

Abstract
A novel series of cyanopyridines 7a-j were synthesized via a one-pot multicomponent reaction of arylidene 4 with ammonium acetate 5 and respective methylaryl/heterylketones 6a-j in ethanol using vanillin as a natural starting material. Moreover, the regioselective alkylation reaction was studied by the treatment of cyanopyridines 7a-f and 7j with CH3I in the presence of K2CO3 in DMF to afford O-methylcyanopyridines 8a-g (major) and N-methylcyanopyridines 9a-g (minor), whereas bipyridine 7h gave bipyridinium iodide salt 10. All of the designed cyanopyridines were evaluated as anti-breast cancer (MCF-7) cell lines via PIM Kinase inhibitory activity, and the results displayed that some of them showed high activities, especially compounds 7h and 8f, which showed excellent activities against MCF-7 with IC50 values of 1.89 and 1.69 μM, respectively, more potent than the reference drug doxorubicin. Mechanistically, compounds 7h and 8f exhibited strong in vitro PIM-1 kinase inhibitory activity with an IC50 of 0.281 and 0.58 μM, respectively, compared to the reference staurosporine. Moreover, compound 7h arrested the tumor cells at the S phase and caused cell death mainly by inducing early and late apoptosis. Molecular docking studies against PIM-1 revealed good binding modes of the synthesized compound and showed agreement with the biological results.
Graphical abstract

Keywords: Cyanopyridines, Regioselective, Anticancer, PIM inhibitors, Molecular docking studies, Breast cancer
Introduction
With an estimated 15 million deaths per year by 2030, cancer poses a significant crisis to global public health and healthcare systems. Initially regarded as a genetic disease, it is now widely recognized that cancer is a complex condition influenced by both genetic and epigenetic factors [1]. The intricate signaling networks involved in cancer necessitate the perturbation of multiple targets simultaneously, as cancer cells can employ diverse compensatory pathways for their survival [2, 3].
PIM-1, a serine/threonine kinase belonging to the PIM (proviral insertion site in Moloney murine leukemia virus) kinase family, is considered a proto-oncogene. This family includes two other isoforms, PIM-2 and PIM-3 [4, 5]. PIM-1 exerts a critical role in cell signaling pathways and various cellular functions, including cell cycle regulation, cell survival, proliferation, apoptosis, and drug resistance. It achieves this by phosphorylating and regulating the activity of numerous proteins involved in these processes [6–9]. PIM-1 has demonstrated higher expression levels in various solid cancers, including prostate, colon, hepatic, pancreatic, and breast cancers, as well as hematological cancers like leukemia, multiple myeloma, and diffuse large B cell lymphomas (DLBCL) [10–16]. It has also been observed in circulating tumor cells (CTCs) from patients with metastatic castration-resistant prostate cancer (mCRPC) [17]. Notably, its expression is absent in benign tumors. Overexpression of PIM-1 has been linked to cancer initiation and progression through three significant mechanisms: inhibiting apoptosis, promoting cell proliferation [18, 19], and promoting genomic instability [20]. In vitro studies have shown that PIM-1 overexpression enhances tumor growth and confers resistance to drug-induced apoptosis in cancer cells [21]. The degree of PIM-1 overexpression correlates with tumor grade and neoplastic transformation [22]. Furthermore, inhibiting these isoforms of kinases in mouse experimental models did not exhibit significant side effects [23]. Moreover, specific, and potent inhibitors of PIM-1 kinase have been identified, which have been shown to induce apoptotic cell death, sensitize cancer cells to chemotherapy, and synergize with other anti-tumor agents.
On the other hand, many of the cyanopyridnes were reported as promising anticancer agents with high PIM-1 kinase inhibitory activity, such as compound A (IC50 = 0.05 μM) [24], compound B (IC50 = 0.94 μM) [25], compound C (IC50 = 0.46 μM) [26], compound D (IC50 = 0.019 μM) [27], compound E (IC50 = 0.63 μM) [28], and compound F (IC50 = 0.13 μM) [29] (Fig. 1). Therefore, PIM-1 kinase represents an attractive therapeutic target [20, 30], and inhibiting it presents an intriguing strategy for treating various tumor types by inducing apoptosis and suppressing proliferation [22].
Fig. 1.

Some of the cyanopyridines have high PIM-1 kinase inhibitory activity
Thus, from the above mention information and in continuation of our previous work [31–36], we designed and synthesized a novel series of cyanopyridines 7a-j via one-pot multicomponent reaction, O-methylcyanopyridines 8a-g, and N-methylcyanopyridines 9a-g via alkylation reaction. The in vitro cytotoxicity of the target compounds were screened against the breast cancer (MCF-7) cell line using the MTT assay method. Furthermore, the PIM-1 kinase inhibitory activity for the most potent cyanopyridines (7h and 8f) was investigated. The most potent compound 7h was investigated for apoptosis and cell cycle progression. The molecular docking was studied against PIM-1 (PDB ID: 2OH4). Finally, the study of the structure–activity relationship (SAR) was determined.
Results and discussion
Chemistry
Herein, we used a simple method for the synthesis of O-alkyl vanillin 3 [37, 38] from vanillin 1 (an available and inexpensive natural product) by stirring it with ethanolic NaOEt to give vanillin sodium salt 2, which was isolated and subjected to react with 2-chloro-N-phenylacetamide as an active halo-compound in dry dimethylformamide at 70 οC. The synthetic O-alkyl vanillin 3 undergoes Knoevenagel condensation reaction when it is reacted with ethyl cyanoacetate in the presence of TEA as a catalyst to afford the arylidene 4 in ethanol (Scheme 1).
Scheme 1.

Synthesis of O-alkyl vanillin 3 and arylidene 4
The chemical structures of the newly synthesized compounds were proven by the spectral (IR, 1H NMR) and elemental analyses. The IR spectrum of vanillin sodium salt 2 showed absorption bands corresponding to CH aromatic at 3052 cm−1, CH aliphatic at 2971, 2936 cm−1, and C = O of the formyl group at 1669 cm−1. Its 1HNMR spectrum showed the disappearance of the OH group and the appearance of two singlet signals corresponding to CHO and OCH3 at 9.31 and 3.71 ppm, as well as one singlet signal at 7.02 ppm and two doublet signals at 7.15 and 6.31 ppm due to aromatic protons with a coupling constant of J = 7.9 Hz. The 13C NMR of vanillin sodium salt 2 showed two signals at 187.1 and 55.1 ppm corresponding to C = Oformyl and OCH3, respectively. As well as the aromatic sp2 carbons are characterized by signals at 170.6, 151.7, 130.9, 118.7, 117.8, and 108.0 ppm.
Moreover, a novel series of cyanopyridines 7a-j were synthesized via one-pot multicomponent reaction (MCR) of arylidene 4 with ammonium acetate 5 and respective methylaryl/heterylketones 6a-j, namely acetophenone, 3-methoxyacetophenone, 4-methoxyacetophenone, 4-chloroacetophenone, 4-bromoacetophenone, 2-acetylfuran, 2-acetylthiophene, 3-acetylpyridine, 4-acetylpyridine, and/or 2-acetylnaphthalene, respectively, in ethanol (Scheme 2).
Scheme 2.

The synthesis of cyanopyridines 7a-j via one-pot multicomponent reaction
The structures of cyanopyridines 7a-j were confirmed by spectral analyses, which showed the absence of CHolefinic group with the appearance of a new NH group and aryl group. For example, the IR spectrum of cyanopyridine 7a showed absorption bands corresponding to NH at 3390 cm−1, CH aromatic at 3076 cm−1, CH aliphatic at 2969 and 2917 cm−1, C≡N at 2217 cm−1, and C = O amide at 1690 cm−1. Its 1H NMR spectrum showed four singlet signals corresponding to 2NH, CH2, and OCH3 at 12.62, 10.02, 4.81, and 3.92 ppm, beside CH of aromatic protons as two singlet signals at 7.42, 6.86 ppm, respectively; three doublets at 7.90, 7.63, and 7.15 with a coupling constant of 6.8, 7.8, and 8.4 Hz, respectively; two triplet signals 7.34, and 7.09 with a coupling constant of 7.3 Hz, and one multiplet 7.57–7.53 ppm. Its 13C NMR revealed the appearance of four signals of two C = Oamide, CH2, and OCH3 at 166.7, 162.6, 68.6, and 56.4 ppm, respectively; and the rest of aromatic sp2 carbons appeared at 159.8, 151.7, 149.8, 149.4, 138.7, 132.9, 131.5, 129.8, 129.3, 129.2, 128.1, 124.2, 121.8, 120.0, 117.2, 114.4, 113.1, 106.6, and 90.3 ppm.
The plausible reaction mechanism for the synthesis of pyridines 7a-j can be explained by the formation of the arylidene 4 via the Knoevenagel condensation reaction of O-alkyl vanillin 3 with ethyl cyanoacetate in basic medium. The arylidene 4 undergoes a nucleophilic addition reaction by attacking the enolate ion of the activated methylaryl/heterylketone 6a-j in the presence of ammonium acetate to afford the intermediate IV, followed by nucleophilic addition of ammonia (produced from the decomposition of CH3CO2NH4) on the C = Oester group to form the intermediate V with the liberation of the EtOH molecule. The intermediate V undergoes the intramolecular cyclization reaction of the amino group to another C = O group via a Michael addition reaction to give intermediate VI, which is easily aromatized by the dehydration and deprotonation to afford the target products (Scheme 3).
Scheme 3.

The suggestion reaction mechanism for the formation of the cyanopyridines 7a-j
Furthermore, the treatment of cyanopyridines 7a-f and 7j with methyl iodide in the presence of potassium carbonate as a catalyst undergoes regioselective alkylation reactions in dry DMF to afford two new series of O-methylcyanopyridines 8a-g and N-methylcyanopyridines 9a-g (Scheme 4). The products were separated by thin layer chromatography using a mixture of chloroform and petroleum ether (9:1) as a suitable eluent; the O-methylcyanopyridines were separated as a major product (Rf = 0.10:0.20), whereas the N-methylcyanopyridines were separated as a minor product (Rf = 0.23:0.47) (see Experimental part).
Scheme 4.

Synthesis of O-methylcyanopyridines 8a-g and N-methylcyanopyridines 9a-g
The IR spectrum of O-methylcyanopyridine 8b (as an example) showed the appearance of an absorption band corresponding to NH, CH aromatic, CH aliphatic, C≡N and C = O amide at 3396, 3067, 2944–2843, 2218, and 1693 cm−1, respectively. Its 1H NMR spectrum showed the disappearance of the NH signal and the appearance of five singlet signals at 10.13, 4.83, 4.15, 3.93, and 3.86 ppm due to NH, CH2, and three OCH3, respectively. In addition, Ar–H protons split into two singlets at 7.44 and 7.34 ppm, one doublet with a coupling constant of 4.2 Hz at 7.65 ppm, and two multiplets at 7.82–7.78 and 7.16–7.10 ppm, respectively. Also, its 13C NMR spectrum showed C = Oamide, CN, CH2, and three OCH3 signals at 166.7, 116.1, 68.4, 56.3, 55.7, and 54.9 ppm, respectively, and the residual signals appeared at 164.7, 160.1, 157.3, 156.4, 149.5, 149.0, 149.4, 138.8, 138.6, 130.4, 129.4, 129.2, 124.1, 121.9, 120.3, 119.9, 116.5, 114.2, 114.1, 113.3, and 92.5 ppm assigned to sp2 aromatic carbons.
Also, the N-methylcyanopyridine of 9b (as another example) showed in the IR spectrum absorption bands corresponding to NH at 3392 cm−1, CH aromatic at 3072 cm−1, CH aliphatic at 2960, 2940, and 2839, C≡N at 2219 cm−1, and C = O amide at 1684 cm−1. Its 1H NMR spectrum revealed the disappearance of one of NH and the appearance of five singlet signals corresponding to NH, CH2, two OCH3, and NCH3 at 10.14, 4.82, 3.89, 3.82, and 3.35 ppm, respectively. Beside the aromatic protons appeared in the range 7.84–6.53 ppm. As well, its 13C NMR revealed the appearance of six signals of C = Oamide, CN, CH2, two OCH3, and NCH3 at 166.6, 116.2, 68.3, 56.2, 55.8, and 35.1 ppm, respectively; and the residual of aromatic sp2 carbons appeared at 161.5, 159.7, 157.6, 154.4, 149.9, 149.3, 138.8, 135.8, 130.4, 129.2, 128.9, 124.1, 121.8, 120.9, 119.8, 117.4, 114.4, 113.9, 112.7, 108.8, and 98.1 ppm. The Dept-135 NMR spectrum of 9b confirmed his structure because it showed the appearance of CH2 in a negative direction at 68.6 ppm and the fourteen Ar–H carbons and two OCH3 and NCH3 appeared in a positive direction at 130.4, 129.2, 124.1, 121.8, 120.9, 119.8, 116.2, 114.4, 113.8, 112.7, 108.8, 56.2, 55.8, and 35.1 ppm, respectively.
On the other hand, the alkylation of bipyridine 7 h, which contains two pyridine rings in the same conditions, undergoes two folds of CH3I on the OH of the cyanopyridine ring and nitrogen on another one, giving only one product, bipyridinium iodide salt 10 (Eq. 1).
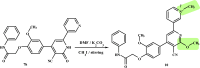 |
1 |
Synthesis of pyridinium iodide salt 10.
The IR spectrum of bipyridinium iodide salt 10 showed the absorption bands corresponding to the NH group at 3178 cm−1, CH aromatic group at 3061 cm−1, CH aliphatic group at 2952, and 2830 cm−1, Cyano group at 2220 cm−1 and C = O amide group at 1689 cm−1. Its 1H NMR spectrum revealed the appearance of five singlet signals at 10.21, 4.86, 4.48, 4.23, and 3.92 ppm due to NH, CH2, N+CH3, and two OCH3, respectively. The aromatic protons were divided into three singlet signals at 9.81, 8.15, and 7.48 ppm, three doublet signals at 9.37, 9.11, and 7.64 ppm with a coupling constant of 8.1, 5.8, and 7.8 Hz, respectively, one triplet at 8.31 ppm with a coupling constant of 7.0 Hz, and two multiplets at 7.40–7.32 and 7.19–7.08 ppm. The 13C NMR spectrum of bipyridinium iodide salt 10 appeared six signals at 166.6, 115.8, 68.5, 56.6, 55.6, and 48.9 for C = Oamidic, CN, CH2, two OCH3, and N+CH3, respectively. The residual signals appeared at 165.1, 157.2, 150.9, 150.0, 149.6, 146.6, 145.1, 143.1, 138.8, 136.5, 129.1, 128.8, 128.2, 124.1, 122.2, 119.9, 115.5, 114.5, 113.5, and 95.2 ppm due to aromatic sp2 carbons.
The structure of compound 10 was confirmed by the NOESY NMR spectrum. The pattern of NOE contacts showed the correlation peaks between the protons of compound 10 which observed strong interaction between OCH3 with H-2, N+CH3 with H-2' (Py) & H-6' (Py), CH2 with NH & H-5, H-3 (Cy-Py) with H-6 & H-2' (Py), NH with H-Ph and H-4' (Py) with H-5' (Py) (see SI, Fig. 2).
Fig. 2.

The correlation peaks between the protons by the NOESY NMR spectrum of compound 10
Biology
Anticancer activity
Anti-proliferative activity:
In the present study, all synthesized cyanopyridine hybrids were subjected to in vitro cytotoxicity testing using the MTT assay method against MCF-7 human cancer. Notably, our findings revealed the existence of distinct groups with differing levels of activity when compared to the reference drug, doxorubicin. Firstly, a group of compounds displaying remarkably potent activity that far surpassed the efficacy of doxorubicin’s IC50 value of 11.49 ± 0.47 µM, the established reference drug. This group exhibited a superior cytotoxic effect, including 3-cyano-2-methoxypyridine derivatives, particularly compounds 8f and 10 containing naphthyl and 3-pyridyl moieties, respectively, which demonstrated the highest cytotoxic activity with IC50 values of 1.69 ± 0.07 µM and 2.13 ± 0.09 µM, respectively. The cyano-2-methoxypyridine derivative with 4-methoxyphenyl group 8c displayed significant antiproliferative activity with IC50 = 3.74 ± 0.15 µM but less than the naphthyl and 3-pyridyl derivatives. Moreover, 3-cyanopyrid-2-one derivatives bearing 3-pyridyl 7h, furan 7f, and thiophenyl 7g groups displayed significant cytotoxicity, with IC50 values of 1.89 ± 0.08 µM, 3.98 ± 16 µM and 1.92 ± 0.08 µM, respectively. Additionally, the 3-cyano-1-methylpyrid-2-one derivative 9d, containing a 4-chlorophenyl group, exhibited notable cytotoxicity, with an IC50 value of 2.05 ± 0.08 µM (Table 1, Fig. 3).
Table 1.
The IC50 (µM) of antiproliferative assays of the target cyanopyridine derivatives 7a-j, 8a-g, 9a-g, and 10
| Ser | Sample | Cytotoxicity IC50/uM, SD ( ±) |
|||
|---|---|---|---|---|---|
| Code | Structure | Ar | M.W (g/mol) | MCF7 | |
| 1 | 7a |  |
C6H5- | 451.47 | 8.0617 ± 0.33 |
| 2 | 7b | 3-MeO-C6H4- | 481.49 | 56.813 ± 2.34 | |
| 3 | 7c | 4-MeO-C6H4- | 481.49 | 7.7251 ± 0.32 | |
| 4 | 7d | 4-Cl-C6H4- | 485.91 | 41.126 ± 1.69 | |
| 5 | 7e | 4-Br-C6H4- | 530.36 | 21.907 ± 0.9 | |
| 6 | 7f | 2-Furyl | 441.43 | 3.9819 ± 0.16 | |
| 7 | 7 g | 2-Thiophenyl | 457.50 | 1.928 ± 0.08 | |
| 8 | 7 h | 3-pyridyl | 452.46 | 1.8937 ± 0.08 | |
| 9 | 7i | 4-pyridyl | 452.46 | 23.95 ± 0.98 | |
| 10 | 7j | 2-Naphthyl | 501.53 | 20.094 ± 0.83 | |
| 11 | 8a |  |
C6H5- | 465.49 | 14.28 ± 0.59 |
| 12 | 8b | 3-MeO-C6H4- | 495.52 | 36.192 ± 1.49 | |
| 13 | 8c | 4-MeO-C6H4- | 495.52 | 3.7466 ± 0.15 | |
| 14 | 8d | 4-Cl-C6H4- | 499.94 | 17.973 ± 0.74 | |
| 15 | 8e | 4-Br-C6H4- | 544.39 | 42.638 ± 1.75 | |
| 16 | 8f | 2-Naphthyl | 515.55 | 1.6966 ± 0.07 | |
| 17 | 8 g | 2-Furyl | 455.46 | 21.251 ± 0.87 | |
| 18 | 9a |  |
C6H5- | 465.49 | 9.1005 ± 0.37 |
| 19 | 9b | 3-MeO-C6H4- | 495.52 | 8.7957 ± 0.36 | |
| 20 | 9c | 4-MeO-C6H4- | 495.52 | 26.07 ± 1.07 | |
| 21 | 9d | 4-Cl-C6H4- | 499.94 | 2.0534 ± 0.08 | |
| 22 | 9e | 4-Br-C6H4- | 544.39 | 18.285 ± 0.75 | |
| 23 | 9f | 2-Naphthyl | 515.55 | 41.421 ± 1.7 | |
| 24 | 9 g | 2-Furyl | 455.46 | 13.199 ± 0.54 | |
| 25 | 10 |  |
– | 608.42 | 2.1387 ± 0.09 |
| *** | Doxorubicin | – | – | 543.52 | 11.495 ± 0.47 |
Fig. 3.

The IC50 (µM) of antiproliferative assays of Doxorubicin and the target compounds against MCF-7 human cancer cell lines
Secondly, another group of compounds demonstrated comparable activity to that of doxorubicin. This finding suggests that these compounds possess a similar level of cytotoxic potential. Including 3-cyanopyrid-2-one derivatives 7a and 7c bearing phenyl and 4-methoxy phenyl respectively and showing IC50 = 8.06 ± 0.33, 7.72 ± 1.69 µM respectively. The 3-cyano-1-methylpyrid-2-one derivatives 9a and 9b containing phenyl and 3-methoxyphenyl respectively, showed similar cytotoxicity with IC50 of 9.10 ± 0.37 and 8.79 ± 0.36 µM. The remaining derivatives displayed varying degrees of cytotoxic activity, their IC50 values ranged from 13.9 to 56.8 µM, indicating moderate to weak efficacy in inhibiting cell growth.
From the results, it is obvious that 3-cyanopyridin-2-one derivatives showed similar activities as the derivatives that alkylated with methyl group on both nitrogen and oxygen groups. It was observed that the aromatic substituent on the cyanopyridine ring exerted varied cytotoxic activity.
Among the various substituents considered, the naphthyl group demonstrated the highest level of cytotoxic activity, followed by the 3-pyridyl group. Subsequently, the 4-chlorophenyl substituent exhibited a lower degree of cytotoxicity, succeeded by the 4-methoxyphenyl, followed by furan and phenyl substituents, followed by the 3-methoxyphenyl substituent, while the remaining substituent showed a varied range of weak to moderate activities. These findings can be succinctly represented in Fig. 4, illustrating the cytotoxicity structure activity relationship (SAR) of the cyanopyridine derivatives.
Fig. 4.

SAR of the cytotoxicity of the synthetic cyanopyridine derivatives
PIM1 Kinase inhibition activity:
In accordance with the findings from the in vitro cytotoxicity study, the evaluation of PIM-1 Kinase inhibition for the most potent derivatives 7h and 8f demonstrated persuasive results as shown in (Table 2, Fig. 5). Specifically, the 3-cyanopyridine hybrid, 8f, with methoxy group at position 2 and 2-naphthyl moiety at position 6, exhibited a comparable IC50 value of 0.58 µM, while the 3-cyanopyridin-2-one hybrid 7h, with 3-pyridyl moiety at position 6, displayed the highest PIM-1 kinase inhibitory activity among all, with almost equipotent potency (IC50 = 0.283 µM) compared to the reference drug Staurosporine, (IC50 = 0.223 µM).
Table 2.
The IC50 (µM) of PIM1 Kinase inhibition assays of the target cyanopyridine derivatives 7h and 9f
| Ser | Compound | PIM1-Kinase | |
|---|---|---|---|
| ID | MW | IC50 (µM) | |
| 1 | 7h | 452.46 | 0.281 ± 0.012 |
| 2 | 8f | 515.55 | 0.58 ± 0.025 |
| *** | Staurosporine | 466.54 | 0.223 ± 0.01 |
Fig. 5.
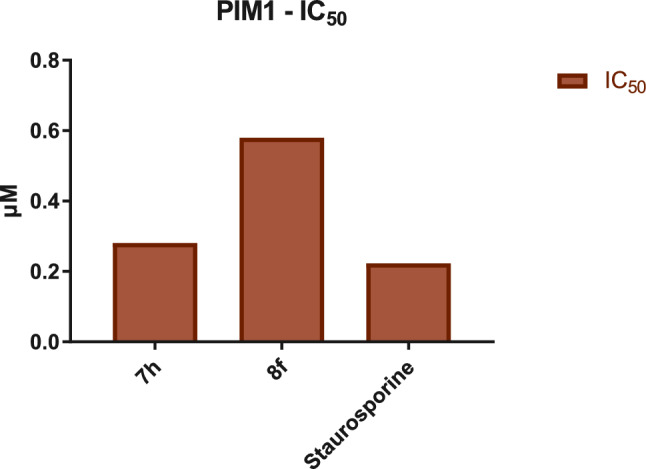
The IC50 (µM) of antiproliferative assays of Staurosporine and the target compounds against PIM1 Kinase enzyme
Thus, it can be inferred that the 3-cyanopyridine nucleus with such promising anti-breast cancer results of the synthesized target compounds, in particular, compound 7h, still represents a significant scaffold in the quest for highly potent PIM-1 kinase inhibitors, which merits more investigation and SAR study.
Flow cytometric cell cycle analysis:
The cell cycle encompasses four distinct phases: G1 phase, S phase (synthesis), G2 phase, and M phase. During G1, cellular amplification and preparation for DNA replication take place. The S phase involves DNA replication and chromatid duplication. In the G2 phase, DNA repair and further growth occur. The M phase is characterized by nuclear division. The investigation of the impact of compound 7h on cell cycle progression and the induction of apoptosis in MCF-7 cells. The MCF-7 cell line was subjected to incubation with the IC50 concentration of compound 7h for 24 h. Subsequently, the cells were stained with PI/Annexin V and analyzed using flow cytometry (BD FASCCalibur). Upon analyzing the results (Table 3, Fig. 6), it was evident that the percentage of cell accumulation in the S phase increased in MCF-7 cells treated with compound 7h after 24 h of incubation. This observation indicates a cell cycle arrest at the S phase, with the percentage of cells accumulated in this phase rising from 25.83% in the control group to 33.61%.
Table 3.
Cell cycle analysis t in MCF-7 cell line treated with 7h compound
| Ser | Sample | DNA content | ||||
|---|---|---|---|---|---|---|
| Code | IC50 uM |
%G0-G1 | %S | %G2/M | Comment | |
| 1 | 7 h/MCF7 | – | 54.28 | 33.61 | 12.11 | Cell growth arrest@ S |
| 2 | Cont.MCF7 | – | 61.03 | 25.83 | 13.14 | – |
Fig. 6.
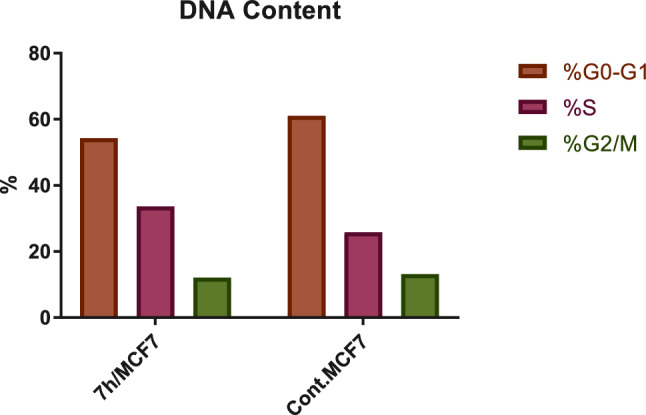
Cell cycle analysis in the MCF-7 cell line treated with compound 7h compared with the control
Compound 7h was chosen to investigate its abilities in inducing cancer cell apoptosis. Cell cycle analysis of MCF-7 after treatment with compound 7h. To corroborate the ability of the compound to induce apoptosis, cells were stained with Annexin V/PI, incubated for 24 h, and analyzed. Analysis of early and late apoptosis showed that compound 7h was positively able to make significant levels of apoptosis with necrosis percent 3.95 (Table 4, Fig. 7).
Table 4.
Cell cycle analysis and apoptosis detection of compound 7h
| Ser | Code | Conc | Apoptosis | Necrosis | ||
|---|---|---|---|---|---|---|
| Total | Early | Late | ||||
| 1 | 7 h/MCF7 | – | 42.18 | 15.42 | 22.81 | 3.95 |
| 2 | Cont.MCF7 | – | 2.29 | 0.41 | 0.27 | 1.61 |
Fig. 7.

Cell cycle analysis and Apoptosis induction analysis using Annexin V/PI of compound 7h and control untreated RPMI8226 cell
Molecular docking studies:
To study the ability of compounds 7h and 8f to bind to the PIM-1 kinase enzyme (PDB entry: 2OBJ), the ligand VRV40 was firstly docked. As illustrated in Fig. 8, ligand VRV400 engaged in the formation of two hydrogen bonds with Phe49 and Lys67 and many hydrophobic interactions (pi-cation, alkyl, and pi-alkyl) with Val52, Ala65, Lys67, Ile104, Leu120, Leu174, Ile185, and Asp186 amino acid residues.
Fig. 8.

Docking and binding mode of VRV400 into the active site of PIM-1 (PDB entry: 2OBJ) (A) 3D structure of VRV400 (cyan) (B) 2D structure of VRV400 (cyan)
Additionally, docking results of compound 7h into the PIM-1 active site evidenced that compound 7h displayed affinity of -10.1 kcal/mol and incorporated in the formation of five hydrogen bonds with Gly48, Phe49, Asp186 (two hydrogen bonds), and Lys67, in addition to numerous hydrophobic interactions such as van der Waals, pi-cation, pi-anion, pi-sigma, and pi-alkyl with Leu44, Phe49, Val52, Ala65, Val126, Lys169, Glu171, Leu174, Ile185, and Asp186 amino acid residues, which reflects its potency on inhibition of PIM-1 kinase enzymes and explains the highest potency of compound 7h against the MCF7 breast cancer cell line, which may be attributed mechanistically to the activity of PIM-1 inhibitory activity (Fig. 9).
Fig. 9.

Docking and binding mode of hybrid 7h into the active site of PIM-1 (PDB entry: 2OBJ) (A) 3D structure of hybrid 7h (blue) (B) 2D structure of hybrid 7h (blue)
Furthermore, docking results of compound 8f into the PIM-1 active site proved that compound 8f showed affinity of -9.9 kcal/mol and involved in the formation of five hydrogen bonds with Gly48, Phe49, Asp186 (two hydrogen bonds), and Lys67, in addition to several hydrophobic interactions such as carbon hydrogen bond, pi-cation, pi-anion, pi-sigma, and pi-alkyl with Leu44, Phe49, Val52, Val126, Lys169, Glu171, Leu174, Ile185, and Asp186 amino acid residues, which reflects its potency on inhibition of the PIM-1 kinase enzymes and explains the highest potency of compound 8f against the MCF7 breast cancer cell line, which may be attributed mechanistically to the activity of PIM-1 inhibitor (Fig. 10).
Fig. 10.

Docking and binding mode of compound 8f into the active site of PIM-1 (PDB entry: 2OBJ) (A) 3D structure of compound 8f (violet); B 2D structure of compound 8f (violet)
Taken together, the docking results were in agreement with the biological study, and it could be concluded that compounds 7h and 8f are entitled to be used as a future template, which deserve further study, which were rapidly identified as an attractive lead candidate for PIM-1 inhibitor hybrids and were extensively profiled.
Conclusion
In summary, the new derivatives of cyanopyridine 7a-j, 8a-g, 9a-g, and 10 have been designed and synthesized utilizing one-pot multicomponent reaction (MCR). Most of the tested compounds displayed good anti-breast cancer (MCF-7) activity; in particular, compounds 7h, and 8f showed potent anticancer activity against MCF-7 via inhibition of PIM-1 kinase. Moreover, compound 7h arrested the tumor cells at the S phase. In addition, compound 7h caused cell death mainly by inducing early and late apoptosis. Taken together, the molecular docking study of 7h and 8f, along with the biological screening could be concluded that the N- and O-alkyl cyanopyridine scaffold is considered a promising scaffold for innovation of potent anticancer candidates, and it merits more investigation and SAR study, which are ongoing in our laboratory.
Experimental
Chemistry
General information
All information about reagents and spectral analyses were showed in Supporting Information.
Synthesis of Sodium 4-formyl-2-methoxyphenolate (2):
To a solution of sodium ethoxide (0.075 g, 3.0 mmol) in 30 mL of ethanol, vanillin (0.5 g, 3.0 mmol) was added portion-wise with stirring in an ice bath for 1h. The solvent was evaporated under vacuum, and the formed precipitate was collected, washed with ether, dried, and used as commercial material. White powder, yield: 0.57 g (99%); mp. > 340 °C; FT-IR (ATR) vmax: 3052 (CHarom.), 2971, 2936 (CHaliph.), 1669 (C = Oformyl) cm−1; 1HNMR (400 MHz, DMSO-d6) δ: 9.31 (s, 1H, CHformyl), 7.15 (d, 1H, J = 7.9 Hz, CHarom.), 7.02 (s, 1H, CHarom.), 6.31(d, 1H, J = 7.9 Hz, CHarom.), 3.71 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 187.1 (C = Oformyl), 170.6, 151.7, 130.9, 118.7, 117.8, 108.0, 55.1 (OCH3) ppm. Anal. Calcd. for C8H7NaO3 (174.13): C, 55.18; H, 4.05%. Found: C, 55.30; H, 4.18%.
3-Methoxy-4-(2-oxo-2-phenylethoxy)benzaldehyde (3):
White crystal, yield 0.80 g (98%), mp. 154–156 oC, was prepared according to literature procedure [38].
Procedure for synthesis of ethyl-3-[4-(2-anilino-2-oxoethoxy)-3-methoxyphenyl]-2-cyanoacrylate (4):
A mixture of compound 3 (0.5 g, 1.75 mmol) and respective active methylene compounds such as: ethyl cyanoacetate (0.19 g, 1.75 mmol) were refluxed in the presence of TEA in 20 mL ethanol for 1h. The formed precipitate was filtered off on hot, washed with ethanol, and crystallized from acetonitrile. Yellow powder, yield: 0.63 g (94%), mp. 138–140 °C; FT-IR (ATR) vmax: 3389 (NH), 3062 (CHarom.), 2983, 2919, 2866 (CHaliph.), 2223 (CN); 1714 (C = Oester); 1691 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.22 (s, 1H, NH), 8.33 (s, 1H, CHolefinic), 7.84 (d, 1H, J = 1.5 Hz, CHarom.), 7.73 (d, 1H, J = 8.5 Hz, CHarom.), 7.62 (d, 2H, J = 7.9 Hz, CHarom.), 7.34 (t, 2H, J = 7.8 Hz, CHarom), 7.15 (d, 1H, J = 8.5 Hz, CHarom.), 7.09 (t, 1H, J = 7.3 Hz, CHarom.), 4.89 (s, 2H, OCH2CO), 4.32 (q, 2H, J = 7.1 Hz, OCH2CH3), 3.87 (s, 3H, OCH3); 1.32 (t, 3H, J = 7.1 Hz, OCH2CH3). Anal. Calcd. for C21H20N2O5 (380.39): C, 66.31; H, 5.30; N, 7.36%. Found: C, 66.45; H, 5.19; N, 7.22%.
General procedure for synthesis of pyridines 7a-j:
To a solution of arylidene 4 (0.5 g, 1.3 mmol) and ammonium acetate 5 (0.51 g, 6.5 mmol) in 30 mL of ethanol, 1.3 mmol of respective methylaryl/heterylketone derivatives 6a-j such as acetophenone (0.16 g), 3-methoxyacetophenone (0.2 g), 4-methoxyacetophenone (0.2 g), 4-chloroacetophenone (0.2 g), 4-bromoacetophenone (0.26 g), 2-acetylfuran (0.14 g), 2-acetylthiophene (0.17 g), 3-acetylpyidine (0.16 g), 4-acetylpyridine (0.16 g) and/or 2-acetyl naphthalene (0.22 g) was added. The reaction mixture was refluxed for 6–8 h. (monitored using TLC). The formed precipitate was filtered off (on hot), washed with water several times, then with ethanol, dried, and crystallized from a mixture of ethanol and DMF (1:1).
2-(4-(3-Cyano-2-oxo-6-phenyl-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-Nphenylacetamide(7a):
Pale yellow powder, yield: 0.53 g (89%), mp. 276–278 °C; FT-IR (ATR) vmax: 3390 (NH), 3076 (CHarom.), 2969, 2917 (CHaliph.), 2217 (CN), 1690 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.62 (s, 1H, NH), 10.02 (s, 1H, NH), 7.90 (d, 2H, J = 6.8 Hz, CHarom.), 7.63 (d, 2H, J = 7.8 Hz, CHarom.), 7.57–7.53 (m, 3H, CHarom.), 7.42 (s, 1H, CHarom.), 7.34 (t, 3H, J = 7.3 Hz, CHarom.), 7.15 (d, 1H, J = 8.4 Hz, CHarom.), 7.09 (t, J = 7.3 Hz, 1H, CHarom.), 6.86 (s, 1H, CHarom.), 4.81 (s, 2H, CH2); 3.92 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 162.6, 159.8, 151.7, 149.8, 149.4, 138.7, 132.9, 131.5, 129.8, 129.3, 129.2, 128.1, 124.2, 121.8, 120.0, 117.2, 114.4, 113.1, 106.6, 90.3, 68.6 (CH2), 56.4 (OCH3). Anal. Calcd. for C27H21N3O4 (451.47): C, 71.83; H, 4.69; N, 9.31%. Found: C, 71.61; H, 4.81; N, 9.50%.
2-(4-(3-Cyano-6-(3-methoxyphenyl)-2-oxo-1,2-dihydropyridin-4-yl)-2methoxyphenoxy)-N-phenylacetamide (7b):
Pale yellow powder, yield: 0.55 g (87%), mp. 244–246 °C; FT-IR (ATR) vmax:3378 (NH), 3065 (CHarom.), 2930, 2842 (CHaliph.)., 2220 (CN), 1690 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.66 (s, 1H, NH), 10.09 (s, 1H, NH), 7.65 (d, 2H, J = 7.7 Hz, CHarom.), 7.47–7.42 (m, 4H, CHarom.), 7.36–7.32 (m, 3H, CHarom.), 7.15–7.08 (m, 3H, CHarom.), 6.88 (s, 1H, CHarom.), 4.82 (s, 2H, CH2), 3.92 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 162.5, 159.9, 159.8, 151.2, 149.8, 149.3, 138.8, 134.1, 130.5, 129.7, 129.2, 124.1, 121.8, 120.4, 119.9, 117.7, 117.2, 114.1, 113.1, 106.5, 98.4, 68.5 (CH2), 56.4 (OCH3), 55.8 (OCH3). Anal. Calcd. for C28H23N3O5 (481.49): C, 69.84; H, 4.81; N, 8.73%. Found: C, 69.96; H, 4.67; N, 8.43%.
2-(4-(3-Cyano-6-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (7c):
Pale yellow powder, yield: 0.56 g (89%), mp. 284–286 °C; FT-IR (ATR) vmax:3336 (NH), 3079 (CHarom.), 2936, 2837 (CHaliph.)., 2213 (CN), 1691 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.44 (s, 1H, NH), 10.00 (s, 1H, NH), 7.90 (s, 2H, CHarom.), 7.64 (s, 2H, CHarom.), 7.41–7.33 (m, 4H, CHarom.), 7.15–7.09 (m, 4H, CHarom.), 6.80 (s, 1H, CHarom.), 4.81 (s, 2H, CH2), 3.93 (s, 3H, OCH3); 3.86 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 162.7, 162.1, 159.8, 149.6, 149.2, 138.6, 129.8, 129.7, 129.3, 124.6, 124.3, 121.7, 119.9, 117.5, 114.8, 113.8, 112.7, 105.6, 105.5, 97.1, 68.2 (CH2), 56.3 (OCH3), 55.9 (OCH3). Anal. Calcd. for C28H23N3O5 (481.49): C, 69.84; H, 4.81; N, 8.73%. Found: C, 69.59; H, 4.63; N, 8.89%.
2-(4-(6-(4-Chlorophenyl)-3-cyano-2-oxo-1,2-dihydropyridin-4-yl)-2methoxyphenoxy)-N-phenylacetamide (7d):
Pale yellow powder, yield: 0.53 g (85%), mp. 276–278 °C; FT-IR (ATR) vmax: 3380 (NH), 3083 (CHarom.), 2926, 2839 (CHaliph.), 2219 (CN), 1686 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.74 (s, 1H, NH), 10.15 (s, 1H, NH), 7.92 (s, 2H, CHarom.), 7.61 (s, 4H, CHarom.), 7.40–7.33 (m, 4H, CHarom.), 7.10 (s, 2H, CHarom.), 6.91 (s, 1H, CHarom.), 4.81 (s, 2H, CH2); 3.90 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 165.5, 158.6, 149.9, 149.7, 149.4, 138.7, 136.4, 130.0, 129.7, 129.5, 129.3, 129.2, 128.9, 124.2, 121.8, 120.0, 114.4, 113.7, 113.1, 93.8, 68.6 (CH2), 56.4 (OCH3). Anal. Calcd. for C27H20ClN3O4 (485.91): C, 66.74; H, 4.15; N, 8.65%. Found: C, 66.56; H, 4.35; N, 8.43%.
2-(4-(6-(4-Bromophenyl)-3-cyano-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxy phenoxy)-N-phenylacetamide (7e):
Pale yellow powder, yield: 0.6 g (86%), mp. 329–331 °C; FT-IR (ATR) vmax: 3381 (NH), 3060 (CHarom.), 2924, 2838 (CHaliph.), 2216 (CN), 1689 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.63 (s, 1H, NH), 10.02 (s,1H, NH), 7.87 (d, 2H, J = 8.0 Hz, CHarom.), 7.74 (d, 2H, J = 8.2 Hz, CHarom.), 7.64 (d, 2H, J = 7.9 Hz, CHarom.), 7.42 (s, 1H, CHarom.), 7.36–7.32 (m, 3H, CHarom), 7.15 (d, 1H, J = 8.4 Hz, CHarom.), 7.09 (t, 1H, J = 7.3 Hz, CHarom.), 6.92 (s, 1H, CHarom.), 4.81 (s, 2H, CH2), 3.92 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 166.2, 163.1, 158.8, 155.6, 149.8, 149.3, 142.3, 138.8, 132.3, 130.2, 129.8, 129.6, 129.2, 125.2, 124.1, 121.8, 119.9, 114.1, 113.0, 93.9, 68.5 (CH2), 56.4 (OCH3). Anal. Calcd. for C27H20BrN3O4 (530.37): C, 61.14; H, 3.80; N, 7.92%. Found: C, 61.35; H, 3.63; N, 7.71%.
2-(4-(3-Cyano-6-(furan-2-yl)-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (7f):
Pale yellow powder, yield: 0.53 g (91%), mp. 296–298 oC; FT-IR (ATR) vmax: 3398 (NH), 3020 (CHarom.), 2930, 2810 (CHaliph.), 2219 (CN), 1636 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.70 (s, 1H, NH), 10.09 (s,1H, NH), 8.00 (s, 1H, CHarom.), 7.65–7.63 (m, 3H, CHarom.), 7.38–7.28 (m, 4H, CHarom.), 7.14–7.09 (m, 2H, CHarom.), 6.84 (s, 1H, CHarom.), 6.78 (s, 1H, CHarom.), 4.82 (s, 2H, CH2), 3.91 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 162.1, 159.7, 149.8, 149.3, 147.3, 146.1, 140.8, 138.7, 129.6, 129.2, 124.2, 121.6, 120.0, 117.2, 114.7, 114.1, 113.6, 112.7, 103.1, 97.6, 68.4 (CH2), 56.3 (OCH3). Anal. Calcd. for C25H19N3O5 (441.43): C, 68.02; H, 4.34; N, 9.52%. Found: C, 68.23; H, 4.19; N, 9.74%.
2-(4-(3-Cyano-2-oxo-6-(thiophen-2-yl)-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (7 g):
Pale yellow powder, yield: 0.54 g (90%), mp. 293–295 °C; FT-IR (ATR) vmax:3399 (NH), 3030 (CHarom.), 2944, 2822 (CHaliph.), 2221(CN), 1638 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.76 (s, 1H, NH), 10.18 (s, 1H, NH), 8.06 (d, 1H, J = 3.7 Hz, CHarom.), 7.87 (d, 1H, J = 3.8 Hz, CHarom.), 7.65 (d, 2H, J = 7.7 Hz, CHarom.), 7.37–7.24 (m, 6H, CHarom.), 7.13–7.07 (m, 2H, CHarom.), 4.83 (s, 2H, CH2), 3.91 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.8, 163.2, 158.8, 149.6, 149.4, 149.3, 147.3, 138.5, 131.7, 129.9, 129.6, 129.4, 129.3, 124.4, 121.6, 120.1, 117.1, 114.2, 112.8, 106.8, 99.8, 68.4 (CH2), 56.3 (OCH3). Anal. Calcd. for C25H19N3O4S (457.50): C, 65.63; H, 4.19; N, 9.18%. Found: C, 65.39; H, 4.36; N, 9.40%.
2-(4-(3-Cyano-2-oxo-6-(pyridin-3-yl)-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (7 h):
Pale yellow powder, yield: 0.51 g (85%), mp. 295–297 °C; FT-IR (ATR) vmax:3388 (NH), 3043 (CHarom.), 2935, 2848 (CHaliph.), 2218 (CN), 1682 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.90 (s, 1H, NH), 10.18 (s,1H, NH), 9.10 (s,1H, CHarom.), 8.73 (d, 1H, J = 3.9 Hz, CHarom.), 8.30 (d, 1H, J = 7.5 Hz, CHarom.), 7.65 (d, 2H, J = 7.7 Hz, CHarom.), 7.59–7.56 (m, 1H, CHarom.), 7.44 (s, 1H, CHarom.), 7.39–7.33 (m, 3H, CHarom.), 7.14–7.08 (m, 2H, CHarom.), 7.02 (s, 1H, CHarom.), 4.84 (s, 2H, CH2), 3.92 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 162.7, 159.6, 151.9, 149.8, 149.3, 148.9, 138.8, 135.8, 129.3, 129.2, 129.1, 129.1, 124.2, 124.1, 121.9, 119.9, 117.2, 113.8, 112.9, 107.5, 98.7, 68.3 (CH2), 56.3 (OCH3). Anal. Calcd. for C26H20N4O4 (452.46): C, 69.02; H, 4.46; N, 12.38%. Found: C, 69.29; H, 4.70; N, 12.15%.
2-(4-(3-Cyano-2-oxo-6-(pyridin-4-yl)-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (7i):
Pale yellow powder, yield: 0.52 g (87%), mp. 280–282 °C; FT-IR (ATR) vmax:3388 (NH), 3050 (CHarom.), 2925, 2830 (CHaliph.), 2218(CN), 1682 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.79 (s, 1H, NH), 10.03 (s,1H, NH), 8.75 (d, 2H, J = 5.0 Hz, CHarom.), 7.91 (d, 2H, J = 4.3 Hz, CHarom.), 7.64 (d, 2H, J = 7.7 Hz, CHarom.), 7.44 (s, 1H, CHarom.), 7.37–7.32 (m, 3H, CHarom.), 7.17–7.08 (m, 3H, CHarom.), 4.82 (s, 2H, CH2), 3.93 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.8, 162.8, 159.6, 150.6, 149.7, 149.5, 149.4, 140.6, 138.3, 129.4, 129.3, 124.5, 122.1, 121.9, 120.2, 116.8, 114.3, 113.0, 108.3, 99.3, 68.4 (CH2), 56.4 (OCH3). Anal. Calcd. for C26H20N4O4 (452.46): C, 69.02; H, 4.46; N, 12.38%. Found: C, 68.18; H, 4.25; N, 12.54%.
2-(4-(3-Cyano-6-(naphthalen-2-yl)-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (7j):
Pale yellow powder, yield: 0.58 g (88%), mp. > 300 °C; FT-IR (ATR) vmax:3388 (NH), 3045 (CHarom.), 2938, 2817 (CHaliph.), 2217 (CN), 1690 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 12.85 (s, 1H, NH), 10.20 (s, 1H, NH), 8.55 (s, 1H, CHarom.), 8.05–8.01 (m, 4H, CHarom.), 7.66 (s, 4H, CHarom.), 7.46–7.35 (m, 4H, CHarom.), 7.15–7.04 (m, 3H, CHarom.), 4.85 (s, 2H, CH2), 3.93 (s, 3H, OCH3). Anal. Calcd. for C31H23N3O4 (501.53): C, 74.24; H, 4.62; N, 8.38%. Found: C, 74.51; H, 4.39; N, 8.56%.
General procedure for synthesis of O-methylcyanopyridines 8a-g, N-methylcyanopyridines 9a-g and bipyridinium iodide salt 10:
To a stirred mixture of pyridines 7a-f, 7h, and 7j (1.1 mmol) and potassium carbonate (0.15 g, 1.1 mmol) in 20 mL of DMF, methyl iodide (0.16 g, 1.2 mmol) was added drop by drop in about half an hour at room temperature. The reaction mixture was continuously stirred overnight and then poured into 50 mL of water. The formed precipitate (two products except compound 10) was filtered off, washed with water, dried, and separated by thin layer chromatography using a mixture of chloroform and petroleum ether (9:1) (Rf8a-g = 0.17, 0.16, 0.20, 0.18, 0.17, 0.19, and 0.10, respectively; Rf9a-g = 0.41, 0.32, 0.47, 0.43, 0.23, 0.32, and 0.27, respectively; and Rf10 = 0.11).
2-(4-(3-Cyano-2-oxo-6-phenyl-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (8a):
White powder, yield: 0.23 g (45%), mp. 194–196 °C; FT-IR (ATR) vmax: 3393 (NH), 3065 (CHarom.), 2944, 2851 (CHaliph.), 2217 (CN), 1691 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.10 (s, 1H, NH), 8.27 (d, 2H, J = 3.7 Hz, CHarom.), 7.83 (s, 1H, CHarom.), 7.65 (d, 2H, J = 7.5 Hz, CHarom.), 7.54 (s, 3H, CHarom.), 7.44 (s, 1H, CHarom.), 7.34 (s, 3H, CHarom.), 7.16 (d, 1H, J = 8.2 Hz, CHarom.), 7.09 (t, 1H, J = 6.5 Hz, CHarom.), 4.82 (s, 2H, CH2), 4.16 (s, 3H, OCH3), 3.93 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 164.9, 157.5, 156.4, 149.5, 149.4, 138.8, 137.2, 131.1, 129.4, 129.3, 129.2, 127.9, 124.1, 121.9, 119.8, 116.1, 114.2, 114.1, 113.2, 92.4, 68.4 (CH2), 56.3 (OCH3), 54.96 (OCH3). Anal. Calcd. for C28H23N3O4 (465.49): C, 72.24; H, 4.98; N, 9.03%. Found: C, 72.43; H, 4.79; N, 9.19%.
2-(4-(3-Cyano-2-methoxy-6-(3-methoxyphenyl)-pyridin-4-yl)-2-methoxy-phenoxy)-N-phenylacetamide (8b):
White powder, yield: 0.21 g (40%), mp. 158–160 °C; FT-IR (ATR) vmax: 3396 (NH), 3067 (CHarom.), 2944, 2911, 2843 (CHaliph.), 2218 (CN), 1693 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.13 (s, 1H, NH), 7.82–7.78 (m, 3H, CHarom.), 7.65 (d, 2H, J = 4.2 Hz, CHarom.), 7.44 (s, 2H, CHarom.), 7.34 (s, 3H, CHarom.), 7.16–7.10 (m, 3H, CHarom.), 4.83 (s, 2H, CH2), 4.15 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 3.86 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 164.7, 160.1, 157.3, 156.4, 149.5, 149.0, 149.4, 138.8, 138.6, 130.4, 129.4, 129.2, 124.1, 121.9, 120.3, 119.9, 116.5, 116.1, 114.2, 114.1, 113.3, 92.5, 68.4 (CH2), 56.3 (OCH3), 55.7 (OCH3), 54.9 (OCH3). Anal. Calcd. for C29H25N3O5 (495.52): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.51; H, 5.22; N, 8.36%.
2-(4-(3-Cyano-2-methoxy-6-(4-methoxyphenyl)pyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (8c):
White powder, yield: 0.21 g (40%), mp. 188–190 °C; FT-IR (ATR) vmax: 3398 (NH), 3070 (CHarom.), 2944, 2914, 2850 (CHaliph.), 2222 (CN), 1693 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.10 (s, 1H, NH), 8.27 (s, 2H, CHarom.), 7.82–7.55 (m, 6H, CHarom.), 7.35 (s, 3H, CHarom.), 7.15 (s, 2H, CHarom.), 4.82 (s, 2H, CH2), 4.16 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 3.93 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 164.8, 161.9, 157.6, 156.4, 156.2, 149.5, 138.8, 131.0, 129.6, 129.2, 127.9, 124.1, 121.9, 121.8, 119.9, 114.7, 114.4, 114.0, 113.4, 92.4, 68.6 (CH2), 56.4 (OCH3), 55.8 (OCH3), 54.9 (OCH3). Anal. Calcd. for C29H25N3O5 (495.52): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.13; H, 5.27; N, 8.63%.
2-(4-(6-(4-Chlorophenyl)-3-cyano-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (8d):
White powder, yield: 0.15 g (30%), mp. 243–245 °C; FT-IR (ATR) vmax:3390 (NH), 3069 (CHarom.), 2947, 2921, 2850 (CHaliph.), 2218 (CN), 1689 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.10 (s, 1H, NH), 8.30 (s, 2H, CHarom.), 7.86 (s, 1H, CHarom.), 7.64–7.60 (m, 4H, CHarom.), 7.44 (s, 1H, CHarom.), 7.34 (s, 3H, CHarom.), 7.16 (d, 1H, J = 7.4 Hz, CHarom.), 7.10 (s, 1H, CHarom.), 4.82 (s, 2H, CH2), 4.15 (s, 3H, OCH3), 3.93 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 164.9, 156.6, 156.2, 149.6, 138.8, 136.1, 135.9, 129.7, 129.5, 129.3, 129.2, 124.1, 122.0, 120.0, 114.6, 114.3, 114.1, 113.6, 113.4, 92.8, 68.8 (CH2), 56.5 (OCH3), 55.0 (OCH3). Anal. Calcd. for C28H22ClN3O4 (499.94): C, 67.27; H, 4.44; N, 8.40%. Found: C, 67.45; H, 4.29; N, 8.58%.
2-(4-(6-(4-Bromophenyl)-3-cyano-2-methoxypyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (8e):
White powder, yield: 0.154 g (30%), mp. 246–248 °C; FT-IR (ATR) vmax:3386 (NH), 3060 (CHarom.), 2994,2920, 2848 (CHaliph.), 2218 (CN), 1692 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.10 (s, 1H, NH), 8.21 (d, 2H, J = 7.2 Hz, CHarom.), 7.84 (s, 1H, CHarom.), 7.73 (d, 2H, J = 7.3 Hz, CHarom.), 7.64 (d, 2H, J = 6.6 Hz, CHarom.), 7.43 (s, 1H, CHarom.), 7.34 (s, 3H, CHarom.), 7.16–7.09 (m, 2H, CHarom.), 4.82 (s, 2H, CH2), 4.14 (s, 3H, OCH3), 3.93 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 164.9, 156.6, 156.4, 149.7, 149.6, 138.8, 136.4, 132.3, 129.9, 129.5, 129.2, 124.8, 124.1, 122.0, 120.0, 115.9, 114.6, 114.1, 113.5, 92.9, 68.7 (CH2), 56.5 (OCH3), 55.0 (OCH3). Anal. Calcd. for C28H22BrN3O4 (544.39): C, 61.77; H, 4.07; N, 7.72%. Found: C, 61.54; H, 4.29; N, 7.50%.
2-(4-(3-Cyano-6-(naphthalen-2-yl)-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxy phenoxy)-N-phenylacetamide (8f):
Pale yellow powder, yield: 0.18 g (35%), mp. 193–195°C; FT-IR (ATR) vmax: 3395 (NH), 3053 (CHarom.), 2942, 2918, 2850 (CHaliph.)., 2217 (CN), 1690 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.10 (s, 1H, NH), 8.89 (s, 1H, CHarom.), 8.41 (d, 1H, J = 8.5, CHarom.), 8.10–8.07 (m, 2H, CHarom.), 8.01–7.99 (m, 2H, CHarom.), 7.66–7.60 (m, 4H, CHarom.), 7.48 (s, 1H, CHarom.), 7.39–7.33 (m, 3H, CHarom.), 7.18 (d, 1H, J = 7.2, CHarom.), 7.10 (t, 1H, J = 7.2, CHarom.), 4.83 (s, 2H, CH2), 4.23 (s, 3H, OCH3), 3.95 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 164.9, 157.4, 156.5, 149.5, 149.4, 138.8, 134.7, 134.5, 134.3, 133.3, 129.5, 129.4, 129.3, 128.9, 128.2, 128.1, 127.2, 124.9, 124.1, 121.9, 119.8, 116.2, 114.4, 114.1, 113.3, 92.4, 68.4 (CH2), 56.3 (OCH3), 55.0 (OCH3). Anal. Calcd. for C32H25N3O4 (515.55): C, 74.55; H, 4.89; N, 8.15%. Found: C, 74.68; H, 4.75; N, 8.34%.
2-(4-(3-Cyano-6-(furan-2-yl)-2-methoxypyridin-4-yl)-2-methoxyphenoxy)-N-phenyl acetamide (8 g):
Pale yellow powder, yield: 0.21 g (40%), mp. 178–180 °C; FT-IR (ATR) vmax: 3387 (NH), 3061 (CHarom.), 2949, 2912, 2846 (CHaliph.), 2213 (CN), 1686 (C = Oamide) cm−1; 1H NMR (400 MHz, CDCl3) δ: 8.71 (s, 1H, NH), 7.53 (d, 2H, J = 7.9 Hz, CHarom.), 7.49 (s, 1H, CHarom.), 7.32 (s, 1H, CHarom.), 7.28 (t, 2H, J = 7.6 Hz, CHarom.), 7.17 -7.13 (m, 3H, CHarom.), 7.06 (t, 1H, J = 7.3 Hz, CHarom.), 7.00 (d, 1H, J = 8.2 Hz, CHarom.), 6.50 (s,1H, CHarom.), 4.62 (s, 2H, CH2), 4.05 (s, 3H, OCH3), 3.94 (s, 3H, OCH3); 13C NMR (100 MHz, CDCl3) δ: 166.2, 165.2, 155.7, 152.5, 149.9, 149.4, 148.6, 144.8, 137.1, 131.3, 129.0, 124.7, 121.6, 120.0, 116.1, 115.7, 112.6, 112.4, 112.2, 111.1, 92.2, 69.9 (CH2), 56.3 (OCH3), 54.5 (OCH3). Anal. Calcd. for C26H21N3O5 (455.46): C, 68.56; H, 4.65; N, 9.23%. Found: C, 68.27; H, 4.42; N, 9.51%.
2-(4-(3-Cyano-1-methyl-2-oxo-6-phenyl-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9a):
Pale yellow powder, yield: 0.15 g (30%), mp. 180–182 °C; FT-IR (ATR) vmax: 3426 (NH), 3060 (CHarom.), 2922, 2852 (CHaliph.), 2220 (CN), 1683 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.29 (s, 1H, NH), 7.65–7.57 (m, 7H, CHarom.), 7.36–7.30 (m, 4H, CHarom.), 7.13–7.06 (m, 2H, CHarom.), 6.51 (s, 1H, CHarom.), 4.83 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.34 (s, 3H, NCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 161.6, 157.6, 154.7, 149.9, 149.3, 138.7, 134.5, 130.6, 129.2, 129.0, 128.9, 124.2, 121.8, 120.3, 119.9, 117.3, 114.0, 112.8, 109.0, 98.2, 68.3 (CH2), 56.3 (OCH3), 35.1 (NCH3). Anal. Calcd. for C28H23N3O4 (465.49): C, 72.24; H, 4.98; N, 9.03%. Found: C, 72.40; H, 4.76; N, 9.21%.
2-(4-(3-Cyano-6-(3-methoxyphenyl)-1-methyl-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9b):
White powder, yield: 0.21 g (40%), mp. 188–190°C; FT-IR (ATR) vmax:3392 (NH), 3072 (CHarom.), 2960, 2940, 2839 (CHaliph.), 2219 (CN), 1684 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.14 (s, 1H, NH), 7.84–7.79 (m, 1H, CHarom.), 7.64 (s, 2H, CHarom.), 7.47–7.45 (m, 1H, CHarom.), 7.36–7.33 (m, 4H, CHarom.), 7.18–7.12 (m, 4H, CHarom), 6.53 (s, 1H, CHarom.), 4.82 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.35 (s, 3H, NCH3); 13C NMR (100 MHz, DMSO-d6) δ: 166.6, 161.5, 159.7, 157.6, 154.4, 149.9, 149.3, 138.8, 135.8, 130.4, 129.2, 128.9, 124.1, 121.8, 120.9, 119.8, 117.4, 116.2, 114.4, 113.9, 112.7, 108.8, 98.1, 68.3 (CH2), 56.2 (OCH3), 55.8 (OCH3), 35.1 (NCH3). Dept-135 NMR (100 MHz, DMSO-d6) δ: 130.4, 129.2, 124.1, 121.8, 120.9, 119.8, 116.2, 114.4, 113.8, 112.7, 108.8, 68.6 (CH2, reversed direction), 56.2 (OCH3), 55.8 (OCH3), 35.1 (NCH3). Anal. Calcd. for C29H25N3O5 (495.52): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.45; H, 5.23; N, 8.32%.
2-(4-(3-Cyano-6-(4-methoxyphenyl)-1-methyl-2-oxo-1,2-dihydro pyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9c):
White powder, yield: 0.21 g (40%), mp. 168–170 °C; FT-IR (ATR) vmax: 3394 (NH), 3075 (CHarom.), 2963, 2940, 2836 (CHaliph.), 2219 (CN), 1684 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.33 (s, 1H, NH), 7.65 (d, 2H, J = 5.3 Hz, CHarom.), 7.57 (d, 2H, J = 7.6 Hz, CHarom.), 7.35–7.33 (m, 4H, CHarom.), 7.10 (s, 4H, CHarom.), 6.48 (s, 1H, CHarom.), 4.84 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.85 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.7, 161.9, 161.0, 157.5, 154.8, 149.7, 149.2, 138.6, 130.6, 129.6, 129.3, 129.0, 126.7, 124.3, 121.7, 119.9, 114.6, 113.8, 112.6, 109.1, 97.5, 68.1 (CH2), 56.2 (OCH3), 55.8 (OCH3), 35.3 (NCH3). Anal. Calcd. for C29H25N3O5 (495.52): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.14; H, 5.26; N, 8.36%.
2-(4-(6-(4-Chlorophenyl)-3-cyano-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9d):
White powder, yield: 0.13 g (25%), mp. 203–205 °C; FT-IR (ATR) vmax: 3335 (NH), 3053 (CHarom.), 2963, 2933, 2848 (CHaliph.), 2214 (CN), 1683 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.08 (s, 1H, NH), 7.64–7.62 (m, 6H, CHarom.), 7.36–7.31 (m, 4H, CHarom.), 7.13–7.07 (m, 2H, CHarom.), 6.54 (s, 1H, CHarom.), 4.81 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.34 (s, 3H, NCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.6, 161.5, 157.5, 153.4, 150.0, 149.4, 138.8, 135.5, 133.4, 130.9, 129.2, 129.2, 128.9, 124.1, 121.8, 119.9, 117.2, 114.1, 112.8, 109.0, 98.5, 68.5 (CH2), 56.3 (OCH3), 35.0 (NCH3). Anal. Calcd. for C28H22ClN3O4 (499.94): C, 67.27; H, 4.44; N, 8.40%. Found: C, 67.45; H, 4.27; N, 8.58%.
2-(4-(6-(4-Bromophenyl)-3-cyano-1-methyl-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9e):
White powder, yield: 0.10 g (20%), mp. 210–212 °C; FT-IR (ATR) vmax: 3426 (NH), 3050 (CHarom.), 2956, 2924, 2852 (CHaliph.), 2219 (CN), 1642 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.31 (s, 1H, NH), 8.23(d, 1H, J = 8.3 Hz, CHarom.), 7.77 (d, 2H, J = 8.0 Hz, CHarom.), 7.64 (d, 2H, J = 7.7 Hz, CHarom.), 7.59 (d, 2H, J = 8.1 Hz, CHarom.), 7.36–7.31 (m, 4H, CHarom.), 7.13–7.11 (m, 1H, CHarom.), 6.61–6.54 (m, 1H, CHarom.), 4.83 (s, 2H, CH2), 4.15 (s, 3H, OCH3), 3.24 (s, 3H, NCH3). Anal. Calcd. for C28H22BrN3O4 (544.39): C, 61.77; H, 4.07; N, 7.72%. Found: C, 61.56; H, 4.24; N, 7.51%.
2-(4-(3-Cyano-1-methyl-6-(naphthalen-2-yl)-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9f):
Pale yellow powder, yield: 0.15 g (30%), mp. 193–195 °C; FT-IR (ATR) vmax: 3391 (NH), 3054 (CHarom.), 2940, 2919, 2854 (CHaliph.), 2217 (CN), 1691 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.08 (s,1H, NH), 8.20 (s, 1H, CHarom.), 8.10 (d, 1H, J = 8.5 Hz, CHarom.), 8.03 (d, 2H, J = 7.4 Hz, CHarom.), 7.73–7.62 (m, 5H, CHarom.), 7.39–7.31 (m, 4H, CHarom.), 7.14–7.07 (m, 2H, CHarom.), 6.66 (s, 1H, CHarom.), 4.81 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.41 (s, 3H, NCH3); 13C NMR (100 MHz, DMSO-d6) δ: 166.6, 161.6, 157.6, 154.6, 150.0, 149.4, 138.8, 133.6, 132.8, 132.0, 129.2, 129.1, 128.9, 128.9, 128.7, 128.2, 128.1, 127.5, 125.8, 124.1, 121.8, 119.9, 117.4, 114.2, 112.9, 109.3, 98.3, 68.5 (CH2), 56.3 (OCH3), 35.3 (NCH3). Anal. Calcd. for C32H25N3O4 (515.55): C, 74.55; H, 4.89; N, 8.15%. Found: C, 74.78; H, 4.71; N, 8.29%.
2-(4-(3-Cyano-6-(furan-2-yl)-1-methyl-2-oxo-1,2-dihydropyridin-4-yl)-2-methoxyphenoxy)-N-phenylacetamide (9 g):
Pale yellow powder, yield: 0.155 g (30%), mp. 170–172°C; FT-IR (ATR) vmax: 3373 (NH), 3105 (CHarom.), 2955, 2921, 2851 (CHaliph.)., 2215(CN), 1683 (C = Oamide) cm−1; 1H NMR (400 MHz, CDCl3) δ: 8.66 (s, 1H, NH), 7.59 (s, 1H, CHarom.), 7.53 (d, 2H, J = 7.9 Hz, CHarom.), 7.30–7.27 (m, 3H, CHarom.), 7.16–7.14 (m, 1H, CHarom.), 7.07 (t, 1H, J = 7.3 Hz, CHarom.), 7.00 (d, 1H, J = 8.3 Hz, CHarom.), 6.85 (d, 1H, J = 3.2 Hz, CHarom.), 6.59 (s, 1H, CHarom.), 6.55 (s, 1H, CHarom.), 4.62 (s, 2H, CH2), 3.95 (s, 3H, OCH3), 3.68 (s, 3H, NCH3); 13C NMR (100 MHz, CDCl3) δ: 166.0, 161.5, 157.0, 149.8, 149.7, 149.0, 145.4, 142.7, 138.1, 130.6, 129.1, 124.7, 121.4, 120.0, 115.8, 115.2, 112.3, 112.2, 112.1, 107.4, 93.9, 69.8 (CH2), 56.3 (OCH3), 34.5 (NCH3). Anal. Calcd. for C26H21N3O5 (455.46): C, 68.56; H, 4.65; N, 9.23%. Found: C, 68.37; H, 4.48; N, 9.45%.
5-cyano-6-methoxy-4-(3-methoxy-4-(2-oxo-2-(phenylamino)ethoxy) phenyl)-1'-methyl-2,3'-bipyridin-1'-iumiodide (10):
Yellow powder, yield: 0.46 g (90%), mp. 284–286 °C; FT-IR (ATR) vmax: 3178 (NH), 3061 (CHarom.), 2952, 2830 (CHaliph.), 2220 (CN), 1689 (C = Oamide) cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 10.21 (s,1H, NH), 9.81 (s, 1H, CHarom.), 9.37 (d, 1H, J = 8.1 Hz, CHarom.), 9.11 (d, 1H, J = 5.8 Hz, CHarom.), 8.31 (t, 1H, J = 7.0 Hz, CHarom.), 8.15 (s, 1H, CHarom.), 7.64 (d, 2H, J = 7.8 Hz, CHarom.), 7.48 (s, 1H, CHarom.), 7.40–7.32 (m, 3H, CHarom.), 7.19–7.08 (m, 2H, CHarom.), 4.86 (s, 2H, CH2), 4.48 (s, 3H, N+CH3), 4.23 (s, 3H, OCH3), 3.92 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ: 166.6, 165.1, 157.2, 150.9, 150.0, 149.6, 146.6, 145.1, 143.1, 138.8, 136.5, 129.1, 128.8, 128.2, 124.1, 122.2, 119.9, 115.8, 115.5, 114.5, 113.5, 95.2, 68.5 (CH2), 56.6 (OCH3), 55.6 (OCH3), 48.9 (N+CH3). Anal. Calcd. for C28H25IN4O4 (608.42): C, 55.27; H, 4.14; N, 9.22%. Found: C, 55.13; H, 4.25; N, 9.08%.
Biology
Anticancer
In vitro anti-proliferative assays.
All cell lines were maintained in RPMI1640 medium containing 10% FBS at 37 °C in 5% CO2 humidified incubator. Cell proliferation assay was determined by the MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5- diphenyl-2H-tetrazolium bromide) method [39]. Briefly, cells were plated in triplicate wells (3–5 × 104 cells/well) of 96-well flat-bottomed plates and incubated overnight prior to drug exposure and then treated with different concentrations of the tested compounds for 48 h. After that, 20 μL of MTT reagent at a final concentration of 0.5 mg/mL was added to each well. Cells were then incubated for 2 h with MTT, after which 100 μL of DMSO solution was added to dissolve the formazan salt resulting from the reduction of MTT and the absorbance was read at 570 nm using an automatic plate reader [39]. The IC50 values were calculated according to inhibition ratios from three independent experiments.
PIM-1 kinase inhibitory activity.
The ability of tested compounds for inhibition of PIM-1 kinase enzyme were measured using Promega PIM-1 kinase assay kit (cat # V4032) [40], ADP-Glo™ Reagent and Tecan –spark reader, four concentration (10, 1, 0.1 and 0.01 µM) of the evaluated compounds were prepared using DEMSO as solvent according to prescribed protocol of the manufacturer (Promega corporation, 2800 woods Hollow Road, Madison, WI 537711–5399 USA) [41]. From resulting data IC50 for each compound were calculated using GraphPad Prism software.
Cell cycle analysis.
Cell cycle analysis MCF-7 cell was seeded into six-well plates at a density of 2 × 105 cell per well and incubated for 24 h. The cell was cultured in RPMI 1640 supplemented with fetal bovine serum (FBS, 10%) and incubated at 37 °C and 5% CO2. The medium was removed and replaced with medium (final DMSO concentration, 1% v/v) containing compound 7h (0.283 µM). After incubation for 24 h and 48 h, the cell layer was trypsinized and washed with cold phosphate buffered saline (PBS) and fixed with 70% ethanol. The fixed cells were rinsed with PBS and then stained with the DNA fluorochrome PI in a solution containing Triton X-100 as well as RNase, keep 15 min at 37 °C according to the instruction manual. Then the samples were analyzed with a FACS Caliber flow cytometer (Becton Dickinson & Co., Franklin Lakes, NJ). The number of cells analyzed for each sample was 10,000 [39].
Apoptosis assay.
The MCF-7 was treated with compound 7h for 24 h. After treatment, the cells were suspended in 0.5 mL of PBS, collected by centrifugation, and fixed in ice-cold 70% (v/ v) ethanol, centrifuged the ethanol-suspended cells for 5 min, suspended in 5 mL PBS and centrifuged for 5 min, re-suspended with 1 mL PI staining solution (0.1 mg/ml RNase) + PE Annexin V (component no. 51-65875X) and kept in dark at 37 °C for 10 min, finally analyzed by flow cytometry using FACS caliber (Becton Dickinson). The cell cycle distributions were calculated using Phoenix Flow Systems and Verity Software House [39].
Molecular docking methodology.
AutoDock Vina v.1.2.0 was used for carrying out the molecular docking [42–44]. For more details see supporting information.
Author Contributions
"B.R.M. Hussein and H.H. Mohammed wrote the main manuscript text and O. A. and M.F.M prepared Figs. 3, 4, 5, 6, 7, 8, 9, 10. All authors reviewed the manuscript."
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stazi G, Fioravanti R, Mai A, Mattevi A, Valente S (2019) Histone deacetylases as an epigenetic pillar for the development of hybrid inhibitors in cancer. Curr Opin Chem Biol 50:89–100. 10.1016/j.cbpa.2019.03.002 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S (2014) Drug resistance in cancer: an overview. Cancers 6(3):1769–1792. 10.3390/cancers6031769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asati V, Mahapatra DK, Bharti SK (2019) PIM kinase inhibitors: structural and pharmacological perspectives. Eur J Med Chem 172:95–108. 10.1016/j.ejmech.2019.03.050 [DOI] [PubMed] [Google Scholar]
- 5.Naguib BH, El-Nassan HB, Abdelghany TM (2017) Synthesis of new pyridothienopyrimidinone derivatives as Pim-1 inhibitors. J Enzyme Inhib Med Chem 32(1):457–467. 10.1080/14756366.2016.1261130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nawijn MC, Alendar A, Berns A (2011) For better or for worse: the role of pim oncogenes in tumorigenesis. Nat Rev Cancer 11(1):23–34. 10.1038/nrc2986 [DOI] [PubMed] [Google Scholar]
- 7.Narlik-Grassow M, Blanco-Aparicio C, Carnero A (2014) The PIM family of serine/threonine kinases in cancer. Med Res Rev 34(1):136–159. 10.1002/med.21284 [DOI] [PubMed] [Google Scholar]
- 8.Le BT, Kumarasiri M, Adams JR, Yu M, Milne R, Sykes MJ, Wang S (2015) Targeting pim kinases for cancer treatment: opportunities and challenges. Future Med Chem 7(1):35–53. 10.4155/fmc.14.145 [DOI] [PubMed] [Google Scholar]
- 9.Warfel NA, Kraft AS (2015) PIM kinase (and Akt) biology and signaling in tumors. Pharmacol therapeut 151:41–49. 10.1016/j.pharmthera.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J (2010) PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 95(6):1004–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo S, Mao X, Chen J, Huang B, Jin C, Xu Z, Qiu S (2010) Overexpression of Pim-1 in bladder cancer. J Exp Clin Cancer Res 29(1):161. 10.1186/1756-9966-29-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drygin D, Haddach M, Pierre F, Ryckman DM (2012) Potential use of selective and nonselective pim kinase inhibitors for cancer therapy: miniperspective. J Med Chem 55(19):8199–8208. 10.1021/jm3009234 [DOI] [PubMed] [Google Scholar]
- 13.Foulks JM, Carpenter KJ, Luo B, Xu Y, Senina A, Nix R, Chan A, Clifford A, Wilkes M, Vollmer D (2014) A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia 16(5):403–412. 10.1016/j.neo.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keane NA, Reidy M, Natoni A, Raab MS, O’Dwyer M (2015) Targeting the pim kinases in multiple myeloma. Blood Cancer J 5(7):e325–e325. 10.1038/bcj.2015.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tursynbay Y, Zhang J, Li Z, Tokay T, Zhumadilov Z, Wu D, Xie Y (2016) Pim-1 kinase as cancer drug target: an update. Biomed rep 4(2):140–146. 10.3892/br.2015.561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szydłowski M, Prochorec-Sobieszek M, Szumera-Ciećkiewicz A, Derezińska E, Hoser G, Wasilewska D, Szymańska-Giemza O, Jabłońska E, Białopiotrowicz E, Sewastianik T (2017) Expression of PIM kinases in reed-sternberg cells fosters immune privilege and tumor cell survival in hodgkin lymphoma. Blood 130(12):1418–1429. 10.1182/blood-2017-01-760702 [DOI] [PubMed] [Google Scholar]
- 17.Markou A, Tzanikou E, Strati A, Zavridou M, Mastoraki S, Bournakis E, Lianidou E (2020) PIM-1 is overexpressed at a high frequency in circulating tumor cells from metastatic castration-resistant prostate cancer patients. Cancers 12(5):1188. 10.3390/cancers12051188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Möröy T, Grzeschiczek A, Petzold S, Hartmann KU (1993) Expression of a Pim-1 transgene accelerates lymphoproliferation and inhibits apoptosis in lpr/lpr mice. Pro Natl Acad Sci 90(22):10734–10738. 10.1073/pnas.90.22.10734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amaravadi R, Thompson CB (2005) The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest 115(10):2618–2624. 10.1172/JCI26273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magnuson NS, Wang Z, Ding G, Reeves R (2010) Why target PIM1 for cancer diagnosis and treatment? Future oncol 6(9):1461–1478. 10.2217/fon.10.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiskus WC, Buckley KM, Rao R, Wang Y, Joshi A, Chong DG, Jillella A, Ustun C, Atajada P, Quadt C, Bhalla KN (2009) Synergistic activity of co-treatment with PIM1 kinase inhibitor SGI-1776 and histone deacetylase inhibitor panobinostat or heat shock protein 90 inhibitor AUY922 against human CML and myeloproliferative neoplasm (MPN) cells. Blood 114(22):2651. 10.1182/blood.V114.22.2651.2651 [Google Scholar]
- 22.Chen WW, Chan DC, Donald C, Lilly MB, Kraft AS (2005) Pim family kinases enhance tumor growth of prostate cancer cells. Mol Cancer Res 3(8):443–451. 10.1158/1541-7786.MCR-05-0007 [DOI] [PubMed] [Google Scholar]
- 23.Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, Berns A (2004) Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol 24(13):6104–6115. 10.1128/MCB.24.13.6104-6115.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheney IW, Yan S, Appleby T, Walker H, Vo T, Yao N, Hamatake R, Hong Z, Wu JZ (2007) Identification and structure–activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg Med Chem Lett 17(6):1679–1683. 10.1016/j.bmcl.2006.12.086 [DOI] [PubMed] [Google Scholar]
- 25.Abouzid KA, Al-Ansary GH, El-Naggar AM (2017) Eco-friendly synthesis of novel cyanopyridine derivatives and their anticancer and PIM-1 kinase inhibitory activities. Eur J Med Chem 134:357–365. 10.1016/j.ejmech.2017.04.024 [DOI] [PubMed] [Google Scholar]
- 26.Mansour B, Salem YA, Attallah KM, El-kawy OA, Ibrahim IT, Abdel-Aziz NI (2023) Cyanopyridinone-and cyanopyridine-based cancer cell Pim-1 inhibitors: design, synthesis, radiolabeling, biodistribution, and molecular modeling simulation. ACS Omega 8:19351–19366. 10.1021/acsomega.2c08304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdelaziz ME, El-Miligy MM, Fahmy SM, Mahran MA, Hazzaa AA (2018) Design, synthesis and docking study of pyridine and thieno [2,3-b] pyridine derivatives as anticancer PIM-1 kinase inhibitors. Bioorg Chem 80:674–692. 10.1016/j.bioorg.2018.07.024 [DOI] [PubMed] [Google Scholar]
- 28.Farrag AM, Ibrahim MH, Mehany ABM, Ismail MMF (2020) New cyanopyridine-based scaffold as PIM-1 inhibitors and apoptotic inducers: synthesis and SARs study. Bioorg Chem 105:104378. 10.1016/j.bioorg.2020.104378 [DOI] [PubMed] [Google Scholar]
- 29.Ibrahim MH, Harras MF, Mostafa SK, Mohyeldin SM, Altwaijry N, Sabour R (2022) Development of novel cyanopyridines as PIM-1 kinase inhibitors with potent anti-prostate cancer activity: synthesis, biological evaluation, nanoparticles formulation and molecular dynamics simulation. Bioorg Chem 129:106122. 10.1016/j.bioorg.2022.106122 [DOI] [PubMed] [Google Scholar]
- 30.Mumenthaler SM, Ng PY, Hodge A, Bearss D, Berk G, Kanekal S, Redkar S, Taverna P, Agus DB, Jain A (2009) Pharmacologic inhibition of Pim kinases alters prostate cancer cell growth and resensitizes chemoresistant cells to taxanes. Mol Cancer Therapeut 8(10):2882–2893. 10.1158/1535-7163.MCT-09-0293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussein BRM, Ali AM (2019) Multicomponent reaction for synthesis of novel 2-tosyloxyphenylpyridines. J Heterocycl Chem 56:1420–1425. 10.1002/jhet.3521 [Google Scholar]
- 32.Ghattas AEBA, Khodairy A, Moustafa HM, Hussein BRM (2017) New heterocyclic compounds derived from 4,6-diamino-3-cyano-2-methylthiopyridine and their biological activity. J Heterocycl Chem 54:879–888. 10.1002/jhet.2649 [Google Scholar]
- 33.Ghattas AEBA, Khodairy A, Moustafa HM, Hussein BRM, Farghaly MM, Aboelez MO (2017) Synthesis, in vitro antibacterial and in vivo anti-inflammatory activity of some new pyridines. Pharm Chem J 51:652–660. 10.1007/s11094-017-1670-8 [Google Scholar]
- 34.Hussein BRM, El-Saghier SM, Allam RM, Mohamed MF, Amer AA (2024) An efficient methodological approach for synthesis of selenopyridines: generation, reactions, anticancer activity, EGFR inhibitory activity and molecular docking studies. Mol Divers. 10.1007/s11030-024-10872-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bass AK, Abdelhafez E, El-Zoghbi M, Mohamed MF, Badr M, Abuo-Rahma GEDA (2021) 3-Cyano-2-oxa-pyridines: a promising template for diverse pharmacological activities. J Adv Biomedical Pharm Sci 4(2):81–86. 10.21608/jabps.2020.52641.1113 [Google Scholar]
- 36.Bass AK, Nageeb ESM, El-Zoghbi MS, Mohamed MF, Badr M, Abuo-Rahma GEDA (2022) Utilization of cyanopyridine in design and synthesis of first-in-class anticancer dual acting PIM-1 kinase/HDAC inhibitors. Bioorg Chem 119:105564. 10.1016/j.bioorg.2021.105564 [DOI] [PubMed] [Google Scholar]
- 37.Petrović MM, Roschger C, Chaudary S, Zierer A, Mladenović M, Marković V, Trifunović S, Joksović MD (2021) Low cytotoxic quinoline-4-carboxylic acids derived from vanillin precursors as potential human dihydroorotate dehydrogenase inhibitors. Bioorg Med Chem Lett 46:128194. 10.1016/j.bmcl.2021.128194 [DOI] [PubMed] [Google Scholar]
- 38.Mohamed SK, Ahsin A, Rehman HM, Mohammed HH, Mague JT, Al-Salahi R, El Bakri Y, Hussein BRM (2024) XRD/DFT, Hirshfeld surface analysis and molecular modelling simulations for unfolding reactivity of newly synthesized vanillin derivatives: excellent optical, NLO and protein binding efficiency. J Biomol Struct Dyn. 10.1080/07391102.2024.2308774 [DOI] [PubMed] [Google Scholar]
- 39.Sobottka SB, Berger MR (1992) Assessment of antineoplastic agents by MTT assay: partial underestimation of antiproliferative properties. Cancer Chemother Pharmacol 30:385–393. 10.1007/BF00689967 [DOI] [PubMed] [Google Scholar]
- 40.Company P (2020) PIM1 Kinase Enzyme System, Available from: https://www.promega.in/products/cell-signaling/kinase-assays-and-kinase-biology/pim1-kinase-enzyme-system/?catNum=V4032. (Accessed 11/2020).
- 41.Alves J, Goueli SA, Zegzouti H (2020) Promega corporation, PIM1 kinase assay. Available from: <https://www.promega.com/-/media/files/resources/protocols/kinase-enzyme-appnotes/pim1-kinase-assay.pdf?la=en>. (Accessed 11/2020).
- 42.Blessy JJ, Sharmila DJS (2015) Molecular simulation of N-acetylneuraminic acid analogs and molecular dynamics studies of cholera toxin-Neu5Gc complex. J Biomol Struct Dyn 33(5):1126–1139. 10.1080/07391102.2014.931825 [DOI] [PubMed] [Google Scholar]
- 43.Trott O, Vina AOA (2010) Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeleke D, Eswaramoorthy R, Belay Z, Melaku Y (2020) Synthesis and antibacterial, antioxidant, and molecular docking analysis of some novel quinoline derivatives. J Chem 1:1324096. 10.1155/2020/1324096 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analysed during the current study.