. Anal. Chem. 72:5172-5177. [DOI] [PubMed] [Google Scholar]
- 154.Le, X. C., M. Ma, X. Lu, W. R. Cullen, H. V. Aposhian, and B. Zheng. 2000. Determination of monomethylarsenous acid, a key arsenic methylation intermediate, in human urine. Environ. Health Perspect. 108:1015-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.LeClerc, G. M., and D. A. Grahame. 1996. Methylcobamide: coenzyme M methyltransferase isozymes from Methanosarcina barkeri. J. Biol. Chem. 271:18725-18731. [DOI] [PubMed] [Google Scholar]
- 156.Lederer, W. H., and R. J. Fensterheim (ed.). 1983. As--industrial, biomedical, and environmental perspectives. Van Nostrand Reinhold Co., Inc., New York, N.Y.
- 157.Lerrigo, A. F. 1932. The biological method for the detection of arsenic. Analyst 57:155-158. [Google Scholar]
- 158.Reference deleted.
- 159.Lindemann, T., A. Prange, W. Dannecker, and B. Neidhart. 1999. Simultaneous determination of arsenic, selenium and antimony species using HPLC/ICP-MS. Fresenius J. Anal. Chem. 364:462-466. [DOI] [PubMed] [Google Scholar]
- 160.Lintschinger, J., I. Koch, S. Serves, J. Feldmann, and W. R. Cullen. 1997. Determination of antimony species with high-performance liquid chromatography using element specific detection. Fresenius J. Anal. Chem. 359:484-491. [Google Scholar]
- 161.Lintschinger, J., O. Schramel, and A. Kettrup. 1998. The analysis of antimony species by using ESI-MS and HPLC-ICP-MS. Fresenius J. Anal. Chem. 361:96-102. [Google Scholar]
- 162.Liu, J., T. B. Gladysheva, L. Lee, and B. P. Rosen. 1995. Identification of an essential cysteinyl residue in the ArsC arsenate reductase of plasmid R773. Biochemistry 34:13472-13476. [DOI] [PubMed] [Google Scholar]
- 163.Ludwig, M. L., and R. G. Matthews. 1997. Structure-based perspectives on B12-dependent enzymes. Annu. Rev. Biochem. 66:269-313. [DOI] [PubMed] [Google Scholar]
- 164.Lunde, G. 1977. Occurrence and transformation of arsenic in the marine environment. Environ. Health Perspect. 19:47-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Maassen, A. 1902. Die biologische Methode Gosio's zum Nachweis des Arsens und die Bildung organischer Arsen-, Selen- und Tellurverbindungen durch Schimmelpilze und Bakterien. Arbeiten Kaiserlichen Gesundheitsamte 18:475-489. [Google Scholar]
- 166.Macy, J. M., J. M. Santini, B. V. Pauling, A. H. O'Neill, and L. I. Sly. 2000. Two new arsenate/sulfate-reducing bacteria: mechanisms of arsenate reduction. Arch. Microbiol. 173:49-57. [DOI] [PubMed] [Google Scholar]
- 167.Maeda, S. 1994. Safety and environmental effects, p. 725-759. In S. Patai (ed.), The chemistry of organic arsenic, antimony and bismuth compounds. Wiley & Sons, Ltd., Chichester, United Kingdom.
- 168.Maeda, S. 1994. Biotransformation of arsenic in the freshwater environment, p. 155-187. In J. O. Nriagu (ed.), Arsenic in the environment, part I. Cycling and characterization. John Wiley & Sons, Inc. New York, N.Y.
- 169.Maeda, S., A, Ohki, K. Miyahara, K. Naka, and S. Higashi. 1992. Metabolism of methylated arsenic compounds by arsenic-resistant bacteria (Klebsiella oxytoca and Xanthomonas sp.). Appl. Organomet. Chem. 6:415-420. [Google Scholar]
- 170.Maeda, S., A, Ohki, K. Miyahara, T. Takeshita, and S. Higashi. 1990. Growth characteristics and arsenic metabolism of two species of arsenic-tolerant bacteria. Appl. Organomet. Chem. 4:245-250. [Google Scholar]
- 171.Marafante, E., M. Vahter, and J. Envall. 1985. The role of the methylation in the detoxification of arsenate in the rabbit. Chem.-Biol. Interact. 56:225-238. [DOI] [PubMed] [Google Scholar]
- 172.McBride, B. C., and T. L. Edwards. 1977. Role of the methanogenic bacteria in the alkylation of arsenic and mercury, p. 1-19. In Biological implications of metals in the environment. ERDA Symposium Series no. 42. U.S. Energy Research and Development Administration, Springfield, Va.
- 173.McBride, B. C., H. Merilees, W. R. Cullen, and W. Pickett. 1978. Anaerobic and aerobic alkylation of arsenic, p. 94-115. In F. E. Brinckman and J. M. Bellama (ed.), Organometals and organometalloids. Occurrence and fate in the environment. American Chemical Society Symposium series 82. American Chemical Society, Washington, D.C.
- 174.McBride, B. C., and R. S. Wolfe. 1971. A new coenzyme of methyl transfer, coenzyme M. Biochemistry 10:2317-2324. [DOI] [PubMed] [Google Scholar]
- 175.McBride, B. C., and R. S. Wolfe. 1971. Biosynthesis of dimethylarsine by a methanobacterium. Biochemistry 10:4312-4317. [DOI] [PubMed] [Google Scholar]
- 176.Merrill, W., and D. W. French. 1964. The production of arsenous gases by wood-rotting fungi. Proc. Minn. Acad. Sci. 31:105-106. [Google Scholar]
- 177.Messens, J., G. Hayburn, A. Desmyter, G. Laus, and L. Wyns. 1999. The essential catalytic redox couple in arsenate reductase from Staphylococcus aureus. Biochemistry 38:16857-16865. [DOI] [PubMed] [Google Scholar]
- 178.Michalke, K., E. B. Wickenheiser, M. Mehring, A. V. Hirner, and, R. Hensel. 2000. Production of volatile derivatives of metal(loid)s by microflora involved in anaerobic digestion of sewage sludge. Appl. Environ. Microbiol. 66:2791-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mohri, T., A. Hisanaga, and N. Ishinishi. 1990. Arsenic intake and excretion by Japanese adults: a 7-day duplication diet study. Food Chem. Toxicol. 28:521-529. [DOI] [PubMed] [Google Scholar]
- 180.Moon, J., Y. S. Kim, J. Y. Lee, S.-J. Cho, H. K. Song, J. H. Cho, B. M. Kim, K. K. Kim, and S. W. Suh. 2000. Crystallization and preliminary X-ray diffraction of Saccharomyces cerevisiae Ygr203p, a homologue of Acr2 arsenate reductase. Acta Crystallog. D Biol. Crystallog. 56:778-780. [DOI] [PubMed] [Google Scholar]
- 181.Mota, J. P. V., M. R. Fernández de la Campa, and A. Sanz-Medel. 1998. Merging zones flow injection for the determination of ultratraces of bismuth by volatile species generation atomic absorption spectrometry using sodium tetraethylborate (III). J. Anal. Atom. Spectrom. 13:431-435. [Google Scholar]
- 182.Mukhopadhyay, R., J. Shi, and B. P. Rosen. 2000. Purification and characterization of Acr2p, the Saccharomyces cerevisiae arsenate reductase. J. Biol. Chem. 275:21149-21157. [DOI] [PubMed] [Google Scholar]
- 183.Nash, M. J., J. E. Maskall, and S. J. Hill. 2000. Methodologies for determination of antimony in terrestrial environmental samples. J. Environ. Monit. 2:97-109. [DOI] [PubMed] [Google Scholar]
- 184.Nriagu, J. O. (ed.). 1994. Arsenic in the environment. Part I. Cycling and characterization. Advances in environmental science and technology, vol. 26. John Wiley & Sons, Inc., New York, N.Y.
- 185.Nriagu, J. O. (ed.). 1994. Arsenic in the environment. Part II. Human health and ecosystem effects. Advances in environmental science and technology, vol. 27. John Wiley & Sons, Inc., New York, N.Y.
- 186.Oden, K. L., T. B. Gladysheva, and B. P. Rosen. 1994. Arsenate reduction mediated by the plasmid-encoded ArsC protein is coupled to glutathione. Mol. Microbiol. 12:301-306. [DOI] [PubMed] [Google Scholar]
- 187.Parris, G. E., and F. E. Brinckman. 1975. Reactions which relate to environmental mobility of arsenic and antimony. I. Quaternization of trimethylarsine and trimethylstibine. J. Org. Chem. 40:3801-3803. [DOI] [PubMed] [Google Scholar]
- 188.Parris, G. E., and F. E. Brinckman. 1976. Reactions which relate to environmental mobility of arsenic and antimony. II. Oxidation of trimethylarsine and trimethylstibine. Environ. Sci. Technol. 10:1128-1134. [DOI] [PubMed] [Google Scholar]
- 189.Pearce, R. B., M. E. Callow, and L. E. Macaskie. 1998. Fungal volatilization of arsenic and antimony and the sudden infant death syndrome. FEMS Microbiol. Lett. 158:261-265. [DOI] [PubMed] [Google Scholar]
- 190.Pergantis, S. A., W. R. Cullen, D. T. Chow, and G. K. Eigendorf. 1997. Liquid chromatography and mass spectrometry for the speciation of arsenic animal feed additives. J. Chromatogr. A 764:211-222. [DOI] [PubMed] [Google Scholar]
- 191.Pergantis, S. A., W. R. Cullen, and G. K. Eigendorf. 1994. Evaluation of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for the analysis of organoarsenic compounds of environmental interest. Biol. Mass Spec. 23:749-755. [Google Scholar]
- 192.Pergantis, S. A., W. Winnick, E. M. Heithmar, and W. R. Cullen. 1997. Investigations of arsine-generating reactions using deuterium-labeled reagents and mass spectrometry. Talanta 44:1941-1947. [DOI] [PubMed] [Google Scholar]
- 193.Petrick, J. S., F. Ayala-Fierro, W. R. Cullen, D. E. Carter, and H. V. Aposhian. 2000. Monomethylarsonous acid (MMAIII) is more toxic than arsenite in Chang human hepatocytes. Toxicol. Appl. Pharmacol. 163:203-207. [DOI] [PubMed] [Google Scholar]
- 194.Pickett, A. W., B. C. McBride, and W. R. Cullen. 1988. Metabolism of trimethylarsine oxide. Appl. Organomet. Chem. 2:479-482. [Google Scholar]
- 195.Pickett, A. W., B. C. McBride, W. R. Cullen, and H. Manji. 1981. The reduction of trimethylarsine oxide by Candida humicola. Can. J. Microbiol. 27:773-778. [DOI] [PubMed] [Google Scholar]
- 196.Pratt, J. M. 1993. Making and breaking the Co-alkyl bond in B12 derivatives, p. 229-286. In H. Sigel and A. Sigel (ed.), Metal ions in biological systems, vol. 29. Marcel Dekker, Inc., New York, N.Y. [Google Scholar]
- 197.Przygoda. G., J. Feldman, and W. R. Cullen. 2001. The arsenic eaters of Styria: a different picture of people who were chronically exposed to arsenic. Appl. Organomet. Chem. 15:457-462. [Google Scholar]
- 198.Puntoni, V. 1917. Arsenioschizomiceti. Ann. Ig. 27:293-303. [Google Scholar]
- 199.Radabaugh, T. R., and H. V. Aposhian. 2000. Enzymatic reduction of arsenic compounds in mammalian systems: reduction of arsenate to arsenite by human liver arsenate reductase. Chem. Res. Toxicol. 13:26-30. [DOI] [PubMed] [Google Scholar]
- 200.Raper, K. B., and C. Thom. 1949. Scopulariopsis, p. 694-704. A manual of the penicillia. Williams & Wilkins, Baltimore, Md.
- 201.Reglinski, J. 1998. Environmental and medicinal chemistry of arsenic, antimony, and bismuth, p.403-440. In N. C. Norman, (ed.), Chemistry of arsenic, antimony and bismuth. Blackie Academic and Professional, London, United Kingdom
- 202.Richardson, B. A. 1994. Sudden infant death syndrome: a possible primary cause. J. Forensic Sci. Soc. 34:199-204. (Erratum, 34:284). [DOI] [PubMed] [Google Scholar]
- 203.Ridley, W. P., L. Dizikes, A. Cheh, and J. M. Wood. 1977. Recent studies on biomethylation and demethylation of toxic elements. Environ. Health Perspect. 19:43-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ridley, W. P., L. Dizikes, and J. M. Wood. 1977. Biomethylation of toxic elements in the environment. Science 197:329-332. [DOI] [PubMed] [Google Scholar]
- 205.Rosen, B. P. 1999. Families of arsenic transporters. Trends Microbiol. 7:207-212. [DOI] [PubMed] [Google Scholar]
- 206.Salvatore, F., E. Borek, V. Zappia, H. G. Williams-Ashman, and F. Schlenk (ed.). 1977. The biochemistry of adenosylmethionine. Columbia University Press, New York, N.Y.
- 207.Sanger, C. R. 1893. On the formation of volatile compounds of arsenic from arsenical wall papers. Proc. Am. Acad. Arts Sci. 29:112-147. [Google Scholar]
- 208.Sanger, C. R. 1893. On chronic arsenical poisoning from wall papers and fabrics. Proc. Am. Acad. Arts Sci. 29:148-177. [Google Scholar]
- 209.Schrauzer, G. N. 1974. Mechanisms of corrin dependent enzymatic reactions, p. 583-628. In W. Herz, H. Grisebach, and G. W. Kirby (ed.), Progress in the chemistry of natural products, vol. 31. Springer-Verlag, Vienna, Austria. [DOI] [PubMed] [Google Scholar]
- 210.Schrauzer, G. N., J. A. Seck, R. J. Holland, T. M. Beckham, E. M. Rubin, and J. W. Sibert. 1972. Reductive dealkylation of alkylcobaloximes, alkylcobalamins, and related compounds: simulation of corrin dependent reductase and methyl group transfer reactions. Bioinorg. Chem. 2:93-124. [Google Scholar]
- 211.Selmer, T., J. Kahnt, M. Goubeaud, S. Shima, W. Grabarse, U. Ermler, and R. K. Thauer. 2000. The biosynthesis of methylated amino acids in the active site region of methyl-coenzyme M reductase. J. Biol. Chem. 275:3755-3760. [DOI] [PubMed] [Google Scholar]
- 212.Shariatpanahi, M., A. C. Anderson, and A. A. Abdelghani. 1982. Uptake and distribution of sodium arsenate by bacterial cells. Trace Subst. Environ. Health 16:170-173. [Google Scholar]
- 213.Shariatpanahi, M., A. C. Anderson, A. A. Abdelghani, and A. J. Englande. 1983. Microbial metabolism of an organic arsenical herbicide, p. 268-277. In T. A. Oxley and S. Barry (ed.), Biodeterioration, vol. 5. John Wiley & Sons, Chichester, United Kingdom. [Google Scholar]
- 214.Shariatpanahi, M., A. C. Anderson, A. A. Abdelghani, A. J. Englande, J. Hughes, and R. F. Wilkinson. 1981. Biotransformation of the pesticide, sodium arsenate. J. Environ. Sci. Health 16:35-47. [DOI] [PubMed] [Google Scholar]
- 215.Shi, J., A. Vlamis-Gardikas, F. Åslund, A. Holmgren, and B. P. Rosen. 1999. Reactivity of glutaredoxins 1, 2, and 3 from Escherichia coli shows that glutaredoxin 2 is the primary hydrogen donor to ArsC-catalyzed arsenate reduction. J. Biol. Chem. 274:36039-36042. [DOI] [PubMed] [Google Scholar]
- 216.Sigel, H., and A. Sigel (ed.). 1993. Metal ions in biological systems. Marcel Dekker, Inc., New York, N.Y.
- 217.Silver, S., and H. Nakahara. 1983. Bacterial resistance to arsenic compounds, p. 190-199. In W. H. Lederer and R. J. Fensterheim (ed.), As--industrial, biomedical, and environmental perspectives. Van Nostrand Reinhold, New York, N.Y.
- 218.Silver, S., and T. K. Misra. 1984. Bacterrial transformation of and resistances to heavy metals, p. 23-46. In G. S. Omenn and A. Hollaender (ed.), Genetic control of environmental pollutants. Plenum Press, New York, N.Y.
- 219.Silver, S., and T. K. Misra. 1988. What DNA sequence analysis tells us about toxic heavy metal resistance, p. 211-242. In P. J. Craig and F. Glockling (ed.), The biological alkylation of heavy elements. Special publication no. 66. Royal Society of Chemistry, London, United Kingdom.
- 219a.Silver, S., and Phung, L. T. 1996. Bacterial heavy metal resistance: new surprises. Annu. Rev. Microbiol. 50:753-789. [DOI] [PubMed] [Google Scholar]
- 220.Slejkovec, Z., A. R. Byrne, T. Stijve, W. Goessler, and K. J. Irgolic. 1997. Arsenic compounds in higher fungi. Appl. Organomet. Chem. 11:673-682. [Google Scholar]
- 221.Smichowski, P., Y. Madrid, and C. Cámara. 1998. Analytical methods for antimony speciation in waters at trace and ultratrace levels. Fresenius J. Anal. Chem. 360:623-629. [Google Scholar]
- 222.Smith, H. R., and E. J. Cameron. 1933. Mold growth test for minute amounts of arsenic. Ind. Eng. Chem. Anal. Ed. 5:400-401. [Google Scholar]
- 223.Sprott, J. 1996. The cot death cover-up. Penguin Books, New York, N.Y.
- 224.Stevens, S. Y., W. Hu, T. Gladysheva, B. P. Rosen, E. R. P. Zuiderweg, and L. Lee. 1999. Secondary structure and fold homology of the ArsC protein from the Escherichia coli arsenic resistance plasmid R773. Biochemistry 38:10178-10186. [DOI] [PubMed] [Google Scholar]
- 225.Stolz, J. F., and R. S. Oremland. 1999. Bacterial respiration of arsenic and selenium. FEMS Microbiol. Rev. 23:615-627. [DOI] [PubMed] [Google Scholar]
- 226.Stupperich, E. 1993. Recent advances in elucidation of biological corrinoid functions. FEMS Microbiol. Rev. 12:349-366. [DOI] [PubMed] [Google Scholar]
- 227.Styblo, M., M. Delnomdedieu, and D. J. Thomas. 1995. Biological mechanisms and toxicological consequences of the methylation of arsenic, p. 407-433. In R. A. Goyer and M. G. Cherian (ed.), Toxicology of metals: biochemical aspects. Springer-Verlag, New York, N.Y.
- 228.Styblo, M., M. Delnomdedieu, and D. J. Thomas. 1996. Mono- and dimethylation of arsenic in rat liver cytosol in vitro. Chem. Biol. Interact. 99:147-164. [DOI] [PubMed] [Google Scholar]
- 229.Styblo, M., L. M. Del Razo, L. Vega, D. R. Germolec, E. L. LeCluyse, G. A. Hamilton, W. Reed, C. Wang, W. R. Cullen, and D. J. Thomas. 2000. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 74:289-299. [DOI] [PubMed] [Google Scholar]
- 230.Styblo, M., S. V. Serves, W. R. Cullen, and D. J. Thomas. 1997. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem. Res. Toxicol. 10:27-33. [DOI] [PubMed] [Google Scholar]
- 231.Styblo, M., and D. J. Thomas. 1995. In vitro inhibition of glutathione reductase by arsenotriglutathione. Biochem. Pharmacol. 49:971-977. [DOI] [PubMed] [Google Scholar]
- 232.Styblo, M., and D. J. Thomas. 1997. Factors influencing in vitro methylation of arsenicals in rat liver cytosol, p. 283-295. In C. O. Abernathy, R. L Calderon, and W. R. Chappell (ed.), Arsenic. Exposure and health effects. Chapman & Hall, London, United Kingdom.
- 233.Summers, A. O., and S. Silver. 1978. Microbial transformation of metals. Annu. Rev. Microbiol. 32:637-672. [DOI] [PubMed] [Google Scholar]
- 234.Tamaki, S., and W. T. Frankenberger. 1989. Environmental biochemistry of arsenic, p. 19. In Document prepared for San Joaquin Valley drainage program under U.S. Bureau of Reclamation contract no. 8-PG-20-11780. San Joaquin Valley Drainage Program, Sacramento, Calif.
- 235.Tamaki, S., and W. T. Frankenberger. 1992. Environmental biochemistry of arsenic. Rev. Environ. Contam. Toxicol. 124:79-110. [DOI] [PubMed] [Google Scholar]
- 236.Thayer, J. S. 1993. Global bioalkylation of the heavy metals, p. 1-36. In H. Sigel and A. Sigel (ed.), Metal ions in biological systems, vol. 29. Marcel Dekker, Inc., New York, N.Y. [Google Scholar]
- 237.Thom, C., and K. B. Raper. 1932. The arsenic fungi of Gosio. Science 76:548-550. [DOI] [PubMed] [Google Scholar]
- 238.Thomas, J. E., and R. D. Rhue. 1997. Volatilization of arsenic in contaminated cattle dipping vat soil. Bull. Environ. Contam. Toxicol. 59:882-887. [DOI] [PubMed] [Google Scholar]
- 239.Thompson, D. J. 1993. A chemical hypothesis for arsenic methylation in mammals. Chem. Biol. Interact. 88:89-114. [DOI] [PubMed] [Google Scholar]
- 240.Tuovinen, O. H., T. M. Bhatti, J. M. Bigham, K. B. Hallberg, O. Garcia, and E. B. Lindström. 1994. Oxidative dissolution of arsenopyrite by mesophilic and moderately thermophilic acidophiles. Appl. Environ. Microbiol. 60:3268-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Turpeinen, R., M. Pantsar-Kallio, M. Häggblom, and T. Kairesalo. 1999. Influence of microbes on the mobilization, toxicity and biomethylation of arsenic in soil. Sci. Total Environ. 236:173-180. [DOI] [PubMed] [Google Scholar]
- 242.Vahter, M., and E. Marafante. 1988. In vivo methylation and detoxication of arsenic, p. 105-119. In P. J. Craig and F. Glockling (ed.). The biological alkylation of heavy elements. Special publication no. 66. Royal Society of Chemistry, London, United Kingdom.
- 243.Vidal, F. V., and V. M. V. Vidal. 1980. Arsenic metabolism in marine bacteria. Mar. Biol. 60:1-7. [Google Scholar]
- 244.Vilanó, M., and R. Rubio. 2001. Determination of arsenic species in oyster tissue by microwave-assisted extraction and liquid chromatography-atomic fluorescence detection. Appl. Organomet. Chem. 15:658-666. [Google Scholar]
- 245.Vogels, G. D., J. T. Keltjens, and C. Van der Drift. 1988. Biochemistry of methane production, p. 707-769. In A. J. B. Zehnder (ed.), Biology of anaerobic microorganisms. Wiley-Interscience, New York, N.Y.
- 246.Warnock, D. W., H. T. Delves, C. K. Campbell, I. W. Croudace, K. G. Davey, E. M. Johnson, and C. Sieniawska. 1995. Toxic gas generation from plastic mattresses and sudden infant death syndrome. Lancet 346:1516-1520. [DOI] [PubMed] [Google Scholar]
- 247.White, R. H. 1990. Biosynthesis of methanopterin. Biochemistry 29:5397-5404. [DOI] [PubMed] [Google Scholar]
- 248.Wildfang, E., R. A. Zakharyan, and H. V. Aposhian. 1998. Enzymatic methylation of arsenic compounds. VI. Characterization of hamster liver arsenite and methylarsonic acid methyltransferase activities in vitro. Toxicol. Appl. Pharmacol. 152:366-375. [DOI] [PubMed] [Google Scholar]
- 249.Wood, J. M. 1988. Mechanism for B12-dependent methyl transfer to heavy elements, p. 62-76. In P. J. Craig and F. Glockling (ed.). The biological alkylation of heavy elements. Special publication no. 66. Royal Society of Chemistry, London, United Kingdom.
- 250.Wood, J. M., A. Cheh, L. J. Dizikes, W. P. Ridley, S. Rakow, and J. R. Lakowicz. 1978. Mechanisms for the biomethylation of metals and metalloids. Fed. Proc. 37:16-21. [PubMed] [Google Scholar]
- 251.Wood, J. M., F. S. Kennedy, and C. G. Rosen. 1968. Synthesis of methyl-mercury compounds by extracts of a methanogenic bacterium. Nature 220:173-174. [DOI] [PubMed] [Google Scholar]
- 252.Woolson, E. A. 1977. Generation of alkylarsines from soil. Weed Sci. 25:412-416. [Google Scholar]
- 253.Woolson, E. A., and P. C. Kearney. 1973. Persistence and reactions of 14C-cacodylic acid in soil. Environ. Sci. Technol. 7:47-50. [Google Scholar]
- 254.Wormser, W., and I. Nir. 1994. Pharmacology and toxicology of organic bismuth, arsenic and antimony compounds, p. 715-723. In S. Patai (ed.), The chemistry of organic arsenic, antimony and bismuth compounds. John Wiley & Sons Ltd., Chichester, United Kingdom.
- 255.Xia, H. H., B. C. Yu Wong, N. J. Talley, and S. K. Lam. 2001. Helicobacter pylori infection: current treatment and practice. Expert Opin. Pharmacother. 2:253-266. [DOI] [PubMed] [Google Scholar]
- 256.Yamauchi, H., T. Kaise, K. Takahashi, and Y. Yamamura. 1990. Toxicity and metabolism of trimethylarsine in mice and hamsters. Fund. Appl. Toxicol. 14:399-407. [DOI] [PubMed] [Google Scholar]
- 257.Yu, R., J. P. Coffman, V. Van Fleet-Stalder, and T. G. Chasteen. 1997. Toxicity of oxyanions of selenium and of a proposed bioremediation intermediate dimethyl selenone. Environ. Toxicol. Chem. 16:140-145. [Google Scholar]
- 258.Zakharyan, R. A., and H. V. Aposhian. 1999. Enzymatic reduction of arsenic compounds in mammalian systems: the rate-limiting enzyme of rabbit liver arsenic biotransformation is MMAV reductase. Chem. Res. Toxicol. 12:1278-1283. [DOI] [PubMed] [Google Scholar]
- 259.Zakharyan, R. A., and H. V. Aposhian. 1999. Arsenite methylation by methylvitamin B12 and glutathione does not require an enzyme. Toxicol. Appl. Pharmacol. 154:287-291. [DOI] [PubMed] [Google Scholar]
- 260.Zakharyan, R. A., F. Ayala-Fierro, W. R. Cullen, D. M. Carter, and H. V. Aposhian. 1999. Enzymatic methylation of arsenic compounds. VII. Monomethyl-arsonous acid (MMAIII) is the substrate for MMA methyltransferase of rabbit liver and human hepatocytes. Toxicol. Appl. Pharmacol. 158:9-15. [DOI] [PubMed] [Google Scholar]
- 261.Zingaro, R. A., and K. J. Irgolic. 1975. The methylation of arsenic compounds. Science 187:765. [DOI] [PubMed] [Google Scholar]
- 262.Zussman, R. A., E. E. Vicher, and I. Lyon. 1961. Arsine production by Trichophyton rubrum. J. Bacteriol. 81:157. [DOI] [PMC free article] [PubMed] [Google Scholar]