

Abstract
Modular type I polyketide synthases (PKSs) comprise a family of enzymes that synthesize a diverse class of natural products with medicinal applications. The biochemical features of these systems include the extension and processing of polyketide chains in a stepwise, stereospecific manner, organized by a series of modules divided into distinct catalytic domains. Previous work revealed that a primary hurdle for utilizing PKS modules to create diverse macrolactones hinges on the selectivity of the thioesterase (TE) domain. Herein, we generated novel hybrid 12-membered macrolactone/lactam ring systems employing unnatural amide-containing hexaketide intermediates in conjunction with an engineered TE S148C mutant from the pikromycin (Pik) biosynthetic pathway. Specifically, unnatural macrocycle (3) was initially formed in severely attenuated yields compared to the native product generated from the natural hexaketide substrate. A stepwise directed evolution campaign generated Pik TE variants with enhanced selectivity for macrocycle formation over hydrolysis. Over three rounds of evolution, a series of mutant Pik TE proteins were identified, and further combinations of beneficial mutations carried from each round produced a composite variant with six-fold enhanced isolated yield of the hybrid macrocycle compared to the parent TE S148C mutant enzyme. This study offers new insights into the range of amino acid residues, both proximal and distal to the active site, that impart improved selectivity and yield against the unnatural polyketide substrate and overcoming a key PKS pathway gatekeeper.
Graphical Abstract

Introduction
Macrocyclic scaffolds are found in many natural products from diverse sources. These commonly include twelve or more atoms in their cyclic framework.1 Synthetic strategies have been developed for macrolactonization, and macrolactamization, which include transition metal catalyzed coupling reactions, click chemistry and ring-closing metathesis.1, 2 A wide variety of these molecules have been developed as pharmaceuticals with over one hundred FDA approved drugs carrying a macrocyclic core (e.g. macrolide and cyclic peptide antibiotics, anthelmintics, anticancer agents, immunomodulators); showing their current medicinal value and continued promise for future drug discovery and development.3, 4 Macrolide antibiotics, including erythromycin and its semi-synthetic derivatives (clarithromycin, azithromycin, among others) act against bacterial pathogens as ribosome inhibitors and have synergistic immunomodulatory activity in the human host.5, 6 The macrolide chemical structure contains a macrolactone ring possessing one or more sugar moieties (amino sugars, deoxy sugars or both) and other functionalities such as hydroxyl groups that are important for their mechanism of action in the bacterial ribosome peptidyl transferase center.7 Frequently, the structural complexity of large-ring macrolactones introduces significant synthetic challenges. Options for solving these include total synthesis, semi-synthesis and chemoenzymatic strategies.8–17 For example, the first total synthesis of erythromycin18–20 in 1981 reported over 50 synthetic steps and an overall yield of less than 1%. These large macrocycles exhibit stereochemical and structural diversity, which often results in laborious, low yielding reaction schemes, possessing a barrier for further diversification or pursuing structure-activity relationship studies. Alternatively, semi-synthesis has been employed to generate widely used antibiotics such as the erythromycin derivatives clarithromycin, azithromycin, telithromycin, and others.5, 9, 12, 21 Despite these approaches to gain synthetic efficiencies and improve overall yields, modifications to the starting material scaffolds are often limited by reactive functional groups and selectivity challenges at several positions within these intricate ring structures.2, 14, 21–25
Chemoenzymatic synthesis encompasses assembly of intermediates from simple building blocks, combined with enzyme catalyzed reactions to obtain efficient production of novel molecules and pharmaceuticals using aqueous reaction conditions, while avoiding toxic reagents, solvents and protecting groups.26–31 Although workable synthetic methodologies exist for macrolactonization, these are often met with unfavorable entropic and enthalpic factors relating to the energetics of intramolecular cyclization.15–17, 32 To address the challenges of total and semi-synthesis, we have been investigating the pikromycin (Pik) biosynthetic pathway thioesterase (TE) domain as a key biocatalyst for macrolide antibiotic discovery.26, 33 Earlier studies revealed how the Pik TE binds its linear substrate and prepares it for cyclization reactions. Moreover, it can accommodate hexa- and heptaketides in its active site through a hydrophobic chamber that include substrate-protein anchoring hydrogen bonds. Remarkably, the TE induces a curled conformation of the linear polyketide through the hydrophobicity of the surrounding residues, coupled with a hydrophilic barrier at the exit site of the enzyme channel. This barrier forces the substrate back into the active site, and positions the nucleophilic hydroxyl group for macrocyclization.34–36
The Pik pathway is comprised of a modular type I PKS that selectively catalyzes key transformations on structurally distinct intermediates generating a 12-membered macrolactone ring, 10-deoxymethynolide (10-DML) and a 14-membered macrolactone ring, narbonolide (NBL). These megasynthases are comprised of modules divided into distinct catalytic domains with specific roles.33, 37 Each round of polyketide elongation is performed by three main domains, the acyltransferase (AT) that selects an extender unit, an acyl carrier protein (ACP) and a ketosynthase (KS) that accepts the polyketide chain from an upstream ACP domain and catalyzes a decarboxylative Claisen condensation reaction. Modules may contain combinations of other domains including ketoreductase (KR), dehydratase (DH) and enoyl reductase (ER) to transform the β-keto group into hydroxyl, alkene or alkane functionalities, respectively.26, 38, 39 At the end of the pathway, a terminal module, typically a TE domain, catalyzes formation and off-loading of the hydrolyzed or cyclized product.39 Developing these enzymes as biocatalysts represents a complementary addition to synthetic chemistry for achieving molecular diversification.40–42 Thus, enhancing our understanding and improving Pik TE catalysis for macrocyclization represents a priority area to explore scalable applications of PKS systems.
Previous studies on PKS TEs from diverse organisms and biosynthetic pathways, as well as engineering efforts to understand hydrolysis and cyclization processes have been reported.39, 43–63 In a broader context, classifications of TE enzyme families based on structure, function, substrate specificity, products generated, and catalytic mechanisms enable their use as biocatalysts for the synthesis of diverse molecules.43, 45, 46, 49, 50, 64 Additionally, various TE evolutionary models for enzyme reactivity and selectivity have provided clues for altering TE catalysis to form a desired product.43, 49 In some cases, TEs can process a wide variety of substrates, while in others, they show a high degree of selectivity for specific structural features, depending on the TE loading or release step considered during the overall off-loading mechanism.43, 49 Thus, we envision that understanding TE function through the investigation of diverse unnatural substrates can shed light on the biocatalytic parameters required for generating a wider range of products with high selectivity and efficiency.
Previously, we demonstrated the ability to form 12-membered macrolactones utilizing unnatural pentaketides in conjunction with the PikAIII-TE fusion protein.40 Depending on substrate structure, the cyclized products were obtained in 9%−66% isolated yields, with competing hexaketide hydrolysis and truncated byproducts formed when using PikAIII-TE WT40 and the PikAIII-TE S148C mutant41. The results indicated that Pik TE has limited substrate flexibility and often functions as a gatekeeper in the processing of unnatural substrates.40–42 Although diverse unnatural macrolactones of different ring sizes have been generated, the introduction of functional groups not typically found in polyketide chain elongation intermediates often results in failed or inefficient cyclization. Thus, we were motivated to deliberately engineer and expand the ability of Pik TE to process structurally variant polyketide substrates in the pursuit of understanding substrate-TE interactions. Protein engineering has been used widely to obtain enzymes with new and finely tuned properties,65–68 but has not been applied to polyketide macrocyclization catalysts. In this work, we employed directed evolution to create and assess a library of Pik TE variants that favor efficient formation of a hybrid macrolactone/lactam derivative (3) over its corresponding hydrolysis product (2) starting from amide hexaketide (1) (Scheme 1).33, 69–71 We identified new variants with enhanced selectivity for macrocyclization, which was demonstrated by increased total turnover numbers (TTNs), initial reaction rates and isolated yields.
Scheme 1.

(A) Native hexaketide substrate (derived from an NBOM hydroxyl protected intermediate)70 against Pik TE WT generates 10-DML. (B) N-H amide hexaketide (1) against Pik TE WT generates hydrolysis product (2) whereas Pik TE S148C catalyzes formation of the hybrid macrolactone/lactam (3) with hydrolysis of 1 to afford 2 still as the major pathway. (C) N-Me amide hexaketide (4) against Pik TE S148C generates hybrid macrolactone/lactam (5) as the major product.
Results
Chemical Synthesis of Amide Hexaketide Substrates 1 and 4
Our earlier studies with the native Pik hexaketide demonstrated an effective chemoenzymatic strategy for the generation of 10-DML when utilizing Pik TE WT (Scheme 1A).71 We then focused on simplifying the native hexaketide and altering the stereochemistry of the nucleophilic hydroxyl group.40, 41 In choosing new hexaketide targets, we were motivated by amide-containing derivatives of erythromycin that show antibiotic activity comparable to N-methylated azithromycin.72–76 Thus, the impact of exchanging the native C-Me functionality to N-H/N-Me, creating an amide group in the chain-elongation intermediate (Scheme 1B) became a central objective. This choice was also made to assess the impact of a single heteroatom replacement in the chain on TE-mediated cyclization. Moreover, amide functionality is present in over half of FDA approved pharmaceuticals, and editing the macrocycle to include a new H-bond donor was also a compelling objective for future bioactivity studies.77–82 Robust methods are available for amide synthesis, including dehydrative condensations between carboxyl and amino groups, utilization of condensation reagents, Beckmann rearrangements, and Schmidt reaction mechanisms.83, 84 Thus, to further pursue diversification of polyketide systems, we designed amide-containing Pik hexaketides to explore TE-mediated cyclization toward hybrid ring systems (Scheme 2).
Scheme 2.

Chemical synthesis of amide hexaketides (1 and 4).
The target amide hexaketides (1 and 4) were synthesized starting with Evan’s aldol reaction85 between (R)-4-benzyl-3-propionyloxazolidin-2-one (7)86, 87 and aldehydes 8 and 9 (generated from (S)-3-(Boc-amino)-2-methylpropionic acid) employing dibutylboron triflate to give 10 and 11. Next, lithium hydroxide/hydrogen peroxide cleavage of the Evan’s auxiliary11, 88 afforded 12 and 13, followed by trimethylsilyl (TMS) diazomethane esterification to make 14 and 15. Subsequent deprotection of N-Boc enabled coupling with 16 to yield amides 17 and 18. Hydrolysis of 17 and 18 with lithium hydroxide followed by thioesterification and final cleavage of the silyl ether11 with hydrofluoric acid (HF) generated the amide hexaketides 1 and 4 (Scheme 2).
Isolation of Novel Hybrid 12-membered Macrolactone/Lactam Rings 3 and 5
With the desired substrates 1 and 4 in hand, we tested them against Pik TE WT and the Pik TE S148C active site mutant.41 Reaction of N-H hexaketide 1 with Pik TE WT results in exclusive formation of the amide hydrolysis product 2 [M+H] = 302 m/z (Scheme 1B), which was isolated in 87% yield from a scale-up reaction (Table S1). Although reaction with Pik TE S148C also resulted in majority of the seco-acid hydrolysis product 2, it was accompanied by the mass expected for the hybrid 12-membered macrolactone/lactam 3 [M+H] = 284 m/z (Scheme 1B), suggesting potential formation of the desired macrocycle (Figure S1). Subsequent scale-up, high-performance liquid chromatography (HPLC) purification and isolation provided a 13% isolated yield of the macrocyclic compound 3 that was confirmed by NMR (Table S2) and X-ray crystallography (Scheme 1B), which verified the molecular architecture and stereochemistry of the product (Figure S30, CCDC Deposition Number 2294580). Thus, the Pik TE S148C active site mutant was able to catalyze formation of the desired 12-membered ring 3, albeit in poor overall yield. In contrast to the N-H amide hexaketide 1, the N-methylated hexaketide variant 4 reacts in a similar manner to the native or near native Pik hexaketide substrates. From LC-MS analysis, reaction with Pik TE WT produces a mixture of seco-acid hydrolysis 6 [M+H] = 316 m/z and hybrid 12-membered macrolactone/lactam 5 [M+H] = 298 m/z products (Table S3, Figure S2). Reaction with Pik TE S148C provides a substantial increase in amount of 12-membered ring, where subsequent scale-up and purification afforded 5 in 75%, which was confirmed by NMR (Table S4) and X-ray crystallography (Scheme 1C, Figure S31, CCDC Deposition Number 2378053). Given the stark difference in production of hybrid 12-membered macrolactone/lactams 3 and 5 from amide hexaketides 1 and 4 using Pik TE S148C, we decided to pursue directed evolution to create additional Pik TE S148C variants with enhanced selectivity for the target macrocycle 3.
Pik TE Directed Evolution Strategy
Our objective for reactions using Pik TE S148C with the amide hexaketide substrate 1 was to obtain incremental improvements in macrocycle formation with a corresponding decrease in hydrolysis products screened via liquid chromatography-mass spectrometry (LC-MS). To guide the first round of evolution, we examined the Pik TE WT X-ray crystal structure34 with a covalent pentaketide affinity label (PDB: 2HFJ), and twenty-one resides were identified within 6 Å of the substrate in the active site region (Figure 1). The crystal structure-substrate mimic complex likely represents a close approximation to the native hexaketide substrate covalently bound to Ser148 in the TE active site showing the Ser148, Asp176, His268 catalytic triad, and revealed the residues likely involved in binding and cyclization to generate macrolactone 3. Next, site saturation mutagenesis was employed to create thousands of Pik TE (S148C) active site region mutants by changing each selected residue one by one to all twenty canonical amino acids.34, 89, 90 Screening 2,016 protein isolates, we identified five single amino acid Pik TEs S148C variants (L29M, R160C, A217T, A217Y, G222V) with improved macrocycle yields. These variants were further confirmed by protein production, purification and enzymatic reactions. The Pik TE S148C with additional single mutations L29M, A217T and A217Y were identified at the expected 6 Å distance from the active site region. However, the corresponding G222V and R160C mutations were located 12 Å and 20 Å, respectively, distal to the active site as unexpected polymerase-generated polymerase chain reaction (PCR) non-silent mutations. Surprisingly, these amino acid changes resulted in enhanced macrolactone ring formation. Next, we explored combinations of beneficial mutations from round 1 (R1), and found that the Pik TE S148C, A217T, G222V variant showed 3-fold enhanced activity (39% macrocycle yield based on LC-MS, Figure S4) compared to the original Pik TE S148C enzyme (Figure 2A). This triple-mutant form of Pik TE was generated by combining S148C, with mutation A217T within 6 Å and the unexpected non-silent mutation G222V located 12 Å from the active site region.
Figure 1.

Pik TE WT (Ser148, Asp176, His268) with covalent pentaketide affinity label (PDB: 2HFJ) (yellow). Twenty-one residues identified within 6 Å of the substrate in the active site region (cyan). Active site catalytic triad (purple).
Figure 2.

(A) Pik TE variants from directed evolution confirmed by scale-up protein production, purification and enzymatic reactions analyzed in LC-MS. (B) Pik TE WT crystal structure (PDB: 2HFJ) with mutations in cyan depicting Pik TE R2 using PyMOL mutagenesis tool. Pentaketide mimic affinity label in yellow and the TE catalytic triad in purple. (C) Fold improvement (% isolated yield) in formation of macrocycle 3 from substrate 1 using parental Pik TE S148C and optimized mutants.
Next, we conducted a second round of evolution focused on improvement of the optimal first round combined variant Pik TE S148C, A217T, G222V (R1). Based on the beneficial G222V we were motivated to pursue random mutagenesis in regions more distal to the active site. To this end, we employed an error-prone polymerase chain reaction (epPCR) strategy to create random mutations across the pikAIV TE gene at a higher mutation rate.90, 91 In round 2 (R2), we screened 2,880 protein isolates and identified sixteen variants with improved production of hybrid macrolactone/lactam 3. Assessing the individual mutations revealed that the distances of mutated amino acid residues relative to the active site region varied from 6 – 20 Å. This outcome demonstrated that critical residues impacting Pik TE selectivity and turnover can reside at relatively remote distances from the active site. The identified variants with improved selectivity included single, double, and triple mutations added to the original Pik TE R1 variant. These were selected for protein production, purification, and confirmation of enhanced biocatalytic activity, similar to our approach during R1. For R2, we generated the combination TE variant comprised of six mutations (Pik TE S148C, A217T, G222V, M271V, Y25C, L126V) with a 5.5-fold improvement (71% macrocycle yield based on LC-MS) (Figure 2B).
We subsequently reiterated epPCR random mutagenesis utilizing the R2 variant for a third round of evolution, following the same parameters as the second round. After screening 1,536 protein isolates, six single amino acid variants (all ~20 Å from active site region) were identified with potentially improved ring formation. However, further scaleup revealed similar yields (71% macrocycle yield based on LC-MS) compared to R2. Although these hits and two combination mutants were generated (including the combination mutant S148C, A217T, G222V, M271V, Y25C, L126V, Q18K, L87Q, A9G, A120V, E294G, G138W from round 3 (R3) containing 12 mutations by combining R2 and the six hits or mutations obtained in R3), none resulted in a yield increase and in some cases, a slight reduction on macrocyclization was observed (Figure 2A).
To demonstrate productivity of the engineered Pik TE variants at a larger scale, we conducted reactions utilizing up to 10 mg of the amide hexaketide substrate 1. HPLC purification and isolation of the desired cyclic product 3 afforded 42% (Pik TE R1), 79% (Pik TE R2) and 69% (Pik TE R3) isolated yields, respectively (Figure 2C). The remaining materials from these biocatalytic reactions included seco-acid 2 and small amounts of the corresponding glycerol ester as indicated by LC-MS (Figure S11). However, these compounds were not further quantified.
To evaluate the potential generality of the beneficial mutations that enabled improved formation of 3, we evaluated the highest yielding variants from each round of the directed evolution campaign against N-methylated hexaketide 4 as well as the O-Me form of the native hexaketide (see SI, compound S9).71 Based on LC-MS AUCs of macrolactone and hydrolysis products, Pik TE S148C provides the highest amount of 12-membered ring 5 from N-methylated hexaketide 4, while the best mutants from R1, R2, and R3 lead to an increase in seco-acid production (Figure S5). Interestingly, the Pik TE S148C, A217T mutant from R1 reflects the reaction selectivity of the Pik TE R1 combination mutant, while the Pik TE S148C, G222V mutant displayed little effect on reaction selectivity with respect to Pik TE S148C. For the O-Me derivative of native the Pik hexaketide, near identical ratios of macrocycle to hydrolysis products were observed for Pik TE S148C and the most highly optimized composite mutants from R1, R2, and R3 (Figure S6).
Total Turnover Numbers (TTNs), Thermal Shift Assay, Initial Rates for Substrate Consumption and Macrolactone Formation and Steady-State Kinetics
Additional analysis of the Pik TE S148C, R1, R2 and R3 mutants, including TTNs, thermal shift assay melting temperatures (Tm’s), and initial rates for the reactions, revealed individual variant’s stability and catalytic performance.92–94 TTN enzyme measurements enabled determination of the number of catalytic events performed by one biocatalyst active site during its lifespan or until its total decay.92, 95, 96 Our results demonstrated an increase in TTN for cyclized product compared to Pik TE S148C (Figure 3A). Notably, for the TTNs over the 18-hour reaction period and 0.2 mol % reduced biocatalyst loading, both Pik TE R2 and R3 retained the highest amount of starting material among the four enzymes, suggesting potential protein deactivation92, 95, 96 (Figure S10). Interestingly, Pik TE R1 and R3 exhibited highly similar TTN values, yet Pik TE R1 showed the lowest remaining substrate compared to R3. We then performed a thermal shift assay using SYPRO orange dye to determine protein decay and stability by thermal denaturation of the mutants.93, 97 The results indicated that both Pik TE WT and the S148C mutant had highly similar Tm values, whereas Pik TE R1 showed a Tm increase of 0.6 °C. Pik TE R2 and R3 showed a decrease in Tm by 5.0 °C and 3.8 °C, respectively, suggesting enzyme denaturation occurs at lower temperatures, which reflects lower protein stability compared to Pik TE WT, S148C and R1 (Figure 3A). These data help explain the results obtained for TTN values, since R1 as the more stable protein had the highest TTN value due to greater stability during the 18-hour TTN reaction time and 0.2 mol % reduced enzyme loading. Pik TE R2 and R3 TTN reactions had considerable amounts of starting material remaining, consistent with the deactivation of the mutant proteins under the TTN experiment conditions (Figure S10).
Figure 3.

(A) Total turnover numbers (TTNs), melting temperatures (Tm, °C), initial rates (μM/min) of hexaketide 1 consumption and macrolactone 3 formation, and enzyme parameters for hexaketide 1 and select Pik TE variants. (B) Reaction time courses: hexaketide 1 and macrolactone 3 HPLC peaks shown as area under the curve (AUC) as a function of time (min) (see Supplemental Information (SI) data for additional details on AUC values and corresponding μM concentrations). N.D. = not determined.
Additionally, a time course analysis was conducted with all four Pik TE mutants, illustrating the differences in selectivity for generating the hybrid macrolactone/lactam 3 and hydrolysis products over three rounds of directed evolution (Figure 3B). Initial rates for substrate consumption were determined for all Pik TEs, revealing similar values across the mutants, suggesting that higher concentrations of protein (1.0 mol %) and substrate have an impact on the reaction rate compared to TTN values. Likewise, initial rates of ring formation were determined, with increasing values for the evolved variants (Figure 3A). Compared to the initial rate for Pik TE S148C, Pik TE R1 has a 3.5-fold improvement, followed by Pik TE R2 and Pik TE R3 with a ~14-fold enhancement on initial rates for cyclization. These results show the increase in selectivity of our evolved Pik TE variants toward macrocyclization (Figure 3B). The data also indicates that despite the TTN results and thermal shift assay showing Pik TE R2 and R3 variants becoming more unstable over time, addition of 1.0 mol % of the enzyme hastens the reaction sufficiently to generate high quantities of the desired macrocyclic product without compromising the % yield.
Further, we performed steady-state kinetics analysis using the higher producing Pik TEs R1, R2, and a select R3 variant (S148C, A217T, G222V, M271V, Y25C, L126V, Q18K, L87Q, A9G, A120V, E294G, G138W) against hexaketide 1. An increase (~2-fold) in kcat/Km values for R2 and R3 TEs compared to Pik TE R1 revealed an improved catalytic efficiency toward cyclization (Figures 3A, S26–28). The kcat/Km values previously determined for the native Pik hexaketide (Scheme 1A) using Pik TE WT and the S148C mutant are 25 and 109 μM−1 min−1, respectively.41 These values are approximately 4 to 18 times higher than the values obtained for the best performing variant (Pik TE R2) against amide-containing hexaketide 1.
Pik TE Variants Cysteine 148 to Serine Reversions
Finally, to query the importance of the Pik TE S148C active site mutation in the Pik TE R1 variant, it was reverted to the WT catalytic triad bearing Ser148. The enzymatic reaction with the amide hexaketide 1 and Pik TE R1 C148Sreversion showed a >10-fold decrease in ring formation (39% to 3% yield; Figure S4) by LC-MS analysis. This result confirms that the S148C mutation is a critical starting point for improved activity in the Pik TE R1 biocatalyst. Moreover, combining the S148C, A217T and G222V variants within the Pik TE R1 revealed synergistic effects of the combined individual mutations. Similarly, we reverted the Pik TE R2 variant back to the C148S WT residue, generating Pik TE R2 C148Sreversion. Testing amide hexaketide 1 against the Pik TE R2 reversion mutant resulted in a decrease from 71% to a 14% yield of the macrocycle (Figure 4A, Figure S4). Despite the attenuated activity of the reversion mutants, which confirmed Pik TE S148C as foundational to success of the current strategy, these results demonstrate gain-of-function for the Pik TE WT (Ser148) and could be a potential future avenue to independently generate the hybrid macrocyclic product 3 through further directed evolution (Figure 4B).
Figure 4.
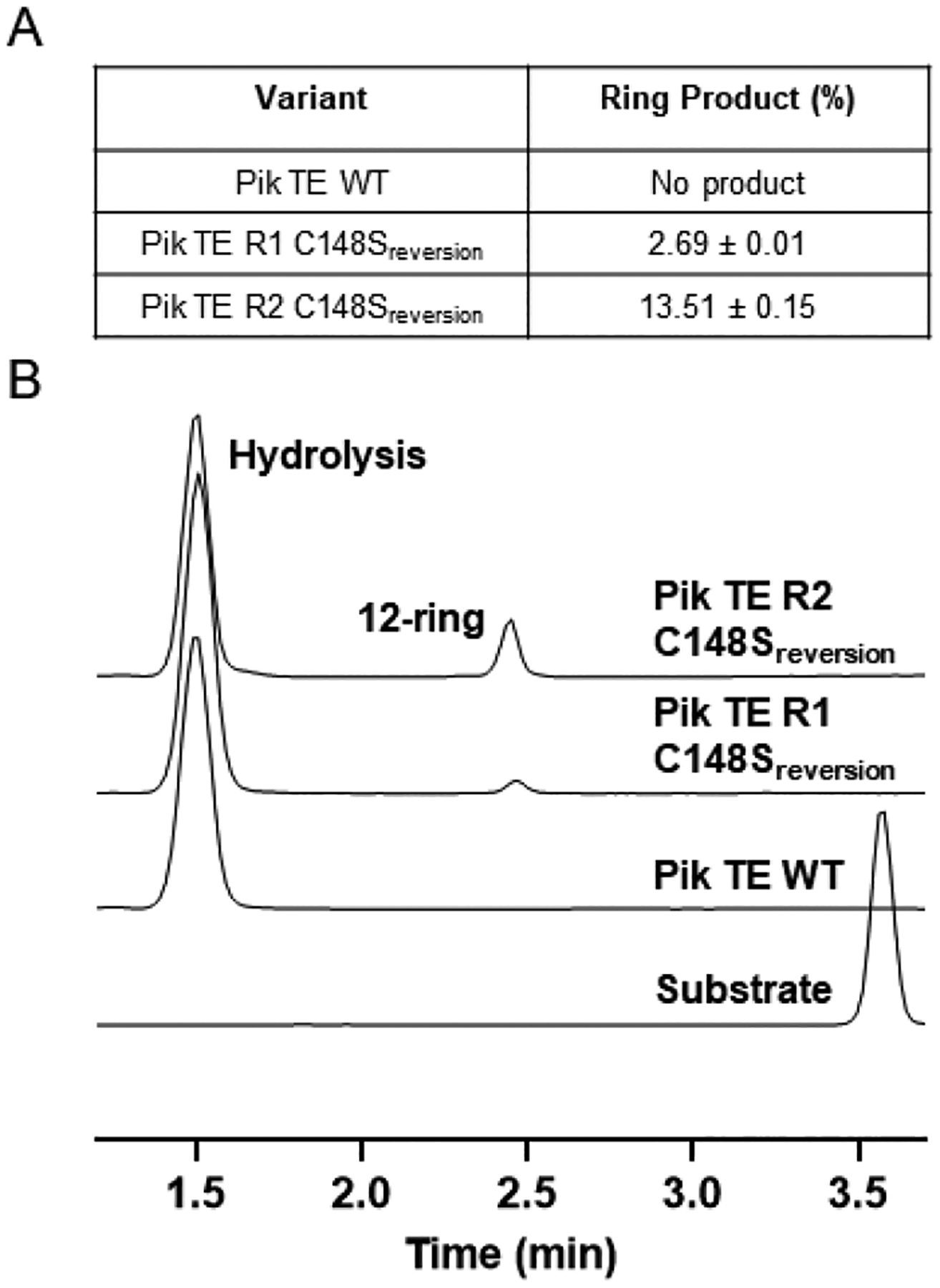
(A) LC-MS yields (%) of 3 and (B) reaction traces for hexaketide 1 and Pik TE WT, Pik TE R1 C148Sreversion and Pik TE R2 C148Sreversion highlighting a gain of function for the WT enzyme.
Discussion
Macrolactonization represents a challenging transformation in organic synthesis, including unfavorable energetics of intramolecular reactions to form rings with more than six atoms.15–17 Although the generation of macrolactones and macrolides by total and semi-synthesis is feasible, limitations to both approaches including the large number of synthetic steps, low yields, difficulties for selective addition of diverse functional groups and the inherent reactivity of large macrocycles represents a compelling area to explore with chemoenzymatic synthesis.2, 4, 9, 12, 21–23 The discovery of modular type I PKSs that assemble complex natural products like erythromycin and pikromycin allowed us to identify pathway enzymes that catalyze macrolactonization.33, 40, 41, 71, 98 In these biosynthetic systems, the PKS TE domain controls the formation of 12- and 14-membered ring structures.39 In earlier studies, Pik TE was shown to be a catalytic bottleneck for the desired ring closing reaction and was identified as a gatekeeper for processing unnatural substrates, hence impeding structural diversification and production of new macrocycles.40–42 This work demonstrates the value of TE directed evolution for macrocycle bioengineering to create and enhance the formation of new macrocyclic compounds.
Previously, the Pik TE S148C mutation revealed enhanced reaction kinetics and gain of function for the processing of an unnatural hexaketide diastereomer, which led to a shift in reaction mechanism between the WT and S148C variant as determined by quantum mechanical (QM) methods.41 In the current study, the S148C mutation is foundational for the generation of the hybrid 12-membered macrolactone/lactam ring 3, whereas the Pik TE WT leads to exclusive formation of the seco-acid amide hexaketide 2. Similar to amide hexaketide 1, production of hybrid 12-membered macrolactone/lactam ring 5 from the corresponding N-methylated variant 4 also substantially benefits from the S148C mutation. With only two rounds of evolution, the Pik TE R2 mutant provided a 6-fold improvement in isolated % yield of 3 compared to the Ser148Cys starting-point enzyme, enabling construction of a novel cyclic ring system with high productivity and selectivity (Figure 2C). The discrepancy of Pik TE R1 and R2 between macrocycle LC-MS % yields (Figure 2A) and isolated % yields (Figure 2C) could be attributed to molecule ionization dependence in LC-MS whereas isolated products are purified and quantified by weighted mass. For Pik TE R3, the variability in isolated % yield could reflect loss of product 3 during reaction workup or HPLC purification.
Our work benefitted from the Pik TE/substrate mimic crystal structure complex,34 which guided the identification of active site residues where site saturation mutagenesis was employed in a first round of evolution (Figure 1). The Pik TE R1 variant contains the A217T mutation, located 6 Å from active site. Interestingly, the A217T mutation enhances the cyclization efficiency of 1 while promoting hydrolysis of substrate 4. Intrigued by this discrepancy, we modeled substrates 1 and 4 into the active sites of Pik TE S148C and Pik TE R1 to better understand the mechanistic rationale behind this point mutation. Notably, the modeled position of the nucleophilic hydroxyl hydrogen of substrate 1 was ~1.6Å closer to the Nε of His268 in Pik TE R1, indicating its important role in the observed increased cyclization of 1 by this mutant (Figure S32). Despite the additional hydrogen-bonding capability of the A217T mutation, the threonine side chain did not form hydrogen bond contacts with the ligand. Instead, A217T appears to reorient the ligand conformation through steric interactions. This observation aligns with our findings that the A217T mutation is specific to 1, as the greater steric hindrance from substrate 4 may negatively impact cyclization (Figure S32). While these observations provide initial insights into the molecular basis of macrocycle formation versus hydrolysis catalyzed by Pik TE mutants generated through directed evolution, we expect future computational approaches will deepen our mechanistic understanding of these enzymes. Finally, although the G222V mutation is 12 Å from the active site region, increasing non-polar or hydrophobic effects of the more distal enzyme regions could induce conformational changes near the active site that promote cyclization and protein stability (Figure 2B). Previous studies have similarly shown that mutating residues relatively distant from the active site can improve enzymatic activity.36, 68, 99–101
The epPCR random mutagenesis strategy enabled us to investigate residues from the entire Pik TE sequence. Over the course of this study, sixteen mutants were identified that improved macrocycle product yields compared to variants generated during the first round of directed evolution. Additional synergistic effects were observed when combining three of the mutations M271V, Y25C, L126V with Pik TE R1 (Figure 2A). Residue Y25 was shown in the Pik TE crystal structure to engage in direct interactions with the 12-membered cyclized product 10-DML.34 Therefore, we propose that Cys25 might promote polar contacts with the unnatural substrate that favor product formation. Mutation M271V is positioned behind the Asp176 and His268 catalytic triad residues 12 Å distal to the structure bound substrate in the active site region, whereas L126V is 20 Å away, and to the opposite side of the triad (Figure 2B).34 These valine mutations in Pik TE R2 could have hydrophobic interactions that create a more hospitable active site pocket and nearby regions of the catalytic site, allowing macrocyclization to occur more readily and excluding water that alleviates competing TE mediated hydrolysis.102
Regarding R3 mutations, all resided 20 Å distal to the active site region located at the surface of the TE and failed to provide higher production of 3, thus revealing the evolutionary limits of our current screen. Our most highly optimized mutant Pik TE R2 (79% isolated yield), contains three valines, two cysteines and one threonine mutations added compared to the Pik TE WT (Figure 2B). Enzyme performance limits with directed evolution have been reported previously.103–105 Although predicting the potential benefits of additional mutations or rounds of evolution can be difficult, exploring existing mutations and understanding their impact on enzyme active site conformation, substrate binding, reaction mechanism, including the effects of individual mutations and combinations for overall protein stability could be informative for future studies.106–108 In addition, complementary or parallel trajectories of individual variants can be pursued to maximally improve enzyme activities.103, 104, 109, 110 Another option is ultra-high-throughput droplet-based microfluidic screening to identify further optimized enzymes.111 TTNs were chosen as these experiments show the timescale of the enzyme activity over the course of its deactivation.92 TTN values for the Pik TE variants were experimentally shown to be Pik TE R1 > Pik TE R3 > Pik TE R2 > Pik TE S148C (Figure 3A). We hypothesized that this trend may be attributed to enzymatic deactivation with increased ring selectivity for R3, and resulting elevated TTN values comparable to R1. Thus, although Pik TE R1 has a lower selectivity for macrocyclization, it remains active for a longer period.112, 113 Our observation of the trend observed between TTN and Tm values for the Pik TE variants could be rationalized from other studies showing enzymes possessing high thermal stability also exhibit superior TTN numbers.92 Improved thermostability typically indicates that the protein retains activity over a longer period of time.114, 115 A study where TTNs were estimated using the half-life of glucose dehydrogenase from Bacillus subtilis and its mutants showcased that in some cases the activity of an enzyme may be increased at the expense of another such as the half-life. One of the glucose dehydrogenase mutant forms was 19% less active than the WT and contained an 80% longer half-life, which resulted in a TTN 49% higher than the WT. Although this might not always be the case, longer half-lives often resulted in higher biocatalyst TTNs.92 The trade-off observed between adding novel or improved enzyme functionalities and encountering stability issues has been observed frequently in protein engineering studies.106, 108, 112, 116 Alternatives to improve the stability of the biocatalysts include rounds of evolution targeted to increasing the thermostability114, 117–119 and protein immobilization strategies.118 Our thermal shift assay analysis revealed that Pik TE % yield improvement for macrocyclization occurred at the cost of protein stability due to lower Tm values from R2 and R3 in comparison to Pik TE WT and the S148C mutant (Figure 3A). Although some degree of enzyme deactivation was observed in the thermal shift assay (Figure 3A), it did not significantly impact the enzymatic reactions at both analytical and scale-up levels. This was mainly due to employing higher enzyme concentrations (1.0 mol %) resulting in complete conversion of the substrate within one hour, overcoming possible stability challenges present in longer reaction times (Figure 3B).
In comparison, the initial rates for substrate consumption by the Pik TE S148C, R1, R2, and R3 mutants showed similar μM/min values, indicating that an increase in protein concentration and substrate had an impact on the rate. Moreover, initial rates for formation of 12-membered ring 3 demonstrated an increase in enzymatic activity and shift in selectivity for the engineered TEs (Figure 3A). In the reaction time courses, Pik TE R2 and R3 showed an initial increase in macrocycle and hydrolysis products at the onset of the reaction and then macrolactone formation surpassed seco-acid formation, which could relate to the enzyme mechanism parameters that influence hydrolysis or cyclization (Figure 3B). In a previous study, we showed that Pik TE WT followed a stepwise addition-elimination mechanism with transient formation of a tetrahedral intermediate. By contrast, Pik TE S148C followed a concerted acyl substitution process, which is lower in energy.41 The new mutations from this directed evolution campaign could have an impact on the biocatalyst energetic barriers in comparison to both Pik TE WT and S148C.34, 35, 102, 120–126 Despite the increased enzymatic efficiency and yield improvements from these engineering efforts of Pik TE, the kcat/Km values remain substantially lower compared to Pik TE WT and the S148C mutant for the native Pik hexaketide. These findings are consistent with the attenuated performance (kcat/Km) of other non-native hexaketides using Pik TE WT and the S148C mutant.41
The Pik TE S148C catalytic triad mutation was also identified as fundamental in Pik TE R1 and Pik TE R2 variants as shown in reversion to the WT Ser148. Restoration of the Ser active site led to a >10-fold decrease in macrocycle formation (39% to 3% LC-MS yield) in R1 and 5-fold decrease for R2 (71% to 14% LC-MS yield) (Figure 4). Previously, the S148C mutation was shown to possess improved reaction kinetics for processing and accepting a hexaketide substrate with non-native hydroxyl group stereochemistry.41
Overall, the current study represents a proof-of-concept approach for relieving the PKS TE bottleneck in the formation of new macrocycles. The results demonstrate that directed evolution improved selectivity and productivity in the catalysis of an unnatural amide-containing hexaketide substrate 1 and offers significant promise for expanding to other substrates with diverse functionalities, aiming to generate novel macrolactones, macrolactams, depsipeptides, and other TE-mediated biosynthetic systems. Moreover, future computational analysis through QM, molecular dynamics (MD) calculations and machine learning (ML) approaches127–132 could provide new mechanistic insights relating to mutations identified in our of biocatalyst variants. These approaches could also predict additional amino acids that modulate selectivity in TEs beyond the amide hexaketide substrate 1.
The identification of catalytic bottlenecks in modular PKS systems has been the subject of considerable interest. Recent work on the role of substrate selectivity of KS domains has revealed its differentiating role toward unnatural substrates for subsequent chain extension.133 The role of the KS domain for large scale combinatorial biosynthesis efforts to create new polyketide metabolites has also been addressed.134 It is evident from these studies and others that the KS and TE domains represent critical targets for further studies.34, 36, 40, 41, 43 Future structural biology, reaction mechanism, and protein engineering studies will be essential to enable broad and efficient access to novel compounds using modular PKSs as biocatalytic tools.
Conclusions
In summary, we have assessed the ability of Pik TE and a library of variants to catalyze formation of hybrid 12-membered macrolactone/lactam rings from two unnatural amide hexaketide substrates. We employed directed evolution to engineer the Pik TE, starting from the previously reported Pik TE S148C mutant41 for an increase in macrocyclization of 13% up to 79% isolated yield from substrate 1. With three rounds of evolution, we generated and screened >6,000 mutants through site saturation mutagenesis focused on the active site region (R1), and random mutagenesis covering the entire protein sequence (R2, R3). We initially identified residues of potential importance for cyclization to 3 using the previously reported Pik TE WT crystal structure with a covalently bound pentaketide mimic,34 which enabled target residues of interest to be identified for directed evolution. We expect that future computational and ML-based approaches41, 127–130 currently underway will deepen our mechanistic understanding of the cyclization process and the impact of select mutations on catalytic productivity. Moreover, our Pik TE variant library will provide an ongoing resource to identify efficient biocatalysts for varied hexaketide/heptaketide substrates with the goal of obtaining novel bioactive macrolides and related molecules.
Supplementary Material
Acknowledgements
We are grateful for support from NIH grant R35 GM118101, a Diversity Supplement to R35 GM118101S, a F31 Fellowship GM143769 (to M.L.A.C.), a Michigan Pioneer Fellowship (to B.J.C.) and the Hans W. Vahlteich Professorship (to D.H.S.). We also thank the UM Rackham Graduate Student Research Grant program. We thank Filipa Pereira for advice and assistance in data analysis, interpretation and graphic design, also to Sean Newmister, Matthew S. Sigman, Austin LeSueur and Hanna D. Clements for helpful scientific discussions. The authors acknowledge Rajani Arora, UM LSI Multimedia and Social Media Specialist, for her contribution to the TOC figure.
Footnotes
Conflicts of interest
The authors declare no competing financial interest.
References
- 1.Saridakis I; Kaiser D; Maulide N, Unconventional Macrocyclizations in Natural Product Synthesis. ACS Cent. Sci 2020, 6 (11), 1869–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu X; Sun D, Macrocyclic Drugs and Synthetic Methodologies Toward Macrocycles. Molecules 2013, 18 (6), 6230–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Driggers EM; Hale SP; Lee J; Terrett NK, The Exploration of Macrocycles for Drug Discovery - An Underexploited Structural Class. Nat. Rev. Drug Discov 2008, 7 (7), 608–624. [DOI] [PubMed] [Google Scholar]
- 4.Cummings MD; Sekharan S, Structure-Based Macrocycle Design in Small-Molecule Drug Discovery and Simple Metrics To Identify Opportunities for Macrocyclization of Small-Molecule Ligands. J. Med. Chem 2019, 62 (15), 6843–6853. [DOI] [PubMed] [Google Scholar]
- 5.Kirst HA, Semi-Synthetic Derivatives of Erythromycin. Prog. Med. Chem 1993, 30, 57–88. [DOI] [PubMed] [Google Scholar]
- 6.Parnham MJ; Haber VE; Giamarellos-Bourboulis EJ; Perletti G; Verleden GM; Vos R, Azithromycin: Mechanisms of Action and their Relevance for Clinical Applications. Pharmacol. Ther 2014, 143 (2), 225–245. [DOI] [PubMed] [Google Scholar]
- 7.Breiner-Goldstein E; Eyal Z; Matzov D; Halfon Y; Cimicata G; Baum M; Rokney A; Ezernitchi AV; Lowell AN; Schmidt JJ; Rozenberg H; Zimmerman E; Bashan A; Valinsky L; Anzai Y; Sherman DH; Yonath A, Ribosome-Binding and Anti-Microbial Studies of the Mycinamicins, 16-Membered Macrolide Antibiotics from Micromonospora griseorubida. Nucleic Acids Res. 2021, 49 (16), 9560–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar CNSSP, Total Synthesis of Macrolides. In Organic Synthesis - A Nascent Relook, Nandeshwarappa BP, Ed. 2019. [Google Scholar]
- 9.Fernandes P; Martens E; Pereira D, Nature Nurtures the Design of New Semi-Synthetic Macrolide Antibiotics. J. Antibiot. (Tokyo) 2017, 70 (5), 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinde PB; Oh HS; Choi H; Rathwell K; Ban YH; Kim EJ; Yang I; Lee DG; Sherman DH; Kang HY; Yoon YJ, Chemoenzymatic Synthesis of Glycosylated Macrolactam Analogues of the Macrolide Antibiotic YC-17. Adv. Synth. Catal 2015, 357 (12), 2697–2711. [Google Scholar]
- 11.Lowell AN; DeMars MD; Slocum ST; Yu FA; Anand K; Chemler JA; Korakavi N; Priessnitz JK; Park SR; Koch AA; Schultz PJ; Sherman DH, Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C-H Functionalization. J. Am. Chem. Soc 2017, 139 (23), 7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jelić D; Antolović R, From Erythromycin to Azithromycin and New Potential Ribosome-Binding Antimicrobials. Antibiotics (Basel) 2016, 5 (3), 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatsuta K, Total Synthesis of the Big Four Antibiotics and Related Antibiotics. J. Antibiot. (Tokyo) 2013, 66 (3), 107–129. [DOI] [PubMed] [Google Scholar]
- 14.Hogan PC; Chen CL; Mulvihill KM; Lawrence JF; Moorhead E; Rickmeier J; Myers AG, Large-Scale Preparation of Key Building Blocks for the Manufacture of Fully Synthetic Macrolide Antibiotics. J. Antibiot. (Tokyo) 2018, 71 (2), 318–325. [DOI] [PubMed] [Google Scholar]
- 15.Marti-Centelles V; Pandey MD; Burguete MI; Luis SV, Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev 2015, 115 (16), 8736–834. [DOI] [PubMed] [Google Scholar]
- 16.Parenty A; Moreau X; Niel G; Campagne JM, Update 1 of: Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev 2013, 113 (1), PR1–PR40. [DOI] [PubMed] [Google Scholar]
- 17.Fürstner A, Lessons from Natural Product Total Synthesis: Macrocyclization and Postcyclization Strategies. Acc. Chem. Res 2021, 54 (4), 861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodward RB; Logusch E; Nambiar KP; Sakan K; Ward DE; Auyeung BW; Balaram P; Browne LJ; Card PJ; Chen CH; Chenevert RB; Fliri A; Frobel K; Gais HJ; Garratt DG; Hayakawa K; Heggie W; Hesson DP; Hoppe D; Hoppe I; Hyatt JA; Ikeda D; Jacobi PA; Kim KS; Kobuke Y; Kojima K; Krowicki K; Lee VJ; Leutert T; Malchenko S; Martens J; Matthews RS; Ong BS; Press JB; Rajanbabu TV; Rousseau G; Sauter HM; Suzuki M; Tatsuta K; Tolbert LM; Truesdale EA; Uchida I; Ueda Y; Uyehara T; Vasella AT; Vladuchick WC; Wade PA; Williams RM; Wong HNC, Asymmetric Total Synthesis of Erythromycin. 1. Synthesis of an Erythronolide A Seco Acid-Derivative via Asymmetric Induction. J. Am. Chem. Soc 1981, 103 (11), 3210–3213. [Google Scholar]
- 19.Woodward RB; Logusch E; Nambiar KP; Sakan K; Ward DE; Auyeung BW; Balaram P; Browne LJ; Card PJ; Chen CH; Chenevert RB; Fliri A; Frobel K; Gais HJ; Garratt DG; Hayakawa K; Heggie W; Hesson DP; Hoppe D; Hoppe I; Hyatt JA; Ikeda D; Jacobi PA; Kim KS; Kobuke Y; Kojima K; Krowicki K; Lee VJ; Leutert T; Malchenko S; Martens J; Matthews RS; Ong BS; Press JB; Rajanbabu TV; Rousseau G; Sauter HM; Suzuki M; Tatsuta K; Tolbert LM; Truesdale EA; Uchida I; Ueda Y; Uyehara T; Vasella AT; Vladuchick WC; Wade PA; Williams RM; Wong HNC, Asymmetric Total Synthesis of Erythromycin. 2. Synthesis of an Erythronolide A Lactone System. J. Am. Chem. Soc 1981, 103 (11), 3213–3215. [Google Scholar]
- 20.Woodward RB; Logusch E; Nambiar KP; Sakan K; Ward DE; Auyeung BW; Balaram P; Browne LJ; Card PJ; Chen CH; Chenevert RB; Fliri A; Frobel K; Gais HJ; Garratt DG; Hayakawa K; Heggie W; Hesson DP; Hoppe D; Hoppe I; Hyatt JA; Ikeda D; Jacobi PA; Kim KS; Kobuke Y; Kojima K; Krowicki K; Lee VJ; Leutert T; Malchenko S; Martens J; Matthews RS; Ong BS; Press JB; Rajanbabu TV; Rousseau G; Sauter HM; Suzuki M; Tatsuta K; Tolbert LM; Truesdale EA; Uchida I; Ueda Y; Uyehara T; Vasella AT; Vladuchick WC; Wade PA; Williams RM; Wong HNC, Asymmetric Total Synthesis of Erythromycin. 3. Total Synthesis of Erythromycin. J. Am. Chem. Soc 1981, 103 (11), 3215–3217. [Google Scholar]
- 21.Seiple IB; Zhang Z; Jakubec P; Langlois-Mercier A; Wright PM; Hog DT; Yabu K; Allu SR; Fukuzaki T; Carlsen PN; Kitamura Y; Zhou X; Condakes ML; Szczypinski FT; Green WD; Myers AG, A Platform for the Discovery of New Macrolide Antibiotics. Nature 2016, 533 (7603), 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Force G; Perfetto A; Mayer RJ; Ciofini I; Leboeuf D, Macrolactonization Reactions Driven by a Pentafluorobenzoyl Group. Angew. Chem. Int. Ed 2021, 60 (36), 19843–19851. [DOI] [PubMed] [Google Scholar]
- 23.Marsault E; Peterson ML, Macrocycles are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem 2011, 54 (7), 1961–2004. [DOI] [PubMed] [Google Scholar]
- 24.Yang M; Wang XW; Zhao JF, Ynamide-Mediated Macrolactonization. ACS Catal. 2020, 10 (9), 5230–5235. [Google Scholar]
- 25.Sengupta S; Mehta G, Macrocyclization via C-H Functionalization: A New Paradigm in Macrocycle Synthesis. Org. Biomol. Chem 2020, 18 (10), 1851–1876. [DOI] [PubMed] [Google Scholar]
- 26.Mortison JD; Sherman DH, Frontiers and Opportunities in Chemoenzymatic Synthesis. J. Org. Chem 2010, 75 (21), 7041–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J; Amatuni A; Renata H, Recent Advances in the Chemoenzymatic Synthesis of Bioactive Natural Products. Curr. Opin. Chem. Biol 2020, 55, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stout CN; Renata H, Reinvigorating the Chiral Pool: Chemoenzymatic Approaches to Complex Peptides and Terpenoids. Acc. Chem. Res 2021, 54 (5), 1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang LC; Deng HP; Renata H, Recent Progress and Developments in Chemoenzymatic and Biocatalytic Dynamic Kinetic Resolution. Org. Process Res. Dev 2022, 26 (7), 1925–1943. [Google Scholar]
- 30.Vanable EP; Habgood LG; Patrone JD, Current Progress in the Chemoenzymatic Synthesis of Natural Products. Molecules 2022, 27 (19), 6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelraheem EMM; Busch H; Hanefeld U; Tonin F, Biocatalysis Explained: From Pharmaceutical to Bulk Chemical Production. React. Chem. Eng 2019, 4 (11), 1878–1894. [Google Scholar]
- 32.Sisco SW; Larson BM; Moore JS, Relaxing Conformational Constraints in Dynamic Macrocycle Synthesis. Macromolecules 2014, 47 (12), 3829–3836. [Google Scholar]
- 33.Hansen DA; Rath CM; Eisman EB; Narayan AR; Kittendorf JD; Mortison JD; Yoon YJ; Sherman DH, Biocatalytic Synthesis of Pikromycin, Methymycin, Neomethymycin, Novamethymycin, and Ketomethymycin. J. Am. Chem. Soc 2013, 135 (30), 11232–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akey DL; Kittendorf JD; Giraldes JW; Fecik RA; Sherman DH; Smith JL, Structural Basis for Macrolactonization by the Pikromycin Thioesterase. Nat. Chem. Biol 2006, 2 (10), 537–542. [DOI] [PubMed] [Google Scholar]
- 35.Giraldes JW; Akey DL; Kittendorf JD; Sherman DH; Smith JL; Fecik RA, Structural and Mechanistic Insights into Polyketide Macrolactonization from Polyketide-Based Affinity Labels. Nat. Chem. Biol 2006, 2 (10), 531–536. [DOI] [PubMed] [Google Scholar]
- 36.McCullough TM; Choudhary V; Akey DL; Skiba MA; Bernard SM; Kittendorf JD; Schmidt JJ; Sherman DH; Smith JL, Substrate Trapping in Polyketide Synthase Thioesterase Domains: Structural Basis for Macrolactone Formation. ACS Catal. 2024, 12551–12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weissman KJ, Uncovering the Structures of Modular Polyketide Synthases. Nat. Prod. Rep 2015, 32 (3), 436–453. [DOI] [PubMed] [Google Scholar]
- 38.Weissman KJ, Genetic Engineering of Modular PKSs: From Combinatorial Biosynthesis to Synthetic Biology. Nat. Prod. Rep 2016, 33 (2), 203–230. [DOI] [PubMed] [Google Scholar]
- 39.Adrover-Castellano ML; Schmidt JJ; Sherman DH, Biosynthetic Cyclization Catalysts for the Assembly of Peptide and Polyketide Natural Products. ChemCatChem 2021, 13 (9), 2095–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansen DA; Koch AA; Sherman DH, Identification of a Thioesterase Bottleneck in the Pikromycin Pathway through Full-Module Processing of Unnatural Pentaketides. J. Am. Chem. Soc 2017, 139 (38), 13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koch AA; Hansen DA; Shende VV; Furan LR; Houk KN; Jimenez-Oses G; Sherman DH, A Single Active Site Mutation in the Pikromycin Thioesterase Generates a More Effective Macrocyclization Catalyst. J. Am. Chem. Soc 2017, 139 (38), 13456–13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koch AA; Schmidt JJ; Lowell AN; Hansen DA; Coburn KM; Chemler JA; Sherman DH, Probing Selectivity and Creating Structural Diversity through Hybrid Polyketide Synthases. Angew. Chem. Int. Ed 2020, 59 (32), 13575–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horsman ME; Hari TP; Boddy CN, Polyketide Synthase and Non-Ribosomal Peptide Synthetase Thioesterase Selectivity: Logic Gate or a Victim of Fate? Nat. Prod. Rep 2016, 33 (2), 183–202. [DOI] [PubMed] [Google Scholar]
- 44.Yuan L; Voelker TA; Hawkins DJ, Modification of the Substrate Specificity of an Acyl-Acyl Carrier Protein Thioesterase by Protein Engineering. Proc. Natl. Acad. Sci. U.S.A 1995, 92 (23), 10639–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cantu DC; Chen YF; Reilly PJ, Thioesterases: A New Perspective Based on their Primary and Tertiary Structures. Protein Sci. 2010, 19 (7), 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caswell BT; de Carvalho CC; Nguyen H; Roy M; Nguyen T; Cantu DC, Thioesterase Enzyme Families: Functions, Structures, and Mechanisms. Protein Sci. 2022, 31 (3), 652–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayer KM; Shanklin J, A Structural Model of the Plant Acyl-Acyl Carrier Protein Thioesterase FatB Comprises Two Helix/4-Stranded Sheet Domains, the N-terminal Domain Containing Residues that Affect Specificity and the C-Terminal Domain Containing Catalytic Residues. J. Biol. Chem 2005, 280 (5), 3621–3627. [DOI] [PubMed] [Google Scholar]
- 48.Wang M; Boddy CN, Examining the Role of Hydrogen Bonding Interactions in the Substrate Specificity for the Loading Step of Polyketide Synthase Thioesterase Domains. Biochemistry 2008, 47 (45), 11793–11803. [DOI] [PubMed] [Google Scholar]
- 49.Hari TP; Labana P; Boileau M; Boddy CN, An Evolutionary Model Encompassing Substrate Specificity and Reactivity of Type I Polyketide Synthase Thioesterases. ChemBioChem 2014, 15 (18), 2656–2661. [DOI] [PubMed] [Google Scholar]
- 50.Argyropoulos P; Bergeret F; Pardin C; Reimer JM; Pinto A; Boddy CN; Schmeing TM, Towards a Characterization of the Structural Determinants of Specificity in the Macrocyclizing Thioesterase for Deoxyerythronolide B Biosynthesis. Biochim. Biophys. Acta 2016, 1860 (3), 486–497. [DOI] [PubMed] [Google Scholar]
- 51.Jing F; Yandeau-Nelson MD; Nikolau BJ, Identification of Active Site Residues Implies a Two-Step Catalytic Mechanism for Acyl-ACP Thioesterase. Biochem. J 2018, 475 (23), 3861–3873. [DOI] [PubMed] [Google Scholar]
- 52.Jing F; Zhao L; Yandeau-Nelson MD; Nikolau BJ, Two Distinct Domains Contribute to the Substrate Acyl Chain Length Selectivity of Plant Acyl-ACP Thioesterase. Nat. Commun 2018, 9 (1), 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang RF; Tao WT; Liu L; Li C; Bai LQ; Zhao YL; Shi T, Insights into Specificity and Catalytic Mechanism of Amphotericin B/Nystatin Thioesterase. Proteins 2021, 89 (5), 558–568. [DOI] [PubMed] [Google Scholar]
- 54.Jiang D; Li Y; Wu W; Zhang H; Xu R; Xu H; Zhan R; Sun L, Identification and Engineering on the Nonconserved Residues of Metallo-Beta-Lactamase-Type Thioesterase to Improve the Enzymatic Activity. Biotechnol. Bioeng 2021, 118 (12), 4623–4634. [DOI] [PubMed] [Google Scholar]
- 55.Little RF; Hertweck C, Chain Release Mechanisms in Polyketide and Non-Ribosomal Peptide Biosynthesis. Nat. Prod. Rep 2022, 39 (1), 163–205. [DOI] [PubMed] [Google Scholar]
- 56.Gokhale RS; Hunziker D; Cane DE; Khosla C, Mechanism and Specificity of the Terminal Thioesterase Domain from the Erythromycin Polyketide Synthase. Chem. Biol 1999, 6 (2), 117–125. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt JJ; Khatri Y; Brody SI; Zhu C; Pietraszkiewicz H; Valeriote FA; Sherman DH, A Versatile Chemoenzymatic Synthesis for the Discovery of Potent Cryptophycin Analogs. ACS Chem. Biol 2020, 15 (2), 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai SC; Miercke LJ; Krucinski J; Gokhale R; Chen JC; Foster PG; Cane DE; Khosla C; Stroud RM, Crystal Structure of the Macrocycle-Forming Thioesterase Domain of the Erythromycin Polyketide Synthase: Versatility from a Unique Substrate Channel. Proc. Natl. Acad. Sci. U.S.A 2001, 98 (26), 14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai SC; Lu HX; Cane DE; Khosla C; Stroud RM, Insights into Channel Architecture and Substrate Specificity from Crystal Structures of Two Macrocycle-Forming Thioesterases of Modular Polyketide Synthases. Biochemistry 2002, 41 (42), 12598–12606. [DOI] [PubMed] [Google Scholar]
- 60.Kao CM; Luo GL; Katz L; Cane DE; Khosla C, Manipulation of Macrolide Ring Size by Directed Mutagenesis of a Modular Polyketide Synthase. J. Am. Chem. Soc 1995, 117 (35), 9105–9106. [Google Scholar]
- 61.Lu H; Tsai SC; Khosla C; Cane DE, Expression, Site-Directed Mutagenesis, and Steady State Kinetic Analysis of the Terminal Thioesterase Domain of the Methymycin/Picromycin Polyketide Synthase. Biochemistry 2002, 41 (42), 12590–12597. [DOI] [PubMed] [Google Scholar]
- 62.Pinto A; Wang M; Horsman M; Boddy CN, 6-Deoxyerythronolide B Synthase Thioesterase-Catalyzed Macrocyclization Is Highly Stereoselective. Org. Lett 2012, 14 (9), 2278–2281. [DOI] [PubMed] [Google Scholar]
- 63.Kohli RM; Walsh CT, Enzymology of Acyl Chain Macrocyclization in Natural Product Biosynthesis. Chem. Commun 2003, (3), 297–307. [DOI] [PubMed] [Google Scholar]
- 64.Swarbrick CMD; Nanson JD; Patterson EI; Forwood JK, Structure, Function, and Regulation of Thioesterases. Prog. Lipid Res 2020, 79, 101036. [DOI] [PubMed] [Google Scholar]
- 65.Kaur J; Sharma R, Directed Evolution: An Approach to Engineer Enzymes. Crit. Rev. Biotechnol 2006, 26 (3), 165–199. [DOI] [PubMed] [Google Scholar]
- 66.Nannemann DP; Birmingham WR; Scism RA; Bachmann BO, Assessing Directed Evolution Methods for the Generation of Biosynthetic Enzymes with Potential in Drug Biosynthesis. Future Med. Chem 2011, 3 (7), 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dalby PA, Strategy and Success for the Directed Evolution of Enzymes. Curr. Opin. Struct. Biol 2011, 21 (4), 473–480. [DOI] [PubMed] [Google Scholar]
- 68.Arnold FH, Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed 2018, 57 (16), 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao H; Bao Z; Zhao H, High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res 2015, 54 (16), 4011–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ye L; Yang C; Yu H, From Molecular Engineering to Process Engineering: Development of High-Throughput Screening Methods in Enzyme Directed Evolution. Appl. Microbiol. Biotechnol 2018, 102 (2), 559–567. [DOI] [PubMed] [Google Scholar]
- 71.Hansen DA; Koch AA; Sherman DH, Substrate Controlled Divergence in Polyketide Synthase Catalysis. J. Am. Chem. Soc 2015, 137 (11), 3735–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pavlovic D; Fajdetic A; Mutak S, Novel Hybrids of 15-Membered 8a- and 9a-Azahomoerythromycin A Ketolides and Quinolones as Potent Antibacterials. Bioorg. Med. Chem 2010, 18 (24), 8566–8582. [DOI] [PubMed] [Google Scholar]
- 73.Stimac V; Alihodzic S; Lazarevski G; Mutak S; Istuk ZM; Fajdetic A; Palej I; Paljetak HC; Marsic N; Padovan J; Tavcar B; Erakovic Haber V, Synthesis and Biological Properties of 4”-O-Acyl Derivatives of 8a-Aza-8a-Homoerythromycin. J. Antibiot. (Tokyo) 2009, 62 (3), 133–144. [DOI] [PubMed] [Google Scholar]
- 74.Hutinec A; Derek M; Lazarevski G; Sunjic V; Paljetak HC; Alihodzic S; Erakovic Haber V; Dumic M; Marsic N; Mutak S, Novel 8a-Aza-8a-Homoerythromycin-4”-(3-Substituted-Amino)Propionates with Broad Spectrum Antibacterial Activity. Bioorg. Med. Chem. Lett 2010, 20 (11), 3244–3249. [DOI] [PubMed] [Google Scholar]
- 75.Pavlovic D; Mutak S, Synthesis and Structure-Activity Relationships of Novel 8a-Aza-8a-Homoerythromycin A Ketolides. J. Med. Chem 2010, 53 (15), 5868–5880. [DOI] [PubMed] [Google Scholar]
- 76.Janas A; Przybylski P, 14-and 15-Membered Lactone Macrolides and their Analogues and Hybrids: Structure, Molecular Mechanism of Action and Biological Activity. Eur. J. Med. Chem 2019, 182, 111662. [DOI] [PubMed] [Google Scholar]
- 77.Lubberink M; Finnigan W; Schnepel C; Baldwin CR; Turner NJ; Flitsch SL, One-Step Biocatalytic Synthesis of Sustainable Surfactants by Selective Amide Bond Formation. Angew. Chem. Int. Ed 2022, 61 (30), e202205054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brown DG; Bostrom J, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have all the New Reactions Gone? J. Med. Chem 2016, 59 (10), 4443–4458. [DOI] [PubMed] [Google Scholar]
- 79.Massolo E; Pirola M; Benaglia M, Amide Bond Formation Strategies: Latest Advances on a Dateless Transformation. Eur. J. Org. Chem 2020, (30), 4641–4651. [Google Scholar]
- 80.Boström J; Brown DG; Young RJ; Keseru GM, Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug Discov 2018, 17 (12), 709–727. [DOI] [PubMed] [Google Scholar]
- 81.Tiz DB; Bagnoli L; Rosati O; Marini F; Santi C; Sancineto L, FDA-Approved Small Molecules in 2022: Clinical Uses and Their Synthesis. Pharmaceutics 2022, 14 (11), 2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jimenez DG; Poongavanam V; Kihlberg J, Macrocycles in Drug Discovery-Learning from the Past for the Future. J. Med. Chem 2023, 66 (8), 5377–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muramatsu W; Hattori T; Yamamoto H, Amide Bond Formation: Beyond the Dilemma Between Activation and Racemisation. Chem. Commun 2021, 57 (52), 6346–6359. [DOI] [PubMed] [Google Scholar]
- 84.Bering L; Craven EJ; Thomas SAS; Shepherd SA; Micklefield J, Merging Enzymes with Chemocatalysis for Amide Bond Synthesis. Nat. Commun 2022, 13 (1), 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Evans DA; Bartroli J; Shih TL, Enantioselective Aldol Condensations. 2. Erythro-Selective Chiral Alol Condensations via Boron Enolates. J. Am. Chem. Soc 1981, 103 (8), 2127–2129. [Google Scholar]
- 86.Gage JR; Evans DA, Diastereoselective Aldol Condensation Using a Chiral Oxazolidinone Auxiliary - (2S*, 3S*)-3-Hydroxy-3-Phenyl-2-Methylpropanoic Acid. Org. Synth 1990, 68, 83–91. [Google Scholar]
- 87.Willwacher J; Kausch-Busies N; Furstner A, Divergent Total Synthesis of the Antimitotic Agent Leiodermatolide. Angew. Chem. Int. Ed 2012, 51 (48), 12041–12046. [DOI] [PubMed] [Google Scholar]
- 88.Beutner GL; Cohen BM; DelMonte AJ; Dixon DD; Fraunhoffer KJ; Glace AW; Lo E; Stevens JM; Vanyo D; Wilbert C, Revisiting the Cleavage of Evans Oxazolidinones with LiOH/H2O2. Org. Process Res. Dev 2019, 23 (7), 1378–1385. [Google Scholar]
- 89.Brandenberg OF; Chen K; Arnold FH, Directed Evolution of a Cytochrome P450 Carbene Transferase for Selective Functionalization of Cyclic Compounds. J. Am. Chem. Soc 2019, 141 (22), 8989–8995. [DOI] [PubMed] [Google Scholar]
- 90.Romney DK; Murciano-Calles J; Wehrmuller JE; Arnold FH, Unlocking Reactivity of TrpB: A General Biocatalytic Platform for Synthesis of Tryptophan Analogues. J. Am. Chem. Soc 2017, 139 (31), 10769–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McIsaac RS; Engqvist MK; Wannier T; Rosenthal AZ; Herwig L; Flytzanis NC; Imasheva ES; Lanyi JK; Balashov SP; Gradinaru V; Arnold FH, Directed Evolution of a Far-Red Fluorescent Rhodopsin. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (36), 13034–13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rogers TA; Bommarius AS, Utilizing Simple Biochemical Measurements to Predict Lifetime Output of Biocatalysts in Continuous Isothermal Processes. Chem. Eng. Sci 2010, 65 (6), 2118–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huynh K; Partch CL, Analysis of Protein Stability and Ligand Interactions by Thermal Shift Assay. Curr. Protoc. Protein Sci 2015, 79, 28.9.1–28.9.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gomes MD; Woodley JM, Considerations when Measuring Biocatalyst Performance. Molecules 2019, 24 (19), 3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bommarius AS, Total Turnover Number - A Key Criterion for Process Evaluation. Chem. Ing. Tech 2023, 95 (4), 491–497. [Google Scholar]
- 96.Kozuch S; Martin JML, “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2 (12), 2787–2794. [Google Scholar]
- 97.Polizzi KM; Bommarius AS; Broering JM; Chaparro-Riggers JF, Stability of Biocatalysts. Curr. Opin. Chem. Biol 2007, 11 (2), 220–5. [DOI] [PubMed] [Google Scholar]
- 98.Mortison JD; Kittendorf JD; Sherman DH, Synthesis and Biochemical Analysis of Complex Chain-Elongation Intermediates for Interrogation of Molecular Specificity in the Erythromycin and Pikromycin Polyketide Synthases. J. Am. Chem. Soc 2009, 131 (43), 15784–15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Payne JT; Poor CB; Lewis JC, Directed Evolution of RebH for Site-Selective Halogenation of Large Biologically Active Molecules. Angew. Chem. Int. Ed 2015, 54 (14), 4226–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mendonca LM; Marana SR, Single Mutations Outside the Active Site Affect the Substrate Specificity in a β-Glycosidase. Biochim. Biophys. Acta 2011, 1814 (12), 1616–1623. [DOI] [PubMed] [Google Scholar]
- 101.Rix G; Watkins-Dulaney EJ; Almhjell PJ; Boville CE; Arnold FH; Liu CC, Scalable Continuous Evolution for the Generation of Diverse Enzyme Variants Encompassing Promiscuous Activities. Nat. Commun 2020, 11 (1), 5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu L; Tao WT; Bai LQ; Kim ES; Zhao YL; Shi T, Why Does Tautomycetin Thioesterase Prefer Hydrolysis to Macrocyclization? Theoretical Study on its Catalytic Mechanism. Cata. Sci. Technol 2019, 9 (22), 6391–6403. [Google Scholar]
- 103.Goldsmith M; Aggarwal N; Ashani Y; Jubran H; Greisen PJ; Ovchinnikov S; Leader H; Baker D; Sussman JL; Goldenzweig A; Fleishman SJ; Tawfik DS, Overcoming an Optimization Plateau in the Directed Evolution of Highly Efficient Nerve Agent Bioscavengers. Protein Eng. Des. Sel 2017, 30 (4), 333–345. [DOI] [PubMed] [Google Scholar]
- 104.Baier F; Hong N; Yang G; Pabis A; Miton CM; Barrozo A; Carr PD; Kamerlin SCL; Jackson CJ; Tokuriki N, Cryptic Genetic Variation Shapes the Adaptive Evolutionary Potential of Enzymes. Elife 2019, 8, e40789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li H; Bao QQ; Zhao JF; Xu YB; Yang SY; Xue WS; Sun Y; Liu YP, Directed Evolution Engineering to Improve Activity of Glucose Dehydrogenase by Increasing Pocket Hydrophobicity. Front. Microbiol 2022, 13, 1044226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tokuriki N; Tawfik DS, Stability Effects of Mutations and Protein Evolvability. Curr. Opin. Struct. Biol 2009, 19 (5), 596–604. [DOI] [PubMed] [Google Scholar]
- 107.Studer RA; Christin PA; Williams MA; Orengo CA, Stability-Activity Tradeoffs Constrain the Adaptive Evolution of RubisCO. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (6), 2223–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Romero PA; Arnold FH, Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell. Biol 2009, 10 (12), 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang GR; Miton CM; Tokuriki N, A Mechanistic View of Enzyme Evolution. Protein Sci. 2020, 29 (8), 1724–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pinto GP; Corbella M; Demkiv AO; Kamerlin SCL, Exploiting Enzyme Evolution for Computational Protein Design. Trends Biochem. Sci 2022, 47 (5), 375–389. [DOI] [PubMed] [Google Scholar]
- 111.Obexer R; Godina A; Garrabou X; Mittl PR; Baker D; Griffiths AD; Hilvert D, Emergence of a Catalytic Tetrad During Evolution of a Highly Active Artificial Aldolase. Nat. Chem 2017, 9 (1), 50–56. [DOI] [PubMed] [Google Scholar]
- 112.Stimple SD; Smith MD; Tessier PM, Directed Evolution Methods for Overcoming Trade-Offs Between Protein Activity and Stability. AIChE J. 2020, 66 (3), e16814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang T; Ye ZX; Lynch MD, “Multiagent” Screening Improves Directed Enzyme Evolution by Identifying Epistatic Mutations. ACS Synth. Biol 2022, 11 (5), 1971–1983. [DOI] [PubMed] [Google Scholar]
- 114.Salazar O; Cirino PC; Arnold FH, Thermostabilization of a Cytochrome p450 Peroxygenase. ChemBioChem 2003, 4 (9), 891–3. [DOI] [PubMed] [Google Scholar]
- 115.Wang Y; Huang W; Sathitsuksanoh N; Zhu Z; Zhang YH, Biohydrogenation from Biomass Sugar Mediated by In Vitro Synthetic Enzymatic Pathways. Chem. Biol 2011, 18 (3), 372–80. [DOI] [PubMed] [Google Scholar]
- 116.Bloom JD; Labthavikul ST; Otey CR; Arnold FH, Protein Stability Promotes Evolvability. Proc. Natl. Acad. Sci. U.S.A 2006, 103 (15), 5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Poor CB; Andorfer MC; Lewis JC, Improving the Stability and Catalyst Lifetime of the Halogenase RebH by Directed Evolution. ChemBioChem 2014, 15 (9), 1286–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prier CK; Soto KC; Forstater JH; Kuhl N; Kuethe JT; Cheung-Lee WL; Di Maso MJ; Eberle CM; Grosser ST; Ho HI; Hoyt E; Maguire A; Maloney KM; Makarewicz A; McMullen JP; Moore JC; Murphy GS; Narsimhan K; Pan WL; Rivera NR; Saha-Shah A; Thaisrivongs DA; Verma D; Wyatt A; Zewge D, Amination of a Green Solvent via Immobilized Biocatalysis for the Synthesis of Nemtabrutinib. ACS Catal. 2023, 13 (12), 7707–7714. [Google Scholar]
- 119.Wu S; Snajdrova R; Moore JC; Baldenius K; Bornscheuer UT, Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed 2021, 60 (1), 88–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Peracchi A, Enzyme Catalysis: Removing Chemically ‘Essential’ Residues by Site-Directed Mutagenesis. Trends Biochem. Sci 2001, 26 (8), 497–503. [DOI] [PubMed] [Google Scholar]
- 121.Chen XP; Shi T; Wang XL; Wang JT; Chen QH; Bai LQ; Zhao YL, Theoretical Studies on the Mechanism of Thioesterase-Catalyzed Macrocyclization in Erythromycin Biosynthesis. ACS Catal. 2016, 6 (7), 4369–4378. [Google Scholar]
- 122.Kokkonen P; Slanska M; Dockalova V; Pinto GP; Sanchez-Carnerero EM; Damborsky J; Klan P; Prokop Z; Bednar D, The Impact of Tunnel Mutations on Enzymatic Catalysis Depends on the Tunnel-Substrate Complementarity and the Rate-Limiting Step. Comput. Struct. Biotechnol. J 2020, 18, 805–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schafer JW; Zoi I; Antoniou D; Schwartz SD, Optimization of the Turnover in Artificial Enzymes via Directed Evolution Results in the Coupling of Protein Dynamics to Chemistry. J. Am. Chem. Soc 2019, 141 (26), 10431–10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Patel KD; d’Andrea FB; Gaudelli NM; Buller AR; Townsend CA; Gulick AM, Structure of a Bound Peptide Phosphonate Reveals the Mechanism of Nocardicin Bifunctional Thioesterase Epimerase-Hydrolase Half-Reactions. Nat. Commun 2019, 10, 3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shi T; Liu LX; Tao WT; Luo SG; Fan SB; Wang XL; Bai LQ; Zhao YL, Theoretical Studies on the Catalytic Mechanism and Substrate Diversity for Macrocyclization of Pikromycin Thioesterase. ACS Catal. 2018, 8 (5), 4323–4332. [Google Scholar]
- 126.Zhou YC; Tao WT; Qi Z; Wei JH; Shi T; Kang QJ; Zheng JT; Zhao YL; Bai LQ, Structural and Mechanistic Insights into Chain Release of the Polyene PKS Thioesterase Domain. ACS Catal. 2022, 12 (1), 762–776. [Google Scholar]
- 127.Kelly SP; Shende VV; Flynn AR; Dan Q; Ye Y; Smith JL; Tsukamoto S; Sigman MS; Sherman DH, Data Science-Driven Analysis of Substrate-Permissive Diketopiperazine Reverse Prenyltransferase NotF: Applications in Protein Engineering and Cascade Biocatalytic Synthesis of (−)-Eurotiumin A. J. Am. Chem. Soc 2022, 144 (42), 19326–19336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang S; DeMars MD; Grandner JM; Olson NM; Anzai Y; Sherman DH; Houk KN, Computational-Based Mechanistic Study and Engineering of Cytochrome P450 MycG for Selective Oxidation of 16-Membered Macrolide Antibiotics. J. Am. Chem. Soc 2020, 142 (42), 17981–17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shende VV; Harris NR; Sanders JN; Newmister SA; Khatri Y; Movassaghi M; Houk KN; Sherman DH, Molecular Dynamics Simulations Guide Chimeragenesis and Engineered Control of Chemoselectivity in Diketopiperazine Dimerases. Angew. Chem. Int. Ed 2023, 62, e2022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clements HD; Flynn AR; Nicholls BT; Grosheva D; Lefave SJ; Merriman MT; Hyster TK; Sigman MS, Using Data Science for Mechanistic Insights and Selectivity Predictions in a Non-Natural Biocatalytic Reaction. J. Am. Chem. Soc 2023, 145 (32), 17656–17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Andorfer MC; Evans D; Yang S; He CQ; Girlich AM; Vergara-Coll J; Sukumar N; Houk KN; Lewis JC, Analysis of Laboratory-Evolved Flavin-Dependent Halogenases Affords a Computational Model for Predicting Halogenase Site Selectivity. Chem. Catal 2022, 2 (10), 2658–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wittmann BJ; Johnston KE; Wu Z; Arnold FH, Advances in Machine Learning for Directed Evolution. Curr. Opin. Struct. Biol 2021, 69, 11–18. [DOI] [PubMed] [Google Scholar]
- 133.Murphy AC; Hong H; Vance S; Broadhurst RW; Leadlay PF, Broadening Substrate Specificity of a Chain-Extending Ketosynthase Through a Single Active-Site Mutation. Chem. Commun 2016, 52 (54), 8373–8376. [DOI] [PubMed] [Google Scholar]
- 134.Ray KA; Lutgens JD; Bista R; Zhang J; Desai RR; Hirsch M; Miyazawa T; Cordova A; Keatinge-Clay AT, Assessing and Harnessing Updated Polyketide Synthase Modules Through Combinatorial Engineering. Nat. Commun 2024, 15 (1), 6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.