

Abstract
The Simplified Molecular Input Line Entry System (SMILES) is one of the most widely adopted molecular representations. However, SMILES notation suffers from limited token diversity and a lack of chemical information within individual tokens. To address these limitations while maintaining its simplicity, we propose a molecular representation method through the hybridization of standard SMILES tokens with Atom-In-SMILES (AIS) tokens, which incorporate local chemical environment information into a single token. This hybrid representation, termed SMI + AIS, combines SMILES and AIS tokens, allowing AIS tokens to differentiate chemical elements based on their chemical context without introducing additional tokens for less frequent elements. Using the SMI + AIS representation, we evaluated its performance by comparing the predefined metric of generated structures in chemical structure generation based on latent space optimization. Compared to standard SMILES, SMI + AIS achieved a 7% improvement in binding affinity and a 6% increase in synthesizability, highlighting its utility in the enhancement of machine learning-based molecular design. Our results demonstrate that the SMI + AIS representation provides a more effective and informative approach to encapsulate chemical context and presents potential for performance enhancement in other machine learning tasks in chemistry.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-01890-7.
Keywords: SMILES, Molecular representation, Generative modeling, Small-molecule drug, Latent space optimization, Drug discovery
Subject terms: Drug discovery, Chemistry, Mathematics and computing
Introduction
AI is actively utilized for exploring large and complex chemical space1,2. To effectively apply AI, existing chemical representations need to evolve to become more AI-friendly. While graph-based machine learning models have been developed to directly learn chemical connectivity3–6, there have also been numerous attempts to represent compounds using string-based methods. These methods represent chemical structures as a sequence of characters, analogous to natural language where artificial intelligence achieves incredible successes. Among these, the Simplified Molecular Input Line Entry System (SMILES) is a string-based representation which is most widely adopted for organic compounds. SMILES encodes molecules as a sequence of atomic symbols and some non-physical characters indicating ring, branch, or stereochemical information. This representation is used in many prominent chemical databases such as GDB and PubChem due to its simplicity7,8
SMILES has been extensively applied in machine learning tasks for predicting molecular properties. The SMILES representation was used to train a VAE model that demonstrates the ability to generate novel chemical compounds with desirable properties such as water-octanol partition coefficient (logP) and the Quantitative Estimate of Drug-likeness (QED)9. This descriptor is also widely utilized in web-based platforms for evaluating absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, further highlighting its versatility and significance in computational chemistry and drug discovery10–13. Despite many application examples for SMILES, SMILES has inherent limitations, including non-unique representations of the same molecule, potential for generating invalid structures, an absence of lattice information for periodic system and insufficient chemical context in individual tokens14–17. To address these issues, the fragment-based tokenization methods derived from SMILES have been proposed for various applications in chemistry18,19. Moreover, alternative representations such as SELFIES and InChI have also received notable attention20,21. InChI provides a unique identifier for each structure and SELFIES guarantees chemical validity of encoded strings, which means any string flows SELFIES syntax should be mapped to a valid chemical structure. Although these new representations overcome some limitations of SMILES, in aspect of token diversity, their token sets are not much different because their token is mapped to a single atom or bond. As a result, token diversity remains inherently restricted to the available atom and bond types, which is significantly simpler than the complexity of chemical space. To overcome extend the token diversity, AIS representation method that enriches molecular tokens by incorporating neighboring atoms and ring information into individual tokens, was introduced22. AIS method demonstrated superior performance in both regression and classification tasks of the MoleculeNet benchmark set for property prediction, outperforming SMILES and SELFIES. This highlights that AIS token provides a more informative and expressive representation with consistently low repetition rates which were measured by the ratio of single-token repetitions to the total sequence.
In this work, we propose a seamless hybridization method to incorporate chemical environment into tokens without losing simplicity of SMILES and validate its suitability in molecular structure generation as proof of concept. We construct a hybrid representation by selectively replacing certain SMILES tokens with the most frequent AIS tokens identified from the ZINC database. This hybridization enables the combination of SMILES tokens with AIS tokens to capture critical chemical environment details while preserving the simplicity and readability of SMILES. In drug discovery, molecular structure generation is a challenging machine learning task that focuses on the De novo design and optimization of molecular structures to achieve specific properties. (e.g., activity, solubility, toxicity, etc23–26.) For small molecule drug design, this structure optimization aims to simultaneously maximize synthetic accessibility and binding affinity. To demonstrate the potential of our method, we conduct structure optimization using a language model-based approach and confirm competitiveness of our new representation method.
Results
SMI + AIS(N) representation
The SMILES descriptor represents molecular structures by serially enumerate atomic symbols (Element token) and some characters (Non-physical token) that indicate connectivities with neighboring atoms. Each element token represents an atom type and indicates whether the atom is a part of aromatic system in certain cases. On the other hand, the AIS representation method adopts SMILES grammar without changes but the element token in the AIS method incorporates not only the atomic symbol but also additional information for chemical environment. Figure 1 shows the composition of an AIS token and an example case for SMILES (SMI) and AIS representations for a molecule. AIS token is delimited by semicolons and describes three key aspects of the atomic environment. The first component denotes the elemental symbol of the central atom. The second component indicates the ring information. When the central atom is not included in a ring, it is expressed as ‘!R’, otherwise ‘R’. The final element is the neighboring atoms connected to the central atom. This additional information in AIS tokens can be categorized precisely into numerous AIS tokens while retaining the SMILES grammar and some non-physical tokens. Although AIS method provides a more detailed distinction between atoms considering their chemical environment, AIS tokens are identical to SMI tokens in that one AIS token corresponds to a single atom. Therefore, SMI tokens are replaceable by AIS tokens without any changes in SMI grammar. This fact allows us to build a hybrid representation of AIS tokens with the SMI method, named SMI + AIS(N) where N represents the number of AIS tokens to be introduced. To build the hybridization token set of SMI + AIS(N) method, all SMI strings in the ZINC database are first converted into their corresponding AIS tokens. Subsequently, the frequency of each AIS token is counted. Based on these frequencies, the top-N most frequent AIS tokens are selected and included in the vocabulary, while all other tokens are represented as standard SMI notation. In this work, we set N as 50, 100, 150, and 200.
Fig. 1.

Overview of SMILES, AIS and their hybridization for a benzoic acid. (Right) The molecular structure of benzoic acid and (Bottom) its string representation using SMILES (SMI) and SMI + AIS methods are illustrated. (Left) Individual SMILES and AIS tokens for the oxygen in carboxyl group are noted. The AIS token includes atomic environmental information (central atom;ring-formation;neighbor atom).
Mitigation of token frequency imbalance
Introduced chemical-environment-aware AIS tokens cause a change in the token frequency distribution. Figure 2 illustrates the frequency distributions of the ZINC database using standard SMILES and various SMI + AIS(N) representations where N is the number of added AIS tokens. When only SMI tokens are used to represent molecules, as shown in Fig. 2a, a huge imbalance in the frequency distribution is observed. As shown in Fig. 2b–e, this huge imbalance is mitigated by the introduction of AIS tokens whose frequencies are represented as green bars. This is because one frequently observed SMILES token (e,g. C) is replaced into many AIS tokens that are distinguished by the chemical environment in the vicinity of the atom (e.g. [cH;R;CC], [c;R;CCC], and [CH3;!R;C], etc.). To determine the token set of AIS + SMI(N), we select the most frequently appearing AIS tokens when the ZINC database is represented with only AIS tokens. The selected AIS tokens effectively lower the frequency of highly populated SMILES tokens. Therefore, most of blue bars in Fig. 2 in the top rank region disappear and, reversely, green bars appear.
Fig. 2.

Token frequency distribution of the database using AIS + SMI(N) representation. The distributions are shown for (a) SMI, (b) SMI + AIS(50), (c) SMI + AIS(100), (d) SMI + AIS(150), and (e) SMI + AIS(200) representations where the numbers in the parenthesis indicate the numbers of added AIS tokens. Red, green, and blue bars indicate non-physical tokens, AIS tokens, and SMI element tokens, respectively.
The replacement of AIS tokens for frequently occurring SMILES tokens is more evident in Table 1. This table presents the frequencies of selected elements and the number of token types used to represent each element in SMI + AIS(N) method. In the standard SMILES representation, the number of token types per element is not proportional to its frequency. For instance, the most frequent element, C is represented by 16 token types, whereas Sn which appears only 1,613 times, is represented by 10 token types. However, this imbalance is mitigated by the introduction of AIS tokens. Relatively common elements (e.g., C, O, and N) are more likely to encounter diverse chemical environments. The SMI + AIS(N) representation utilizes AIS tokens to distinguish the element in different chemical environments, resulting in an increase in the number of token types to represent the element. In contrast, the SMILES representation uses only a limited number of tokens for each element. The numbers in parentheses in Table 1 indicate the percentage of AIS token frequency relative to the total frequency of the elements. In the SMI + AIS(N) representation, elements that encounter diverse chemical environments are represented with many AIS tokens, and thus the proportion of AIS tokens relative to the total occurrence of these elements is also higher. Conversely, for elements with fewer chemical environments (e.g., halogen atoms), the number of token types does not increase significantly as N increases, and the percentage of AIS tokens remains very low. This indicates that the addition of AIS tokens selectively incorporates tokens considering frequencies of chemical context without unnecessary tokens, which can negatively impact training of machine learning model. The overuse of token types can exacerbate data sparsity, especially for uncommon atomic species. Notably, it is important to set an appropriate a token set to yield the best performance of machine learning. In the SMI + AIS(N) representation, the total token set can be effectively managed by selecting an optimal value for N, resulting in more balanced token distribution is obtained. For ZINC database, 100–150 is a reasonable N to represent chemical structures effectively.
Table 1.
Frequency and Numbers of Token Types for Representative Elements in ZINC database using SMILES and SMI + AIS(N) methods.
| Element | Frequency | Number of token types (Percentage of the case represented by AIS tokens, %) |
||||
|---|---|---|---|---|---|---|
| SMILES | SMI + AIS(50) |
SMI + AIS(100) |
SMI + AIS(150) |
SMI + AIS(200) |
||
| C | 183,860,954 | 16 | 46 (68) | 78 (73) | 114 (74) | 145 (74) |
| O | 27,270,229 | 8 | 13 (10) | 16 (11) | 17 (11) | 24 (11) |
| N | 26,022,928 | 11 | 19 (8) | 32 (1) | 38 (10) | 46 (10) |
| Xa | 6,137,030 | 7 | 10 (2) | 10 (2) | 11 (2) | 11 (2) |
| S | 4,581,307 | 12 | 15 (1) | 17 (2) | 22 (2) | 24 (2) |
| P | 21,704 | 11 | 11 (0) | 11 (0) | 11 (0) | 11 (0) |
| B | 10,316 | 7 | 7 (0) | 7 (0) | 7 (0) | 7 (0) |
| Si | 4,601 | 5 | 5 (0) | 5 (0) | 5 (0) | 5 (0) |
| Sn | 1,613 | 10 | 10 (0) | 10 (0) | 10 (0) | 10 (0) |
aX: Halogen (including F, Cl, Br, and I).
Overview of molecular structure generation
To investigate the effect of SMI + AIS(N) representation in a machine learning task, we conduct molecular structure generation which is a challenging problem in many different fields of chemistry9,23,27,28. Figure 3 shows a workflow of chemical structure generation task. We investigate the optimization performance using two reference methods (SMILES, and SELFIES) and our hybrid method (SMI + AIS). SMILES and SELFIES representations are well-established baseline methods for evaluating chemical language models29. Many different strategies to optimize chemical structure have been proposed23–26,30. Here, we employ latent space optimization with Bayesian Optimization (BO) to generate molecular structure having high synthetic accessibility and strong binding affinity. The trained encoder converts initial molecular structures to latent vectors and BO finds candidate vectors based on objective values of initial compounds. (Details for training of CVAE encoder & decoder and selection of initial compound are explained in Methods section) The decoder translates the latent vectors suggested from BO to string representations. The overall molecular structure generations are iteratively performed by repeatedly conducting BO with generated chemical structures from previous BO. For each step, we check the validity of generated representations and calculate objective values (Obj. Val. in Fig. 3) The object value is defined as  where BA and SA refer to binding affinity score and synthetic accessibility score, respectively. A low (large negative) BA indicates strong binding affinity, and a low SA indicates ease of synthesis. In the process of objective value computations, some of generated strings are not matched to chemical structure due to syntax errors. For those cases, a huge negative value (-100) is set as the objective value to discourage additional exploration of the associated latent space where a latent vector is not translated into a normal chemical structure. Otherwise, the objective values are computed from SA and BA. By iteratively updating latent vectors, BO yields chemical compound having high objective values.
where BA and SA refer to binding affinity score and synthetic accessibility score, respectively. A low (large negative) BA indicates strong binding affinity, and a low SA indicates ease of synthesis. In the process of objective value computations, some of generated strings are not matched to chemical structure due to syntax errors. For those cases, a huge negative value (-100) is set as the objective value to discourage additional exploration of the associated latent space where a latent vector is not translated into a normal chemical structure. Otherwise, the objective values are computed from SA and BA. By iteratively updating latent vectors, BO yields chemical compound having high objective values.
Fig. 3.

Workflow of molecular structure generation. The ‘Calculate Objectives’ step computes the objective value, designed as 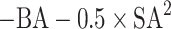 where BA and SA indicate binding affinity and synthetic accessibility score, respectively. The ‘Bayesian Opt’ step generates candidate vectors and ‘Syntax Validation Check’ step verifies grammar of all generated string representations using RDKit. ‘Encoder’ and ‘Decoder’ refer to the components of conditional variational autoencoder that convert string to latent vector and back, respectively. The Bayesian optimization is iteratively performed incorporating updated information from the generated molecular representations.
where BA and SA indicate binding affinity and synthetic accessibility score, respectively. The ‘Bayesian Opt’ step generates candidate vectors and ‘Syntax Validation Check’ step verifies grammar of all generated string representations using RDKit. ‘Encoder’ and ‘Decoder’ refer to the components of conditional variational autoencoder that convert string to latent vector and back, respectively. The Bayesian optimization is iteratively performed incorporating updated information from the generated molecular representations.
Performance of molecular structure generation using various representations
For the aforementioned molecular structure generation task, we investigate the performances of two reference representations (SMILES and SELFIES) and our hybrid representation with varying numbers of AIS tokens. The optimization process began with the random selection of five compounds from BindingDB31 for the target protein. The initial molecular structures for PDK4 protein are shown in Fig. 4a. To ensure a fair comparison, we repeated the optimization process independently 10 times under identical conditions using the same set of initial compounds because the latent space optimization process is inherently stochastic. Molecular structure generations were performed using four proteins: PDK4, 5-HT1B, PARP1, and CK1d. For simplicity, we first discuss and analyze the results for PDK4, while the results for other proteins are provided in Figs. S1–S3.
Fig. 4.

Molecular structure generation results for PDK4 target with various representation methods. (a) Initial structures and their properties (BA, SA, and MW indicates binding affinity, synthetic accessibility and molecular weights, respectively) (b) The distributions of objective values of Top-1 structures obtained from 10 independently performed optimizations. Left, mid and right subplot shows the optimized Top-1 results in 1, 3, 5 iterations. The red line denoted the maximum objective value among the initial compounds. (c) The 10 Top-1 structures and their molecular properties derived from 10 independent optimizations with SMI + AIS(100) representation. The red and green colors in (a) and (c) indicates acetamide and piperidine, respectively.
Figure 4b illustrates the distribution of objective value from ten-independent optimization tasks aimed at maximizing SA and BA for PDK4. The subplots on the left, middle and right show the Top-1 objective values distribution at 1, 3, and 5 iterations, respectively. The red line indicates the maximum objective value among the initial compounds. All representation methods in Fig. 4b demonstrate a consistent upward trend in the Top-1 objective values with continuing iterations. Additionally, most of Top-1 scores at 5th iteration exceed the maximum objective values of the initial compounds, confirming that the optimization process improves structural performance across all representations. However, the extent of improvement varies between representation methods.
The SMI + AIS(100) representation achieves the highest objective values, while SMILES and SELFIES show only modest improvements. We believe this is due to the ability of SMI + AIS(N) to distinguish chemical context more precisely through 100 additional AIS token types which are frequently observed in ZINC database. This interpretation is supported by the validity ratios in Table S1. For example, the validity ratio of SMI (defined as the proportion of generated strings that are successfully converted to valid compounds without syntax errors and for which SA and BA are calculable) is approximately 40%. In contrast, SMI + AIS showed significant improvements in validity ratios as N increases. However, with SMI + AIS(200), the validity ratio is saturated, leading to slightly lower optimization performance compared to SMI + AIS(150). This decline is likely attributed to the excessive introduction of infrequent tokens, which seem insufficiently trained and could interfere with the latent space optimization. SELFIES, while exhibiting the highest validity ratio, fails to generate chemically meaningful structures with desirable BA and SA scores. We speculate that this limitation stems from the unique grammar of SELFIES, in which the meaning of a token is highly context-dependent. Because of this sensitivity, minor variations in the latent space can result in significant and often undesirable structural alterations.
Figure 4c presents the chemical structures of the Top-1 cases from 10 optimization runs. Some molecular fragments were observed not only in the initial molecular structures but also in the optimized molecules. This suggests that, even without explicitly applying fragment constraints during the optimization process, the model can preserve chemically meaningful structural motifs, potentially enhancing its practical applicability in drug design. Notably, the retained molecular fragments (acetamide and piperidine) have been frequently reported in other drugs and are highlighted in red and green, respectively. Additionally, the molecular weights (MW) of the visualized structures are approximately 400 amu, consistent with the conditions set in the CVAE model. This trend is observed not only in the Top-1 structures but also across all optimized structures, as shown in Fig. S4. While the MW distribution of the training database is relatively broad with dual peaks around 330 and 410 amu, that of the optimized compounds from SMI + AIS(N) exhibits a narrow Gaussian distribution clearly centered near 400 amu, which indicates the condition in generative modeling works well. The visualized structures exhibit BA scores between − 10 and − 9 and SA scores between 2.0 and 2.3.
We provide a more detailed analysis of the impact of molecular representations on optimization by displaying BA and SA of all generated structures in a 2D density map shown in Fig. 5. To clearly visualize the success of multi-target (BA and SA) optimization, we present the results of a single molecular structure generation that yield maximum Top-1 score among 10 independent optimizations. Initial compounds’ BA and SA values are visualized as red crosses and those of Top-1 structures are marked as black stars. The SAs from the optimization with SMI shows lower values (more synthesizable) than those from the SELFIES case but BA values are not much different. However, SMI + AIS(100) show noticeably low BA and SA simultaneously, which means many synthetically feasible molecules with strong binding affinity are generated from the optimization result. These improvements are consistently observed with SMI + AIS(N) as well as other target systems and the distributions from the optimization with other SMI + AIS representations are visualized in Figs. S5–S8. This difference in optimization results shows that the choice of representation can have a significant impact not only the model training but also on the applications of a trained model, and that the SMI + AIS representation can be a one of competitive candidates.
Fig. 5.

Synthetic accessibility (SA) and binding affinity (BA) of generated compounds targeting PDK4. The plots show 2D density maps of molecules generated with (a) SELFIES, (b) SMI, and (c) SMI + AIS(100). Red crosses represent the scores of initial compounds, while black stars indicate the scores of Top-1 optimized structures.
Molecular structure generation results for representative protein targets
To validate transferability of our representation to other protein targets, we carried out structure generations using four representative protein targets (PDK4, 5-HT1B, PARP1, and CK1d). Figure 6 shows the objective value distributions of Top-1, Top-10 and Top-100 structures from 10 independently performed optimizations. In case of the Top-1 distribution for 5-HT1B, there is a slight difference between SMI and SMI + AIS(100). However, for other targets, significant improvements of objective values are observed. These observations demonstrate that our proposed hybrid method is target-independent and can be widely applied to discover and prioritize numerous molecules with desirable properties.
Fig. 6.

The distributions of Top-k objective values from the optimizations using 4 different protein targets. (a) Top-1, (b) Top-10, and (c) Top-100 results from 10 independent molecular generations are displayed. The green- and orange-filled regions denote the distributions of objective values from optimizations with SMI and SMI + AIS(100), respectively.
Discussion
SMILES is one of the most widely used molecular string representations for small organic compounds due to its simplicity. Nonetheless, it has several limitations, including non-unique representations, the possibility of invalid string, and the lack of chemical information within individual tokens. To address these issues, various alternative string-based representations have been proposed, such as SELFIES20, InChI21, and DeepSMILES14. These approaches aim to overcome the constraints of SMILES, but SMILES is still commonly adopted in many molecular databases7,31–33.
In this work, we introduce a hybrid representation method, termed SMI + AIS, which combines SMILES with Atom-Informed SMILES (AIS). This hybrid representation utilizes AIS tokens that are frequently observed in a training dataset. By extending SMILES token set, we construct fine-grained representation for ZINC database while maintaining compatibility with SMILES syntax. The SMI + AIS(N) method provides a systematic way to control the number of additional token types while ensuring compatibility with standard SMILES. Its token set is constructed as the union of conventional SMILES tokens and frequently used AIS tokens, which offer enhanced atom-level differentiation by considering the chemical environment. By employing the SMI + AIS(N) representation, the token imbalance inherent in traditional SMILES is mitigated.
We applied this hybrid method to improve the performance of machine learning models for molecular structure generation task. Latent space optimization using SMI + AIS representation generated chemical structures that are both more synthetically plausible and exhibit stronger binding affinities to protein targets compared to results from SELFIES and standard SMILES. Specifically, structures derived from SMI + AIS representation achieved approximately a 12% improvement over standard SMILES and a 28% improvement over SELFIES based on the median of Top-1 objective values. Consistent performance enhancements were observed across four different protein families.
Our proposed method offers both expanded token diversity and seamless compatibility with string-based chemical language models. However, SMI + AIS method still captures insufficient context for stereochemistry consistent with SMILES. Additionally, this study focused on molecular generation due to limitations in computing power and time, future work will include diverse benchmarking tests and experimental validation. We hope that this method will be further explored and validated in broader scientific context.
Conclusions
In this work, we propose a seamless hybridization of SMILES by incorporating additional tokens that can distinguish atoms considering their chemical environment. The proposed representation, referred to as SMI + AIS(N), offers notable advantages: (1) Each token encodes more comprehensive information on the chemical environment compared to traditional SMILES; (2) Bidirectional conversion with the standard SMILES is strictly preserved; (3) The number of additional token types is systematically and easily controlled. To validate utility of SMI + AIS(N) for machine learning tasks, we applied it to a molecular structure generation task aimed at optimizing both chemical synthesizability and binding affinity to a target protein. Compared to the other two baseline representation methods (standard SMILES, and SELFIES), SMI + AIS(N) representation achieves the highest scores in both synthesizability and binding affinity to protein targets.
Beyond performance in specific tasks, chemically expressive and structured language representations can offer insights into the kinds of chemical principles that AI models are capable of learning. Improving their interpretability could not only enhance model performance but also contribute to a deeper scientific understanding, potentially inspiring novel ideas for experts in chemistry. This work may contribute to the development of interpretable and chemically informed language representations by providing a more expressive encoding of molecular structures.
Methods
Conditional autoencoder
Encoder and decoder models are responsible for mapping strings to latent vectors and back for latent space optimization (The overview of our model architecture as shown in Fig. S9). The encoder used in this study is a typical BERT encoder that consist of entity, positional embeddings and multi-head attention layers. The last layer of encoder yields mean and standard deviation vectors on latent space. The decoder transforms the sampled latent vectors with the condition vector into token indices using gated recurrent units (GRU). The encoder is stacked with 4 multi-head attention layers and each layer utilizes 8 heads. For decoder, 4 stacked GRU layers are used. For all tested representations, conditional autoencoder model was trained under identical conditions for 20 epochs. The hyperparameters used for training the conditional autoencoder are summarized in Table S2.
Protein targets
The molecular structure generation is carried out within the binding pocket of the protein, which makes the selection of target proteins critical. The protein structures used for optimization are chosen to investigate applicability on various types of binding pockets. We choose four protein targets to design small-molecule drug, pyruvate dehydrogenase kinases 4 (PDK4), 5-hydroxytryptamine receptor 1B (5-HT1B), Poly (ADP-ribose) polymerase 1 (PARP1) and Casein kinase I isoform delta (CK1d). The protein structures of PDK4, 5-HT1B, PARP1, and CK1d used in this study were obtained from the Protein Data Bank (PDB) with the respective PDB IDs 4V26, 4IAQ, 6I8M, and 4TN634–36. All of these structures are co-crystallized structures available in the PDB database. Our targets cover a variety of conformations of binding pockets from narrow and deep-positioning to shallow and open sites. PDK4 and PARP1 possess binding sites suitable for binding small and flexible molecules37,38, while CK1d has an accessible and broad conformation which has been reported to accommodate various chemical scaffolds39. We also adopted the 5-HT1B as a representative GPCR family in which the binding pockets are mainly embedded within cell membrane and defined by a shallow and open conformation with hydrophobic residues35,40,41.
Latent space optimization
To explore the chemical space, we employ Bayesian Optimization on latent space constructed by training a CVAE. The optimizer is designed to maximize objective values, defined as  by searching optimal mean and standard deviation vectors which are the outputs of encoder. The binding affinity is calculated as a docking score from the qvina02 program42. Because the docking simulation rely on stochastic processes therefore, we perform docking simulations 10 times for each ligand to set binding affinity. For the docking simulation, we define the binding pocket of the target based on a ligand position in the PDB structure and set a pocket size to 25 Å in all directions. Further details in the docking simulation are included in Table S3. The SA score indicates how difficult the ligand is synthesized and it is freely accessible in RDKit package43,44.
by searching optimal mean and standard deviation vectors which are the outputs of encoder. The binding affinity is calculated as a docking score from the qvina02 program42. Because the docking simulation rely on stochastic processes therefore, we perform docking simulations 10 times for each ligand to set binding affinity. For the docking simulation, we define the binding pocket of the target based on a ligand position in the PDB structure and set a pocket size to 25 Å in all directions. Further details in the docking simulation are included in Table S3. The SA score indicates how difficult the ligand is synthesized and it is freely accessible in RDKit package43,44.
For BO, we use the BoTorch, an open-source library built on PyTorch for efficient Monte–Carlo method45. A total of 800 candidate vectors are derived from BO and the corresponding objective values are computed at each optimization iteration. The set of candidate vectors and the objective values are updated, and BO recommend a set of candidate vector again based on the updated information.
The key hyperparameters for latent space optimization and average wall time for a single structure optimization are presented in Tables S4 and S5, respectively.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by Inha University Research Grant and National Research Foundation of Korea (NRF-2022M3E5F308567821).
Author contributions
M.Y and S.C proposed the main idea and initiated the research. S.C and H.H designed, developed, and evaluated the method. H.H conducted the analysis. S.C and H.H interpreted the results and wrote the paper with editing by M.Y. All authors read and approved the file manuscript.
Data availability
We use ZINC database which is composed of commercially available chemical compounds for virtual screening in drug discovery46. After pre-processing the SMILES strings in the ZINC database through canonicalization and duplicate removal, a total of 9 million compounds are retained for model training. Then, the database are split randomly in a ratio of 8:1:1 as train:valid:test. These datasets and source code can be downloaded from https://github.com/herim-han/AIS-Drug-Opt.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Min Sun Yeom, Email: ms.yeom@namuict.co.kr.
Sunghwan Choi, Email: sunghwanchoi@inha.ac.kr.
References
- 1.Fialkowski, M. et al. Architecture and evolution of organic chemistry. Angew. Chem. Int. Ed.44, 7263–7269. 10.1002/anie.200502272 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Lipkus, A. H. et al. Recent changes in the scaffold diversity of organic chemistry as seen in the CAS registry. J. Org. Chem.84, 13948–13956. 10.1021/acs.joc.9b02111 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Coley, C. W. et al. Convolutional embedding of attributed molecular graphs for physical property prediction. J. Chem. Inf. Model57, 1757–1772. 10.1021/acs.jcim.6b00601 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Kojima, R. et al. kGCN: A graph-based deep learning framework for chemical structures. J. Cheminform.12, 32. 10.1186/s13321-020-00435-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo, Z., Guo, K., Nan, B. et al. Graph-Based Molecular Representation Learning (2022). 10.48550/ARXIV.2207.04869
- 6.Rong, Y., Bian, Y., Xu, T. et al. Self-Supervised Graph Transformer on Large-Scale Molecular Data (2020). 10.48550/ARXIV.2007.02835
- 7.Ruddigkeit, L., Van Deursen, R., Blum, L. C. & Reymond, J.-L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model52, 2864–2875. 10.1021/ci300415d (2012). [DOI] [PubMed] [Google Scholar]
- 8.Kim, S. et al. PubChem substance and compound databases. Nucleic Acids Res.44, D1202–D1213. 10.1093/nar/gkv951 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci.4, 268–276. 10.1021/acscentsci.7b00572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.7, 42717. 10.1038/srep42717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu, L. et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res.52, W422–W431. 10.1093/nar/gkae236 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu, Y. et al. admetSAR3.0: a comprehensive platform for exploration, prediction and optimization of chemical ADMET properties. Nucleic Acids Res.52, W432–W438. 10.1093/nar/gkae298 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swanson, K. et al. ADMET-AI: A machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics40, btae416. 10.1093/bioinformatics/btae416 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Boyle, N. & Dalke, A. DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures (2018). 10.26434/chemrxiv.7097960.v1
- 15.Daylight Chemical Information Systems Inc.; 4. SMARTS-A Language for Describing Molecular Patterns (2007).
- 16.Bucior, B. J. et al. Identification schemes for metal-organic frameworks to enable rapid search and cheminformatics analysis. Cryst. Growth Des.19, 6682–6697. 10.1021/acs.cgd.9b01050 (2019). [Google Scholar]
- 17.Xiao, H. et al. An invertible, invariant crystal representation for inverse design of solid-state materials using generative deep learning. Nat. Commun.14, 7027. 10.1038/s41467-023-42870-7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu, J.-N. et al. t-SMILES: A fragment-based molecular representation framework for de novo ligand design. Nat. Commun.15, 4993. 10.1038/s41467-024-49388-6 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aksamit, N., Tchagang, A., Li, Y. & Ombuki-Berman, B. Hybrid fragment-SMILES tokenization for ADMET prediction in drug discovery. BMC Bioinform.25, 255. 10.1186/s12859-024-05861-z (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krenn, M., Häse, F., Nigam, A. et al. Self-Referencing Embedded Strings (SELFIES): A 100% Robust Molecular String Representation (2019). 10.48550/ARXIV.1905.13741
- 21.Heller, S. R. et al. InChI, the IUPAC international chemical identifier. J. Cheminform.7, 23. 10.1186/s13321-015-0068-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ucak, U. V., Ashyrmamatov, I. & Lee, J. Improving the quality of chemical language model outcomes with atom-in-SMILES tokenization. J. Cheminform.15, 55. 10.1186/s13321-023-00725-9 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abeer, A. N. M. N. et al. Multi-objective latent space optimization of generative molecular design models. Patterns5, 101042. 10.1016/j.patter.2024.101042 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korshunova, M. et al. Generative and reinforcement learning approaches for the automated de novo design of bioactive compounds. Commun. Chem.5, 129. 10.1038/s42004-022-00733-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maziarka, Ł et al. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminform.12, 2. 10.1186/s13321-019-0404-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin, B., Park, S., Bak, J. & Ho, J. C. Controlled Molecule Generator for Optimizing Multiple Chemical Properties (2020). 10.48550/ARXIV.2010.13908 [DOI] [PMC free article] [PubMed]
- 27.Chen, S. et al. Design of target specific peptide inhibitors using generative deep learning and molecular dynamics simulations. Nat. Commun.15, 1611. 10.1038/s41467-024-45766-2 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen, Y. et al. Deep generative model for drug design from protein target sequence. J. Cheminform.15, 38. 10.1186/s13321-023-00702-2 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leon, M. et al. Comparing SMILES and SELFIES tokenization for enhanced chemical language modeling. Sci. Rep.14, 25016. 10.1038/s41598-024-76440-8 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang, M. et al. RELATION: A deep generative model for structure-based de novo drug design. J. Med. Chem.65, 9478–9492. 10.1021/acs.jmedchem.2c00732 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Liu, T. et al. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res.35, D198–D201. 10.1093/nar/gkl999 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fink, T. & Reymond, J.-L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model47, 342–353. 10.1021/ci600423u (2007). [DOI] [PubMed] [Google Scholar]
- 33.Blum, L. C. & Reymond, J.-L. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc.131, 8732–8733. 10.1021/ja902302h (2009). [DOI] [PubMed] [Google Scholar]
- 34.Moore, J. D. et al. VER-246608, a novel pan-isoform ATP competitive inhibitor of pyruvate dehydrogenase kinase, disrupts Warburg metabolism and induces context-dependent cytostasis in cancer cells. Oncotarget5, 12862–12876. 10.18632/oncotarget.2656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang, C. et al. Structural basis for molecular recognition at serotonin receptors. Science340, 610–614. 10.1126/science.1232807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papeo, G. et al. Discovery of stereospecific PARP-1 inhibitor isoindolinone NMS-P515. ACS Med. Chem. Lett.10, 534–538. 10.1021/acsmedchemlett.8b00569 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tso, S.-C. et al. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem.289, 4432–4443. 10.1074/jbc.M113.533885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baptista, S. J. et al. Novel PARP-1 inhibitor scaffolds disclosed by a dynamic structure-based pharmacophore approach. PLoS ONE12, e0170846. 10.1371/journal.pone.0170846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sunkari, Y. K., Meijer, L. & Flajolet, M. The protein kinase CK1: Inhibition, activation, and possible allosteric modulation. Front. Mol. Biosci.9, 916232. 10.3389/fmolb.2022.916232 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasmussen, S. G. F. et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature477, 549–555. 10.1038/nature10361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katritch, V., Cherezov, V. & Stevens, R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol.53, 531–556. 10.1146/annurev-pharmtox-032112-135923 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alhossary, A., Handoko, S. D., Mu, Y. & Kwoh, C.-K. Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics31, 2214–2216. 10.1093/bioinformatics/btv082 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform.1, 8. 10.1186/1758-2946-1-8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.RDKit: Open-source cheminformatics. http:www.rdkit.org
- 45.Balandat, M., Karrer, B., Jiang, D. R. et al. BoTorch: A Framework for Efficient Monte-Carlo Bayesian Optimization (2019). 10.48550/ARXIV.1910.06403
- 46.Sterling, T. & Irwin, J. J. ZINC 15: Ligand discovery for everyone. J. Chem. Inf. Model55, 2324–2337. 10.1021/acs.jcim.5b00559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We use ZINC database which is composed of commercially available chemical compounds for virtual screening in drug discovery46. After pre-processing the SMILES strings in the ZINC database through canonicalization and duplicate removal, a total of 9 million compounds are retained for model training. Then, the database are split randomly in a ratio of 8:1:1 as train:valid:test. These datasets and source code can be downloaded from https://github.com/herim-han/AIS-Drug-Opt.