

Abstract
The use of next-generation sequencing technologies, such as exome and genome sequencing, in research and clinical care has transformed our understanding of the molecular architecture of genetic kidney diseases. Whilst the capability to identify and rigorously assess genetic variants and their relationship to disease has significantly advanced in the last decade, the curation of clinically relevant relationships between genes and specific phenotypes has received less attention, yet it underpins accurate genomic test interpretation. In this review we discuss the need to accurately define gene-disease relationships in nephrology and provide a framework for critically appraising genetic and experimental evidence. We describe existing international programs that provide expert curation of gene-disease relationships and discuss sources of discrepancy as well as efforts at harmonisation. Further, we highlight the need for alignment of disease and phenotype terminology to ensure robust and reproducible curation of knowledge. These collective efforts to support evidence-based translation of genomic sequencing into practice across clinical, diagnostic and research settings are critical to delivering the promise of precision medicine in nephrology, providing more patients with timely diagnoses, accurate prognostic information, and access to targeted treatments.
Introduction
Chronic kidney disease is a major cause of morbidity and mortality worldwide, with an estimated prevalence of 9%1 and up to 13–16% of adults having evidence of kidney disease2.3 Both common and rare genetic variation contributes significantly to disease burden. Over 600 genes have now been implicated in the development of monogenic forms of kidney disease, across a broad range of clinical presentations, including glomerulopathies, tubulopathies, cystic kidney disorders, complement-mediated diseases and congenital malformations (Figure 1). It is estimated that monogenic diseases are responsible for 50% of children4–6 and 10% of adults3,6–8 affected by kidney disease. Further, for those undergoing kidney transplantation, an underlying genetic cause of kidney disease is present in up to 20%9,10. This substantial expansion in understanding of genetic causes for kidney disease has unfurled synergistically, with growth in knowledge of underlying kidney biology, in some instances, enabling new mechanistic insights, which in turn have begun to translate into targeted therapies with APOL1-associated Kidney Disease11 and Autosomal Dominant Polycystic Kidney Disease12 being example.
Fig. 1|. Examples of genetic kidney disorders.
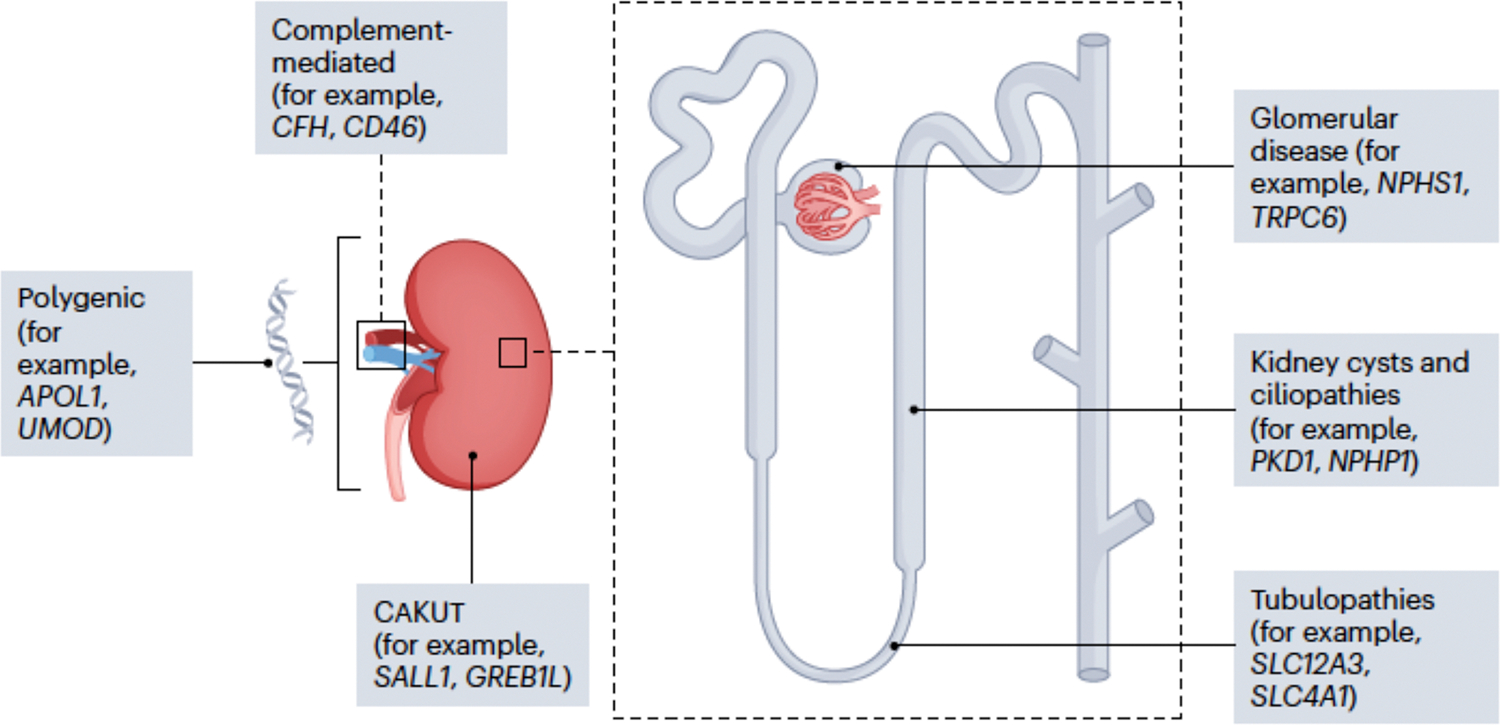
More than 600 genes have been implicated in the pathogenesis of kidney disorders, including monogenic glomerular, cystic and tubular disorders, as well as congenital anomalies of the kidney and urinary tract (CAKUT), complement-mediated disorders and polygenic contributors to chronic kidney dysfunction and failure.
Genomic sequencing technologies, such as exome and genome sequencing, have fuelled this rapid growth in knowledge over the past decade. Genomic sequencing is now increasingly transitioning from the research environment to routine clinical care, forming a virtuous cycle to continuously improve patient care13. Large cohort studies in different countries have established evidence for diagnostic and clinical utility14–18 as well as cost-effectiveness19–21 of genomic sequencing in patients with kidney disease. The clinical impacts of a precise molecular diagnosis are wide-ranging and include informing prognosis, transplant and treatment decisions as well as providing accurate reproductive counselling and cascade testing for family members. With the emergence of precision treatments targeting the underlying molecular mechanisms of disease, a timely molecular diagnosis is increasingly a pre-requisite to treatment access22,23.
However, a robust evidence base is needed for accurate genomic data interpretation in the clinical setting. Knowledge about which genes cause relevant diseases, patterns of inheritance and disease mechanisms all form the foundation of accurate variant interpretation. International standards and global collaboration are key to the timely, evidence-based implementation of genomics into healthcare, including for those affected by kidney disease. Yet wide variability in diagnostic laboratory and clinical practice, particularly in selecting which genes to analyse in specific clinical scenarios, exists and hampers high quality, consistent patient care.
In this review, we describe the impact of genomics on the delineation of gene-disease relationships in nephrology. We outline the principles for critically assessing evidence about gene-disease relationships and summarize current international efforts in gene curation, particularly in relation to kidney disease. Finally, we explore the need to align disease terminology with underlying genetic aetiology and to accurately define gene-disease relationships in a way that is dynamic and up to date. Altogether, accomplishing this will help ensure effective knowledge translation across research, clinical and diagnostic settings.
Sequencing DNA and interpreting genetic variation: a primer
The technical ability to accurately determine the sequence of nucleotides in DNA, thus providing a read out of the genetic code, underpins all efforts to understand the genetic determinants of disease at the gene and gene variant level. Over the past 50 years, numerous DNA sequencing techniques have emerged, progressively enabling larger amounts of DNA to be sequenced at diminishing costs and with increasing speeds24,25. Traditional (or first-generation) DNA sequencing techniques, such as Sanger sequencing, enable relatively short fragments of DNA, such as single genes to be sequenced robustly and accurately and were the first to be widely adopted in research and clinical practice (Table 1).
Table 1|.
Genetic and genomic sequencing techniques
| Approach | Advantages | Disadvantages |
|---|---|---|
| Traditional genetic testing approaches | ||
| Karyotype | Unbiased, genome-wide assessment Can detect changes in chromosome number (for example, trisomies) and structural variants (for example, deletions, duplications, translocations and inversions) |
Low resolution (3–10Mb) Can only detect large chromosomal abnormalities |
| Microarray | Unbiased, genome-wide assessment Can detect changes in chromosome number and some structural variants (CNVs: deletions, duplications) |
Moderate resolution (50–100kb) Cannot detect balanced structural variants (for example, translocations) or small variants (for example, SNVs) |
| FISH | Detects and locates a specific DNA sequence directly (for example, recurrent CNV) Rapid results |
Biased, requires a priori hypothesis Limited repertoire of readily available probes |
| MLPA | Detects CNVs directly, ranging from exon-level to whole chromosome | Biased, requires a priori hypothesis |
| Sanger sequencing | Detects causative SNV directly | Biased, requires a priori hypothesis Long and expensive process of potentially sequential testing in genetically heterogeneous disorders (for example, nephronophthisis) |
| Next-generation sequencing approaches | ||
| Gene panel testing | Customizable to allow optimization for genes of interest Allows detection of multiple variant classes Ease of interpretation Low chance of incidental findings |
Biased, requires a priori hypothesis Can quickly become out of date as new gene–disease associations are discovered, requiring re-design |
| Exome sequencing | Single test for all genes (both known and undescribed disease genes) Can limit analysis to genes of interest (for example, virtual gene panel) or perform unbiased, hypothesis-free testing for diagnosis and discovery High coverage allows detection of mosaic variants |
Does not reliably identify all classes of genomic variation or non-coding variation Increased complexity of interpretation Possibility of incidental findings |
| Genome sequencing (short-read) | Can limit analysis to genes of interest or perform unbiased, hypothesis-free testing for diagnosis and discovery Performs well for identifying all classes of genomic variation |
High complexity in interpretation, particularly of non-coding variants Challenges with data handling Possibility of incidental findings Relatively high cost Relatively low sequencing depth and limited detection of mosaic variants |
| Genome sequencing (long-read) | Better detection of structural variants, repeat expansions and variants in complex genomic regions | Currently expensive and lower-throughput than short-read sequencing Methodology less well established than for short-read sequencing, limited availability in the diagnostic setting |
| RNA sequencing | As an adjunct to DNA sequencing, allows interpretation of variant effect (for example, variants that disrupt splicing or gene expression) | RNA requires specialized handling and is susceptible to degradation Use is dependent on access to disease-relevant tissue Limited availability in the diagnostic setting |
| Optical genome mapping | High-throughput, genome-wide assessment of structural chromosomal variants including aneuploidies, insertions, deletions, duplications, inversions, translocations and repeat expansions or contractions | Emerging technology, not readily available in the clinical setting |
CNV, copy number variant; FISH, fluorescent in situ hybridization; MLPA, multiplex ligation-dependent probe amplification; SNV, single-nucleotide variant.
Sanger and shotgun sequencing enabled the first draft of the human genome sequence in 200126,27; however, this was achieved at a cost exceeding USD$3 billion and took over 10 years. In response to these challenges, next-generation sequencing (NGS) methods, also referred to as massively parallel sequencing or high-throughput sequencing, were developed, allowing the simultaneous production of sequencing data at the genome scale at ever decreasing cost. Rapid uptake of the technology in the research and, increasingly, the clinical setting over the past 10 years has been facilitated by large government investments worldwide28, aimed at accelerating discovery while building infrastructure, capacity and capability to support healthcare system implementation.
The massively parallel nature of NGS enables the entire genome (GS) or the entire exome (the protein coding region; ES) of an individual to be sequenced in a single assay (Table 1). This allows an unbiased approach to the diagnosis of Mendelian disorders. By comparing sequence reads to the human reference genome, variants causative of disease can be identified29. ES and GS generate vast amounts of data and the key challenge no longer lies in data generation but in the accurate interpretation of the identified variants, and distinguishing causative variants from the background of more than four million non-pathogenic or disease-unrelated variants. 30
Genomic data interpretation is complex and typically involves multiple steps, beginning with variant annotation and filtering in order to prioritise potentially causative ‘variants of interest’, before then classifying the relationship of a given variant in a gene to a given disease and inheritance mechanism according to a standard framework. Classified variants are then assessed in relation to the clinical and phenotypic information of the individual prior to reporting. Accurate genomic data interpretation is highly reliant on the global sharing of data and knowledge31. Examples of common databases used in genomic interpretation are provided in Table 2.
Table 2|.
Gene and variant curation resources
| Resource | Key features | Link |
|---|---|---|
| Population genetic variation | ||
| gnomAD | Originally launched in 2014 as ExAC Combines exome and genome sequencing data from a variety of large-scale sequencing projects, producing summary data of variant frequencies It excludes cohorts with severe paediatric disease v4 release contains 730,947 exomes and 76,215 genomes |
https://gnomad.broadinstitute.org/ |
| Disease-associated genetic variation | ||
| ClinVar | Open public archive of variants classified in the context of human disease, with supportive evidence. As of March 2024, contains more than 4 million records submitted by over 2,700 diagnostic laboratories, research groups and other consortia | https://www.ncbl.nlm.nlh.gov/clinvar/ |
| HGMD | Manually collates genetic variants associated with human disease reported in peer-reviewed publications Public and subscription-only versions available |
http://www.hgmd.org |
| LOVD | Free, open-source database used to establish thousands of locus-specific curated variant databases for specific genes as part of a federated network | https://www.lovd.nl |
| DECIPHER | Interactive, web-based database that shares candidate diagnostic variants and phenotypic data from individuals with genetic disorders and incorporates a suite of other tools to aid genomic variant interpretation | https://www.deciphergenomics.org |
| Gene–disease associations | ||
| OMIM | Continuously updated compendium of human genes and associated phenotypes, biocurated at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine Entries are based on peer-reviewed publications Each OMIM entry is given a unique six-digit number ‘?’ denotes provisional gene–disease relationships, generally those reported in a single family |
https://omim.org/ |
| ClinGen | Central resource dedicated to defining the clinical relevance of genes and variants, funded by the NIH Validity of gene–disease relationships is assessed by trained biocurators and reviewed by disease-specific expert committees, which are convened on a volunteer basis and have wide international participation Uses an objective, points-based system to assign the strength of evidence into one of seven categories, ranging from Refuted to Definitive |
https://clinlcalgenome.org |
| Orphanet | Contains Information on rare diseases and orphan drugs, provides medical terminology dedicated to rare diseases, the Orphanet nomenclature of rare diseases (also known as ORPHA codes) Gene–disease relationships are catalogued, without a formal assessment of the evidence | www.orpha.net |
| Genomics England PanelApp | Originally developed by Genomics England to support the delivery of the 100,000 Genomes Project, PanelApp now supports the NHS Genomic Medcine Service Contains assessments of the validity of gene–disease relationships, with genes assigned to virtual panels to support diagnostic analysis for specific clinical indications Evidence of the validity of gene–disease relationships is rated using a traffic light system, with genes receiving a Green rating used in diagnostic analysis Evidence is crcwdsourced from a wide range of contributors but assessed by an internal team of biocurators |
https://panelapp.genomicsengland.co.uk |
| PanelApp Australia | PanelApp Australia is managed by Australian Genomics, and is used by Australian diagnostic laboratories, clinicians and researchers to establish and maintain consensus virtual panels for use in genomic analysis Evidence of the validity of gene–disease relationships is rated using a traffic light system, with genes receiving a Green rating used in diagnostic analysis Evidence is crowdsourced but assessed by an internal team of biocurators |
https://panelapp.agha.umccr.org |
| Gene Curation Coalition | Centralized database that brings together groups performing gene curation, with the aim of harmonizing terminology and facilitating consistent assessment of gene–disease validty assessments | https://thegencc.org |
| MatchMaker Exchange | Connects multiple other databases focused on the discovery of new candidate gene–disease relationships by enabling interested parties to match based on gene name and establish collaborations | https://www.matchmakerexchange.org |
ClinGea, Clinical Genome Resource; ExAC, Exome Aggregation Consortium; gnomAD, Genome Aggregation Database; HGMD, Human Gene Mutation Database; LOVD, Leiden Open Variation Database; NHS, UK National Health Service; NIH, National Institutes of Health; OMM, Online Mendelian Inheritance in Man.
The prioritisation of potentially causative variants for more in-depth evaluation relies on a combination of two key analytical approaches: genotype-driven and phenotype-driven32. Genotype-driven analysis approaches aim to identify variants that are highly likely to be disease-causing based on their properties. This includes predicted loss-of-function variants (e.g. frameshift, truncating); variants previously reported in the medical literature or databases as pathogenic/likely pathogenic; and variants that fit proposed models of inheritance (e.g. biallelic variants in a gene causing autosomal recessive disease). Genotype-driven approaches are unbiased and are particularly useful in detecting unanticipated genetic diagnoses, e.g. in patients with unusual or complex presentations or those with multiple diagnoses. Genotype-driven analysis is also the primary approach used in analyses directed at discovering novel gene-disease associations.
Phenotype-driven analysis approaches are complimentary and focus on specific genes that are relevant to a patient’s phenotype, e.g. genes known to be associated with haematuria. This allows for comprehensive review of variants that are highly relevant to the clinical question. One common strategy for phenotype-driven analysis is the design and application of virtual gene panels based on common clinical indications for testing (e.g. cystic kidney disease).
Currently, analysis of genomic data, particularly in patients with clinical features predominantly affecting single organ systems, such as those with kidney disease, typically first targets analysis to genes of clinical interest. If a diagnosis is not obtained, the approach may then be broadened to genotype-driven strategies to identify candidate variants, revealing unanticipated diagnoses or identifying novel gene-disease relationships. Both strategies are limited by current knowledge and methods to detect and interpret genetic variation, meaning for example that even when whole genome data is available, analysis is still limited to the clinically interpretable fraction of the available data, for example coding variants in genes currently associated with disease.
Once variant(s) of interest are identified, an extensive manual review is undertaken to classify the relationship of the variant to disease causality. This assessment typically follows well-recognised standards for the classification of sequence variants that incorporate multiple lines of evidence to assess causality at the gene and the variant level30. The most commonly used evidence framework, developed by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP), considers multiple categories of evidence and assigns each a weight based on the strength that type of evidence represents. These are then combined using a set of rules to determine the final variant classification in one of five categories: pathogenic, likely pathogenic, variant of uncertain significance, likely benign and benign. The lines of evidence considered in variant classification fall into two main classes: human observational data and functional/predictive data.
Lines of evidence considered as part of human observational data all pertain to the phenotype, or lack thereof, displayed by individuals with the variant under assessment, and therefore consider the effect of the variant at the organism-level. For example, the presence of a variant in population variation databases, such as gnomAD, at frequencies inconsistent with the known incidence and severity of the disease, would be considered as ‘strong’ evidence for the variant being benign. On the other hand, the observation of the same variant in multiple, unrelated individuals with the same disease, that is not observed in unaffected individuals, constitutes ‘strong’ evidence for the variant being pathogenic. A variant co-segregating with disease in a family, provides support towards pathogenicity, equivalent to the number of co-segregations observed, however, a variant that is present in unaffected family members or that is absent in affected family members provides support towards benignity.
Lines of evidence considered as part of functional and predictive data, on the other hand, instead consider the expected or observed impact of the variant on the protein and the consequences that has on its’ function. With knowledge of the molecular mechanism underlying a given disease, the unique biology of a given gene, and of general biological concepts, the expected impact of a variant can be predicted. For example, frameshift or truncating variants that are expected to be the target of nonsense-mediated-decay of messenger RNA will lead to an absent protein, and therefore garner strong support towards pathogenicity when loss-of-function is the known mechanism of disease. The consequence of other types of variants, such as missense variants, can be less straightforward to discern, but can be informed by in silico tools that predict whether or not the impact will be damaging, providing support towards either pathogenicity or benignity. Functional studies that allow the consequence of a variant to be observed directly then add further weight towards either pathogenicity or benignity, with strength equivalent to the validity of the cellular, organoid, or animal model used.
Multiple refinements to the ACMG/AMP criteria have now been developed to further elaborate and provide guidance on application in the context of specific types of evidence33,34 or in specific genes35–37.
Evidence to confidently classify variants as disease-causing or benign is often lacking, leading to many rare variants observed as part of diagnostic testing being classified as ‘variants of uncertain significance’ (VUS). VUS currently account for over a third of all variants submitted to databases such as ClinVar38, and are present on a third of laboratory reports in the absence of a causative variant being identified39. Nevertheless, in 2020, the National Human Genome Research Institute (NHGRI) made ten ‘bold predictions’, including that the designation of ‘VUS’ will be obsolete by 203040. Progressing towards this goal over the next few years will leverage further development in the standards used for variant classification, with the release of new ACMG/AMP guidelines anticipated in the next year; improvements in computational variant effect predictor performance; the large-scale application of multiplexed variant effect assays; and increased global data sharing38.
The methods, data and knowledge used to identify and assess variants are still changing dynamically. Unlike other common types of diagnostic investigations, genomic sequencing data are unique in that they can be stored and reanalysed over time. Such reanalysis has repeatedly been shown to increase diagnostic yields by 10–15% if performed at 2–3 yearly intervals41,42. While reanalysis currently largely relies on manual processes43, automation is expected to enable this to be systematically scaled up in the near future.
Gene-disease relationships in the genomic era
ES and GS have had a profound impact on elucidating the genetic basis of Mendelian disorders and on the rate of discovery of novel disease genes. Almost 36% of all published Mendelian disease gene discoveries have been made in the last ~10 years using NGS-based approaches44,45. This has been mirrored in the field of kidney genetics. Since the discovery that variants in the COL4A5 gene cause X-linked Alport syndrome in 199046, characterized by hearing loss, eye abnormalities, and glomerular phenotypes, variants in over 100 genes have been associated with glomerular disease alone47.
In addition to accelerating discovery, unbiased sequencing approaches have also challenged our traditional understanding of inheritance patterns and the natural history of conditions caused by variants in established disease genes. For example, while variants in PKD1 are classically associated with autosomal dominant polycystic kidney disease48, autosomal recessive forms of PKD1-related disease are now also recognised, typically presenting with very early, including prenatal, onset of renal cysts49,50. Conversely, while variants in IFT140 were originally associated with an autosomal recessive multi-system ciliopathy disorder, including nephronophthisis51, more recently a relationship with adult-onset, autosomal dominant disorder characterised by large kidney cysts and limited kidney insufficiency, has emerged52.
The improved understanding of underlying genetic and molecular mechanisms has also challenged existing disease nosologies which have historically relied on clinical and phenotypic features to define disease entities. For example, variants in CLCN5 are now known to underlie Dent disease (OMIM#300009), as well as X-linked nephrocalcinosis (OMIM#310468), X-linked recessive hypophosphataemic rickets (OMIM#300554), and low-molecular-weight proteinuria with nephrocalcinosis (OMIM#308990). These disorders are now considered to be part of a single disease entity, with variable phenotypic severity.
The growth of databases aggregating genomic sequencing data that allows us to define the prevalence of variants in a normal population, such as gnomAD53 (Table 2), together with the emergence of systematic approaches to evaluate the pathogenicity of variants30, has called into question some historical gene-disease relationships. For example, variants in SIX5 were first associated with branchio-oto-renal syndrome in 200754 and have been reported in at least six individuals54,55. However, the reported variants are now recognised as being present in population databases at frequencies that are inconsistent with causality for a monogenic disorder. For example, one of the original variants reported, p.(Ala158Thr) is present in 475 individuals in gnomAD v4.0. Further, many of the variants originally reported had no evidence of pathogenicity supplied and/or an alternate cause of the disease was later reported56. Two independent SIX5 mouse models had cataracts and no ear or kidney abnormalities, further calling the gene-disease relationship into question57,58.
A precise molecular diagnosis in individuals with kidney disease has direct impact on clinical care59. It can inform transplant and treatment decisions, and can obviate the need for invasive kidney biopsies, particularly in children14,15,19. It also enables accurate reproductive counselling; provides access to reproductive options such as prenatal testing and preimplantation genetic diagnosis; and facilitates cascade testing for family members, including informing choice of living related kidney donors. Variant information is also increasingly used to provide more accurate prognostic information, for example in polycystic kidney disease60,61 and in Alport syndrome62,63. Stratification based on expected progression of disease is likely to become increasingly important for accessing clinical trials.
Importantly for improving patient outcomes, these new insights into genetic aetiologies and molecular mechanisms of kidney disease are increasingly leading to precision therapies. In the most common genetic kidney disease, Autosomal Dominant Polycystic Kidney Disease (ADPKD), the identification in 1985 of the genetic locus64 was followed by the complete description of the PKD1 gene and encoded protein65, and elucidation of targetable disease pathways66. This then led to the identification of a potential targeted compound (tolvaptan)67, positive phase 3 randomised control trials68,69 and eventual regulatory approval by the United States Food and Drug Administration (FDA) in 201870. For the much rarer condition atypical haemolytic uraemic syndrome (aHUS), the formalisation of classification and nomenclature for the broader disease spectrum71 was followed by description of variants in complement regulatory genes underpinning aHUS72. Alignment with a previously described anti-C5 antibody73 then resulted in successful case reports74,75 and cohort studies76 with ensuing FDA approval in 201177. More recently, precision therapies for NPHS2-related Nephrotic Syndrome78,79, ADTKD (MUC1)80 and APOL1-associated kidney disease81 have been based upon fundamental gene and disorder pathobiology descriptions82,83,84, and have generated much hope for imminent translation into pre-clinical and clinical trials.
These examples across the spectrum of ultra-rare as well as more common genetic kidney diseases demonstrate how fundamental understanding of gene-phenotype relationships is the basis on which unravelling of disease mechanisms and identification of potential treatments rests.
Gene-disease relationships: the need for evidence
Information about gene-disease relationships underpins the ability to provide accurate and timely molecular diagnosis to patients, which in turn informs prognosis, recurrence risk counselling, and increasingly, access to precision treatments. The rapid growth in knowledge about gene-disease relationships needs to be translated quickly into laboratory practice to avoid missing the opportunity for improved diagnostic outcomes. Processes for rapid integration of knowledge benefit both the analysis of data from new patients, as well as the reanalysis of existing data, with the growth of new gene-disease relationships having consistently been identified as the single largest contributor to increased diagnostic yields over time41. However, the processes for incorporating new knowledge also need to be evidence-based as reporting variants in genes with only speculative evidence for association with disease risks misdiagnosis and inappropriate patient and family management.
Genomic testing for patients with clinical features predominantly affecting single organ systems, such as those with kidney disease, frequently uses a phenotype-driven, ‘virtual’ gene panel approach, which targets analysis of exome or genome data to genes most likely to be clinically relevant. Virtual panel design and maintenance relies on accurate and up-to-date knowledge of gene-disease relationships and associated phenotypes. However, comparisons of panel content between different diagnostic providers typically reveal a high degree of variability85,86. Such wide discrepancies raise serious concerns about variability in diagnostic outcomes for patients tested through different providers and resultant variability in downstream clinical care.
Principles of assessing the validity of gene-disease relationships
The development and consistent application of evidence-based frameworks for the assessment of the validity of gene-disease relationships is therefore an essential component of precision diagnostics. Determining the validity of the gene-disease relationship is a prerequisite for being able to interpret the significance of gene variants: if a gene is not convincingly linked to disease, the pathogenicity or otherwise of variants cannot be determined30.
Typically, new gene-disease relationships are proposed based on two types of evidence: genetic data and supportive experimental data87,88, Figure 2. In rare monogenic disorders, including those affecting the kidney, novel gene discovery in the genomic era has primarily been driven by the identification of gene candidates in single families. This typically then prompts a search for additional families either in large cohorts or through sharing of candidate gene information on platforms such as the MatchMaker Exchange89, a federated network that connects rare disease databases around the globe to facilitate connecting researchers looking for cases with similar phenotypes and variants in the same candidate gene.
Fig. 2|. From clinical assessment to diagnosis.

Accurate genomic diagnosis relies on building virtuous cycles between research and clinical care, which help to understand the contribution of genetic variation to kidney disease through data sharing, functional genomics and continuous critical appraisal of the evidence for gene–disease and variant–disease associations. For example, clinical trials, kidney donor assessment and unexplained kidney failure. CMA, chromosomal microarray; WES, whole exome sequencing; WGS, whole genome sequencing.
Generally, the greater the number of unrelated families identified with variants in a gene and a specific phenotype, the greater the confidence of the relationship between gene and disease. This requires critical assessment of all the clinical, variant and pedigree information. For example, some variant types, such as those leading to loss of function of the gene (e.g. frameshift and truncating variants) can more confidently be expected to be disease-causing than other variant types such as missense variants. Such assessments though, must also consider whether the variant type is consistent with the anticipated mechanism of disease. Where only a single individual in the family is affected, demonstrating that a variant has occurred de novo adds further evidence for causation. Where multiple individuals are affected however, there needs to be sufficient number of family members with and without disease tested to be certain that the variant of interest segregates with disease and is not a chance observation. The strength of additional lines of evidence, such as the predictive and functional data evaluated as part of variant pathogenicity classification, should also be considered carefully.
Secondly, candidate genes typically also have a demonstrated role in biological processes that plausibly result in the disease phenotype. Such experimental evidence should also be subject to critical appraisal, with some study types providing more compelling evidence than others. For example, the gene may be expressed in the relevant tissue (e.g. kidney) and may be known to interact with other genes already implicated in renal disease. A much more compelling line of evidence, however, is the generation of cell- or animal-based experimental systems where disruption of the gene recapitulates the human phenotype.
Ideally, multiple lines of evidence, preferably from independent studies are required to unequivocally establish gene-disease relationship. Gene curation is a laborious process, which relies on information being extracted from a variety of sources containing unstructured data, such as publications (including supplementary materials), online databases and other repositories. Machine learning approaches, such as natural language processing, hold promise in assisting expert curators in identifying and extracting pertinent information to accelerate evidence appraisal.
The general principles for evaluating the strength of evidence for gene-disease relationships outlined here have been developed to be broadly applicable for rare monogenic disorders. For disorders with known variable penetrance and/or expressivity, such as congenital anomalies of the kidney and urinary tract, systematic modifications can appropriately account for these varied scenarios. Different paradigms are needed though to assess evidence pertaining to genetic contributors, at the gene and/or variant level, for disorders such as atypical haemolytic uraemic syndrome and for polygenic disorders such as steroid sensitive nephrotic syndrome.
International efforts to provide expert curation of gene-disease relationships
Many groups internationally provide public databases of curated gene-disease relationships. The oldest and perhaps best known of these is the Online Mendelian Inheritance in Man (OMIM). Other well-known public databases include Orphanet, Genomics England PanelApp, PanelApp Australia, and the Clinical Genome Resource (ClinGen; Table 2). These databases have historically been established to serve different purposes and have different strengths. Only some of these resources (ClinGen, Genomics England PanelApp, PanelApp Australia) are directly aimed at supporting diagnostic genomic testing85,88,90. Others such as Orphanet primarily serve as catalogues of reported gene-disease relationships without an overlay of critical assessment of the evidence. These differences in approach, combined with curation efforts historically occurring in silos have resulted in the emergence of multiple standards and terminologies used to describe the validity of gene-disease relationships. For example, PanelApp has three categories to delineate the strength of gene-disease relationship, whereas ClinGen has seven, OMIM has two, and Orphanet one. This accounts for many of the apparent discrepancies when the content of these resources is compared, creating confusion for clinical and diagnostic users.
Harmonization of these independent efforts is now underway through the international Gene Curation Coalition (GenCC)91. The GenCC brings together public databases and diagnostic laboratories that have agreed to share their internally curated gene-disease validity assessments. The aims of the GenCC are twofold. Firstly, to develop standardized terminology for gene-disease validity assessment, including how to describe inheritance, allelic requirement, and mechanism of disease, for example whether disease is caused by mono- and/or bi-allelic variants as outlined in the earlier examples of PKD1 and IFT14092. The use of mutually agreed structured terminology to describe gene-disease relationships allows harmonisation across multiple resources, enables greater degrees of automation in genomic data analysis, and aids robust variant curation, classification, and reporting.
Secondly, the GenCC provides a central public depository of gene-disease curations from multiple groups. This allows for discrepancies to be readily identified, with the GenCC facilitating a process of discordance resolution between groups. Such data sharing and harmonization efforts have already proven to be highly effective in improving evidence-based diagnostic practice, both in relation to variant curation93 and to gene-disease validity and virtual panel curation85. As of December 2023, the GenCC database contains 18,504 submissions about 4,888 unique genes from 12 groups performing gene-disease curation, including ClinGen, Orphanet, Genomics England PanelApp and PanelApp Australia (Table 2). Over time, these efforts will be key to achieving consistency internationally, reducing duplication of effort and variability while increasing the quality and timeliness of gene curation.
ClinGen Kidney Clinical Domain Working Group
The Clinical Genome Resource (ClinGen) is an initiative funded by the National Institutes for Health (NIH), which aims to drive collaborative efforts in improving understanding of genetic variation and improving clinical care. ClinGen has developed widely used resources to support both variant- and gene-level curation. In the area of gene curation, the ClinGen Gene Curation Working group has established standards for assigning the level of evidence of a gene-disease relationship using a semi-quantitative framework88. Where genes are associated with multiple conditions, additional guidance has also been developed to support decision-making on ‘lumping and splitting’ disease entities prior to commencing gene curation94. For example, clinical entities that were once thought distinct may now be considered to be part of a spectrum of a single disease entity if they have been shown to be caused by variants in the same gene, with the same pattern of inheritance and disease mechanism. The ClinGen Gene-Disease Validity framework is designed to guide the assessment of both the quantity and quality of genetic and experimental evidence supporting a gene-disease relationship (Figure 3). The semi-quantitative approach, with defined points assigned based on the quality of each piece of evidence promotes objectivity, transparency and consistency between curations. More points are assigned to genetic evidence (0–12), compared with experimental evidence (0–6), to a maximum score of 18.
Fig. 3|. Process of ClinGen gene curation.

The ClinGen gene curation process encompasses gathering case-level and experimental data, which are then assessed critically using a semi-quantitative framework, prior to expert review and final classification. The final scores assigned are used to classify the strength of the association between the variant and kidney disease.
Genetic evidence can be derived from case-level or case-control data. Case-level data refers to studies describing individuals or families. Each case evaluated is assessed for the robustness of the genetic evidence presented, including inheritance information, variant consequence, evidence of pathogenicity, and segregation with disease in family members if relevant. Case-control data on the other hand refers to studies using statistical analyses to compare variants in cases and controls. Here, evaluation focuses on the quality of the study design including the method used for variant detection, statistical power, and the presence of possible bias and confounding factors. The framework also encourages contradictory evidence to be sought, most commonly new data on the presence of variants in large population databases such as gnomAD at implausibly high frequencies, which may not have been available at the time of the original study.
A variety of experimental studies are also considered under the framework, including those assessing biochemical function, protein interactions, expression, functional alteration, model systems and rescue experiments. Studies reporting functional data from in vivo systems such gene knockout and rescue experiments are assigned more points compared with studies reporting data from cell-based systems such as protein interactions and expression.
The total score from genetic and experimental evidence leads to a suggested clinical validity and can fall into one of seven categories: Definitive, Strong, Moderate, Limited, Disputed, Refuted or No Reported Evidence. For example, a Limited classification requires at least one plausible disease-causing variant to be proposed as being associated with a disease with or without experimental data (0–6 points). By contrast, the Strong rating is typically applied when there are numerous unrelated individuals reported with strong variant-level evidence, coupled with gene-level evidence from different types of experimental data (12–18 points). The Definitive rating is reserved for strong gene-disease assertions that have been upheld over time in the research and clinical setting, with no contradictory evidence emerging for at least three years.
Gene-disease relationship curations are performed by volunteer biocurators who receive dedicated training in the use of the framework and have, or hope to gain, disease area-specific expertise. Prepared curations are then reviewed in dedicated meetings where the evidence is presented and critically appraised by an international panel of gene, disease, and curation experts to establish consensus on the final classification outcome. Final classifications and the supporting evidence used are publicly available on the ClinGen website.
As of December 2023, ClinGen has 46 active gene curation expert panels (GCEPs) across 15 clinical domains including kidney, cardiovascular, haematological, cancer, immunological, metabolic and neurological disorders95. Many GCEPs have already published their assessments, providing valuable guidance to the clinical, research and diagnostic communities and establishing a strong evidence-based foundation for the implementation of genomic medicine in a broad range of disease areas96–99. In one notable example, the Brugada syndrome GCEP assessed 21 genes reported to be associated with the condition and commonly included in diagnostic gene panels100. Only one gene, SCN5A, was found to have Definitive evidence for relationship with Brugada syndrome, and the remaining 20 genes were classified as Disputed, highlighting the value of systematically assessing evidence for gene-disease association prior to clinical use.
The Kidney Clinical Domain Working Group was established in 2020. It has convened international, multidisciplinary expert groups to appraise the clinical validity of gene-disease relationships across five disease areas, spanning the full spectrum of genetic kidney disease: glomerulopathies, tubulopathies, cystic and ciliopathy disorders, complement-mediated kidney diseases, and congenital anomalies of the kidney and urinary tract (Figure 1). It is anticipated that these working groups will evaluate approximately 600 gene-disease relationships. Curation is underway, with some expert groups such as those assessing genes associated with tubulopathies anticipating completion in 2024.
In addition to gene curation, ClinGen also promotes evidence-based practice in variant curation. The most widely used resource is the ClinVar database which accepts depositions of variant data and annotations from a wide range of contributors, including diagnostic laboratories, researchers, expert groups, healthcare providers, patients and other linked databases such as OMIM. As of December 2023, ClinVar contains 3,633,065 variant classification submissions from 2,716 submitters. The development and wide use of this resource have encouraged standardization of variant description and interpretation, as well as the identification and reclassification of variants that have been misinterpreted historically101. To support activities aimed at improving variant classification, ClinGen Clinical Domain Working Groups also establish Variant Curation Expert Panels (VCEPs). These use expert knowledge about individual genes and associated diseases to develop specifications to the ACMG/AMP criteria30 where necessary and apply these refined criteria to a selection of commonly reported and difficult to interpret variants. These are then given ‘three star’ ratings in the ClinVar database to highlight that the variant pathogenicity assertion has been reviewed by a panel of international experts36. Since December 2018, ClinGen has been recognised by the Food and Drug Administration of the United States of America for the quality of its variant resources, the first database to have such recognition. The Kidney Clinical Domain Working Group has established two VCEPs, one related to Polycystic Kidney Disease and the second related to Alport syndrome; the two most common genetic kidney diseases.
Disease naming in the genomics era: time for change?
As the molecular basis for many kidney diseases is elucidated and genomics moves to be commonplace in the nephrology clinic, it will be critical for disease nomenclature and ontology systems to accurately reflect the underlying biology of these disorders and be aligned with contemporary knowledge of disease pathogenesis. At present, inaccuracies and inconsistencies in disease naming can hinder clinical recognition and impact patients by restricting access to appropriate testing, monitoring, and treatment. For example, variants in COL4A3 cause both Alport syndrome 3A, autosomal dominant (MIM#104200) and Haematuria, benign familial, 2 (MIM#620320). However, these two conditions are more appropriately viewed as part of a single spectrum, with the attendant need to provide lifelong surveillance for the development of kidney failure if disease-causing genetic variants are identified.
Both genetic and non-genetic disorders have historically been named to reflect the observable clinical features (single feature or spectrum), pathological findings, or physiological processes or anatomical systems affected, though many diseases have also been named after medical practitioners, patients, ethnic groups or geographical locations linked to the first report in Western medical literature. Naming based on clinical features allows seemingly similar disorders to be considered together which can help guide clinical care, even in the absence of genetic testing to determine precise aetiology102. However, such names generally provide little insight into the underlying pathophysiology which can inform prognosis and management in a more nuanced way. A diagnosis of autosomal dominant polycystic kidney disease, for example, can have very different implications in terms of the chance of developing end-stage kidney failure at an early age depending on whether it’s caused by variants in PKD1, PKD2 or IFT140. Names based solely on clinical features may also require change over time as understanding of the disease evolves.
Genetic diseases, however, can be considered as comprising two components: the phenotype, representing the clinical entity, as well as the genotype, representing the aetiology,with both elements being relevant to diagnosis, counselling, treatment and prognosis. It has therefore been proposed that disease naming for genetic disorders should reflect both aspects via a dyadic naming approach103 in order to provide a name that is medically informative, meaningful, and unique, while simultaneously allowing grouping of related disorders.
The ClinGen Kidney Disease CDWG, in coordination with the ClinGen Disease Naming Working Group and with input from diverse representatives across the global renal community59, have worked to suggest consensus principles to guide genetic nephropathy naming and propose a two-tiered, dyadic naming model that would be appropriate for most Mendelian kidney disorders. In this proposed model, kidney diseases with an established Mendelian basis (a gene-disease relationship classified as moderate or stronger) would have a two-part core name that could be augmented with additional, specific descriptive terms, where applicable. The name for all disorders would follow the format of ‘core disease name - gene name’, whereby the core disease name describes the primary disorder, and the name of the causative gene is appended following this description. The core disease name selected should be reflective of the overall pathology of the disorder, not just the specific phenotype observed at a given point in time and should be kept consistent across closely related disorders to support clinical recognition and patient identity. The gene name is suggested to follow the core disease name, rather than precede it, to support consistency for patients before and after the genetic cause is identified and to be inclusive of patients without an established molecular diagnosis. For some diseases, where well-established, robustly characterized differences (e.g., in epidemiology, inheritance pattern, presentation, management, or outcome) are helpful to differentiate related disorders, additional disease distinguishing phenotype and/or genotype descriptors should also be prepended or appended to the two-part core name. Examples of proposed dyadic names for genetic kidney disorders are presented in Table 3.
Table 3|.
Examples of proposed renamed disease entities
| Gene Name | Historical disease names | Proposed disease name |
|---|---|---|
| PKD1 | PKD1 (MIM#173900) ADPKD |
ADPKD–PKD1 |
| UMOD | Familial juvenile hyperuricaemic nephropathy Uromodulin-associated kidney disease Medullary cystic kidney disease Tubulointerstitial kidney disease, autosomal-dominant, 1 (MIM#162000) |
ADTKD–UMOD |
| CRB2 | FSGS 9 (MIM#616220) Ventriculomegaly with cystic kidney disease (MIM#219730) |
Autosomal-recessive glomerulopathy, with or without ventriculomegaly and renal cysts–CRB2 |
| LMX1B | Nail-patella-like renal disease FSGS 10 (MIM#256020) | Isolated glomerulopathy–LMX1B |
| WNK1 | Pseudohypoaldosteronism, type IIC (MIM#614492) | Familial hypertension with hyperkalaemia–WNK1 |
ADPKD, autosomal-dominant polycystic kidney disease; ADTKD, autosomal-dominant tubulointerstitial kidney disease; FSGS, focal segmental glomerulosclerosis.
In addition to having clear and simple disease names which are easy to remember, provide insight regarding aetiology, and are easily explained to patients and across disciplines, there is growing appreciation for assigning unambiguous numerical disease identifiers that are easily computable and remain stable over time, even if disease entities are further refined and gene names change. Examples of commonly used numerical disease identifier systems in rare disease include OMIM and Orphanet. The MONDO disease ontology is increasingly preferentially used in this context and maps to other existing terminologies including the widely used International Classification of Diseases (ICD) codes104. In addition, MONDO’s development is coordinated with the Human Phenotype Ontology (HPO), which describes the phenotypic features of diseases. As such, MONDO provides a unifying system to describe disease entities that supports data integration by computational systems, and is continuously updated via manual curation and through synchronization with external resources105.
Changing disease names can have widespread and sometimes unexpected ramifications across the research, clinical, diagnostic, and patient communities. In particular, changing well-known and widely accepted disease names, such as Alport syndrome, should be minimized to avoid creating confusion and disrupting communities that connect through disease terminology. Changes must therefore only be considered in a highly collaborative, consultative, and consistent manner with active engagement from all stakeholders relevant to a given disease. Ongoing and open dialogue is critical to progress in this area, and to ensuring that any resultant change is implemented in a manner that generates positive impact through consensus and pragmatism.
Conclusions
Expertly curated information about genes, their function and relationship to kidney disease is the foundation of accurately interpreting genomic data and delivering the promise of personalised medicine through timely diagnosis, improved prognostication and targeted treatments. The success of these endeavours relies on partnership, broad data sharing and the publication of detailed clinical, molecular and experimental evidence to support assertions for new gene-disease relationships in nephrology. The importance of these evidence-based approaches also extends to existing gene-disease relationships and broad engagement with these concepts and processes among the nephrology community is needed. Global collaboration will be key in scaling critical review and appraisal of gene- and variant-level evidence and in maintaining reliable repositories of iteratively curated expert knowledge to ensure equitable diagnostic outcomes and to enhance the clinical utility of genomic testing.
Key points.
Genomic sequencing technologies are transforming our understanding of genetic kidney disease.
Evidence based frameworks, such as the ACMG/AMP guidelines, are critical to appraisal of individual genetic variants.
Curation of clinically relevant relationships between genes and specific phenotypes underpins accurate genomic test and evidence evaluation.
Several collaborative initiatives are undertaking gene-disease curation in genetic kidney disease within the international Gene Curation Coalition, including ClinGen, Genomics England PanelApp, OMIM, Orphanet, and PanelApp Australia
ClinGen uses a semi-quantitative framework with gene-disease relationship classifications determined collaboratively between disease and curation experts on the basis of critical appraisal of human and experimental data.
As the molecular basis of genetic kidney diseases is elucidated, disease nomenclature will need to evolve, moving towards a two-tiered, dyadic naming model, represented as “core disease name – gene name”and augmented with additional terms as applicable, applied consistently for most Mendelian kidney diseases.
Acknowledgements
The authors acknowledge the support and contributions of the ClinGen Kidney Disease Clinical Domain Working Group and its associated Gene Curation and Variant Curation Expert Panels.
AJM is supported by a Queensland Health Advancing Clinical Research Fellowship. ABB is supported by the ClinGen NIH grant U24 HG006834 (to Broad/Geisinger).
Footnotes
Competing interests
The authors declare no competing interests.
References:
- 1.Chronic Kidney Disease Collaboration, Global Burden of Disease. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chadban SJ, et al. Prevalence of kidney damage in Australian adults: The AusDiab kidney study. J Am Soc Nephrol 14, S131–138 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Coresh J, et al. Prevalence of chronic kidney disease in the United States. JAMA 298, 2038–2047 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Fletcher J, McDonald S, Alexander SI, Australian & New Zealand Pediatric Nephrology, A. Prevalence of genetic renal disease in children. Pediatr Nephrol 28, 251–256 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Hildebrandt F Decade in review--genetics of kidney diseases: Genetic dissection of kidney disorders. Nat Rev Nephrol 11, 635–636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vivante A & Hildebrandt F Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12, 133–146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cocchi E, Nestor JG & Gharavi AG Clinical Genetic Screening in Adult Patients with Kidney Disease. Clin J Am Soc Nephrol 15, 1497–1510 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallett A, et al. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis 9, 98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrezenmeier E, et al. The underestimated burden of monogenic kidney disease in adults waitlisted for kidney transplantation. Genet Med 23, 1219–1224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snoek R, et al. Genetics-first approach improves diagnostics of ESKD patients <50 years old. Nephrol Dial Transplant 37, 349–357 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Sedor JR APOL1 Kidney Disease: Discovery to Targeted Therapy in 10 Years. Clin J Am Soc Nephrol 19, 126–128 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornec-Le Gall E, Alam A & Perrone RD Autosomal dominant polycystic kidney disease. Lancet 393, 919–935 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Mallett AJ, Knoers N, Sayer J & Stark Z Clinical versus research genomics in kidney disease. Nat Rev Nephrol 17, 570–571 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Groopman EE, et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N Engl J Med 380, 142–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayasinghe K, et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet Med 23, 183–191 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanudisastro HA, et al. Australia and New Zealand renal gene panel testing in routine clinical practice of 542 families. NPJ Genom Med 6, 20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahl NK, et al. The Clinical Utility of Genetic Testing in the Diagnosis and Management of Adults with Chronic Kidney Disease. J Am Soc Nephrol 34, 2039–2050 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Claus LR, Snoek R, Knoers N & van Eerde AM Review of genetic testing in kidney disease patients: Diagnostic yield of single nucleotide variants and copy number variations evaluated across and within kidney phenotype groups. Am J Med Genet C Semin Med Genet 190, 358–376 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jayasinghe K, et al. Cost-Effectiveness of Targeted Exome Analysis as a Diagnostic Test in Glomerular Diseases. Kidney Int Rep 6, 2850–2861 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, et al. Genomic testing for suspected monogenic kidney disease in children and adults: A health economic evaluation. Genet Med 25, 100942 (2023). [DOI] [PubMed] [Google Scholar]
- 21.Becherucci F, et al. A Clinical Workflow for Cost-Saving High-Rate Diagnosis of Genetic Kidney Diseases. J Am Soc Nephrol 34, 706–720 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrelfs SF, et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N Engl J Med 384, 1216–1226 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Germain DP, et al. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol Genet Metab 137, 49–61 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Heather JM & Chain B The sequence of sequencers: The history of sequencing DNA. Genomics 107, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shendure J, et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Venter JC, et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Stark Z, et al. Integrating Genomics into Healthcare: A Global Responsibility. Am J Hum Genet 104, 13–20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilissen C, Hoischen A, Brunner HG & Veltman JA Unlocking Mendelian disease using exome sequencing. Genome Biol 12, 228 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rehm HL, et al. GA4GH: International policies and standards for data sharing across genomic research and healthcare. Cell Genom 1(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Austin-Tse CA, et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom Med 7, 27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh R, et al. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat 39, 1525–1530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abou Tayoun AN, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat 39, 1517–1524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oza AM, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat 39, 1593–1613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel MJ, et al. Disease-specific ACMG/AMP guidelines improve sequence variant interpretation for hearing loss. Genet Med 23, 2208–2212 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly MA, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med 20, 351–359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fowler DM & Rehm HL Will variants of uncertain significance still exist in 2030? Am J Hum Genet 111, 5–10 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rehm HL, et al. The landscape of reported VUS in multi-gene panel and genomic testing: Time for a change. Genet Med 25, 100947 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunter C & Green ED To boldly go: Unpacking the NHGRI’s bold predictions for human genomics by 2030. Am J Hum Genet 110, 1829–1831 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai P, et al. Recommendations for next generation sequencing data reanalysis of unsolved cases with suspected Mendelian disorders: A systematic review and meta-analysis. Genet Med 24, 1618–1629 (2022). [DOI] [PubMed] [Google Scholar]
- 42.Robertson AJ, et al. Re-analysis of genomic data: An overview of the mechanisms and complexities of clinical adoption. Genet Med 24, 798–810 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Fehlberg Z, Stark Z & Best S Reanalysis of genomic data, how do we do it now and what if we automate it? A qualitative study. Eur J Hum Genet (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bamshad MJ, Nickerson DA & Chong JX Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am J Hum Genet 105, 448–455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baxter SM, et al. Centers for Mendelian Genomics: A decade of facilitating gene discovery. Genet Med 24, 784–797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barker DF, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227 (1990). [DOI] [PubMed] [Google Scholar]
- 47.Li AS, Ingham JF & Lennon R Genetic Disorders of the Glomerular Filtration Barrier. Clin J Am Soc Nephrol 15, 1818–1828 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell 78, 725 (1994). [PubMed] [Google Scholar]
- 49.Rossetti S, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int 75, 848–855 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durkie M, Chong J, Valluru MK, Harris PC & Ong ACM Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genet Med 23, 689–697 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perrault I, et al. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet 90, 864–870 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senum SR, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet 109, 136–156 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoskins BE, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet 80, 800–804 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hwang DY, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int 85, 1429–1433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krug P, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum Mutat 32, 183–190 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Klesert TR, et al. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet 25, 105–109 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Sarkar PS, et al. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat Genet 25, 110–114 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Participants KC Genetics in chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 101, 1126–1141 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cornec-Le Gall E, et al. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol 27, 942–951 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang YH, et al. Refining Genotype-Phenotype Correlation in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol 27, 1861–1868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson JT, et al. Genotype-phenotype correlations for COL4A3-COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci Rep 12, 2722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bekheirnia MR, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol 21, 876–883 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reeders ST, et al. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature 317, 542–544 (1985). [DOI] [PubMed] [Google Scholar]
- 65.Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell 81, 289–298 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Gattone VH 2nd, Wang X, Harris PC & Torres VE Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9, 1323–1326 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Yamamura Y, et al. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J Pharmacol Exp Ther 287, 860–867 (1998). [PubMed] [Google Scholar]
- 68.Torres VE, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367, 2407–2418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torres VE, et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N Engl J Med 377, 1930–1942 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Chebib FT, et al. A Practical Guide for Treatment of Rapidly Progressive ADPKD with Tolvaptan. J Am Soc Nephrol 29, 2458–2470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Besbas N, et al. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int 70, 423–431 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Kavanagh D, Richards A & Atkinson J Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med 59, 293–309 (2008). [DOI] [PubMed] [Google Scholar]
- 73.Thomas TC, et al. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol 33, 1389–1401 (1996). [DOI] [PubMed] [Google Scholar]
- 74.Nurnberger J, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med 360, 542–544 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Gruppo RA & Rother RP Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med 360, 544–546 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Mache CJ, et al. Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 4, 1312–1316 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidtko J, Peine S, El-Housseini Y, Pascual M & Meier P Treatment of atypical hemolytic uremic syndrome and thrombotic microangiopathies: a focus on eculizumab. Am J Kidney Dis 61, 289–299 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Kuzmuk V, et al. A small molecule chaperone rescues keratin-8 mediated trafficking of misfolded podocin to correct genetic Nephrotic Syndrome. Kidney Int (2023). [DOI] [PubMed] [Google Scholar]
- 79.Ding WY, et al. Adeno-associated virus gene therapy prevents progression of kidney disease in genetic models of nephrotic syndrome. Sci Transl Med 15, eabc8226 (2023). [DOI] [PubMed] [Google Scholar]
- 80.Dvela-Levitt M, et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 178, 521–535 e523 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Egbuna O, et al. Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants. N Engl J Med 388, 969–979 (2023). [DOI] [PubMed] [Google Scholar]
- 82.Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24, 349–354 (2000). [DOI] [PubMed] [Google Scholar]
- 83.Kirby A, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45, 299–303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Genovese G, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stark Z, et al. Scaling national and international improvement in virtual gene panel curation via a collaborative approach to discordance resolution. Am J Hum Genet 108, 1551–1557 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yuskaitis CJ, Sheidley BR & Poduri A Variability Among Next-Generation Sequencing Panels for Early-Life Epilepsies. JAMA Pediatr 172, 779–780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.MacArthur DG, et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strande NT, et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am J Hum Genet 100, 895–906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boycott KM, Azzariti DR, Hamosh A & Rehm HL Seven years since the launch of the Matchmaker Exchange: The evolution of genomic matchmaking. Hum Mutat 43, 659–667 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin AR, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet 51, 1560–1565 (2019). [DOI] [PubMed] [Google Scholar]
- 91.DiStefano MT, et al. The Gene Curation Coalition: A global effort to harmonize gene-disease evidence resources. Genet Med 24, 1732–1742 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roberts AM, et al. Towards robust clinical genome interpretation: developing a consistent terminology to characterize Mendelian disease-gene relationships - allelic requirement, inheritance modes and disease mechanisms. Genet Med, 101029 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tudini E, et al. Shariant platform: Enabling evidence sharing across Australian clinical genetic-testing laboratories to support variant interpretation. Am J Hum Genet 109, 1960–1973 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thaxton C, et al. Lumping versus splitting: How to approach defining a disease to enable accurate genomic curation. Cell Genom 2(2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Milko LV, et al. Development of Clinical Domain Working Groups for the Clinical Genome Resource (ClinGen): lessons learned and plans for the future. Genet Med 21, 987–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Welch CL, et al. Defining the clinical validity of genes reported to cause pulmonary arterial hypertension. Genet Med 25, 100925 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Riggs ER, et al. Clinical validity assessment of genes frequently tested on intellectual disability/autism sequencing panels. Genet Med 24, 1899–1908 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walsh R, et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur Heart J 43, 1500–1510 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jordan E, et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 144, 7–19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hosseini SM, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 138, 1195–1205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rehm HL, et al. ClinGen--the Clinical Genome Resource. N Engl J Med 372, 2235–2242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rasmussen SA, Hamosh A & curators O What’s in a name? Issues to consider when naming Mendelian disorders. Genet Med 22, 1573–1575 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Biesecker LG, et al. A dyadic approach to the delineation of diagnostic entities in clinical genomics. Am J Hum Genet 108, 8–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Organization WH International Classification of Diseases, Eleventh Revision (ICD-11). Vol. 2024 (2019). [Google Scholar]
- 105.Vasilevsky NA, et al. Mondo: Unifying diseases for the world, by the world. medRxiv, 2022.2004.2013.22273750 (2022). [Google Scholar]