

Abstract
Sequential adaptive trial designs can help accomplish the goals of personalized medicine, optimizing outcomes and avoiding unnecessary toxicity. Here we describe the results of incorporating a promising antibody-drug conjugate, datopotamab-deruxtecan (Dato-DXd) in combination with programmed cell death-ligand 1 inhibitor, durvalumab, as the first sequence of therapy in the I-SPY2.2 phase 2 neoadjuvant sequential multiple assignment randomization trial for high-risk stage 2/3 breast cancer. The trial includes three blocks of treatment, with initial randomization to different experimental agent(s) (block A), followed by a taxane-based regimen tailored to tumor subtype (block B), followed by doxorubicin-cyclophosphamide (block C). Subtype-specific algorithms based on magnetic resonance imaging volume change and core biopsy guide treatment redirection after each block, including the option of early surgical resection in patients predicted to have a high likelihood of pathologic complete response, which is the primary endpoint assessed when resection occurs. There are two primary efficacy analyses: after block A and across all blocks for six prespecified HER2-negative subtypes (defined by hormone receptor status and/or response-predictive subtypes). In total, 106 patients were treated with Dato-DXd/durvalumab in block A. In the immune-positive subtype, Dato-DXd/durvalumab exceeded the prespecified threshold for success (graduated) after block A; and across all blocks, pathologic complete response rates were equivalent to the rate expected for the standard of care (79%), but 54% achieved that result after Dato-DXd/durvalumab alone (block A) and 92% without doxorubicin-cyclophosphamide (after blocks A + B). The treatment strategy across all blocks graduated in the hormone-negative/immune-negative subtype. No new toxicities were observed. Stomatitis was the most common side effect in block A. No patients receiving block A treatment alone had adrenal insufficiency. Dato-DXd/durvalumab is a promising therapy combination that can eliminate standard chemotherapy in many patients, particularly the immune-positive subtype. ClinicalTrials.gov registration: NCT01042379.
INTRODUCTION
Breast cancer is the most common cancer in women and the second leading cause of death from cancer both in the US and worldwide.1,2 Despite recent advances in therapy, there is a need to optimise treatment strategies that not only maximise survival, but also reduce long-term treatment-related toxicities. This is particularly true for aggressive breast cancer subtypes in which many patients are cured of their disease, but at the expense of long-lasting, serious adverse effects, including peripheral neuropathy and adrenal insufficiency. And patients receiving anthracycline-based chemotherapy, have small but real risk of death from therapy related leukaemia, as well as long term cardiac toxicity.3
The I-SPY trial consortium has, for over a decade, worked to accelerate the development of new therapeutics for early breast cancer, focusing on rapid identification of biologically targeted regimens most likely to be successful in phase III trials. The I-SPY2 trial was among the first and is now widely considered the archetype multicentre adaptive platform trial.4,5 I-SPY2 focused on patients with stage II and III molecularly high-risk breast cancer, with the early endpoint of pathologic complete response (pCR), a strong correlate of event free survival that predicts good long-term outcomes.6,7 The trial evaluated multiple agents in parallel, using an adaptive randomisation algorithm that preferentially assigned patients to study arms that exhibited efficacy in the same breast cancer subtype. After continuously enrolling for 14 years and evaluating 23 agents or combinations, rates of pCR in some breast cancer subtypes using standard of care continued to improve such that over 70% of patients will achieve pCR. This has the unintended consequence of serving as a deterrent to patient enrolment in a clinical trial evaluating unproven, experimental therapies. However, shifting the goals towards identifying agents with similar effectiveness, but with reduced toxicities, provides tremendous value to, and gains renewed interest by patients and their providers. The new iteration of the trial, I-SPY2.2, was designed to address these issues.
I-SPY2.2 retains the approach of its predecessor, as a signal-finding trial focused on identifying biologically targeted agents/regimens with strong efficacy signals that indicate a high probability of success in phase III trials. The trial utilizes a Sequential Multiple Assignment Randomised Trial (SMART) design8,9 to permit multiple serial randomisations/assignments based upon a patient’s treatment response combined with a platform approach. This hybrid design retains our ability to rapidly assess the efficacy of multiple agents in parallel, while offering participants the reassurance that should they have poor response to an experimental therapy, they can switch to a treatment optimised for their specific tumour subtype based on both the standard of care and I-SPY2’s experience over 2118 patients. Additional design features also provide opportunities to minimise exposure to ineffective or redundant therapy once patients are predicted to have reached the trial endpoint of predicted complete pathologic response (pCR) as a means of minimising toxicity. Importantly, the SMART design provides the statistical framework that allows not only evaluation of the effectiveness of the individual therapy, but also the efficacy of strategies comprising multiple sequentially applied treatments.
I-SPY2.2’s SMART design consists of three serial, neoadjuvant treatment ‘blocks’ (Figure 1A). In Block A, participants are randomised to receive one of several experimental regimens (without paclitaxel) -- novel combinations that have promising preclinical and clinical data, but are currently untested in the curative setting. Block B consists of treatment with an ‘optimised’ subtype-specific treatment. Block C consists of rescue therapy, of anthracycline chemotherapy at a minimum. The clinical goal is to get each patient to pathologic complete response with the minimum required amount of therapy, to limit exposure to potential toxicities.
Figure 1. Study design and CONSORT diagram.

(A) I-SPY2.2 study schematic. Participants receive up to three round (or ‘blocks’) of treatment, depending upon response assessed by MRI. Block A is a platform trial design that randomises participants to one of several arms testing experimental agents or combinations. Patients not responding to treatment in Block A, receive paclitaxel-based standard of care therapy in Block B assigned (or randomised) based on their response-predictive subtype. Those not responding in Block B proceed to treatment in Block C and receive a minimum of AC chemotherapy. All patients complete treatment with definitive surgery. Patients who meet the threshold to have a high probability of RCB 2/3 disease at 6 weeks (based on MRI at 0,3,6 weeks in A or after 6 weeks in B) have the option of skipping the remained of treatment in that block (early treatment redirection to next block). (B) CONSORT diagram. Enrolment period was defined as date of first screening consent from arm to date of arm closure to randomisation (6/27/2022 – 9/1/2023). Numbers of patients that adhered to the protocol specified treatment recommendations for early de-escalation to surgery were also shown.
Beginning in Block A, response to therapy is assessed in real time every 3–6 weeks with breast MRI-using an imaging biomarker, functional tumour volume (FTV) to determine the rate of response to therapy.10 At the completion of the full course of therapy (typically 12 weeks), a tumour bed and/or lymph node biopsy is performed to evaluate for residual cancer cells. An algorithm, ‘preRCB,’ uses MRI FTV and tumour biopsy information to predict the likelihood of pCR.10,11 Patients with sufficiently high likelihood are given the option to forgo additional neoadjuvant therapy and undergo surgical resection, forgoing remaining treatment blocks. If either tumour bed or lymph node (in those with positive nodes at presentation) biopsies are positive for residual cancer, or pre-RCB is below a predefined threshold, the patient is recommended to continue on trial and proceed to Block B. This process is repeated following treatment in Block B, resulting in some patients going to surgery after treatment in Block B and others proceeding to Block C.
Importantly, as an additional means of minimising patient exposure to unnecessary therapy, Blocks A and B each utilize FTV from serial MRI to assess for response at the mid-point of treatment.12 Participants meeting prespecified thresholds in ΔFTV (that predict a >92% chance of high residual disease burden by the end of the treatment) are recommended to forgo the remainder of the current therapy and proceed to the next treatment block.
Breast cancer subtypes in I-SPY2.2 are defined using response predictive subtypes (RPS), a classification system developed from I-SPY2 that refines breast cancer subtypes according to their response to available classes of treatments.13 RPS incorporate a tumour’s immune, DNA repair, and HER2/Luminal phenotype, improving the prediction of response and/or resistance to a variety of different targeted treatment options. MammaPrint® and Blueprint® assessments are used to select for eligible molecularly high-risk patients14–16 and transcriptomic assays are used to determine each patient’s RPS at screening.
Here, we report on one of the first Block A arms evaluated in I-SPY2.2, using a combination of anti-PD-L1 immune checkpoint inhibitor durvalumab and the antibody drug conjugate (ADC) datopotamab deruxtecan (Dato-DXd). Durvalumab is a human IgG1κ mAb that blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1) and is engineered to reduce antibody-dependent cellular toxicity. Durvalumab is FDA-approved in solid tumour types and has been previously evaluated in breast cancer, including in the I-SPY2 trial.17–19 Dato-DXd is an ADC comprised of a recombinant humanized anti-trophoblast cell surface protein 2 (TROP2) immunoglobulin 1 (IgG1) monoclonal antibody, MAAP-9001a, covalently conjugated to a drug-linker, MAAA-1162a via thioether bonds.20 The released drug, MAAA-1181a, inhibits deoxyribonucleic acid (DNA) topoisomerase I and leads to apoptosis of target cells. Data from the Phase I TROPION-PanTumor01 trial encompassing all solid tumours, showed promising results in a subset of 85 patients with heavily pretreated metastatic breast cancer, particularly those with HR+/HER2− BC and TNBC.21
Both preclinical and clinical data suggest that topoisomerase I inhibitors can enhance the antitumour efficacy of immunotherapy.22,23 In the phase IB/II BEGONIA trial for first-line treatment of metastatic triple-negative breast cancer, the combination of durvalumab and Dato-DXd demonstrated an objective overall response rate of 79%, irrespective on PD-L1 expression.24 In the early-stage breast cancer setting, the TROPION-Breast03 is ongoing and evaluating the combination of Dato-DXd with or without durvalumab in patients with Stage I-III triple-negative breast cancer and residual disease after completion of standard of care neoadjuvant treatment.25
Here, we report final results of Dato-DXd combined with durvalumab in HER2-negative patients in the I-SPY2.2 trial, detailing the safety and efficacy of the combination alone (after Block A) and as part of a multi-stage treatment strategy (all subtype-specific treatment strategies spanning Blocks A through C).
RESULTS
Enrolment and Baseline Characteristics
Between September 2022 and August 2023,108 patients were randomised to I-SPY2.2’s Dato-DXd and durvalumab Block A arm. Enrolment in the arm was halted when accrual reached 100, and patients that had already signed consent were allowed to proceed to treatment. Patients who withdrew, did not go to surgery, or went off protocol therapy are assigned non-pCR, and reported as having exited the trial after the block of treatment they last received. Two patients declined treatment consent, resulting in 106 patients receiving the allocated therapy, forming the modified intent to treat (ITT) primary analysis population (Figure 1B). Baseline characteristics of the modified ITT population are shown in Table 1. The arm only included female participants; median age at screening was 50 (range 25–77). The diverse population included 59.4% White, 11.3% Asian, and 8.5% Black and 12.3% Hispanic/Latino.
Table 1:
Patient demographics in the datopotamab-durvalumab arm
| Overall (N=106) | |
|---|---|
| Age (Years) at Screening | |
| Mean (SD) | 50.5 (13.2) |
| Median [Min, Max] | 50.0 [25.0, 77.0] |
| Ethnicity | |
| Hispanic/Latino | 13 (12.3%) |
| Not Hispanic/Latino | 81 (76.4%) |
| Missing | 12 (11.3%) |
| Race | |
| Asian | 12 (11.3%) |
| Black | 9 (8.5%) |
| White | 63 (59.4%) |
| Missing | 22 (20.8%) |
| Subtype | |
| HER2-Immune-DRD+ | 11 (10.4%) |
| HER2-Immune+ | 47 (44.3%) |
| HR-HER2-Immune-DRD- | 23 (21.7%) |
| HR+HER2-Immune-DRD- | 25 (23.6%) |
| HR | |
| Positive | 42 (39.6%) |
| Negative | 64 (60.4%) |
Of the 106 patients initiating treatment in Block A, 42 proceeded to surgery, while 64 continued to treatment in Block B. Of those 64, 39 patients proceeded to surgery and 25 continued to Block C. Across the complete dynamic treatment strategy (all individuals, e.g. Block A +/− Block B +/− Block C), a total of 53 pCRs (50%) were observed across the four RPS subtypes. When separated by timing of when pCR occurred, 25/53 (47.1% of all pCRs), 22/53 (41.5%), and 6/53 (11.3%) were achieved after Blocks A, B, and C, respectively (Supplementary Figure 1). Fifteen patients switched treatment early from Block A to B; and 4 patients switched early from Blocks B to C.
All patients enrolled on the arm had HER2− breast cancer and 64 (60.4%) had hormone receptor negative breast cancer (HR−). Figure 2A shows how hormone receptor (HR) status maps to RPS classifications used in I-SPY2.2. The most common subtype was Immune+, accounting for 47 (44.3%) participants; approximately one-third of HR-positive patients are classified as RPS Immune+.
Figure 2. Efficacy of Dato-DXd + durvalumab treatment.
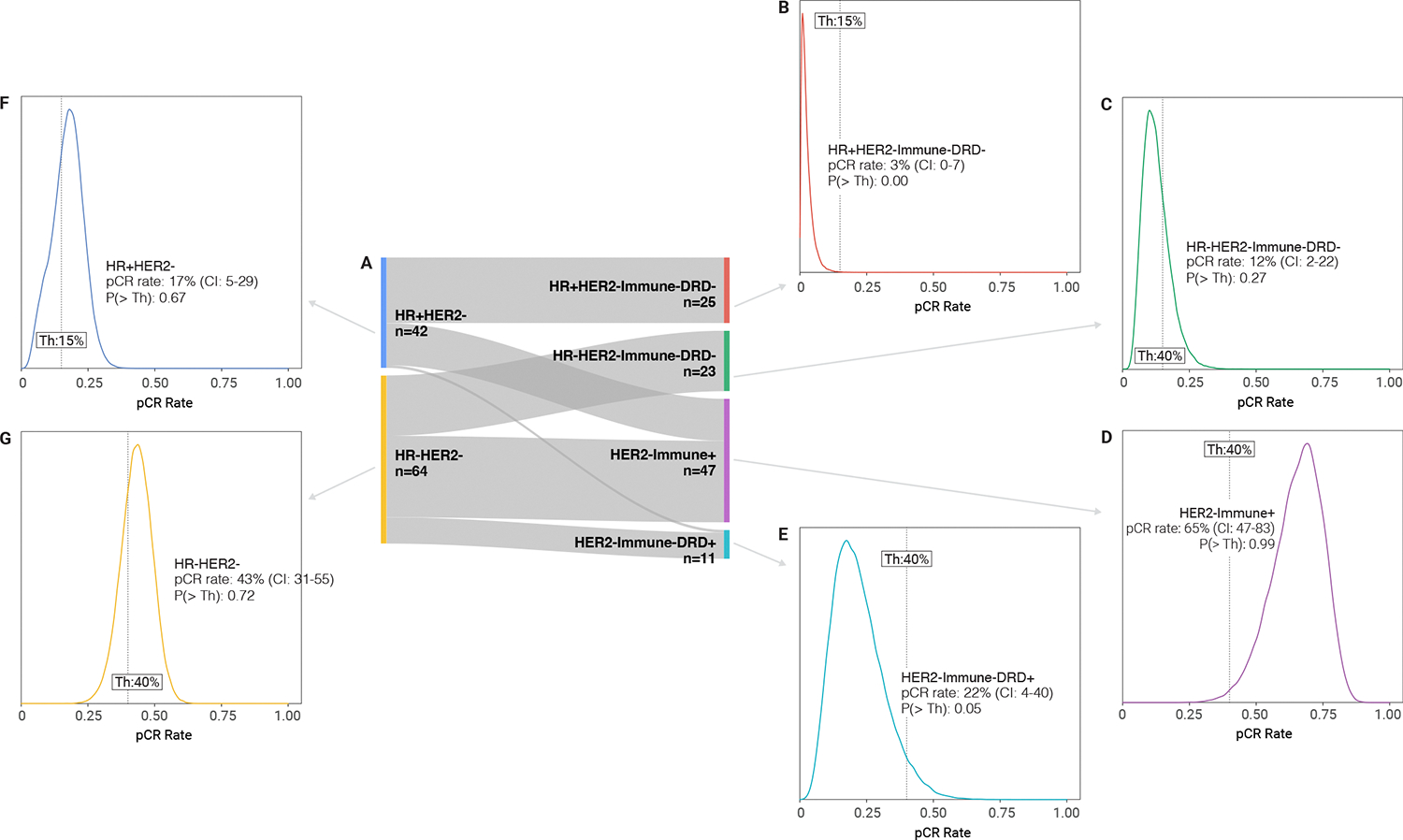
(A) Sankey diagram illustrating how the response-predictive subtypes (RPS) relate to standard HR/HER2 subtypes in the trial, with number of patients of each subtype; (B-G) Posterior probability distributions of pCR rate following treatment in Block A with Dato-DXd + durvalumab in each of the four RPS subtypes and HER2− subtypes. Shown is the mean pCR rate with 95% confidence interval (CI), along with the probability [P(> Th)] that the agent is greater than a preset pCR rate threshold set for a specific subtype. If the PTH for the agent is greater than 85% for the threshold (dotted vertical line), it is said to ‘graduate.’
Dato-DXd and durvalumab efficacy (Block A)
Because I-SPY2.2 is a signal-finding trial, pCR rates after Block A are compared to predefined, subtype-specific thresholds (shown in the vertical line in the probability distribution plots in Figure 2). Agents are said to ‘graduate’ (i.e. recommended for phase II evaluation) if the probability that the pCR rate exceeds the subtype specific threshold [P(>Th)] is >85%. For HR+HER2−Immune−DRD− and HR−HER2−Immune−DRD− subtypes, where the expected pCR rates with standard-of-care are 8–20%, the ‘graduation’ threshold for Block A alone was set at 15%; HER2−Immune+ and HER2−Immune−DRD+ subtypes that expected pCR rates of 40–80%), the threshold was set at 40%.
The Block A combination of Dato-DXd and durvalumab graduated only in the HER2−Immune+ subtype, with an estimated pCR rate of 65% (CI: 47%–83%) and P(>Th)>0.99 probability of being superior to the pCR rate threshold of 40% (Figure 2 and Supplementary Table 1). This modelled rate is based on pCR and MRI data. Specifically, for patients who did not go to surgery after Block A and achieved pCR at later blocks, MRI data is used to impute the probability of pCR at Block A and inform the estimated pCR rate. There were 20 pCRs observed in HER-Immune+ participants of 47 receiving Block A treatment. The combination did not graduate in any of the other RPS, or in HR+HER2− or HR−HER2− subtypes.
Efficacy of the treatment strategy (Blocks A through C)
Patients that proceeded to Block B were assigned RPS-directed therapy with taxane-based regimen; in Block C, treatment was a minimum of anthracycline chemotherapy. This led to five individual treatment strategies specific to each RPS (in Supplementary Table 2). The randomisation in Block B for the DRD+ subtype was to carboplatin plus a taxane plus or minus pembrolizumab. The subtype was small and several patients without pembrolizumab received it off study protocol (this strategy not shown).
As a comparator to assess the relative efficacy of the sequences of treatments making up the subtype-specific treatment strategies, we constructed a series of subtype-specific dynamic controls modeled from data of patients in I-SPY2. Precise details of their construction are provided in the methods. Briefly, we estimated pCR rates of candidate comparators selected from previously tested I-SPY2 arms containing elements of current standard-of-care, including the veliparib + carboplatin arm,26 the pembrolizumab arm,27 and the historical control of paclitaxel followed by AC, using a Bayesian covariate adjusted model. We then combined weighted posterior distributions of the candidates into a single posterior distribution. The resulting probability distributions serve as ‘dynamic controls’ against which the various treatment strategies are compared (filled grey distributions, Figure 3). Modelled pCR rates for treatment strategies are estimated using a Bayesian model informed by the timing of pCR assessment and pCR status. Modelled treatment strategies are said to graduate if the probability they are superior to the dynamic control P(>DC) is >85%. Figure 3 shows resulting probability distributions following treatment strategies for each RPS subtype, and the HR−HER2− population.
Figure 3. Efficacy of treatment strategies spanning Blocks A-C.
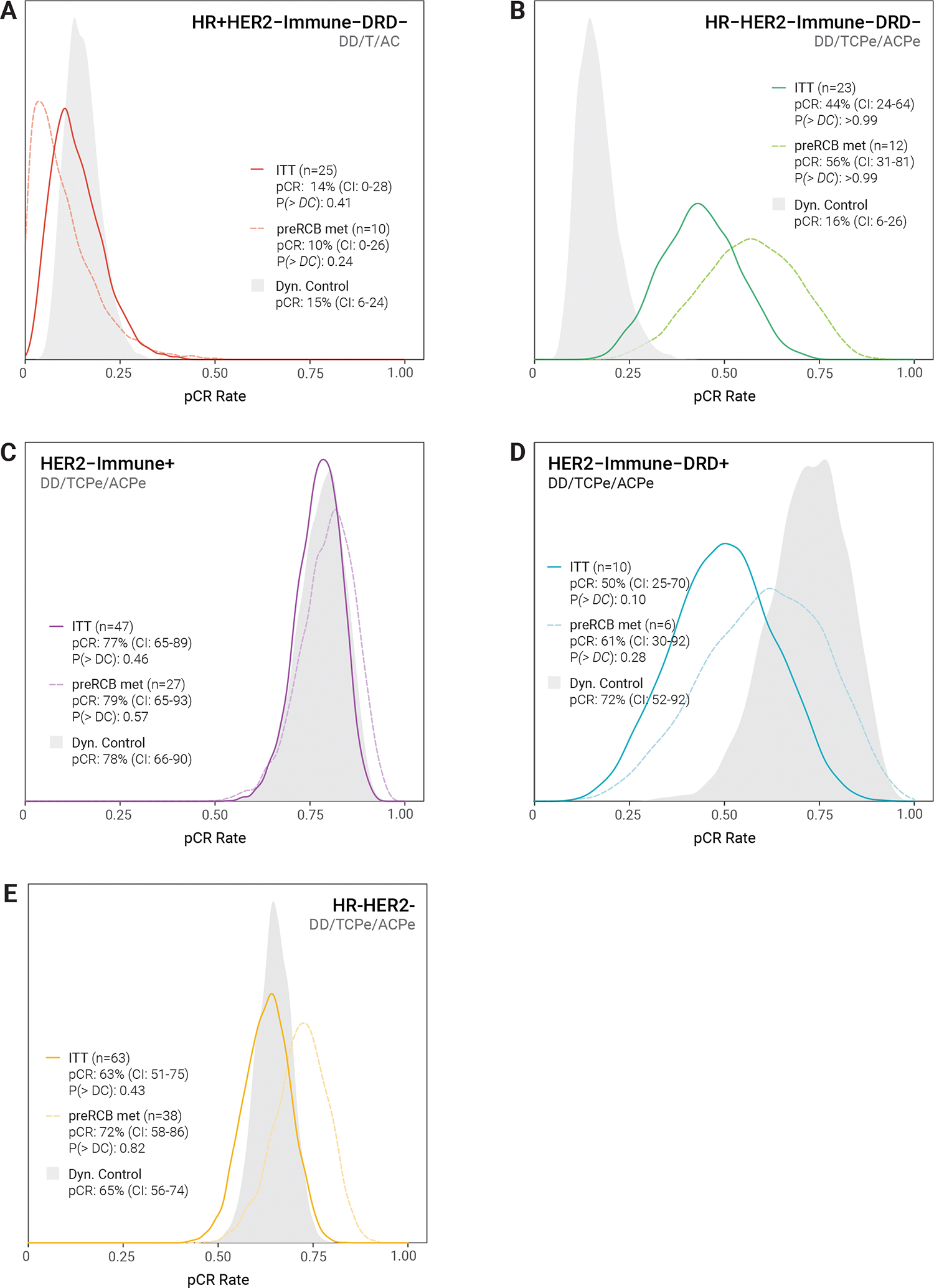
Posterior pCR rate distributions are shown for each RPS subtype (and HR−HER2−) and associated treatment strategy (DD = Dato-DXd + durvalumab; T=paclitaxel; C=carboplatin; Pe=pembrolizumab; AC=doxorubicin/cyclophosphamide) along with the mean pCR rate and 95% confidence interval (CI).The intent-to-treat population is the solid line. The dashed line represents a sensitivity analysis that considers only patients who adhered to preRCB recommendations following Block A and B treatment to either proceed to surgery. P(>DC) is the probability that the pCR rate of the treatment strategy is superior to a subtype-specific dynamic control (filled grey distribution) that was predefined using cumulative data from I-SPY2 reflecting current best-in-subtype treatment. The threshold for graduation of a treatment strategy is P(> DC) ≥ 0.85.
Participants with the Immune+ subtype had the highest pCR rate, with 37 of 47 patients (79%) reaching pCR. However, there was no statistically significant difference [P(>DC) 0.46] between the modelled pCR rate for the treatment strategy (77%, CI: 65–89%) and the dynamic control (78%, CI: 66–90%). The majority of the pCRs in this RPS were in Block A alone: 20 of the 37 Immune+ pCRs (54.1%) in the treatment strategy were achieved at the end of Block A (Figure 4 and Supplementary Table 4). Fourteen (37.8%) of the pCRs were achieved after Block B, resulting in 91.9% of the pCRs being achieved after Blocks A+B. Three additional patients (8.1%) achieved a pCR after Block C. Thus, although the pCR rates for the entire treatment strategy was similar to the dynamic control, almost all Immune+ patients achieved pCR without AC, which patients received as part of the dynamic control.
Figure 4. Treatment blocks in which pCRs occurred in the HER2−Immune+ subtype.

Of the 47 HER2−Immune+ patients receiving Dato-DXd + durvalumab in Block A, 20 proceeded directly to surgery, forgoing subsequent treatment; all of these patients had pathology-confirmed pCR (RCB 0). 54% of all cases achieving pCR in this subtype did so after the course of the Dato-DXd + durvalumab combination alone. Of the 27 patients receiving to Block B’s paclitaxel-based treatment, 14 of 19 who proceeded to surgery following treatment had pathology-confirmed pCR; 92% of all pCRs recorded in this subtype were achieved without the use of AC chemotherapy. Of the remaining 8 patients who received AC chemotherapy in Block C, 3 additional pCRs were recorded.
In HR−Immune−DRD− patients, 9 of 23 (39%) achieved pCR, with a modelled rate of 44% (CI: 24–64%). The treatment strategy outperformed the corresponding dynamic control’s mean pCR rate of 16% [CI: 6–26%, P(>DC) >0.99]. 2 of 9 (22%) of HER2−HR−Immune−DRD− pCRs occurred after Block A alone, 6 of 9 (66.6%) pCRs were achieved after Block B (one additional pCR after Block C).
Rates of pCR were lower in HR+Immune-DRD− (2/25, 8%) and Immune-DRD+ (5/10, 50%) subtypes, where modelled pCR rates did not outperform the dynamic control (Figure 3, Supplementary Table 3). Note that several HR+Immune-DRD+ patients randomised to receive TC without Pe in Block B received Pe off study protocol and were assigned non-pCR as having exited the trial after Block A (i.e. their last on-study treatment) for analysis. This resulted in only 4 Immune-DRD+ patients entering Block B, with 3 randomised to TCPe and 1 to TC in Block B.
Among all HR−/HER2− tumours (triple-negative), 39 of 63 patients (62%) reached pCR, with modelled pCR of 63% (CI: 51–75%) failing to outperform the dynamic control pCR rate of 65% (CI: 56–74%) [(P>DC)=0.42]. Notably, 21 of the 39 (54%) HR−HER2− achieved a pCR with Block A alone. Fifteen (38.4%) of the pCRs were achieved after Block B, resulting in 92.3% of the pCRs being achieved after Blocks A+B. Three additional patients (7.7%) achieved a pCR after Block C.
Additional information can be gleaned from looking at residual cancer burden (RCB) scores derived from surgical specimens,28 a secondary endpoint of I-SPY2.2 (Supplementary Figure 2). Three patients in the HER2−Immune+ subtype had RCB-1, indicating minimal residual disease after treatment completion. An additional 25% of patients with HER2−HR+ (luminal B) disease had RCB-1, for a total of 35% achieving complete or near pCR. Five patients in the HR−/HER2−group achieved RCB-1. In the HR−Immune−DRD− population, two patients achieved RCB-1. The planned secondary endpoint of event-free survival is not reported in this manuscript
Sensitivity Analysis of Efficacy
Although the I-SPY2.2 protocol recommends patients proceed to either surgery or the next treatment block based on pre-RCB assessment, the final decision was at the discretion of the treating physician. At the completion of Block A, 42 of 106 patients went to surgery and forwent additional treatment, despite the fact that only 21of the 42 met pre-RCB criteria (4 who met criteria proceeded to Block B - Figure 1B). Similarly, of the 64 patients treated in Block B, only 19 of the 39 who proceeded to surgery following treatment met the predefined pre-RCB criteria. Twenty-five patients advanced to Block C, nineteen of whom did not meet pre-RCB criteria for early surgery at the end of Block A or Block B (Figure 1B). Given that a significant number of patients prematurely ended treatment against the protocol-recommended pre-RCB criteria, we performed a sensitivity analysis for efficacy including only those individuals who adhered to the protocol’s pre-RCB guidelines.
As shown in Figure 3 (dotted lines), adherence to the pre-RCB recommendations generally resulted in modelled pCR rates exceeding those from the ITT analysis, except in the HR+Immune-DRD− population that received Dato-DXd/paclitaxel/AC. Adherence to pre-RCB protocol recommendations did not result in differences that were large enough to change the graduation status of the treatment strategy in any of the subtypes, however, the improvements in pCR rates in the HR−HER2−Immune−DRD− (44 to 56%) separated the probability distribution from the dynamic control even further. The shift in the HER2−Immune-DRD+ (50–61%) distribution resulted in performance that was not statistically different from the dynamic control.
Safety and toxicity
The incidence of adverse events (AE) that occurred in at least 20% of patients in Block A and then at any point during the treatment strategy are shown in Figure 5. The most prominent adverse events observed in patients treated in Block A were nausea (83.9%), stomatitis (77.3%), fatigue (75.5%), rash (64.2%), constipation (62.2%), alopecia (62.2%), and ocular issues (43.4%). These AEs were predominantly at grade 1 or 2 severity. Notably, five patients experienced grade 3 stomatitis (4.7%) and one was diagnosed with persistent grade 4 stomatitis (1.0%), with the majority of these events persisting at the time of surgery. During Block A, 2 patients (1.9%) discontinued Dato-DXd and 5 patients (4.7%) discontinued durvalumab due to due adverse events (Supplementary Table 5).
Figure 5. Incidence of most frequently observed adverse events.
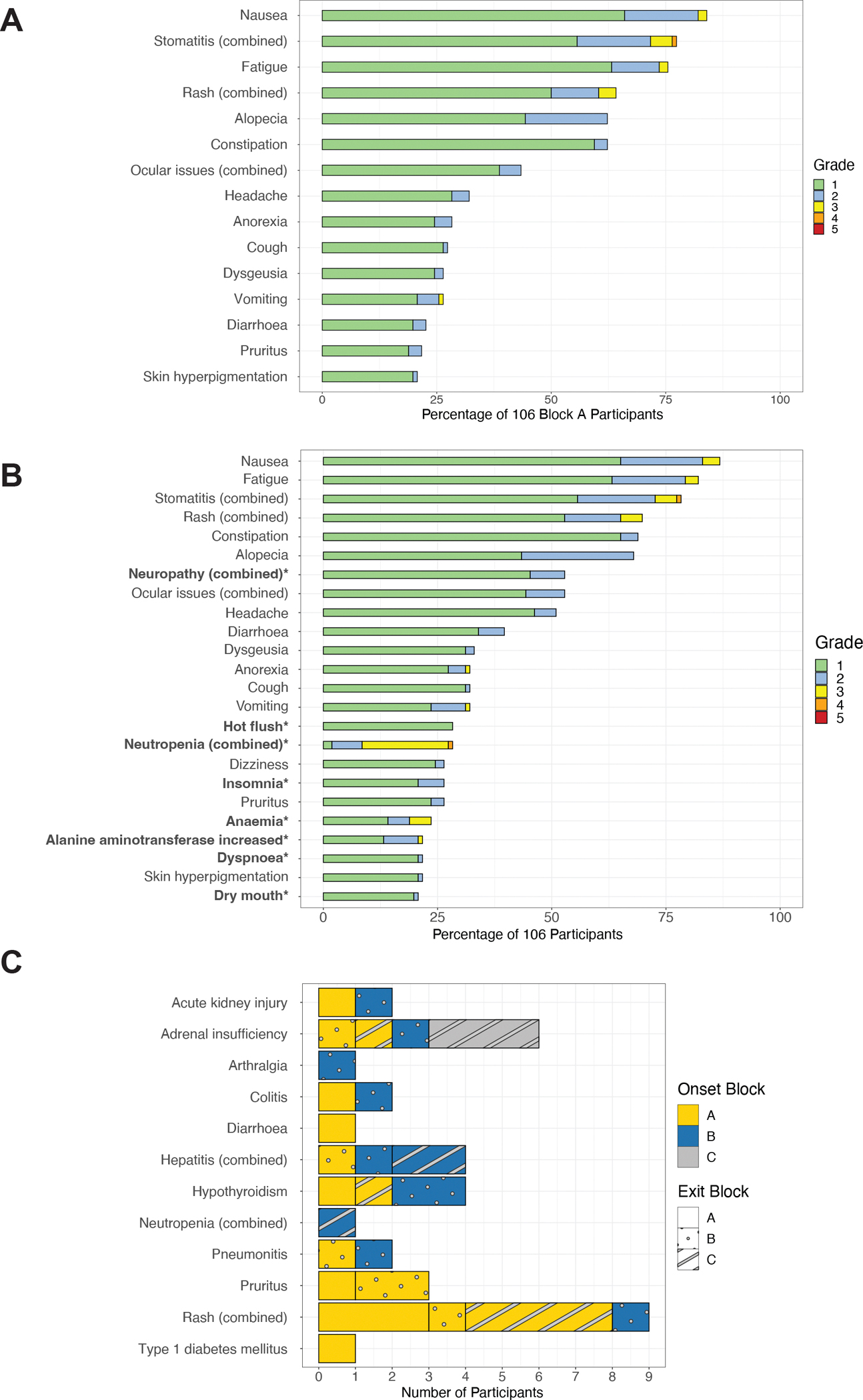
(A) Incidence of AE of any grade that were experienced by greater than 20% of participants during Block A treatment with Dato-DXd + durvalumab (n=106); (B) Incidence of AE of any grade that were experienced by greater than 20% of patients during any block of treatment (n=106); Note that all grade 3/4 adverse events observed in the study are represented here. The adverse events that were associated only with Blocks B and C are bolded. (C) Immune-related adverse events experienced by participants in any treatment block. Adrenal insufficiency (AdI) events were reported (12) and adjudicated by 2 expert endocrinologists as described in the methods and 7 (6.6%) were determined to be adrenal insufficiency, irrespective of subtype of block of onset. The events are annotated by the block of onset for each irAE event (color) and the Block at which each patient with an irAE exited their neoadjuvant treatment (fill pattern). Note that 48 patients in total received pembrolizumab + paclitaxel + carboplatin in Block B; 16 received AC + pembrolizumab in Block C.
AEs listed as “combined” refer to iMedDRA combined terms as follows: Stomatitis (Combined): stomatitis, oropharyngeal pain, mouth ulceration, mouth injury, oral pain, gingival pain; Rash (Combined): rash, rash maculo-papular, dermatitis, rash pustular, skin disorder, dermatitis acneiform, rash pruritic, rash erythematous, eczema, urticaria, and rash macular; Ocular events (Combined): keratitis, eye pain, vision blurred, eye irritation, photophobia, eye pruritus, dry eye, photopsia, conjunctivitis, eye infection, eyelid irritation, corneal ulcer, uveitis, corneal opacity, eye disorder; Neuropathy (Combined): peripheral sensory neuropathy, peripheral motor neuropathy, neuropathy peripheral; Neutropenia (combined): febrile neutropenia, neutropenia, and neutrophil count decreased.
Across the entire treatment strategy, additional adverse events emerged during Blocks B and C, shown in bold, including neuropathy (45.2% grade 1 and 7.5% grade 2), hot flushes (28.3% grade 1), neutropenia (1.9% grade 1, 6.6% grade 2, 18.9% grade 3, and 0.9% grade 4), insomnia (20.8% grade 1 and 5.7% grade 2), and anaemia (14.1% grade 1, 4.7% grade 2, and 4.7% grade 3).
We examined immune-related adverse events (irAEs), based on the block of onset and the block where the patient exited treatment (Figure 5C). The most common reported irAE was rash (N=9), of which nearly all started in Block A. A total of 12 adrenal insufficiency events were reported. After adjudication by two endocrinologists, 6 were determined to meet criteria for Adrenal insufficiency. Only two of these events began in Block A. Notably, no patients who went to the OR early after Block A experienced an adrenal insufficiency event.
There were 3 cases of pneumonitis. One grade 2 event occurred after one dose of Dato+durva. The patient stopped Block A treatment and went on to receive Block B without pembrolizumab. One grade 2 event occurred in Block B. This patient had discontinued pembrolizumab and carboplatin before ILD onset due to acute kidney injury. There was one grade 1 event during Block B that had no impact on study therapy. .
DISCUSSION
The results of the Dato-DXd and durvalumab arm of I-SPY2.2 demonstrate that improved tumour classification and real-time MRI-based assessment of response can improve the ability to determine who is most likely to benefit from new targeted therapies. Here, patients are profiled using RPS at screening, in contrast to using standard oestrogen and progesterone receptor, and HER2. MRI and biopsy data permits assessment of on-treatment response, identifying patients who may reach pCR without the need to endure a longer course and/or the additional toxicities associated with taxanes and anthracyclines.
The Immune+ subtype, comprising 47/106 (44%) patients treated, exceeded the predefined threshold for graduation in Block A. Immune+ patients had excellent overall modelled rates of pCR (77%) for the treatment strategy of DatoDxD+durvalumab/paclitaxel+carboplatin+pembrolizumab/AC+pembrolizumab (DD/TCPe/ACPe). Importantly, of the 37 Immune+ pCRs, 54%, occurred after Block A and 92% after Blocks A and B, meaning that the vast majority of Immune+ patients achieved pCR without receiving anthracycline chemotherapy, instead of the standard of care treatments given in the Keynote 522 regimen for TNBC29 or ddAC-T regimen for HR+/HER2− breast cancer.
In addition, using RPS, we identified immune-enriched/responsive HR+ patients who would not have otherwise been eligible for treatment with immunotherapy if classified by the standard HR/HER2 subtypes that restrict the use of immune checkpoint inhibitors to TNBC only. Using RPS, 38% of all HR+ patients were Immune+ by RPS. With the results of the KN-756 trial30 and the ongoing SWOG S2206 trial (NCT06058377), there is increasing interest in selecting the patients with HR+HER2− early-stage breast cancer who may benefit from the addition of immunotherapy to neoadjuvant treatment.
The treatment strategy graduated in the HR−HER2−Immune−DRD− subtype, with 9 of 23 (39%) achieving pCR, with probability of exceeding the 16% mean pCR rate of the dynamic control >0.99. While the number of patients in this subgroup was small, this is may be an important finding, as historically this subgroup contains some of the hardest to treat breast cancers with the worst rates of pCR, as they tend to be “cold” tumours with immune-desert tumour microenvironments and overall poor survival outcomes. There was not enough evidence after Block A alone; however, the covariate model used in the trial tend to underestimate pCR rates in small subtypes with poor expected response. After the entire strategy, the HR−HER2−Immune−DRD− subtype clearly graduated, outperforming the dynamic control. Two of the pCRs occurred after Block A alone, and 8/9 (89%) occurred after Block B. There were 2 patients (20%) who had minimal residual disease (RCB-1). Dato-DXd is highly active in this subtype, and these results warrant further investigation, with either Dato alone or the combination with Durva, perhaps using additional cycles of therapy. A neoadjuvant study with 8 cycles is currently ongoing.
The HR−Immune−DRD− subtype and 31/47 (66%) of the Immune+ subtype fall under the larger group of HR−HER2−, traditionally reported as triple-negative breast cancer, which also demonstrated high rates of pCR with the Dato-DXd and durvalumab treatment strategy: 62%, which improved to 72% in the sensitivity analysis, with an 82% chance of being better than the dynamic control.
The most prominent adverse events due to the Dato-DXd and durvalumab combination were nausea (83.9%), stomatitis (77.3%), fatigue (75.5%), and rash (64.2%). Events were predominantly grade 1 and resolved after cessation of the drug. Of note, there were 5 cases of grade 3 stomatitis (4.7%) and 1 case of grade 4 stomatitis (1.0%), despite the use of dexamethasone mouthwash (provided to and recommended for use 4x daily by participants). Additional treatment-related toxicities appeared in patients treated in Blocks B and C, including neuropathy and cytopenia, the former of which were very low in response to Dato-DXd and durvalumab, but occurred in over half of patients following paclitaxel administration. Peripheral neuropathy is a hallmark of taxanes, occurring at higher rates in Black/African American women, with no effective prevention or treatment available other than dose modification. This highlights the value of evaluating experimental neoadjuvant treatments alone rather than on a background of taxanes, if we are to identify effective agents with reduced side-effect burdens that patients fervently desire.
Rash (n=9) was the most common immune-related adverse event (irAE) observed. IrAEs are of particular interest in patients with early-stage breast cancer -- while many require only minimal intervention and resolve with discontinuation of treatment, more serious irAEs, such as pneumonitis, adrenal insufficiency, and type 1 diabetes, can have serious long-term consequences. The onset of irAEs can vary significantly, most commonly developing within 3 months of starting treatment, but can also present after treatment discontinuation.31 Adrenal insufficiency (as adjudicated by endocrinologists) was experienced by 6 participants to date (irAEs are assessed as part of ongoing follow-up). Minimising the number of doses of immunotherapy patients receive may serve to reduce the risk of serious irAEs, as is the approach taken in I-SPY2.2 trial. The strategy of de-escalating immunotherapy is also being evaluated in the ongoing OptimICE-PCR trial (NCT05812807), which seeks to determine if individuals with pCR after standard treatment for early-stage triple-negative breast cancer benefit from additional adjuvant pembrolizumab. Importantly, only two experienced adrenal insufficiency with onset at Block A, and none who exited after Block A had such an event.
The I-SPY2.2 design enables a personalized approach to therapy and anticipates the way in which we would ideally treat patients clinically. All patients do not respond the same, but in most adjuvant and neoadjuvant trials, all patients receive the same amount of therapy regardless of response. In this design, we use the predicted achievement of pCR as a way to lower the amount of therapy. Dato-DXd plus durvalumab and Dato-DXd alone were the first two arms in I-SPY2.2, and not all investigators followed treatment redirection recommendations based on the algorithm used in the trial. It is a limitation of the current study that 41 patients out of 106 went to the operating room early, when the pre-RCB assessment had not predicted all of these patients to have a high likelihood of pCR. Only 4 patients underwent additional treatment when preRCB indicated pCR had been achieved. We are conducting a detailed analysis of the reasons for these trial deviations. Patient and investigators were anxious to show a benefit from shorter course of therapy, and there is an increasing desire to avoid anthracycline treatment on the part of patients and physicians. This was the first arm of the new trial design, and adjustments to the protocol and return of results have been made to ensure timely provision of data. Provider feedback and additional training and education for treating physicians appears to have reduced early redirection to surgery in patients that do not meet pre-RCB criteria. We are developing new patient materials to explain that the goal of the strategy is to get patients to pCR and to try to minimise toxicity when possible and appropriate.
Another important limitation this study is the lack of a concurrent control population. Going forward, we have modified the design to include a concurrently randomised control arm. We used RPS to assign therapy, as our data strongly suggest that this subtyping can substantially improve pCR rates. However, this limits the comparisons that can be made to outside trials where the RPS are not available. The graduation of the Immune−DRD− strategy when Dato-DXd is included could represent an under-representation of response in the dynamic control, which did not include a paclitaxel + carboplatin + pembrolizumab combination. Although the numbers are small, both Dato-DXd and Dato-DXd plus Durva regimens showed graduation in this subtype, with clear benefit during Block A alone. A final limitation is that the RPS classification leads to small numbers in some of the subtypes, and the design inherently results in reduced numbers of patients entering each subsequent treatment block. Given the small numbers in the Immune-DRD+ positive subtype, we are reconsidering how to better classify these patients to avoid very small subsets.
The Dato-DXd and durvalumab arm of I-SPY2.2 represents one of the first examples of I-SPY’s new SMART design that features a number tools developed over the past several years, including RPS, FTV response assessment, preRCB, and combined iMedDRA toxicity reporting, and collection of patient reported outcomes. The trial is ongoing - learning and improving based on each specific patient’s experience and treatment outcome. The results from the Dato-DXd and durvalumab arm warrant further investigation in the randomised controlled trial setting in the Immune+ and HR−Immune−DRD− subtypes, which appear to be the most likely to benefit from this combination. The 35% of luminal B patients with no or minimal residual disease may warrant investigation of a longer course of therapy.
The ability to tailor treatment to biology and the observed response represents an exciting advance in personalized therapy. We have demonstrated that it is possible to conduct a randomised trial that enables individualization of care within the trial. This is a patient-centric feature that makes patients want to participate in these studies. The lessons from this arm and every arm we evaluate continue to inform us and allow us to learn and improve the design.
METHODS
Study Design
I-SPY2.2, targeting early breast cancer, is a hybrid design consisting of a Sequential Multiple Assignment Randomised Trial (SMART)8 that integrates a phase II adaptive platform approach for early breast cancer at high risk of recurrence (clinicaltrials.gov identifier NCT01042379). The design enables the development of novel agents and combination treatments, while permitting treatment re-direction within the trial, based upon treatment response assessed by magnetic resonance imaging (MRI). As shown in Figure 1, the trial consists of three ‘Blocks’ of treatment:
Treatment Block A.
Treatment Block A uses the platform approach to evaluate multiple novel agents in parallel. The master protocol of I-SPY2.2 allows agents to leave or enter Block A investigation using protocol amendments. Block A agents are administered as single agents (or combinations) without paclitaxel. MRI is used to assess treatment response longitudinally by assessing change in functional tumour volume (FTV) from pre-treatment baseline and predict the probability of pCR.10 Following completion of a 12-week regimen, participants with MRI-predicted pCR probability above prespecified thresholds whose core biopsy at 12-weeks also show no residual cancer at the primary site, are offered the ability to proceed directly to surgery, forgoing treatment in the remaining two treatment Blocks. Participants not meeting the combined MRI and biopsy criteria then proceed to treatment Block B.
Treatment Block B.
Block B offers participants treatment with a standard of care regimen predicted to yield the highest rate of pathologic complete response (pCR) based on the response-predictive subtype (RPS) classification developed as part of I-SPY2.13 Six RPS phenotypes are used, based upon HR, HER2, immune-responsive, DNA repair deficiency and intrinsic luminal-ness biomarker signatures. In the current arm, only HER2-negative subtypes were eligible for randomisation: HR+Immune−DRD−, HR−Immune−DRD−, Immune+, Immune-DRD+. Regimens in Block B typically incorporate paclitaxel. As with Block A, participants whose MRI predicted pCR probability is above threshold after a 12-week course of treatment and who had no residual disease found in a post-treatment core biopsy may proceed directly to surgery, while those not meeting the MRI and biopsy combined criteria are offered rescue therapy in Block C.
Treatment Block C.
In Block C, participants are assigned to receive a minimum of chemotherapy with standard of care doxorubicin/cyclophosphamide over an 8 or 12 week treatment cycle.
Early treatment switching.,
To minimise toxicity of ineffective treatment, in Blocks A and B, participants with <30% FTV reduction from baseline at 3-weeks and subsequently show <65% FTV reduction from baseline at 6 weeks may forgo the remaining treatment cycles in that Block and proceed to the next treatment block.12 based on an algorithm that predicts a high chance (>90%) of having substantial residual (RCB2/3) disease if they remained on treatment.
Endpoint.
The primary endpoint of I-SPY2.2 is pCR, which is assessed using the residual cancer burden (RCB) method.28,32 Efficacy is evaluated within several clinical signatures, include traditional HR/HER2 subtypes as well as in each of the RPS classification for: each novel agent following completion of Block A treatment; and for each unique sequence of treatments received across Blocks A, B, and C.
Safety is assessed using the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) v5.0.33 This is augmented by patient-reported adverse events using the patient reported outcomes version of CTCAE (PRO-CTCAE),34 PROMIS35 quality of life questionnaire, and FACT-GP536 single item question about the impact of adverse events.
Secondary endpoints include RCB score and class32, 3- and 5-year event-free survival (EFS), and distant relapse-free survival (DRFS). All participants were followed for long-term outcomes.
Dynamic Control.
Because clinical standards of breast cancer care evolve over time, and the I-SPY trial is continuously open and enrolling, we realize our comparator (control) arm needs to update as well to appropriately reflect these ongoing clinical advances. To address this in I-SPY2.2, we utilize a ‘dynamic control’ in which we identified a set of candidate comparator arms for each subtype from previously tested I-SPY2 agents which include elements of newer clinical standards (e.g. checkpoint inhibitors, carboplatin, dual HER2-targeting in combination with paclitaxel followed by AC). Candidate comparators forming the dynamic control for each subtype are shown in Table S6.
Using a Bayesian covariate adjusted model (described in detail in the statistical analysis section) and data from 1818 I-SPY2 patients enrolled between March 2010 and April 2022 across 20 arms, we estimated the posterior distribution of pCR rates of each candidate comparator arm. The dynamic control is defined as the weighted posterior distribution of each candidate comparator arm, where the weight is the probability that it is the best arm among the candidate comparator arms within the dynamic control. This weighting forms a single posterior distribution for the dynamic control against which I-SPY2.2 treatment strategies are compared.
Randomisation.
When Dato-DXd + Durvalumab was open to enrollment, participants were randomised with equal probability to open arms at Block A for their subtype. The number of open Block A arms in each subtype varied over the course of Dato-DXd and durvalumab’s enrollment. Of note, the Dato-DXd monotherapy arm was also open for enrollment during the same period; randomisation to the combination vs. monotherapy arms was at equal probability. In Block B, participants with the Immune-DRD+ subtype were randomised 1:1 to receive one of two Block B treatment courses: paclitaxel plus carboplatin, or paclitaxel plus carboplatin and pembrolizumab. For all other subtypes, Block B treatment was assigned to a single available course of treatment for this block. Simple randomisation is performed using an R script implementing a pseudo-random number generator when each patient enters the trial, taking as input a random number seed, randomisation probabilities to each arm (for Block A or B) and subtype information. An initial random number seed was provided for the first draw; and the random number seed was updated following each subsequent randomisation and stored for the next input. The randomisation script is called upon completion of relevant data entry and case report forms into the OpenClinica EDC system and integrated with the EDC via a Java wrapper.
Efficacy Assessment.
Like I-SPY2, I-SPY2.2 is a signal-finding trial that aims to rapidly identify agents or treatment sequences that are likely to be successful in phase III trials in responsive subtypes. I-SPY2.2 assesses efficacy in two different ways: 1) Evaluating the efficacy of experimental agents/combinations immediately following completion of treatment in Block A; and 2) Evaluating the efficacy of total treatment administered to individuals, in the form of each unique treatment strategy that occurs across the different agents used in Blocks A through C.
In assessing efficacy of the experimental agent in Block A alone, agents deemed good candidates for further evaluation are said to ‘graduate’ if there is an 85% probability that their pCR rate exceeds a pre-determined fixed pCR rate threshold. These thresholds are selected to reflect a level of activity considered clinically meaningful for a novel regimen when given alone without standard chemo/targeted therapies. There are two thresholds, one for subtypes with high response rates to previously evaluated I-SPY2 agents, set at 40%, and one for subtypes with lower response rates, set at 15%.
In assessing efficacy across all blocks, graduation is determined by a comparison to a dynamic control based on the pCR rates expected if participants had been given standard of care or the previous best-in-class sequence from I-SPY2. An agent graduates if the probability that the estimated pCR rate of the sequence is superior to the dynamic control is greater than or equal to 85% in any given clinical signature (subtype).
Both efficacy analyses are modified intent-to-treat analyses where all participants receiving on-study therapy are considered evaluable. Participants who withdrew prior to surgery or receive non-protocol therapy prior to surgery are classified as non-pCR and their block of exit is classified as the last block of on-study therapy they received.
Efficacy assessment begins after 20 patients have been enrolled onto an arm; the pCR rates are monitored by the Data Safety Monitoring Board as enrollment in the arm accumulates. For agents open to HER2− (or HER2+) patients, a maximum sample size of 100 was preset based on simulations of the trial operating characteristics for the efficacy analysis of Block A alone. An arm exits the trial upon reaching maximum accrual. However, similar to I-SPY2, if the graduation threshold in the efficacy analysis across all blocks is reached within at least one subtype prior to maximum accrual, the arm is a candidate for exiting the trial early.
Eligibility
I-SPY2.2 is open to women and men aged ≥18 years with anatomic stage II or III breast cancer whose primary tumours are ≥2.5 cm by clinical exam or ≥2.0 cm by imaging. Participants must have an Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1.37 Those with MammaPrint® low-risk HR-positive, HER2-negative were excluded from participation due to their low risk of recurrence.15 Only HER2-negative participants were eligible for randomisation to the Dato-DXd and durvalumab arm. Refer to the study protocol for additional enrollment criteria. Participants did not receive compensation for trial participation.
Trial Oversight
I-SPY2.2 was designed and conducted by I-SPY Consortium members, patient advocates and investigators. AstraZeneca provided funds and study drugs (datopotamab deruxtecan and durvalumab), but played no role in the study design, collection/analysis of data, or in manuscript preparation. The study was approved by Wake Forest School of Medicine, who acted as the central IRB, and a Data Safety Monitoring Board met monthly to review patient safety and study progress. All patients provided written informed consent for screening and treatment. The I-SPY2.2 trial complies with all local and national regulations regarding the use of human study participants and was conducted in accordance to the criteria set by the Declaration of Helsinki. The authors of this manuscript vouch for the accuracy and completeness of the data reported.
Treatments
Block A treatment in the Dato-DXd and durvalumab (DD) arm consisted of intravenous 6 mg/kg Dato-DXd along with 1120mg intravenous durvalumab on day 1 of each 3-week cycle for up to 4 cycles.
Participants in Block B were prescribed one of three 12-week regimens depending on their RPS classification: paclitaxel (T) only [for HR+Immune−DRD−], paclitaxel (T) plus concurrent carboplatin(C) [for HER2−Immune−DRD+], or concurrent pembrolizumab (Pe), paclitaxel and carboplatin [for HR−HER2−Immune−DRD−, HER2−Immune+, HER2−Immune-DRD+]. Pembrolizumab was administered at 200mg intravenous every 3 weeks for 4 cycles. The target dose of intravenous carboplatin was AUC 1.5 weekly. Paclitaxel was administered intravenous 80mg/m2 weekly for 12 weeks. For the first paclitaxel infusion, 20 mg dexamethasone was administered; if no infusion reaction was noted, dexamethasone was reduced to 10 mg then discontinued if no further reactions. Institutions were allowed to individualize steroid dosing. Order of administration in Block B was pembrolizumab, then paclitaxel, and then carboplatin. All participants reaching Block C were prescribed four cycles of 60 mg/m2 intravenous doxorubicin (A) and 600 mg/m2 cyclophosphamide (C), administered every 2 or 3 weeks (dose dense or standard q3week AC). For certain subtypes, Block C therapy also included four cycles of 200mg iv pembrolizumab every 3 weeks over a total of 12 weeks.
This created the following treatment strategies dependent upon RPS subtype: DD/T/AC in HR+HER2−Immune−DRD−; DD/TCPe/ACPe in HR−HER2−Immune−DRD−; DD/TCPe/ACPe in HER−2Immune+; DD/TCPe/ACPe and DD/TC/ACPe in HER2−Immune−DRD+. The HER2−Immune−DRD+subtype was small. The efficacy of both strategies (DD/TCPe/ACPe and DD/TC/ACPe) are shown for completeness in supplemental table S3; but only the DD/TCPe/ACPe strategy are shown in Figure 3 and considered in additional analyses (e.g. timing of pCR and RCB class distribution). Of note, the above RPS-based treatment assignments result in 63/64 HR−HER2− patients (across RPS) receiving treatments consistent with strategy DD/TCPe/ACPe (enabling efficacy evaluation across all blocks for this subtype).
Dose reductions and toxicity management were specified in the protocol. Adverse events were collected according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 5.0.
Following completion of neoadjuvant therapy, definitive surgery by lumpectomy or mastectomy was performed at the discretion of the treating surgeon. Sentinel node dissection was allowed in node-negative participants, with axillary node dissection in node-positive patients according to NCCN and local practice guidelines.38 Adjuvant treatment was not mandated by the trial, and was left to the discretion of the treating oncologist.
Assessments
Specimens.
16-gauge core needle biopsies from the primary tumour were collected at screening, and at the completion of Block A and Block B (when applicable) and prepared according to standard I-SPY methods;19 surgical specimens were prepared in similar fashion. Whole blood for plasma and buffy coat aliquots were collected (when applicable) at screening (baseline), and 3, 6, and 12-weeks following start of treatment in each of Blocks A and B, and again prior to surgery. All unused portions of specimens are banked for future research.
Serial MRI.
MRI were performed at screening (baseline), after 3, 6, and 12-weeks of treatment in each of Blocks A and B (when applicable), and prior to surgery. Another MRI at the discretion of the treating physician could be scheduled at 6-weeks following start of Block C treatment. For each contrast-enhanced examination, FTV and change in FTV from baseline (ΔFTV) were calculated using previous published methods.10
MammaPrint.
MammaPrint® classification was used for eligibility assessment and in subtype classification. Using baseline specimens, participants were classified as MammaPrint (Agendia, Amsterdam, Netherlands) High risk (MP1) or MammaPrint ultra-high risk (MP2), using a pre-defined threshold applied to the 70-gene risk score evaluated on Agilent 44K arrays.14,15 The threshold used is equivalent to the median cut point of I-SPY1 participants (−0.154 in the original I-SPY1 dataset) who fit the eligibility criteria for I-SPY2.
Response-Predictive Subtype Assignment:
Response predictive subtypes are defined by combining clinical biomarkers: hormone receptor (HR) status and HER2 status and classifications from three expression-based signatures computed from Agilent arrays (Agendia, Amsterdam, Netherlands): BluePrint, ImPrint, and RePrint. Participants were classified using the 80-gene molecular subtyping test BluePrint into Luminal versus Basal or HER2 subtypes.13 The ImPrint signature was developed based on data from the I-SPY2 trial as a predictive biomarker of immunotherapy response as previously described13 and used to classify patients into Immune+ (i.e. predicted sensitive) versus Immune−(predicted insensitive) subsets. The 60-gene RePrint signature, also developed from I-SPY2 trial data to predict sensitivity to DNA repair deficiency (DRD) targeting agents, was used to classify patients into DRD+ (i.e. predicted sensitive) vs DRD− (i.e. predicted insensitive) subsets. These five biomarkers are combined to form six phenotypic response predictive subtypes (RPS) with differential responsiveness to previously evaluated I-SPY2 agents: HR+HER2−Immune−DRD−, HR−HER2−Immune−DRD−, HER2−Immune+, HER2−Immune−DRD+, HER2+non-Luminal, HER2+Luminal.
Residual Cancer Burden (RCB).
pCR, the primary endpoint was assessed on surgical specimens using the Residual Cancer Burden method, equivalent to RCB 0.32 All new sites go through a quality assurance process and review of 2 cases with the chairs of the pathology working group.
Adverse events – adrenal insufficiency adjudication.
Reported adverse events were adjudicated and validated by the I-SPY2 safety chairs (HSR, RN) in collaboration with QLH. irAEs were validated and adjudicated using published guidelines with confirmation of adrenal insufficiency by two independent endocrinologists.39,40
Return of Results
The central IRB chair set up a process whereby results of investigational diagnostics (conducted under an IDE) were able to be shared with patients. The IRB reviews and approves the proposed test results to share and the patient friendly report in which the information is returned. Return of results (ROR) are used for reporting information to patients that affect their care. This includes the RPS subtypes used for randomisation, the MRI functional tumour volumes (FTV) and the recommendations for continuing treatment or escalating to the next block of therapy, at 6 weeks, as well as the preRCB report and recommendation for whether they have met the threshold for having a high probability of having a compete response, triggering a recommendation to proceed to surgical resection, or if not meeting criteria, to continue on to the next block of therapy.
Statistical Analysis
Efficacy of the Dato-DXd and durvalumab arm was assessed within 6 pre-specified HER2− signatures defined by clinical HR/HER2 status and/or the response predictive subtypes (RPS).
Efficacy Assessment of Block A alone.
For each Block A agent, we leveraged existing I-SPY2 statistical methodologies to model the posterior distribution of pCR rates after Block A within each signature to compare it against a fixed pCR rate threshold as described below.
The Bayesian model used has two main components: (1) a core covariate-adjusted Bayesian logistic regression and (2) an MRI imputation model.
The Core Logistic Regression Model:
Probability distributions of pCR rates are estimated using a Bayesian covariate-adjusted logistic model with marker statuses (HR, HER2, Immune, BPLuminal, and DRD) as covariates for each eligible signature.
Let be the indicator for the pCR response of patient at Block A. Covariates represent the HR, HER2, Immune, DRD, and BluePrint statuses of patient (with 1 indicating positive and 0 negative for HR, HER2, Immune and DRD, and “HER2orBasal” and “Luminal” for BluePrint). Label the treatment arm assigned to patient . The full model is
The model’s components are as follows:
The terms capture the effect of being in a particular subgroup defined by (HR, HER2, Immune, DRD, BluePrint) status.
The terms are the treatment effects within each of the (HR, HER2, Immune, DRD, BluePrint) subgroups.
We set to ensure parameter identifiability.
The term represents the effect of being on a particular treatment arm for all patients.
In addition to the core logistic regression structure of the above terms we include imputation of the pCR status based on longitudinal MRI data over time. The MRI imputation component of the model is described below.
MRI Imputation Model:
If all the pCR data yi were available, then this is a standard Bayesian logistic regression model that may be fit using Markov Chain Monte Carlo (MCMC) methods. When pCR data for some patients are not available, we use imputation based on the patients for whom pCR status is known. Of note, patients who did not go to surgery at the end of Block A but had a positive 12-week biopsy or did not achieve a pCR at later treatment blocks are considered non-pCR (not missing) for analysis. We then use the imputed values in the logistic regression model.
For modelling missing pCR, we employ a simple imputation model that exploit MRI tumour volume measurements during treatment. We fit this separate imputation logistic regression model as described below to assess the probability of achieving pCR based on the available MRI information. The imputation model is used for modelling missing pCR results, while the previous model describes the modelling of the likelihood of pCR given baseline factors and treatment arm.
Define to be the tumour volumes at baseline, 3 weeks, 6 weeks and 12 weeks. Then the tumour volume reduction at visit is for .
Negative values of correspond to a decrease in tumour volume, with indicating that the tumour is not detectable by MRI at visit . To formulate a multiple imputation model for PCR status in cases where pCR is not available, we discretize the range of values into categories, for . The mapping between and is provided in Supplementary Table 7.
For each time point , we use data from all patients for whom both the pCR response and MRI assessment are available, and we fit this model:
where is the parameter corresponding to the category , where (see Supplemental Table 7), for patient at time . We impose the monotonicity constraint for to reflect the condition that greater tumour reduction cannot be less likely to lead to a pCR.
At each MCMC iteration, if the pCR status for patient is missing, a mixture distribution is formed with the predicted probability of where is the latest time point for which, we have a tumour volume measurement for patient .
Distribution of Molecular Tumour Subclasses:
The population incidence rates across the 32 disease subclasses defined by HR, HER2, Immune, DRD and BluePrint is modelled as a categorical distribution (i.e., multinomial distribution with 32 outcomes) over the subtypes indexed by , with . We use a Dirichlet prior
which results in a posterior distribution
where is the number of patients observed in subclass c.
pCR Modelling for Signatures.
The posterior distribution of the pCR rate for signatures is created from the pCR rates by subtypes contained in each signature and weighted by the prevalence parameters as follows.
Let be the probability of pCR for treatment arm in disease subclass . If represents ), where for the (HR, HER2, Immune, DRD and BluePrint) value, then
Let be the probability of for treatment arm a for signature . Then
(The notation means subclass is included in signature .)
Prior Distributions: Prior distributions for coefficients in the core logistic model are taken as normally distributed with standard deviation of 1. Prior means for the treatment effects coefficients are assumed to be 0. Prior means for the marker coefficients in the core logistic model and for the MRI imputation coefficients were obtained using the set of n=1818 I-SPY2 patients with full pCR. Since only the 3- and 12-week MRI were collected in I-SPY2, the 6-week MRI imputation mean was linearly interpolated. The distribution of coefficients for each MRI timepoint was taken as an ordered normal distribution with standard deviation of 0.2.
Efficacy Assessment of Block A agent in the context of a treatment strategy (across all blocks).
Efficacy assessment after Block A alone is myopic in that it does not consider the performance of the Block A regimen as part of a treatment strategy. For instance, a Block A agent may potentiate the effect of a Block B agent to give a highly effective treatment strategy, even though it did not meet our threshold for success based on the efficacy assessment of Block A alone. Thus, we will also evaluate efficacy of Block A agents in the context of a treatment strategy.
The efficacy analysis of treatment strategies (across all blocks) uses pCR data and timing of surgery (e.g., after Block A, B, or C) to estimate the pCR rate. A treatment strategy is a “path”: administer treatment at Block A, if the participant proceeds to Block B, then administer treatment , and if the participant proceeds to Block C, administer treatment . There are three ways a participant can be consistent this treatment strategy: (i) if they receive at Block A and went to surgery after Block A, (ii) if they receive at Block A and then at Block B and went to surgery after Block B, and (iii) if they receive , , and at their respective blocks. The pCR rate of a treatment strategy is estimated using a combination of five Bayesian models (described below) from data of patients whose treatment is consistent with the treatment strategy.
Bayesian Model for PCR Rate of Strategy.
Because treatment options are subtype-specific and we expect PCR rates to depend on the subtype, we independently conduct this algorithm for each subtype. We describe it in general below. Analysis is performed only for 5 of the 6 pre-specified signatures (each RPS and HR−HER2−), as HR+HER2− patients did not share any common Block B treatment option across response predictive subtypes.
Let be the indicator the subject elects to receive surgery at Block A; similarly define for Block B. Next, let be the indicator that a subject has a PCR at Block A given they went to surgery; similarly define and . Let and indicate treatments at blocks A and B, respectively.
Consider the following probabilities:
Using these probabilities, we define the probability of a PCR in the trial given treatment strategy :
This posterior distribution is based on the posterior distributions of , , , , and . For each of these, we assume prior distributions and update the posteriors with the observed data. For example, if 50 people have been assigned treatment and 35 of those people have , then the posterior for .
Of note, different sets of patients contribute to each of the five Bayesian models: Participants who receive at Block A contribute to estimation of and those participants who also proceeded to surgery at Block A contribute to estimating . Similarly, participants who receive at Block A and at Block B informs and those who also proceed to surgery at Block B contribute to estimating . Participants who receive at Block A, at Block B, and at Block C contribute to estimation of .
Efficacy analysis of Block A alone was performed using R version 4.1.3 and STAN 2.21.0 via the RSTAN package (2.21.5), which implements the NUTS sampler. Efficacy analysis of treatment strategy was performed using R version 4.2.2 via the stats package (4.2.2).
Supplementary Material
ACKNOWLEDGEMENTS
Research reported in this manuscript was supported by the National Cancer Institute of the National Institutes of Health under award number P01CA210961. The authors wish to acknowledge the generous support of the study sponsors and CRO, Quantum Leap Healthcare Collaborative (QLHC, 2013 to present) and the Foundation for the National Institutes of Health (2010 to 2012)[LE]. The authors sincerely appreciate the ongoing support for the I-SPY2.2 Trial from the Safeway Foundation, the William K. Bowes, Jr. Foundation, Give Breast Cancer the Boot, QLHC and the Breast Cancer Research Foundation [LE]. Sincere thanks to all the patients who have volunteered to participate in I-SPY2. AstraZeneca provided funds and study drugs (datopotamab deruxtecan and durvalumab). With the exception of the study sponsor, QLHC, the funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors wish to thank Denise Wolf and Jessica Gibbs for their important contributions to this work.
COMPETING INTERESTS
RAS reports institutional research funding from OBI Pharmaceuticals, Quantum Leap Healthcare Collaborative, AstraZeneca and Gilead; serves on AstraZeneca and Stemline Advisory Boards; Gilead Speaker’s Bureau; consultancy role with Quantum Leap Healthcare Collaborative. MST reports institutional research funding from Lilly, Gilead Sciences, Phoenix Molecular Design, AstraZeneca, Regeneron, Merck and Novartis. CY reports institutional research grant from NCI/NIH; salary support and travel reimbursement from Quantum Leap Healthcare Collaborative; US patent titled, “Breast cancer response prediction subtypes,” (No. 18/174,491); and University of California Inventor Share. RN reports research funding from Arvinas, AstraZeneca, BMS, Corcept Therapeutics, Genentech/Roche, Gilead, GSK, Merck, Novartis, OBI Pharma, OncoSec, Pfizer, Relay, Seattle Genetics, Sun Pharma, Taiho; advisory roles with AstraZeneca, BeyondSpring, Daiichi Sankyo, Exact Sciences, Fujifilm, GE, Gilead, Guardant Health, Infinity, iTeos, Merck, Moderna, Novartis, OBI, Oncosec, Pfizer, Sanofi, Seagen, Stemline. HSR reports institutional research support from AstraZeneca, Daiichi Sankyo, Inc., F. Hoffmann-La Roche AG/Genentech, Inc., Gilead Sciences, Inc., Lilly; Merck & Co., Novartis Pharmaceuticals Corporation, Pfizer, Stemline Therapeutics, OBI Pharma, Ambrx, Greenwich Pharma; advisory and consulting roles with Chugai, Puma, Sanofi, Napo, and Mylan. MD reports research grants from NIH/NCI and NIH/NIA, and contracts from PCORI. AJC reports institutional research funding from Merck, Amgen, Puma, Seagen, Pfizer, Olema; advisory roles with Astra Zeneca and Genentech. ESR reports grants from V Foundation, NIH, Susan G. Komen; institutional research funding from GSK, Seagen, Pfizer, Lilly; consulting and honoraria from Novartis, Merck, Seagen, Astrazeneca, Lilly; Cancer Awareness Network Board member and support from ASCO, NCCN. JCB reports institutional research funding from Eli Lilly and SymBioSis; participation on the Data Safety Monitoring Committee of Cairn Surgical; honoraria from PER, PeerView, OncLive, EndoMag and Up-To-Date. CO reports consulting fees from AstraZeneca, Guardant Health, and Jazz Pharmaceuticals. KMK reports advisory and consultant roles for Eli Lilly, Pfizer, Novartis, Astra Zeneca, Daiichi Sankyo, Puma, 4D Pharma, Oncosec, Immunomedics, Merck, Seagen, Mersana, Menarini Silicon Biosystems, Myovant, Takeda, Biotheranostics, Regor, Gilead, Prelude Therapeutics, RayzeBio, eFFECTOR Therapeutics, and Cullinan Oncology; and reports institutional research funding from Genentech/Roche, Novartis, Eli Lilly, AstraZeneca, Daiichi Sankyo, and Ascentage. ADE reports support from Scorpion, Infinity and Deciphera. CV reports institutional research funding from Pfizer, Seagen, H3 Biomedicine/Eisai, AstraZeneca, CytomX; research funding to previous institution from Genentech, Roche, Pfizer, Incyte, Pharmacyclics, Novartis, TRACON Pharmaceuticals, Innocrin Pharmaceuticals, Zymeworks, H3 Biomedicine; advisory and consulting roles with Guidepoint, Novartis, Seagen, Daiichi Sankyo, AstraZeneca, CardinalHealth; unpaid consulting with Genentech; participation in non-CME activity with Gilead, AstraZeneca. NW reports participation on Gilead Advisory Board; honoraria from Onclive, NCCN and Total Health Conferencing. KSA reports institutional research funding from AstraZeneca, Daiichi-Sankyo, Seattle Genetics, Quantum Leap Healthcare Collaborative; IDSM committee at Seattle Genetics. ASC reports institutional research funding from Novartis and Lilly. CF reports honoraria from Curio Oncology and OncLive; institutional research funding from Eli Lily. CI reports institutional research funding from Tesaro/GSK, Seattle Genetics, Pfizer, AstraZeneca, BMS, Genentech, Novartis and Regeneron; consultancy roles with AstraZeneca, Genentech, Gilead, ION, Merck, Medscape, MJH Holdings, Novartis, Pfizer, PUMA, Seagen; royalties from Wolters Kluwer (UptoDate); McGraw Hill (Goodman and Gillman). AT owns stock at Johnson & Johnsons, Gilead, Bristol Myers Squib; participation on Pfizer Advisory Board: AstraZeneca; reports institutional research funding from Merck and Sanofi; royalties from Up-to-Date. JT serves as institutional PI for clinical trial with Intuitive Surgical; Editor Lead for ABS, CGSO, SCORE, Breast Education Committee Track Leader, ASCO SESAP 19 and Breast Co-Chair, ACS. KY received research support unrelated to this work and paid to the institution from Pfizer, Gilead, Seagen, Dantari Pharmaceuticals, Treadwell Therapeutics, and Relay Therapeutics; support from American Cancer Society IRG grant # IRG-19–230-48-IRG, UC San Diego Moores Cancer Center, Specialized Cancer Center support grant NIH/NCI P30CA023100, Curebound Discovery Award (2023,2024). LA reports advisory and consultancy roles with Pfizer, and AstraZeneca. KG reports participation in Advisory Board with honoraria to institution for AstraZeneca, Novartis, Puma Biotechnologies, Eli Lilly, Gilead, Exact Sciences, Neogenomics and Tersera Therapeutics. FMH reports consultancy for Novartis. TS reports honoraria from Hologic. ALA, PB, and PN are employees of Quantum Leap Healthcare Collaborative. GLH reports institutional research grant from NIH (1R01CA255442). WFS reports shares of IONIS Pharmaceuticals and Eiger Biopharmaceuticals; received consulting fees from AstraZeneca; is a cofounder with equity in Delphi Diagnostics; issued patents for: (1) a method to calculate residual cancer burden, and (2) genomic signature to measure sensitivity to endocrine therapy. JP reports honoraria from Methods in Clinical Research - Faculty SCION Workshop; support from ASCO and Advocate Scholarship; AACR - SSP Program; VIVLI, U Wisc SPORE - EAB, QuantumLEAD - COVID DSMB, PCORI - Reviewer and I-SPY Advocate Lead. PP reports institutional research funding from Genentech/Roche, Fabre-Kramer, Advanced Cancer Therapeutics, Caris Centers of Excellence, Pfizer, Pieris Pharmaceuticals, Cascadian Therapeutics, BOLT, Byondis, Seagen, Orum Therapeutics, Carisma Therapeutics; consulting fees from Personalized Cancer Therapy, OncoPlex Diagnostics, Immunonet BioSciences, Pfizer, HERON, Puma Biotechnology, Sirtex, CARIS Lifesciences, Juniper, Bolt Biotherapeutics, Abbvie; honoraria from Dava Oncology, OncLive/MJH Life Sciences, Frontiers - Publisher, SABCS, ASCO; Speakers’ Bureau: Genentech/Roche (past); patents United States Patent no. 8,486,413, United States Patent no. 8,501,417, United States Patent no. 9,023,362, United States Patent no. 9,745,377; uncompensated roles with Pfizer, Seagen and Jazz. AD reports institutional research funding from Novartis, Pfizer, Genentech and Neogenomics; Program Chair, Scientific Advisory Committee, ASCO. DY reports research funding from NIH/NCI P30 CA 077598, P01 CA234228–01 and R01CA251600; consulting fees from Martell Diagnostics; and honoraria and travel for speaking at the “International Breast Cancer Conference.” LJvV is a founding advisor and shareholder of Exai BIo; part-time employee and owns stock in Agendia. NMH reports institutional research finding from NIH. LJE reports funding from Merck & Co.; participation on an advisory board for Blue Cross Blue Shield; and personal fees from UpToDate; unpaid board member of QLHC. All other authors declare no competing interests.
Footnotes
CODE AVAILABILITY
The statistical code used in this clinical trial is available to other investigators upon approval of the I-SPY Data Access and Publications Committee. Investigators interested in accessing the code complete an application at https://www.quantumleaphealth.org/for-investigators/clinicians-proposal-submissions/ and include a brief description of the intended use. Access to the code may be granted upon approval of the request, subject to compliance with ethical guidelines and applicable institutional and regulatory requirements.
clinicaltrials.gov identifier NCT01042379
DATA AVAILABILITY
De-identified subject level data and/or clinical specimens are made available to members of the research community upon approval of the I-SPY Data Access and Publications Committee. Details of the application and review process are available at: https://www.quantumleaphealth.org/for-investigators/clinicians-proposal-submissions/. I-SPY aims to make complete patient-level clinical datasets available for public access within 6 months of publication, as the data is complex and requires significant annotation to ensure its usability.
REFERENCES
- 1.Siegel RL, Giaquinto AN & Jemal A Cancer statistics, 2024. CA: A Cancer J. Clin. 74, 12–49 (2024). [DOI] [PubMed] [Google Scholar]
- 2.Giaquinto AN et al. Breast Cancer Statistics, 2022. CA: A Cancer J. Clin. 72, 524–541 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Swain SM, Whaley FS & Ewer MS Congestive heart failure in patients treated with doxorubicin. Cancer 97, 2869–2879 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Harrington D & Parmigiani G I-SPY 2 — A Glimpse of the Future of Phase 2 Drug Development? New Engl J Med 375, 7–9 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Das S & Lo AW Re-inventing drug development: A case study of the I-SPY 2 breast cancer clinical trials program. Contemp Clin Trials 62, 168–174 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Cortazar P et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014). [DOI] [PubMed] [Google Scholar]
- 7.I-SPY2 Trial Consortium et al. Association of Event-Free and Distant Recurrence–Free Survival With Individual-Level Pathologic Complete Response in Neoadjuvant Treatment of Stages 2 and 3 Breast Cancer. Jama Oncol 6, 1355–1362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavori PW & Dawson R Introduction to dynamic treatment strategies and sequential multiple assignment randomization. Clin Trials 11, 393–399 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kidwell KM SMART designs in cancer research: Past, present, and future. Clin Trials 11, 445–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W et al. Predicting breast cancer response to neoadjuvant treatment using multi-feature MRI: results from the I-SPY 2 TRIAL. Npj Breast Cancer 6, 63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W et al. Abstract P4–02-10: MRI models by response predictive subtype for predicting pathologic complete response. Cancer Res 83, P4–02-10-P4–02–10 (2023). [Google Scholar]
- 12.Onishi N et al. Abstract P3–03-01: Functional tumor volume at 3 and 6-week MRI as an indicator of patients with inferior outcome after neoadjuvant chemotherapy. Cancer Res 82, P3–03-01-P3–03–01 (2022). [Google Scholar]
- 13.Wolf DM et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: Predictive biomarkers across 10 cancer therapies. Cancer Cell 40, 609–623.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piccart M et al. 70-gene signature as an aid for treatment decisions in early breast cancer: updated results of the phase 3 randomised MINDACT trial with an exploratory analysis by age. Lancet Oncol. 22, 476–488 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Cardoso F et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N. Engl. J. Med. 375, 717–729 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Göker E et al. Treatment response and 5-year distant metastasis-free survival outcome in breast cancer patients after the use of MammaPrint and BluePrint to guide preoperative systemic treatment decisions. Eur. J. Cancer 167, 92–102 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Loibl S et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Annals of Oncology 30, 1279–1288 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Foldi J et al. Neoadjuvant durvalumab plus weekly nab-paclitaxel and dose-dense doxorubicin/cyclophosphamide in triple-negative breast cancer. Npj Breast Cancer 7, 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pusztai L et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 39, 989–998.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okajima D et al. Datopotamab deruxtecan (Dato-DXd), a novel TROP2-directed antibody-drug conjugate, demonstrates potent antitumor activity by efficient drug delivery to tumor cells. Mol. Cancer Ther. 20, molcanther.0206.2021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardia A et al. Datopotamab Deruxtecan in Advanced or Metastatic HR+/HER2− and Triple-Negative Breast Cancer: Results From the Phase I TROPION-PanTumor01 Study. J. Clin. Oncol. 42, 2281–2294 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenzie JA et al. The Effect of Topoisomerase I Inhibitors on the Efficacy of T-Cell-Based Cancer Immunotherapy. JNCI: J. Natl. Cancer Inst. 110, 777–786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwai T et al. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and up-regulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti-PD-L1 antibodies. Oncotarget 9, 31411–31421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmid P Datopotamab derutecan (Dato-Dxd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): Updated results from BEGONIA, a phase Ib/II study. Annals of Oncology 34, (2023). [Google Scholar]
- 25.Bardia A et al. TROPION-Breast03: a randomized phase III global trial of datopotamab deruxtecan ± durvalumab in patients with triple-negative breast cancer and residual invasive disease at surgical resection after neoadjuvant therapy. Ther. Adv. Méd. Oncol. 16, 17588359241248336 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rugo HS et al. Adaptive Randomization of Veliparib–Carboplatin Treatment in Breast Cancer. New Engl J Med 375, 23–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanda R et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer. Jama Oncol 6, 676–684 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yau C et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: a multicentre pooled analysis of 5161 patients. Lancet Oncol. 23, 149–160 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid P et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 382, 810–821 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Cardoso F et al. LBA21 KEYNOTE-756: Phase III study of neoadjuvant pembrolizumab (pembro) or placebo (pbo) + chemotherapy (chemo), followed by adjuvant pembro or pbo + endocrine therapy (ET) for early-stage high-risk ER+/HER2− breast cancer. Ann. Oncol. 34, S1260–S1261 (2023). [Google Scholar]
- 31.Schneider BJ et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 39, 4073–4126 (2021). [DOI] [PubMed] [Google Scholar]
METHODS ONLY REFERENCES
- 32.Symmans WF et al. Measurement of Residual Breast Cancer Burden to Predict Survival After Neoadjuvant Chemotherapy. J Clin Oncol 25, 4414–4422 (2007). [DOI] [PubMed] [Google Scholar]
- 33.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) | Protocol Development | CTEP. https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm (2021). [Google Scholar]
- 34.Basch E et al. Development of the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). Jnci J National Cancer Inst 106, dju244–dju244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacob S et al. Use of PROMIS to capture patient reported outcomes over time for patients on I-SPY2. J. Clin. Oncol. 41, 611–611 (2023). [Google Scholar]
- 36.Pearman TP, Beaumont JL, Mroczek D, O’Connor M & Cella D Validity and usefulness of a single-item measure of patient-reported bother from side effects of cancer therapy. Cancer 124, 991–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oken MM et al. Toxicity and Response Criteria of the Eastern-Cooperative-Oncology-Group. American Journal of Clinical Oncology-Cancer Clinical Trials 5, 649–655 (1982). [PubMed] [Google Scholar]
- 38.Beltran PJ et al. Ganitumab (AMG 479) Inhibits IGF-II–Dependent Ovarian Cancer Growth and Potentiates Platinum-Based Chemotherapy. Clin Cancer Res 20, 2947–2958 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brahmer JR et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 9, e002435 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haanen J et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up ☆. Ann. Oncol. 33, 1217–1238 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
De-identified subject level data and/or clinical specimens are made available to members of the research community upon approval of the I-SPY Data Access and Publications Committee. Details of the application and review process are available at: https://www.quantumleaphealth.org/for-investigators/clinicians-proposal-submissions/. I-SPY aims to make complete patient-level clinical datasets available for public access within 6 months of publication, as the data is complex and requires significant annotation to ensure its usability.