

Abstract
Background
The need to execute a sequence of events in an orderly and timely manner is central to many biological processes, including cell cycle progression and cell differentiation. For self-perpetuating systems, such as the cell cycle oscillator, delay times between events are defined by the network of interacting proteins that propagates the system. However, protein levels inside cells are subject to genetic and environmental fluctuations, raising the question of how reliable timing is maintained.
Results
We compared the robustness of different mechanisms for encoding delay times to fluctuations in protein expression levels. Gradual accumulation and gradual decay of a regulatory protein have an equivalent capacity for defining delay times. Yet, we find that the former is highly sensitive to fluctuations in gene dosage, while the latter can buffer such perturbations. In particular, a positive feedback where the degrading protein auto-enhances its own degradation may render delay times practically insensitive to gene dosage.
Conclusion
While our understanding of biological timing mechanisms is still rudimentary, it is clear that there is an ample use of degradation as well as self-enhanced degradation in processes such as cell cycle and circadian clocks. We propose that degradation processes, and specifically self-enhanced degradation, will be preferred in processes where maintaining the robustness of timing is important.
Background
Protein levels within cells are subject to genetic and environmental variations, but mechanisms have evolved that buffer cellular processes against those fluctuations [1]. Quantitative analysis has indicated that the need to ensure robustness can largely restrict the design of the underlying network [2-4]. Maintaining a reliable sequence of events appears straightforward in cases where the completion of one event directly triggers the next [5]. Often, however, temporal cascades are propagated by a self-sustained biochemical network, which functions even in the absence of feedback signals [6]. For example, the cell cycle is governed by an autonomous oscillator, although this oscillator is executively sensitive to checkpoint signals that may halt its progression [7]. Similarly, while the circadian timing is synchronized by light or temperature, it oscillates as well under constant conditions [8]. The prevalence of self-sustained networks that coordinate temporal cascades suggests that at least in certain cases not only is the temporal order important, but also the relative timing of events needs to be maintained. However, whether mechanisms that code for delay times can also buffer those times against fluctuations has not yet been examined.
Strategies for coding delay times can be classified into two main categories, which are based on the accumulation or on the decay of some regulatory protein, or of its active form (Fig. 1A–B). A typical cascade employs both strategies, but it is not clear which is rate limiting for defining the timing. For example, in the budding yeast cell cycle, Cln3 accumulation is followed by Sic1 degradation, with both processes required for the G1/S transition and the initiation of START [9-12].
Figure 1.

Strategies for coding delay times. A schematic description of each strategy is shown on the top panel, while the respective protein dynamics is shown at the bottom panel. The solid black line corresponds to some reference system, while the dashed black line corresponds to a system in which production rates were reduced two-fold. The time to reach the threshold (taken here as 10% of Pmax) is also shown. It can be seen that the delay time sensitivity is largest for accumulation and smallest for non-linear decay. Moreover, the location of the threshold is limited; threshold of 90% (light grey) will never be crossed by a perturbed system with η of less than 0.9. a, Accumulation strategy. In this case gene production is initiated at t = 0. Once a certain threshold is reached, downstream genes would be affected. b, Degradation strategy. In this case, protein production is stopped at t = 0. Once protein levels have decayed below a certain threshold, target genes would be affected. c, Same as b, except that degradation is non-linear with n = 2.
In this work, we compare the two strategies for coding delay times with respect to their capacity to buffer fluctuations in gene dosage.
Results and discussion
Consider a protein P that is present at a low level Plow. Accumulation of P can be initiated either by enhancing its production or by inhibiting its degradation. In either case, P will start accumulating toward a new steady state Pmax. Subsequent events will follow once P has passed some threshold PT, defined for example by its affinity to target genes promoters if P is a transcription activator. The corresponding delay time, T0, is defined by the time it takes to accumulate this threshold level PT of proteins. Alternatively, delay time can be encoded by an analogous system, where P decays from Pmax toward Plow, activating subsequent events once its levels are reduced below the threshold PT.
Comparing the robustness of time coding strategies
The above two strategies appear to be equivalent for defining delay times. For example, in the absence of feedback, where each protein is degraded independently, both accumulation and degradation follow analogous exponential profiles (Table 1). To keep the equivalence of the two profiles, we assume, for example, that the two situations are characterized by the same degradation rate α. Thus, the only difference between the accumulation and decay situations resides in the production rate, which is either enhanced or repressed at the onset of the respective dynamics1. Evidently, accumulation times are precisely the same as degradation times (Table 1); for example, the time to accumulate or degrade 90% of Pmax is given by α-1log (10). For simplicity we also assume that Plow = 0, although our results do not depend on this assumption as long as P low is much lower than the threshold level PT (Additional file 1).
Table 1.
Comparison of models
| Linear Modela | Non Linear Modela,b | ||
| Accumulation | Decay | Non Linear Decayd | |
| Model |
|
||
| Solution | P = Pmax (1 - e-αt) | P = Pmax e-αt | |
| T0 Unperturbed delay time |
|||
| T1 Perturbedc delay time |
> η-1T0 (η < 1) <η-1T0 (η > 1) |
||
|
Delay time sensitivity |
≥ |η-1 - 1| | 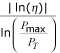 |
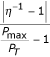 |
a Pmax ![]()
b T0, T1 and δt are presented for n = 2
c Peturbation: v0 → ηv0
![]()
We examined the sensitivity of the delay times to fluctuations in the production rate of P. Since production rate correlates with gene dosage, it is likely to be mostly sensitive to gene-specific perturbations. Perturbation was implemented by changing the production rate of P, v0, by some factor η. Consequently, the delay time T0, coded by the time to accumulate or degrade the protein level from its initial value to the threshold level PT , is changed. We denote this perturbed time by T1. The sensitivity of the delay time to this change in production rate was defined by the relative change in the delay time:
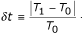
Delay times encoded by decay display a significantly lower sensitivity
Despite their apparent equivalence, we found that the accumulation and decay strategies differ greatly in their capacities to buffer fluctuations in production rate. In fact, for most cases, delay times encoded by decay display a significantly lower sensitivity (Fig. 2 and Table 1). For example, while a two-fold reduction in production rate (η = 1/2) increases delay times by at least 100% in the case of accumulation, it will cause only a 15% (if PT = 0.01 Pmax) or 30% (if PT = 0.1 Pmax) decrease in the case of degradation.
Figure 2.

Delay-time sensitivities for different η and different threshold positions. a, η = 1/2. It can be seen that for all threshold positions the sensitivity of the delay time is smallest for non-linear decay and largest for accumulation. b, η = 2. Also here the sensitivity of the delay time is smallest for non-linear decay and largest for accumulation for most threshold positions. Note that the situation is reversed for high threshold levels corresponding to high sensitivity in all cases (fig. 1). c-e, Delay time sensitivity as a function of η and PT for the cases of accumulation (c), decay (d) and non-linear decay (e). The logarithm of the delay time sensitivity is shown: log (|η - 1|/|δt|) for decay and log (|η-1 - 1|/|δt|) for accumulation. δt was normalized by η-1 for decay and by η-1-1 for accumulation, which correspond to δt in the non-buffered case in which T1 = ηT0 for decay and T1 = η-1T0 for accumulation. Thus, blue represents a non-buffered system. Red represents a buffered system.
The reason underlying this differential behaviour may not be immediately apparent. Within both strategies, changing protein production rate impacts the dynamics in two principal ways. First, it alters the initial rate (v0) by which a protein accumulates or degrades. Second, it modifies the maximal level Pmax (Fig. 1A–B). The key difference between the two systems resides in the initial conditions: in the case of accumulation, the initial condition, and thus the amount of protein that needs to accumulate in order to reach the threshold, remains fixed. Consequently, increased velocity necessarily shortens the time to reach the threshold. In contrast, in the case of degradation, the initial condition, and thus the distance to the threshold, is modified as well. Indeed, this change in initial condition partially balances the change in velocity.
Moreover, this combination of effects leads to a completely different behaviour of the delay time sensitivity, δt. Whereas in the case of accumulation, perturbation in production rate can be mathematically approximated by rescaling time by a constant factor, in the case of degradation such a perturbation is captured by introducing a constant shift in time (see expressions for T1 in Table 1 and Additional file: 1 2.1.2). This difference is due to the fact that production rate enters the equation explicitly in the case of accumulation, but only implicitly, through the initial conditions, in the case of decay. Importantly, this distinct behaviour is not restricted to the linear model, but is in fact applicable also to a general model that includes arbitrary feedback interactions (Additional file: 1 2.2). Consequently, within the accumulation strategy, the dependence of delay times on perturbation will be at best linear with perturbation size, irrespective of possible feedbacks.
Thus, despite the apparent equivalence of the accumulation and degradation strategies, they differ greatly in their capacity to buffer delay times against perturbations in gene dosage. Still, even in the case of degradation, buffering capacity in the absence of feedbacks is limited by realistic dynamical range of the degrading protein. For example, in order to achieve <8% sensitivity to a two-fold change, protein levels have to degrade over four orders of magnitude. This need to increase the dynamical range in order to improve robustness reflects the fact that delay time sensitivity depends on the degradation rate at early times: the faster this initial decay, the greater the robustness (Additional file: 1 3.2.1). However, in the absence of feedback, the system is characterized by a uniform decay rate, so that increasing this initial degradation implies an overall faster decay of P during the given delay time.
Non-linear degradation enhances robustness
One possible way to overcome this interplay between robustness and dynamical range is to introduce a feedback that enhances degradation specifically at early times, while maintaining moderate decay rate during the rest of the time. This will be the case, for example, if the degrading protein functions to enhance its own degradation, either directly or by changing the activity of a third protein. Indeed, a similar feedback mechanism was recently shown to enhance the spatial robustness of morphogen gradients [13].
To rigorously examine the possible impact of an auto-induced degradation on buffering capacity, we extended the linear model to include non-linear degradation (Table 1). In contrast to the exponential dynamics found in the linear system, here the system decays as a power-law in time. Examining the delay time sensitivity, we observed a significantly improved robustness (Fig. 2). For example, for moderate non-linearity, with n = 2, two-fold reduction in production rate will decrease the delay time by merely 1%, compared to 15% in the absence of feedback (for PT = 0.01 Pmax,) Moreover, increasing this coefficient of non-linearity further enhances the robustness (Fig. 2).
The robustness of timing requires fast initial degradation coupled with slower degradation afterwards. In particular, degradation needs to be rapid when protein concentration is above Pmax * η min. Nonlinear degradation enables, in principle, such flexible degradation rates. However, we note that there is an upper limit to degradation rate (see next section).
Auto-regulated degradation is implied in various stages of the cell cycle, such as the transition from S phase to mitosis and the exit from mitosis [14,15]. The degradation of the budding yeast Cdc20 exemplifies this. It begins degrading in late M phase, just before exit from mitosis, and continues throughout G1 phase [16]. This degradation is self-enhanced as Cdc20 itself is an activator of APC-dependent proteolysis through the subunits Cdc23 and Cdc27 [17]. However, degradation in those stages is commonly assumed to occupy only a small portion of the transition time, with most of the delay defined by protein accumulation. It may be that autonomous timing of those stages is less crucial, since the transitions are completely dependent on checkpoint mechanisms that survey the successful completion of the critical events occurring during those cell-cycle stages. Alternatively, protein decay may actually occupy a longer portion of the transition time, or other feedback mechanisms exist but have not yet been identified.
Cell degradation machinery sets a lower limit on time variability
For robust measurement of time, the time for degradation of protein concentrations above Pmax * η min (denoted by δ, different from the sensitivity δt) needs to be short. However, this degradation time is bounded from below by the maximum degradation rate of the cell machinery (in this section we assume that the protein is being degraded rather than being modified):
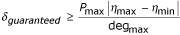
Where δguaranteed is worst case δ. ηmax , ηmin are worst cases for η and degmax is maximum degradation rate [molecules/s].
An order of magnitude estimate of this limit is given, based on a work by Shibatani and Ward [18], which assayed for 20S rat proteasome activity. The 20S proteasome complex is found in all eukaryotic cells and constitutes 0.5–1% of the soluble protein in the cell.
Shibatani and Ward have measured degradation rates in vitro, activating the proteasome with sodium dodecyl sulfate (SDS). The maximum degradation rate measured was 20 nmol/h for 0.07 nmol proteasome. I.e., each proteasome complex degraded roughly 300 molecules per hour. The proteasome composes 0.5–1% of the soluble proteins in the cell (by mass). It is a very heavy complex, about 700 kDa, 14-fold greater than average protein mass, which is roughly 50 kDa. Hence, there are about 1400–2800 proteins per each proteasome complex in the cell. Estimating protein number in the cell as 106-107 (molecules) gives ~ 103 proteasome units. Each unit is capable of degrading 300 molecules in 1 hour, giving degmax ~ 100 [proteins/s].
Assuming Pmax ~ 103 molecules, and fluctuations of the same order (e.g., η = 2), δguaranteed = 10 seconds. Processes sufficiently longer than δguaranteed can be measured accurately: even relaxing some of the assumptions (degradation dedicated to single protein, larger Pmaxetc.), will enable timing of many processes with good accuracy. For example, yeast cell cycle is about 120 minutes, circadian clock is 24 hours – 103-104 fold longer than δguaranteed.
Perturbation to production vs. degradation rates
Our discussion focused on the robustness to fluctuations in production rate (vo) while assuming degradation rate (α) to be relatively stable. Since the degradation machinery plays a crucial role in numerous cellular processes, it is reasonable to assume that its abundance is under a tight regulation, which also limits the noise in degradation rates of individual proteins. Moreover, gene dosage perturbations to production rate are of large magnitude compared to other sources of noise. We expect, as consequence, that mechanisms for buffering against production rate perturbations will be abundant.
Different buffering mechanisms will need to be utilized in the alternative situations where fluctuations in degradation rate dominate. One possible scheme that could reduce the effect of fluctuations in degradation relies on in-cis degradation, where each molecule promotes its own degradation. Such a mechanism was recently reported in the context of cell-cycle timing, where S-phase can only start after UbcH10 undergoes in-cis degradation [19]. Alternatively, delay time could be coded by the linear phase of accumulation, before degradation comes into effect. In this case, the delay time is given by To = PT/vo and does not depend on the degradation rate.
More generally, one may envision other noise characteristics, each dictating its own limitations; for example both production rate and degradation rate might be perturbed together (e.g. temperature effect). The threshold PT might be perturbed together with production rate (Additional file: 1 7) or any other perturbation characteristics. Different buffering mechanisms may need to be tuned for these different perturbations types, which could be analyzed using the framework presented in this paper.
Conclusions
Ensuring the robustness of timing may be of particular importance in order to support crosstalk amongst several processes that are executed in parallel. In such cases, not only the successful completion of events, but also maintaining the coordination, is important. This need may be of particular relevance during development of multicellular organisms, where multiple differentiation processes often proceed in parallel. Our identification of mechanisms that are able to maintain such robustness of timing may provide a new framework for examining the robustness of the long-range cascades that underlie those processes.
Our discussion focused on timing mechanisms that rely on the accumulation or degradation of a single protein component. While such mechanisms can serve as independent timers, more often they present an elementary unit in a more complex cascade. For example, models of cell cycle regulation propose that delay times are generated through the activation of some intermediate components, leading to a delayed negative feedback [20,21]. Further work is required to define how the properties of the full cascade are determined from the properties of its elementary units, and what additional constraints are required for proper coupling of different elementary units.
Methods
Figures were generated using Matlab simulations.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
NR performed the analysis and drafted the manuscript. SW performed the analysis and drafted the manuscript. NB conceived of the study, participated in its design and drafted the manuscript. All authors read and approved the final manuscript.
Note
1 The results are unchanged if we keep production rate fixed and vary the degradation rate. This will become clear later (see expressions for delay time sensitivity in table 1).
Supplementary Material
Supplementary Information
Acknowledgments
Acknowledgements
We thank members of our group for useful discussions. This work was supported by the Minerva and by the ISF. N. B. is the incumbent of the Soretta and Henry Shapiro career development chair.
Contributor Information
Noa Rappaport, Email: noa.rappaport@weizmann.ac.il.
Shay Winter, Email: shay.winter@weizmann.ac.il.
Naama Barkai, Email: naama.barkai@weizmann.ac.il.
References
- Kerszberg M. Noise, delays, robustness, canalization and all that. Curr Opin Genet Dev. 2004;14:440–445. doi: 10.1016/j.gde.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Eldar A, Dorfman R, Weiss D, Ashe H, Shilo BZ, Barkai N. Robustness of the BMP morphogen gradient in Drosophila embryonic patterning. Nature. 2002;419:304–308. doi: 10.1038/nature01061. [DOI] [PubMed] [Google Scholar]
- Von Dassow G, Odell GM. Design and constraints of the Drosophila segment polarity module: Robust spatial patterning emerges from intertwined cell state switches. J Exp Zool. 2002;294:179–215. doi: 10.1002/jez.10144. [DOI] [PubMed] [Google Scholar]
- Barkai N, Leibler S. Robustness in simple biochemical networks. Nature. 1997;387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Murray AW, Kirschner MW. Dominoes and clocks: the union of two views of the cell cycle. Science. 1989;246:614–621. doi: 10.1126/science.2683077. [DOI] [PubMed] [Google Scholar]
- Haase SB, Reed SI. Evidence that a free-running oscillator drives G1 events in the budding yeast cell cycle. Nature. 1999;401:394–397. doi: 10.1038/43930. [DOI] [PubMed] [Google Scholar]
- Edery I. Circadian rhythms in a nutshell. Physiol Genomics. 2000;3:59–74. [Google Scholar]
- Stuart D, Wittenberg C. CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 1995;9:2780–2794. doi: 10.1101/gad.9.22.2780. [DOI] [PubMed] [Google Scholar]
- Tyers M, Tokiwa G, Futcher B. Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. Embo J. 1993;12:1955–1968. doi: 10.1002/j.1460-2075.1993.tb05845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Eldar A, Rosin D, Shilo BZ, Barkai N. Self-enhanced ligand degradation underlies robustness of morphogen gradients. Dev Cell. 2003;5:635–646. doi: 10.1016/S1534-5807(03)00292-2. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Hoyt MA. Eliminating all obstacles: regulated proteolysis in the eukaryotic cell cycle. Cell. 1997;91:149–151. doi: 10.1016/S0092-8674(00)80396-7. [DOI] [PubMed] [Google Scholar]
- Goh PY, Lim HH, Surana U. Cdc20 protein contains a destruction-box but, unlike Clb2, its proteolysisis not acutely dependent on the activity of anaphase-promoting complex. Eur J Biochem. 2000;267:434–449. doi: 10.1046/j.1432-1327.2000.01014.x. [DOI] [PubMed] [Google Scholar]
- Prinz S, Hwang ES, Visintin R, Amon A. The regulation of Cdc20 proteolysis reveals a role for APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr Biol. 1998;8:750–760. doi: 10.1016/S0960-9822(98)70298-2. [DOI] [PubMed] [Google Scholar]
- Shibatani T, Ward WF. Sodium dodecyl sulfate (SDS) activation of the 20S proteasome in rat liver. Arch Biochem Biophys. 1995;321:160–166. doi: 10.1006/abbi.1995.1381. [DOI] [PubMed] [Google Scholar]
- Rape M, Kirschner MW. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 2004;432:588–595. doi: 10.1038/nature03023. [DOI] [PubMed] [Google Scholar]
- Novak B, Tyson JJ. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J Cell Sci. 1993;106:1153–1168. doi: 10.1242/jcs.106.4.1153. [DOI] [PubMed] [Google Scholar]
- Ciliberto A, Petrus MJ, Tyson JJ, Sible JC. A kinetic model of the cyclin E/Cdk2 developmental timer in Xenopus laevis embryos. Biophys Chem. 2003;104:573–589. doi: 10.1016/S0301-4622(03)00060-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information