

Abstract
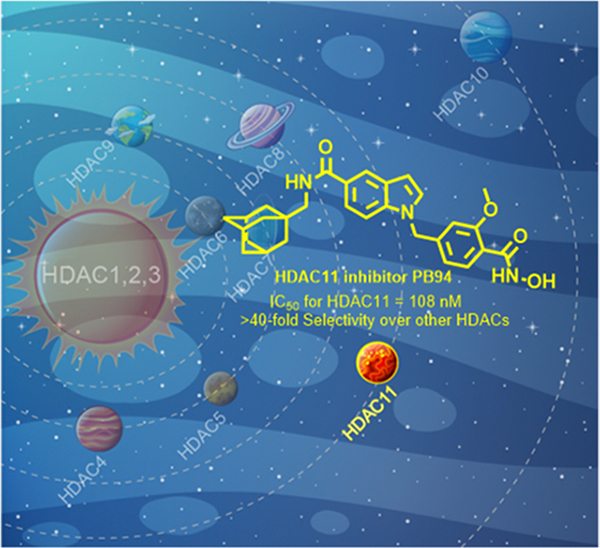
Recent studies have shown that the epigenetic protein histone deacetylase 11 (HDAC11) is highly expressed in the brain and critically modulates neuroimmune functions, making it a potential therapeutic target for neurological disorders. Herein, we report the development of PB94, which is a novel HDAC11 inhibitor. PB94 exhibited potency and selectivity against HDAC11 with IC50 = 108 nM and >40-fold selectivity over other HDAC isoforms. Pharmacokinetic/pharmacodynamic evaluation indicated that PB94 possesses promising drug-like properties. Additionally, PB94 was radiolabeled with carbon-11 as [11C]PB94 for positron emission tomography (PET), which revealed significant brain uptake and metabolic properties suitable for drug development in live animals. Furthermore, we demonstrated that neuropathic pain was associated with brain upregulation of HDAC11 and that pharmacological inhibition of HDAC11 by PB94 ameliorated neuropathic pain in a mouse model. Collectively, our findings support further development of PB94 as a selective HDAC11 inhibitor for neurological indications, including pain.
Graphical Abstract
INTRODUCTION
Histone lysine residue acetylation/deacetylation represents an important epigenetic modification that affects gene expression without DNA sequence modification.1 Histone deacetylases (HDACs) are a class of amide hydrolases that catalyze a variety of substrates, including histones and other proteins.2 Eleven Zn2+-dependent HDACs have been found in mammals, including class I (HDAC1, HDAC2, HDAC3, HDAC8), class IIa (HDAC4, HDAC5, HDAC7, HDAC9), class IIb (HDAC6, HDAC10), and class IV (HDAC11). Each of these isoforms has distinct functions in epigenetic regulation.3–5 Among them, HDAC11 is the most recently identified member of the class IV HDAC,6 with accumulating evidence suggesting its connection with many diseases.7–10 HDAC11 has higher expression levels than other HDACs in the brain, and it has been implicated in neurologic diseases such as multiple sclerosis,11 schizophrenia,12 and Huntington’s disease.13
Neuropathic pain is the secondary damage to the central or peripheral nervous system that affects millions of people.14–16 It has been demonstrated that HDACs play a crucial role in neuropathic pain.17 Several studies have shown that treatment with class-I or class-II HDAC inhibitors in animal models could attenuate neuropathic pain symptoms by targeting overexpressed HDACs and enhancing histone acetylation in the superficial layers of the spinal cord.18–21 As HDAC11 demonstrates significant homology to class I HDACs with 30% of the shared amino acid sequence,6 it indicates that selectively inhibiting HDAC11 could alleviate neuropathic pain.
Only a few HDAC11-selective inhibitors have been reported in recent years, yet none exhibited brain penetrability, nor have they been investigated in animal models of neurological disorders.7 For example, FT895 is the first reported hydroxamic acid-based selective HDAC11 inhibitor that showed anticancer activity.22 Later, Son et al. reported a series of potent HDAC11inhibitors bearing a long alkyl chain group.23 Although the pharmacokinetic (PK) characteristics and biological activities of these reported HDAC11 inhibitors require further investigation, it can be inferred from their chemical structures and physicochemical properties that they are unlikely to effectively cross the blood–brain barrier (BBB) and exert pharmacological effects in the brain. Previous studies revealed that most HDAC inhibitors contain three moieties: “Cap”, “Linker”, and the zinc-binding group (ZBG). Development of isoform-selective inhibitors can be achieved through the modification of each moiety. However, most HDAC inhibitors consist of a highly polar zinc-binding group, which results in unfavorable physicochemical properties for blood–brain barrier (BBB) penetration and limited therapeutic efficiency in neurologic disorders. In our previous work, we obtained a series of class I and HDAC6 inhibitors by introducing adamantane as the Cap group into the molecule and developed brain-permeable PET robes [11C]Martinostat and [18F]Bavarostat for brain HDACs imaging.24,25 The lipophilic adamantane Cap group balances molecules’ lipophilicity, which consequently improves brain penetration. In addition, literature surveys indicate that N-heterobicyclic moieties are commonly utilized in the development of selective HDAC inhibitors.26–28 As such, we developed an indol-adamantane-based hydroxamic acid 4a (Figure 1) and found that it exhibits selectivity against HDAC6 and HDAC11. This finding indicated that 4a could serve as a structural basis for developing brain-permeable selective HDAC11 inhibitors. In this study, we describe the development and characterization of selective HDAC11 inhibitor PB94, which was identified by structural optimization of 4a. We determined that PB94 has significant potency and selectivity toward HDAC11, and we also evaluated its drug-like properties, including pharmacokinetic/pharmacodynamic studies and phenotypic activity profile testing. We further radiolabeled PB94 with carbon-11 as [11C]PB94 to examine its pharmacokinetics in vivo using positron emission tomography (PET). Furthermore, cell-based assays using mouse microglia BV2 cells were conducted to investigate the neuroinflammatory effects of PB94. Moreover, a neuropathic pain model was used to determine the therapeutic efficacy of PB94 in neuropathic pain. Our studies set the stage for further development of PB94 as a potential therapeutic agent for CNS-related disorders as well as the development of brain-permeable HDAC11-selective inhibitors.
Figure 1.

Structures of Bavarostat, 4a and the selective HDAC11 inhibitor PB94.
RESULTS AND DISCUSSION
Synthesis and HDACs Inhibitory Activity Evaluation of Analogues 4a–j.
As our previous studies indicated that the substituents on the benzyl linker might lead to a remarkable impact on HDAC potency and selectivity,29,30 we first synthesized a series of 4a analogues bearing different substituents on the benzyl linker. A total of 10 analogues were obtained via the synthetic route outlined in Scheme 1. The intermediate (2) was prepared by coupling 1H-indole-5-carboxylic acid (1) with adamantan-1-ylmethanamine. Several commercially available benzyl bromides were reacted with intermediate (2) to form the corresponding ester intermediates 3a–j, which were converted to the desired hydroxamates 4a–j in the presence of aqueous NH2OH/NaOH solution. The inhibitory ability of analogues 4a–j to HDACs was tested, using selective class I and II HDAC inhibitor trichostatin A (TSA) as a reference compound. As illustrated in Table 1, these analogues displayed moderate to good inhibitory potency against HDAC11. The structure–activity relationship (SAR) revealed that introducing a substituent on the phenyl linker resulted in improved HDAC11 inhibitory activity and selectivity compared with compound 4a, which does not have any substituent on the phenyl linker. Among all of the analogues, compound 4j, which bears a 3′-bromine atom, displayed the most potent inhibitory activity against HDAC11 (IC50 = 13.7 nM), while compound 4e (PB94) bearing a methoxy group on the 3′-position had significant HDAC11 potency and the highest HDAC11 selectivity (IC50 = 108 nM and over 40-fold over other HDAC isoforms, Figure 1c). Notably, the introduction of a fluorine atom on the phenyl linker (4b and 4c) resulted in enhanced HDAC11 potency while retaining potent HDAC6 potency.
Scheme 1. Synthetic Routes of Analogues 4a–ja.

aReagents and conditions: (i) adamantan-1-ylmethanamine, EDCI, dichloromethane, room temperature (rt), 6–8 h, yield: 62%; (ii) benzyl bromides, NaH, tetrahydrofuran (THF), rt, overnight, yield: 46.2–62.5%; (iii) NaOH, 50 wt % aq NH2OH, THF/MeOH (1:1), 0 °C to rt, rt, 30 min, yield: 90–95%.
Table 1.
Full of 11 HD AC Isoforms Profiling of Synthetic Compounds 4a–ja
| enzymatic inhibitory activity (IC50, μM) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HDAC isoforms | |||||||||||
| cpd. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|
| |||||||||||
| 4a | 0.88 ± 0.07 | 1.90 ± 0.14 | 0.98 ±0.11 | 1.09 ± 0.21 | >10 | 0.018 ± 0.005 | 1.64 ± 0.25 | 1.95 ± 0.31 | 5.25 ± 1.05 | >10 | 0.48 ± 0.12 |
| 4b | 0.19 ± 0.02 | 1.02 ± 0.04 | 0.69 ± 0.03 | 1.12 ± 0.04 | >10 | 0.008 ± 0.003 | 0.4 ± 0.01 | 0.54 ± 0.04 | 2.09 ± 0.44 | 0.23 ± 0.01 | 0.087 ± 0.01 |
| 4c | 0.49 ± 0.06 | 3.40 ± 0.12 | 0.18 ± 0.03 | 0.11 ± 0.02 | 0.50 ± 0.07 | 0.004 ± 0.002 | 0.05 ± 0.01 | 1.39 ± 0.11 | 0.51 ± 0.04 | 0.60 ± 0.04 | 0.079 ± 0.01 |
| 4d | >10 | >10 | 1.33 ± 0.20 | 3.04 ± 0.41 | 1.88 ±0.11 | 0.64 ± 0.16 | 0.47 ± 0.06 | 0.16 ± 0.03 | 2.84 ± 0.31 | >10 | 0.093 ± 0.01 |
| 4e (PB94) | >10 | >10 | >10 | >10 | >10 | 4.18 ± 0.33 | 7.09 ± 0.52 | 8.44 ± 0.59 | >10 | >10 | 0.11 ± 0.01 |
| 4f | >10 | >10 | >10 | >10 | >10 | 0.41 ± 0.03 | >10 | 2.06 ± 0.32 | >10 | >10 | 0.35 ± 0.01 |
| 4g | >10 | >10 | 8.12 ± 0.87 | 1.60 ± 0.02 | 7.25 ± 0.36 | 0.09 ± 0.01 | 1.21 ± 0.02 | >10 | >10 | >10 | 0.16 ± 0.01 |
| 4h | >10 | >10 | >10 | >10 | 6.04 ± 0.59 | 0.44 ± 0.04 | 4.03 ±0.11 | >10 | >10 | >10 | 0.25 ± 0.02 |
| 4i | >10 | >10 | 2.03 ± 0.04 | 1.20 ± 0.08 | 5.12 ± 0.22 | 0.22 ± 0.02 | 0.57 ± 0.02 | 0.41 ± 0.03 | 4.53 ± 0.78 | 9.05 ± 0.9 | 0.14 ± 0.01 |
| 4j | 7.32 ± 0.66 | >10 | 2.23 ± 0.44 | 1.37 ± 0.02 | 3.51 ± 0.63 | 0.17 ± 0.02 | 0.61 ± 0.08 | 8.38 ± 1.01 | 9.31 ± 0.92 | 8.97 ± 1.1 | 0.013 ± 0.01 |
| TSA | 0.001 ± 0.001 | 0.003 ± 0.001 | 0.003 ± 0.001 | 1.21 ± 0.03 | 1.38 ± 0.02 | 0.002 ± 0.001 | 0.44 ± 0.01 | 0.24 ± 0.01 | 5.37 ± 0.46 | 0.001 ± 0.001 | 9.08 ± 0.88 |
IC50 values are the mean ± SD of two experiments obtained from curve-fitting of a 12-point enzymatic assay starting from 10 μM with 3-fold serial dilution against all 11 HDAC isoforms; trichostatin A (TSA) was used as a positive control.
Synthesis and HDACs Inhibitory Activity Evaluation of Analogues 7a–b, 10a–b, 12, and 13.
Based on the SAR results, we established that the methoxy group on the 3′-position of the phenyl linker is a key factor for HDAC11 potency and selectivity. In order to explore whether the Cap group and ZBG of analogues have any effect on the potency and selectivity against HDAC11, we next synthesized compounds 7a–b and 10a–b, which lack the indole moiety on the Cap group. Previous studies demonstrated that the hydrazide groups could be unitized to replace the hydroxamate ZBG to improve inhibitors’ pharmacokinetic properties and HDAC selectivity.31,32 Therefore, we additionally synthesized trifluoromethyl-bearing (12) analogues. As the synthetic routes show in Scheme 2, to synthesize 7a–b, the adamantanamines (5a–b) were employed to react with methyl-4-(bromomethyl)-2-methoxybenzoate to afford ester intermediates 6a–b and 9a–b. The resulting ester intermediates were converted to hydroxamates (7a–b, 10a–b) under the above-mentioned condition. To prepare analogues 12, ester intermediate 3e was hydrolyzed to afford 11, which was further transformed to the desired analogues 12.
Scheme 2. Synthetic Routes of Analogues 7a–b, 10a–ba.

aReagents and conditions: (i) 3-methoxy-4-(methoxycarbonyl)benzoic acid, 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), dichloromethane, rt, 6–8 h, yield: 52.1–69.2%; (ii) NaOH, 50 wt % aq NH2OH, THF/MeOH (1:1), 0 °C to rt, rt, 30 min, yield: 90–95%; (iii) 4-formyl-2-methoxybenzoate, NaBH4, MeOH, rt, 4 h, yield: 64.3–85%; (iv) formic acid, formalin, rt, 24 h, yield: 85–88.2%; (v) LiOH (aq), rt, 1 h, 95%; (vi) HATU, N,N-diisopropylethylamine (DIPEA), dimethylformamide (DMF), rt, 12 h, yield: 49.2%.
The inhibitory activities of the new compounds against HDAC6 and HDAC11 were evaluated. As the data shown in Table 2, compounds without indol moiety in the Cap group display weak inhibitory activity against HDAC11 and no significant activity against HDAC6, suggesting that the indol moiety is essential for structural modification. On the other hand, the analogues bearing a hydrazide group failed to show improved HDAC11 binding affinity, with moderate potency (IC50 = 4.2 and 3.1 μM, respectively) and minimal selectivity against HDAC11.
Table 2.
Inhibitory Activities of Compounds 7a–b, 10a–b, 12, and 13 against HDAC6 and HDAC11a
| Cpd. | Structure | Enzymatic inhibitory activity (IC50, μM) |
|
|---|---|---|---|
| HDAC6 | HDAC11 | ||
|
| |||
| 7a |
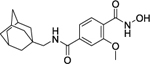
|
>10 | 8.89 ± 0.69 |
| 7b |

|
>10 | 6.89 ± 0.72 |
| 10a |

|
>10 | >10 |
| 10b |

|
>10 | 4.21 ± 0.55 |
| 12 |

|
>10 | 2.61 ± 0.42 |
| TSA | - | 0.002 ± 0.001 | 9.08 ± 1.1 |
IC50 values are the mean ± SD of two experiments obtained from curve-fitting of a 12-point enzymatic assay starting from 10 μM with 3-fold serial dilution against all 11 HDAC isoforms; trichostatin A (TSA) was used as a positive control.
PB94 Inhibits HDAC11 in Cells.
With the encouraging results that PB94 showed in the enzymatic assays, we further tested the HDAC11 inhibitory activity of PB94 in HEK293T cells. SHMT2 was identified as a defatty acylation substrate of HDAC11 in cells.33 Thus, we treated HEK293T cells with PB94 to test whether it could upregulate the fatty acylation level of SHMT2 via HDAC11 inhibition. HEK293T was incubated with Alk14 (an alkyne-tagged myristic acid analogue) and PB94 at different concentrations for 3 h. Then, the Alk14-labeled SHMT2 was conjugated with biotin using click chemistry, pulled down with streptavidin, and measured by Western blot.33,34 A known HDAC11 inhibitor, TD034,23 was used as the reference compound. As a result, PB94 significantly increased the fatty acylation level of SHMT2 at a concentration of 20 μM (Figure 2), indicating it has HDAC11 inhibitory activity in cells.
Figure 2.
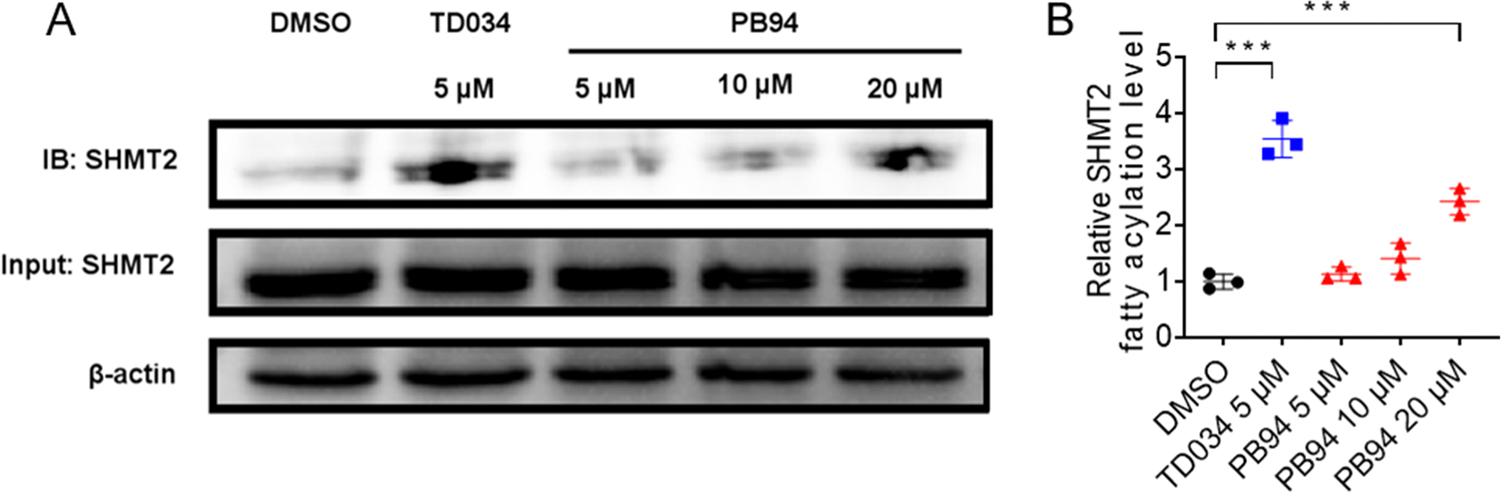
PB94 inhibits HDAC11 in HEK293T cells. (A) Representative Western blot images show that PB94 increases endogenous fatty acylation levels of SHMT2 in HEK293T cells at the concentration of 20 μM. (B) PB94 (20 μM) and reference compound TD034 (5 μM) significantly increase endogenous fatty acylation levels of SHMT2 in HEK293T cells. Signal intensity was quantified by ImageJ, and the signal of the control group (DMSO) without inhibitor treatment was set as 1.0. *p-value < 0.1; **p-value < 0.05; ***p-value < 0.01.
In Vitro ADME Evaluation and In Vivo Pharmacokinetic Profiling of PB94.
Next, several in vitro ADME assessments were carried out to evaluate the drug-like profiles of PB94. Data shown in Table 3 indicate that PB94 possesses good metabolic stability in human liver microsomal and mouse plasma, with half-lives (t1/2) of 54.6 and 133.8 min, respectively. In addition, no significant inhibitory effects were observed on cytochrome P450 enzymes (CYPs) 1A2, 2C19, and 2D6 at 10 μM of PB94. The pharmacokinetic (PK) profiles of PB94 were assessed by administrating 10 mg/kg PB94 intravenously (i.v) and orally (p.o) in mice. Results show that PB94 had suitable PK properties with a half-life of 5.3 h by p.o. administration. Of note, PB94 showed less favorable bioavailability when by oral administration (11.2%). Moreover, to evaluate the brain permeability of PB94, in vivo brain/plasma pharmacokinetic studies were performed by IP administering PB94 at 1 mg/kg in C57BL/6 mice. As a result, PB94 exhibited good brain permeability, with brain/plasma ratios of 2.3 at 30 min and 2.2 at 4 h postinjection.
Table 3.
ADME/PK Studies of PB94a
| in vitro ADMET profiles | |||||
|---|---|---|---|---|---|
|
| |||||
| hepatocyte stability (t1/2, min) | human | 54.6 | |||
| plasma stability (t1/2, min) | mouse | 133.8 | |||
| CYP inhibition (%@10 μM) | 1A2 | 33.5 | |||
| 2C9 | 78.6 | ||||
| 2C19 | 37.8 | ||||
| 2D6 | 37.1 | ||||
| plasma protein binding (%) | human | 99.7% | |||
|
| |||||
| mouse PK studies | |||||
|
| |||||
| parameters | units | i.v. dose of 10 mg/kg | p.o. dose of 10 mg/kg | ||
|
| |||||
| C max | μg/L | 2481.18 | 141.1 | ||
| T max | h | 0.08 | |||
| AUC(0–t) | μg/L*h | 1985.71 | 226.7 | ||
| T 1/2 | h | 5.1 | 5.3 | ||
| MRT(0–∞) | h | 1.1 | 6.5 | ||
| F | 11.2% | ||||
|
| |||||
| brain/plasma PK studies | |||||
|
| |||||
| route | dose (mg/kg) | time (h) | brain concn. (ng/mL) | plasma concn. (ng/mL) | brain/plasma ratio |
|
| |||||
| IP | 1 | 0.5 | 14.4 | 6.31 | 2.3 |
| IP | 1 | 1 | 9.58 | 6.31 | 1.5 |
| IP | 1 | 4 | 2.07 | 0.93 | 2.2 |
ADME studies and in vivo PK profiling were conducted by HD Biosciences (China) Co., Ltd. Data are expressed as the mean of three repeat experiments.
Binding Selectivity Evaluation and In Vitro Phenotypic Activity Profile of PB94.
We further tested the off-target binding in a panel of 46 targets [National Institute on Mental Health-Psychoactive Drug Screening Program (PDSP)] and observed no significant off-target binding at 10 μM (details presented in Table S2). The in vitro activity profile of PB94 in the BioMAP Diversity PLUS panel of 12 human primary cell-based systems (Eurofins Discovery) was carried out. As a result, PB94 did not exhibit any cytotoxic effects at tested concentrations (0.37–3.3 μM) but exhibited antiproliferative activity to human primary endothelial cells, T cells, B cells, and coronary artery smooth muscle cells at 3.3 μM (Figure S1). Based on the cellular activity profile of PB94 in the testing panel, a comparative analysis of the biological activities of known bioactive agents in the BioMAP reference database was used to predict the safety, efficacy, and function of PB94. As a result, there are 22 common and 51 differentiating activities observed in the overlaid bioactivity fingerprint of PB94 and reference benchmark vorinostat [a pan-inhibitor of HDAC1, HDAC2 and HDAC3 (Class I), HDAC7 (Class II), and HDAC11 (Class IV)]35 (Figure 3A). Interestingly, PB94 showed similar pharmacological activities to a selective HDAC1 inhibitor parthenolide (Figure 3B), which can selectively inhibit HDAC1 without affecting other class I/II HDACs and modulate NF-κB-mediated inflammatory responses.36 Additionally, mechanism HeatMAP analysis (Figure 3C) showed that PB94 is modestly active with three inflammation-related biomarkers, including IL-10, sPGE2, MCP1, and MIG, indicating the potential anti-inflammatory property of PB94.
Figure 3.
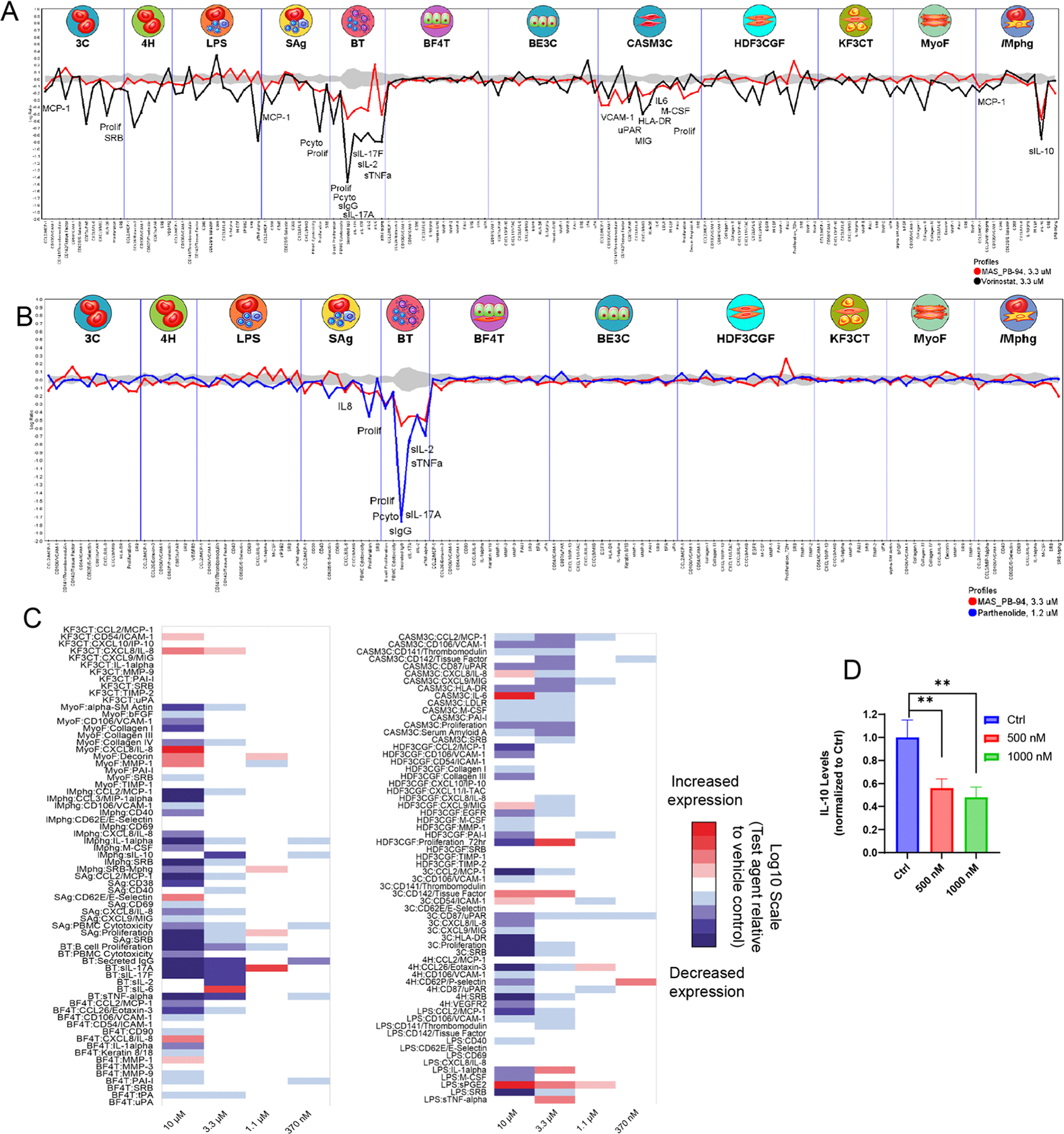
Phenotypic activity profile of PB94. (A) Reference benchmark overlay of PB94 (3.3 μM) and vorinostat (3.3 μM); (B) top database search result for PB94 (3.3 μM) is parthenolide (1.2 μM); (C) HeatMAP analysis for PB94. HeatMAP analysis of the 148 biomarker readouts (rows) within the Diversity PLUS Panel by PB94 in comparison to 19 consensus mechanism class profiles (columns); (D) mechanism of action by which PB94 affects neuroinflammatory events was characterized by a focus on IL-10 using mouse microglia BV2 cells. BV2 cells were utilized to assess inflammatory changes induced by lipopolysaccharides (LPS). Cells were treated with LPS (10 ng/mL) alone or in combination with 500 or 1000 nM PB94 for 24 h. The medium was then applied to the MSD-cytokine assay to measure levels of IL-10. mean ± standard error of the mean (SEM); t-test; n = 4 for each group.
Cell-Based Assay of PB94 Using Mouse Microglia BV2 Cells.
Previous findings showed that HDAC11 is characterized by immune regulatory functions involving regulation of IL-10.37 Thus, we performed mechanistic studies and asked whether PB94 may affect neuroinflammatory events with a focus on IL-10 using mouse microglial BV2 cells. Mouse microglia BV2 cells were induced by the well-characterized and previously reported immune-stimulating molecule lipopolysaccharides (LPS), alone or in combination with PB94, to assess inflammatory changes as a function of PB94. As a result, PB94 significantly reduced IL-10 expression in LPS (10 ng/mL) treated cells (Figure 3D), indicating the inflammation regulatory activity of PB94.
PET/CT Imaging Studies in Rodents of [11C]PB94.
To investigate the pharmacokinetics of PB94 in vivo, we radiolabeled PB94 with carbon-11 and carried out PET imaging studies with [11C]PB94 in rodents. [11C]PB94 was prepared through a two-step reaction using 3g as the precursor (Figure 4A). Next, 100–150 μCi [11C]PB94 was injected into C57BL/6 mice (male, n = 4) through the tail vein for a 60 min dynamic PET imaging followed by a 10 min computed tomography (CT). The representative PET/CT images focused on the mice brain (coronal, sagittal, and axial, summed from 0 to 60 min), and time–activity curves (TACs) of eight brain regions of interest are shown in Figure 4B–D. As expected, [11C]PB94 exhibited significant blood–brain barrier (BBB) penetration and fast brain uptake, with the maximum %ID/cc (percent injected dose per cc tissue) of 2.8 in the whole brain at the first few minutes postinjection. Regional brain analysis was carried out using the FUSION module (Ma-Benveniste-Mirrione) in PMOD (PMOD 4.003, PMOD Technologies Ltd., Zurich, Switzerland). Heterologous distribution of radioactivity was observed in eight ROIs, indicating the heterologous expression level of HDAC11 in brain regions. Of note, relatively high radioactivity uptake was found in the striatum, cortex, and amygdala, while the cerebellum and brain stem showed lower radioactivity uptake.
Figure 4.

Biodistribution studies of PB94 using PET imaging of [11C]PB94. (A) Radiosynthesis of [11C]PB94 using 3a as the precursor. Reagents and conditions: (1) K2CO3, DMF, 100 °C, 3.0 min RCY: 24–19% (non-decay-corrected, n = 3); (2) 0.6 M NaOH, NH2OH (50 wt % in H2O), MeOH/THF (1:1), 10 min. RCY: 66–70% (non-decay-corrected). Molar activity: 122 GBq/μmol (EOB); (B) representative mice PET/CT images of [11C]PB94 focus on the mice brain (coronal, sagittal, and axial, summed from 0 to 60 min), and mice body PET/CT images at 5, 15, 30, and 60 min postinjection in mice (n = 4 for each time point); (C, D) time–activity curves of [11C]PB94 in mice brain regions of interest, including cortex, cerebellum, brain stem, thalamus, hypothalamus, striatum, hippocampus, and amygdala; (E) biodistribution of [11C]PB94 in peripheral organs in mice at five different time points (1, 5, 15, 30, and 60 min).
We further explored the biodistribution of [11C]PB94 in several peripheral organs of mice including the heart, liver, lung, kidney, and spleen. Four time points (5, 10, 30, and 60 min) after [11C]PB94 injection were selected to investigate the uptake, distribution, and clearance of PB94 in the organs of interest. As shown in Figure 4E, high radioactivity (%ID/cc > 5) was seen in the blood-rich organs, including the heart, liver, and kidney, after [11C]PB94 injection. In the kidney, the radioactivity accumulated moderately and peaked at 15 min, while the radio signals decreased gradually in other organs. The high accumulation of radioactivity in the liver and kidney suggested the hepatobiliary and urinary excretion of [11C]PB94.
Safety Profile of PB94.
To investigate the safety profile of PB94, we carried out an acute toxicity assay in mice. The result showed that there is no observed adverse effect after treatment with BP94 at 200 mg/kg. The biochemical analysis showed that BP94 administration did not change the important kidney and liver function indexes, such as uric acid and alanine aminotransferase, as well as electrolytes including Na+, K+, and Cl−. The blood analysis showed that BP94 administration did not change the blood count indexes, including red blood cells, white blood cells, and blood platelets (Figure S2A). And there are no significant differences in body weight between the control (Figure S2B). In addition, no pathological abnormality of the main organs, including the heart, liver, spleen, lung, or kidney, was observed (Figure S2C). Collectively, BP94 is well tolerated in vivo.
HDAC11 Protein Level Increased in Neuropathic Pain.
The promising drug-like properties of PB94 support its therapeutic potential for neuropathic pain. To examine the potential roles of HDAC11 in a pathological status, we established a chronic constriction injury (CCI) mouse model of neuropathic pain.38 The primary somatosensory cortex has been previously identified as a key brain region implicated in pain processing.39 Therefore, we assessed HDAC11 expression in the primary somatosensory cortex after CCI injury by quantitative polymerase chain reaction (qPCR). Notably, the level of HDAC11 protein significantly increased in the cortex compared with Sham mice 14 days after surgery (Figure 5A).
Figure 5.

Pharmacological Inhibition of HDAC11 by PB94 attenuated neuropathic pain in CCI mice. (A) qPCR analysis indicated HDAC11 expression level was increased in the cortex at 14 days after CCI (Sham vs CCI, n = 3, unpaired t-test); (B) mechanical paw withdrawal thresholds (PWTs) significantly decreased in the surgery paw compared with the contralateral paw in CCI mice; (C) the dose–response of PB94 on nociceptive behavior in mice at 14 days after CCI surgery. P-values indicate the difference in the threshold between 10 mg/kg group and CCI group (n = 8, two-way ANOVA followed by Bonferroni post hoc analyses); (D) the von Frey test. P-values indicate the difference in the latency between the CCI + PB94 group and the Sham group (n = 8, two-way ANOVA followed by Bonferroni post hoc analyses); (E) the Hargreaves test. P-values indicate the difference in the threshold between the CCI + PB94 group and the Sham group (n = 8, two-way ANOVA followed by Bonferroni post hoc analyses); (F) mouse body weight change during the treatment.
PB94 Ameliorated Neuropathic Pain in a Chronic Constrictive Injury Mouse Model.
The increase of HDAC11 in the primary somatosensory cortex raised intriguing possibilities that HDAC11 is critically implicated in pain and that targeting HDAC11 might provide therapeutic effects. To test this, we administered PB94 in mice that underwent CCI to assess the development of nociceptive behavior. In one set of experiments, as shown in Figure 5B, the hind paw mechanical withdrawal thresholds ipsilateral to the injury side decreased after surgery and remained at low levels from day 3 to day 14. In contrast, no significant change in mechanical pain threshold was observed in the contralateral paw. At day 14 after CCI injury, the mice were treated with PB94 at different doses and mechanical withdrawal thresholds were examined. Single-dose injection of PB94 was able to increase mechanical withdrawal thresholds (Figure 5C). Notably, the effect of PB94 appeared to be dose-dependent. At 10 mg/kg, these effects lasted for more than 3 h. As such, 10 mg/kg was chosen to be administered twice daily for another set of experiments. As shown in Figure 5D,E, PB94 (10 mg/kg) was able to significantly alleviate mechanical and thermal pain-like behaviors during the entire experimental period of 14 days. Importantly, no significant side effects were noticed during PB94 treatment. Mouse body weight remained similar between saline or PB94-treated animals (Figure 5F).
To interrogate potential mechanisms that are linked to the analgesic effect of PB94, brain samples of mice that received 14 days of PB94 treatment were stained for Iba-1, a microglia marker. Previously, microglia-mediated neuroinflammation has been shown to be critical for the development of neuropathic pain, including pain in the CCI model.40 The hindlimb region of the primary somatosensory cortex (S1HL) and anteroposterior region (VPL) of the thalamus, two brain regions that are important for pain processing,41 exhibited a lower number of Iba-1 positive cells in the “PB94 + CCI” group than the “vehicle + CCI” group (Figure 6), suggesting the role of PB94 in neuroinflammation modulation.
Figure 6.

Pharmacological inhibition of HDAC11 by PB94 attenuates the CCI-induced microgliosis in mice. (A) Representative Iba1 staining of Sham, PB94 + CCI, vehicle + CCI. Images were taken at 4× (scale bar represents 500 μm); Iba1 + cells in the boxed regions of S1HL and VPL were analyzed. (Scale bar represents 50 μm); (B) S1HL and VPL microglia counts significantly increased in mice that underwent CCI surgery. P-values indicate the difference in the microglia counts between CCI + Vehicle and CCI + PB94 (n = 3, unpaired t-test). Data are represented as mean ± SEM *p < 0.05.
CONCLUSIONS
In this work, we described the development, characterization, and therapeutic effects of a novel brain-permeable HDAC11-selective inhibitor PB94. Enzyme inhibition evaluations showed that PB94 has good HDAC11 potency and selectivity. In vitro ADME and in vivo brain-plasma pharmacokinetic studies reveal that PB94 exhibited negative CYP inhibition, good metabolic stability, and favorable brain permeability. In addition, PET imaging with [11C]PB94 in rodents showed good brain uptake of PB94 and a heterologous distribution of radioactivity in brain regions and organs of interest. Furthermore, we found HDAC11 upregulation in the somatosensory cortex of CCI mice, and a single-dose treatment of PB94 at 10 mg/kg attenuated mechanical hypersensitivity in mice with CCI. A battery of tests was used to assess the analgesic effect of PB94 on pain-like behaviors, which led to findings supporting its effectiveness at 10 mg/kg. Collectively, our studies support the further development of PB94 as a promising drug candidate for the treatment of neuropathic pain and potentially other CNS-related disorders.
EXPERIMENTAL SECTION
All commercially available chemical reagents and solvents were ordered from commercial suppliers in ACS-grade purity or higher and directly used without further purification. Tubastatin A was purchased from MedChemExpress. NMR spectra were recorded on a JEOL JNM-ECZ500R spectrometer at room temperature [500 MHz (1H), 471 MHz (19F), and 126 MHz (13C)]. Chemical shifts were given in δ values (ppm), using tetramethylsilane (TMS) as the internal standard. Mass spectrometry data were recorded on an Agilent 6310 ion trap mass spectrometer (ESI source) connected to an Agilent 1200 series HPLC with a quaternary pump, vacuum degasser, diode-array detector, and autosampler. Analytical HPLC was carried out on an Agilent 1200 series under the following conditions:: column (Agilent C18, 1.8 μm, 2.1 × 100 mm), mobile phase = 0.1% TFA in water/0.1% TFA in acetonitrile at a flow rate of 1.0 mL/min for 11 min, gradient method. The purities of all tested compounds were >95%, as determined by analytical HPLC.
All animal studies were carried out at Massachusetts General Hospital (PHS Assurance of Compliance No. A3596-01). The Subcommittee on Research Animal Care (SRAC) serves as the Institutional Animal Care and Use Committee (IACUC) of the Massachusetts General Hospital (MGH). SRAC reviewed and approved all procedures detailed in this paper.
[11C]CO2 (1.2 Ci) was obtained via the 14N (p, α) 11C reaction on nitrogen with 2.5% oxygen, with 11 MeV protons (Siemens Eclipse cyclotron, Siemens Healthcare GmbH, Erlangen, Germany), and trapped on molecular sieves in a TRACERlab FX-MeI synthesizer (General Electric, GE Healthcare, Boston, MA). [11C]CH4 was obtained by the reduction of [11C]CO2 in the presence of Ni/hydrogen at 350 °C and recirculated through an oven containing I2 to produce [11C]CH3I via a radical reaction.
Chemistry.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1H-indole-5-carboxamide (2).
To a mixture of 1H-indole-5-carboxylic acid (1, 1 g, 6.2 mmol) and EDCI (1.02 g, 6.2 mmol) in dichloromethane (30 mL) was added adamantan-1-ylmethanamine (1.4 g, 1.4 mmol). The mixture was stirred at room temperature for 6–8 h, and the reaction was monitored by thin layer chromatography (TLC). After the complete consumption of compound 1, the residue was concentrated under vacuum and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with H2O (2 × 30 mL) and brine (15 mL), dried over sodium sulfate, filtered, and concentrated under a vacuum. The crude product was purified via Combi-Flash column chromatography (EtOAc/hexane = 0–100%) to afford compound 2 in 62% yield. 1H NMR (500 MHz, Chloroform-d) δ 8.44 (s, 1H), 8.08 (d, J = 1.7 Hz, 1H), 7.65 (dd, J = 8.6, 1.7 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.27 (t, J = 2.7 Hz, 1H), 6.62 (t, J = 2.7 Hz, 1H), 6.20 (t, J = 6.5 Hz, 1H), 3.19 (d, J = 6.1 Hz, 2H), 1.21–1.97 (m, 3H), 1.72 (dt, J = 12.5, 3.2 Hz, 3H), 1.67–1.63 (m, 3H), 1.58 (br s, 6H). MS (ESI+): 309.2 (M + H)+
General Procedure for the Synthesis of Compounds 3a–j.
To a mixture of 2 (100 mg, 0.32 mmol) and NaH (15.5 mg, 0.64 mmol) in THF (5 mL) was added the corresponding benzoic bromine derivative (0.38 mmol). The mixture was stirred at room temperature overnight. After the complete consumption of compound 2, the residue was concentrated under vacuum and extracted with EtOAc (3 × 30 mL), and the combined organic layers were washed with H2O (2× 30 mL) and brine (15 mL), dried over sodium sulfate, filtered, and concentrated under vacuum. The crude product was purified via Combi-Flash column chromatography (EtOAc/hexane = 0–100%) to afford compounds 3a–j.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)benzoate (3a).
White solid; Yield: 57.1%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.65 (dd, J = 8.6, 1.8 Hz, 1H), 7.22 (s, 1H), 7.17 (d, J = 3.1 Hz, 1H), 6.65 (dd, J = 8.1, 2.1 Hz, 2H), 6.62 (s, 1H), 6.18 (t, J = 6.3 Hz, 1H), 5.36 (s, 2H), 3.86 (s, 3H), 3.18 (d, J = 6.2 Hz, 2H), 2.02–1.97 (m, 3H), 1.72 (s, 3H), 1.64 (s, 3H), 1.58–1.54 (m, 6H). MS (ESI+): 457.3 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-3-fluorobenzoate (3b).
White solid; Yield: 52.4%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.10 (d, J = 1.7 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.61 (dd, J = 8.6, 1.7 Hz, 1H), 7.18 (d, J = 3.1 Hz, 1H), 6.65 (dd, J = 8.1, 2.1 Hz, 2H), 6.61 (s, 1H), 6.18 (t, J = 6.3 Hz, 1H), 5.35 (s, 2H), 3.85 (s, 3H), 3.74 (s, 3H), 3.18 (d, J = 6.2 Hz, 2H), 2.02–1.97 (m, 3H), 1.70 (s, 3H), 1.63 (s, 3H), 1.57 (d, J = 2.9 Hz, 6H). MS (ESI+): 475.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-fluorobenzoate (3c).
White solid; Yield: 58.8%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.74 (s, 1H), 7.68 (dd, J = 8.6, 1.8 Hz, 1H), 7.27 (d, J = 3.8 Hz, 1H), 6.65 (dd, J = 8.1, 2.1 Hz, 2H), 6.61 (s, 1H), 6.18 (t, J = 6.3 Hz, 1H), 5.38 (s, 2H), 3.87 (s, 3H), 3.19 (d, J = 6.2 Hz, 2H), 2.12–1.99 (m, 3H), 1.74 (s, 3H), 1.66–1.64 (m, 3H), 1.57–1.54 (m, 6H). MS (ESI+): 475.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-3-methoxybenzoate (3d).
White solid; Yield: 62.5%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.11 (s, 1H), 7.73 (s, 1H), 7.68 (dd, J = 8.6, 1.5 Hz, 1H), 7.27 (d, J = 3.2 Hz, 1H), 6.66 (dd, J = 7.1, 2.1 Hz, 2H), 6.64 (t, J = 2.3 Hz, 1H), 6.16 (t, J = 6.3 Hz, 1H), 5.36 (s, 2H), 3.87 (s, 3H), 3.77 (s, 3H), 3.15 (d, J = 6.2 Hz, 2H), 2.11–1.96 (m, 3H), 1.77–1.74 (m, 3H), 1.65–1.62 (m, 3H), 1.56–1.54 (m, 6H). MS (ESI+): 487.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-methoxybenzoate (3e).
White solid; Yield: 55.3%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.77 (s, 1H), 7.61 (dd, J = 7.8, 1.4 Hz, 1H), 7.24 (d, J = 1.2 Hz, 1H), 6.65 (dd, J = 7.5, 2.2 Hz, 2H), 6.64 (t, J = 1.8 Hz, 1H), 6.12 (t, J = 6.3 Hz, 1H), 5.32 (s, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 3.14 (d, J = 6.2 Hz, 2H), 2.13–1.97 (m, 3H), 1.75–1.72 (m, 3H), 1.66–1.63 (m, 3H), 1.55–1.51 (m, 6H). MS (ESI+): 487.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-methylbenzoate (3f).
White solid; Yield: 50.5%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.76 (s, 1H), 7.63 (dd, J = 7.8, 1.5 Hz, 1H), 7.25 (d, J = 1.8 Hz, 1H), 6.65 (dd, J = 7.1, 1.2 Hz, 2H), 6.61 (t, J = 1.8 Hz, 1H), 6.12 (t, J = 6.3 Hz, 1H), 5.32 (s, 2H), 3.86 (s, 3H), 3.13 (d, J = 6.8 Hz, 2H), 2.22 (s, 3H), 1.92 (s, 3H), 1.77–1.74 (m, 3H), 1.66–1.64 (m, 3H), 1.55–1.52 (m, 6H). MS (ESI+): 471.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-hydroxybenzoate (3g).
White solid; Yield: 46.2%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.76 (d, J = 7.2, 2.5 Hz, 1H), 7.62 (dd, J = 7.1, 1.5 Hz, 1H), 7.24 (s, 1H), 6.62 (dd, J = 7.1, 1.2 Hz, 2H), 6.59 (t, J = 1.8 Hz, 1H), 6.17 (t, J = 6.3 Hz, 1H), 5.35 (s, 2H), 3.86 (s, 3H), 3.14 (d, J = 6.8 Hz, 2H), 2.22–1.94 (m, 3H), 1.76–1.74 (m, 3H), 1.66–1.61 (m, 3H), 1.56–1.51 (m, 6H).MS (ESI+): 473.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-ethoxybenzoate (3h).
White solid; Yield: 52.8%. 1H NMR (500 MHz, Chloroform-d) δ 1H NMR (500 MHz, Chloroform-d) δ 8.11 (s, 1H), 7.74 (d, J = 7.6, 1.5 Hz, 1H), 7.62 (dd, J = 7.1, 1.2 Hz, 1H), 7.23 (s, 1H), 6.62 (dd, J = 7.5, 1.2 Hz, 2H), 6.61 (s, 1H), 6.15 (t, J = 6.5 Hz, 1H), 5.32 (s, 2H), 3.85 (s, 3H), 3.68–3.61 (m, 2H), 3.15–3.11 (m, 2H), 2.23–1.98 (m, 3H), 1.76–1.72 (m, 5H), 1.66–1.62 (m, 3H), 1.58–1.54 (m, 6H). MS (ESI+): 501.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-chlorobenzoate (3i).
White solid; Yield: 58.8%. 1H NMR (500 MHz, Chloroform-d) δ 8.09 (s, 1H), 7.75 (s, 1H), 7.67 (dd, J = 8.1, 1.2 Hz, 1H), 7.26 (d, J = 3.8 Hz, 1H), 6.66 (dd, J = 8.1, 2.1 Hz, 2H), 6.60 (s, 1H), 6.18 (t, J = 6.3 Hz, 1H), 5.37 (s, 2H), 3.86 (s, 3H), 3.18 (d, J = 6.1 Hz, 2H), 2.13–1.98 (m, 3H), 1.75 (s, 3H), 1.67–1.63 (m, 3H), 1.58–1.55 (m, 6H). MS (ESI+): 492.2 (M + H)+.
Methyl-4-((5-((((3r,5r,7r)-adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-bromobenzoate (3j).
White solid; Yield: 60.7%. 1H NMR (500 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.73 (s, 1H), 7.67 (dd, J = 8.1, 1.5 Hz, 1H), 7.27 (dd, J = 3.7 Hz, 1.2 Hz 1H), 6.65 (t, J = 8.1 Hz, 2H), 6.62 (s, 1H), 6.17 (t, J = 6.3 Hz, 1H), 5.38 (s, 2H), 3.86 (s, 3H), 3.18 (d, J = 6.2 Hz, 2H), 2.13–1.97 (m, 3H), 1.73 (s, 3H), 1.65–1.60 (m, 3H), 1.56–1.51 (m, 6H). MS (ESI+): 536.1 (M + H)+.
General Procedure for the Synthesis of Compounds 4a–j.
Solid NaOH (374 mg, 9.36 mmol) was dissolved in a 50% aqueous solution of NH2OH (4 mL) at 0 °C. A solution of compound 3a–j (2.34 mmol) in 1:1 THF/MeOH (6 mL) was added dropwise with vigorous stirring. Upon complete addition, the ice bath was removed, and the reaction was allowed to stir for 15 min. The reaction was then neutralized with 2 N HCl solution, and the mixture was concentrated under a vacuum. The crude product was purified by Combi-Flash reverse column chromatography (ACN/H2O = 0–100%) to yield the compounds 4a–j after lyophilization.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4a).
White solid; Yield: 62.8%. 1H NMR (500 MHz, DMSO-d6) δ 10.82–10.72 (m, 1H), 9.31 (s, 1H), 8.03–7.96 (m, 2H), 7.66–7.57 (m, 2H), 7.54–7.51 (m, 2H), 7.05 (d, J = 10.2 Hz, 1H), 6.88 (s, 1H), 6.59 (t, J = 5.3 Hz, 1H), 5.48 (d, J = 7.3 Hz, 2H), 2.95 (d, J = 6.3 Hz, 2H), 1.87 (d, J = 4.2 Hz, 3H), 1.64 (br s, 3H), 1.55 (br s, 3H), 1.46 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 164.3, 152.1, 142.5, 137.8, 131.5, 130.2, 123.6, 121.8, 118.4, 113.8, 112.5, 110.7, 102.6, 100.4, 54.1, 51.6, 48.2, 42.7 (m, 2C), 37.2 (2C), 35.3, 34.9, 28.3 (2C), 27.8, 25.3. HRMS (ESI) for C28H31N3O3 [M + H]+ calcd 458.2399, found: 458.2402.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(2-fluoro-4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4b).
White solid; Yield: 52.5%. 1H NMR (500 MHz, DMSO-d6) δ 10.88 (br s, 1H), 9.67 (br s, 1H), 8.11 (s, 2H), 7.66–7.61 (m, 2H), 7.54 (s, 2H), 7.15 (d, J = 7.8 Hz, 1H), 6.98 (s, 1H), 6.67 (t, J = 7.3 Hz, 1H), 5.45 (d, J = 7.8 Hz, 2H), 2.94 (d, J = 6.2 Hz, 2H), 1.79 (d, J = 4.2 Hz, 3H), 1.65 (s, 3H), 1.55 (m, 3H), 1.45–1.43 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 162.5, 151.1, 142.6, 136.3, 130.4, 130.1, 123.2, 122.7, 118.3, 112.5, 111.9, 111.6, 102.4, 101.9, 54.3, 51.2, 47.2, 45.7 (m, 2C), 37.2 (3C), 34.4, 29.6 (2C), 28.4, 23.8. HRMS (ESI) for C28H30FN3O3 [M + H]+ calcd 476.2305, found: 476.2309.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(3-fluoro-4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4c).
White solid; Yield: 57.9%. 1H NMR (500 MHz, DMSO-d6) δ 10.92–10.79 (m, 1H), 9.15 (s, 1H), 8.10 (q, J = 3.8 Hz, 2H), 7.63–7.56 (m, 2H), 7.44 (dt, J = 20.4, 7.3 Hz, 2H), 7.03 (d, J = 10.5 Hz, 1H), 6.98 (d, J = 7.9 Hz, 1H), 6.58 (t, J = 4.3 Hz, 1H), 5.48 (d, J = 7.3 Hz, 2H), 2.95 (d, J = 6.3 Hz, 2H), 1.88 (d, J = 4.2 Hz, 3H), 1.61 (d, J = 10.5 Hz, 3H), 1.55 (d, J = 12.6 Hz, 3H), 1.46 (d, J = 5.6 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 161.4, 152.6, 140.8, 137.2, 131.7, 130.8, 125.3, 122.7, 118.5, 115.7, 113.8, 110.6, 104.5, 100.7, 53.9, 51.0, 47.1, 45.5 (m, 2C), 38.5 (3C), 36.3, 29.7 (2C), 28.6, 24.8. HRMS (ESI) for C28H30FN3O3 [M + H]+ calcd 476.2305, found: 476.2311.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(4-(hydroxycarbamoyl)-2-methoxybenzyl)-1H-indole-5-carboxamide (4d).
White solid; Yield: 44.9%. 1H NMR (500 MHz, DMSO-d6) δ 11.14 (s, 1H), 9.00 (s, 1H), 8.12–8.08 (m, 2H), 7.59 (dd, J = 8.8, 1.7 Hz, 1H), 7.47 (d, J = 3.1 Hz, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.35 (d, J = 1.6 Hz, 1H), 7.17 (dd, J = 7.9, 1.6 Hz, 1H), 6.67 (d, J = 7.9 Hz, 1H), 6.56 (d, J = 3.3 Hz, 1H), 5.38 (s, 2H), 3.88 (s, 3H), 2.95 (d, J = 6.3 Hz, 2H), 1.90–1.87 (m, 3H), 1.62 (d, J = 12.2 Hz, 3H), 1.55 (d, J = 12.0 Hz, 3H), 1.46 (d, J = 2.9 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 161.0, 151.7, 141.2, 137.5, 131.2, 130.8, 126.3, 121.7, 117.5, 116.9, 115.8, 110.2, 108.5, 102.2, 54.9, 52.4, 47.1, 45.5 (m, 3C), 37.1 (3C), 38.8, 29.0 (2C), 28.7, 22.9. HRMS (ESI) for C29H33N3O4 [M + H]+ calcd 488.2505, found: 476.2508.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(4-(hydroxycarbamoyl)-3-methoxybenzyl)-1H-indole-5-carboxamide (4e, PB94).
White solid; Yield: 67.5%; 1H NMR (500 MHz, DMSO-d6) δ 10.51 (s, 1H), 9.00 (s, 1H), 8.12–8.05 (m, 2H), 7.62–7.54 (m, 2H), 7.47 (d, J = 8.6 Hz, 1H), 7.40 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 1.4 Hz, 1H), 6.64 (dd, J = 7.8, 1.4 Hz, 1H), 6.57 (d, J = 3.1 Hz, 1H), 5.44 (s, 2H), 3.74 (s, 3H), 2.95 (d, J = 6.3 Hz, 2H), 1.90–1.86 (m, 3H), 1.64–1.59 (m, 3H), 1.55 (d, J = 11.5 Hz, 3H), 1.46 (d, J = 3.0 Hz, 6H).13C NMR (126 MHz, DMSO-d6) δ 162.3, 156.2, 140.1, 138.9, 130.9, 130.5, 121.4, 120.8, 119.2, 112.3, 111.1, 110.1, 102.7, 100.9, 56.1, 51.0, 49.6, 40.7 (m, 3C), 37.1 (3C), 34.9, 28.3 (3C), 25.4. MS (ESI+): 488.2 (M + H)+. HRMS (ESI) for C29H33N3O4[M + H]+, 488.2535; found, 488.2539. Melting point: 184.7–193.2 °C.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(4-(hydroxycarbamoyl)-3-methylbenzyl)-1H-indole-5-carboxamide (4f).
White solid; Yield: 54.1%. 1H NMR (500 MHz, DMSO-d6) δ 11.14 (s, 1H), 9.00 (s, 1H), δ 8.12 (s, 1H), 7.75 (s, 1H), 7.64 (dd, J = 7.8, 1.2 Hz, 1H), 7.26 (d, J = 1.8 Hz, 1H), 6.75 (dd, J = 7.1, 1.2 Hz, 2H), 6.63 (t, J = 1.5 Hz, 1H), 6.12 (t, J = 6.3 Hz, 1H), 5.52 (s, 2H), 3.13 (d, J = 6.8 Hz, 2H), 2.32 (s, 3H), 1.92 (s, 3H), 1.76–1.73 (m, 3H), 1.66–1.62 (m, 3H), 1.55–1.53 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 161.1, 158.6, 144.5, 138.4, 132.7, 131.0, 126.7, 122.7, 119.4, 113.1, 110.7, 109.4, 102.8, 100.4, 57.1, 54.1, 49.7, 42.6 (m, 3C), 37.6 (3C), 33.4, 34.9, 28.7 (3C), 25.8. HRMS (ESI) for C29H33N3O3 [M + H]+ calcd 472.2555, found: 472.2558.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(3-hydroxy-4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4g).
White solid; Yield: 40.5%. 1H NMR (500 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.22 (s, 1H), 8.07 (dt, J = 7.5, 3.5 Hz, 2H), 7.57 (dd, J = 8.6, 2.2 Hz, 1H), 7.49 (t, J = 2.7 Hz, 1H), 7.44 (ddd, J = 14.5, 8.4, 2.0 Hz, 2H), 6.53 (t, J = 2.7 Hz, 1H), 6.39–6.33 (m, 1H), 6.28 (s, 1H), 5.27 (d, J = 2.0 Hz, 2H), 2.95 (dd, J = 6.4, 2.1 Hz, 2H), 1.89 (s, 3H), 1.62 (d, J = 12.2 Hz, 3H), 1.58–1.51 (m, 3H), 1.47 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 164.3, 152.1, 142.5, 137.8, 131.5, 130.2, 123.6, 121.8, 118.4, 113.1, 112.5, 110.7, 102.6, 100.4, 54.1, 51.6, 48.2, 42.7 (m, 2C), 37.2 (2C), 35.3, 34.9, 28.3 (2C), 27.8, 25.3. HRMS (ESI) for C29H33N3O4 [M + H]+ calcd 474.2348, found: 474.2347.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(3-ethoxy-4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4h).
White solid; Yield: 44.3%. 1H NMR (500 MHz, DMSO-d6) δ 8.10–8.07 (m, 2H), 7.61–7.56 (m, 2H), 7.47 (d, J = 8.7 Hz, 1H), 7.38 (d, J = 7.9 Hz, 1H), 7.00 (d, J = 1.4 Hz, 1H), 6.66 (dd, J = 7.7, 1.4 Hz, 1H), 6.56 (d, J = 3.2 Hz, 1H), 5.42 (s, 2H), 4.01 (q, J = 6.9 Hz, 2H), 2.95 (d, J = 6.4 Hz, 2H), 1.89–1.88 (m, 3H), 1.62 (d, J = 11.9 Hz, 3H), 1.55 (d, J = 12.0 Hz, 3H), 1.46 (d, J = 2.9 Hz, 6H), 1.26 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 162.4, 155.9, 140.3, 138.7, 132.4, 130.1, 124.9, 122.8, 118.4, 114.4, 112.5, 110.2, 102.8, 100.6, 54.2, 51.6, 48.2, 41.6 (m, 2C), 37.2 (2C), 35.3, 35.9, 28.4 (2C), 28.1, 25.5. HRMS (ESI) for C30H35N3O4 [M + H]+ calcd 502.2661, found: 502.2664.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(3-chloro-4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4i).
White solid; Yield: 50.4%. 1H NMR (500 MHz, DMSO-d6) δ 10.94 (s, 1H), 9.12 (s, 1H), 8.09 (s, 1H), 7.76 (s, 1H), 7.72 (dd, J = 8.2, 1.4 Hz, 1H), 7.22 (d, J = 3.8 Hz, 1H), 6.65 (dd, J = 8.1, 2.1 Hz, 2H), 6.60 (s, 1H), 6.15 (t, J = 6.5 Hz, 1H), 5.38 (s, 2H), 3.18 (d, J = 6.5 Hz, 2H), 2.12–1.97 (m, 3H), 1.76 (s, 3H), 1.67–1.64 (m, 3H), 1.58–1.56 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 162.3, 151.3, 142.5, 137.7, 131.4, 130.1, 122.5, 120.5, 118.4, 116.5, 112.4, 111.7, 102.5, 100.2, 54.1, 51.6, 48.2, 42.4 (m, 2C), 38.5 (2C), 35.1, 33.9, 28.8 (2C), 27.5, 25.1. HRMS (ESI) for C28H30ClN3O3 [M + H]+ calcd 493.1946, found: 493.1945.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(3-bromo-4-(hydroxycarbamoyl)benzyl)-1H-indole-5-carboxamide (4j).
White solid; Yield: 40.6%. 1H NMR (500 MHz, DMSO-d6) δ 10.84 (s, 1H), 9.18 (s, 1H), 8.10 (d, J = 6.2 Hz, 2H), 7.60 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 7.5 Hz, 2H), 7.28–7.23 (m, 1H), 7.14 (d, J = 8.0 Hz, 1H), 6.58 (t, J = 3.2 Hz, 1H), 5.46 (s, 2H), 2.95 (d, J = 6.3 Hz, 2H), 1.89–1.87 (m, 2H), 1.61 (d, J = 11.9 Hz, 3H), 1.55 (d, J = 12.5 Hz, 3H), 1.46 (d, J = 3.1 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 161.8, 150.5, 144.2, 137.5, 136.9, 132.5, 121.2, 120.7, 119.5, 116.1, 114.4, 111.9, 102.2, 101.1, 54.4, 51.7, 45.2, 44.6, 42.3, 38.6 (2C), 35.8, 33.5, 28.7 (2C), 25.5, 25.7. HRMS (ESI) for C28H30BrN3O3 [M + H]+ calcd 537.1450, found: 537.1452.
General Procedure for the Synthesis of Compounds 6a–b.
To a solution of 3-methoxy-4-(methoxycarbonyl)benzoic acid (300 mg, 1.4 mmol) in DMF (20 mL) were added 2-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, 597.0 mg, 1.57 mmol, 1.1 equiv) and triethylamine (433.3 mg, 4.28 mmol, 2 equiv). Adamantan-1-ylmethanamine (259.5 mg, 1.57 mmol, 1.1 equiv) or 2-(adamantan-1-yl)ethan-1-amine (281.5 mg, 1.57 mmol, 1.1 equiv) was then added after the reaction mixture was stirred at room temperature for 30 min. After the completion of the reaction, the DMF was evaporated under reduced pressure, and the residue was diluted with EtOAc and water. The organic layers were combined, washed with brine, separated, dried over Na2SO4, filtered, and concentrated in vacuo. The oily residue was purified by flash column chromatography to yield the corresponding intermediates.
General Procedure for the Synthesis of Compounds 8a–b.
4-Formyl-2-methoxybenzoate (1g, 5.15 mmol), adamantan-1-ylmethanamine (1.02 g, 6.18 mmol, 1.2 equiv), and 2-(adamantan-1-yl)ethan-1-amine (1.11 g, 6.18 mmol, 1.2 equiv) were dissolved in MeOH (25 mL). The mixture was stirred at room temperature for 2 h. Sodium borohydride (0.49 g, 7.73 mmol, 1.5 equiv) was then added, and the suspension was stirred overnight at room temperature. After completion of the reaction, MeOH was evaporated under reduced pressure, and the residue was diluted with EtOAc and water. The organic layers were combined, washed with brine, separated, dried over Na2SO4, filtered, and concentrated in vacuo. The oily residue was purified by flash column chromatography to yield corresponding intermediates.
General Procedure for the Synthesis of Compounds 9a–b.
8a (750.7 mg, 2.1 mmol) or 8b (721.3 mg, 2.1 mmol) was refluxed in 10 mL of formic acid and 10 mL of formalin for 24 h. The reaction mixture was neutralized with a saturated sodium bicarbonate solution and partitioned between water and ethyl acetate. The aqueous layer was extracted twice more with ethyl acetate, and the combined organic layers were washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography to yield 9a or 9b as a colorless oil.
General Procedure for the Synthesis of Compounds 7a–b, 10a–b.
Solid NaOH (374 mg, 9.36 mmol) was dissolved in a 50% aqueous solution of NH2OH (4 mL) at 0 °C. A solution of ester intermediates 6a–b and 9a–b (2.34 mmol) in 1:1 THF/MeOH (6 mL) was added dropwise with vigorous stirring. Upon complete addition, the ice bath was removed, and the reaction was allowed to stir for 15 min. The reaction was then neutralized with 2 N HCl solution, and the mixture was concentrated under vacuum. The crude product was purified by Combi-Flash reverse column chromatography (ACN/H2O = 0–100%) to yield the compounds 7a–b, and 10a–b after lyophilization.
N4-(Adamantan-1-ylmethyl)-N1-hydroxy-2-methoxyterephthalamide (7a).
White solid; Yield: 95%. 1H NMR (400 MHz, DMSO-d6): δ = 10.70 (s, 1H), 9.14 (s, 1H), 8.39 (s, J = 6.0 Hz 1H), 7.60 (d, J = 8.0 Hz, 1H), 3.89 (s, 3H), 3.30 (m, 2H), 1.94 (s, 3H), 1.68–1.58 (m, 6H), 1.50 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ = 166.3, 163.1, 156.9, 138.4, 130.1, 125.3, 119.7, 110.0, 56.3, 51.1, 40.6, 37.0, 34.9, 28.2. HRMS (ESI) for C20H26N2O4 [M + H]+ calcd 359.1965, found: 359.1959.
N4-(2-(Adamantan-1-yl)ethyl)-N1-hydroxy-2-methoxyterephthalamide (7b).
White solid; Yield: 90% 1H NMR (400 MHz, DMSO-d6): δ = 10.70 (s, 1H), 9.14 (s, 1H), 8.48 (s, 1H), 7.58 (d, J = 8.0 Hz 1H), 7.49 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 3.88 (s, 3H), 3.31–3.26 (m, 2H), 1.94 (s, 3H), 1.70–1.61 (m, 6H), 1.53 (m, 6H), 1.36–1.32 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ = 165.5, 163.0, 156.9, 138.2, 130.1, 125.3, 119.6, 110.9, 56.3, 43.7, 42.3, 37.1, 34.7, 32.0, 28.5. HRMS (ESI) for C21H28N2O4 [M + H]+ calcd 373.2122, found: 373.2118.
4-(((Adamantan-1-ylmethyl)(methyl)amino)methyl)-N-hydroxy-2-methoxybenzamide (10a).
White solid; Yield: 95%. 1H NMR (400 MHz, DMSO-d6): δ = 10.54 (s, 1H), 9.04 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.09 (s, 1H), 7.00 (s, J = 8.0 Hz, 1H), 3.83 (s, 3H), 3.55 (d, 2H), 2.19 (s, 3H), 2.12 (s, 2H), 1.93 (s, 3H), 1.69–1.58 (m, 6H), 1.51–1.48 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ = 163.5, 157.0, 145.1, 130.3, 121.0, 120.6, 111.8, 70.7, 64.5, 55.9, 45.9, 41.1, 37.2, 35.3, 28.3. HRMS (ESI) for C21H30N2O3 [M + H]+ calcd 359.2329, found: 359.2324.
4-(((2-(Adamantan-1-yl)ethyl)(methyl)amino)methyl)-N-hydroxy-2-methoxybenzamide (10b).
White solid; Yield: 90%. 1H NMR (400 MHz, DMSO-d6): δ = 10.56 (s, 1H), 9.06 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.02 (s, 1H), 6.94 (d, J = 8.0 Hz, 1H), 3.83 (s, 3H), 3.48 (s, 2H), 2.51 (s, 2H), 2.33–2.37(m, 2H), 2.11 (s, 3H), 1.90 (s, 3H), 1.67–1.57 (m, 6H), 1.47 (m, 6H), 1.29–1.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ = 163.5, 157.1, 144.3, 130.2, 121.1, 120.9, 111.9, 61.8, 56.0, 51.6, 42.5, 42.4, 41.3, 37.1, 32.0, 28.5. HRMS (ESI) for C22H32N2O3 [M + H]+ calcd 373.2486, found: 373.2424.
4-((5-((((3r,5r,7r)-Adamantan-1-yl)methyl)carbamoyl)-1H-indol-1-yl)methyl)-2-methoxybenzoic Acid (11).
To a solution of 3e (100 mg, 0.21 mmol) in THF/H2O was added LiOH powder (24.6 mg, 1.05 mmol) at room temperature. The mixture was stirred for 30 min at room temperature. After completion of the reaction. The organic solution was removed under vacuum, and the resultant solution was adjusted to pH = 7. The reaction was extracted with DCM. The organic layers were collected and purified by column chromatography to afford 11. White solid; Yield: 95%. 1H NMR (400 MHz, DMSO-d6): δ = 9.12 (s, 1H), 8.34 (s, 1H), 7.15 (d, J = 6.0 Hz, 1H), 7.02 (s, 1H), 6.75 (s, J = 8.0 Hz, 1H), 6.64–6.57 (m, 1H), 3.83 (s, 3H), 3.47–3.33 (m, 2H), 2.12–1.96 (m, 3H), 1.69–1.58 (m, 6H), 1.57–1.21 (m, 6H). MS (ESI+): 473.2 (M + H)+.
N-(((3r,5r,7r)-Adamantan-1-yl)methyl)-1-(4-(2-propylhydrazine-1-carbonyl)benzyl)-1H-indole-5-carboxamide (12).
To a round-bottom flask with compound 11 (100 mg, 0.21 mmol), HATU (119.8 mg, 0.32 mmol), and DIPEA (40.7 mg, 0.32 mmol) in DMF (30 mL) was added 1-chloro-2-propylhydrazine (45.6 mg, 0.42 mmol). The resulting mixture was stirred at room temperature for 12 h. Dichloromethane was added and the mixture was washed with saturated NaHCO3 water (3 × 30 mL) and brine (2 × 30 mL) and dried over anhydrous Na2SO4. Volatiles were removed under a vacuum to get the crude residue, which was purified via Combi-flash chromatography to afford 12 as a white solid powder. Yield: 49.2%. 1H NMR (400 MHz, DMSO-d6): δ = 8.11 (s, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.19 (s, 1H), 7.20–7.17 (m, 2H), 6.72–6.67 (m, 1H), 6.66–6.56 (m, 1H), 6.20–6.18 (m, 1H), 5.36 (s, 2H), 4.44 (s, 1H), 3.70 (s, 3H), 3.17–3.21 (m, 2H), 2.02–1.76 (m, 4H), 1.72–1.65 (m, 9H), 1.61–1.57 (m, 16H), 0.74–0.78 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ = 163.5, 157.0, 145.1, 130.3, 121.0, 120.6, 111.8, 70.7, 64.5, 55.9, 45.9, 41.1, 37.2, 35.3, 28.3. HRMS (ESI) for C31H38N4O2 [M + H]+ calcd 499.3028, found: 499.3032.
Radiosynthesis of [11C]PB94.
[11C]methyl iodide ([11C]CH3I) was trapped in a TRACERlab FX-M synthesizer reactor (General Electric) preloaded with a solution of precursor 3g in anhydrous DMF (2.0 mg/mL, 0.3 mL) and K2CO3 (5 mg). The mixture was stirred at 100 °C for 3 min. Then, the reaction solution was cooled to room temperature, quenched with 0.1% TFA in water (1.5 mL), and purified by reversed-phase semipreparative HPLC (Column: Agilent Eclipse XDB-C18, 5 μm, 250 mm × 9.4 mm, flow rate = 5.0 mL/min, mobile phase = 0.1% TFA in water/0.1% TFA in acetonitrile, 30/70, v/v). The desired fraction ([11C]3g) was collected and then loaded onto a solid-phase exchange (SPE) C-18 cartridge, rinsed with H2O (5 mL). The C-18 cartridge was eluted with 0.6 M NaOH in MeOH/THF (1:1, 1.0 mL) into a vial containing 100 μL of hydroxylamine aqueous solution (50 wt % in H2O). After stirring at room temperature for 5 min, 1 mL of 3 M HCl was added to the mixture. The resulting mixture was injected into a reversed-phase semipreparative HPLC for purification (Column: Agilent Eclipse XDB-C18, 5 μm, 250 mm × 9.4 mm, flow rate = 5.0 mL/min, mobile phase = 0.1% TFA in water/0.1% TFA in acetonitrile, 52/48, v/v). [11C]PB94 was collected and reformulated for PET animal imaging studies. The average time required for the synthesis from the end of cyclotron bombardment to the end of synthesis was approximately 35–40 min. The average radiochemical yield was 6.2–9% (non-decay-corrected to trapped [11C]CH3I, n = 3). Chemical and radiochemical purities were ≥98% with a specific activity of 120 GBq/μmol (EOB). For more analytical data, refer to the Supporting Information (Figure S2).
HDAC 1–11 Enzyme Inhibition Assays.
The HDAC inhibition assay of target compounds was carried out at Nanosyn (Santa Clara CA, United States) using the electrophoretic mobility shift assay. Full-length human recombinant HDAC proteins were expressed in the baculoviral system and purified by affinity chromatography. The peptide substrates were used: FAM-RHKK(Ac)-NH2 for HDAC3, HDAC6, and HDAC8; FITC-H3K27(Ac)-NH2 for HDAC1, HDAC2, and HDAC10; and FAM-RHKK (trifluor-Ac)-NH2 for HDAC4, HDAC5, HDAC7, HDAC9, and HDAC11. Test compounds were diluted in 100% DMSO using 3-fold dilution steps. The final compound concentration in the assay ranged from 10 μM to 0.056 nM. Compound, enzymes (Table S1), and substrate were combined in reaction buffer (100 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES; pH 7.5], 25 mM KCl, 0.1% bovine serum albumin, 0.01% Triton X-100) at 25 °C and quenched by the addition of termination buffer (100 mM HEPES [pH 7.5], 0.01% Triton X-100, 0.05% sodium dodecyl sulfate). The fluorescence intensity of the electrophoretically separated deacetylated product and substrate peptide was measured and analyzed using the LabChip 3000 microfluidic electrophoresis instrument (PerkinElmer/Caliper Life Sciences). The 50% inhibitory concentration (IC50) values of inhibitors were determined by fitting the percent inhibition curves with a 4-parameter dose–response model using XLfit 4 software (IDBS). Reference compounds, TSA were tested in an identical manner.
Defatty Acylation of SHMT2 Assays.
The procedure of defatty acylation of SHMT2 assays refers to a previous method.23,42 Briefly, HEK293T cells were incubated with 50 μM Alk14 and inhibitors at various concentrations for 3 h. The cells were harvested and lysed in SDS lysis buffer (50 mM triethanolamine, 150 mM NaCl, and 4% SDS, pH 7.4) with a 1:100 protease inhibitor cocktail and 1:1000 nuclease for 15 min. Then, the cell lysates were diluted with HEPES buffer (50 mM HEPES, 150 mM NaCl, 1% NP-40, pH 7.4), and concentrated for 30 min at 21000g. Meanwhile, magnetic streptavidin beads (10 μg) were suspended in a HEPES buffer (100 μL), after which Biotin-N3 (5 μL, 5 mM in DMF) was added. The mixture was shaken at 37 °C for 30 min, and then the supernatant was removed; the beads were washed with HEPES buffer (1 × 100 μL). The mixture was shaken for 1 h at 37 °C, and the supernatant was removed. Hydroxylamine in a HEPES buffer (100 μL, 0.5 M) was added, and the mixture was then shaken for 30 min. The supernatant was removed, and the beads were washed with HEPES buffer (3 × 500 μL). The remaining beads were incubated at 95 °C with 20 μL of 4% SDS lysis buffer and 4 μL of 6× loading buffer for 10 min. The eluants were further analyzed by SDS-PAGE and Western blot for SHMT2.
qPCR Analysis.
A one-step real-time PCR was performed using an iTaq Universal SYBR Green One-Step Kit (BioRad 1725150) in a Thermofisher 7500 fast thermocycler. The primer sequences are as follows:
GAPDH: CATCACTGCCACCCAGAAGACTG (forward) and ATGCCAGTGAGCTTCCCGTTCAG (reverse); HDAC11: ATGGGGCAAGGTGATCAACT (forward) and AGGACCACTTCAGCTCGTTG (reverse). Mouse brain tissue (contralateral somatosensory cortex) was harvested 1 week after Sham or CCI surgery, followed by RNA isolation using Trizol. Melting curve and Ct value were used for quantification. Gapdh was used as the internal control for normalization of each sample.
PET/CT Imaging in Rodents.
The PET/CT imaging studies in rodents in this work were referred to our previous work.43,44 Micro-PET/CT imaging was performed in two groups: male C57BL/6 mice (6 months old), used for general brain imaging and blocking studies. After intravenous injection of [11C]PB94 (3.7–5.6 Mbq per animal), a 60 min dynamic PET acquisition was performed followed by a 10 min CT scan. PET data were reconstructed using a 3D-MLEM method, resulting in a full width at half-maximum resolution of 1 mm. These files were imported and analyzed using PMOD (PMOD 4.01, PMOD Technologies Ltd., Zurich, Switzerland).
Chronic Constriction Injury (CCI).
The mice were anesthetized with 2.5–3% isoflurane in an anesthesia induction chamber. The left lower extremity was prepared with an alcohol swab. A 0.5–1 cm incision was made using a blade on the left lower extremity. The left side of the sciatic nerve was exposed in the midthigh. Four ligatures using 6.0 mm chronic gut sutures were loosely placed around the exposed sciatic nerve with a 1.0–1.5 mm interval between each ligature. Skin incision was closed with two 6–0 Vicryl sutures (Ethicon, Somerville, NJ)
Mechanical Withdrawal Threshold.
Mice (C57/BL6, male, 6-month-old) were habituated and placed on a platform with a clear chamber individually 30 min daily for 3 consecutive days. Mechanical paw withdrawal thresholds (PWTs) were carried out using calibrated manual Von Frey filaments (Stoelting, Kiel, Wl). A Von Frey monofilament was applied to the plantar surface of the hind paw, and a logarithmic scale of force was delivered for 2 s. The positive response was recorded when the mice were withdrawn or shacked their paws during the stimulation. A negative response was followed by testing with the next larger filament. The “up-down” methodological approach was used to calculate the mechanical sensitivity. All CCI mice were tested before surgery (baseline) and 3, 5, 7, 10, and 14 days after surgery. On day 21, all of the mice received a final test for dose–response evaluation: PB94 treatment. PB94 was dissolved in DMSO/Tween80/saline (1/1/8, v/v/v). On day 21 after surgery, CCI mice were randomly divided into three groups (n = 4 for each group) and treated with an IP injection of 3, 6, and 10 mg/kg, respectively. PWTs of CCI mice were measured every 30 min during a 3 h period after PB94 administration.
Hind Paw Withdrawal Latency.
Mice were placed on a preheated glass platform (28–29 °C) and clear Plexiglas cubicles to acclimate to the testing room 30 min daily for 3 consecutive days before the testing. A radiant heat source emitted from underneath the glass and focused on the middle of the hindpaws of mice. Paw withdrawal latency was defined as the time (seconds) from the initiation of heat exposure to hind paw withdrawal. A cutoff time was set at 20 s to avoid tissue damage.
Iba-1 Staining.
Mice were transcranially perfused with ice-cold phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. Extracted mouse brains were stored at 4 °C with 4% PFA fixation for 2 days. After being washed in 1× PBS, brains were sliced at 40 μm thickness using a Leica vibratome (VT 1000s). The desired slices were bathed in a blocking buffer containing 0.03% Tween-20 and 5% BSA for 1 h at room temperature. The primary antibody (Iba1; 1:1000; Wako) in PBS was incubated at 4 °C overnight. After three times of 1× PBS washing, the slices were incubated with the second antibody of antirabbit Alexa488 (1:2000; Invitrogen) for 1 h at room temperature. Using an Olympus microscope, images were taken at 4× and 20× magnification for analysis. Images were analyzed by using ImageJ (NIH open-source software).
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by Cure Alzheimer’s Fund. The authors are grateful to the Lab staff in Athinoula A. Martinos Center Radiopharmacy for assistance in radiochemistry.
ABBREVIATIONS USED
- HDACs
histone deacetylases
- ZBG
zinc-binding group
- CCI
chronic constriction injury
- PET
positron emission tomography
- AD
Alzheimer’s disease
- BBB
brain–blood barrier
- CNS
central nervous system
- CT
computed tomography
- DMSO
dimethyl sulfoxide
- EOB
end of bombardment
- EOS
end of synthesis
- HPLC
high-performance liquid chromatography
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01491.
Molecular formula string (CSV)
Acute toxicity assays of PB94; in vitro metabolic stability evaluation of PB94; mice brain/plasma PK studies of PB94; the off-target binding of PB94 (Table S2); and 1H NMR, 13C NMR, HRMS, and HPLC spectra for the synthesized compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.3c01491
Contributor Information
Ping Bai, Department of Respiratory and Critical Care Medicine, Targeted Tracer Research and Development Laboratory, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; Institute of Respiratory Health, Targeted Tracer Research and Development Laboratory, Frontiers Science Center for Disease-Related Molecular Network and Precision Medicine Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; The Research Units of West China, Chinese Academy of Medical Sciences, West China Hospital, Chengdu, Sichuan 610041, China; State Key Laboratory of Respiratory Health and Multimorbidity, West China Hospital, Chengdu, Sichuan 610041, China.
Yan Liu, Athinoula A. Martinos Center for Biomedical Imaging, Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Liuyue Yang, Department of Anesthesia, Critical Care and Pain Medicine Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Weihua Ding, Department of Anesthesia, Critical Care and Pain Medicine Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Prasenjit Mondal, Genetics and Aging Research Unit, McCance Center for Brain Health, MassGeneral Institute for Neurodegenerative Disease, Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Na Sang, Department of Respiratory and Critical Care Medicine, Targeted Tracer Research and Development Laboratory, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; Institute of Respiratory Health, Targeted Tracer Research and Development Laboratory, Frontiers Science Center for Disease-Related Molecular Network and Precision Medicine Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; The Research Units of West China, Chinese Academy of Medical Sciences, West China Hospital, Chengdu, Sichuan 610041, China; State Key Laboratory of Respiratory Health and Multimorbidity, West China Hospital, Chengdu, Sichuan 610041, China.
Gang Liu, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, People’s Republic of China.
Xiaoxia Lu, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, People’s Republic of China.
Thanh Tu Ho, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Yanting Zhou, Department of Respiratory and Critical Care Medicine, Targeted Tracer Research and Development Laboratory, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; Institute of Respiratory Health, Targeted Tracer Research and Development Laboratory, Frontiers Science Center for Disease-Related Molecular Network and Precision Medicine Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; The Research Units of West China, Chinese Academy of Medical Sciences, West China Hospital, Chengdu, Sichuan 610041, China; State Key Laboratory of Respiratory Health and Multimorbidity, West China Hospital, Chengdu, Sichuan 610041, China.
Rui Wu, Department of Respiratory and Critical Care Medicine, Targeted Tracer Research and Development Laboratory, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; Institute of Respiratory Health, Targeted Tracer Research and Development Laboratory, Frontiers Science Center for Disease-Related Molecular Network and Precision Medicine Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; The Research Units of West China, Chinese Academy of Medical Sciences, West China Hospital, Chengdu, Sichuan 610041, China; State Key Laboratory of Respiratory Health and Multimorbidity, West China Hospital, Chengdu, Sichuan 610041, China.
Vishal C. Birar, Athinoula A. Martinos Center for Biomedical Imaging, Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States
Moses Q. Wilks, Gordon Center for Medical Imaging, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States
Rudolph E. Tanzi, Genetics and Aging Research Unit, McCance Center for Brain Health, MassGeneral Institute for Neurodegenerative Disease, Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States
Hening Lin, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States; Howard Hughes Medical Institute; Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Can Zhang, Genetics and Aging Research Unit, McCance Center for Brain Health, MassGeneral Institute for Neurodegenerative Disease, Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Weimin Li, Department of Respiratory and Critical Care Medicine, Targeted Tracer Research and Development Laboratory, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; Institute of Respiratory Health, Targeted Tracer Research and Development Laboratory, Frontiers Science Center for Disease-Related Molecular Network and Precision Medicine Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China; The Research Units of West China, Chinese Academy of Medical Sciences, West China Hospital, Chengdu, Sichuan 610041, China; State Key Laboratory of Respiratory Health and Multimorbidity, West China Hospital, Chengdu, Sichuan 610041, China.
Shiqian Shen, Department of Anesthesia, Critical Care and Pain Medicine Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Changning Wang, Athinoula A. Martinos Center for Biomedical Imaging, Department of Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts 02129, United States.
REFERENCES
- (1).Bird A Perceptions of epigenetics. Nature 2007, 447 (7143), 396–398. [DOI] [PubMed] [Google Scholar]
- (2).Seto E; Yoshida MJ Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor Perspect. Biol. 2014, 6 (4), No. a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ruijter AJ; van Gennip AH; Caron HN; Kemp S; van Kuilenburg AB Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shahbazian MD; Grunstein MJ Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [DOI] [PubMed] [Google Scholar]
- (5).Ruijter AJ; Gennip AH; Caron HN; Kemp S; Kuilenburg AB Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370 (3), 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gao L; Cueto MA; Asselbergs F; Atadja P Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277 (28), 25748–25755. [DOI] [PubMed] [Google Scholar]
- (7).Liu S-S; Wu F; Jin Y-M; Chang W-Q; Xu T-MJB pharmacotherapy, HDAC11: a rising star in epigenetics. Biomed. Pharmacother. 2020, 131, No. 110607. [DOI] [PubMed] [Google Scholar]
- (8).Buglio D; Khaskhely NM; Voo KS; Martinez-Valdez H; Liu Y-J; Younes AJB HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood 2011, 117 (10), 2910–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Deubzer HE; Schier MC; Oehme I; Lodrini M; Haendler B; Sommer A; Witt OJ HDAC11 is a novel drug target in carcinomas. Int. J. Cancer Res. 2013, 132 (9), 2200–2208. [DOI] [PubMed] [Google Scholar]
- (10).Sun L; Marin de Evsikova C; Bian K; Achille A; Telles E; Pei H; Seto E Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine 2018, 33, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sun L; Telles E; Karl M; Cheng F; Luetteke N; Sotomayor EM; Miller RH; Seto EJ Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 2018, 1 (5), No. e201800039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bryant DT; Landles C; Papadopoulou AS; Benjamin AC; Duckworth JK; Rosahl T; Benn CL; Bates GP Disruption to schizophrenia-associated gene Fez1 in the hippocampus of HDAC11 knockout mice. Sci. Rep. 2017, 7 (1), No. 11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kumar V; Kaur S; Kapil L; Singh C; Singh A HDAC11: A novel inflammatory biomarker in Huntington’s disease. EXCLI J. 2022, 21, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nickel FT; Seifert F; Lanz S; Maihöfner CJ Mechanisms of neuropathic pain. Eur. Neuropsychopharmacol. 2012, 22 (2), 81–91. [DOI] [PubMed] [Google Scholar]
- (15).Colloca L; Ludman T; Bouhassira D; Baron R; Dickenson AH; Yarnitsky D; Freeman R; Truini A; Attal N; Finnerup NB; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3 (1), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Campbell JN; Meyer RA Mechanisms of neuropathic pain. Neuron 2006, 52 (1), 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Descalzi G; Ikegami D; Ushijima T; Nestler EJ; Zachariou V; Narita MJ Epigenetic mechanisms of chronic pain. Trends Neurosci. 2015, 38 (4), 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bai G; Wei D; Zou S; Ren K; Dubner RJ Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol. Pain 2010, 6, 1744–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Denk F; Huang W; Sidders B; Bithell A; Crow M; Grist J; Sharma S; Ziemek D; Rice AS; Buckley NJ; McMahon SB HDAC inhibitors attenuate the development of hypersensitivity in models of neuropathic pain. Pain 2013, 154 (9), 1668–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sanna MD; Guandalini L; Romanelli MN; Galeotti NJ Behavior, The new HDAC1 inhibitor LG325 ameliorates neuropathic pain in a mouse model. Pharmacol. Biochem. Behav. 2017, 160, 70–75. [DOI] [PubMed] [Google Scholar]
- (21).Matsushita Y; Araki K; Omotuyi O; Mukae T; Ueda HJB HDAC inhibitors restore C-fibre sensitivity in experimental neuropathic pain model. Br. J. Pharmacol. 2013, 170 (5), 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Martin MW; Lee JY; Lancia DR Jr; Ng PY; Han B; Thomason JR; Lynes MS; Marshall CG; Conti C; Collis A; et al. Discovery of novel N-hydroxy-2-arylisoindoline-4-carboxamides as potent and selective inhibitors of HDAC11. Bioorg. Med. Chem. Lett. 2018, 28 (12), 2143–2147. [DOI] [PubMed] [Google Scholar]
- (23).Son SI; Su D; Ho TT; Lin H Garcinol Is an HDAC11 Inhibitor. ACS Chem. Biol. 2020, 15 (11), 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang C; Schroeder FA; Wey H-Y; Borra R; Wagner FF; Reis S; Kim SW; Holson EB; Haggarty SJ; Hooker JM In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57 (19), 7999–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Strebl MG; Campbell AJ; Zhao W-N; Schroeder FA; Riley MM; Chindavong PS; Morin TM; Haggarty SJ; Wagner FF; Ritter T; Hooker JM HDAC6 brain mapping with [18F] bavarostat enabled by a Ru-mediated deoxyfluorination. ACS Cent. Sci. 2017, 3 (9), 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kozikowski AP; Shen S; Pardo M; Tavares MT; Szarics D; Benoy V; Zimprich CA; Kutil Z; Zhang G; Bařinka C; et al. Brain penetrable histone deacetylase 6 inhibitor SW-100 ameliorates memory and learning impairments in a mouse model of fragile X syndrome. ACS Chem. Neurosci. 2019, 10 (3), 1679–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shen S; Picci C; Ustinova K; Benoy V; Kutil Z; Zhang G; Tavares MT; Pavlicek J; Zimprich CA; Robers MB; Van Den Bosch L; Barinka C; Langley B; Kozikowski AP Tetrahydroquinoline-Capped Histone Deacetylase 6 Inhibitor SW-101 Ameliorates Pathological Phenotypes in a Charcot-Marie-Tooth Type 2A Mouse Model. J. Med. Chem. 2021, 64 (8), 4810–4840. [DOI] [PubMed] [Google Scholar]
- (28).Butler KV; Kalin J; Brochier C; Vistoli G; Langley B; Kozikowski AP Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132 (31), 10842–10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bai P; Mondal P; Bagdasarian FA; Rani N; Liu Y; Gomm A; Tocci DR; Choi SH; Wey HY; Tanzi RE; Zhang C; Wang C Development of a potential PET probe for HDAC6 imaging in Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12 (10), 3891–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Watson PR; Bai P; Wang C; Cragin AD; Hooker JM; Christianson DW Aromatic Ring Fluorination Patterns Modulate Inhibitory Potency of Fluorophenylhydroxamates Complexed with Histone Deacetylase 6. Biochemistry 2022, 61 (18), 1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yue K; Sun S; Jia G; Qin M; Hou X; Chou CJ; Huang C; Li X First-in-Class Hydrazide-Based HDAC6 Selective Inhibitor with Potent Oral Anti-Inflammatory Activity by Attenuating NLRP3 Inflammasome Activation. J. Med. Chem. 2022, 65 (18), 12140–12162. [DOI] [PubMed] [Google Scholar]
- (32).Frühauf A; Meyer-Almes FJ Non-Hydroxamate Zinc-Binding Groups as Warheads for Histone Deacetylases. Molecules 2021, 26 (17), No. 5151, DOI: 10.3390/molecules26175151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cao J; Sun L; Aramsangtienchai P; Spiegelman NA; Zhang X; Huang W; Seto E; Lin H HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (12), 5487–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Charron G; Zhang MM; Yount JS; Wilson J; Raghavan AS; Shamir E; Hang HC Robust fluorescent detection of protein fatty-acylation with chemical reporters. J. Am. Chem. Soc. 2009, 131 (13), 4967–4975. [DOI] [PubMed] [Google Scholar]
- (35).Duvic M; Vu JJE Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16 (7), 1111–1120. [DOI] [PubMed] [Google Scholar]
- (36).Gopal YV; Arora TS; Van Dyke MW biology, Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem. Biol. 2007, 14 (7), 813–823. [DOI] [PubMed] [Google Scholar]
- (37).Villagra A; Cheng F; Wang HW; Suarez I; Glozak M; Maurin M; Nguyen D; Wright KL; Atadja PW; Bhalla K; Pinilla-Ibarz J; Seto E; Sotomayor EM The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10 (1), 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bennett GJ; Xie Y-K A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33 (1), 87–107. [DOI] [PubMed] [Google Scholar]
- (39).Condylis C; Lowet E; Ni J; Bistrong K; Ouellette T; Josephs N; Chen JL Context-Dependent Sensory Processing across Primary and Secondary Somatosensory Cortex. Neuron 2020, 106 (3), 515–525.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Inoue K; Tsuda M Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19 (3), 138–152. [DOI] [PubMed] [Google Scholar]
- (41).Zhu X; Tang HD; Dong WY; Kang F; Liu A; Mao Y; Xie W; Zhang X; Cao P; Zhou W; Wang H; Farzinpour Z; Tao W; Song X; Zhang Y; Xue T; Jin Y; Li J; Zhang Z Distinct thalamocortical circuits underlie allodynia induced by tissue injury and by depression-like states. Nat. Neurosci. 2021, 24 (4), 542–553. [DOI] [PubMed] [Google Scholar]
- (42).Son SI; Cao J; Zhu CL; Miller SP; Lin H Activity-Guided Design of HDAC11-Specific Inhibitors. ACS Chem. Biol. 2019, 14 (7), 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bai P; Lan Y; Wang H; Liu Y; Striar R; Yuan G; Afshar S; Zagaroli JS; Tocci DR; Langan AG; Wang C Synthesis and characterization of a positron emission tomography imaging probe selectively targeting the second bromodomain of bromodomain protein BRD4. Bioconjugate Chem. 2021, 32 (8), 1711–1718. [DOI] [PubMed] [Google Scholar]
- (44).Bai P; Lan Y; Patnaik D; Wang H; Liu Y; Chen Z; Yuan G; Afshar S; Striar R; Zagaroli JS; Tocci DR; Langan AG; Haggarty SJ; Wang C Design, synthesis, and evaluation of thienodiazepine derivatives as positron emission tomography imaging probes for bromodomain and extra-terminal domain family proteins. J. Med. Chem. 2021, 64 (19), 14745–14756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.