

Abstract
CD33 rs3865444 polymorphism is closely associated with the risk of Alzheimer’s disease (AD), and CD33 is part of the sialic acid-binding Ig-superfamily of lectins (SIGLECs). Immunostaining experiments in previous studies have confirmed the expression of CD33 in human brain microglial cells, and an increase in CD33 mRNA expression in the brain microglial cells of patients with cognitive impairment has been observed. The minor allele CD33 rs3865444 (A) has a protective effect against Alzheimer’s disease pathology and is associated with reduced CD33 expression and clearance of amyloid-beta plaques. The risk allele CD33 rs3865444 (C) can cause abnormal activation of microglial cells, thereby inducing neuroinflammation, accompanied by an increase in metabolic levels. We hypothesize that the CD33 rs3865444 polymorphism may affect the coupling between glucose metabolism and neuronal activity, thereby influencing individual cognitive trajectories and the progression of cognitive impairment. In this study, we included 107 patients with mild cognitive impairment, among whom the limbic-orbital frontal cortex glucose–oxygen coupling (G/O) coefficient of the CD33 rs3865444 CC group was significantly reduced. Additionally, the results of the mediation analysis showed that the glucose–oxygen coupling coefficient completely mediated the effect of the CD33 rs3865444 polymorphism on the rate of clinical dementia rating increase.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-01927-x.
Keywords: Functional MRI (fMRI), Mild cognitive impairment, Glycometabolism, Oxygen metabolism, CD33 rs3865444, Cognitive trajectory
Subject terms: Computational biology and bioinformatics, Computational neuroscience, Alzheimer's disease
Introduction
The characteristic feature of neurodegenerative diseases is the damage and loss of function of neurons, leading to cognitive decline, with Alzheimer’s disease (AD) being the most common among them. The pathological features of such diseases include the aggregation and deposition of pathological proteins (tau, amyloid-beta) in neurons or extracellular spaces, accompanied by dysfunction of microglial cells. The loss of microglial function leads to the inability to timely clear extracellular amyloid plaques, resulting in toxic effects on neurons1–3 and inducing abnormal and detrimental activation of microglia4,5. This abnormal activation of microglia leads to increased brain glucose metabolism and the generation of reactive oxygen species, causing damage to synapses and neurons, thereby accelerating the progression of cognitive impairment6,7. CD33 encodes sialic acid-binding immunoglobulin-like lectin (Siglec-3). CD33 is closely associated with cellular adhesion processes, inhibition of cytokine release, endocytosis, immune cell growth8, and regulation of Tlr4 signaling9. Previous studies have shown that the protective allele (A) of CD33 rs3865444 is associated with reduced amyloid plaque burden in the brains of individuals with Alzheimer’s disease (AD)10. CD33, an immunomodulatory receptor expressed on microglia, is a susceptibility factor for AD. Located on chromosome 19, the CD33 gene produces two isoforms—long (hCD33M) and short (hCD33m)—whose expression is influenced by single nucleotide polymorphisms and associated with AD risk. Specifically, in carriers of the rs3865444 C allele, the transcriptional ratio of hCD33M to hCD33m is approximately 9:1, whereas in carriers of the AD-protective rs3865444 A allele, the ratio shifts to 7:311. Cells expressing the A allele exhibit reduced hCD33M protein levels11,12. Furthermore, transgenic animal models carrying the rs3865444 A allele show higher expression of hCD33m compared to those carrying the C allele13. Functionally, hCD33m interacts with intracellular signaling molecules to modulate cellular processes such as phagocytosis, migration, and proliferation, while hCD33M, predominantly localized on the cell surface, binds extracellular ligands to regulate immune signaling and inhibit phagocytic activity14. The risk allele (C) of CD33 rs3865444 is associated with reduced clearance capacity of amyloid-beta plaques, and increased AV45 Standardized Uptake Value Ratios (SUVR) values in human brain PET imaging12. Elevated hCD33M levels in the brain are associated with cognitive impairment15 and the increased pathology of neurodegenerative diseases16. Therefore, gene therapy targeting CD33 can diminish amyloid-beta plaques and neuroinflammation17. Under normal circumstances, there is a certain correlation between glucose metabolism and neuronal activity18, and abnormal activation of microglial cells can lead to enhanced glucose metabolism6. Thus, we hypothesize that the glucose-oxygen coupling coefficient can serve as an imaging biomarker for CD33 rs3865444 variation and may predict the progression of cognitive impairment in patients. The development of modern imaging techniques has enabled the visualization of glucose metabolism levels using fluorodeoxyglucose positron emission tomography (FDG PET), and FDG PET signals are closely associated with abnormal microglial cell activation6. In addition to the requirement for glucose, neuronal activity is also accompanied by oxygen consumption, and the amplitude of low frequency fluctuation (ALFF) reflects regional spontaneous blood oxygen level-dependent (BOLD) signal activity intensity. In this study, ALFF (Eq. 1) was used to represent oxygen metabolism.
In this study, we propose the use of the glucose oxygen coupling (G/O, Eq. 2) coefficient to represent the consistency between glucose metabolism and neuronal activity, aiming to better capture changes in the G/O coefficient caused by pathologies such as brain microglial cells. We segmented the FDG PET and normalized whole-brain ALFF distribution maps onto the Schaefer2018_1000Parcels_17Networks brain atlas, calculating the G/O coefficients for 17 brain networks (Fig. 1). Subsequently, participants were divided into two groups based on the CD33 rs3865444 SNP, and networks with significant differences in G/O coefficients between the CD33 rs3865444 CC group and the CD33 rs3865444 CA/AA group were identified (Fig. 1). Additionally, all participants’ follow-up records of ADAS scores and CDR scores were obtained, and based on this, changes in cognitive trajectories between the two groups of patients were compared. Finally, to further clarify the relationship between CD33 rs3865444 SNP, G/O coefficients, and cognitive trajectories, mediation analysis (Eqs. 3, 4, 5) was conducted on the three variables to determine whether the G/O coefficient mediated the effect of CD33 rs3865444 SNP on cognitive trajectories.
Fig. 1.

The experimental workflow of this study is illustrated in the figure. First, preprocessing was conducted on images of various modalities. Subsequently, the G/O coefficient for each network was computed based on the Schaefer 2018_1000Parcels_17Networks brain atlas, and differences in cognitive trajectories among groups were compared. Finally, mediation analysis was performed to determine the relationship between CD33 rs3865444, the G/O coefficient, and cognitive trajectories.
Materials and methods
The single nucleotide polymorphism (SNP) data and multimodal brain imaging data used in this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). The data collection process involved in this study was conducted in accordance with relevant guidelines and regulations. The experimental protocols were approved by the Institutional Review Board of Hangzhou Dianzi University (IRB-2020001) and the Ethics Committee of Beijing Hospital (2022BJYYEC-375-01).
Sample cohort
This study recruited 107 patients with mild cognitive impairment (MCI) from the ADNI database to participate in the research. Participants underwent amyvid (AV45) positron emission tomography (PET) and fluorodeoxyglucose (FDG) PET scans, as well as functional magnetic resonance imaging (fMRI) and T1-weighted MRI scans within one year. In the participant cohort, efforts were made to minimize the time interval between the selected individuals’ FDG PET and AV45 PET images, with fMRI and MRI images chosen to be closest to the FDG PET scanning date, and demographic information for the imaging data is provided in Table 1. The genotype data for the CD33 rs3865444 gene of the participants included in the study were obtained from the Genome-Wide Association Study (GWAS) database in the ADNI archive, with participants undergoing genomic sequencing using the Illumina Omni 2.5 M chip. Details of the SNP data detection methods can be found in the ADNI Genetics Core19. Using the plink command to extract the genotype of rs3865444 from all samples, the results showed that there were 48 participants carrying at least one A allele and 59 samples with the rs3865444 CC genotype. Individuals with homozygous ApoE ε4 have been excluded from this study. The Institutional Review Board authorized ADNI investigators for studies involving human participants. All ADNI sites obtained approval from their respective institutional review boards, and all ADNI project participants provided written informed consent. Access to ADNI data is managed according to the guidelines of the ADNI Data Sharing and Publications Committee.
Table 1.
ADNI sample characteristics.
| rs3865444 CC |
rs3865444 CA |
rs3865444 AA |
p-value | |
|---|---|---|---|---|
| Age | 72.76 ± 4.44 | 72.56 ± 6.70 | 74.23 ± 5.24 | 0.86 |
| Gender (M/F) | 28/31 | 14/28 | 4/2 | 0.18 |
| Education | 16.17 ± 1.86 | 16.38 ± 2.08 | 16.00 ± 0.67 | 0.88 |
| ApoE ε4 pos (1)/neg (0) | 30/29 | 19/23 | 2/4 | 0.53 |
M male, F female, pos positive, neg negative.
Image acquisition and preprocessing
The imaging protocol for structural MRI data of patients in the ADNI Image and Data Archive (IDA) is as follows: A three-dimensional T1-weighted magnetization-prepared rapid acquisition gradient-echo (MPRAGE) sequence was used, with 1 × 1 × 1 mm resolution and a repetition time value of 2300 ms. Functional MRI scans were performed using a three-dimensional echo-planar imaging (EPI) sequence with a voxel resolution of 3.4 mm and a repetition time/echo time/flip angle set to 3000/30/90°. Each participant received a total of 140 volumes of resting-state fMRI data. To obtain amyloid PET images of patients’ brains, 18F-AV45 was injected first, followed by scanning four frames of 5 min each 50–70 min post-injection. For 18F-FDG PET images, six frames of 5 min each were scanned 30 min after the injection of 5.0 mCi 18F-FDG.
This study employed various software processing pipelines for image data analysis. The petsurfer command within the freesurfer software was used to compute the metabolic intensity and amyloid plaque accumulation in various brain regions of the brain atlas. Preprocessing of resting-state functional MRI (fMRI) data was conducted using SPM12, which included temporal correction, motion correction, registration, segmentation, standardization, and other processes20. To obtain data specific to brain functional regions, it is necessary to segment structural MRI images using a brain atlas. In this study, we utilized the Schaefer2018_1000Parcels_17Networks atlas for this purpose21. Standardized Uptake Value Ratios (SUVR) were calculated on segmented anatomical regions of interest (ROIs), which were subsequently normalized to the mean SUVR value relative to the pons22.
Measurement of glucose-oxygen coupling
Functional MRI images mainly collect the blood oxygen level-dependent (BOLD) signal, but they do not directly reflect the intensity of neuronal activity and require frequency-domain transformation23,24. Currently, it is generally believed that the frequency range of neuronal activity signals is 0.01 to 0.08 Hz; signals below 0.01 Hz are linear drift, and those above 0.08 Hz are respiratory or heartbeat noise20,25. Here, we extract signals in the frequency band of 0.01 to 0.08 Hz, using the amplitude of low-frequency fluctuations (ALFF) to indicate the neuronal metabolic level, with the specific formula as follows:
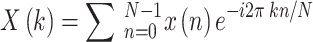 |
1 |
where N represents the number of sampling points and k represents frequency (0 ≦ k ≦ N-1).
Calculate the square root of the power spectrum and the mean of the power spectrum in the time domain to obtain the average value. Finally, normalize the ALFF values using the mean, obtain the ALFF value for each voxel, and generate a three-dimensional heatmap.
In this study, we calculated the glucose-oxygen coupling coefficients for all network modules, using freesurfer commands to compute FDG SUVR and ALFF values for all brain regions within 17 network modules. Subsequently, we computed the correlation coefficients between FDG SUVR and ALFF values for all brain regions within each network module26. The specific formula is as follows:
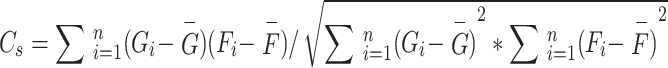 |
2 |
where  represents the glucose-oxygen coupling coefficients of the network module s, n represents the number of brain regions in each network module,
represents the glucose-oxygen coupling coefficients of the network module s, n represents the number of brain regions in each network module,  represents the FDG SUVR of the region i,
represents the FDG SUVR of the region i, 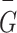 represents the average FDG SUVR of all regions in the network module,
represents the average FDG SUVR of all regions in the network module, 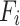 represents the ALFF of the region i, and
represents the ALFF of the region i, and  represents the average ALFF of all regions in the network module.
represents the average ALFF of all regions in the network module.
Cognitive impairment trajectory assessment
All patients underwent clinical dementia assessments. The main clinical assessments included the Alzheimer’s Disease Assessment Scale (ADAS)27 and the Clinical Dementia Rating Scale (CDR)28. The ADAS scale primarily evaluates memory, orientation, attention, and praxis, with a total score ranging from 1 to 5 for each item. The ADAS scale can accurately identify the severity of mild cognitive impairment and mid-stage Alzheimer’s disease. The Clinical Dementia Rating (CDR) is utilized as a tool for evaluating cognitive function in the elderly, particularly for assessing Alzheimer’s disease clinically. The CDR offers certain advantages over other cognitive assessment scales. It is relatively easy to use in practical clinical settings, featuring a questionnaire format that is easy to administer and relatively brief, making it suitable for long-term tracking of patients’ cognitive status. The CDR assesses cognitive function across six different domains, including memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care29. Higher scores on both scales indicate more severe cognitive impairment. The total score of the ADAS scale is 60 points. This study evaluates the combined score of ADAS-cog and ADAS-non-cog. The CDR score is obtained by summing the subscores, with a maximum total score of 21 points. Each participant in this study underwent at least four assessments using the ADAS scale and the CDR scale, with follow-up intervals of one year. The four consecutive ADAS and CDR follow-up records, including the baseline, were used for this study.
Statistical analysis
This study utilized the R software (v4.0.5) for data analysis, with all statistical tests being two-tailed. A comparison was made between the CD33 rs3865444 CC group and the CD33 rs3865444 CA/AA group, with a confidence interval set at 95%. When examining whether the CD33 rs3865444 polymorphism affects glucose-oxygen coupling, the influence of patients’ age, sex, education level, and ApoEε4 status was simultaneously considered. The results were represented using box plots, where the median of each group was depicted by the center line of the box, and the upper and lower edges of the box represented the first and third quartiles (Q1 and Q3) of the data for each group. The cognitive trajectory is represented by the slope of the clinical scale scores over time. The impact of the CD33 rs3865444 polymorphism on cognitive resilience was assessed by comparing the slopes of ADAS scores and CDR scores among participants in each group. To investigate the relationship between rs3865444 SNP, glucose-oxygen coupling coefficients, and cognitive trajectories, we employed mediation analysis to examine whether the association between CD33 rs3865444 polymorphism and the rate of change in ADAS scores and CDR scores is mediated by glucose-oxygen coupling coefficients30. The specific formula is as follows:
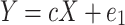 |
3 |
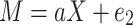 |
4 |
 |
5 |
where c represents the total effect of rs3865444 SNP on cognitive trajectories, a represents the relationship between rs3865444 SNP and the mediator variable glucose-oxygen coupling coefficients, c′ represents the direct effect of rs3865444 SNP on cognitive trajectories after controlling for the glucose-oxygen coupling coefficients, and 10,000 bootstrap simulations were conducted to test the confidence of the results.
Sensitivity analysis was performed using the bootstrap method to examine the relationship between G/O coefficients across all brain regions and the ADAS slope.
 |
6 |
To improve the reliability of the results, this study employed 1000 iterations of bootstrapping.
Results
The relationship between CD33 rs3865444 and G/O coefficients
During the image preprocessing stage, FDG SUVR and ALFF values of all brain regions within each network module from the Schaefer 2018 1000Parcels 17Networks brain atlas were obtained. The correlation coefficient between FDG SUVR values and ALFF values within each brain region of the module was used to represent the glucose-oxygen coupling coefficient of each network module18. Participants were initially divided into two groups based on CD33 rs3865444 polymorphism, and the FDG PET and ALFF images of carriers of CD33 rs3865444 CC and carriers of CD33 rs3865444 CA/AA were rendered and visualized in three dimensions (Fig. 2A,B). From the rendering results, the distribution of FDG SUVR values in both groups appeared relatively uniform, indicating that the differences in glucose metabolism levels across various brain regions were not very pronounced. However, the distribution of ALFF values in the two groups of individuals appeared more scattered, suggesting more pronounced differences in neuronal oxygen metabolism intensities across various brain regions (Fig. 2A,B). To further explore whether the rs3865444 SNP is associated with the correlation between glucose metabolism and oxygen metabolism, we calculated the glucose-oxygen coupling coefficients for carriers of the CD33 rs3865444 CC allele and carriers of the CD33 rs3865444 CA/AA alleles across all networks. The global G/O coefficients of the whole brain were visualized using Freeview in three dimensions (Fig. 3). From a global perspective, initial observations revealed relatively pronounced G/O differences in certain regions of the Salience Network, Limbic, and Default Mode Network in the brains of the two groups of individuals (Fig. 3). Carriers of the CD33 rs3865444 CA/AA alleles exhibited significantly higher G/O coefficients in the limbic-orbital frontal cortex compared to carriers of the CD33 rs3865444 CC allele (Fig. 4A,B, p < 0.01). Although the CD33 rs3865444 SNP also had notable effects on the G/O coefficients in certain regions of the Salience Network and Default Mode Network, after Bonferroni correction, only the correlation between the G/O coefficients in the limbic-orbital frontal cortex and the CD33 rs3865444 SNP remained significant (Fig. 4B, p < 0.01). To further ensure the reliability of the results, we performed sensitivity analyses to identify key brain regions (Eq. 6). A multivariate linear regression analysis of the G/O coefficients across all brain regions and the rs3865444 SNP revealed that the limbic-OFC and default mode networks exhibited higher sensitivity (Table 2). Similarly, a multivariate linear regression analysis of the G/O coefficients across all brain regions and the ADAS slope indicated that the limbic-OFC G/O coefficient had the highest sensitivity (Table 3). Other networks in the brain atlas mainly included the visual system, somatosensory system, and dorsal attention network. These networks exhibited similar trends in G/O coefficients to those described above, with most networks of CD33 rs3865444 CA/AA allele carriers having higher G/O coefficients but without significant statistical differences (Supplementary Table S4).
Fig. 2.

The impact of CD33 rs3865444 polymorphism on FDG SUVR and ALFF distributions. (A) Distribution of FDG SUVR and ALFF per voxel across the whole brain in the CD33 rs3865444 CC group. (B) Distribution of FDG SUVR and ALFF per voxel across the whole brain in the CD33 rs3865444 CA/AA group. Left: left hemisphere; Right: right hemisphere.
Fig. 3.

The impact of CD33 rs3865444 polymorphism on G/O distributions. (A) The distribution of G/O coefficients for each network across the whole brain in the CD33 rs3865444 CC group. (B) The distribution of G/O coefficients for each network across the whole brain in the CD33 rs3865444 CA/AA group. Left: left hemisphere; Right: right hemisphere.
Fig. 4.

The impact of the CD33 rs3865444 SNP on G/O coefficients, glucose metabolism levels, amyloid pathology, and intergroup differences in G/O coefficients within the spectrum of the aforementioned variables was assessed. (A) The anatomical location of the limbic-orbital frontal cortex in the brain. (B) Box plots illustrate the differences in limbic-orbital frontal cortex G/O between carriers of CD33 rs3865444 CC (n = 59) and carriers of CD33 rs3865444 CA/AA (n = 48). (C) Box plots depict the differences in limbic-orbital frontal cortex FDG SUVR between carriers of CD33 rs3865444 CC (n = 59) and carriers of CD33 rs3865444 CA/AA (n = 48). (D) Box plots illustrate the differences in limbic-orbital frontal cortex ALFF between carriers of CD33 rs3865444 CC (n = 59) and carriers of CD33 rs3865444 CA/AA (n = 48). (E) Box plots display the differences in limbic-orbital frontal cortex AV45 SUVR between carriers of CD33 rs3865444 CC (n = 59) and carriers of CD33 rs3865444 CA/AA (n = 48). (F) Box plots illustrate the differences in limbic-orbital frontal cortex G/O between carriers of CD33 rs3865444 CC (n = 47) and carriers of CD33 rs3865444 CA/AA (n = 40) (amyloid-negative cohort). (G) Box plots depict the differences in limbic-orbital frontal cortex FDG SUVR between carriers of CD33 rs3865444 CC (n = 47) and carriers of CD33 rs3865444 CA/AA (n = 40) (amyloid-negative cohort). (H) Box plots illustrate the differences in limbic-orbital frontal cortex ALFF between carriers of CD33 rs3865444 CC (n = 47) and carriers of CD33 rs3865444 CA/AA (n = 40) (amyloid-negative cohort). (I) Box plots display the differences in limbic-orbital frontal cortex AV45 SUVR between carriers of CD33 rs3865444 CC (n = 47) and carriers of CD33 rs3865444 CA/AA (n = 40) (amyloid-negative cohort). Comparison analysis was conducted using t-tests, with significance levels denoted as: *p < 0.05, **p < 0.01, ***p < 0.001.
Table 2.
Sensitivity analysis of G/O coefficients across all brain regions and the CD33 rs3865444 SNP (bootstrapping 1000). Specific names of ROI are shown in table S6 in the supplementary material.
| ROI Index | Original coefficient | Std. Error | Sensitivity |
|---|---|---|---|
| R1 | 0.13 | 0.29 | 0.45 |
| R2 | 0.037 | 0.27 | 0.14 |
| R3 | 0.13 | 0.27 | 0.48 |
| R4 | − 0.11 | 0.27 | 0.41 |
| R5 | 0.21 | 0.32 | 0.66 |
| R6 | − 0.023 | 0.24 | 0.10 |
| R7 | 0.42 | 0.30 | 1.40 |
| R8 | 0.13 | 0.22 | 0.59 |
| R9 | 0.37 | 0.21 | 1.76 |
| R10 | − 0.050 | 0.16 | 0.31 |
| R11 | 0.081 | 0.27 | 0.30 |
| R12 | − 0.070 | 0.24 | 0.29 |
| R13 | 0.047 | 0.19 | 0.25 |
| R14 | 0.096 | 0.31 | 0.31 |
| R15 | − 0.80 | 0.33 | 2.42 |
| R16 | 0.096 | 0.19 | 0.51 |
| R17 | 0.21 | 0.18 | 1.17 |
Table 3.
Sensitivity analysis of G/O coefficients across all brain regions and the slope of clinical dementia scores (bootstrapping 1000). Specific names of ROI are shown in table S6 in the supplementary material.
| ROI Index | Original coefficient | Std. Error | Sensitivity |
|---|---|---|---|
| R1 | − 0.72 | 0.72 | 1.00 |
| R2 | − 0.46 | 0.66 | 0.70 |
| R3 | 0.19 | 0.71 | 0.27 |
| R4 | 0.60 | 0.70 | 0.86 |
| R5 | − 0.29 | 0.79 | 0.37 |
| R6 | − 0.27 | 0.58 | 0.47 |
| R7 | 0.68 | 0.77 | 0.88 |
| R8 | 0.51 | 0.58 | 0.88 |
| R9 | − 1.60 | 0.55 | 2.91 |
| R10 | − 0.44 | 0.40 | 1.10 |
| R11 | − 0.44 | 0.66 | 0.67 |
| R12 | − 0.60 | 0.59 | 1.02 |
| R13 | 0.61 | 0.48 | 1.27 |
| R14 | − 0.26 | 0.81 | 0.32 |
| R15 | 0.49 | 0.89 | 0.55 |
| R16 | 0.30 | 0.50 | 0.60 |
| R17 | 0.0062 | 0.45 | 0.01 |
The relationship between CD33 rs3865444 SNP and single-modal imaging
We further analyzed whether the CD33 rs3865444 SNP is associated with levels of glucose metabolism, oxygen metabolism, and amyloid plaque accumulation, controlling for age, gender, education level, and ApoE ε4 status. Initially, we compared the FDG SUVR, ALFF, and AV45 SUVR values of the limbic-orbital frontal cortex region, where significant differences in G/O coefficients were observed. The results indicated no significant differences between the rs3865444 CC group and the rs3865444 CA/AA group (Fig. 4C–E). To further investigate the impact of the rs3865444 SNP on G/O coefficients in amyloid-negative populations, we selected participants with AV45 SUVR < 1.031 and compared the differences in FDG SUVR, ALFF, AV45 SUVR, and G/O coefficients between individuals with the rs3865444 CC genotype and those with the rs3865444 CA/AA genotype. The results showed that the G/O coefficients in the CD33 rs3865444 CC group were significantly lower than those in the rs3865444 CA/AA group (Fig. 4F, p < 0.05). Similarly, there were no significant differences in FDG SUVR, ALFF, and AV45 SUVR between the two groups in the amyloid-negative population (Fig. 4G–I). In addition, we conducted separate analyses for APOE4 non-carriers and carriers. The results showed no significant differences in metabolic levels or limbic-OFC G/O coefficients between the rs3865444 CC and rs3865444 CA/AA groups in the APOE4 non-carrier population (Supplementary S2A,C). However, among APOE4 carriers, the rs3865444 CC group exhibited higher metabolic levels, and the rs3865444 CA/AA group showed significantly higher limbic-OFC G/O coefficients compared to the CC group (Supplementary S2B, D, p < 0.05), a difference not observed in APOE4 non-carriers. These findings suggest that APOE4 may modulate the effect of the CD33 rs3865444 SNP, and their interaction may contribute to alterations in metabolic levels. Furthermore, there were no significant differences in plasma Abeta40 and plasma Abeta42 levels between the rs3865444 CC group and the rs3865444 CA/AA group (Fig. 5A,B). We also compared the Abeta40/Abeta42 ratio and reached the same conclusion (Fig. 5C). The metabolic and amyloid pathology results of other networks are provided in the supplementary materials. The FDG SUVR values of various networks in the rs3865444 CC group are similar to those in the rs3865444 CA/AA group, while the mean AV45 SUVR values of various networks in the rs3865444 CC group are generally higher than those in the rs3865444 CA/AA group, but without significant differences (Supplementary Table S1, S3). Regarding neuronal activity, the mean ALFF values of other brain networks in carriers of the rs3865444 CA/AA allele are generally higher than those in the rs3865444 CC group, but the differences are not pronounced (Supplementary Table S2). Table S5 in the supplementary materials analyzes the effect of ApoEε4 on the G/O coefficients of all network modules. The results show that the presence of ApoEε4 heterozygosity does not impact the G/O coefficients of the various brain network modules. Correlation analysis shows that as AV45 SUVR values increase, the growth rates of ADAS scores (β = 3.02, p < 0.001, Fig. 5D) and CDR scores also rise (β = 0.79, p < 0.001, Fig. 5E). Conversely, there is a significant negative correlation between the G/O coefficient and AV45 SUVR (β = − 0.24, p < 0.05, Fig. 5F).
Fig. 5.

(A) Box plots depict the differences in plasma Abeta40 between carriers of CD33 rs3865444 CC and carriers of CD33 rs3865444 CA/AA. (B) Box plots depict the differences in plasma Abeta42 between carriers of CD33 rs3865444 CC and carriers of CD33 rs3865444 CA/AA. (C) Box plots depict the differences in Abeta40/Abeta42 between carriers of CD33 rs3865444 CC and carriers of CD33 rs3865444 CA/AA. (D) The association between the AV45 SUVR of the limbic-orbital frontal cortex and the ADAS score growth rate. (E) The association between the AV45 SUVR of the limbic-orbital frontal cortex and the CDR score growth rate. (F) The association between the G/O coefficient of the limbic-orbital frontal cortex and the AV45 SUVR. Correlations in the scatter plot are represented by least squares regression lines, with shaded areas indicating confidence intervals.
The CD33 rs3865444 SNP affects cognitive trajectories
We hypothesized that longitudinal changes in clinical dementia scores are associated with CD33 rs3865444 alleles. Prior immunostaining has confirmed CD33 expression in human brain microglia, and there is a positive correlation between CD33 mRNA expression, Alzheimer’s disease status, and the rs3865444 CC allele11,32. We speculate that CD33 rs3865444 polymorphism may be associated with pathological microglial overactivation, thereby influencing individual rates of cognitive decline. Compared to individuals carrying the CD33 rs3865444 CC allele, those carrying the CD33 rs3865444 CA/AA alleles exhibit a slower rate of increase in clinical dementia scores, indicating a slower rate of cognitive decline. To further validate the hypothesis regarding CD33 rs3865444 polymorphism, we used the follow-up records of ADAS scores and CDR scores as dependent variables and follow-up time as the independent variable. Linear regression analysis was performed on both, yielding the rates of change over time for these scores. ADAS and CDR scores reflect the severity of cognitive impairment, with higher scores indicating more severe conditions. In the analysis of ADAS scores, the rate of increase in ADAS scores was higher in the CD33 rs3865444 CC allele group compared to the CD33 rs3865444 CA/AA group (+ 0.62 points/year, − 0.071 points/year, Fig. 6A). These trends were largely consistent with the longitudinal analysis of CDR scores, where carriers of the CD33 rs3865444 CC allele exhibited a higher rate of increase in CDR scores compared to patients with CD33 rs3865444 CA/AA alleles (+ 0.24 points/year, + 0.036 points/year, Fig. 6B). This suggests that the CD33 rs3865444 CA/AA alleles modulate the risk of Alzheimer’s disease, leading to a slower progression of cognitive impairment symptoms in individuals carrying these alleles. Additionally, we analyzed the baseline status of participants for both scores. There was no significant difference in baseline ADAS scores between the CD33 rs3865444 CA/AA group and the CD33 rs3865444 CC group (Fig. 6C), and a similar pattern was observed for baseline CDR scores (Fig. 6D). The amyloid-negative population also exhibited a consistent pattern. The results indicate that the rs3865444 CC group exhibited a higher rate of increase in ADAS (+ 0.39 points/year, Fig. 6E) and CDR (+ 0.20 points/year, Fig. 6F) scores compared to the rs3865444 CA/AA group (ADAS: -0.14 points/year, Fig. 6E, CDR: − 0.041 points/year, Fig. 6F). However, the baseline levels of ADAS and CDR scores did not show significant differences between the two groups (Fig. 6G,H).
Fig. 6.

Differential cognitive decline based on the CD33 rs3865444 SNP. (A) ADAS (higher score is worse) longitudinal analysis. (B) CDR (higher score is worse) longitudinal analysis. (C) ADAS baseline score. (D) CDR baseline score. (E) ADAS (higher score is worse) longitudinal analysis (amyloid-negative cohort). (F) CDR (higher score is worse) longitudinal analysis (amyloid-negative cohort). (G) ADAS baseline score (amyloid-negative cohort). (H) CDR baseline score (amyloid-negative cohort).
G/O mediates the effect of CD33 rs3865444 on the cognitive trajectory
The previous research findings indicate that the CD33 rs3865444 SNP can affect cognitive trajectory and G/O coefficients. The CD33 rs3865444 CC allele may lead to accelerated cognitive decline and decreased G/O coefficients. The relationship among these factors warrants further investigation. To explore whether the influence of CD33 rs3865444 SNP on cognitive trajectory is mediated by G/O coefficients (Eqs. 3, 4, 5), we employed mediation analysis with 10,000 bootstrap iterations, controlling for age, gender, education level, and ApoE ε4 status. Supplementary Fig. S1A shows that hippocampal volumes between the two groups are very similar, with no significant differences (p = 0.83). There is no correlation between hippocampal volume and the G/O coefficient (p = 0.41, Fig. S1B). First, the rate of increase in ADAS scores was significantly higher in individuals from the CD33 rs3865444 CC group compared to the CD33 rs3865444 CA/AA group (p < 0.05, Fig. 7A). Based on analysis of the limbic-orbital frontal cortex region, there was a significant negative correlation between the G/O coefficients of this brain region and the rate of change in ADAS scores over time (β = − 1.33, p < 0.01, Fig. 7B), indicating that higher G/O coefficients in the limbic-orbital frontal cortex region are associated with a slower progression of cognitive impairment, supporting our hypothesis. However, after introducing G/O coefficients as a mediator, the direct effect of CD33 rs3865444 SNP on the rate of change in ADAS scores over time was no longer significant (β = − 0.48, Fig. 7C), while the significant correlation between G/O coefficients and ADAS slope persisted (β = − 1.09, p < 0.05, Fig. 7C), indicating that the relationship between CD33 rs3865444 SNP and ADAS slope is fully mediated by the G/O coefficients of the limbic-orbital frontal cortex region. In the amyloid-negative cohort, there was no significant difference in the rate of ADAS score increase between the rs3865444 CC group and the rs3865444 CA/AA group (Fig. 7D). However, the limbic-OFC G/O coefficient remained significantly negatively correlated with the rate of ADAS score increase (Fig. 7E, p < 0.05). After introducing the G/O coefficient as a mediator, the correlation between the G/O coefficient and the ADAS slope was no longer significant (β = − 0.84, Fig. 7F), though the significance of the correlation between the limbic-OFC G/O coefficient (p = 0.082) and the ADAS slope was stronger than that of the rs3865444 SNP (p = 0.15).
Fig. 7.

The G/O coefficient mediates the effect of the CD33 rs3865444 SNP on cognitive trajectories. (A) The box plot shows the difference in ADAS score growth rate between CD33 rs3865444 CC carriers (n = 59) and CD33 rs3865444 CA/AA carriers (n = 48). (B) The association between the G/O coefficient of the limbic-orbital frontal cortex and the ADAS score growth rate. (C) The G/O coefficient of the limbic-orbital frontal cortex mediates the effect of CD33 rs3865444 SNP on the ADAS score growth rate. (D) The box plot shows the difference in ADAS score growth rate between CD33 rs3865444 CC carriers (n = 47) and CD33 rs3865444 CA/AA carriers (n = 40) (amyloid-negative cohort). (E) The association between the G/O coefficient of the limbic-orbital frontal cortex and the ADAS score growth rate (amyloid-negative cohort). (F) The G/O coefficient of the limbic-orbital frontal cortex mediates the effect of CD33 rs3865444 SNP on the ADAS score growth rate (amyloid-negative cohort). Correlations in the scatter plot are represented by least squares regression lines, with shaded areas indicating confidence intervals. Path weights are shown as correlation coefficients, where *p < 0.05, **p < 0.01, and ***p < 0.001. The significance of the mediating effect was determined through bootstrapping with 10,000 iterations. c represents the direct association between CD33 rs3865444 SNP and ADAS score growth rate, while c’ represents the correlation after introducing the mediating variable.
Discussion
The main purpose of this experiment is to investigate whether CD33 rs3865444 polymorphism affects the glucose-oxygen coupling coefficients in specific brain regions, thereby altering individual cognitive trajectories. First, we calculated the G/O coefficients for each network based on the 1000 parcels of the Schaefer2018_17Networks, and after Bonferroni correction, we found that the glucose-oxygen coupling level in the limbic-orbital frontal cortex region of the rs3865444 CA/AA group was significantly higher than that of the rs3865444 CC group. Similarly, consistent results were observed in the amyloid-negative population (Fig. 4). This suggests a significant relationship between CD33 rs3865444 polymorphism and G/O coefficients in specific brain regions, providing preliminary evidence that G/O coefficients can serve as imaging biomarkers for inflammation-related genetic variations. To further validate our hypothesis, we collected follow-up records of ADAS scores and CDR scores for all participants and analyzed the longitudinal changes in these clinical scores. Initially, there were no significant differences in the baseline levels of both scores between the rs3865444 CA/AA group and the rs3865444 CC group. However, the rate of change over time in ADAS scores was lower in the rs3865444 CA/AA group compared to the rs3865444 CC group, with similar results observed in the analysis based on CDR scores (Fig. 6). This suggests that at a macro level, CD33 rs3865444 polymorphism influences individual cognitive trajectories, possibly associated with CD33-induced neuroinflammatory pathology. Finally, we analyzed the relationship between CD33 rs3865444 polymorphism, G/O coefficient, and cognitive trajectories. The rate of ADAS score increase in the rs3865444 CC group was significantly higher than in the rs3865444 CA/AA group, while a significant negative correlation existed between the rate of ADAS score increase and the G/O coefficient (Fig. 7). Introducing the G/O coefficient of the limbic-orbital frontal cortex as a mediating variable into the regression analysis rendered the impact of CD33 rs3865444 polymorphism on individual cognitive trajectories non-significant (Fig. 7). In conclusion, our study demonstrates that CD33 rs3865444 polymorphism influences the progression rate of cognitive impairment, mediated through the G/O coefficient, which holds significant implications for the diagnosis and prognosis of neurodegenerative diseases.
For our initial finding, there was an association between the CD33 rs3865444 CA/AA genotypes and higher glucose-oxygen coupling coefficients, indicating that the G/O coefficient could serve as an imaging biomarker for CD33 variation. Importantly, we performed three-dimensional visualization of cortical glucose metabolism and neuronal activity levels between the CD33 rs3865444 CA/AA and CC groups, showing overall consistency in the distribution of FDG SUVR and ALFF values with some regional deviations. Subsequently, we computed the G/O coefficients for each network, revealing group differences only in the limbic-orbital frontal cortex G/O coefficient after Bonferroni correction. The sensitivity analysis also confirmed this conclusion. Previous studies have shown that both the cognitive resilience group and the cognitive decline group tend to be spatially segregated along limbic regions, with some research further supporting this spatial separation33. Moreover, the relationship between CD33 rs3865444 polymorphism and G/O coefficient appeared to be independent of FDG SUVR and ALFF values overall. Some studies have shown no significant correlation between brain glucose metabolism and amyloid protein in patients with cognitive impairment34,35. However, some studies have found a significant correlation between brain glucose metabolism and amyloid levels in different brain regions among normal elderly subjects and AD patients36. This study found a pattern similar to previous research18, showing a significant negative correlation between AV45 SUVR and the G/O coefficient (Fig. 5F). This may be due to amyloid accumulation inducing microglial activation in rs3865444 CC individuals, leading to increased brain glucose metabolism, which subsequently affects the G/O coefficient. However, this association was not apparent in groups with low AV45 SUVR and FDG SUVR levels, consistent with previous findings. Previous studies have identified neuroinflammatory pathology as a key factor in the onset of Alzheimer’s disease (AD)37. It is now established that certain genes can modulate the onset and progression of AD, deepening our understanding of the pathogenesis of AD. Specifically, many known AD-related genes are involved in microglial pathways, indicating a strong correlation between these genes and AD, which has been supported by numerous studies38. Genome-wide association studies (GWAS) have identified several single nucleotide polymorphisms (SNPs) associated with AD that are related to microglial cells, primarily involving genes such as APOE, TREM2, CD33, and ABCA739,40. However, further research is needed to elucidate how these variations influence the course of AD. CD33 has been identified as one of the major risk genes for AD and has been confirmed in multiple genetic studies41–44. It is important to note that this study primarily focuses on the impact of human CD33 on cognitive trajectories, and there are several key differences between human and murine CD3314. In addition to structural and functional distinctions, current research indicates a crucial difference in that mice lack AD-associated variants of CD33, namely the CD33M and CD33m protein isoforms. Therefore, studying transgenic mice expressing human CD33 isoforms is essential45. Findings show that CD33M increases Aβ levels, promotes the formation of more diffuse plaques, and reduces the number of disease-associated microglia, whereas CD33m facilitates plaque compaction, enhances microglial-plaque interactions, and exerts a protective role against Alzheimer’s disease45. Aβ accumulation activates microglia5, and PET imaging studies have confirmed widespread amyloid deposition and microglial activation in AD patients, with the level of activation negatively correlated with cognitive function4. Future use of combined amyloid and microglial PET imaging may help evaluate the efficacy of anti-amyloid therapies. Additionally, PET-based studies have shown that microglial activation in neurodegenerative diseases is associated with increased glucose uptake and FDG signal intensity6. A recent study that integrated brain tissue analysis with fMRI imaging successfully mapped mitochondrial respiratory capacity across the human brain, providing important insights for future research46. Building on this approach, researchers can segment brain tissue using the Schaefer2018_17Networks atlas and quantify the levels of the two CD33 isoforms, CD33M and CD33m, within each segmented region. This would allow for characterization of the spatial distribution of CD33 isoforms across brain regions and their associations with fMRI features, offering a functional network-based understanding of the regional heterogeneity and diversity of CD33 isoforms. Furthermore, integrating single-cell sequencing techniques could enable the investigation of isoform-specific expression patterns across different glial cell subtypes, thereby expanding the understanding of the pathogenic mechanisms and molecular underpinnings of CD33 polymorphisms.
Therefore, based on the above research foundation, we propose using the coupling between glucose metabolism levels and neuronal activity intensity to describe the neuropathology induced by the CD33 rs3865444 SNP. The transmission of neuronal electrical signals in the cerebral cortex underlies the maintenance of complex cognitive functions and working memory in the brain, a process inevitably accompanied by glucose metabolism47–49. Hence, there is a certain correlation between glucose metabolism and neuronal fluctuations18,50. However, the activation of astrocytes associated with neuroinflammation and AD can lead to enhanced metabolism, thereby affecting FDG SUVR values. Our second finding validates these viewpoints6,23,24. CD33 rs3865444 significantly impacts individual cognitive trajectories, and this effect is mediated through G/O, indicating that the risk allele of CD33 rs3865444 may disrupt the coupling between glucose metabolism and neuronal activity, thereby affecting patients’ cognitive recovery ability and the progression of cognitive impairment (Fig. 7), with cognitive trajectories represented by the rate of change in ADAS scores and CDR scores over time. Participants in the CD33 rs3865444 CC group exhibited higher rates of increase in ADAS scores and CDR scores, with a significant correlation observed between G/O coefficients and ADAS score slopes. This is consistent with previous research findings, and there was no significant difference observed in the baseline status of ADAS scores and CDR scores, indicating the potential of CD33 rs3865444 to predict the progression of early mild cognitive impairment patients, with the effect of CD33 rs3865444 mediated through the G/O coefficients of the limbic-orbital frontal cortex (Figs. 6 and 7).
This study also has some limitations. Firstly, the number of participants in this study constitutes a moderate sample size, and further research on a larger scale is needed to validate our conclusions. Secondly, it is noteworthy that the current research results indicate that the G/O coefficient mediates the impact of the CD33 rs3865444 SNP on cognitive trajectories. However, some other pathological processes may also accelerate cognitive decline, some of which do not involve activation of microglia and can induce neuronal death51–53. Nevertheless, we believe that the current results are not contradictory to these pathological processes; neuroinflammation is a key factor in neurodegenerative changes, and imaging biomarkers based on this have significant implications. Lastly, consideration should be given to the comorbid pathological conditions; the CD33 gene is closely associated with synaptopathy, which is related to the onset of many brain diseases, such as schizophrenia54, autism55, and bipolar affective disorder56. Incorporating more comprehensive scales during clinical assessments may help to mitigate the impact of comorbid conditions.
In summary, our study findings reveal an association between CD33 rs3865444 SNP and the cognitive trajectories of individuals with mild cognitive impairment, mediated through the G/O coefficient of the limbic-orbital frontal cortex. This discovery provides a potential new avenue for the clinical diagnosis of Alzheimer’s disease or other neurodegenerative disorders, where the G/O coefficient can serve as an indicator for monitoring the efficacy of novel cognitive impairment treatment strategies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by the Key Program of the Natural Science Foundation of Zhejiang Province (CN) under Grant No. LZ24F010007, and the National Natural Science Foundations of China (CN) under grant No. 62271177. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Author contributions
J.S. designed the project and supervised the overall research; Z.Y. collected data, performed experiments and analysis; Q.Z. and J.C. co-designed the research; Y.X., and L.W. participated in data collection or analysis. Z.Y. and J.S. wrote the manuscript.
Data availability
The imaging data for this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. Access to the data can be obtained by registering and adhering to the data usage agreement at the IDA database (adni.loni.usc.edu). Upon reasonable request, additional information can be provided by the corresponding author.
Declarations
Competing interests
The authors declare no competing interests.
Ethical statement
This study was approved by the institutional review board (IRB) at Hangzhou Dianzi University (IRB-2020001) and the ethics committee at Beijing Hospital (2022BJYYEC-375-01). The Alzheimer’s Disease Neuroimaging Initiative has obtained informed consent from participants as research subjects, with support from the ADNI Executive Committee and representatives of the National Institute on Aging. The study investigators maintain and be responsible for determining how to use the participants’ data for future research. All links related to the participants’ identities have been removed from the data before sharing. Methods were carried out in accordance with relevant guidelines and regulations.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Walsh, D. M. et al. Naturally secreted oligomers of amyloid Β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 416, 535–539 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Cirrito, J. R. et al. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron. 48, 913–922 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Wang, J-Z. & Liu, F. Microtubule-associated protein Tau in development, degeneration and protection of neurons. Prog. Neurobiol.85, 148–175 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Edison, P. et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11 C](R) PK11195-PET and [11 C] PIB-PET study. Neurobiol. Dis.32, 412–419 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Solito, E. & Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol.3, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiang, X. et al. Microglial activation States drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci. Transl. Med.13, eabe5640 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Friker, L. L. et al. β-amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell. Rep.30, 3743–3754.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crocker, P. R., Paulson, J. C. & Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol.7, 255–266 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Ishida, A. et al. Negative regulation of Toll-like receptor-4 signaling through the binding of glycosylphosphatidylinositol-anchored glycoprotein, CD14, with the Sialic acid-binding lectin, CD33. J. Biol. Chem.289, 25341–25350 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griciuc, A. et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 78, 631–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malik, M. et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci.33, 13320–13325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradshaw, E. M. et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci.16, 848–850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacherjee, A. et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ 1–42 phagocytosis in microglia. Mol. Neurodegener. 16, 1–22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eskandari-Sedighi, G., Jung, J. & Macauley, M. S. CD33 isoforms in microglia and Alzheimer’s disease: friend and foe. Mol. Aspects Med.90, 101111 (2023). [DOI] [PubMed] [Google Scholar]
- 15.Karch, C. M. et al. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One. 7, e50976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker, D. G. et al. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging. 36, 571–582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griciuc, A. et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet.29, 2920–2935 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding, C. et al. Coupling relationship between glucose and oxygen metabolisms to differentiate preclinical Alzheimer’s disease and normal individuals. Hum. Brain. Mapp.42, 5051–5062 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saykin, A. J. et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimer’s Dement.11, 792–814 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franzmeier, N. et al. Resting-state connectivity of the left frontal cortex to the default mode and dorsal attention network supports reserve in mild cognitive impairment. Front. Aging Neurosci.9, 264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaefer, A. et al. Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb. Cortex. 28, 3095–3114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe, V. J. et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J. Nucl. Med.50, 878–886 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aiello, M. et al. Relationship between simultaneously acquired resting-state regional cerebral glucose metabolism and functional MRI: a PET/MR hybrid scanner study. Neuroimage. 113, 111–121 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Marchitelli, R. et al. Simultaneous resting-state FDG-PET/fMRI in alzheimer disease: relationship between glucose metabolism and intrinsic activity. Neuroimage. 176, 246–258 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Biswal, B., Zerrin Yetkin, F., Haughton, V. M. & Hyde, J. S. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn. Reson. Med.34, 537–541 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Makowski, D., Ben-Shachar, M. S., Patil, I. & Lüdecke, D. Methods and algorithms for correlation analysis in R. J. Open. Source Softw.5, 2306 (2020). [Google Scholar]
- 27.Mohs, R. C. et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s disease assessment scale that broaden its scope. Alzheimer Dis. Assoc. Disord. 11, 13–21 (1997). [PubMed] [Google Scholar]
- 28.O’Bryant, S. E. et al. Staging dementia using clinical dementia rating scale sum of boxes scores: a Texas Alzheimer’s research consortium study. Arch. Neurol.65, 1091–1095 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris, J. C. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the alzheimer type. Int. Psychogeriatr.9, 173–176 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Zhao, X., Lynch, J. G. Jr & Chen, Q. Reconsidering Baron and Kenny: Myths and truths about mediation analysis. J. Consum. Res.37, 197–206 (2010). [Google Scholar]
- 31.Pemberton, H. G. et al. Quantification of amyloid PET for future clinical use: a state-of-the-art review. Eur. J. Nucl. Med. Mol. Imaging. 49, 3508–3528 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raj, T. et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet.23, 2729–2736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duong, M. T. et al. Dissociation of Tau pathology and neuronal hypometabolism within the ATN framework of Alzheimer’s disease. Nat. Commun.13, 1495 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furst, A. J. et al. Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiol. Aging. 33, 215–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furst, A. J. & Lal, R. A. Amyloid-β and glucose metabolism in Alzheimer’s disease. J. Alzheimers Dis.26, 105–116 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Shin, J. et al. Resting-state glucose metabolism level is associated with the regional pattern of amyloid pathology in Alzheimer′ s disease. Int. J. Alzheimer’s Dis.2011, 759780 (2011). [DOI] [PMC free article] [PubMed]
- 37.Zhao, L. CD33 in Alzheimer’s disease—biology, pathogenesis, and therapeutics: a mini-review. Gerontology. 65, 323–331 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Ransohoff, R. M. & El Khoury, J. Microglia in health and disease. Cold Spring Harb. Perspect. Biol.8, a020560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Efthymiou, A. G. & Goate, A. M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J. & Pocock, J. M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol.36, 74–81 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet.43, 436–441 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dos Santos, L. R. et al. Validating GWAS variants from microglial genes implicated in Alzheimer’s disease. J. Mol. Neurosci.62, 215–221 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Cukier, H. et al. Exome sequencing of extended families with Alzheimer’s disease identifies novel genes implicated in cell immunity and neuronal function. J. Alzheimer’s Dis. Parkinson.7 (2017). [DOI] [PMC free article] [PubMed]
- 44.Li, X. et al. CD33 rs3865444 polymorphism contributes to Alzheimer’s disease susceptibility in Chinese, European, and North American populations. Mol. Neurobiol.52, 414–421 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Eskandari-Sedighi, G. et al. Alzheimer’s disease associated isoforms of human CD33 distinctively modulate microglial cell responses in 5XFAD mice. Mol. Neurodegener. 19, 42 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mosharov, E. V. et al. A human brain map of mitochondrial respiratory capacity and diversity. Nature. 1–10 (2025). [DOI] [PubMed]
- 47.Liu, Y. et al. Impaired long distance functional connectivity and weighted network architecture in Alzheimer’s disease. Cereb. Cortex. 24, 1422–1435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fell, J. & Axmacher, N. The role of phase synchronization in memory processes. Nat. Rev. Neurosci.12, 105–118 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Gallinat, J. et al. Hippocampal glutamate concentration predicts cerebral theta oscillations during cognitive processing. Psychopharmacology187, 103–111 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Dong, Q-Y. et al. Glucose metabolism in the right middle Temporal gyrus could be a potential biomarker for subjective cognitive decline: a study of a Han population. Alzheimers Res. Ther.13, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan, M., Rutten, B. P. & Kim, M. O. MST1 regulates neuronal cell death via JNK/Casp3 signaling pathway in HFD mouse brain and HT22 cells. Int. J. Mol. Sci.20, 2504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pombo, C. M., Iglesias, C., Sartages, M. & Zalvide, J. B. MST kinases and metabolism. Endocrinology160, 1111–1118 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Qu, J. et al. MST1 suppression reduces early brain injury by inhibiting the NF-κB/MMP-9 pathway after subarachnoid hemorrhage in mice. Behav. Neurol.2018. (2018). [DOI] [PMC free article] [PubMed]
- 54.Isomura, R., Kitajima, K. & Sato, C. Structural and functional impairments of polysialic acid by a mutated polysialyltransferase found in schizophrenia. J. Biol. Chem.286, 21535–21545 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang, X., Li, L., Chai, X. & Liu, J. The association between ST8SIA2 gene and behavioral phenotypes in children with autism spectrum disorder. Front. Behav. Neurosci.16, 929878 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Müller-Miny, L. et al. Association of polysialic acid serum levels with schizophrenia spectrum and bipolar disorder-related structural brain changes and hospitalization. Sci. Rep.13, 2085 (2023). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The imaging data for this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. Access to the data can be obtained by registering and adhering to the data usage agreement at the IDA database (adni.loni.usc.edu). Upon reasonable request, additional information can be provided by the corresponding author.