

ABSTRACT
Background
An unmet clinical need requires the discovery of new treatments for men facing advanced prostate cancer. Aberrant glycosylation is a universal feature of cancer cells and plays a key role in tumour growth, immune evasion and metastasis. Alterations in tumour glycosylation are closely associated with prostate cancer progression, making glycans promising therapeutic targets. Fucosyltransferase 8 (FUT8) drives core fucosylation by adding α1,6‐fucose to the innermost GlcNAc residue on N‐glycans. While FUT8 is recognised as a crucial factor in cancer progression, its role in prostate cancer remains poorly understood.
Methods & Results
Here, we demonstrate using multiple independent clinical cohorts that FUT8 is upregulated in high grade and metastatic prostate tumours, and in the blood of prostate cancer patients with aggressive disease. Using novel tools, including PhosL lectin immunofluorescence and N‐glycan MALDI mass spectrometry imaging (MALDI‐MSI), we find FUT8 underpins the biosynthesis of malignant core fucosylated N‐glycans in prostate cancer cells and using both in vitro and in vivo models, we find FUT8 promotes prostate tumour growth, cell motility and invasion. Mechanistically we show FUT8 regulates the expression of genes and signalling pathways linked to prostate cancer progression. Furthermore, we find that fucosylation inhibitors can inhibit the activity of FUT8 in prostate cancer to suppress the growth of prostate tumours.
Conclusions
Our study cements FUT8‐mediated core fucosylation as an important driver of prostate cancer progression and suggests targeting FUT8 activity for prostate cancer therapy as an exciting area to explore.
Keywords: core fucosylation, fucosylation inhibitors, fucosyltransferase 8 (FUT8), glycans, prostate cancer, therapeutics, tumour growth
1. Introduction
Prostate cancer is the second most common cancer in men worldwide resulting in 375,000 deaths annually [1, 2]. Localised prostate cancer is largely curable and has a 5‐year survival rate of more than 99%, but for advanced prostate cancer only 32% of patients will still be alive after 5 years [3]. The growth of prostate tumours is driven by androgen receptor (AR) signalling and initial therapeutic options for advanced prostate cancer are hormone‐based therapies such as anti‐androgens [4, 5, 6]. Androgen deprivation therapy (ADT) typically leads to tumour shrinkage, but tumours inevitably relapse into the lethal form of the disease, termed castration‐resistant prostate cancer (CRPC), where the acquisition of resistance mechanisms mean tumours persist despite low androgen conditions [7, 8]. Patients with CRPC can be managed with second generation AR inhibitors (enzalutamide, abiraterone, apalutamide, or darolutamide), chemotherapy, immunotherapy, poly‐ADP ribose polymerase (PARP) inhibitors or radium‐233 for bone metastases [3, 9, 10, 11, 12]. However, resistance to these treatments is very common and once CRPC occurs the median survival rate for patients is only 9–30 months, meaning there is an urgent unmet clinical need to develop new therapeutic interventions [13, 14].
Altered glycosylation is a hallmark of cancer that is closely linked to a malignant phenotype [15, 16, 17]. Cancer‐associated glycans can directly impact key processes supporting tumour growth, metastasis and immune evasion and are an area of innovation in the search for new cancer therapies [18, 19, 20]. A widely occurring cancer‐associated change in glycosylation is altered fucosylation [17]. Fucosylation is a type of glycosylation where fucose residues are attached to glycans and can be divided into either terminal or core fucosylation. A family of 13 fucosyltransferase (FUT) enzymes catalyse fucosylation [21, 22, 23, 24]. Core fucosylation is catalysed by α1,6 fucosyltransferase 8 (FUT8) which transfers fucose to the innermost GlcNAc of N‐linked glycoproteins by an α1,6 linkage [22, 25, 26]. FUT8 is the only FUT enzyme responsible for core fucosylation, and as most other fucosyltransferases are functionally redundant this makes FUT8 and core fucosylation unique [27, 28, 29, 30, 31]. The therapeutics field has long benefited from ablating the FUT8 gene in antibody producing cells, and it is well established that this can enhance the effector functions of antibodies [32]. FUT8‐mediated core fucosylation also plays an important role in cancer biology, and upregulation of FUT8 has been identified in numerous cancer types including lung [33, 34, 35], liver [36], colorectal [37, 38, 39], thyroid [40], melanoma [41], pancreatic [42], ovarian [43, 44], breast [45, 46, 47, 48] and prostate cancer [49, 50]. In lung cancer, FUT8 can globally modify surface antigens, receptors and adhesion molecules and knockdown of FUT8 in aggressive cell lines inhibits in vivo tumour growth and metastasis [33]. A systems biology approach recently identified core fucosylation as a crucial factor in the aggressive behaviour of melanoma cells and showed FUT8 is a key driver of melanoma metastasis [41]. Similarly, in breast cancer, a network of core fucosylated glycoproteins with functional roles in breast cancer progression have been identified and linked to metastasis [45, 47]. For prostate cancer, core fucosylation of prostate specific antigen (PSA) has been widely investigated as a biomarker for aggressive disease [51, 52, 53, 54, 55] and serum fucosylated haptoglobin is known to be upregulated in high grade disease [56].
FUT8 has previously been reported as upregulated in high grade and metastatic prostate cancer and is linked increased cell motility and the development of CRPC [49, 50], however these studies were based on cell lines and small numbers of clinical samples and did not specifically investigate the in vivo functional role of FUT8. Furthermore, FUT8 is yet to be investigated as a potentially important clinical target in prostate cancer. Here we monitor FUT8 levels in > 1500 clinical samples across multiple patient cohorts and verify that FUT8 is upregulated in high grade and metastatic prostate tumours and in the blood of prostate cancer patients with aggressive prostate disease. Using novel tools, including PhosL lectin immunofluorescence and N‐glycan MALDI mass spectrometry imaging (MALDI‐MSI), we find FUT8 regulates the expression of malignant core fucosylated N‐glycans in prostate cancer cells, and using both in vitro and in vivo models, we show FUT8 can promote prostate tumour growth and increase cell migration and invasion. Using RNA‐sequencing, we reveal FUT8 controls the expression of genes and proteins linked to disease progression. Furthermore, we reveal that the action of FUT8 can be targeted in vivo using fucosyltransferase inhibitors. Our study identifies FUT8‐mediated core fucosylation as an important player in aggressive prostate cancer and highlights the targeting of FUT8 activity as a promising new strategy for prostate cancer therapy.
2. Methods
2.1. Cell Culture
Cell culture of cells was as described previously [57]. PC3 (CRL‐1435), DU145 (HTB‐81) and CW‐22Rv1 (CRL‐250) cells were obtained from ATCC (CRL‐1435 and CRL‐2505). All cells were cultured at 37°C, 5% CO2 in a humidified incubator, and passaged with trypsin every 3–4 days. Stable cell lines were created using lentiviral transduction. For FUT8 knockdown, shRNA lentiviral particles were purchased from Santa Cruz (FUT8 shRNA sc‐45757‐V and Control shRNA sc‐108080). Transductions were carried out according to the manufacturer's instructions using MOI = 5. For FUT8 overexpression, Lentifect purified lentiviral particles were purchased from Tebu‐Bio (FUT8 LPP‐A1604‐Lv242‐050 and negative control 217LPP‐NEG‐Lv242‐025‐C). Transductions were carried out according to the manufacturer's instructions using MOI = 5. Cell lines were authenticated using DNA STR analysis and tested every 3 months for mycoplasma contamination.
2.2. Real‐Time PCR
Total RNA was isolated from cultured cell lines using a RNeasy Mini Kit (Qiagen 205,411) and treated with DNase 1 (Ambion, AM2222). cDNA synthesis was performed using a Superscript VILO cDNA synthesis kit (Invitrogen, 15,596–026) according to the manufacturer's instructions. Real‐time quantitative PCR (RT‐qPCR) was carried out as previously described [57, 58]. Briefly, cDNA was tested in triplicate using SYBR Green PCR Master Mix (Invitrogen, 4,309,155) using the QuantStudio 7 Flex Real‐Time PCR System (Life Technologies). All samples were normalised using the average of three reference genes (actin, tubulin and GAPDH). Primer sequences are provided in Table S1.
2.3. Lectin Immunofluorescence
Lectin immunofluorescence was as described previously [59]. Cells were cultured in Lab‐TekII Chamber Slides (Thermo Scientific, 154,453) in complete media. After 72 h, cells were washed with PBS before permeabilization and fixation with ice‐cold absolute methanol for 10 min at −20°C. Next, slides were washed with PBS and blocked with 1X Carbo‐Free Blocking Solution (1X CFB) (Vector Laboratories, SP‐5040‐125) for 1 h at room temperature. Slides were incubated for 3 h at room temperature with 1:1000 biotinylated PhoSL (a novel lectin from the mushroom Pholiota squarrosa which specifically recognises core‐fucose [60], kindly gifted to us by Professor Ethan Goddard). Subsequently, cells were stained with Streptavidin AZDye 647 (Abcam, ab272190) for 1 h at room temperature in dark conditions. Finally, slides were washed with PBS and stained with Hoechst (Thermo Scientific, 62,249) for 15 min at room temperature. Cells were mounted using ProLong Gold Antifade reagent (Thermo Fisher, P36930). Images were acquired and processed with the ZEISS Axio Imager 3.
2.4. Immunocytochemistry
Immunocytochemistry assays were performed as described previously [61]. Briefly, cells were cultured for 72 h in complete media in Lab‐TekII Chamber Slides (Thermo Scientific, 154,453). Cells were then washed with PBS before permeabilization and fixation with ice‐cold absolute methanol at −20°C for 10 min. Slides were washed with PBS and blocked with 10% goat serum (Abcam, ab7481) for 1 h at room temperature. Slides were incubated overnight at 4°C with FUT8 rabbit polyclonal antibody (Sigma, HPA043410), PTGES3 mouse monoclonal antibody (Proteintech, 67,736‐1‐Ig), IGFBP5 rabbit polyclonal antibody (Proteintech, 55,205‐1‐AP) or IL1B rabbit polyclonal antibody (Proteintech, 16,806‐1‐AP) followed by 1 h incubation with appropriate secondary antibodies. Finally, slides were washed with PBS and stained with Hoechst (Thermo Scientific, 62,249) for 15 min at room temperature. Images were acquired and processed with the ZEISS Axio Imager 5.
2.5. Immunohistochemistry
For immunohistochemistry analysis of FUT8 protein levels in prostate cancer tissue microarrays (TMAs), antigen retrieval was performed by pressure cooking for 90 s in 10 mM citrate pH 6.0 (Sigma‐Aldrich, C9999) followed by staining with FUT8 antibody (Sigma HPA043410, 1:100). Nuclei were counterstained with haematoxylin (Sigma‐Aldrich, 51,275). Slides were scanned using an Aperio CS2 (Leica biosystems) and the levels of FUT8 were assessed using the cytoplasmic v2 algorithm. In some cases, an epithelia mask was included to identify the epithelia in the sample before running the cytoplasmic v2 algorithm. The FUT8 antibody was validated using formalin‐fixed paraffin‐embedded (FFPE) cell pellets with knockdown of FUT8 (Figure S2).
2.6. ELISA Assays
Human FUT8 sandwich ELISA kits were purchased from Cambridge Bioscience (RayBioTech, ELH‐FUT8). Samples and standards were assayed in duplicate according to the manufacturer's protocol. Conditioned media samples were prepared as described previously [58, 61].
2.7. Clinical Samples
2.7.1. RNA‐Sequencing Data
FUT8 mRNA levels in the TCGA Firehouse Legacy cohort [62] and the Cancer Cell 2018 cohort [63] were analysed using cBioportal [64, 65] as described previously [58] (Figures S1A,B).
2.7.2. RNA From Clinical Tissue
FUT8 mRNA levels were monitored using real‐time qPCR in four previously published patient cohorts [57, 58, 66, 67] (Figure 1A–D).
FIGURE 1.

FUT8 is upregulated in high grade and metastatic prostate tumours. (A–D) FUT8 gene expression levels were detected in clinical samples using real‐time quantitative PCR (RT‐qPCR). (A) FUT8 mRNA levels were significantly higher in prostate cancer relative to benign prostate hyperplasia (BPH) (n = 12, unpaired t‐test, p < 0.01, **). (B) FUT8 mRNA was monitored in a cohort of 33 BPH and 16 prostate cancer samples using real‐time PCR. FUT8 levels were higher in prostate cancer relative to BPH (n = 49, unpaired t‐test, p < 0.01, **). (C) Higher FUT8 expression was also detected in a sub‐group of prostate tumours with ‘metastatic’ biology compared to tumours with a ‘non‐metastatic’ phenotype [67] (n = 20, unpaired t‐test, p < 0.05, *). (D) FUT8 gene expression levels were also significantly increased in metastatic prostate cancer relative to localised disease (n = 20, unpaired t‐test, p < 0.01, **). (E) Immunohistochemistry (IHC) analysis of FUT8 protein levels in a previously published tissue microarray (TMA) [58, 61]. The levels of FUT8 were significantly higher in both Gleason grade 7 tumours (including both 3 + 4 and 4 + 3 tumours) and Gleason grade 8–10 tumours compared to Gleason grade 6 tumours (n = 80, unpaired t test, p = 0.0029 ** and p < 0.0001, ****). Scale bar is 300 μm. (F) Immunohistochemistry analysis of a previously published 125 case TMA [68, 69] to compare FUT8 levels in localised prostate cancer tumours and in prostate cancer tissues presenting with metastasis (all biopsy samples were taken from the primary site). FUT8 levels are significantly higher in metastatic tumours compared to localised tumours (n = 125, unpaired t test, p = 0.0084, **). Scale bar is 200 μm.
2.7.3. Prostate Cancer Tissue Microarrays (TMAs)
FUT8 protein levels were monitored using immunohistochemistry in two previously published prostate cancer TMAs, including a 96 case TMA (US Biomax, PR1921b) [58, 61] (Figure 1E) and an intermediate density TMA comprising 125 cases of advanced prostate cancer presenting with either localised prostate cancer or prostate patients presenting with metastasis (all biopsy samples were taken from the primary site) [68, 69] (Figure 1F).
2.7.4. Blood Samples
The plasma samples tested in Figure 2A were collected by the Exeter Clinical Research Facility tissue bank (Ref: STB20) during standard routine National Health Service (NHS) clinical practice and spun (at least 30 min after collection) at 4500 × g for 10 min. The separated plasma was removed, aliquoted, and stored at −80 °C. Written informed consent for the use of biological samples was provided by all patients. The patient plasma samples tested in Figure 2B were collected with ethical permission from Castle Hill Hospital (Cottingham, Hull) (ethics number: 07/H1304/121) and prepared using Histopaque (Sigma‐Aldrich, 1077) as per the manufacturer's instructions (samples were spun at 600 × g for 15 min at room temperature). Use of patient tissue was approved by the local research ethics committees. Patients gave informed consent, and all patient samples were anonymised. The serum samples analysed in Figure 2C,D were kindly provided by Dr. Colm Morrissey (University of Washington) via the Prostate Cancer Biorepository Network (PCBN). The serum samples in Figure 2C were taken from patients who had prostatectomies. For Figure 2D, the pre‐ADT samples were taken from prostate cancer patients undergoing radical prostatectomies prior to any hormonal therapy. Matched post‐ADT samples were taken from the same patients 1–4 months after beginning either Lupron or LH therapy. Our study was peer reviewed and approved by PCBN. Samples were prepared and stored using standard protocols and all patients gave informed consent.
FIGURE 2.
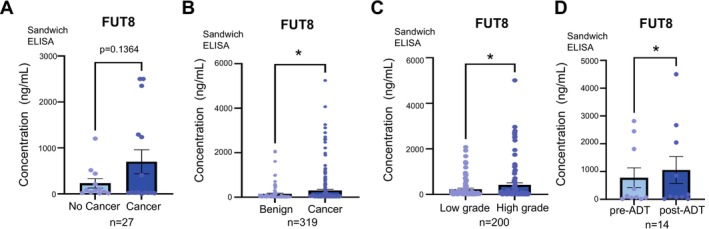
FUT8 protein levels are increased in the blood of patients with aggressive prostate cancer. (A‐D) Detection of FUT8 protein in blood samples from patients with prostate cancer using sandwich ELISA assays. (A) FUT8 levels were 3.02‐fold higher in plasma samples from patients with prostate cancer compared to patients given a no‐cancer diagnosis (n = 27, unpaired t test, p = 0.109). (B) The levels of FUT8 protein were 2.09‐fold higher in plasma samples from men with prostate cancer compared to men diagnosed with BPH (n = 319, unpaired t test, p = 0.0457, *). (C) FUT8 levels were 1.86‐fold increased in serum samples from patients with high grade prostate cancer (Gleason grade 8–9) compared to patients with low grade prostate cancer (Gleason grade 6–7) (n = 200, unpaired t test, p < 0.0218, *). (D) Analysis of FUT8 levels in matched serum samples from 7 men with prostate cancer taken before and after ADT. FUT8 serum levels significantly increase after ADT (n = 14, paired t test, p = 0.047, *).
2.8. N‐Glycan MALDI‐MSI
FFPE tissue was prepared for N‐glycan Matrix‐assisted laser desorption/ionisation mass spectrometry imaging (MALDI‐MSI) using previously published protocols for antigen retrieval, enzyme and matrix applications by solvent sprayer (M5, HTX Imaging, Durham, NC), and analysis on a timsTOF fleX QTOF mass spectrometer (Bruker Corp, Germany) [70, 71, 72]. A modification to the standard protocol was spraying a combined mixture of PNGaseF PRIME (100 mg/tissue) and Endoglycosidase F3 PRIME (10 mg/tissue, Endo F3) (N‐Zyme Scientifics, Doylestown, PA) to release N‐glycans. Endo F3 specifically cleaves core fucosylated N‐glycans, leaving a GlcNAc‐Fuc still attached to the protein, and a − 349 m/z glycan product [73]. Spectra and tissue images were annotated in SCiLS Lab software (v. 2024a) by matching peaks against an in‐house N‐glycan database [70, 72].
2.9. Lectin Flow Cytometry
Cells were cultured for 72 h in complete media containing DMSO (vehicle control) or the indicated concentrations of Fucotrim I. Cells were washed with PBS and harvested with trypsin and centrifugation (500 × g, 5 min at room temperature). The cells were washed twice with 1X Carbo‐Free Blocking Solution (1X CFB) (Vector labs, SP‐5040‐125) then resuspended in 100 μL of 1:2000 FITC‐conjugated AAL lectin (Vector Labs, FL‐1391‐1) or LCA lectin (Vector Labs, FL‐1041‐5) in 1X CFB and incubated for 30 min at 4°C. Cells were washed with PBS twice before being resuspended in 500 μL PBS with 1 μg/mL propidium iodide. 10,000 events per sample were acquired on a BD LSRFortessa Cell Analyser (BD Biosciences). Data was analysed using the FCS Express Flow Cytometry Analysis Software (the plots shown are representative of three biological repeats).
2.10. Proliferation Assays
WST1 and colony formation assays were performed as described previously [74]. For fucosylation inhibitor experiments, cells were pre‐treated with respective inhibitor/concentration for 3 days followed by 24‐h serum‐starvation.
2.11. Migration Assays
Cells were seeded into a 24‐well plate at a concentration of 2.5 × 105 cells/well and cultured until a confluent monolayer was observed. Then, media was removed from each well and the monolayer was scratched horizontally across the middle section using a P1000 pipette tip. Residues from the scratch were removed by washing with PBS before adding fresh media (10% FBS) to the wells. Scratches were imaged using a light microscope with a 4X objective magnification at time 0 and every 8 h, until a maximum incubation time of 72 h. The area of scratch was defined and quantified using ImageJ software (v 1.53 s).
2.12. Invasion Assays
Assays were conducted in collagen‐coated Oris Pro 384‐well microplates (Platypus Technologies, PRO384CMACC5) as per the manufacturer's instructions. Cells were seeded in triplicate at 10 × 105 cells/well and incubated for 2 h. Post migratory images were taken at 48 h. The area of the detection zone was measured both pre‐ and post‐invasion using ImageJ and the average percent closure was calculated.
2.13. Mouse Models
2.13.1. PC3 Tumour Xenografts
3 × 106 PC3 cells with FUT8 knockdown were implanted into the subcutaneous space of the right flank of 8‐week old Naval Medical Research Institute (NMRI) nude mice (n = 6 mice/group). The mice were randomised into control or treatment groups before cancer cell inoculation. Cells were injected in a volume of 50 μL of cell culture media and Matrigel in a 1:1 mixture. Animals were weighed and tumour volumes were monitored by calliper measurement three times a week by an unblinded researcher until the first animal met a humane endpoint (defined as tumour volume reaching 1000 mm3).
2.13.2. CWR22RV1 Tumour Xenografts
Male CD‐1 nude mice (Charles Rivers) were inoculated at 7 weeks of age with 1 × 107 CWR22RV1 cells with FUT8 overexpression by unilateral subcutaneous injection into the right flank (n = 10 mice/group). The mice were randomised into control or treatment groups before cancer cell inoculation. Cells were injected in a volume of 100 μL cell culture media and Matrigel in a 1:1 mixture. Animals were weighed and tumour volumes were monitored by calliper measurement three times a week by a blinded researcher until the first animal met a humane endpoint (defined as tumour volume reaching 1000 mm3). Tumours with ulceration were excluded from the analysis.
2.13.3. SGN‐2FF Study
CD‐1 nude mice (Charles Rivers) were randomised to start treatment with either 150 mg/kg fucosylation inhibitor SGN‐2FF (Cambridge Bioscience, HY‐107366) or water via oral gavage daily 7 days prior to implantations (n = 10 mice/group). On day 7, CWR22Rv1 cells were subcutaneously injected into the right flank of 7‐week‐old CD‐1 nude mice (1.0 × 107 cells in 50 μL Matrigel/media). Daily treatment by oral gavage for both groups continued for the duration of the study. Tumour size was measured up to 5 times weekly using callipers.
For subcutaneous xenograft models tumours were removed from the flank and prepared for histological analysis. Tumours were fixed in 10% neutral buffered formalin (Sigma HT501128‐4 L) for 24 h. After 24 h, tissue was washed in 70% ethanol to remove the residual formalin and to stop fixation before storing in fresh 70% ethanol for up to one month prior to processing. All animal experiments were approved by the Newcastle Ethical Review Committee and performed under a UK Home Office licence (PPL: PP5794374, Huw Thomas and PIL: I65375803, Kayla Bastian). All mice once obtained were housed with unrestricted access to food and water and maintained on a constant 12 h light–dark cycle.
2.14. RNA‐Sequencing
RNA sequencing data can be accessed on the GEO repository (submission GSE280132). RNA was extracted from cell lines transduced with negative‐control lentiviral particles or with stable FUT8 overexpression/knockdown particles with 3 biological repeats per experimental condition. Samples were prepared as described previously [74]. Overexpression samples and their respective controls were sequenced using an Illumina NextSeq 550, giving approximately 18 million single reads per sample. Knockdown samples and their respective controls were sequenced using an Illumina NovaSeq 6000 instrument, giving approximately 21 million single reads per sample. All data analyses were performed in Galaxy version 22.01 [75]. Quality control was performed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and reads were trimmed with Cutadapt [76]. Reads were mapped to hg38 using HISAT2 [77] and quantified with featureCounts [78]. Differential gene expression analysis was performed using limma‐voom [79] and a volcano plot was generated with ggplot2 [80]. Gene ontology (GO) analysis was performed with goseq [81] applying a significance threshold of adjusted p‐value < 0.05 for differentially expressed genes. Gene Set Enrichment Analysis (GSEA) was performed with the package EGSEA [82]. Normalised count matrix values were used to create a heatmap with gplots [83].
2.15. Statistical Analysis
Statistics were performed using the GraphPad Prism software (version 9.4.1). Data are presented as the mean of three independent samples ± standard error of the mean (SEM). Statistical significance is indicated as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
3. Results
3.1. The Fucosyltransferase Enzyme FUT8 Is Upregulated in High Grade and Metastatic Prostate Tumours
FUT8 has previously been identified as upregulated in prostate cancer tumours and linked with the development of high‐grade disease [49]. However, this study relied on cell lines and a relatively small number of clinical samples. Here, we monitor FUT8 expression at both the gene and protein level in 8 independent cohorts (comprising > 1500 clinical samples) and confirm upregulation of FUT8 in aggressive high grade prostate tumour tissue. Analysis of RNA sequencing data from The Cancer Genome Atlas Prostate Adenocarcinoma (TCGA PRAD) cohort [62] revealed FUT8 levels are significantly higher in Gleason grade 7 and 8+ tumours, compared to Gleason grade 6 tumours (n = 595, p < 0.001, p < 0.01) (Figure S1A). Similarly, in the Cancer Cell 2018 cohort [63], FUT8 levels were significantly higher in Gleason 8+ tumours compared to Gleason 6/7 prostate tumours (n = 118, p < 0.05) (Figure S1B). Previous studies have suggested that FUT8 can be repressed by androgens [84, 85], and consistent with this, we detected an increase in FUT8 levels in clinical samples from patients treated with ADT and a decrease in FUT8 levels in prostate cancer cells stimulated with androgens (Figure S1C–G). Real‐time quantitative PCR detected upregulation of the FUT8 gene in prostate cancer relative to benign prostate hyperplasia (BPH) gland (n = 12, p < 0.01) (Figure 1A) which was further validated in a larger independent patient cohort (n = 49, p < 0.01) (Figure 1B). In additional cohorts of patients with prostate cancer, FUT8 mRNA was upregulated in prostate cancers with a ‘metastatic’ signature compared to tumours with ‘non‐metastatic’ biology [67] (n20, p < 0.05) (Figure 1C) and in metastatic prostate tumours compared to localised disease (n = 20, p < 0.05) (Figure 1D). Next, to test if FUT8 is also upregulated at the protein level in high grade prostate tumours, we used immunohistochemistry (IHC) to monitor FUT8 protein levels in two previously published prostate cancer tissue microarrays (TMAs) [58, 61, 68, 69]. We confirmed the specificity of our FUT8 immunohistochemistry via detection of protein depletion in Formalin Fixed Paraffin embedded (FFPE) cell pellets (Figure S2). FUT8 protein levels were significantly higher in Gleason grade 7 and Gleason grade 8–10 (8+) tumours compared to Gleason grade 6 tumours (p < 0.01 and p < 0.0001) (Figure 1E) and in patients with metastasis compared to patients with localised disease (p = 0.0084) (Figure 1F). Taken together, our data suggest FUT8 is upregulated at both the gene and protein level in high grade prostate tumours and in patients with metastatic disease.
3.2. FUT8 Protein Levels Are Elevated in the Blood of Patients With Prostate Cancer
Shedding of glycosyltransferases from cells has previously been reported [86, 87, 88, 89] and we recently identified upregulation of the glycosyltransferase enzymes GALNT7 and ST6GAL1 in the blood of prostate cancer patients [58, 61]. We thus hypothesised that the FUT8 enzyme might also be detectable in serum/plasma samples from men with prostate cancer. Using pre‐validated sandwich ELISA assays (Figure S3), we monitored FUT8 protein levels in blood samples from men with prostate cancer. First, we tested FUT8 levels in plasma samples from 27 men with suspected prostate cancer. FUT8 levels were 3‐fold higher in plasma samples taken from men later diagnosed with prostate cancer compared to men given a ‘no cancer’ diagnosis (n = 27, p = 0.1364) (Figure 2A). Next, we monitored FUT8 plasma levels in 319 men diagnosed with either benign disease or prostate cancer. FUT8 protein levels were 2.1‐fold higher in men with prostate cancer compared to men with benign disease (n = 319, p < 0.05) (Figure 2B). We also detected higher levels of FUT8 protein in serum samples from men with high grade prostate cancer (Gleason grade 8–9) compared to low grade disease (Gleason grade 6–7) (n = 200, p < 0.0218) (Figure 2C). Finally, consistent with FUT8 being repressed by androgens, we detected significantly higher levels of FUT8 in matched serum samples taken from patients after ADT (n = 14, p = 0.002) (Figure 2D). Taken together, our findings show in addition to being upregulated in high grade prostate tumour tissue, the levels of FUT8 are also significantly higher in the blood of patients with aggressive disease.
3.3. FUT8 Promotes Prostate Tumour Growth, Cell Motility and Invasion
The data presented above show a link between FUT8 and aggressive prostate cancer.
Previous studies have linked FUT8 to the in vitro growth and motility of prostate cancer cells [84, 85, 90]. However, the impacts of FUT8 on prostate cancer biology have not yet been investigated in vivo. To address this, we created prostate cancer cells with stable overexpression (upregulation) or knockdown (downregulation) of FUT8 (Figure S3) and used these to study the effects of FUT8 on prostate cancer cell behaviour. Our findings show overexpression of FUT8 in CWR22Rv1 cells promotes proliferation and colony formation in vitro, whereas knockdown of FUT8 in PC3 and DU145 cells has the opposite effect (Figure S4). Next, using sub‐cutaneous xenograft models, we found that overexpression of FUT8 increased the growth of CWR22Rv1 tumours by 2.23 fold (p = 0.1993) (Figure 3A) whereas knockdown of FUT8 significantly suppressed the growth of PC3 tumours (p = 0.0055) (Figure 3B). Furthermore, in vitro assays showed that FUT8 can promote prostate cancer cell migration and invasion (Figure 3C–F). Taken together, the above data suggest that upregulation of FUT8 is linked to a more aggressive prostate cancer cell phenotype.
FIGURE 3.

Upregulation of FUT8 in prostate cancer cells promotes tumour growth, migration and invasion. (A) Upregulation of FUT8 in CWR22Rv1 cells increases the growth of subcutaneous xenograft tumours. 1 × 107 cells were injected into the flank of CD‐1 nude mice. Tumour size was measured every 3–4 days using callipers. Over 15 days the CWR22V1 tumours with overexpression of FUT8 were 2.23 folf bigger (n = 16, unpaired t test, p = 0.1993). Representative tumour images from each group are shown. (B) Knockdown of FUT8 using shRNA significantly reduces the growth of PC3 tumours in a subcutaneous xenograft model. 3 × 106 PC3 cells were injected into the flank of NMRI mice. Tumour size was measured every 3–4 days using callipers. Over 40 days, the growth of PC3 tumours with knockdown of FUT8 was significantly reduced (n = 12, unpaired t test, p = 0.0055, **). (C‐F) Upregulation of FUT8 in CWR22Rv1 cells promotes cell migration (unpaired t text, p = 0.0092, **) and invasion (unpaired t test, p = 0.0156, *). Knockdown of FUT8 in PC3 cells decreases prostate cancer cell migration (unpaired t test, p = 0.0102, *) and invasion (unpaired t test, p = 0.0113, *). Scale bar is 20 μm.
3.4. FUT8 Regulates Core Fucosylation of N‐Glycans in Prostate Cancer Cells
The above data links FUT8 to high grade prostate cancer and a more aggressive tumour phenotype. To test if altered FUT8 expression changes the cell surface core fucosylation of prostate cancer cells, we utilised cell lines with knockdown or overexpression of FUT8.
(Figure S3). We then monitored recognition by the core fucose specific lectin Pholiota squarrosa (PhoSL), which binds exclusively to core α‐1,6‐fucosylated N‐glycans (and not other types of fucosylated oligosaccharides) [60, 91, 92]. Patterns of PhoSL immunofluorescence revealed that knockdown of FUT8 correlated with reduced levels of core fucosylation (Figure 4A and Figure S5A), whereas upregulation of FUT8 correlated with increased levels of core fucose (Figure 4B). This finding was confirmed via N‐glycan Matrix‐assisted laser desorption/ionisation mass spectrometry imaging (MALDI‐MSI) [71, 93]. Using an enzyme that specifically cleaves core‐fucosylated N‐glycans, endoglycosidase F3 [73], we show that increased levels of FUT8 in prostate tumours correlates with a core fucosylated N‐glycan structural theme (Figure 4C). Using PhoSL immunofluorescence, we also detected an abundance of core fucosylated N‐glycans in clinical prostate cancer tissue (Figure S5B). Together, this data indicates that upregulation of the fucosyltransferase FUT8 underpins the biosynthesis of malignant core fucosylated N‐glycans in prostate cancer cells.
FIGURE 4.

FUT8 mediates core fucosylation of N‐glycans in prostate cancer cells. (A,B) Detection of core fucosylated N‐glycans using PhoSL immunofluorescence. (A) PC3 cells with knockdown of FUT8 and have reduced levels of core fucosylated N‐glycans (unpaired t test, p = 0.0227, *) (B) CWR22Rv1 cells with overexpression of FUT8 have increased levels of core fucosylated N‐glycans (unpaired t test, p = 0.0005, ***). Scale bar = 10 μM. Corrected total cell fluorescence (CTCF) indicates a significant decrease in PhoSL binding intensity with FUT8 knockdown, while overexpression of FUT8 significantly increases PhoSL binding intensity. (C) Analysis of FUT8 protein and core‐fucosylated N‐glycans in CWR22Rv1 xenograft tumours (from the experiment shown in Figure 2A) using immunohistochemistry and N‐glycan Matrix‐assisted laser desorption/ionizationmass spectrometry imaging (MALDI‐MSI) to identify core‐fucosylated N‐glycans. Images show the spatial distribution of core fucosylated bi‐antennary N‐glycan (1773.581 m/z), tri‐antennary N‐glycan (1825.5961 m/z) and the complex core fucosylated tetra‐antennary N‐glycan (2190.7632 m/z). EndoF3 cleavage induced a shift of 349.137 amu. Glycan nomenclature: Blue square indicates GlcNAc, yellow circle indicates galactose, green circle indicates mannose, red triangle indicates fucose, and purple diamond indicates sialic acid. Scale bar is 5 mm.
3.5. Upregulation of FUT8 Alters Oncogenic Genes and Proteins in Prostate Cancer Cells
The findings presented above suggested overexpression of FUT8 is linked to high grade prostate cancer and can promote an aggressive cell phenotype. Next, to search for signalling networks regulated by FUT8 target glycoproteins, we used RNA‐sequencing to identify genes that change with either knockdown or overexpression of FUT8. Bioinformatic analysis identified 381 significant differentially expressed genes when FUT8 is overexpressed in CWR22Rv1 cells and 3519 significant differentially expressed genes when. FUT8 was depleted in PC3 cells (adjusted p‐value < 0.05, Log2FC 0.58) (Figure 5A, Figure S6 and Tables S2 and S3). Interestingly, Gene Ontology analysis revealed CWR22Rv1 cells overexpressing FUT8 have enrichment in ‘ossification’, ‘bone mineralisation’ and ‘regulation of osteoblast differentiation’, whereas PC3 cells with knockdown of FUT8 have enrichment in pathways related to the ‘immune system’, ‘cell migration’ and ‘adhesion’ (Figure 5B,C, Figure S6 and Tables S4 and S5). Validation at the protein level in prostate cancer cells confirmed a correlation between FUT8 and levels of insulin‐like growth factor binding protein‐5 (IGFBP5) (which is linked to prostate cancer progression [94]), interleukin 1 beta (IL1B) (a cytokine linked to an immune suppressive microenvironment [95]) and Prostaglandin E synthase 3 (PTGES3) (an AR regulator that promotes cell proliferation [96]) (Figure 5D–F). Furthermore, analysis of the TCGA PRAD cohort [62] revealed a significant correlation between expression of FUT8 and genes for IGFBP5, IL1B and PTGES3 in clinical prostate cancer tissue (Figure 5G). These findings provide novel insights into molecular mechanisms important for prostate cancer progression and point towards targeting FUT8 and/or its associated glycoproteins as novel targets for prostate cancer therapeutics.
FIGURE 5.
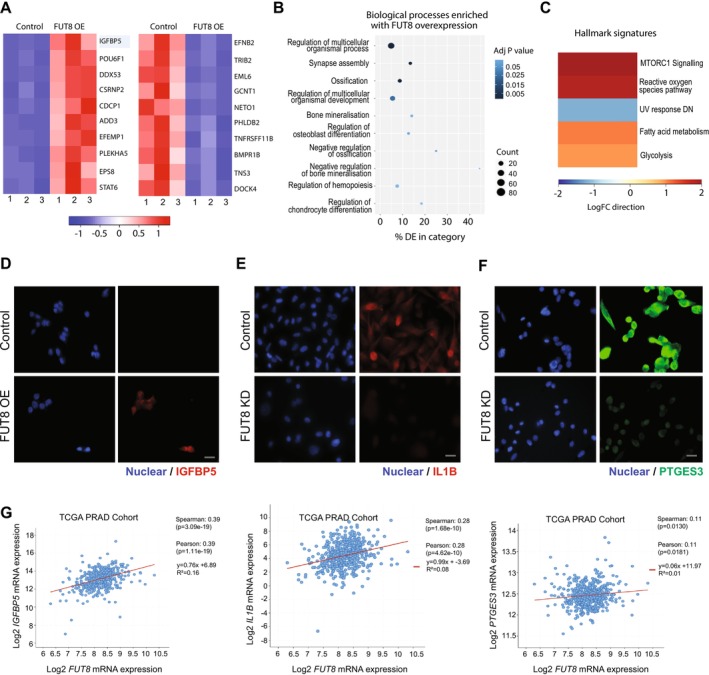
FUT8 regulates oncogenic genes and proteins in prostate cancer cells. RNA‐sequencing analysis of CWR22Rv1 cells with overexpression of FUT8 identified 381 differentially expressed genes (adjusted p‐value < 0.05, Log2FC 0.58) (Table S2). (A) Heatmap to illustrate the top 10 upregulated and 10 ten downregulated differentially expressed genes. (B, C) Gene Ontology and gene set enrichment analyses of genes regulated by FUT8 revealed CWR22Rv1 cells overexpressing FUT8 have enrichment in ‘ossification’, ‘bone mineralisation’ and ‘regulation of osteoblast differentiation’. (D–F) Validation at the protein level using immunocytochemistry shows (D) IGFBP5 is upregulated when FUT8 is overexpressed in CWR22Rv1 cells and (E, F) Knockdown of FUT8 downregulates IL1B and PTGES3 in PC3 cells. Scale bar is 20 μm. (G) Analysis of the TCGA PRAD cohort shows a significant correlation between the FUT8 gene and levels of IGFBP5, IL1B and PTGES3 in clinical prostate cancer tissue.
3.6. Targeting FUT8 Activity With Fucosyltransferase Inhibitors Suppresses Prostate Tumour Growth
To assess whether inhibition of FUT8 in prostate cancer cells will be clinically useful, we next chose to investigate whether systemic treatment with SGN‐2FF (a cell permeable fucosyltransferase inhibitor which has shown promising effects on tumour cells, immune cells, and the tumour microenvironment [97, 98, 99, 100, 101, 102]) can inhibit the in vivo growth of prostate tumours in mice. Daily oral gavage treatment with SGN‐2FF significantly suppressed the growth of CWR22Rv1 xenografts over 21 days (Figure 6A,B) and this was consistent with inhibition of core fucosylated N‐glycans in tumours (detected via MALDI‐MSI, where following EndoF3 treatment there was a shift of 349 m.u. for detected core fucosylated species) (Figure 6C). Although a Phase I clinical trial with SGN‐2FF for advanced solid tumours produced a significant drop in tumour burden, the study was terminated due to safety concerns (NCT 02952989) [104]. However, the efficacy of SGN‐2FF and its promise as a cancer therapeutic has inspired the development of new fucosylation inhibitors with higher potency than SGN‐2FF, including the SGN‐2FF derivatives A2FF1P and B2FF1P, and the metabolic inhibitors Fucotrim I and II [91, 105, 106]. Previously, we showed these compounds can effectively shut down the synthesis of fucosylated glycans in prostate cancer cells to remodel the prostate cancer glycome with only minor apparent side effects on other glycan types [74]. Using concentrations previously optimised by us for use on prostate cancer cells [74], we next tested if potent metabolic fucosylation inhibitors can inhibit the activity of FUT8 in prostate cancer. We show that treatment with A2FF1P, B2FF1P, or Fucotrim I/II significantly reduces the proliferation and survival of CWR22Rv1 cells with upregulation of FUT8 (Figure 6D,E and Figure S7). As our previous study identified Fucotrim I as having the highest efficacy for prostate cancer cells [74], we next chose to test this inhibitor using additional prostate cancer models. Treatment with Fucotrim I suppressed the growth of PC3 cells to a similar level as FUT8 knockdown (Figure 6D,E). Furthermore, our findings show Fucotrim I can block fucose incorporation and suppress colony formation in mouse prostate cancer cell lines (Figure 6F,G and Figure S8). Taken together, our data shows that blocking fucosylation inhibits the growth of prostate tumours and highlights the potential therapeutic use of fucosylation inhibitors (once modifications render them more targeted towards cancer cells) to block the malignant action of FUT8 in prostate cancer.
FIGURE 6.

Targeting FUT8‐mediated core fucosylation in prostate cancer with fucosylation inhibitors suppresses tumour growth. (A) CWR22Rv1 cells were subcutaneously injected into the flank of 7‐week‐old CD‐1 nude mice. 7 days prior to implantations mice were randomised to start treatment with either 150 mg/kg fucosylation inhibitor SGN‐2FF or water via oral gavage daily (n = 10 mice/group). Tumour size was measured every 3–4 days using callipers. (B) Tumour volume (mm3) was significantly reduced in the SGN‐2FF treated mice after 21 days (Welch's t‐test for tumour volume on Day 21, p = 0.0034, **). Representative images of tumours are shown. (C) Analysis of CWR22Rv1 xenograft tumours (from experiment shown in Figure 6B) using N‐glycan MALDI‐MSI to identify core‐fucosylated N‐glycans. Images show the spatial distribution of core fucosylated bi‐antennary N‐glycan (1773.581 m/z), tri‐antennary N‐glycan (1825.5961 m/z) and the complex core fucosylated tetra‐antennary N‐glycan (2190.7632 m/z). EndoF3 cleavage induced a shift of 349.137 amu. Glycan nomenclature: blue square indicates GlcNAc, yellow circle indicates galactose, green circle indicates mannose, red triangle indicates fucose, and purple diamond indicates sialic acid. Scale bar is 5 mm. (D) WST‐1 cell proliferation assays show FUT8 overexpression significantly increases the proliferation of CWR22RV1 cells (unpaired t‐test, p < 0.0001, ****), and this is suppressed by treatment with 30 μM of Fucotrim I over 72 h (unpaired t‐test, p < 0.0001, ****). WST‐1 cell proliferation assays also show FUT8 knockdown significantly reduces the proliferation of PC3 cells (unpaired t‐test, p < 0.0001, ****) and by treatment with 30 μM of Fucotrim I for 72 h (unpaired t‐test, p = 0.0012, ***). (E) Colony formation assays show FUT8 overexpression significantly increases the ability of CWR22RV1 cells to survive and grow in colonies over 14 days (unpaired t‐test, p < 0.0001, ****), and this is suppressed by treatment with 30 μM Fucotrim I (unpaired t test, p < 0.0001, ****). PC3 cells with knockdown of FUT8 have reduced colony formation over 14 days (unpaired t‐test, p = 0.019, **). PC3 cells treated with 30 μM Fucotrim I for 14 days have reduced ability to survive and grow in colonies over 14 days (unpaired t‐test, p = 0.0015, ***). (F) Inhibition of fucosylation in TRAMPC2 and RM1 mouse prostate cancer cells Fucotrim I detected using LCA lectin flow cytometry (which recognises core fucosylated N‐glycans [103]. Cells were treated with a range of concentrations of Fucotrim I from 1 nM to 128 μM for 72 h. The mean fluorescence intensities were normalized to a DMSO control. (G) Colony formation assays show treatment with 64 μM Fucotrim significantly reduced cell colony formation for both TRAMPC2 cells (unpaired t‐test, p < 0.0001, ****) and RM1 cells (unpaired t‐test, p < 0.0001, ****) over 7 days.
4. Discussion
Altered core fucosylation mediated by FUT8 is a key change in tumour glycan patterns that contributes to cancer growth, metastasis, and immune evasion [22, 25, 91, 107]. In this study, we measured the levels of FUT8 in > 1500 clinical samples across multiple patient cohorts and verify upregulation of FUT8 in high grade tumours and in patients with metastasis, and further show that the levels of blood borne FUT8 are also increased in patients with aggressive disease. Our findings show FUT8 underpins the synthesis of malignant core fucosylated N‐glycans in prostate cancer cells and functionally links FUT8 with prostate tumour growth and the regulation of genes and pathways implicated in disease progression. Furthermore, we find that blocking the activity of FUT8 using fucosylation inhibitors can suppress the growth of prostate tumours. Based on these findings, we propose FUT8‐mediated core fucosylation regulates pro‐oncogenic mechanisms involved in prostate cancer progression and this can likely be exploited for therapeutic usage.
Targeting aberrant fucosylation holds huge potential for cancer research, and given the critical roles of FUT8 in tumour pathology, it is poised to be a druggable target for new cancer therapies [107]. The fucosylation inhibitor SGN‐2FF has demonstrated promising anti‐cancer effects on tumour cells, immune cells and the tumour microenvironment [22, 74, 97, 98, 99, 100, 102, 108]. However, although a Phase I clinical trial with SGN‐2FF produced promising findings for advanced solid tumours, the study was terminated early due to safety issues (NCT 02952989) [104]. Since then, a range of additional fucosylation inhibitors have been developed [109], and have begun to show promise for treating cancer [74]. Here, we find that blocking fucosylation using SGN‐2FF can suppress the in vivo growth of prostate tumours. Furthermore, using in vitro assays, we find treatment with next generation fucosylation inhibitors (which reach higher effective concentrations within the cell [98, 99]) can inhibit the activity of FUT8 in prostate cancer cells. Our study provides proof‐of principle data to show metabolic inhibitors of fucosylation can be used to target FUT8‐mediated core fucosylation to reduce prostate cancer cell growth and highlights the potential to utilise these type of inhibitors as new therapies for prostate cancer.
As all fucosyltransferases use GDP‐fucose as a substrate, inhibiting global fucosylation will block all fucose containing glycans (and not just those containing core fucose) which could lead to unwanted off target side effects. Recent advances in deciphering the crystal structure of FUT8 [29, 107] have led to the development of selective FUT8 inhibitors using rationally optimised compounds in combination with virtual screening techniques. FDW028 is highly selective small‐molecule inhibitor of FUT8 identified through virtual screening and chemical refinement to bind the GDP‐fucose pocket that exhibits potent anti‐tumour activity by defucosylation, has demonstrated in vivo efficacy when applied locally near tumours, and can prolong the survival of mice with metastatic colorectal cancer [110, 111]. Manabe et al. have reported a GDP‐dependent covalent inhibitor that functions in cells without mimicking the donor substrate [112], and Gilormini et al. have introduced β‐carbafucose, a non‐selective metabolic inhibitor that targets several fucosyltransferases, including FUT8. Notably, β‐carbafucose treatment led to a marked increase in antibody‐dependent cellular cytotoxicity (ADCC) by producing afucosylated IgG in vivo, which is highly desirable for therapeutic antibody enhancement [113]. To date, no compensatory mechanisms have been described that restore core α1,6‐fucosylation in the absence of FUT8. This enzyme appears to be functionally unique, and its loss leads to profound phenotypic effects, including disrupted receptor signalling and developmental defects, without evidence of redundancy by other fucosyltransferases [114, 115]. In published in vivo studies, no significant toxic effects have been observed with the use of FDW028. Specifically, treated mice exhibited no notable changes in body weight, and no cytotoxicity was detected in cell‐based assays, suggesting a favourable tolerability profile at therapeutic doses [110]. Further studies will of course be needed to assess long‐term safety, but current data are encouraging. Moving forward, we anticipate these specific FUT8 inhibitors will be relevant for prostate cancer therapy and are candidates for further investigation.
Core fucosylation is reported to occur in 20–90% of proteins, including cytokines, receptors and immune checkpoint molecules, influencing their cellular function and playing an important role in the tumour immune microenvironment (TIME) [25, 91]. Studies have identified a role for FUT8 mediated core fucosylation in regulating epidermal growth factor receptor (EGFR) [85, 116], transforming growth factor beta receptor 1 (TGFBR1) [33], E‐cadherin [39, 117], and the immune checkpoint molecules programmed cell death receptor 1 (PD‐1) and B7‐H3 [46, 118]. A recent study utilised MALDI‐IMS to document N‐glycome alterations in tumours and revealed that while total core fucosylation levels do not change in prostate cancer relative to normal prostate tissue, it is likely the specific N‐glycans being core fucosylated are the important factor [72]. Here, we show upregulation of FUT8 correlates with the expression of oncogenic proteins, including IGFBP5, IL1B and PTGES3, which have been functionally linked to disease progression [94, 95, 96]. Our findings are consistent with a previous study which revealed FUT8 is a master regulator of cell surface receptors in aggressive prostate tumours and can promote cell survival in androgen depleted conditions [85]. While the full repertoire of specific N‐glycoproteins modified by FUT8 in prostate cancer are likely still to be fully discovered, it is clear the role of FUT8 is multi‐faceted (and likely involves the regulation of cell signalling receptors, cytokines and immune checkpoint molecules). Aberrant core fucosylation of tumours has been functionally implicated in tumour immune evasion and metastasis [22, 25, 41] but this has not yet been investigated for prostate cancer. Our data identifies correlations between FUT8 levels and pathways including ‘regulation of osteoblast differentiation’ and ‘immune response’. Metastasis to bone is common in prostate cancer and osteoblasts (bone‐forming cells) are implicated in this process [119]. FUT8 maintains high expression and protein stability of immune checkpoint molecules (including PD1, PDL1, PDL1 and B7H3) meaning there is a functional link between FUT8 activity and the suppressive state of the tumour microenvironment [46, 118, 120], and FUT8 is now a central target for cancer immunotherapy [91]. Future studies could investigate how upregulation of FUT8 in aggressive prostate cancer impacts both bone metastasis and the tumour immune microenvironment (including regulation of osteoblasts and immune checkpoint molecules). Further deciphering the biological effects mediated by FUT8 in prostate cancer could lead to new strategies targeting the FUT8 immune checkpoint axis to improving anti‐tumour immune responses in patients with cancer.
In summary, we report FUT8 is upregulated in high grade prostate tumours, and this is linked to a more aggressive tumour phenotype. Mechanistically, we show FUT8 regulates malignant core fucosylated N‐glycans on prostate cancer cells and is correlated with the expression of oncogenic proteins and pathways linked to disease progression. Furthermore, we find FUT8‐mediated core fucosylation can be targeted using metabolic fucosylation inhibitors, and that this suppresses the growth of prostate tumours. Our study cements FUT8 as an important driver of prostate cancer progression and points to the need for further characterisation of core fucosylation in prostate tumours. Given the critical roles of FUT8 in prostate cancer biology, it is poised to be a druggable target for cancer therapy. Moving forward, we propose that both global fucosylation and small molecule inhibitors of FUT8 are relevant to patients with prostate cancer and should be explored as new therapeutic avenues.
Author Contributions
K.B., M.O.‐M., K.H., E.A.V., H.S., O.H. and Z.P. performed in vitro experiments. K.B., E.S. and H.T. performed the in vivo studies. F.F., B.K., P.M., J.M., M.C., L.H. and N.J.M. contributed to clinical sample collection. K.B. and L.W. performed IHC on tissue sections. K.B. and L.W. scored pathology sections. K.B., G.G. and R.R.D. performed N‐glycan MALDI‐IMS. K.H., M.O.‐M. and K.B. carried out bioinformatics analyses. E.D.G.‐B. provided PhoSL lectin for use in the study and assisted with study design. E.R., J.F.A.P. and N.E. provided Fucotrim I for use in the study and assisted with study design and data analysis. K.B., J.M., M.O.‐M., R.R.D., R.H.G. and E.S. designed, analysed and interpreted the study. J.M. wrote the manuscript and created the figures. R.R.D., D.J.E., R.H., R.H.G., T.J.B., R.R.D. and N.W. contributed to critical review and paper writing. J.M. conceived the study and is senior author and corresponding author. All authors read the manuscript, agreed with the content, and were given the opportunity to provide input.
Conflicts of Interest
J.M. and E.S. are shareholders of GlycoScoreDx Ltd. and have filed patents related to this work (GB Patent GB2,594,103 and US Patent App. 17/780,508). J.F.A.P. and E.R. are shareholders of and employed by GlycoTherapeutics B.V. T.J.B. is a shareholder of and scientific advisor of GlycoTherapeutics B.V.; J.F.A.P. and T.J.B. are shareholders of Synvenio B.V. Radboud University and Radboudumc have filed patent applications related to Fucotrim I and Fucotrim II. All other authors declare no conflicts of interest.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Figure S7.
Figure S8.
Data S1.
Acknowledgements
This work was funded by Prostate Cancer UK and the Bob Willis Fund through Research Innovation Awards [RIA16‐ST2‐011 and RIA21‐ST2‐006], the Medical Research Council [MC/PC/18057], Prostate Cancer Research and the Mark Foundation for Cancer Research (grant references 6961 and 6974). This work was supported by an ERC‐Stg, (GlycoEdit, 758913) awarded to T.J.B. The research was supported/funded by the NIHR Exeter Clinical Research Facility. The opinions given in this paper do not necessarily represent those of the NIHR, the NHS or the Department of Health. This work is also supported by the Department of Defence Prostate Cancer Research Program, DOD Award No W81XWH‐18‐2‐0013, W81XWH‐18‐2‐0015, W81XWH‐18‐2‐0016, W81XWH‐18‐2‐0017, W81XWH‐18‐2‐0018 and W81XWH‐18‐2‐0019 PCRP Prostate Cancer Biorepository Network (PCBN). The authors would like to thank urology surgeon Mr. Matthew Simms and tissue procurement officer Dr. Vincent Mann for help collecting clinical samples.
Funding: This work was supported by Prostate Cancer UK, RIA16‐ST2‐011, RIA21‐ST2‐006, Medical Research Council, MC/PC/18057, and Prostate Cancer Research and the Mark Foundation for Cancer Research, 6961 and 6974.
Data Availability Statement
The data that supports the findings of this study are available in the [Link], [Link] of this article.
References
- 1. Sung H., Ferlay J., Siegel R. L., et al., “Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,” CA: A Cancer Journal for Clinicians 71, no. 3 (2021): 209–249. [DOI] [PubMed] [Google Scholar]
- 2. Bray F., Laversanne M., Sung H., et al., “Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,” CA: A Cancer Journal for Clinicians 74, no. 3 (2024): 229–263. [DOI] [PubMed] [Google Scholar]
- 3. Archer Goode E., Wang N., and Munkley J., “Prostate Cancer Bone Metastases Biology and Clinical Management (Review),” Oncology Letters 25, no. 4 (2023): 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nuhn P., De Bono J. S., Fizazi K., et al., “Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration‐Resistant Prostate Cancer in the Era of Precision Oncology,” European Urology 75, no. 1 (2019): 88–99. [DOI] [PubMed] [Google Scholar]
- 5. Mateo J., Fizazi K., Gillessen S., et al., “Managing Nonmetastatic Castration‐Resistant Prostate Cancer,” European Urology 75, no. 2 (2019): 285–293. [DOI] [PubMed] [Google Scholar]
- 6. Karen E. L., Jennifer M., and David J. E., “Androgen receptor and prostate cancer,” AIMS Molecular Science 3, no. 2 (2016): 280–299. [Google Scholar]
- 7. Rodrigues D. N., Boysen G., Sumanasuriya S., Seed G., Marzo A. M., and de Bono J., “The Molecular Underpinnings of Prostate Cancer: Impacts on Management and Pathology Practice,” Journal of Pathology 241, no. 2 (2017): 173–182. [DOI] [PubMed] [Google Scholar]
- 8. Zong Y. and Goldstein A. S., “Adaptation or Selection—Mechanisms of Castration‐Resistant Prostate Cancer,” Nature Reviews Urology 10, no. 2 (2013): 90–98. [DOI] [PubMed] [Google Scholar]
- 9. Rice M. A., Malhotra S. V., and Stoyanova T., “Second‐Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer,” Frontiers in Oncology 9 (2019): 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vellky J. E. and Ricke W. A., “Development and Prevalence of Castration‐Resistant Prostate Cancer Subtypes,” Neoplasia 22, no. 11 (2020): 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adashek J. J., Jain R. K., and Zhang J., “Clinical Development of PARP Inhibitors in Treating Metastatic Castration‐Resistant Prostate Cancer,” Cells 8, no. 8 (2019): 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amaral T. M., Macedo D., Fernandes I., and Costa L., “Castration‐Resistant Prostate Cancer: Mechanisms, Targets, and Treatment,” Prostate Cancer 2012 (2012): 327253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong L., Zieren R. C., Xue W., de Reijke T. M., and Pienta K. J., “Metastatic Prostate Cancer Remains Incurable, Why?,” Asian Journal of Urology 6, no. 1 (2019): 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cerasuolo M., Maccarinelli F., Coltrini D., et al., “Modeling Acquired Resistance to the Second‐Generation Androgen Receptor Antagonist Enzalutamide in the TRAMP Model of Prostate Cancer,” Cancer Research 80, no. 7 (2020): 1564–1577. [DOI] [PubMed] [Google Scholar]
- 15. Munkley J. and Elliott D. J., “Hallmarks of Glycosylation in Cancer,” Oncotarget 7, no. 23 (2016): 35478–35489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vajaria B. N. and Patel P. S., “Glycosylation: A Hallmark of Cancer?,” Glycoconjugate Journal 34, no. 2 (2017): 147–156. [DOI] [PubMed] [Google Scholar]
- 17. Pinho S. S. and Reis C. A., “Glycosylation in Cancer: Mechanisms and Clinical Implications,” Nature Reviews. Cancer 15, no. 9 (2015): 540–555. [DOI] [PubMed] [Google Scholar]
- 18. Smith B. A. H. and Bertozzi C. R., “The Clinical Impact of Glycobiology: Targeting Selectins, Siglecs and Mammalian Glycans,” Nature Reviews. Drug Discovery 20, no. 3 (2021): 217–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mereiter S., Balmana M., Campos D., Gomes J., and Reis C. A., “Glycosylation in the Era of Cancer‐Targeted Therapy: Where Are we Heading?,” Cancer Cell 36, no. 1 (2019): 6–16. [DOI] [PubMed] [Google Scholar]
- 20. Costa A. F., Campos D., Reis C. A., and Gomes C., “Targeting Glycosylation: A New Road for Cancer Drug Discovery,” Trends Cancer 6, no. 9 (2020): 757–766. [DOI] [PubMed] [Google Scholar]
- 21. Becker D. J. and Lowe J. B., “Fucose: Biosynthesis and Biological Function in Mammals,” Glycobiology 13, no. 7 (2003): 41R–53R. [DOI] [PubMed] [Google Scholar]
- 22. Bastian K., Scott E., Elliott D. J., and Munkley J., “FUT8 Alpha‐(1,6)‐Fucosyltransferase in Cancer,” International Journal of Molecular Sciences 22, no. 1 (2021): 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J., Hsu H. C., Mountz J. D., and Allen J. G., “Unmasking Fucosylation: From Cell Adhesion to Immune System Regulation and Diseases,” Cell Chemical Biology 25, no. 5 (2018): 499–512. [DOI] [PubMed] [Google Scholar]
- 24. Miyoshi E., Moriwaki K., and Nakagawa T., “Biological Function of Fucosylation in Cancer Biology,” Journal of Biochemistry 143, no. 6 (2008): 725–729. [DOI] [PubMed] [Google Scholar]
- 25. Shi M., Nan X. R., and Liu B. Q., “The Multifaceted Role of FUT8 in Tumorigenesis: From Pathways to Potential Clinical Applications,” International Journal of Molecular Sciences 25, no. 2 (2024): 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uozumi N., Yanagidani S., Miyoshi E., et al., “Purification and cDNA Cloning of Porcine Brain GDP‐L‐Fuc:N‐Acetyl‐Beta‐D‐Glucosaminide alpha1‐‐>6fucosyltransferase,” Journal of Biological Chemistry 271, no. 44 (1996): 27810–27817. [DOI] [PubMed] [Google Scholar]
- 27. Miyoshi E., Noda K., Yamaguchi Y., et al., “The alpha1‐6‐Fucosyltransferase Gene and Its Biological Significance,” Biochimica et Biophysica Acta 1473, no. 1 (1999): 9–20. [DOI] [PubMed] [Google Scholar]
- 28. Schneider M., Al‐Shareffi E., and Haltiwanger R. S., “Biological Functions of Fucose in Mammals,” Glycobiology 27, no. 7 (2017): 601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. García‐García A., Ceballos‐Laita L., Serna S., et al., “Structural Basis for Substrate Specificity and Catalysis of α1,6‐Fucosyltransferase,” Nature Communications 11, no. 1 (2020): 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Q. and Wang L. X., “Mammalian Alpha‐1,6‐Fucosyltransferase (FUT8) is the Sole Enzyme Responsible for the N‐Acetylglucosaminyltransferase I‐Independent Core Fucosylation of High‐Mannose N‐Glycans,” Journal of Biological Chemistry 291, no. 21 (2016): 11064–11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang X., Inoue S., Gu J., et al., “Dysregulation of TGF‐beta1 Receptor Activation Leads to Abnormal Lung Development and Emphysema‐Like Phenotype in Core Fucose‐Deficient Mice,” Proceedings of the National Academy of Sciences of the United States of America 102, no. 44 (2005): 15791–15796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamane‐Ohnuki N., Kinoshita S., Inoue‐Urakubo M., et al., “Establishment of FUT8 Knockout Chinese Hamster Ovary Cells: An Ideal Host Cell Line for Producing Completely Defucosylated Antibodies With Enhanced Antibody‐Dependent Cellular Cytotoxicity,” Biotechnology and Bioengineering 87, no. 5 (2004): 614–622. [DOI] [PubMed] [Google Scholar]
- 33. Chen C. Y., Jan Y. H., Juan Y. H., et al., “Fucosyltransferase 8 as a Functional Regulator of Nonsmall Cell Lung Cancer,” Proceedings of the National Academy of Sciences of the United States of America 110, no. 2 (2013): 630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Honma R., Kinoshita I., Miyoshi E., et al., “Expression of Fucosyltransferase 8 Is Associated With an Unfavorable Clinical Outcome in Non‐Small Cell Lung Cancers,” Oncology 88, no. 5 (2015): 298–308. [DOI] [PubMed] [Google Scholar]
- 35. Li F., Zhao S., Cui Y., et al., “α1,6‐Fucosyltransferase (FUT8) Regulates the Cancer‐Promoting Capacity of Cancer‐Associated Fibroblasts (CAFs) by Modifying EGFR Core Fucosylation (CF) in Non‐Small Cell Lung Cancer (NSCLC),” American Journal of Cancer Research 10, no. 3 (2020): 816–837. [PMC free article] [PubMed] [Google Scholar]
- 36. Noda K., Miyoshi E., Uozumi N., et al., “Gene Expression of α1‐6 Fucosyltransferase in Human Hepatoma Tissues: A Possible Implication for Increased Fucosylation of α‐Fetoprotein,” Hepatology 28, no. 4 (1998): 944–952. [DOI] [PubMed] [Google Scholar]
- 37. Muinelo‐Romay L., Vázquez‐Martín C., Villar‐Portela S., Cuevas E., Gil‐Martín E., and Fernández‐Briera A., “Expression and Enzyme Activity of Alpha(1,6)fucosyltransferase in Human Colorectal Cancer,” International Journal of Cancer 123, no. 3 (2008): 641–646. [DOI] [PubMed] [Google Scholar]
- 38. Noda M., Okayama H., Kofunato Y., et al., “Prognostic Role of FUT8 Expression in Relation to p53 Status in Stage II and III Colorectal Cancer,” PLoS One 13, no. 7 (2018): e0200315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osumi D., Takahashi M., Miyoshi E., et al., “Core Fucosylation of E‐Cadherin Enhances Cell‐Cell Adhesion in Human Colon Carcinoma WiDr Cells,” Cancer Science 100, no. 5 (2009): 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ito Y., Miyauchi A., Yoshida H., et al., “Expression of alpha1,6‐Fucosyltransferase (FUT8) in Papillary Carcinoma of the Thyroid: Its Linkage to Biological Aggressiveness and Anaplastic Transformation,” Cancer Letters 200, no. 2 (2003): 167–172. [DOI] [PubMed] [Google Scholar]
- 41. Agrawal P., Fontanals‐Cirera B., Sokolova E., et al., “A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis,” Cancer Cell 31, no. 6 (2017): 804–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tada K., Ohta M., Hidano S., et al., “Fucosyltransferase 8 Plays a Crucial Role in the Invasion and Metastasis of Pancreatic Ductal Adenocarcinoma,” Surgery Today 50, no. 7 (2020): 767–777. [DOI] [PubMed] [Google Scholar]
- 43. Lv X., Song J., Xue K., et al., “Core Fucosylation of Copper Transporter 1 Plays a Crucial Role in Cisplatin‐Resistance of Epithelial Ovarian Cancer by Regulating Drug Uptake,” Molecular Carcinogenesis 58, no. 5 (2019): 794–807. [DOI] [PubMed] [Google Scholar]
- 44. Takahashi T., Ikeda Y., Miyoshi E., Yaginuma Y., Ishikawa M., and Taniguchi N., “alpha1,6fucosyltransferase Is Highly and Specifically Expressed in Human Ovarian Serous Adenocarcinomas,” International Journal of Cancer 88, no. 6 (2000): 914–919. [DOI] [PubMed] [Google Scholar]
- 45. Tu C. F., Wu M. Y., Lin Y. C., Kannagi R., and Yang R. B., “FUT8 Promotes Breast Cancer Cell Invasiveness by Remodeling TGF‐β Receptor Core Fucosylation,” Breast Cancer Research 19, no. 1 (2017): 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang Y., Zhang H. L., Li Z. L., et al., “FUT8‐Mediated Aberrant N‐Glycosylation of B7H3 Suppresses the Immune Response in Triple‐Negative Breast Cancer,” Nature Communications 12, no. 1 (2021): 2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tu C. F., Li F. A., Li L. H., and Yang R. B., “Quantitative Glycoproteomics Analysis Identifies Novel FUT8 Targets and Signaling Networks Critical for Breast Cancer Cell Invasiveness,” Breast Cancer Research 24, no. 1 (2022): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yue L., Han C., Li Z., et al., “Fucosyltransferase 8 Expression in Breast Cancer Patients: A High Throughput Tissue Microarray Analysis,” Histology and Histopathology 31, no. 5 (2016): 547–555. [DOI] [PubMed] [Google Scholar]
- 49. Wang X., Chen J., Li Q. K., et al., “Overexpression of α (1,6) Fucosyltransferase Associated With Aggressive Prostate Cancer,” Glycobiology 24, no. 10 (2014): 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Höti N., Yang S., Hu Y., Shah P., Haffner M. C., and Zhang H., “Overexpression of α (1,6) Fucosyltransferase in the Development of Castration‐Resistant Prostate Cancer Cells,” Prostate Cancer and Prostatic Diseases 21, no. 1 (2018): 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fujita K., Hatano K., Tomiyama E., et al., “Serum Core‐Type Fucosylated Prostate‐Specific Antigen Index for the Detection of High‐Risk Prostate Cancer,” International Journal of Cancer 148, no. 12 (2021): 3111–3118. [DOI] [PubMed] [Google Scholar]
- 52. Hatano K., Yoneyama T., Hatakeyama S., et al., “Simultaneous Analysis of Serum α2,3‐Linked Sialylation and Core‐Type Fucosylation of Prostate‐Specific Antigen for the Detection of High‐Grade Prostate Cancer,” British Journal of Cancer 126, no. 5 (2022): 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gilgunn S., Conroy P. J., Saldova R., Rudd P. M., and O'Kennedy R. J., “Aberrant PSA Glycosylation—A Sweet Predictor of Prostate Cancer,” Nature Reviews Urology 10, no. 2 (2013): 99–107. [DOI] [PubMed] [Google Scholar]
- 54. Llop E., Ferrer‐Batalle M., Barrabes S., et al., “Improvement of Prostate Cancer Diagnosis by Detecting PSA Glycosylation‐Specific Changes,” Theranostics 6, no. 8 (2016): 1190–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Halldórsson S., Hillringhaus L., Hojer C., et al., “Development of a First‐In‐Class Antibody and a Specific Assay for α‐1,6‐Fucosylated Prostate‐Specific Antigen,” Scientific Reports 14, no. 1 (2024): 16512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fujita K., Shimomura M., Uemura M., et al., “Serum Fucosylated Haptoglobin as a Novel Prognostic Biomarker Predicting High‐Gleason Prostate Cancer,” Prostate 74, no. 10 (2014): 1052–1058. [DOI] [PubMed] [Google Scholar]
- 57. Munkley J., Li L., Krishnan S. R. G., et al., “Androgen‐Regulated Transcription of ESRP2 Drives Alternative Splicing Patterns in Prostate Cancer,” eLife 8 (2019): 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scott E., Hodgson K., Calle B., et al., “Upregulation of GALNT7 in Prostate Cancer Modifies O‐Glycosylation and Promotes Tumour Growth,” Oncogene 42, no. 12 (2023): 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goode E. A., Orozco‐Moreno M., Hodgson K., et al., “Sialylation Inhibition Can Partially Revert Acquired Resistance to Enzalutamide in Prostate Cancer Cells,” Cancers (Basel) 16, no. 17 (2024): 2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kobayashi Y., Tateno H., Dohra H., et al., “A Novel Core Fucose‐Specific Lectin From the Mushroom Pholiota Squarrosa,” Journal of Biological Chemistry 287, no. 41 (2012): 33973–33982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scott E., Archer Goode E., Garnham R., et al., “ST6GAL1‐Mediated Aberrant Sialylation Promotes Prostate Cancer Progression,” Journal of Pathology 261, no. 1 (2023): 71–84. [DOI] [PubMed] [Google Scholar]
- 62. Cancer Genome Atlas Research N , “The Molecular Taxonomy of Primary Prostate Cancer,” Cell 163, no. 4 (2015): 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gerhauser C., Favero F., Risch T., et al., “Molecular Evolution of Early‐Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories,” Cancer Cell 34, no. 6 (2018): 996–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cerami E., Gao J., Dogrusoz U., et al., “The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data,” Cancer Discovery 2, no. 5 (2012): 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gao J., Aksoy B. A., Dogrusoz U., et al., “Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal,” Science Signaling 6, no. 269 (2013): pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Munkley J., Vodak D., Livermore K. E., et al., “Glycosylation Is an Androgen‐Regulated Process Essential for Prostate Cancer Cell Viability,” eBioMedicine 8 (2016): 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Walker S. M., Knight L. A., McCavigan A. M., et al., “Molecular Subgroup of Primary Prostate Cancer Presenting With Metastatic Biology,” European Urology 72, no. 4 (2017): 509–518. [DOI] [PubMed] [Google Scholar]
- 68. Nouri M., Massah S., Caradec J., et al., “Transient Sox9 Expression Facilitates Resistance to Androgen‐Targeted Therapy in Prostate Cancer,” Clinical Cancer Research 26, no. 7 (2020): 1678–1689. [DOI] [PubMed] [Google Scholar]
- 69. Hodgson K., Orozco‐Moreno M., Goode E. A., et al., “Sialic Acid Blockade Inhibits the Metastatic Spread of Prostate Cancer to Bone,” eBioMedicine 104 (2024): 105163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Powers T. W., Neely B. A., Shao Y., et al., “MALDI Imaging Mass Spectrometry Profiling of N‐Glycans in Formalin‐Fixed Paraffin Embedded Clinical Tissue Blocks and Tissue Microarrays,” PLoS One 9, no. 9 (2014): e106255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Drake R. R., Powers T. W., Norris‐Caneda K., Mehta A. S., and Angel P. M., “In Situ Imaging of N‐Glycans by MALDI Imaging Mass Spectrometry of Fresh or Formalin‐Fixed Paraffin‐Embedded Tissue,” Current Protocols in Protein Science 94, no. 1 (2018): e68. [DOI] [PubMed] [Google Scholar]
- 72. Wallace E. N., West C. A., McDowell C. T., et al., “An N‐Glycome Tissue Atlas of 15 Human Normal and Cancer Tissue Types Determined by MALDI‐Imaging Mass Spectrometry,” Scientific Reports 14, no. 1 (2024): 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. West C. A., Liang H., Drake R. R., and Mehta A. S., “New Enzymatic Approach to Distinguish Fucosylation Isomers of N‐Linked Glycans in Tissues Using MALDI Imaging Mass Spectrometry,” Journal of Proteome Research 19, no. 8 (2020): 2989–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Orozco‐Moreno M., Visser E. A., Hodgson K., et al., “Targeting Aberrant Sialylation and Fucosylation in Prostate Cancer Cells Using Potent Metabolic Inhibitors,” Glycobiology 33, no. 12 (2023): 1155–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Galaxy C. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update,” Nucleic Acids Research 50, no. W1 (2022): W345–W351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Martin M., Cutadapt Removes Adapter Sequences from High‐Throughput Sequencing Reads, vol. 17 (EMBnet, 2011), 3–10. [Google Scholar]
- 77. Kim D., Langmead B., and Salzberg S. L., “HISAT: A Fast Spliced Aligner With Low Memory Requirements,” Nature Methods 12, no. 4 (2015): 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liao Y., Smyth G. K., and Shi W., “featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features,” Bioinformatics 30, no. 7 (2013): 923–930. [DOI] [PubMed] [Google Scholar]
- 79. Law C. W., Chen Y., Shi W., and Smyth G. K., “Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA‐Seq Read Counts,” Genome Biology 15, no. 2 (2014): R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Valero‐Mora P. M., “ggplot2: Elegant Graphics for Data Analysis,” Journal of Statistical Software Book Reviews 35, no. 1 (2010): 1–3.21603108 [Google Scholar]
- 81. Young M. D., Wakefield M. J., Smyth G. K., and Oshlack A., “Gene Ontology Analysis for RNA‐Seq: Accounting for Selection Bias,” Genome Biology 11, no. 2 (2010): R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alhamdoosh M., Ng M., Wilson N. J., et al., “Combining Multiple Tools Outperforms Individual Methods in Gene Set Enrichment Analyses,” Bioinformatics 33, no. 3 (2017): 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Warnes G. R., Bolker B., Bonebakker L., et al., gplots: Various R Programming Tools for Plotting Data. R Package Version, vol. 3 (R package version, 2015). [Google Scholar]
- 84. Hoti N., Yang S., Hu Y., Shah P., Haffner M. C., and Zhang H., “Overexpression of Alpha (1,6) Fucosyltransferase in the Development of Castration‐Resistant Prostate Cancer Cells,” Prostate Cancer and Prostatic Diseases 21, no. 1 (2018): 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hoti N., Lih T. S., Pan J., et al., “A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance,” Cancers (Basel) 12, no. 2 (2020): 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sun X., Mahajan D., Chen B., Song Z., and Lu L., “A Quantitative Study of the Golgi Retention of Glycosyltransferases,” Journal of Cell Science 134, no. 20 (2021): 258564. [DOI] [PubMed] [Google Scholar]
- 87. Lichtenthaler S. F., Lemberg M. K., and Fluhrer R., “Proteolytic Ectodomain Shedding of Membrane Proteins in Mammals‐Hardware, Concepts, and Recent Developments,” EMBO Journal 37, no. 15 (2018): e99456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hirata T., Takata M., Tokoro Y., Nakano M., and Kizuka Y., “Shedding of N‐Acetylglucosaminyltransferase‐V Is Regulated by Maturity of Cellular N‐Glycan,” Communications Biology 5, no. 1 (2022): 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hait N. C., Maiti A., Wu R., et al., “Extracellular Sialyltransferase st6gal1 in Breast Tumor Cell Growth and Invasiveness,” Cancer Gene Therapy 29, no. 11 (2022): 1662–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang X., Chen J., Li Q. K., et al., “Overexpression of Alpha (1,6) Fucosyltransferase Associated With Aggressive Prostate Cancer,” Glycobiology 24, no. 10 (2014): 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mao C., Li J., Feng L., and Gao W., “Beyond Antibody Fucosylation: Alpha‐(1,6)‐fucosyltransferase (Fut8) as a Potential New Therapeutic Target for Cancer Immunotherapy,” Antimicrobial Therapy 6, no. 2 (2023): 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yamasaki K., Yamasaki T., and Tateno H., “The Trimeric Solution Structure and Fucose‐Binding Mechanism of the Core Fucosylation‐Specific Lectin PhoSL,” Scientific Reports 8, no. 1 (2018): 7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. McDowell C. T., Lu X., Mehta A. S., Angel P. M., and Drake R. R., “Applications and Continued Evolution of Glycan Imaging Mass Spectrometry,” Mass Spectrometry Reviews 42, no. 2 (2023): 674–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Miyake H., Pollak M., and Gleave M. E., “Castration‐Induced Up‐Regulation of Insulin‐Like Growth Factor Binding Protein‐5 Potentiates Insulin‐Like Growth Factor‐I Activity and Accelerates Progression to Androgen Independence in Prostate Cancer Models,” Cancer Research 60, no. 11 (2000): 3058–3064. [PubMed] [Google Scholar]
- 95. Wang D., Cheng C., Chen X., et al., “IL‐1beta Is an Androgen‐Responsive Target in Macrophages for Immunotherapy of Prostate Cancer,” Advanced Science (Weinheim) 10, no. 17 (2023): 2206889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li H., Melnyk J. E., Fu B. X. H., et al., “Abstract B066: Genome‐Wide CRISPR Screens Identify PTGES3 as a Druggable AR Modulator,” Cancer Research 83, no. 11_Supplement (2023): B066. [Google Scholar]
- 97. Li J., Guillebon A. D., Hsu J. W., et al., “Human fucosyltransferase 6 enables prostate cancer metastasis to bone,” British Journal of Cancer 109, no. 12 (2013): 3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Okeley N. M., Alley S. C., Anderson M. E., et al., “Development of Orally Active Inhibitors of Protein and Cellular Fucosylation,” Proceedings of the National Academy of Sciences of the United States of America 110, no. 14 (2013): 5404–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhou Y., Fukuda T., Hang Q., et al., “Inhibition of Fucosylation by 2‐Fluorofucose Suppresses Human Liver Cancer HepG2 Cell Proliferation and Migration as Well as Tumor Formation,” Scientific Reports 7, no. 1 (2017): 11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Carrascal M. A., Silva M., Ramalho J. S., et al., “Inhibition of Fucosylation in Human Invasive Ductal Carcinoma Reduces E‐Selectin Ligand Expression, Cell Proliferation, and ERK1/2 and p38 MAPK Activation,” Molecular Oncology 12, no. 5 (2018): 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Okeley N. M., Heiser R. A., Zeng W., et al., “Abstract 5551: SGN‐2FF: A Small‐Molecule Inhibitor of Fucosylation Modulates Immune Cell Activity in Preclinical Models and Demonstrates Pharmacodynamic Activity in Early Phase 1 Analysis,” Cancer Research 78, no. 13_Supplement (2018): 5551. [Google Scholar]
- 102. Disis M. L., Corulli L. R., Gad E. A., et al., “Therapeutic and Prophylactic Antitumor Activity of an Oral Inhibitor of Fucosylation in Spontaneous Mammary Cancers,” Molecular Cancer Therapeutics 19, no. 5 (2020): 1102–1109. [DOI] [PubMed] [Google Scholar]
- 103. Tateno H., Nakamura‐Tsuruta S., and Hirabayashi J., “Comparative Analysis of Core‐Fucose‐Binding Lectins From Lens Culinaris and Pisum sativum Using Frontal Affinity Chromatography,” Glycobiology 19, no. 5 (2009): 527–536. [DOI] [PubMed] [Google Scholar]
- 104. Do K. T., Chow L. Q. M., Reckamp K., et al., “First‐In‐Human, First‐In‐Class, Phase I Trial of the Fucosylation Inhibitor SGN‐2FF in Patients With Advanced Solid Tumors,” Oncologist 26, no. 11 (2021): 925–e1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pijnenborg J. F. A., Visser E. A., Noga M., et al., “Cellular Fucosylation Inhibitors Based on Fluorinated Fucose‐1‐Phosphates*,” Chemistry 27, no. 12 (2021): 4022–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pijnenborg J. F. A., Rossing E., Merx J., et al., “Fluorinated Rhamnosides Inhibit Cellular Fucosylation,” Nature Communications 12, no. 1 (2021): 7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lv Y., Zhang Z., Tian S., Wang W., and Li H., “Therapeutic Potential of Fucosyltransferases in Cancer and Recent Development of Targeted Inhibitors,” Drug Discovery Today 28, no. 1 (2023): 103394. [DOI] [PubMed] [Google Scholar]
- 108. Okeley N. M., Heiser R. A., Zeng W., et al., “SGN‐2FF: A Small‐Molecule Inhibitor of Fucosylation Modulates Immune Cell Activity in Preclinical Models and Demonstrates Pharmacodynamic Activity in Early Phase 1 Analysis,” Cancer Research 78, no. 13_Supplement (2018): 5551. [Google Scholar]
- 109. Rossing E., Pijnenborg J. F. A., and Boltje T. J., “Chemical Tools to Track and Perturb the Expression of Sialic Acid and Fucose Monosaccharides,” Chemical Communications (Cambridge, England) 58, no. 87 (2022): 12139–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang M., Zhang Z., Chen M., et al., “FDW028, a Novel FUT8 Inhibitor, Impels Lysosomal Proteolysis of B7‐H3 via Chaperone‐Mediated Autophagy Pathway and Exhibits Potent Efficacy Against Metastatic Colorectal Cancer,” Cell Death & Disease 14, no. 8 (2023): 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lv Y., Zhang Z., Wang M., et al., “Discovery of Novel FUT8 Inhibitors With Promising Affinity and In Vivo Efficacy for Colorectal Cancer Therapy,” Bioorganic Chemistry 149 (2024): 107492. [DOI] [PubMed] [Google Scholar]
- 112. Manabe Y., Takebe T., Kasahara S., et al., “Development of a FUT8 Inhibitor With Cellular Inhibitory Properties,” Angewandte Chemie (International Ed. in English) 63, no. 52 (2024): e202414682. [DOI] [PubMed] [Google Scholar]
- 113. Gilormini P. A., Thota V. N., Fers‐Lidou A., et al., “A Metabolic Inhibitor Blocks Cellular Fucosylation and Enables Production of Afucosylated Antibodies,” Proceedings of the National Academy of Sciences of the United States of America 121, no. 27 (2024): e2314026121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang X., Gu J., Miyoshi E., Honke K., and Taniguchi N., “Phenotype Changes of Fut8 Knockout Mouse: Core Fucosylation Is Crucial for the Function of Growth Factor Receptor(s),” Methods in Enzymology 417 (2006): 11–22. [DOI] [PubMed] [Google Scholar]
- 115. Guo H. and Abbott K. L., “Functional Impact of Tumor‐Specific N‐Linked Glycan Changes in Breast and Ovarian Cancers,” Advances in Cancer Research 126 (2015): 281–303. [DOI] [PubMed] [Google Scholar]
- 116. Liu Y. C., Yen H. Y., Chen C. Y., et al., “Sialylation and Fucosylation of Epidermal Growth Factor Receptor Suppress Its Dimerization and Activation in Lung Cancer Cells,” Proceedings of the National Academy of Sciences of the United States of America 108, no. 28 (2011): 11332–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hu P., Shi B., Geng F., Zhang C., Wu W., and Wu X. Z., “E‐Cadherin Core Fucosylation Regulates Nuclear Beta‐Catenin Accumulation in Lung Cancer Cells,” Glycoconjugate Journal 25, no. 9 (2008): 843–850. [DOI] [PubMed] [Google Scholar]
- 118. Okada M., Chikuma S., Kondo T., et al., “Blockage of Core Fucosylation Reduces Cell‐Surface Expression of PD‐1 and Promotes Anti‐Tumor Immune Responses of T Cells,” Cell Reports 20, no. 5 (2017): 1017–1028. [DOI] [PubMed] [Google Scholar]
- 119. Logothetis C. J. and Lin S. H., “Osteoblasts in Prostate Cancer Metastasis to Bone,” Nature Reviews. Cancer 5, no. 1 (2005): 21–28. [DOI] [PubMed] [Google Scholar]
- 120. Zhang N., Li M., Xu X., et al., “Loss of Core Fucosylation Enhances the Anticancer Activity of Cytotoxic T Lymphocytes by Increasing PD‐1 Degradation,” European Journal of Immunology 50, no. 11 (2020): 1820–1833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Figure S7.
Figure S8.
Data S1.
Data Availability Statement
The data that supports the findings of this study are available in the [Link], [Link] of this article.