

Abstract
Human noroviruses (HuNoVs) are the leading cause of acute gastroenteritis worldwide in all age groups and cause significant disease and economic burden globally. To date, no approved vaccines or antiviral therapies are available to treat or prevent HuNoV illness. Several candidate vaccines are in clinical trials, although potential barriers to successful development must be overcome. Recently, significant advances have been made in understanding HuNoV biology owing to breakthroughs in virus cultivation using human intestinal tissue-derived organoid (or enteroid) cultures, advances in structural biology technology combined with epitope mapping and increased metagenomic sequencing. New and unexpected strain-specific differences in pandemic versus non-pandemic virus structures, replication properties and virus–host interactions, including host factors required for susceptibility to infection and pathogenesis, are discussed.
Introduction
Human noroviruses (HuNoVs) are a leading cause of gastroenteritis and are estimated to cause 677 million cases of acute gastroenteritis globally each year1. The greatest disease burden is in children under 5 years of age, but persons of all ages are affected. The global economic burden is significant, with an estimated US$4.2 billion in direct healthcare costs and $60.3 billion in societal costs annually2. Norovirus-associated illness is characterized by vomiting and diarrhoea, and although disease is usually self-limited, chronic diarrhoea leading to malnutrition can occur in immunocompromised persons3. These important pathogens have been difficult to study because they remained noncultivatable for almost 50 years. This barrier was overcome with HuNoV cultivation in human intestinal enteroids, providing new tools to study virus biology and pathogenesis. In addition, advances in the structural biology of HuNoVs has provided new insights into virus replication and immunity. In this Review, we highlight recent progress in the field, along with developments in the evaluation of candidate vaccines. We summarize information about HuNoV classification and highlight new information about HuNoV biology that has been discovered in the past 5 years using the replication system, including strain-specific differences in cell entry and innate immune responses. We describe neutralization assays combined with monoclonal antibodies, nanobodies and structural biology techniques to define epitopes and mechanisms of neutralization, as well as targets for antiviral development. The status of vaccine evaluation is presented, along with potential barriers to successful vaccine development.
Genetic classification and epidemiology
Norovirus is one of 11 genera in the family Caliciviridae, and its members infect a variety of mammalian species. The Norovirus genome consists of a single-stranded, positive-sense RNA of approximately 7.5 kb in length, with a viral protein (VPg) covalently linked to the 5′ end and a polyadenylated tail at the 3′ end (Fig. 1). The HuNoV genome contains three open reading frames (ORFs). ORF1 encodes a precursor polyprotein that is post-translationally cleaved by the viral protease into six non-structural proteins (NSPs): p48 (NS1/2), p41 (NS3), p22 (NS4), VPg (NS5), protease (NS6) and RNA-dependent RNA polymerase (RdRP) (NS7) (Fig. 1a,b). ORF2 and ORF3 are translated from a subgenomic RNA that encodes the major (VP1) and minor (VP2) structural proteins4.
Fig. 1 |. Human norovirus genome, entry, replication and structure.

a, The human norovirus (HuNoV) genome (approximately 7.5 kb) is a viral protein (VPg)-linked positive-sense RNA with a poly(A) tail consisting of three open reading frames (ORFs). Replication of the HuNoV consists of several stages (1–8): (1) cell attachment involving interactions with histoblood group antigen (HBGA); (2) endocytic internalization; (3) disassembly; (4) ribosomal translation of the three ORFs; (5) cleavage of the polyprotein encoded by ORF1 by the viral protease (Pro) into six non-structural proteins (NSPs), synthesis of structural proteins VP1 and VP2 encoded by ORF2 and ORF3, respectively; (6) replication of the genomic and subgenomic RNA by the viral RNA-dependent RNA polymerase (RdRP); (7) encapsidation of the progeny VPg–RNA into capsids formed of VP1 and VP2 that are synthesized from subgenomic viral RNA; and the final stage of (8) viral capsid release. b, Among the NSPs, Pro and the RdRP are structurally the best characterized and have been the targets for developing a variety of classes of small molecule inhibitors. c, The T = 3 capsid of the HuNoV formed by 180 VP1 subunits in three quasi-equivalent positions (A, B and C) is shown along the icosahedral 3-fold axis with surrounding 5-fold axes denoted. The subunits A and B forming A/B dimers surrounding the 5-fold axes are shown in cyan and blue, whereas the C subunits, which form C–C dimers at the icosahedral 2-fold axes, are shown in green. Cartoon representations of the bent (A–B) and flat (C–C) dimers are shown on the right with protruding (P) and shell (S) domains and the N-terminal arm (NTA) denoted in the A/B dimer. d, HBGA (solid spheres in red) binding at the P2 subdomains of VP1 dimer (cyan and blue) in GI (left) and GII (right).
The Norovirus genus is further subdivided into genogroups and genotypes using phylogenetic analysis of the complete amino acid sequence of VP1 (refs. 5,6). Defined genogroups (and genotypes) have at least two complete VP1 sequences from geographically distinct locations. There are currently ten recognized genogroups (GI–GX), with two additional tentative (non-assigned) genogroups (GNA1 and GNA2); human viruses are found in GI, GII, GIV, GVIII and GIX genogroups. These genogroups are further subdivided into 48 recognized genotypes, with three non-assigned genotypes (two in GII and one in GIV) (Fig. 2a). Variants have been recognized for some genotypes and represent epidemiologically relevant phylogenetic subclusters within the genotype5,6.
Fig. 2 |. Phylogenetic trees of norovirus genogroups and genotypes.

a, VP1 amino acid sequences and b, RNA-dependent RNA polymerase (RdRP) nucleotide sequences using previously published sequences6,13 reflect current classification systems (Supplementary methods). The natural hosts affected by each genotype are colour coded. c, GII.4 strains undergo epochal evolution and emergent variants are shown by their time of circulation8,9,16,127,230. The Sydney variant has two major P types (P31 for the 2012-like strains and P16 for the 2015-like strains).
Recombination between ORF1 and ORF2 allows the association of multiple different ORF1s with a single capsid genotype and led to the development of a second classification schema using partial nucleotide sequences of the viral RdRP to assign P types (Fig. 2b). There are eight P genogroups (corresponding to GI–GVII and GX capsid genogroups) and two tentative P genogroups containing 60 P types and an additional 14 tentative P types6. The GVIII and GIX genogroup viruses have GII P types. Two genotyping tools are publicly available: Norovirus Typing Tool and the Human Calicivirus Typing Tool.
The most common genotype that has caused infection and illness for more than two decades is GII.4 and in most years represents more than 50% of identified strains7–9. Other genotypes may emerge globally and predominate for a year or two (such as GII.2 and GII.17)10–12, but they have not persisted like the GII.4 viruses13. There can also be local and regional differences in the circulating genotypes over time8,13. The reasons for emergence and persistence are multifactorial and include RdRP fidelity, recombination, binding affinity for histoblood group antigens (HBGAs), population susceptibility and immunity. Norovirus genomic replication is accomplished by the viral RdRP, and the low fidelity of the enzyme leads to mutations in the genome that can accumulate over time based upon virus fitness and other environmental factors14. Among non-GII.4 strains, the nucleotide changes are largely synonymous, leading to minimal changes in the capsid protein sequence and suggesting that there are evolutionary constraints on VP1 evolution for these viruses15. By contrast, GII.4 strains have mutations that occur in one or more antigenic sites, leading to the potential for immune escape and the emergence of new variants. This has occurred at least nine times over the past three decades (Fig. 2c), with the most recent being the emergence of the San Francisco variant16. Recombination events leading to association of the capsid with a new ORF1 (such as P16) may also influence the emergence of new variants13,17.
Host genetic factors also influence susceptibility to norovirus infections. HuNoVs recognize and bind to HBGAs in a strain-specific manner (Table 1), and there is substantial variability in HBGA expression that is genetically determined. Persons who do not express the appropriate HBGAs on their cell surface are resistant to infection with certain strains18–20. Mutations in the capsid protein may also increase both the breadth of recognition and stronger binding affinity to HBGAs, as described for newly emerging GII.17 and GII.4 strains21–24.
Table 1 |.
Association of histoblood group binding characteristics with infections observed in controlled human infection models and epidemiological studies
| Genotype (variant) | Saliva binding assay | Oligosaccharide binding assay | Human studies (CHIM and epi) | Refs. |
|---|---|---|---|---|
| GI.1 | Binds A and H; no binding to B or secretor-negative | Binds A, H and Le-b; no binding to B or Le-a | CHIM: infects persons expressing either A, B, H or Le-b, secretors only | 18,19,73,173–176 |
| GI.3 (A) | Binds A and H and Le-a; no binding to B or Le-b | Not reported for this variant | Epi: infects persons expressing either A, B, H, Le-a or Le-b | 177,178 |
| GII.2 | Binds B and in presence of bile acid binds A; no binding to A, H or secretor-negative | No binding to A, H, Le-a or Le-b (B glycan binding not reported) | CHIM: infects persons expressing either A, B, H, Le-a or Le-b, secretors and non-secretors | 74,173,179–181 |
| GII.3 | Binds A and B; does not bind H or secretor-negative | Binds A, B, H and Le-b; does not bind Le-a | Epi: infects secretors and non-secretors | 175,176,182,183 |
| GII.4 (2002/Farmington Hills) | Binds A, B and H; does not bind secretor-negative | Binds B and H; does not bind A, Le-a or Le-b | CHIM: infects secretors and non-secretors, with secretors at higher risk than non-secretors | 126,184 |
| GII.4 (2004/Hunter) | Binds A, B, H and Le-b; no binding to secretor-negative except saliva from one asymptomatically infected Le-a non-secretor | Binds A, B, H and Le-b; does not bind Le-a | Epi: infects secretors and non-secretors, with secretors at higher risk than non-secretors; infects Le-b and Le-a | 21,185 |
| GII.4 (2006b/Den Haag) | Binds A, B, H and Le-positive; does not bind Le-negative non-secretor | Binds A, B, H and Le-b; does not bind Le-a | Epi: infects persons expressing A, B, H, Le-a and Le-b in one study; in another 100% of infected are secretors | 21,182,186 |
| GII.4 (2009/New Orleans) | Binds A, B and H; non-secretor not reported | Binds A, B, H, Le-a and Le-b | Epi: 100% of infected are secretors | 22,186,187 |
| GII.4 (2012/Sydney) | Binds A, B and H; non-secretor not reported | Binds A, B, H, Le-a and Le-b | Epi: 100% of infected are secretors | 22,186–188 |
| GII.6 (B) | Binds A, B and H; no binding to non-secretor | Binds A and H; no binding of Le-a or Le-b (B glycan binding not reported) | Epi: infects persons expressing either A, B, H, Le-a or Le-b, with secretors at higher risk than non-secretors | 189,190 |
| GII.7 | Binds B, H and secretor-negative; may bind A | Binds B, Le-a and Le-b; does not bind A or H | Epi: infects secretors and non-secretors | 176,177,186 |
| GII.17 | Binds A, B and H; no binding to secretor-negative | Not reported for this genotype | Epi: infects persons expressing either A, B, H, Le-a, or Le-b, with secretors at higher risk than non-secretors | 191,192 |
A, A glycan; B, B glycan; CHIM, controLLed human infection model; Epi, epidemiological; H, H glycan; Le-a, Lewis a glycan; Le-b, Lewis b glycan.
Animal models of replication
The in vivo growth of HuNoVs has been examined in several animal species. HuNoV will infect the enterocytes of the small intestine in gnotobiotic pigs25 and calves26, and modest virus-specific IgG and IgA responses are observed. However, these models are cumbersome, expensive and limited by the number of animals that can be used in an experiment. BALB/c mice deficient in the recombination activation gene 1 (Rag1) or Rag2 and common gamma chain (γc) (Rag-γc) support the replication of GII.4 HuNoV in many tissues after intraperitoneal injection27, and they have been evaluated to assess in vivo antiviral activity28. To date, replication of non-GII.4 strains has not been reported in this model. Zebrafish larvae have also been used as a replication model to evaluate candidate antivirals29. Microinjection of virus into the larval yolk sac 3 days post-fertilization leads to virus replication for a number of different genotypes, and the addition of candidate drugs with significant antiviral activity to the water containing the larvae can significantly reduce virus levels in the larvae29,30. Virus inactivation also has been assessed in this model, and injection of virus into the yolk sac of the embryo (6 h post-fertilization) can simplify the inoculation31,32.
Nonhuman primates have also been evaluated for their ability to be infected by HuNoVs. Early studies indicated that rhesus monkeys33,34 and chimpanzees35 could be asymptomatically infected after oral inoculation. A later report indicated that pig-tailed macaques can be infected and develop diarrhoea after norovirus inoculation36, but there have been no follow-up reports in this model. Intravenous administration of Norwalk virus (GI.1) to chimpanzees also led to infection, but the model is no longer available owing to restriction on working with chimpanzees37,38. Further investigation using the rhesus macaque model demonstrated asymptomatic infection with several different HuNoV genotypes following oral inoculation. The study found virus antigen and RNA in jejunal epithelial cells, and prolonged faecal shedding associated with changes in the virus genetic sequence over time38. The animal model is proposed as a model for the evaluation of norovirus vaccines and therapeutics.
Norovirus replication and antiviral targets
Once HuNoV binds to HBGAs, the virus enters cells and replication occurs in the cytoplasm. Details of how HuNoV replication is dependent on many of the NSPs has been predicted from studies of animal norovirus replication in a variety of cultured cells. In addition to functioning as a primer to initiate replication, norovirus VPg also functions as an mRNA cap substitute by interacting with the cap-binding eIF4G-complex during translation39–41. Genomic replication and subgenomic RNA synthesis occur following translation and the initial round of viral protein synthesis. When all the viral proteins are synthesized and genome replication is complete, the viral RNA progeny is packaged into icosahedral particles. VP2 is likely to play a critical role in this process (Fig. 1a).
The NSPs of norovirus and other caliciviruses share sequence motifs with picornaviruses, indicating functional and structural similarities between the two families. HuNoV protease and RdRP (similarly to their picornavirus counterparts) are well characterized structurally and functionally, and have been targets for drug development because of their critical role in replication42–44. Different classes of effective small molecule inhibitors have been discovered for the HuNoV protease and RdRP (Fig. 1b; reviewed elsewhere44). Interestingly, studies in a recent preprint have shown that the small molecule inhibitors that target GI protease are less effective against the GII.4 proteases because of significant differences in the substrate binding pockets and their catalytic sites despite sharing the same polypeptide fold. This may pose a problem in developing inhibitors effective against both GI and GII proteases45. Recent findings that norovirus RdRP induces liquid-like phase-separated condensates as replication hubs during infection may open avenues for discovering a new class of antivirals46. In contrast to the protease and RdRP, structural and functional information of other NSPs encoded by ORF1 and their role in the virus replication, particularly in HuNoVs, remain less clear47. Although the precise role in the virus replication cycle is yet to be ascertained, HuNoV p22 (NS4), which has a membrane association domain and highly conserved endoplasmic reticulum export signal motif, is implicated in Golgi apparatus disassembly and inhibition of the cellular secretory pathway48. p41 (NS3), which has signature SF3 helicase motifs, exhibits helicase and RNA chaperone activities49 and induces intracellular membrane alterations to form replication organelles together with p22 (NS4)50, as well as apoptosis through both caspase-8-dependent and caspase-9-dependent pathways51.
Development of a cultivation system for human norovirus
After the Norwalk virus was discovered in 1972, numerous efforts to cultivate the virus using routine tissue culture cells and human fetal organ cultures were unsuccessful, including a large collaborative study using multiple cell cultures52–54. By contrast, transfected HuNoV genomic RNA is infectious in several human cultured cell lines (Caco-2 and Huh-7 or Huh-7.5.1) that are otherwise nonpermissive to infection, and virus particles can be produced at low levels, but only one round of RNA replication occurs55. In vivo, biopsies from experimentally infected volunteers and immunocompromised individuals (including transplant recipients or a chronically infected individual with common variable immunodeficiency) showed histopathology or viral antigen staining in the small intestine and little evidence of histopathology in the colon52,56–60.
Murine noroviruses (MNVs) replicate in immune and tuft cells, and a few HuNoV strains exhibit modest replication (10-fold to 50-fold increases of RNA) in human B cell lines61–63. Studies using unfiltered, unprocessed stool as inocula showed that bacterial surface-expressed HBGAs are important factors for successful virus replication. HBGA-expressing Enterobacter cloacae added to B cell cultures restored the infectivity of filtered HuNoV-positive stool filtrate, whereas a non-HBGA-expressing bacterium did not61. Reproducibility of this system has been challenging62. HuNoV also replicates in primary human B cells, with replication levels comparable to those in transformed B cells, and the virus triggers B cell activation in vitro, a property attributed to NS1/264. B cell-deficient patients can be infected and shed HuNoV at high titres, indicating that extensive viral replication must occur in host cells other than B lymphocytes65. These studies led to a proposal that there is a dual tropism of HuNoV66,67. Implications of virus uptake into immune cells in vitro to virus pathogenesis in humans remain to be determined29.
Robust and reproducible cultivation system in human tissue-derived intestinal enteroids
The studies described above suggest that replication of HuNoV in vitro depends on the ability to mimic the differentiation stage of intestinal epithelial cells and perhaps the luminal microenvironment in a cell culture system. The development of nontransformed tissue stem cell-derived human intestinal enteroids (HIEs; also called tissue-derived organoids)68 allowed the successful cultivation of HuNoVs69 (Fig. 3a). These nontransformed, multicellular cultures are physiologically active and exhibit circadian rhythms. Thus, the HIE model closely resembles the architecture, biology and physiology of the human small intestinal epithelium and can represent the heterogeneity of the human population70. Successful HuNoV infection of enterocytes requires differentiated cultures71, virus particles are produced from infected enterocytes and are detectable by antigen staining (Fig. 3b,c,d). Replication occurs in 2D cultures plated as monolayers or on transwells; subsequent studies have shown that bile acid and ceramide are two components in bile that allow replication of bile acid-dependent HuNoV strains69,72 (Fig. 3e,f).
Fig. 3 |. The human intestinal enteroid culture system for human norovirus.
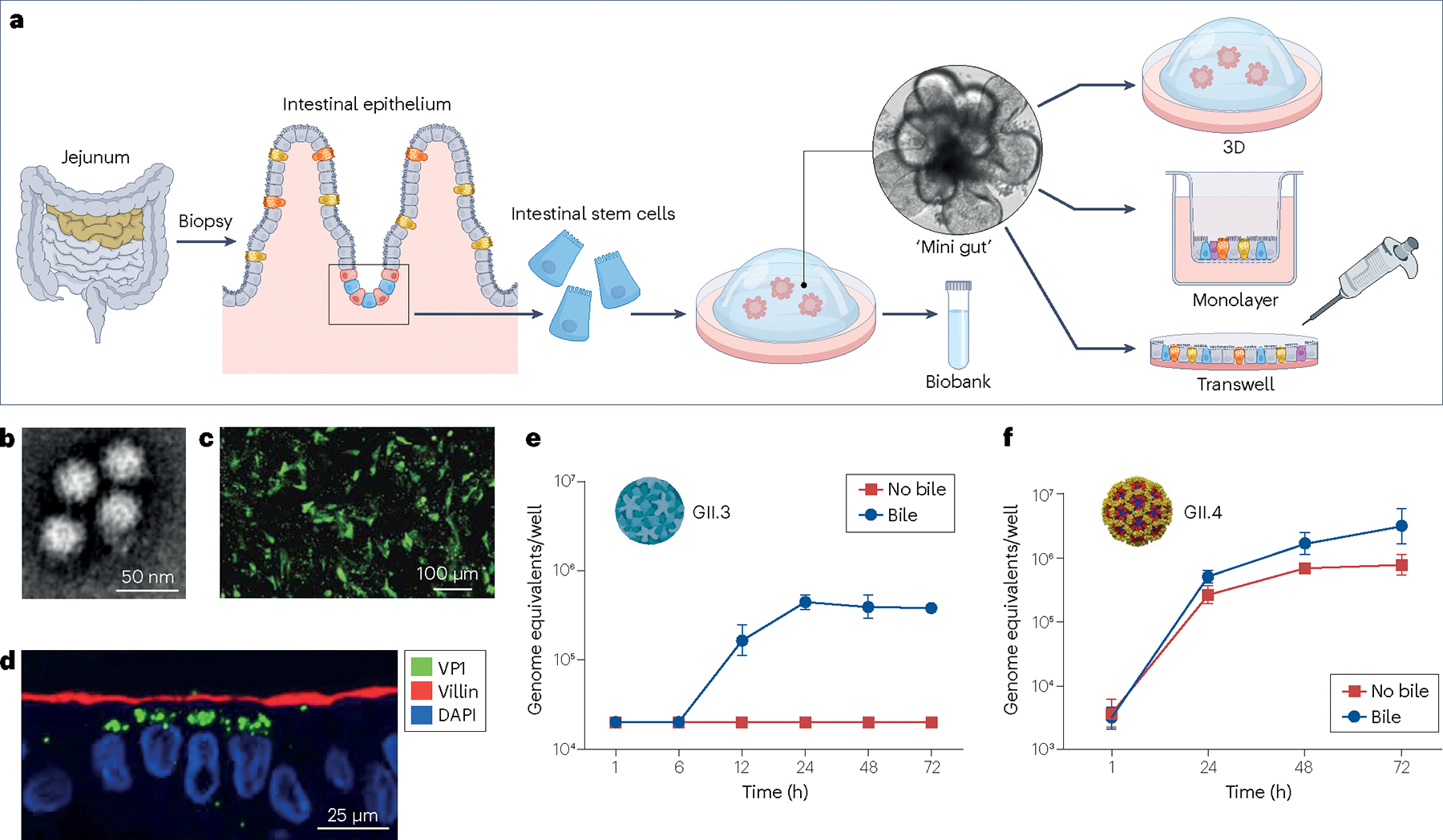
a, Intestinal enteroids are produced from intestinal tissue samples that contain intestinal stem cells that will grow and self-assemble into 3D cultures within several days of being cultivated in media containing Matrigel and growth factors that support proliferation of the stem cells68. After expansion of the ‘mini-gut’ cultures, some are frozen down to create a biobank. Removal of growth factors from the media of proliferating cultures induces the stem cells to differentiate into all the multiple cell types present in the mature intestinal epithelium including absorptive (enterocytes) and secretory cells (enteroendocrine, goblet and Paneth cells) and these cultures can be plated in different formats (3D, on transwells or 2D on plastic wells for infection studies. Inside each 3D mini-gut culture, enterocytes form the brush border surrounding a single luminal compartment, which receives secretions from the other cells70. b, Virus produced from infected human intestinal enteroids (HIEs) visualized by negative stain electron microscopy. c, Immunofluorescence detects the VP1 capsid protein in infected HIEs plated on 96 wells. d, Immunofluorescence detects the VP1 capsid protein (green), villin (red) and nuclei (4′,6-diamidino-2-phenylindole (DAPI), blue) in infected HIEs plated on transwells. e, A time course illustrates the kinetics of RNA replication and shows bile is required for GII.3 replication. f, A time course illustrates the kinetics of RNA replication and shows bile enhances GII.4 replication. Parts b–f adapted with permission from ref. 69, AAAS.
The dose of inoculum required to produce infection in 50% of inoculated tissue culture wells (TCID50) varies for each virus strain (approximately 1.2 × 102 genomic equivalents (ge) for a GII.4/Sydney/2012 strain and approximately 2.0 × 104 for a GII.3 strain in initial studies in the J2 HIE line)69. These values are similar to the human infectious dose 50 observed in a controlled human infection model (CHIM) for GI.1 Norwalk virus (2.8 × 103 ge among secretor-positive persons)73 and for GII.2 Snow Mountain virus (5 × 105 ge)74. The TCID50 is both viral inoculum and cell line dependent75–77. GII.4 viruses generally replicate better than GI viruses77–80. Advancements of the HIE system include identification of a commercial medium that enhances replication and the production of genetically modified cultures with lower innate immune responses, leading to cultivation of more viruses (currently 41 viruses from stools including 4 GI genotypes, 11 GII genotypes and 8 GII.4 variants), an increase in virus yields77,79 and easier adoption of the system in many laboratories. To date, HuNoV replication has not been detected in enteroendocrine cells (EECs) in HIE cultures, even though these cells have been shown to contain viral genomes in immunocompromised individuals60. Whether this reflects a low number of EECs or a lack of a specific type of EEC in HIE cultures remains to be determined. HIEs from infants75 or genetically modified HIEs, which inducibly express neurogenin81 and express higher numbers of EECs, can be tested to further explore possible EEC HuNoV tropism.
Human norovirus replication in human induced pluripotent stem cell-derived intestinal organoids
Another human intestinal organoid (HIO) culture system made from directed differentiation of embryonic or pluripotent stem cells produces intestinal epithelial cultures surrounded by mesenchymal cells82,83. A secretor-positive HIO (that is, an HIO with a functional fucosyltransferase 2 gene) supported limited GII.4 virus replication, and the limited replication was postulated to be due to higher numbers of immature enterocytes in the HIOs that are less permissive to infection compared with enterocytes in tissue-derived HIEs84. Other HIO cultures report good virus propagation85. Recently, induced pluripotent stem cell-derived HIOs, transplanted into mice to induce the maturation of the cultures86,87, were used to create tissue-derived ‘transplanted HIEs’ that support the replication of several HuNoV strains at levels similar to those of standard HIE cultures77.
Biological relevance of the human intestinal enteroid cultivation system
HIEs retain the phenotypic characteristics of the donor and have intestinal anatomical segment specificity. For example, HIE lines from individuals lacking a functional FUT2 gene, termed secretor-negative, are resistant to infection by many HuNoV strains, whereas GII.3 strains can infect secretor-positive and some secretor-negative HIE lines69. Genetic modification of HIEs has confirmed that FUT2 expression is both necessary and sufficient for most HuNoV strains but it is not essential for at least one GII.3 HuNoV strain88. There are rare exceptions of infections in secretor-negative persons by GII.4 HuNoV; this may occur because the microbiome of an individual modifies the HBGA phenotype of enterocytes89.
HIEs also vary in their responses according to intestinal segment of origin, culture conditions and plating format (for instance 3D or 2D cultures on hard plastic versus glass or transwells) and cellular differentiation status90. Virus replication occurs in HIE cultures from all segments of the small intestine (duodenum, jejunum and ileum), but not the colon, using cultures from these segments that have been established from the same donor as well as from different donors77,79. These data confirm the intestinal tropism that had been predicted from the early volunteer studies52.
Human norovirus replication reveals unexpected strain-specific differences in entry into human intestinal enteroids
GII.4 HuNoVs replicate well in secretor-positive HIEs. By contrast, a GII.3 HuNoV strain failed to grow until exogenous bile was added to the culture medium to mimic the extracellular milieu of the small intestine69 (Fig. 3e). Although GII.4 strains grow without bile, their replication is enhanced in the presence of bile (Fig. 3f). Hydrophobic bile acids are an active component in bile that are required for replication of GII.3 HuNoVs, and bile acid-mediated replication correlates with the hydrophobicity of the bile acid72. The conjugated bile acid glycochenodeoxycholic acid (GCDCA) is effective and soluble and so is now used in most HIE replication studies. Several other HuNoV strains (GI.1, GII.1, GII.3, GII.6 and GII.17) are now known to be bile acid-dependent79,91. The effects of GCDCA on GII.3 infection have been evaluated in jejunal HIEs (Fig. 4a, left panel). The effects of GCDCA are mediated through a G protein-coupled receptor (sphingosine-1-phosphate receptor 2 (S1PR2)) and do not involve GCDCA binding to the virus, the detergent properties of GCDCA or interactions with the classic farnesoid × receptor or Takeda G protein-coupled receptor 5. GCDCA acts early during infection, inducing multiple cellular processes including the induction of endocytosis leading to GII.3 uptake followed by endosomal acidification. Ceramide is generated at the apical cell surface through the action of acid sphingomyelinase (ASM), an endosomal/lysosomal enzyme. GII.3 infection is reduced by inhibitors of S1PR2, endosomal acidification, ASM activity and the lysosomal exocytosis of ASM72. These data suggest a model of GII.3 infection whereby the virus binds to a cellular receptor and, in conjunction with a bile acid such as GCDCA acting through S1PR2 and ASM, ceramide-rich lipid rafts are formed containing the virus and its receptor. The virus is then internalized via endocytosis and the virus or its genome is released to the cytoplasm after endosomal acidification. GII.3 also infects duodenal and ileal HIEs in a similar GCDCA-dependent manner that is decreased by S1PR2 inhibition, suggesting that GII.3 infection throughout the small intestine occurs in a similar fashion92.
Fig. 4 |. Distinct entry pathways and replication of bile acid-dependent GII.3 and bile acid-independent GII.4 into human intestinal enteroids.

a, Each virus uses a different entry pathway. GII.3 requires sphingosine-1-phosphate receptor 2 (S1PR2) and bile acid to induce endocytosis (left panel), whereas GII.4 causes initial membrane injury and wound repair mechanisms to induce clathrin-independent carrier (CLIC)-mediated endocytosis (right panel). Common elements include initial binding to histoblood group antigens (HBGAs), putative receptor clustering in lipid rafts, endosomal acidification and lysosomal exocytosis that results in ceramide (Cer) appearance on the cell surface. b, The table highlights strain-specific differences in the biology of GII.3 and GII.4 viruses related to the requirement for FUT2 gene expression69,88, cell entry72,94, bile acid dependence69,72, acid sphingomyelinase involvement72,94, effects of JAK1/JAK2 inhibition100, effects of knockout of innate immune factors99 and host transcriptional responses99,100,231,232. ASM, acid sphingomyelinase; Gal-3, Galectin-3; IFN, interferon; SM, sphingomyelinase; V-ATPase, vacuolar-type ATPase.
Structural studies suggest that some bile acids can interact with the major capsid protein VP1 of certain HuNoVs (GII.10 virus-like particles (VLPs) and the P domain of GII.1, GII.10 and GII.19)93. The GII.1 virus can replicate in HIEs in the presence of bile acids, but the requirements for GII.10 and GII.19 viruses have not been tested to our knowledge. VLPs of GI.1 and GII.4 viruses that replicate in HIEs and the P domains of GI.1, GII.3, GII.4 and GII.17 do not bind bile acids. Bile acid binding to some HuNoV particles may have biological consequences as this is essential to allow GII.1 virus to bind to HBGAs, and GII.10 binding to HBGAs is enhanced by bile acid binding93. GII.2 and GII.12 VLPs also require bile acids to bind HBGAs, although these particles have not been tested for binding by structural studies91. These studies suggest direct particle binding to bile acids may be another strain-specific property of HuNoVs but how this influences virus infectivity remains to be determined.
GII.4 entry into HIEs does not involve bile acid-induced endocytosis that is utilized by the GII.3 virus (Fig. 4a, left panel). Instead, it involves a dynamin-independent, clathrin-independent carrier (CLIC) endocytosis pathway and wound repair mechanisms94 (Fig. 4a, right panel). GII.4 VLPs, but not the P domain alone, interact with the endosomal sorting complexes required for the transport protein ALIX on epithelial cells to induce plasma membrane wounding, which then leads to lysosomal exocytosis and additional membrane repair mechanisms. Effectors of the CLIC pathway, including cholesterol, Cdc42, ADP-ribosylation factor 1 and Galectin-3 (Gal-3), then mediate GII.4 cell entry in conjunction with endosomal acidification. Although individual components of the CLIC pathway have been associated with the entry of several other viruses into a range of different cell types, the use of all of the mediators has not been described previously for viruses. However, bacterial Shiga and cholera toxins, and certain lectins, can interact with surface glycolipids to cause narrow membrane bending, the formation of tubular endocytic pits and subsequent clathrin-independent uptake into cells. Interactions between cargo and glycosphingolipids drive the formation of deep and narrow membrane invaginations both in cells and model membranes from which CLICs are generated, similar to what has been described in vivo in mouse enterocytes95. Similarly, the formation of tubular invaginations in giant unilamellar vesicles treated with GII.4 VLPs is consistent with the HIE membrane changes observed during GII.4 entry96. Whether other viruses use this pathway to infect HIEs remains to be investigated.
Diversity in strain-specific abrogation of the host response
The HuNoV replication system in HIEs is limited by the lack of indefinite serial passaging of virus, suggesting a role for uncharacterized host factors in restricting replication. Innate immune responses, including the synthesis and secretion of interferons and their downstream effectors, interferon-stimulated genes (ISGs), form a first line of defence against many viral infections97,98. Two studies of the epithelial response to HuNoV replication in HIEs using RNA sequencing show that the induced host innate immune response is primarily driven by a type III interferon response (with some induction of interferon-β (IFNβ) but not IFNα) that leads to ISG induction and restricts HuNoV replication in unexpected strain-specific ways99,100. Interferon induction requires live virus and it appears concomitantly with replicating viral RNA. HuNoV replication is sensitive to exogenous treatment of HIEs with type I interferon (IFNα or IFNβ) or type III interferon. The intrinsic host response results in upregulation of hundreds of genes which are detectable by 10 h post-infection (hpi), but not at 6 hpi, and which continue to increase at 24 hpi (the latest time point examined)99. Most upregulated genes are ISGs such as IFI144L, MX1, IFIT3, STAT1, IFIT1, IFITM1, ISG15, OASL and CXCL10, that were detected in both studies. Transcription enrichment analysis identified STAT1 and STAT2 binding sites as highly enriched in the promoter regions of genes whose levels of expression were significantly upregulated, suggesting that the JAK–STAT signalling pathway is activated following HuNoV infection100. Pharmacological inhibition of the Janus kinase (JAK1 or JAK2) with ruxolitinib increases replication of both GII.3 and GII.4, indicating that interferon-deficient enteroids are more permissive for HuNoV replication and that the innate response limits virus replication100. Attenuation of the interferon pathways by using a decoy receptor for type I and type III interferons as well as the production of five interferon-deficient HIEs that lack type I or type III interferon receptors (STAT1, MAVS and STAT1/STAT2) and exhibit attenuated ISG induction, failed to alter or enhance GII.4 replication. This unexpected result indicates that GII.4 has mechanisms to antagonize the innate immune response, and this may partially explain why these viruses predominate and are recognized as pandemic viruses. In contrast to GII.4, GII.3 replication is increased 10-fold to 100-fold in STAT1 knockout and type I interferon receptor knockout HIEs, and the enhanced replication is accompanied by reduced ISG responses. GII.3 virus can also spread in STAT1 knockout cultures that exhibit a lower TCID50 when compared with the parental HIE line99. These studies indicate that GII.3 HuNoV replication is restricted by endogenous host interferon pathways and that inhibition of STAT1 increases viral replication and spreading. The discovery of differences in sensitivity to intrinsic interferon pathways between two HuNoV strains adds to the growing list of strain-specific biological behaviours of HuNoVs99. GII.3 replication is also increased in HIEs when interferon induction is reduced by the expression of interferon antagonists, but GII.4 replication was not reported100. The mechanisms by which the ISGs and other antiviral proteins restrict virus replication remains to be determined. Several long non-coding RNAs are also upregulated in GII.4-infected HIEs but their roles in infection also remain uncharacterized99.
Human norovirus structure — hollows and protrusions
Although, as noted earlier, HIE systems are helpful in studying many aspects of HuNoV replication, there are still challenges in these systems to successfully propagate and obtain virus in sufficient quantities for structural and biochemical studies, which have relied on VLPs produced by the co-expression of VP1 and VP2 (ref. 101). In place of authentic viruses, VLPs have been invaluable in understanding the structural, immunological and biological aspects of many strains of HuNoVs94. The X-ray crystallographic structure of Norwalk virus (GI.1) VLPs, determined in 1999, provided an atomic-level description of the capsid architecture of a norovirus102. The structure revealed that the approximately 40 nm diameter capsid exhibits T = 3 icosahedral symmetry formed by 180 molecules of the major capsid protein VP1 (Fig. 1c). Subsequently, structure determination of a variety of animal caliciviruses, including feline calicivirus (FCV)103,104, San Miguel Sea Lion virus105 and MNV106–108, showed similar capsid architecture.
Briefly, in these capsid structures, the 180 subunits of VP1 as three quasi-equivalent subunits — A, B and C — are assembled as 60 A–B dimers at the 5-fold axes and 30 C–C dimers at the icosahedral 2-fold axes, such that there are prominent hollows at the 5-fold and 6-fold axes. Each VP1 subunit has a modular domain organization consisting of an internal N-terminal arm (NTA) and two distinct domains termed shell (S) and protruding (P) domains, separated by a flexible hinge102,109. The S domains of the VP1 dimers interact closely to form a smooth shell held together by the NTA, located beneath the shell, adapting to the quasi-equivalent environments of T = 3 symmetry. The VP1 dimers exhibit bent (A–B) and flat (C–C) conformations that are necessary for inducing the correct curvature of the shell (Fig. 1c). The P domain, with a distinctive fold observed only in calicivirus structures, projects from the S domain and is divided into P1 and P2 subdomains. The P1 subdomain (located closer to the S domain) is formed by the N-terminal and C-terminal regions of the P domain, whereas the P2 subdomain (which is distally located) is formed by the intervening region. The hypervariable P2 subdomain is involved in recognizing cellular receptors, as shown by structural studies of FCVs103,104,110 and MNVs111,112, and in recognizing the polymorphic HBGAs in the case of HuNoVs113 (Fig. 1d).
Notably, the minor capsid protein VP2, which is presumed to be present in small amounts, is not observed in any calicivirus capsid structures determined to date. However, the cryogenic electron microscopy (cryo-EM) structure of FCV in complex with its receptor fJAM-A103 revealed that the N-terminal α-helical region of VP2 forms a portal-like assembly projecting out from one of the hollows at a 6-fold axis, which is likely to be triggered by the receptor interaction. This finding suggests a potential role for VP2 in genome uncoating and delivery, akin to the receptor-triggered extrusion of VP4 and the N terminus of VP1 implicated in membrane penetration and genome delivery in picornaviruses114,115. Earlier studies have also suggested that VP2 in HuNoV confers stability to the capsid and that it interacts with the NTA of the VP1 (refs. 116,117). It is possible that any trigger, such as a receptor interaction, disengages VP2, leading to its extrusion to facilitate genome release.
Histoblood group antigen binding: prevalence and evolution of human norovirus
The recombinantly expressed P domain, which readily forms a dimer and adopts an identical conformation as found in the capsid structure, has been extensively used in structural and biochemical characterization of the interactions between HuNoVs and HBGAs102,118–120. All these studies have shown that HuNoVs bind to HBGAs through the hypervariable P2 subdomain of VP1 (refs. 101,117–119) and exhibit strain-specific HBGA binding patterns121–123. The HBGA binding sites in GI and GII are distinct (Fig. 1d). In contrast to GI viruses, in which each subunit of the VP1 dimer has an HBGA binding site, in GII viruses the residues from both units form the two HBGA binding sites in the dimers. Another important distinction with implications for strain predominance is that the HBGA binding in GI HuNoV is predominantly mediated by interactions with the terminal galactose (Gal). By contrast, in most GII viruses, especially in GII.4, the binding is mediated by the primary secretor-fucose (SeFuc) of the HBGA. As SeFuc is a common moiety in all HBGA families (ABH and Lewis glycans)19,124, GII viruses can potentially bind to all of them and, thus, infect a larger population. By contrast, the HBGA binding profile is relatively restricted in GI viruses. Some epochally emergent GII.4 variants, particularly those circulating after 2002, show strong binding to di-fucosyl Lewis HBGAs because of sequence alterations in one of the solvent-accessible loops (between residues 390 and 400, referred to as loop 2) in the P2 subdomain to form a binding site for the Lewis fucose (LeFuc) of the HBGA125. Compared with the SeFuc binding site in GII HuNoVs, which is exceptionally well conserved, the LeFuc binding site is only moderately conserved and thus could contribute to the evolution of GII.4 HuNoVs. Surrounding the highly conserved SeFuc HBGA sites, GII.4 variants exhibit several sequence changes, particularly in the loop regions, that can modulate HBGA binding affinities between the GII.4 variants. Several reports have shown how these changes alter the HBGA binding profiles and the binding affinities in the GII.4 variants21,22,126. Although these studies provide general trends and support the notion that sequence changes influence binding affinities, they show some inconsistencies between the observed HBGA profiles for certain GII.4 variants (Table 1). Modelling these sequence variations in the context of the P domain structure has shown that they also profoundly alter the surface topography and electrostatic landscape of the P2 subdomain. This is consistent with observed strain-dependent alterations in antigenicity120,125, which forms the basis for identification of these variations into eight distinct antigenic sites127. A coordinated interplay between differential HBGA binding specificities, affinities and antigenic variations is proposed to allow the escape from host immunity and drive the evolution of HuNoVs121. Interestingly, there appears to be temporary stagnation in the evolution of GII.4 after the emergence of the 2012 Sydney variant. Failure to establish sustained immunity for this variant in adults may have allowed this strain to continue its dominance128.
Capsid plasticity — resting and raised conformation states
As observed first in MNVs108,129–131, recent structural studies107,109,132–134 of HuNoV VLPs have shown that VP1, in the context of the T = 3 icosahedral capsid, can exist in two distinct conformations: the ‘resting’ and the ‘raised’ conformations. Such a transformation without affecting the structure of either the P or S domains is facilitated by the conformationally flexible hinge. In the resting conformation, the lower portion of the P domain closely interacts with the S domain, whereas in the raised conformation, the P domain is rotated and raised above the S domain. This is driven either by the removal of stabilizing ions, as in the case of GII.4 VLPs109, or by an increase in pH, as in the case of GII.3 VLPs107,129,130,132. In addition to this plasticity in the capsid, VLPs, particularly GII.4, deviate from the T = 3 icosahedral symmetry and exhibit icosahedral assemblies with different T numbers. Cryo-EM structures of VLPs of some of the GII.4 variants (1994, 2006b and 2012) have shown the capsid with an increased diameter of 50 nm with T = 4 icosahedral symmetry consisting of 120 VP1 dimers that have adopted a raised conformation132,133. Production of VLPs, with or without the co-expression of VP2, as well as possible differences in the expression systems and other factors, may influence the structural assembly of VP1. Negative-stain electron microscopy images of the native GII.4 virions, however, have shown a diameter of approximately 40 nm consistent only with T = 3 icosahedral symmetry.
The crystallographic structure of the VLP of a 2002 strain of GII.4 (HOV) produced by the co-expression of VP1 and VP2 has shown a capsid organization with all the structural features consistent with T = 3 icosahedral symmetry109. This crystal structure was determined without imposing icosahedral symmetry and it also shows how hinge flexibility imparts inherent plasticity to the capsid within the confines of the icosahedral structure to adapt to crystal packing forces. A new finding in this structure is the presence of a cation at the subunit interface at the middle of the P domain dimer. This was also observed in the cryo-EM structure of this GII.4 VLP, which was concurrently determined to exclude the possibility that bound ion in the crystal structure is a crystallization artefact. Removal of this ion resulted in significant conformational changes consistent with transitioning from an ion-bound resting state to the raised state. As in MNV and FCV, recent studies of GII.4 HuNoV have implicated the resting and raised conformations in entry and antibody neutralization mechanisms109,135 (Fig. 5).
Fig. 5 |. Mechanisms of neutralization of human norovirus by antibodies.

a, Neutralization of GI.1 by the human antibody 5I2 (yellow) by directly competing with the histoblood group antigen (HBGA) binding site in the protruding (P) domain dimer (grey). b, Neutralization of GI.1 by the human antibody 10E9 (purple) by directly competing with the HBGA binding site in the P domain dimer (grey). c, Human antibody A1431 (blue) binds to an epitope slightly away from the HBGA binding site in the GII.4 P domain dimer (grey) and neutralizes GII.4 by clumping or crosslinking particles. d, Resting and raised conformations of the P domain (shell (S), P1, and P2 subdomains are shown in blue, red, and yellow, respectively). e, Fragment antigen-binding region (Fab) of the human antibody NORO-320 (yellow) targets capsid plasticity by binding an epitope in the GII.4 P domain (grey) that is exposed only in the raised conformation and probably neutralizes the virus by blocking co-receptor interactions or VP2 externalization. f, The llama nanobody M4 (green) also binds to an epitope exposed only when the P domain (grey) transits to the raised confirmation and neutralizes by compromising capsid integrity.
Antibody neutralization mechanisms — targeting histoblood group antigen binding and capsid plasticity
Mechanisms by which a neutralizing antibody ablates virus infectivity typically involve inhibiting interactions critical for the cell entry process136–138. This includes: (1) clumping the particles owing to the bivalent binding of neutralizing antibody to particles, thereby disabling their ability to interact with cellular receptors; (2) sterically blocking access to the receptor by binding close to the receptor binding site; (3) inhibiting a conformational change in the capsid or the capsid protein necessary for receptor interactions; and (4) inducing disassembly of the capsid or compromising the capsid integrity. For HuNoVs, several mouse139 and human-derived antibodies140–142, and llama-derived single-chain antibodies (nanobodies) have been studied for their ability to neutralize virus infection143,144. Prior to having a cell culture system for HuNoVs, surrogate HBGA blockade assays were routinely used instead of neutralization assays73,126,141. With the establishment of HIE cell culture systems69, antibodies can now be evaluated directly for their ability to neutralize HuNoVs belonging to different genogroups and genotypes. One antibody, 5I2, isolated from peripheral blood mononuclear cells of individuals infected with GI.1 HuNoV145 shows potent HBGA blockade activity and is highly specific to GI.1 (ref. 142). Structural studies of the Fab of 5I2 in complex with the GI.1 P domain show that the antibody binds very close to the HBGA binding site, indicating that the mechanism of neutralization is by sterically blocking the HBGA (Fig. 5a). Further, the epitope that is recognized is only conserved in the GI.1 and not in other GI HuNoVs, thereby providing a clear understanding of how sequence and structural changes mediate escape from Fab 5I2 neutralization in other genotypes142.
For GII.4 HuNoVs, however, neutralization by sterically blocking HBGA binding may be effective for a particular variant146 (Fig. 5b), but this mechanism may not be effective for multiple variants because of the significant sequence variability surrounding the HBGA binding site. Recent studies have uncovered antibody neutralization mechanisms that do not involve direct HBGA blockade but instead target the ability of VP1 to exist in resting and raised conformations (Fig. 5d). Although the IgG and IgA forms of human NORO-320 inhibit HBGA binding and neutralize GII.4 HuNoV infectivity, the Fab of this antibody does not block HBGA binding, yet it neutralizes infectivity as effectively as the IgG or IgA forms140. The crystal structure of NORO-320 Fab in complex with GII.4 P domain has shown that the antibody recognizes an epitope distant from the HBGA binding site close to the bottom end of the P domain140 (Fig. 5e). This epitope is highly conserved among the GII HuNoVs, accounting for the broad specificity of this antibody. In the context of the capsid structure, Fab-320 binding causes severe steric clashes with the neighbouring subunits, but these clashes are relieved when VP1 is in the raised conformation, suggesting that with Fab-320 binding the VP1 is locked into the raised conformation. When modelled into the capsid structures, the binding of the Fab-320 to the raised conformation has shown that it points towards the hollow region of the capsid, masking the S domain to some extent. The neutralization mechanism, therefore, is likely to be one or a combination of the following: (1) restricting the capsid dynamics that may be essential for optimizing receptor interactions; (2) blocking access to cellular ligands such as ALIX, Gal-3 and TSG101, some of which bind to the S domain94; and (3) hindering the extrusion of VP2 portal potentially that is required for genome delivery similar to FCV103. However, by contrast, the IgG form of NORO-320 neutralizes infection by clumping the particles through bivalent binding, as is evident from dynamic light scattering (DLS) experiments of GII.4 VLPs with NORO-320. A similar mechanism, by clumping or crosslinking particles, has been implicated for another human antibody, A1431, which also binds to an epitope away from the HBGA binding site141 (Fig. 5c).
More recently, yet another mechanism of neutralizing HuNoV infectivity was discovered with a norovirus-specific llama-derived nanobody M4 (ref. 135). This targets a highly conserved region distant from the HBGA binding site close to the S–P interacting region (Fig. 5f) and neutralizes multiple GII.4 variants with high potency in HIEs. As with the epitope for NORO-320, this epitope is accessible only when the P domain is in the raised conformation. DLS experiments with GII.4 VLPs have shown that M4 effectively disassembles the particles, suggesting that the neutralization mechanism involves accessing the epitope by altering the conformational dynamics of the capsid and triggering the disassembly of the capsid. Another nanobody, Nano-25, is suggested to neutralize GII.4 infection by a similar mechanism144. A puzzling observation from M4 studies is that although this nanobody binds the GII.3 P domain and dissembles the GII.3 VLPs as effectively as it does GII.4 VLPs, M4 fails to neutralize GII.3 virus, suggesting that the GII.3 virus is more stable than the GII.3 VLP and, by contrast, GII.4 virus and VLPs do not differ in their stabilities and are equally vulnerable to M4 binding. Overall, this indicates that the genome, along with VP2 in the GII.3 virion, probably contributes differentially to the increased stability of the capsid, underscoring how capsid stability and plasticity variations between genotypes may influence the mechanism of neutralization135.
Vaccines in clinical development
There are no vaccines currently licensed for the prevention of norovirus infection or illness, but several vaccines are in clinical development (Table 2), and others are in preclinical development. All vaccines in clinical development are based upon targeting the major capsid protein, VP1, using three different strategies for protein expression. The most frequent approach is the expression of VP1 as a recombinant protein that form VLPs. Protein production is achieved using baculovirus, yeast (Pichia pastoris and Hansenula polymorpha) and plant (Nicotiana benthamiana) expression systems, and in most cases the protein is delivered intramuscularly in combination with an aluminium-based adjuvant. The second approach is the use of a recombinant, replication-incompetent adenovirus-vectored vaccine delivered orally. The vaccine abortively infects intestinal epithelial cells, leading to local expression and both a local and a systemic immune response. The third approach delivers mRNA encoding the VP1 protein encapsulated in a lipid nanoparticle, which when delivered intramuscularly also leads to expression of the viral capsid protein within the vaccine recipient.
Table 2 |.
VP1-based norovirus vaccine candidates in clinical development
| Company | Vaccine platform (expression system) | Adjuvant | Administration route | Genotypes | Development stage | Refs. |
|---|---|---|---|---|---|---|
| Anhui Zhifei Longcom Biologic Pharmacy Co., Ltd | Protein (Pichia pastoris) | Aluminium hydroxide | Intramuscular | GI.1, GII.3, GII.4, GII.17 | Phase I/II | NCT04563533 (ref. 193) and NCT06524947 (ref. 194) |
| Chengdu Kanghua Biological products Co., Ltd | Protein (Pichia pastoris) | Aluminium hydroxide | Intramuscular | GI.1, GII.2, GII.3, GII.4, GII.6, GII.17 | Phase I planned | NCT05805618 (ref. 195) |
| Hillevax, Inc. (nee Takeda Vaccines, Inc., Ligocyte Pharmaceuticals, Inc.) | Protein (Baculovirus) | Aluminium hydroxide | Intramuscular | GI.1, GII.4 | Phase II | NCT01168401 (refs. 148,196–198), NCT01609257 (refs. 150,199–201), NCT02038907 (refs. 202,203), NCT02142504 (refs. 147.204,205), NCT02153112 (refs. 153,154,206), NCT02475278 (refs. 149,207,208), NCT02661490 (ref. 209), NCT02669121 (refs. 151,210,211), NCT03039790 (ref. 212), NCT05281094 (ref. 206), NCT05972733 (ref. 213) and NCT06007781 (ref. 214) |
| Icon Genetics GmbH | Protein (Nicotiana benthamiana) | None | Intramuscular | GI.4, GII.4 | Phase I | NCT05508178 (refs. 160,161,215) |
| Moderna | mRNA lipid nanoparticle | None | Intramuscular | GI.3, GII.3, GII.4 | Phase I | NCT05992935 (ref. 216) and NCT06592794 (ref. 217) |
| National Vaccine and Serum Institute, China | Protein (Hansenula polymorpha) | Aluminium hydroxide | Intramuscular | GI.1, GII.4 | Phase III. | NCT04188691 (ref. 218), NCT04941261 (ref. 219) and NCT05916326 (ref. 220) |
| Vaxart, Inc. | Recombinant virus vector (replication-deficient, adenovirus type 5) | Double-stranded RNA hairpin | Oral | GI.1, GII.4 | Phase II | NCT02868073 (refs. 221,222), NCT03125473 (ref. 223), NCT03897309 (ref. 224), NCT04854746 (ref. 225), NCT04875676 (ref. 226), NCT05212168 (refs. 158,227), NCT05213728 (ref. 228) and NCT05626803 (ref. 229) |
The vaccine that has had the greatest clinical evaluation to date is HIL-214, an aluminium-adjuvanted, bivalent VLP vaccine containing the GI.1 Norwalk virus strain and a consensus GII.4 VLP derived from what at the time were considered three distinct variants (2002/Lanzhou (now reclassified as the 2006a/Yerseke-like strain), 2006a/Yerseke and 2006b/Den Haag). In adults, a single dose elicits a rapid anamnestic HBGA-blocking antibody response that is detectable as early as 4 days after vaccination and peaks within 10 days after vaccination147. HBGA-blocking antibody responses are similar in older adults when compared with younger adults148 and mucosal IgA responses also occur149. A proof-of-concept CHIM study using a GII.4/2002 strain as the challenge virus was too underpowered to demonstrate a significant reduction in norovirus-associated illness150, but a field efficacy study demonstrated vaccine efficacy of 62% (95% confidence interval, 21–82%) in preventing moderate-to-severe norovirus-associated gastroenteritis due to any strain and significant protection against a heterotypic GII.2 strain151. HBGA-blocking antibody responses to heterologous strains have also been observed, although to a lower level than to vaccine strains, providing a potential explanation for the observed heterotypic protection152. The vaccine is also immunogenic in children 1–8 years of age and in children 6–12 months old153,154. Younger children (<4 years) had increased HBGA-blocking antibody responses to both antigens when a second dose was administered 1 month after the first. Although older children (4–8 years) also had higher antibody levels to the GI.1 antigen after a second dose, the second vaccine dose less consistently increased their GII.4 antibody responses153. A two-dose, placebo-controlled, field efficacy trial in children that is administering the bivalent VLP vaccine at 5 months and 6–7 months of age is underway (NCT05281094)155.
Another vaccine evaluated in several clinical trials is a recombinant, non-replicating, adenovirus type 5-vectored vaccine administered orally as a tablet. Both GI.1 and GII.4 vaccine tablets have been developed and evaluated156. The initial clinical study with the GI.1 construct found it to be safe and well tolerated, with both serum HBGA-blocking and mucosal antibody responses being induced157. Other studies, reported in abstracts or media releases, have shown that immune responses in older adults are at a level similar to those of younger adults. Co-administration of the GI.1 and GII.4 vaccines induced immune responses without evidence of interference and the GI.1 vaccine reduced homologous norovirus infection by 21% in a CHIM, a reduction similar to one reported previously for an intranasally administered GI.1 VLP vaccine158,159.
Other vaccine candidates are also in clinical development, but there is much less information available about their safety and immunogenicity. All vaccines under development are multivalent and contain from two to six genotypes. A GII.4 strain is included in all vaccine candidates, but the GI component varies and additional GII strains are included in some (Table 2). An unadjuvanted bivalent VLP vaccine containing GI.4 and GII.4 capsid proteins was safe and immunogenic, inducing HBGA-blocking antibody responses to the vaccine strains as well as cross-reactive IgG responses to several heterotypic (non-vaccine) strains160,161. A phase III clinical trial is planned for a yeast-expressed bivalent VLP vaccine containing GI.1 and GII.4 strains (NCT05916326)162, but no information is yet available on the results of the phase I and II trials of the vaccine.
Future perspectives
This is an exciting time for HuNoV research. Future studies are expected to discover the cell receptor or receptors for HuNoVs, which are predicted to differ between strains. New intestinal biology is being discovered from studies on HuNoV entry into and characterization of the glycolipids and glycoproteins on HIEs163,164. This should allow simpler culture systems to be developed. Understanding mechanisms of host restriction is also expected to lead to improved culture systems and insight into why some HuNoV strains persist globally. Both epidemiological and vaccine studies have demonstrated that induction of protective immunity against norovirus-associated illness is possible. Unanswered questions that need to be assessed in clinical trials include the breadth of immunity induced by vaccination against single genotypes, the importance of priming and past infection history on induction of immunity to non-vaccine strains, the optimal number of strains to include in a vaccine, the duration of immunity after vaccination and the mechanism or mechanisms of protective immunity165. Vaccines have focused on VP1 as an immunogen, but antibody responses to NSPs also occur166,167, and the potential of these as targets for vaccination remains to be assessed. The recent revelation from the structural109 and antibody neutralization studies140 that GII.4 HuNoV capsid exhibits inherent plasticity that the antibodies can target for neutralization suggests that capsid dynamics are likely to play a critical role in cell entry. These observations raise an important question: should this dynamic feature of the capsid be considered when developing capsid-based vaccine strategies? The discovery that a single nanobody neutralizes multiple GII.4 epochal variants135 suggests that nanobodies, given their small size, high stability, strong antigen-binding affinity and water solubility, might be tested to be used alone as an immunotherapeutic or in conjunction with vaccines to improve therapeutic efficiency. The demonstration that immunoglobulin preparations with neutralizing activity against the infecting norovirus successfully eradicated infection in a chronically infected, immunocompromised patient supports the rationale for developing immunotherapeutics168.
Finally, small molecule antivirals and associated therapies are being developed and evaluated in HIEs and other replication systems29,76,169. Currently, these efforts focus primarily on HuNoV RdRPs and proteases, and whether they will lead to the discovery of broadly active compounds remains to be seen. In the case of HuNoV proteases, structural differences observed between GI and GII proteases170 may have to be dealt with for such an endeavour. Pharmacological targeting of the liquid-like condensates induced by HuNoV RdRP46 as putative replication hubs can also be an attractive possibility in the future, as suggested for other viral systems, cancer and neurodegenerative diseases171,172. Further understanding of the structure and function of other HuNoV NSPs, notably p41 with its demonstrated helicase activity49, could lead to new targets for drug development.
Supplementary Material
Acknowledgements
The authors’ work referenced in this Review was funded in part by Public Health Service grant P01-AI057788 to M.K.E., R.L.A. and B.V.V.P., and P30 DK 056338 (to H. El-Serag), which supports the Texas Medical Center Digestive Diseases Center and the GEMS Core. The authors thank their norovirus research team for the input, particularly B. Vijayalakshmi Ayyar for help with Figures 3 and 4, and Urvy Mudgal, for help with Figures 1, 2 and 5.
Footnotes
Competing interests
R.L.A., M.K.E. and B.V.V.P. have grant support from Hillevax, Inc., and R.L.A. and M.K.E. are consultants for that company. Baylor College of Medicine (R.L.A. and M.K.E. as inventors) has a patent for norovirus growth in human intestinal enteroids. M.K.E. has a patent on methods and reagents to detect and characterize Norwalk virus and related viruses. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41579-024-01144-9.
Related links
Human Calicivirus Typing Tool: https://calicivirustypingtool.cdc.gov/
Norovirus Typing Tool: https://www.rivm.nl/mpf/norovirus/typingtool
References
- 1.Pires SM et al. Aetiology-specific estimates of the global and regional incidence and mortality of diarrhoeal diseases commonly transmitted through food. PLoS ONE 10, e0142927 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartsch SM, Lopman BA, Ozawa S, Hall AJ & Lee BY Global economic burden of norovirus gastroenteritis. PLoS ONE 11, e0151219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bok K & Green KY Norovirus gastroenteritis in immunocompromised patients. N. Engl. J. Med. 367, 2126–2132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorne LG & Goodfellow IG Norovirus gene expression and replication. J. Gen. Virol. 95, 278–291 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Kroneman A et al. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 158, 2059–2068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chhabra P et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 100, 1393–1406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannon JL et al. Global trends in norovirus genotype distribution among children with acute gastroenteritis. Emerg. Infect. Dis. 27, 1438–1445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Beek J et al. Molecular surveillance of norovirus, 2005–16: an epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 18, 545–553 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Siebenga JJ et al. Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001–2007. J. Infect. Dis. 200, 802–812 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Chan MCW et al. Global spread of norovirus GII.17 Kawasaki 308, 2014–2016. Emerg. Infect. Dis. 23, 1359–1354 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwok K et al. Increased detection of emergent recombinant norovirus GII.P16-GII.2 strains in young adults, Hong Kong, China, 2016–2017. Emerg. Infect. Dis. 23, 1852–1855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chhabra P et al. Increased circulation of GII.17 noroviruses, six European countries and the United States, 2023 to 2024. Eur. Surveill. 29, 2400625 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kendra JA, Tohma K & Parra GI Global and regional circulation trends of norovirus genotypes and recombinants, 1995–2019: a comprehensive review of sequences from public databases. Rev. Med. Virol. 32, e2354 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bull RA et al. Comparison of the replication properties of murine and human calicivirus RNA-dependent RNA polymerases. Virus Genes. 42, 16–27 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Parra GI et al. Static and evolving norovirus genotypes: implications for epidemiology and Immunity. PLoS Pathog. 13, e1006136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chhabra P et al. Emergence of novel norovirus GII.4 variant. Emerg. Infect. Dis. 30, 163–167 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tohma K, Lepore CJ, Ford-Siltz LA & Parra GI Phylogenetic analyses suggest that factors other than the capsid protein play a role in the epidemic potential of GII.2 norovirus. mSphere 2, 00187–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindesmith L et al. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9, 548–553 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Hutson AM, Airaud F, LePendu J, Estes MK & Atmar RL Norwalk virus infection associates with secretor status genotyped from sera. J. Med. Virol. 77, 116–120 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Thorven M et al. A homozygous nonsense mutation (428G→A) in the human secretor (FUT2) gene provides resistance to symptomatic norovirus (GGII) infections. J. Virol. 79, 15351–15355 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Rougemont A et al. Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 85, 4057–4070 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang Y et al. Evolution of the interactions between GII.4 noroviruses and histo-blood group antigens: insights from experimental and computational studies. PLoS Pathog. 17, e1009745 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Estienney M et al. Epidemiological impact of GII.17 human noroviruses associated with attachment to enterocytes. Front. Microbiol. 13, 858245 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tenge VR et al. Glycan recognition in human norovirus infections. Viruses 13, 2066 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheetham S et al. Pathogenesis of a genogroup II human norovirus in gnotobiotic pigs. J. Virol. 80, 10372–10381 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Souza M, Azevedo MS, Jung K, Cheetham S & Saif LJ Pathogenesis and immune responses in gnotobiotic calves after infection with the genogroup II.4-HS66 strain of human norovirus. J. Virol. 82, 1777–1786 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taube S et al. A mouse model for human norovirus. mBio 4, 00450–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolawole AO, Rocha-Pereira J, Elftman MD, Neyts J & Wobus CE Inhibition of human norovirus by a viral polymerase inhibitor in the B cell culture system and in the mouse model. Antivir. Res. 132, 46–49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Dycke J et al. A robust human norovirus replication model in zebrafish larvae. PLoS Pathog. 15, e1008009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Dycke J et al. Infection of zebrafish larvae with human norovirus and evaluation of the in vivo efficacy of small-molecule inhibitors. Nat. Protoc. 16, 1830–1849 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Tan MTH, Gong Z & Li D Use of zebrafish embryos to reproduce human norovirus and to evaluate human norovirus infectivity decay after UV treatment. Appl. Environ. Microbiol. 89, e0011523 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan MTH et al. The globally re-emerging norovirus GII.2 manifests higher heat resistance than norovirus GII.4 and Tulane virus. J. Appl. Microbiol. 132, 2441–2449 (2022). [DOI] [PubMed] [Google Scholar]
- 33.Cubitt WD Human, small round structured viruses, caliciviruses and astroviruses. Baillieres Clin. Gastroenterol. 4, 643–656 (1990). [DOI] [PubMed] [Google Scholar]
- 34.Rockx BH, Bogers WM, Heeney JL, van Amerongen G & Koopmans MP Experimental norovirus infections in non-human primates. J. Med. Virol. 75, 313–320 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Wyatt RG et al. Experimental infection of chimpanzees with the Norwalk agent of epidemic viral gastroenteritis. J. Med. Virol. 2, 89–96 (1978). [DOI] [PubMed] [Google Scholar]
- 36.Subekti DS et al. Experimental infection of Macaca nemestrina with a Toronto Norwalk-like virus of epidemic viral gastroenteritis. J. Med. Virol. 66, 400–406 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Bok K et al. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proc. Natl Acad. Sci. USA 108, 325–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rimkute I et al. A non-human primate model for human norovirus infection. Nat. Microbiol. 9, 776–786 (2024). [DOI] [PubMed] [Google Scholar]
- 39.Chung L et al. Norovirus translation requires an interaction between the C terminus of the genome-linked viral protein VPg and eukaryotic translation initiation factor 4G. J. Biol. Chem. 289, 21738–21750 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daughenbaugh KF, Fraser CS, Hershey JW & Hardy ME The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 22, 2852–2859 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodfellow I The genome-linked protein VPg of vertebrate viruses — a multifaceted protein. Curr. Opin. Virol. 1, 355–362 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y, Galasiti Kankanamalage AC, Chang KO & Groutas WC Recent advances in the discovery of norovirus therapeutics. J. Med. Chem. 58, 9438–9450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prasad BV et al. Antiviral targets of human noroviruses. Curr. Opin. Virol. 18, 117–125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao B et al. Norovirus protease structure and antivirals development. Viruses 13, 2069 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pham S et al. Conformational flexibility is a critical factor in designing broad-spectrum human norovirus protease inhibitors. Preprint at bioRxiv 10.1101/2024.09.16.613336 (2024). [DOI] [PMC free article] [PubMed]
- 46.Kaundal S et al. RNA-dependent RNA polymerase of predominant human norovirus forms liquid-liquid phase condensates as viral replication factories. Sci. Adv. 10, eadp9333 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campillay-Veliz CP et al. Human norovirus proteins: implications in the replicative cycle, pathogenesis, and the host immune response. Front. Immunol. 11, 961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp TM et al. Secretory pathway antagonism by calicivirus homologues of Norwalk virus nonstructural protein p22 is restricted to noroviruses. Virol. J. 9, 181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li TF et al. Human norovirus NS3 has RNA helicase and chaperoning activities. J. Virol. 92, e01606–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doerflinger SY et al. Membrane alterations induced by nonstructural proteins of human norovirus. PLoS Pathog. 13, e1006705 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yen JB et al. Identification and characterization of human norovirus NTPase regions required for lipid droplet localization, cellular apoptosis, and interaction with the viral P22 protein. Microbiol. Spectr. 9, e0042221 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blacklow NR et al. Acute infectious nonbacterial gastroenteritis: etiology and pathogenesis. Ann. Intern. Med. 76, 993–1008 (1972). [DOI] [PubMed] [Google Scholar]
- 53.Kapikian AZ The discovery of the 27-nm Norwalk virus: an historic perspective. J. Infect. Dis. 181, S295–S302 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duizer E et al. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85, 79–87 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Guix S et al. Norwalk virus RNA is infectious in mammalian cells. J. Virol. 81, 12238–12248 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schreiber DS, Blacklow NR & Trier JS The mucosal lesion of the proximal small intestine in acute infectious nonbacterial gastroenteritis. N. Engl. J. Med. 288, 1318–1323 (1973). [DOI] [PubMed] [Google Scholar]
- 57.Dolin R, Levy AG, Wyatt RG, Thornhill TS & Gardner JD Viral gastroenteritis induced by the Hawaii agent. Jejunal histopathology and serologic response. Am. J. Med. 59, 761–768 (1975). [DOI] [PubMed] [Google Scholar]
- 58.Karandikar UC et al. Detection of human norovirus in intestinal biopsies from immunocompromised transplant patients. J. Gen. Virol. 97, 2291–2300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Estes MK et al. Human norovirus cultivation in nontransformed stem cell-derived human intestinal enteroid cultures: success and challenges. Viruses 11, 638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Green KY et al. Human norovirus targets enteroendocrine epithelial cells in the small intestine. Nat. Commun. 11, 2759 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones MK et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346, 755–759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones MK et al. Human norovirus culture in B cells. Nat. Protoc. 10, 1939–1947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilen CB et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360, 204–208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mirabelli C et al. Human norovirus triggers primary B cell immune activation in vitro. mBio 13, e0017522 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown JR, Gilmour K & Breuer J Norovirus infections occur in B-cell-deficient patients. Clin. Infect. Dis. 62, 1136–1138 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Wobus CE The dual tropism of noroviruses. J. Virol. 92, e01010–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhar S & Jones MK In vitro replication of human norovirus. Viruses 11, 547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sato T et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Ettayebi K et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sato T & Clevers H Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Zou WY et al. Human intestinal enteroids: new models to study gastrointestinal virus infections. Methods Mol. Biol. 1576, 229–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murakami K et al. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc. Natl Acad. Sci. USA 117, 1700–1710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Atmar RL et al. Determination of the 50% human infectious dose for Norwalk virus. J. Infect. Dis. 209, 1016–1022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rouphael N et al. Dose-response of a norovirus GII.2 controlled human challenge model inoculum. J. Infect. Dis. 226, 1771–1780 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adeniyi-Ipadeola GO et al. Infant and adult human intestinal enteroids are morphologically and functionally distinct. mBio 15, e0131624 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lewis MA et al. Standardization of an antiviral pipeline for human norovirus in human intestinal enteroids demonstrates nitazoxanide has no to weak antiviral activity. Antimicrob. Agents Chemother. 67, e0063623 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ettayebi K et al. Insights into human norovirus cultivation in human intestinal enteroids. mSphere 9, e0044824 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Costantini V et al. Human norovirus replication in human intestinal enteroids as a model to evaluate virus inactivation. Emerg. Infect. Dis. 24, 1453–1464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ettayebi K et al. New insights and enhanced human norovirus cultivation in human intestinal enteroids. mSphere 6, e01136–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carmona-Vicente N et al. Human intestinal enteroids platform to assess the infectivity of gastroenteritis viruses in wastewater. Water Res. 255, 121481 (2024). [DOI] [PubMed] [Google Scholar]
- 81.Chang-Graham AL et al. Human intestinal enteroids with inducible neurogenin-3 expression as a novel model of ut hormone secretion. Cell Mol. Gastroenterol. Hepatol. 8, 209–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spence JR et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCracken KW, Howell JC, Wells JM & Spence JR Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 6, 1920–1928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang D et al. Human intestinal organoids express histo-blood group antigens, bind norovirus VLPs, and support limited norovirus replication. Sci. Rep. 7, 12621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sato S et al. Human norovirus propagation in human induced pluripotent stem cell-derived intestinal epithelial cells. Cell. Mol. Gastroenterol. Hepatol. 7, 686–688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watson CL et al. An in vivo model of human small intestine using pluripotent stem cells. Nat. Med. 20, 1310–1314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Finkbeiner SR et al. Generation of tissue-engineered small intestine using embryonic stem cell-derived human intestinal organoids. Biol. Open 4, 1462–1472 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haga K et al. Genetic manipulation of human intestinal enteroids demonstrates the necessity of a functional fucosyltransferase 2 gene for secretor-dependent human norovirus infection. mBio 11, e00251–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruvoen-Clouet N et al. Increase in genogroup II.4 norovirus host spectrum by CagA-positive Helicobacter pylori infection. J. Infect. Dis. 210, 183–191 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Criss ZK II et al. Drivers of transcriptional variance in human intestinal epithelial organoids. Physiol. Genomics 53, 486–508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tenge VR et al. Bile goes viral. Viruses 13, 998 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tenge V et al. Bile acid-sensitive human norovirus strains are susceptible to sphingosine-1-phosphate receptor 2 inhibition. J. Virol. 98, e0202023 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kilic T, Koromyslova A & Hansman GS Structural basis for human norovirus capsid binding to bile acids. J. Virol. 93, e01581–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ayyar BV et al. CLIC and membrane wound repair pathways enable pandemic norovirus entry and infection. Nat. Commun. 14, 1148 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ivashenka A et al. Glycolipid-dependent and lectin-driven transcytosis in mouse enterocytes. Commun. Biol. 4, 173 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rydell GE, Svensson L, Larson G, Johannes L & Romer W Human GII.4 norovirus VLP induces membrane invaginations on giant unilamellar vesicles containing secretor gene dependent α1,2-fucosylated glycosphingolipids. Biochim. Biophys. Acta 1828, 1840–1845 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Koyama S, Ishii KJ, Coban C & Akira S Innate immune response to viral infection. Cytokine 43, 336–341 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Schoggins JW et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lin SC et al. Human norovirus exhibits strain-specific sensitivity to host interferon pathways in human intestinal enteroids. Proc. Natl Acad. Sci. USA 117, 23782–23793 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hosmillo M et al. Norovirus replication in human intestinal epithelial cells is restricted by the interferon-induced JAK/STAT signaling pathway and RNA polymerase II-mediated transcriptional responses. mBio 11, e00215–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang X, Wang M, Graham DY & Estes MK Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66, 6527–6532 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Prasad BV et al. X-ray crystallographic structure of the Norwalk virus capsid. Science 286, 287–290 (1999). [DOI] [PubMed] [Google Scholar]
- 103.Conley MJ et al. Calicivirus VP2 forms a portal-like assembly following receptor engagement. Nature 565, 377–381 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Ossiboff RJ, Zhou Y, Lightfoot PJ, Prasad BV & Parker JS Conformational changes in the capsid of a calicivirus upon interaction with its functional receptor. J. Virol. 84, 5550–5564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen R, Neill JD, Estes MK & Prasad BVV X-ray structure of a native calicivirus: structural insights into antigenic diversity and host specificity. Proc. Natl Acad. Sci. USA 103, 8048–8053 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Williams AN et al. Multiple signals in the gut contract the mouse norovirus capsid to block antibody binding while enhancing receptor affinity. J. Virol. 95, e0147121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Song C et al. Dynamic rotation of the protruding domain enhances the infectivity of norovirus. PLoS Pathog. 16, e1008619 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sherman MB et al. Bile salts alter the mouse norovirus capsid conformation: possible implications for cell attachment and immune evasion. J. Virol. 93, e00970–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hu L et al. Atomic structure of the predominant GII.4 human norovirus capsid reveals novel stability and plasticity. Nat. Commun. 13, 1241 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bhella D & Goodfellow IG The cryo-electron microscopy structure of feline calicivirus bound to junctional adhesion molecule A at 9-angstrom resolution reveals receptor-induced flexibility and two distinct conformational changes in the capsid protein VP1. J. Virol. 85, 11381–11390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nelson CA et al. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl Acad. Sci. USA 115, E9201–E9210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Smith HQ & Smith TJ The dynamic capsid structures of the noroviruses. Viruses 11, 235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Venkataram Prasad BV et al. Structural basis of glycan interaction in gastroenteric viral pathogens. Curr. Opin. Virol. 7, 119–127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Danthi P, Tosteson M, Li QH & Chow M Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J. Virol. 77, 5266–5274 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shah PNM et al. Cryo-EM structures reveal two distinct conformational states in a picornavirus cell entry intermediate. PLoS Pathog. 16, e1008920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vongpunsawad S, Venkataram Prasad BV & Estes MK Norwalk virus minor capsid protein VP2 associates within the VP1 shell domain. J. Virol. 87, 4818–4825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bertolotti-Ciarlet A, White LJ, Chen R, Prasad BV & Estes MK Structural requirements for the assembly of Norwalk virus-like particles. J. Virol. 76, 4044–4055 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cao S et al. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 81, 5949–5957 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Choi JM, Hutson AM, Estes MK & Prasad BV Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl Acad. Sci. USA 105, 9175–9180 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shanker S et al. Structural analysis of determinants of histo-blood group antigen binding specificity in genogroup I noroviruses. J. Virol. 88, 6168–6180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shanker S et al. Structural features of glycan recognition among viral pathogens. Curr. Opin. Struct. Biol. 44, 211–218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Singh BK, Leuthold MM & Hansman GS Human noroviruses’ fondness for histo-blood group antigens. J. Virol. 89, 2024–2040 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tan M & Jiang X Norovirus-host interaction: multi-selections by human histo-blood group antigens. Trends Microbiol. 19, 382–388 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marionneau S et al. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 122, 1967–1977 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shanker S et al. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: implications for epochal evolution. J. Virol. 85, 8635–8645 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lindesmith LC et al. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5, e31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Parra GI et al. Minimal antigenic evolution after a decade of norovirus GII.4 Sydney_2012 circulation in humans. J. Virol. 97, e0171622 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lindesmith LC et al. Immune imprinting drives human norovirus potential for global spread. mBio 13, e0186122 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Katpally U, Wobus CE, Dryden K, Virgin HWT & Smith TJ Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J. Virol. 82, 2079–2088 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Katpally U et al. High-resolution cryo-electron microscopy structures of murine norovirus 1 and rabbit hemorrhagic disease virus reveal marked flexibility in the receptor binding domains. J. Virol. 84, 5836–5841 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sherman M et al. The reversible activation of norovirus by metal ions. J. Virol. 98, e0173523 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Devant JM & Hansman GS Structural heterogeneity of a human norovirus vaccine candidate. Virology 553, 23–34 (2021). [DOI] [PubMed] [Google Scholar]
- 133.Jung J et al. High-resolution cryo-EM structures of outbreak strain human norovirus shells reveal size variations. Proc. Natl Acad. Sci. USA 116, 12828–12832 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Devant JM, Hofhaus G, Bhella D & Hansman GS Heterologous expression of human norovirus GII.4 VP1 leads to assembly of T = 4 virus-like particles. Antivir. Res. 168, 175–182 (2019). [DOI] [PubMed] [Google Scholar]
- 135.Salmen W et al. A single nanobody neutralizes multiple epochally evolving human noroviruses by modulating capsid plasticity. Nat. Commun. 14, 6516 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Burton DR Antiviral neutralizing antibodies: from in vitro to in vivo activity. Nat. Rev. Immunol. 23, 720–734 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reading SA & Dimmock NJ Neutralization of animal virus infectivity by antibody. Arch. Virol. 152, 1047–1059 (2007). [DOI] [PubMed] [Google Scholar]
- 138.Touabi L, Aflatouni F & McLean GR Mechanisms of rhinovirus neutralisation by antibodies. Viruses 13, 360 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tohma K, Ford-Siltz LA, Kendra JA & Parra GI Dynamic immunodominance hierarchy of neutralizing antibody responses to evolving GII.4 noroviruses. Cell Rep. 39, 110689 (2022). [DOI] [PubMed] [Google Scholar]
- 140.Alvarado G et al. Broadly cross-reactive human antibodies that inhibit genogroup I and II noroviruses. Nat. Commun. 12, 4320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lindesmith LC et al. Sera antibody repertoire analyses reveal mechanisms of broad and pandemic strain neutralizing responses after human norovirus vaccination. Immunity 50, 1530–1541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shanker S et al. Structural basis for norovirus neutralization by an HBGA blocking human IgA antibody. Proc. Natl Acad. Sci. USA 113, E5830–E5837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koromyslova AD et al. Nanobody-mediated neutralization reveals an Achilles heel for norovirus. J. Virol. 94, e00660–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Koromyslova AD & Hansman GS Nanobody binding to a conserved epitope promotes norovirus particle disassembly. J. Virol. 89, 2718–2730 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sapparapu G et al. Frequent use of the IgA isotype in human B cells encoding potent norovirus-specific monoclonal antibodies that block HBGA binding. PLoS Pathog. 12, e1005719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Koromyslova AD, Morozov VA, Hefele L & Hansman GS Human norovirus neutralized by a monoclonal antibody targeting the histo-blood group antigen pocket. J. Virol. 93, e02174–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Atmar RL et al. Rapid responses to 2 virus-like particle norovirus vaccine candidate formulations in healthy adults: a randomized controlled trial. J. Infect. Dis. 214, 845–853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Treanor JJ et al. A novel intramuscular bivalent norovirus VLP vaccine candidate — reactogenicity, safety and immunogenicity in a phase I trial in healthy adults. J. Infect. Dis. 210, 1763–1771 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Atmar RL et al. An exploratory study of the salivary immunoglobulin A responses to 1 dose of a norovirus virus-like particle candidate vaccine in healthy adults. J. Infect. Dis. 219, 410–414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bernstein DI et al. Norovirus vaccine against experimental human GII.4 virus illness: a challenge study in healthy adults. J. Infect. Dis. 211, 870–878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sherwood J et al. Efficacy of an intramuscular bivalent norovirus GI.1/GII.4 virus-like particle vaccine candidate in healthy US adults. Vaccine 38, 6442–6449 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Haynes J et al. In depth breadth analyses of human blockade responses to norovirus and response to vaccination. Viruses 11, 392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vesikari T et al. Immunogenicity of a bivalent virus-like particle norovirus vaccine in children from 1 to 8 years of age: a phase 2 randomized, double-blind study. Vaccine 40, 3588–3596 (2022). [DOI] [PubMed] [Google Scholar]
- 154.Lopez P et al. Immunogenicity and tolerability of a bivalent virus-like particle norovirus vaccine candidate in children from 6 months up to 4 years of age: a phase 2 randomized, double-blind trial. Hum. Vaccin. Immunother. 19, 2204787 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05281094 (2024). [DOI] [PubMed]
- 156.Braun MR, Flitter BA, Sun W & Tucker SN An easy pill to swallow: oral recombinant vaccines for the 21st century. Curr. Opin. Immunol. 84, 102374 (2023). [DOI] [PubMed] [Google Scholar]
- 157.Kim BR et al. Incidence and characteristics of norovirus-associated benign convulsions with mild gastroenteritis, in comparison with rotavirus ones. Brain Dev. 40, 699–706 (2018). [DOI] [PubMed] [Google Scholar]
- 158.Vaxart. Vaxart announces topline data from the phase 2 challenge study of its monovalent norovirus vaccine candidate. Vaxart, Inc. https://investors.vaxart.com/news-releases/news-release-details/vaxart-announces-topline-data-phase-2-challenge-study-its (6 September 2023). [Google Scholar]
- 159.Atmar RL et al. Norovirus vaccine against experimental human Norwalk Virus illness. N. Engl. J. Med. 365, 2178–2187 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Leroux-Roels I et al. A randomized, double-blind, placebo-controlled, dose-escalating phase I trial to evaluate safety and immunogenicity of a plant-produced, bivalent, recombinant norovirus-like particle vaccine. Front. Immunol. 13, 1021500 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Waerlop G et al. Immune responses in healthy adults elicited by a bivalent norovirus vaccine candidate composed of GI.4 and GII.4 VLPs without adjuvant. Front. Immunol. 14, 1188431 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05916326 (2023). [DOI] [PubMed]
- 163.Rimkute I et al. Histo-blood group antigens of glycosphingolipids predict susceptibility of human intestinal enteroids to norovirus infection. J. Biol. Chem. 295, 15974–15987 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nilsson J et al. N-glycoproteomic analyses of human intestinal enteroids, varying in histo-blood group geno- and phenotypes, reveal a wide repertoire of fucosylated glycoproteins. Glycobiology 34, cwae029 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ramani S, Estes MK & Atmar RL Correlates of protection against norovirus infection and disease-where are we now, where do we go? PLoS Pathog. 12, e1005334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Matsui SM et al. The isolation and characterization of a Norwalk virus-specific cDNA. J. Clin. Invest. 87, 1456–1461 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Su L et al. Mapping human norovirus antigens during infection reveals the breadth of the humoral immune response. NPJ Vaccines 8, 87 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.van Kampen JJA et al. Clinical and In vitro evidence favoring immunoglobulin treatment of a chronic norovirus infection in a patient with common variable immunodeficiency. J. Infect. Dis. 226, 1781–1789 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hayashi T, Kobayashi S, Hirano J & Murakami K Human norovirus cultivation systems and their use in antiviral research. J. Virol. 98, e0166323 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Viskovska MA et al. GII.4 norovirus protease shows pH-sensitive proteolysis with a unique Arg-His pairing in the catalyticsite. J. Virol. 93, e01479–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wheeler RJ Therapeutics-how to treat phase separation-associated diseases. Emerg. Top. Life Sci. 4, 307–318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zou Y et al. New insights into the important roles of phase seperation in the targeted therapy of lung cancer. Cell Biosci. 13, 150 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Harrington PR, Lindesmith L, Yount B, Moe CL & Baric RS Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J. Virol. 76, 12335–12343 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hutson AM, Atmar RL, Marcus DM & Estes MK Norwalk virus-like particle hemagglutination by binding to H histo-blood group antigens. J. Virol. 77, 405–415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Huang P et al. Norovirus and histo-blood group antigens: demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J. Virol. 79, 6714–6722 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shirato H et al. Noroviruses distinguish between type 1 and type 2 histo-blood group antigens for binding. J. Virol. 82, 10756–10767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yazawa S et al. Blood group substances as potential therapeutic agents for the prevention and treatment of infection with noroviruses proving novel binding patterns in human tissues. PLoS ONE 9, e89071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nordgren J, Kindberg E, Lindgren PE, Matussek A & Svensson L Norovirus gastroenteritis outbreak with a secretor-independent susceptibility pattern, Sweden. Emerg. Infect. Dis. 16, 81–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mallory ML, Lindesmith LC, Brewer-Jensen PD, Graham RL & Baric RS Bile facilitates human norovirus interactions with diverse histoblood group antigens, compensating for capsid microvariation observed in 2016–2017 GII.2 strains. Viruses 12, 989 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lindesmith L et al. Cellular and humoral immunity following Snow Mountain virus challenge. J. Virol. 79, 2900–2909 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lindesmith LC et al. Virus-host interactions between nonsecretors and human norovirus. Cell Mol. Gastroenterol. Hepatol. 10, 245–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jin M et al. Two gastroenteritis outbreaks caused by GII noroviruses: host susceptibility and HBGA phenotypes. PLoS ONE 8, e58605 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yoda T et al. No crucial amino acid changes in the predicted histo blood group antigen-binding sites of norovirus genotype GII.4 capsid between non-secretors and secretors origin might suggest an alternative route of infection or existence of coincidental molecules. J. Med. Microbiol. 64, 1544–1547 (2015). [DOI] [PubMed] [Google Scholar]
- 184.Frenck R et al. Predicting susceptibility to norovirus GII.4 by use of a challenge model involving humans. J. Infect. Dis. 206, 1386–1393 (2012). [DOI] [PubMed] [Google Scholar]
- 185.Carlsson B et al. The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 Norovirus infection. PLoS ONE 4, e5593 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Currier RL et al. Innate susceptibility to norovirus infections influenced by FUT2 genotype in a United States pediatric population. Clin. Infect. Dis. 60, 1631–1638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Uusi-Kerttula H, Tamminen K, Malm M, Vesikari T & Blazevic V Comparison of human saliva and synthetic histo-blood group antigens usage as ligands in norovirus-like particle binding and blocking assays. Microbes Infect. 16, 472–480 (2014). [DOI] [PubMed] [Google Scholar]
- 188.Tu LT et al. Genetic susceptibility to norovirus GII.4 Sydney strain infections in Taiwanese children. Pediatr. Infect. Dis. J. 36, 353–357 (2017). [DOI] [PubMed] [Google Scholar]
- 189.Cong X et al. Functional and structural characterization of norovirus GII.6 in recognizing histo-blood group antigens. Virol. Sin. 38, 56–65 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sharma S et al. Secretor status is associated with susceptibility to disease in a large GII.6 norovirus foodborne outbreak. Food Environ. Virol. 12, 28–34 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang XF et al. An outbreak caused by GII.17 norovirus with a wide spectrum of HBGA-associated susceptibility. Sci. Rep. 5, 17687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dai YC et al. Characterization of antigenic relatedness between GII.4 and GII.17 noroviruses by use of serum samples from norovirus-infected patients. J. Clin. Microbiol. 55, 3366–3373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04563533 (2024). [DOI] [PubMed]
- 194.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT06524947 (2024). [DOI] [PubMed]
- 195.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05805618 (2023). [DOI] [PubMed]
- 196.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01168401 (2018). [DOI] [PubMed]
- 197.Lindesmith LC et al. Broad blockade antibody responses in human volunteers after immunization with a multivalent norovirus VLP candidate vaccine: immunological analyses from a phase I clinical trial. PLoS Med. 12, e1001807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ramani S et al. B-cell responses to intramuscular administration of a bivalent virus-like particle human norovirus vaccine. Clin. Vaccin. Immunol. 24, e00571–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01609257 (2019). [DOI] [PubMed]
- 200.Atmar RL et al. Serological correlates of protection against a GII.4 norovirus. Clin. Vaccin. Immunol. 22, 923–929 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sundararajan A et al. Robust mucosal-homing antibody-secreting B cell responses induced by intramuscular administration of adjuvanted bivalent human norovirus-like particle vaccine. Vaccine 33, 568–576 (2015). [DOI] [PubMed] [Google Scholar]
- 202.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02038907 (2017). [DOI] [PubMed]
- 203.Leroux-Roels G et al. Safety and immunogenicity of different formulations of norovirus vaccine candidate in healthy adults: a randomized, controlled, double-blind clinical trial. J. Infect. Dis. 217, 597–607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02142504 (2017). [DOI] [PubMed]
- 205.Atmar RL et al. Persistence of antibodies to 2 virus-like particle norovirus vaccine candidate formulations in healthy adults: 1-year follow-up with memory probe vaccination. J. Infect. Dis. 220, 603–614 (2019). [DOI] [PubMed] [Google Scholar]
- 206.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02153112 (2019). [DOI] [PubMed]
- 207.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02475278 (2018). [DOI] [PubMed]
- 208.Atmar RL et al. Comparison of microneutralization and histo-blood group antigen-blocking assays for functional norovirus antibody detection. J. Infect. Dis. 221, 739–743 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02661490 (2020). [DOI] [PubMed]
- 210.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02669121 (2021). [DOI] [PubMed]
- 211.Atmar RL et al. A bivalent human norovirus vaccine induces homotypic and heterotypic neutralizing antibodies. J. Infect. Dis. 229, 1402–1407 (2024). [DOI] [PubMed] [Google Scholar]
- 212.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03039790 (2022). [DOI] [PubMed]
- 213.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05972733 (2024). [DOI] [PubMed]
- 214.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT06007781 (2023). [DOI] [PubMed]
- 215.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05508178 (2022). [DOI] [PubMed]
- 216.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05992935 (2024). [DOI] [PubMed]
- 217.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT06592794 (2024). [DOI] [PubMed]
- 218.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04188691 (2021). [DOI] [PubMed]
- 219.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04941261 (2021). [DOI] [PubMed]
- 220.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05916326 (2021). [DOI] [PubMed]
- 221.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02868073 (2018). [DOI] [PubMed]
- 222.Kim L et al. Safety and immunogenicity of an oral tablet norovirus vaccine, a phase I randomized, placebo-controlled trial. JCI Insight 3, e121077 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03125473 (2018). [DOI] [PubMed]
- 224.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03897309 (2022). [DOI] [PubMed]
- 225.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04854746 (2023). [DOI] [PubMed]
- 226.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04875676 (2024). [DOI] [PubMed]
- 227.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05212168 (2024). [DOI] [PubMed]
- 228.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05213728 (2024). [DOI] [PubMed]
- 229.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT05626803 (2024). [DOI] [PubMed]
- 230.Siebenga JJ et al. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 81, 9932–9941 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chan JCM, Mohammad KN, Zhang LY, Wong SH & Chan MC Targeted profiling of immunological genes during norovirus replication in human intestinal enteroids. Viruses 13, 155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lin L et al. Replication and transcriptionomic analysis of human noroviruses in human intestinal enteroids. Am. J. Transl. Res. 11, 3365–3374 (2019). [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.