

Abstract

We present five three-dimensional (3D) coordination polymers (CPs) based on the formate ligand, [NaM(HCOO)3(H2O)]n with M = Co2+ and Ni2+ and [KM(HCOO)3]n with M = Mn2+, Co2+, and Ni2+, introducing three new nuclear structures with the P21 space group for [NaCo(HCOO)3(H2O)]n and [NaNi(HCOO)3(H2O)]n, and P6322 Sohncke SG with chiral nuclear structure for [KNi(HCOO)3]n, along two centric C2/c isomorphs [KMn(HCOO)3]n and [KCo(HCOO)3]n. Magnetic measurements indicate that antiferromagnetic interactions predominate in the five CPs, with averaged antiferromagnetic zJ/kB mean values from −1.18 to −94.9 K. Moreover, magnetic long-range order (LRO) at low temperatures is evidenced by the magnetic susceptibility and heat capacity measurements. Furthermore, single-crystal and powder neutron diffraction experiments were performed to elucidate the magnetic structure, confirming the antiferromagnetic ordering with possible spin canting, thus understanding these systems’ magnetic exchange pathway topology.
Short abstract
We report five three-dimensional formate-based coordination polymers (CPs) with long-range magnetic ordering. A magneto-thermal study reveals general antiferromagnetic behaviors corroborated by neutron diffraction experiments, which evidenced global AF magnetic structures for the five CPs.
Introduction
Materials based on covalently extended coordination entities, or CPs, have been widely studied for more than half a century because they can present attractive properties in catalysis, magnetism, and nonlinear optical properties (NLO), among others.1−6 Furthermore, the rational design of CPs allows us to correlate the structural features with the target properties, making this an active and attractive field of research.7−10 In this sense, nuclear chirality (absence of any mirror symmetry) is a structural characteristic that can raise interesting material properties and enhance other ones already present in the material, such as catalysts in enantioselective synthesis11−13 and NLO activity used as sensors.14−17
On the other hand, magnetic chirality refers to the chiral symmetry of magnetic structures, inducing a preferred rotation of the magnetization.18 Although it could be present in centrosymmetric nuclear structures,19−22 magnetic chirality can also be induced from chiral or noncentrosymmetric nuclear structures as this symmetry feature promotes antisymmetric magnetic interactions (Dzyaloshinskii-Moriya interactions (DMIs)), which play a crucial role in noncollinear magnetic orderings.18,23−25 The interplay between nuclear and magnetic chirality has been previously studied, with the aim of developing new materials for data storage, spintronic, and quantum computing.26−28
The choice of the ligand is a crucial step in the rational design to achieve the desired symmetry features and properties. Regarding this point, carboxylate ligands are attractive candidates in the design of CPs as they can act as both chelating and bridging ligands, affording one-, two-, or 3D networks. Additionally, they can be present simultaneously, conferring a nonlinear linking character to the ligand, therefore adding more complexity to the nuclear structure. More about the bridging character, this group can link two or more cations in different ways, and it has been reported that some of these bridging configurations can be correlated to the nature of magnetic interaction between cations in octahedral symmetry where the magnetic orbitals are the eg level, as has been reported for Cu2+ compounds.29−31 Herein, the syn–syn and anti–anti modes are related to antiferromagnetic interactions as they promote direct overlapping, while the syn–anti is associated with ferromagnetic coupling, as in this case, the direct overlapping is inhibited by the geometry of the bridge (Figure 1).
Figure 1.

dx2–y2 and dz2 orbital disposition in some carboxylate bridge modes.
This magneto-structural correlation can also be evidenced in the work of Jo̷rgensen et al.,32 which reports the magnetic structure of the [Ni(HCOO)2(H2O)2]n 3D CPs elucidated by neutron diffraction.
The formate anion is an interesting building block when it comes to designing magnetic CPs because it is the smallest of its kind, with R = H. This structural character results in compressed frameworks and, thus, shorter distances between spin centers, which can enhance the magnetic interactions, achieving long-range magnetic ordering (LRO).
Moreover, the literature indicates that the arrangement of this organic ligand around the metal cations can lead to acentric or even polar nuclear structures.33−38 An interesting example of this ligand’s structural plasticity is Duan et al.’s work39 about the structural transition of the polymorph [KCo(HCOO)3]n from the Sohncke P6322 space group to the C2/c centric system.
Also, the effect of cation size on the crystallographic phase evolution in [AB(HCOO3)]n (A = Rb, Cs; B = Mn, Co, Ni) and its magnetic properties have been reported by Bovill and Saines.40 Wang et al. studied the derivative with A = NH4.41
On the other hand, the absence of an inversion center in the structure opens the possibility of presenting DMIs between the magnetic cations. In this regard, Ichiraku et al.42 anticipated, by using magnetometry characterization, a chiral soliton lattice originated by a DMI in the {[NH4][Mn(HCOO)3]}n compound. To contribute to the characterization of these magnetic materials, neutron diffraction is a powerful tool for elucidating their magnetic structures. To this date, several examples of successful magnetic structure determinations on formate-based CPs can be found, such as {[CH3NH3][Co(HCOO)3]}n in the work of Mazzuca et al.43 or the incommensurate magnetic structure of {[CH3NH3][Ni(HCOO)3]}n reported by Cañadillas-Delgado et al.44 Greenfield et al.45 reported the magnetic study of the formate-based CP [Co3(HCOO)5Cl(H2O)2]n, where powder neutron diffraction determined a complex arrangement of three helical chains with a triangular arrangement, which led to a homospin topological ferrimagnet and, therefore, to understanding the isothermal magnetization data.
Considering the possibility of synthesizing CPs with acentric or chiral nuclear structures, which could lead to antisymmetric interactions in formate-based frameworks with magnetic LRO, herein, we report the characterization of five 3D CPs based on the formate ligand and Mn2+, Co2+, and Ni2+ cations. Compounds 1 [NaCo(HCOO)3(H2O)]n and 2 [NaNi(HCOO)3(H2O)]n present chiral nuclear structures in the P21 space group, achieved without adding any chiral induction agent, while compound 5 [KNi(HCOO)3]n crystallizes in the Sohncke space group P6322. We carried out a magneto-structural analysis, including X-ray diffraction, magnetometry, and heat capacity measurements, to comprehend their macroscopic magnetic behavior and evidencing magnetic LRO in all the samples. On this basis, we performed powder and single-crystal neutron diffraction experiments to determine the magnetic structure of the five synthesized CPs.
Materials and Characterization
All reagents and solvents were used without any further purification
Synthesis of [NaM(HCOO)3(H2O)]n (M = Co2+ or Ni2+, compounds 1 and 2, respectively)
A 5 mL methanolic solution of 0.625 mmol of CoCl2·6H2O (0.148g) or NiCl2·6H2O (0.148 g) was added dropwise to a solution of 1.30 mmol of NaSCN (0.105 g) in methanol at constant stirring. After 5 min, this solution was immediately added dropwise to a 10 mL methanolic buffer of 13.25 mmol of HCOOH (0.500 mL, 98%) and 5 mmol of HCOONa (0.340 g) and stirred for 5 min. Then, the solution was left undisturbed, and after 2 weeks, deep red crystals were recovered for [NaCo(HCOO)3(H2O)]n (1), and after 1 month, light green powder was recovered for [NaNi(HCOO)3(H2O)]n (2).
Synthesis of [KM(HCOO)3]n (M = Mn2+, Co2+, or Ni2+, compounds 3, 4, and 5, Respectively)
For compound 3, a 5 mL methanolic solution of 0.625 mmol of MnCl2·4H2O (0.124 g) was added dropwise to a solution of 1.30 mmol of KSCN (0.120 g) in methanol at constant stirring, then filtered, and kept filtrated. Meanwhile, 10 mL of a methanolic buffer of 13.25 mmol of HCOOH (0.500 mL, 98%) and 5 mmol of HCOOK (0.340 g) was stirred for 5 min and poured in a 20 mL glass test tube. Then, about 1 mL of MeOH was smoothly added to the surface of the buffer, and later, the MnCl2/KSCN filtered solution was added dropwise to the tube, aiming for a liquid–liquid diffusion system.
Compounds 4 and 5 were prepared when 5 mL of methanolic solution of 0.625 mmol of CoCl2·6H2O (0.148 g) or NiCl2·6H2O (0.148 g) was added dropwise to a solution of 1.30 mmol of KSCN (0.120 g) in methanol at constant stirring. After 5 min, this solution was immediately added dropwise to a 10 mL methanolic buffer of 13.25 mmol of HCOOH (0.500 mL, 98%) and HCOOK (0.340 g) and stirred for 5 min. Then, the solution was left undisturbed; after 1 week, the light pink powder was recovered for [KCo(HCOO)3]n (4), and after 3 weeks, light green crystals were recovered for [KNi(HCOO)3]n (5).
A single crystal of 1, 3, 4, and 5 was directly picked up from the reaction media and glued on a glass capillary using epoxy resin. X-ray diffraction data were collected at room temperature on a BRUKER APEX II diffractometer and processed with the APEX3 program suite, using Mo–Kα as the X-ray wavelength. Frame integration and data reduction were carried out with the program SAINT, and SADABS was employed for multiscan-type absorption corrections. Using the Olex246 package, the structures were solved with the ShelXT47 structure solution program using Dual Space Methods and refined with the ShelXL48 refinement package, using least-squares minimization based on F2. Crystallographic data details on data collection and refinement parameters of the crystal structure are summarized in Table 1. Structure drawings have been elaborated with Crystal Impact’s Diamond 4 software.49 Additional data concerning the crystals and the refinement parameters are detailed in the Supporting Information.
Table 1. Crystallographic Data for Compounds 1–5.
| Compound | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| formula | [NaCo(HCOO)3(H2O)2]n | [NaNi(HCOO)3(H2O)2]n | [KMn(HCOO)3]n | [KCo(HCOO)3]n | [KNi(HCOO)3]n |
| Mw [g mol–1] | 252.99 | 252.77 | 229.09 | 233.09 | 232.85 |
| space group | P21 | P21 | C2/c | C2/c | P6322 |
| radiation | X-ray MoKα | 2.45 Å neutrons | X-ray MoKα | X-ray MoKα | X-ray MoKα |
| T [K] | 296 | 40 | 296 | 296 | 296 |
| a [Å] | 7.3872(3) | 7.3312(4) | 10.8160(2) | 10.7158(12) | 7.0144(4) |
| b [Å] | 7.3982(3) | 7.3138(10) | 9.0584(17) | 8.957(1) | 7.0144(4) |
| c [Å] | 7.9147(4) | 7.8155(5) | 7.0808(13) | 6.8711(8) | 8.2698 (4) |
| β [°] | 116.680(1) | 116.745(4) | 95.175(4) | 95.491(3) | 90 |
| V [Å3] | 386.50(3) | 374.22(8) | 690.90(20) | 656.47(13) | 352.38(4) |
| Z | 2 | 2 | 4 | 4 | 2 |
| ρ [g cm–3] | 2.010 | 2.083 | 2.203 | 2.358 | 2.195 |
| μ [mm–1] | 2.272 | 4.900 | 2.490 | 3.224 | 3.321 |
| F(000) | 234 | 69 | 452 | 460 | 232 |
| sinΘ/λ | 0.0708–0.7188 | 0.0107–0.3778 | 0.0720–0.7213 | 0.0729–0.7151 | 0.0823–0.7201 |
| Rint | 0.0184 | 0.0529 | 0.0239 | 0.0241 | 0.0183 |
| R1 | 0.0134 | 0.0810 | 0.0186 | 0.0192 | 0.0238 |
| wR2 | 0.0327 | 0.0927 | 0.0499 | 0.0470 | 0.0670 |
| GoF | 1.060 | 3.400 | 1.109 | 1.097 | 1.194 |
Magnetic measurements were performed in polycrystalline samples using a Quantum Design Dynacool Physical Properties Measurement System (PPMS) equipped with a Vibrating Sample Magnetometer (VSM). The dc-susceptibility data were collected under an applied magnetic field of 1 kOe in the 1.8–300 K temperature range, except for compound 5, which was measured with 10 kOe, because its magnetic signal was much lower. The isothermal magnetization measurements were performed for 1 and 2 at 2 K, sweeping the applied magnetic field from 0 to ± 90 kOe. Pascal’s constants were considered for diamagnetic corrections.50
Heat capacity measurements were performed using a PPMS equipped with a previously calibrated puck calorimeter at zero applied magnetic field in the temperature range of 1.8 to 35 K. The powder sample was pressed to form a pellet, and then a very small amount of grease (Appiezon N) was used to make proper thermal contact between the pellet and a sample platform. Sample response was obtained from the subtraction of the grease signal from the total contribution of heat capacity inside the calorimeter.
A single-crystal neutron diffraction experiment was performed on a 2 × 1 x 1 mm3 crystal of compound 1 mounted on a vanadium pin on a four-circle D19 diffractometer at the Institute Laue Langevin (ILL), Grenoble (France). The experiment was carried out using a constant wavelength of 1.45 Å and a closed-circuit displex cooling device. Full data acquisitions were made at 4 and 20 K and were used for the nuclear and magnetic refinements. Unit cell determinations were performed using PFIND and DIRAX programs, and processing of the raw data was applied using RETREAT, RAFD19, and Int3D programs.51−53 The data were corrected for the absorption of the low-temperature device using the D19ABSCAN program.54Table S1 contains crystallographic data for the refinement of compound 1 at 20 K.
Powder Neutron diffraction experiments were performed on different instruments at several neutron sources: (i) the cold-neutron diffractometer DMC with a wavelength of 2.45 Å55 and the thermal high-resolution diffractometer HRPT working with 1.49 Å,56 both at the SINQ at the Paul Scherrer Institute (PSI); (ii) the time-of-flight diffractometer POWGEN at the Spallation Neutron Source (SNS) from Oak Ridge National Laboratory (ORNL);57 and (iii) the thermal high-intensity two-axis diffractometer D20 at the ILL58 in Grenoble, (France), using a wavelength of 1.88 Å. Here, we will show only data collected at DMC, for compounds 2 and 5, POWGEN for compound 3, and D20 for compound 4. Table S2 contains crystallographic data for compounds 2–5.
In all of the powder neutron diffraction experiments, the samples were contained in a 6 mm cylindrical vanadium can and placed inside an Orange cryostat. Patterns were acquired above and below the magnetic order temperature previously determined by dc-susceptibility and heat capacity measurements to obtain the structure of both nuclear and magnetic phases for the studied compounds. The data analysis was carried out by performing Rietveld refinement using FULLPROF59 code and the WINPLOTR60 software. We employed the representational analysis proposed by Bertaut61 to determine and label all the possible magnetic structures compatible with their respective space groups and propagation vectors using BasIreps code, included in the Fullprof suite. We also employed the k-SUBGROUPSMAG program from the Bilbao Crystallographic Server (BCS)62−65 to determine the magnetic space groups.
Results and Discussion
Structural Description
[NaM(HCOO)3(H2O)]n, M = Co2+ (1) or Ni2+ (2). 1 and 2 are isostructural crystallizing in a monoclinic unit cell with the P21 Sohncke space group. The crystallographic parameters are given in Table 1. The asymmetric unit comprises an M2+ cation, three formate anions, a sodium ion, and a water molecule. The transition metal cation, M2+, is surrounded by six formate anions reaching a hexacoordination (MO6) with mean distances of dmean(Co–Ofor) = 2.094 (2) Å and dmean(Ni–Ofor) 2.094 (9) Å. Additionally, we calculated the bond valence sum66 (BVS) for Co2+ and Ni2+ ions obtaining values of 2.03 and 1.89, respectively, very close to what is expected (2). Table S3 summarizes the values for all of the compounds.
Moreover, neutron diffraction experiments for 1 at 20 K reveals that dmean(Co–Ofor) = 2.079 (24) Å, indicating similarity, respects the RT structural refinement. More details about neutron diffraction experiments shall be discussed in the following section.
The six formate ligands bridge the M2+ cations in an anti–anti way with two different coordination modes, giving more complexity to the structure. Thus, considering the M2+ and Na+ cations, four of the formates present an anti–anti bridging (μ3–η1:η2) coordination mode (Figure 2B,C), coordinating one M2+ cation by an oxygen atom and one M2+ and Na+ cation by the second oxygen atom, with d(Na–Ofor) = 2.468 (2) Å for 1 and 2.319 (9) Å for 2. Also, it is worth noticing that two of the formate ligands possess the pair M2+/Na+ on the formate plane (in-plane, Figure 2B), while in the other two, the pair M2+/Na+ is located perpendicular to the formate plane (out-of-plane, Figure 2C). These arrangements gave in-plane M–M distances of 5.998 (1) and 5.964 (3) Å for 1 and 2, respectively, while in the out-of-plane, shorter M–M distances of 5.622 (2) Å for 1 and 5.729 (3) Å for 2, respectively, were obtained.
Figure 2.

(A) Asymmetric unit of 1. Coordination modes of formate anion in 1 and 2: (B) μ3-η1:η2 “in-plane”; (C) μ3-η1:η2 “out-of-plane” and (D) μ3-η2:η2.
The remaining two formate anions present an anti–anti chelating–bridging (μ3–η2:η2) coordination mode (Figure 2D), where each oxygen atom coordinates one M2+ and Na+ cation, obtaining Na–Ofor bonds in the range of 2.496 (2)–2.596 (2) Å for 1 and 2.477 (12)–2.657 (12) Å for 2 and M–M distances of 5.844 (1) Å for 1 and 5.585 (3) Å for 2.
In 1 and 2, two subnets are identified considering the M2+ or Na+ cations. Thus, a 2D substructure composed of M4 rhombohedral metal cores is formed by assembling four formate ligands that can be visualized along the ab plane in Figure 3A. The lamellar subnets are stacked between them along the (1 0 1) direction but displaced with respect to the other (Figure 3C). A view along the c-axis locates one M2+ cation of the one M4 core in the center of the rhombohedral arrangement belonging from the above and underneath layers (Figure S1). A second subnet is formed by the Na+ cations that are surrounded by three formate anions, interacting by pairs in a monodentate (Figures 2B,C) and chelating mode (Figure 2D), besides two water molecules, giving place to a hexacoordinated environment (NaO6), with a high distorted octahedral geometry. Then, a second subnet can be identified considering the Na+ cations and the water molecules, arising a zigzag chain that grows along the b-axis (Figure 3B), with Na–OW bond lengths in the range of 2.352 (2)–2.520 (2) Å for 1 and 2.167 (11)–2.566 (9) Å for 2. In this subnet, the Na–OW–Na angle is 138.40 (7)° and 138.72 (6)° for 1 and 2, respectively, while the angle of the OW–Na–OW angle is 107.76 (6)° and 115.23 (1)° for 1 and 2, respectively. Both angles define the zigzag conformation of the Na+ chains.
Figure 3.

(A) 2D subnet of 1 along the ab plane; (B) sodium–water 1D subnet of 1 viewed from the bc plane; (C) two-layer arrangement of 1 along the b-axis perspective, and (D) 3D arrangement for compound 1.
The interaction between the M2+ and Na+ subnets is a product of the coordination of formate ligands in an anti–anti bridging (μ3–η1:η2) and anti–anti chelating–bridging (μ3–η2:η2) coordination mode, as described above (Figure 2D), but also by the hydrogen bond interactions between the water molecules and the formate ligands. Interesting to note is that the HW–Ofor distances are 2.088 (22) Å for 1 and 1.857 (9) Å for 2, in the case of out-of-plane formate ligand, and 1.971 (32) Å for 1 and 1.907 (10) Å for 2, in the case of in-plane formate ligand. The value of the HW–Ofor noncovalent interactions allows us to infer that the water molecules are relatively fixed in the structure, restricting the thermal fluctuations and then fixing the hydrogen positions (Figure S2). Furthermore, the slight difference between the hydrogen bonds could be related to the in-plane and out-of-plane conformations that acquire the formate ligands in the structures of 1 and 2.
Also, it is important to remark that the position of the M2+ cations on the whole net gives place to a corrugated shape in the layers, where a wave shape has a displacement relative to each other, raising hexagonal cavities between the layers (Figure 3C). Figure 3D shows a complete view of these two new 3D networks, [NaM(HCOO)3(H2O)]n, with M = Co2+ or Ni2+.
[KM(HCOO)3]n M = Mn2+ (3) or Co2+ (4) have been reported previously.39,67,68 Both compounds are isostructural, crystallizing in a monoclinic unit cell with the C2/c space group. The lattice parameters of 3 and 4 are found in Table 1. A brief description of the structures is given to understand the magneto-structural correlation better.
In 3 and 4, the cations are coordinated only by formate anions, having an asymmetric unit composed of a hexacoordinated M2+ cation (MO6), three formate ligands, and a potassium ion. The M2+ cation possesses a slightly distorted octahedral environment (Figure 4A), and a dmean(Mn–O) of 2.177 (1) Å and a dmean(Co–O) of 2.099 (1) Å for 3 and 4, respectively. BVSs were calculated, obtaining values of 2.11 for the Mn2+ ion in 3 and 1.99 for the Co2+ ion 4, in both cases close to what is expected (2) (Table S3).
Figure 4.

(A) Asymmetric unit of compound 3, (B) syn–anti coordination mode, and (C) anti–anti coordination mode for the formate ligand in compounds 3 and 4.
Considering the M2+ and K+ cations, the formate ligands are characterized by presenting two bridging coordination modes, syn–anti bridging μ4–η2:η2 and anti–anti chelating–bridging μ5–η3:η3. For the syn–anti μ4–η2:η2, the plane formed by both M–O–K moieties is coplanar with the plane of the formate ligand, with M–O distances in the range of 2.1678 (10) and 2.1762 (10) Å for 3 and 2.095 (1) and 2.101 (1) Å for 4, while the M–M bond lengths achieved are 5.749 (1) Å and 5.645 (1) Å for 3 and 4, respectively. On the other hand, in the anti–anti conformation, the M–O–K moieties are adopting a perpendicular disposition with respect to the formate ligand plane, with an expected longer M–O bond distance of 2.188 (1) Å for 3 and 2.101 (1) Å for 4, affording M–M distances of 6.191 (1) Å for 3 and 6.082 (1) Å for 4.
Similar to compounds 1 and 2, a two-dimensional subnet based on M4 rhombohedral units of the M2+ cation and the syn–anti formate bridge is visualized on the bc crystal plane, forming a lamellar arrangement. Meanwhile, the potassium ions form a second 1D substructure, which can be understood as a zigzag chain along the c crystallographic axis. Herein, the anti–anti formate ligand has a linking role between the cations.
Therefore, the tridimensional polymer structure arises as the zigzag potassium chains are located between the M2+/HCOO layers, arranging tubular cavities that grow along the c axis, with a diameter of ∼4.2 Å, where the hydrogen atoms from both types of formate ligands point to the center of it (Figure 5C).
Figure 5.

(A) M2+/HCOO layers, (B) K+/HCOO chains, (C) two-layer arrangement along the a-axis perspective, and (D) 3D arrangement for compound 3.
Compound 5, [KNi(HCOO)3]n, crystallizes in a hexagonal unit cell with a noncentrosymmetric Sohncke space group P6322 (Table 1). This unreported CP is isostructural to the {[NH4+][M(HCOO)3]}n (M = Mn2+, Co2+, Ni2+) presented in 2007 by Wang et al.41 and to the alkali/nickel [ANi(HCOO3)]n (A = Rb+, Cs+) structures of Bovill and Saines.40 The asymmetric unit comprises a hexacoordinated nickel cation, three formate ligands, and a potassium cation. The hexacoordination of the nickel ion is reached by six formate ligands with the same d(Ni–O) = 2.064 (3) Å, but the octahedral geometry is evidently distorted due to the O–Ni–O angles. The BVS calculated value for the Ni2+ ion is 1.98, in agreement with the expected value (2). Potassium ion is also hexacoordinated with a geometry that could be described as a trigonal antiprism with all K–O distances of 2.843 (3) Å (Figure S3A).
For the Ni2+ cations, the formate ligands are acting in an anti–anti bridging μ4-η2:η2 coordination mode, while the potassium ions are bridged in a syn–syn manner (Figure S3B). The four coordinated cations do not share the same plane, where the K–O–Ni triangles formed in each oxygen atom of the ligand possess a dihedral angle between each other of 50.41 (5)° (Figure S3C). Moreover, the formate bridge affords Ni–Ni distances of 5.787 (1) Å, which would be interesting later from the magnetic property perspective.
Two subnets arise from the 3D arrangement. On one hand, 2D subnet-based M4 units of Ni2+/HCOO are identified on the bc crystallographic plane (Figure 6A), where each cation is connected to other four nickel centers by four formate ligands, forming a layer with corrugated shape, similar to compounds 1 and 2. Meanwhile, 1D substructures composed of potassium ions linked by formate ligands grow along the c crystallographic axis, where the cations are disposed linearly (Figure 6B). The layers are connected by a pair of formate anions in the cis position, giving rise to stacking ordering along the b-axis. The three-dimensional arrangement of the CP is completed when the 1D subnet is identified inside the hexagonal cavities that remain between the Ni2+/HCOO layers (Figure 6C,D).
Figure 6.

(A) Ni2+/HCOO 2D substructure, (B) K+/HCOO 1D substructure, (C) 3D arrangement of two Ni+/HCOO layers, and (D) General view of the 3D arrangement for compound 5.
Magneto-thermal Characterization
dc-Magnetic susceptibility and isothermal magnetization
measurements
were performed to evaluate the magnetic behavior of compounds 1, 2, and 5 and to corroborate the
magnetic behavior of compounds 3 and 4,
previously reported. Heat capacity measurements were also performed
to elucidate the phase transition in compounds 1–5. Figure 7 shows the magnetic dc-magnetic susceptibility (χM) curves measured for compounds 1–5 as a function of the temperature, in the range
of 2 to 50 K. χM(T) plots in the range of 2–300 K is depicted on Figure S4. The χM(T) for compound 1 shows a Curie–Weiss
behavior from 300 to ∼50 K (see insert Figure 7A). At 16.5 K, χM shows an abrupt rise from 6.26 × 10–2 to 1.09 cm3 mol–1, leaving this value
relatively constant to 2 K (Figure 7A), becoming more prominent by using magnetic fields
of less intensity (Figure S5). This abrupt
rise of χM agrees with the peak
observed at TN = 15.2 K at the heat capacity
versus temperature curve, indicating the presence of magnetic LRO,
shown in Figure 8A.
The value of the asymptotic Curie temperature (θCW = −47.5 (7) K) extracted from the fit of χM–1 vs T curve
indicates the presence of antiferromagnetic (AF) interactions on average
of zJ1/kB= −38.0 (6) K, (zJ/kB = 3θCW/S(S+1)). The experimental fitted Curie constant (3.43 (1) cm3 K mol–1) for 1 is higher than the
expected for isotropic ions with S = 3/2 and g = 2, indicating the anisotropic nature of the Co2+ cations. The values of the fitted parameters are listed in Table 2. The theoretical
values of the Curie constant are listed in Table 2 under two different approaches: (i) spin
only approach in which the effective magnetic moment is read as 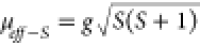 with g = 2 and (ii) weak
spin orbit coupling approximation, in which orbital and spin angular
moments are considered independently. Under this approach, some authors
define the effective magnetic moment as
with g = 2 and (ii) weak
spin orbit coupling approximation, in which orbital and spin angular
moments are considered independently. Under this approach, some authors
define the effective magnetic moment as 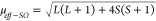 . Considering
a
. Considering
a 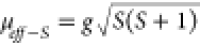 and allowing to vary the Landé g-factor in the fit, we arrive for compound 1 to the
value 2.71 (1), also listed in Table 2.
and allowing to vary the Landé g-factor in the fit, we arrive for compound 1 to the
value 2.71 (1), also listed in Table 2.
Figure 7.

DC magnetic susceptibility versus temperature (main) and fit of the inverse of magnetic susceptibility (insert) and for compounds (A) 1, (B) 2, (C) 3, (D) 4, and (E) 5.
Figure 8.

(A) Heat capacity versus temperature measurements for compounds 1–5. Ordering temperatures are written in the peaks. (B) Magnetization versus magnetic field cycles for compounds 1 (red y-axis) and 2 (green y-axis).
Table 2. Magnetic Parameters of 1–5, Obtained from the Fit of the χM–1(T) Curves.
| compound | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Cexp [cm3 K mol–1] | 3.43(1) | 1.37(1) | 4.41(1) | 3.65(1) | 1.23(1) |
| Ctheo-S [cm3 K mol–1] | 1.875 | 1.00 | 4.375 | 1.875 | 1.00 |
| Ctheo-SO [cm3 K mol–1] | 3.38 | 2.49 | 4.38 | 3.38 | 2.49 |
| g-factor | 2.71(1) | 2.34(1) | 2.01(1) | 2.79(1) | 2.22(1) |
| TN [K] | 15.2 | 30.1 | 3.9 | ∼2.0 | 23.1 |
| θCW [K] | –47.5(7) | –63.3(6) | –3.45(40) | –14.4(4) | –43.1(2) |
| zJ/kB [K] | –38.0(6) | –94.9(1) | –1.18(2) | –11.5(4) | –64.7(3) |
Isothermal magnetization versus magnetic field cycles at 2 K for compound 1 was also measured up to 90 kOe. They are depicted in Figure 8B in the range of −15 to 15 kOe. The curve abruptly increases at low fields and then, after decreasing the slope, continues monotonically increasing until 90 kOe. Some small hysteresis of 540 Oe was detected with a remnant magnetization of Mrca. 0.11 μB, enlightening the presence of a weak ferromagnetic component.
The shape of the χM at low temperatures, together with the hysteresis plots, M(H), could be understood with the presence of some weak ferromagnetic component coming from a slightly canted AF LRO of the Co2+ cations. An estimation of the canting angle (α) for 1 considering the remnant magnetization Mr and the value of the effective magnetic moment extracted from the Curie constant gives a value of α ca. 3.4°.
A similar behavior is observed for compound 2; however, the increment of χM is observed around ca. 31 K, going from 1.26 × 10–2 to 0.107 cm3 mol–1, and as was observed for 1, it reaches higher values as the intensity of the magnetic field decreases (Figure S6). The heat capacity measurement shows a peak at TN = 30.1 K (Figure 8A) that is in agreement with the χM curves. The fit of χM–1(T) for compound 2 gave Cexp and θCW values of 1.37 (1) cm3 K mol–1 and −63.3 (6) K, respectively. It conduces to a g value of 2.34 (1) K for the Ni2+ and an AF average zJ2/kB = −94.9 (1) K (using the same approximation as that for 1). Also, very small hysteresis (∼25 Oe) and remanence (Mr ∼1 × 10–2 μB) are detected for 2 with values one order of magnitude smaller than in 1. For compound 2, a canting angle of 0.5° is estimated.
Susceptibility versus temperature for compound 3 indicates a predominant paramagnetic behavior from room temperature down to ca. 5 K. Then, it abruptly rises at 3.6 K, in agreement with previous reports,67 reaching a maximum value of 0.409 cm3 mol–1 (Figure 7C), which remains constant to 1.8 K. Similar to compounds 1 and 2, this abrupt increase represents a weak ferromagnetic behavior, studied through a hysteresis loop (Figure S7), obtaining an HC ∼100 Oe and a magnetization remanence of Mr∼0.006 μB, being difficult to estimate a canting angle with this very small value. Moreover, the anomaly in the χM curve could indicate some magnetic LRO in agreement with the anomaly at TN = 3.9 K observed in the heat capacity versus temperature curves (Figure 8A) for compound 3. The susceptibility curve shows a small hump (just before TN) possibly related to a crossover from low-dimensional (1D/2D) to 3D magnetic ordering. The fit of χM–1(T) for compound 3 gives Cexp and θCW, respectively, 4.41 (1) cm3 K mol–1 and −3.45 (40) K, from a g value of ∼2.01 for the Mn2+ and an AF average of zJ3/kB = −1.18 (2) K.
For compound 4, the magnetic susceptibility versus temperature plot shows a paramagnetic behavior from room temperature down to 2 K, where a maximum value of 0.776 cm3 mol–1 is observed at 2.0 K (Figure 7D), indicating that an AF LRO could probably appear below 2 K. The heat capacity curve for 4 suggests that a peak is developing for temperatures close to TN∼2 K, although it is not clearly visible because the lowest limit of the experimental temperature is 1.8 K. The analysis of the χM–1(T) measurement at high temperature gives values for Cexp, θCW, and zJ4/kB of 3.65 (1) cm3 K mol–1, −14.4 (4) K, and −11.5 (4) K, respectively. Also, the difference between Cexp and Ctheo−S for 4 could be understood by the anisotropy of the Co2+ cation, and it agrees with the value obtained for 1 (g-factor = 2.79 (1) for 4). The values obtained for Cexp and Msat (∼2.5 μB at 2 K, Figure S8A) agree with the work of Duan et al.;39 however, they found a larger theta value (−24.8 K), which is almost double that of our observation.
The magnetic susceptibility as a function of temperature for compound 5 presents a Curie–Weiss behavior from room temperature until ca. 24 K, where a peak is reached with a value of 1.56 × 10–2 emu mol–1 (Figure 7E). This temperature also coincides with the temperature at which a peak in the heat capacity versus temperature is observed for 5 (Figure 8A), indicating a magnetic LRO of AF nature. Curie–Weiss fitting on the inverse susceptibility plot drops Cexp = 1.23 (1) cm3 K mol–1, higher than the expected value (1.00 cm3 K mol–1) but agrees with the behavior of Cexp of nickel-based compound 2, acquiring an estimated g-factor of 2.22 (1). Additionally, the same analysis affords a θCW = −43.1 (2) K and zJ5/kB = −64.7 (3) K, confirming the AF nature of the exchange between the nickel ions on 5, being both values very close to the obtained for 1, observing that a similar behavior is present in both Ni2+ CPs. In Table 2 are listed the parameters obtained from the analysis of the χM–1(T) curves at high temperatures. Isothermal magnetization measurement at 2 K shows no saturation with Mmax values lower than the expected for a Ni2+ (2.0 μB), in agreement with strong AFM coupling between the ions (Figure S8B).
Moreover, a difference in the lattice heat capacity is observed between compounds 1 and others. We do not have a clear explanation at this moment, but we attribute it to the presence of water molecules in the structure because, in fact, for compound 1, after relatively short times in air exposition, some dehydration transformations are observed, not present in other compounds.
Neutron Diffraction Analysis
Neutron diffraction measurements were performed to investigate the magnetic structure of compounds 1–5. Compound 1 was measured on a single-crystal D19 diffractometer at the ILL using a wavelength of 1.45 Å at 20 K (paramagnetic phase) and at 4 K (ordered phase). At the lowest temperature, the increase of intensity detected in three (0 k 0) reflections with odd k is indicative of some magnetic LRO with a propagation vector k = (0 0 0). By using representational analysis (with the help of BasIreps), we decomposed the magnetic representation ΓM, for the Co2+ cation located at the 2a Wyckoff position of the space group P21, as a direct sum of the irreducible representations (irreps) mΓ1 and mΓ2 of the little group of k (Gk = P21) as shown in Table 3. Both irreps mΓ1 and mΓ2 are real and one-dimensional.
Table 3. Irreducible Representations Decomposition, Fourier Coefficients, Site Symmetry, Magnetic Space Groups, and Fitted Magnetic Moment Components for Cation Sites in Compounds 1–5, which Give the Best Agreement for Rietveld Refinement in the Low-Temperature Patternsa.

In red, the irreps that fits the data.
A simple calculation of the magnetic structure factor (Mq) and the magnetic interaction vector (M⊥q) for the (0 k 0) lines with k-odd for the space group P21 shows that for mΓ2 (Table S4), the magnetic interaction vector is null M⊥(0k0) = 0, and therefore, any magnetic structure described with mΓ2 will never explain the magnetic contribution experimentally observed in (0 1̅ 0), (0 3̅ 0), and (0 5̅ 0), in the ordered phase of 1. Consequently, the only possibility points to a magnetic structure labeled with mΓ1 that allows an AF component perpendicular to the b-axis and a ferromagnetic component parallel to the b-axis. In our neutron diffraction data, we detect only the AF component with a value of 2.63 (35) μB. The ferromagnetic component along b will never be observed in (0 k 0), and within the precision of our experiment, it is indetectable in other lines. However, symmetry allows it, and therefore, it could explain the weak ferromagnetic signal observed at the magnetic susceptibility curve measured for 1. On the other hand, the magnetic moment fitted is comparable to the 3 μB expected for a S = 3/2 as Co2+ with g = 2. Figure S9 contains a plot of the Rietveld refinement of the data.
Additionally, using the k-SUBGROUPSMAG tool from the BCS, we determined the possible magnetic subgroups associated with the parent group P21 and compatible with k = (0 0 0). In this sense, the magnetic subgroup consistent with the irrep mΓ1 is P21.1 (No. 4.7).
The magnetic structure can be described as AF corrugated layers in the ab plane, detailed previously and depicted in Figures 3A and 9B, in which the magnetic moment points toward the a-axis, coupled antiferromagnetically along the c-axis (Figure 9B). This is consistent with the magnetic coupling expected for the anti–anti bridges inside the corrugated lamellar architectures. Furthermore, considering that formate bridges connect the corrugated layers in an anti–anti manner, an AF interaction should also be expected between the layers.
Figure 9.

Magnetic structures for compounds 1 (A,B) and 2 (C,D).
Compound 2 was measured at the DMC instrument at the SINQ facility of the Paul Scherrer Institute, using a constant wavelength of 2.45 Å for the measurements at 40 and 1.6 K. A slight increase in the diffraction peaks (1 0 0) and (1̅ 0 1) around 6.5 Å and (0 0 1) at 7 Å in the d-spacing in the low-temperature pattern is indicative of some magnetic LRO with propagation vector k = (0 0 0). This can be observed in Figure 10A. As both compounds 2 and 1 are isostructural, we can use the same decomposition of ΓM obtained previously for 1.
Figure 10.

For compound 2.(A) Diffractograms measured at 40 K (red), 1.6 K (blue), and difference (green). The magnetic signal is indexed with k = (0 0 0). (B) Rietveld refinement of the 1.6 K diffractogram. The experimental points are in red, the fit is the black continuous line, and the difference between data and fit is the blue line. The green lines signal the positions of the Bragg lines for the nuclear and magnetic phases. The lower green lines correspond to the impurity of [NaNi(HCOO)3]n.
The Rietveld refinement of the pattern in the ordered phase of compound 2 shows a better agreement using mΓ1 (Figure 10B), which describes an AF ordering between the nickel ions along the ac plane with a ferromagnetic component along b (see Table 4). The fitting reveals magnetic moment components of my = 0 and mx = mz = 1.25 (10) with a total magnetic moment of 1.8 (2) μB, close to the 2 μB expected for this cation. Like 1, the magnetic space group for 2 is P21.1 (No. 4.7). A neglectable impurity, impossible to be refined, possibly [NaNi(HCOO)3]n, indexed with the space group P6322 (a = 6.6673 Å c = 8.237 Å), is detected in the powders of compound 2 (see Figure 10B). Like its cobalt isomorph, 1, this 2D AF corrugated structure is coupled antiferromagnetically along the c axis, probably also due to the anti–anti way of the formate bridge that connects the layers in 2. In Figure 9C,D, the magnetic structure of 2 is schematized.
Compound 3 was measured at 4 K in the paramagnetic phase and at 2 K in the ordered phase in the time-of-flight instrument POWGEN (see Figure 11A). In the low-temperature pattern, several nuclear Braggs peaks increase their intensity, e.g., (1 1 0), (0 0 1), (2 0 1), and (0 2 1), which is indicative of magnetic LRO indexed with a propagation vector k = (0 0 0) and in agreement with the susceptibility and heat capacity measurements. The magnetic representation for Mn2+ at the 4c Wyckoff position of the C2/c space group can be decomposed as a direct sum of two irreps (see Table 3 and Table S5). For compound 3, the Rietveld analysis of the ordered phase pattern has better agreement with the symmetry described by mΓ+1, which allows an AF ordering with magnetic moments contained in the ac plane, and ferromagnetism along the b axis (see Table 3), which allows the canting along the y-axis, similar to compounds 1 and 2. This is in agreement with the stair-step behavior of the magnetic susceptibility values near the ordering temperature. The magnetic space group C2/c (No. 15.85) describes the symmetry labeled by mΓ+1.
Figure 11.

For compound 3. (A) Diffractograms measured at 4 K (red) and 2 K (blue) and differences (green). The magnetic signal is indexed with k = (0 0 0). (B) Rietveld refinement of the 2 K diffractogram. The experimental points are in red, the fit is the black continuous line, and the difference between data and fit is the blue line. The green lines are signaling the positions of the Bragg lines for the nuclear and magnetic phases.
The best fit of the new magnetic contribution is reached with mx = −mz = 2.73 (2) and my = 0. It gives an averaged magnetic moment for manganese ions of 3.9 (3) μB, which is lower than the expected value for a S = 5/2 ion (5 μB). This can be due to the fact that measurements are done at temperature, T = 2 K, close to its Néel temperature, TN = 3.9 K, and therefore, the magnetic moments could be still strongly affected by the thermal fluctuations. For compound 3, the magnetic structure can be described by layers along the bc plane in which the magnetic moments are ordered antiferromagnetically along the (1 0 1) direction. These layers are coupled ferromagnetically along the a-axis (see Figure 12A and Figure S10). The magnetic moment has components only along the x and z axes, affording an AF ordering in the ac plane of the system, specifically in the Mn/μ4-HCOO layers described previously, allowing us to infer that the syn–anti formate bridges play an important role in the magnetic interactions of the system.
Figure 12.

Magnetic structure of compounds (A) 3 and (B) 4.
Compound 4 was measured with λ = 1.88 Å at the D20 instrument of the ILL. Diffractograms were collected at the paramagnetic phase (50 K) and the ordered phase (1.6 K), as determined from heat capacity measurements. In the low-temperature pattern, the appearance of new peaks and the increase of intensity in other indexed Bragg’s reflections (Figure 13A) indicate magnetic LRO. The new magnetic signal is indexed with a propagation vector k = (0 1 0), indicating that the new magnetic symmetry has lost the centering translation (1/2 1/2 0) of the parent group C2/c. The Co2+ cations are located at the Wyckoff position (4c) of the C2/c space group, and the propagation vector is k = (0 1 0). The magnetic representation can be decomposed for these constraints as described in Table 3 and Table S6. The fit of the magnetic contribution for compound 4 has better agreement using mY–1, different from the representation used for the isomorphous compound 3 (mΓ+1). Under this irrep, the magnetic moment on the atoms related by the centering translation (1/2 1/2 0) invert their sign. This symmetry describes an AF ordering between the 4 Co2+ atoms in the unit cell and does not allow any ferromagnetic component (see the Fourier coefficients in Table 3). The magnetic space group is P2/c.1′c [C2/c] (No. 13.74).
Figure 13.

For compound 4.(A) Diffractograms measured at 50 K (red) and 1.6 K (blue) and differences (green). The magnetic signal is indexed with k = (0 1 0). (B) Rietveld refinement for the 1.6 K diffractogram. The experimental points are in red, the fit is the black continuous line, and the difference between data and fit is the blue line. The green lines are signaling the positions of the Bragg lines.
The best adjustment of the Rietveld refinement for the 1.6 K pattern is reached with mx = −mz = 1.491 (2) and my = 0 describing magnetic moments along the (1 0 1̅) direction, with a modulus of 2.1 (3) μB. The magnetic structure of 4 can be explained as 2D ferromagnetic layers along the bc plane (with magnetic moments pointing along the (1 0 1̅) axis and in concordance with the lamellar Co/μ4-HCOO substructure. These ferromagnetic layers are coupled antiferromagnetically along the a-axis (see Figure 12B and Figure S11).
In agreement with its manganese isomorph, the Co2+ ion in compound 4 also has a magnetic moment with components in the a and c axes, arising from an AF arrangement in the Co/μ4-HCOO substructure. However, in this case, the layers acquire a ferromagnetic arrangement between each other, as the magnetic moments of the cations along the b-axis possess the same orientation.
The difference between the magnetic structures of 3 and 4 can be explained if one considers the arrangement of the formate bridges and the magnetic orbitals of manganese and cobalt cations. In the case of 3, the manganese ion possesses its five d orbitals with a magnetic character, so it is difficult to establish a magneto-correlation between the carboxylate bridge conformation and the nature of the coupling pathway. This complex mixture of five magnetic orbitals and either both syn–anti and anti–anti conformations lead to a complicated competition between ferromagnetic and AF couplings, which are probably suppressing each other and consequently explaining the low zJ value obtained from the asymptotic Curie temperature (Table 2).
In contrast to the previous situation, compound 4 has two of the three magnetic orbitals belonging to the eg level. The magnetic orbital distribution can be approximated to the scheme of Figure 1. In this case, the syn–anti layers induce ferromagnetic ordering, while the anti–anti bridges cause AF interactions, in agreement with what is proposed for the carboxylate linking configurations.29−32
Diffractograms at the paramagnetic phase (30 K) and the ordered phase (1.6 K) were collected in the DMC instrument using a constant wavelength of 2.45 Å for compound 5. At low temperatures, no new peaks are observed, but just a small increase of intensity of the large d-spacing (low angle) Bragg’s lines is evidenced (Figure 14A), affording us to assume a propagation vector k = (0 0 0). For the Ni2+ cation at the 2d Wyckoff position of the P6322 space group, the magnetic representation is decomposed as a direct sum as expressed in Table 3 and Table S7.
Figure 14.

For compound 5. (A) Diffractograms measured at 40 K (red) and 1.6 K (blue) and differences (green). The magnetic signal is indexed with k = (0 0 0). (B) Rietveld refinement for the 1.6 K diffractogram. The experimental points are in red, the fit is the black continuous line, and the difference between data and fit is the blue line. The green lines are signaling the positions of the Bragg lines.
The low-temperature pattern was fitted by mΓ3 (in red in Table 3), which allowed us to reach the best agreement. This irrep describes the magnetic moments in an AF ordering only along the c axis, which agrees with the negative value of theta from the susceptibility measurements.
Rietveld refinement (Figure 14B) delivers a magnetic moment of 1.47 (9) μB at 1.6 K, smaller than the expected value for an ion with S = 1 as Ni2+ (2.0 μB). In agreement with compounds 3 and 4, this AF ordering grows along the 2D subnet defined by the M4 Ni2+/HCOO units in the ac plane. Similar to compound 3, these AF layers are ordered in an antiparallel fashion along the b axis, allowing the CP to acquire a global AF structure in concordance with the magneto-thermal analysis (Figure 15).
Figure 15.

Magnetic structure for compound 5.
The antiparallel ordering inside the layers in compound 5 is in agreement with the proposed model about the influence of the carboxylate bridge on the magnetic coupling nature for eg magnetic orbitals. In this case, the anti–anti bridges inside layers promotes the AF interactions between the Ni2+, which have (t2g)6(eg)2 electronic configuration. In the same sense, the coupling between two ions from different layers also has an AF character as the formate bridge has also an anti–anti fashion.
The same observation can be made for compounds 1 and 2, where only anti–anti bridges are present in the nuclear structure, both inside and between the layers. For both cases, we only see AF interactions between the ions, in agreement with the proposed model.
On the other hand, the magnetic symmetry determined in compounds 1, 2, and 3 with the neutron diffraction experiments, allows some ferromagnetic component along the b-axis, in agreement with their macroscopic magnetic measurements, whereas for compounds 4 and 5, their magnetic structures are purely AF. However, the formates studied here, even if they do not have an inversion center, do not develop any incommensurate structure, like in the {[CH3NH3][Ni(HCOO)3]}n derivative,44 or helical structure like in the [Co3(HCOO)5Cl(H2O)2]n.45 Moreover, the compounds here described cannot host any chiral soliton lattice, like the one anticipated for the {[NH4][Mn(HCOO)3]}n compound.44
Conclusions
To determine the ordered arrangement of the magnetic spin within 3D lattices, we synthesized five transition metal formate CPs, three of which were synthesized here for the first time, [NaCo(HCOO)3(H2O)2]n (1), [NaNi(HCOO)3(H2O)2]n (2), and [KNi(HCOO)3]n (5), crystallizing in Sohncke space groups. A complete structural, magnetic, and thermal characterization has been done, allowing us to obtain the nuclear structure and determine that compounds 1, 2, and 5 present chiral structures, unlike the compounds [KMn(HCOO)3]n (3) and [KCo(HCOO)3]n (4). Interestingly, only in compounds 3 and 4, the 2D subnet formed by the 3d cations bridged by formate anions are in a syn–anti coordination mode. In the five 3D networks, AF interactions are predominant, and the estimation of the Neél temperatures, which are in good agreement with the maximum observed through the heat capacity measurements, permits us to infer the existence of magnetic long-range ordering in the reported lattices.
Neutron diffraction in powders and single crystals at different temperatures in the ordered and paramagnetic phases allowed information on the magnetic structure for each studied lattice, including the values of the transition metal-ordered magnetic moments. The magnetic structures of each compound at low temperatures have been classified according to the irreps theory, and the magnetic space groups have also been assigned. The compounds 1, 2, 3, and 5 are characterized by presenting AF interactions through the 2D substructure, while the interaction between the lamellas are AF for 1 and 2 and FM for 3 and 5. In contrast, compound 4 is characterized by an FM interaction within the 2D substructure (bc plane) and AF between the lamellas.
This fine crystallographic study and the complete description of the magnetic structure allowed us to understand the role of the different coordination modes of the formate ligand for the assembly of the 3D networks and to generate new materials where ferro- or AF interactions between the metallic cations could be present. Therefore, our approach to obtain antisymmetric magnetic interaction and therefore noncollinear magnetic structures by using formate chiral ligand has been validated through the determination of the magnetic structures of each compound.
Acknowledgments
The authors acknowledge the financial support of CONICYT-FONDEQUIP/PPMS/EQM130086-UNAB and FONDEQUIP 140060, French-Chilean International Research project “Cooperation in Inorganic Chemistry” (IRP-CoopIC), Financiamiento Basal CEDENNA and Laboratorio de Análisis de Sólidos (L.A.S-UNAB). Grant no. PID2022-138492NB-I00-XM4, funded by MCIN/AEI/10.13039/501100011033, and no. E11-23R/M4, funded by Diputación General de Aragón (Spain), supported this work. F.R.S. acknowledges the ANID Ph.D. grant no. 21202331 and the financial support of the Universidad de Santiago de Chile through the POC2023_FQYB1 instrument. V.P.G. and D.V.Y. thank ANID FONDECYT Regular No. 1211394 and No. 1201249. A portion of this research used resources at the SNS, a DOE Office of Science User Facility operated by the ORNL (IPTS-30327). This work is also partly based on experiments performed at the Swiss SNS SINQ (experiment nos. 20222494 and 20222402), from the Paul Scherrer Institut and on experiments performed at the Institut Laue Langevin (proposals 5-31-2930 and DIR-267).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.4c04737.
Additional information on the refinement of single-crystal and powder diffraction data, supplementary magnetization measurements, complete description of the magnetic representations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Novio F. Design of Targeted Nanostructured Coordination Polymers (NCPS) for Cancer Therapy. Molecules 2020, 25 (15), 3449–3465. 10.3390/molecules25153449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwamura N.; Konno T. Heterometallic Coordination Polymers as Heterogeneous Electrocatalysts. Inorg. Chem. Front. 2021, 8 (10), 2634–2649. 10.1039/D1QI00112D. [DOI] [Google Scholar]
- Chelebaeva E.; Larionova J.; Guari Y.; Ferreira R. A. S.; Carlos L. D.; Paz F. A. A.; Trifonov A.; Guérin C. Luminescent and Magnetic Cyano-Bridged Coordination Polymers Containing 4d 4f Ions: Toward Multifunctional Materials. Inorg. Chem. 2009, 48 (13), 5983–5995. 10.1021/ic900378d. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Liu G.; Shi L.; Liu H.; Wang T.; Ye J. Engineering Coordination Polymers for Photocatalysis. Nano Energy 2016, 22, 149–168. 10.1016/j.nanoen.2016.01.029. [DOI] [Google Scholar]
- Hasegawa Y.; Nakanishi T. Luminescent Lanthanide Coordination Polymers for Photonic Applications. RSC Adv. 2015, 5 (1), 338–353. 10.1039/C4RA09255D. [DOI] [Google Scholar]
- Wang J. L.; Wang X. Y.; Wang Y. H.; Hu X. Y.; Lian J. R.; Guan Y. L.; Chen H. Y.; He Y. J.; Wang H. S. Room-Temperature Preparation of Coordination Polymers for Biomedicine. Coord. Chem. Rev. 2020, 411, 213256 10.1016/j.ccr.2020.213256. [DOI] [Google Scholar]
- Chen Y. C.; Guo F. S.; Liu J. L.; Leng J. D.; Vrábel P.; Orendáč M.; Prokleška J.; Sechovský V.; Tong M. L. Switching of the Magnetocaloric Effect of MnII Glycolate by Water Molecules. Chem. - A Eur. J. 2014, 20 (11), 3029–3035. 10.1002/chem.201304423. [DOI] [PubMed] [Google Scholar]
- Näther C.; Wöhlert S.; Boeckmann J.; Wriedt M.; Jeß I. A Rational Route to Coordination Polymers with Condensed Networks and Cooperative Magnetic Properties. Zeitschrift fur Anorg. und Allg. Chemie 2013, 639 (15), 2696–2714. 10.1002/zaac.201300274. [DOI] [Google Scholar]
- Zhao J. P.; Han S. De; Zhao R.; Yang Q.; Chang Z.; Bu X. H. Tuning the Structure and Magnetism of Heterometallic Sodium(1+)-Cobalt(2+) Formate Coordination Polymers by Varying the Metal Ratio and Solvents. Inorg. Chem. 2013, 52 (6), 2862–2869. 10.1021/ic301936k. [DOI] [PubMed] [Google Scholar]
- Du M.; Li C. P.; Liu C. S.; Fang S. M. Design and Construction of Coordination Polymers with Mixed-Ligand Synthetic Strategy. Coord. Chem. Rev. 2013, 257 (7–8), 1282–1305. 10.1016/j.ccr.2012.10.002. [DOI] [Google Scholar]
- Dybtsev D. N.; Bryliakov K. P. Asymmetric Catalysis Using Metal-Organic Frameworks. Coord. Chem. Rev. 2021, 437, 213845 10.1016/j.ccr.2021.213845. [DOI] [Google Scholar]
- Artem’ev A. V.; Fedin V. P. Metal—Organic Frameworks in Asymmetric Catalysis: Recent Advances. Russ. J. Org. Chem. 2019, 55 (6), 800–817. 10.1134/S1070428019060101. [DOI] [Google Scholar]
- Nickerl G.; Henschel A.; Grünker R.; Gedrich K.; Kaskel S. Chiral Metal-Organic Frameworks and Their Application in Asymmetric Catalysis and Stereoselective Separation. Chem.-Ing.-Technol. 2011, 83 (1–2), 90–103. 10.1002/cite.201000188. [DOI] [Google Scholar]
- Thoonen S.; Hua C. Chiral Detection with Coordination Polymers. Chem. - An Asian J. 2021, 16 (8), 890–901. 10.1002/asia.202100039. [DOI] [PubMed] [Google Scholar]
- Sun J.; Liu L.; Cheng F.; Hu M.; Qin W.; Huang R.; Wu G. A Homochiral Lead(II) Double-Stranded Helical Coordination Network as Luminescent Sensor for Iron(III). J. Solid State Chem. 2019, 277 (May), 769–772. 10.1016/j.jssc.2019.05.049. [DOI] [Google Scholar]
- Zhang Y.; Lu L.; Zhu M.; Wang A.; Englert U. A Zn-Coordination Polymer with Serine-Derived Backbone and Its Use as Bifunctional Luminescence Sensor for Ce(III) and Cu(II). J. Solid State Chem. 2022, 306, 122717 10.1016/j.jssc.2021.122717. [DOI] [Google Scholar]
- Yang Y.; Da Costa R. C.; Fuchter M. J.; Campbell A. J. Circularly Polarized Light Detection by a Chiral Organic Semiconductor Transistor. Nat. Photonics 2013, 7 (8), 634–638. 10.1038/nphoton.2013.176. [DOI] [Google Scholar]
- Cheong S.-W.; Xu X. Magnetic Chirality. npj Quantum Mater. 2022, 7, 1. 10.1038/s41535-022-00447-5. [DOI] [Google Scholar]
- Koehler W. C.; Cable J. W.; Wilkinsin M. K.; Wollan E. O. Magnetic Structures of Holmium. I. The Virgin State. Phys. Rev. 1966, 151 (2), 414. 10.1103/PhysRev.151.414. [DOI] [Google Scholar]
- Ghimire N. J.; Dally R. L.; Poudel L.; Jones D. C.; Michel D.; Magar N. T.; Bleuel M.; McGuire M. A.; Jiang J. S.; Mitchell J. F.; Lynn J. W.; Mazin I. I. Competing Magnetic Phases and Fluctuation-Driven Scalar Spin Chirality in the Kagome Metal YMn6Sn6. Sci. Adv. 2020, 6 (51), 1–7. 10.1126/sciadv.abe2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanh N. D.; Nakajima T.; Yu X.; Gao S.; Shibata K.; Hirschberger M.; Yamasaki Y.; Sagayama H.; Nakao H.; Peng L.; Nakajima K.; Takagi R.; Arima T. H.; Tokura Y.; Seki S. Nanometric Square Skyrmion Lattice in a Centrosymmetric Tetragonal Magnet. Nat. Nanotechnol. 2020, 15 (6), 444–449. 10.1038/s41565-020-0684-7. [DOI] [PubMed] [Google Scholar]
- Hirschberger M.; Nakajima T.; Gao S.; Peng L.; Kikkawa A.; Kurumaji T.; Kriener M.; Yamasaki Y.; Sagayama H.; Nakao H.; Ohishi K.; Kakurai K.; Taguchi Y.; Yu X.; Arima T. H.; Tokura Y. Skyrmion Phase and Competing Magnetic Orders on a Breathing Kagomé Lattice. Nat. Commun. 2019, 10 (1), 1–9. 10.1038/s41467-019-13675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet V.; Loire M.; Ballou R. Magnetic chirality as probed by neutron scattering. Eur. Phys. J.: Spec. Top. 2012, 213, 5. 10.1140/epjst/e2012-01661-8. [DOI] [Google Scholar]
- Han M.-G.; Garlow J.; Du K.; Cheong S.-W.; Zhu Y. Chirality Reversal of Magnetic Solitons in Chiral Cr1/3TaS2. Appl. Phys. Lett. 2023, 123 (2), 022405 10.1063/5.0163385. [DOI] [Google Scholar]
- Zorko A.; Pregelj M.; Potočnik A.; Van Tol J.; Ozarowski A.; Simonet V.; Lejay P.; Petit S.; Ballou R. Role of Antisymmetric Exchange in Selecting Magnetic Chirality in Ba 3NbFe 3Si 2O 14. Phys. Rev. Lett. 2011, 107 (25), 2–6. 10.1103/PhysRevLett.107.257203. [DOI] [PubMed] [Google Scholar]
- Sanchez D. S.; Belopolski I.; Cochran T. A.; Xu X.; Yin J. X.; Chang G.; Xie W.; Manna K.; Süß V.; Huang C. Y.; Alidoust N.; Multer D.; Zhang S. S.; Shumiya N.; Wang X.; Wang G. Q.; Chang T. R.; Felser C.; Xu S. Y.; Jia S.; Lin H.; Hasan M. Z. Topological Chiral Crystals with Helicoid-Arc Quantum States. Nature 2019, 567 (7749), 500–505. 10.1038/s41586-019-1037-2. [DOI] [PubMed] [Google Scholar]
- Aiello C. D.; Abendroth J. M.; Abbas M.; Afanasev A.; Agarwal S.; Banerjee A. S.; Beratan D. N.; Belling J. N.; Berche B.; Botana A.; Caram J. R.; Celardo G. L.; Cuniberti G.; Garcia-Etxarri A.; Dianat A.; Diez-Perez I.; Guo Y.; Gutierrez R.; Herrmann C.; Hihath J.; Kale S.; Kurian P.; Lai Y. C.; Liu T.; Lopez A.; Medina E.; Mujica V.; Naaman R.; Noormandipour M.; Palma J. L.; Paltiel Y.; Petuskey W.; Ribeiro-Silva J. C.; Saenz J. J.; Santos E. J. G.; Solyanik-Gorgone M.; Sorger V. J.; Stemer D. M.; Ugalde J. M.; Valdes-Curiel A.; Varela S.; Waldeck D. H.; Wasielewski M. R.; Weiss P. S.; Zacharias H.; Wang Q. H. A Chirality-Based Quantum Leap. ACS Nano 2022, 16 (4), 4989–5035. 10.1021/acsnano.1c01347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y.; Nakashima T.; Yamada M.; Yuasa J.; Rapenne G.; Kawai T. Hierarchical Emergence and Dynamic Control of Chirality in a Photoresponsive Dinuclear Complex. J. Phys. Chem. Lett. 2018, 9 (9), 2151–2157. 10.1021/acs.jpclett.8b00690. [DOI] [PubMed] [Google Scholar]
- Jia L. H.; Liu Z. L.; Zhu L.; Liu W.; Yao K. L. Magnetic Studies of a Syn-Anti Triatomic Carboxylate-Bridging Chainlike Copper(II) Complex Exhibiting Ferromagnetic Exchange. J. Chem. Phys. 2007, 127 (6), 1–7. 10.1063/1.2756827. [DOI] [PubMed] [Google Scholar]
- Das A. K.; De A.; Yadav P.; Lloret F.; Mukherjee R. CuII2, CuII4 and CuII6 Complexes with 3-(2-Pyridyl)Pyrazolate. Structure, Magnetism and Core Interconversion. Polyhedron 2019, 171, 365–373. 10.1016/j.poly.2019.07.023. [DOI] [Google Scholar]
- Zurowska B. Structural and Magnetic Characterization of Cu-Picolinate and Cu-Quinaldinate and Their Mixed Complexes with Water or Halides. Inorg. Chim. Acta 2014, 418, 136–152. 10.1016/j.ica.2014.04.007. [DOI] [Google Scholar]
- Jo̷rgensen M. R. V.; Christensen M.; Schmo̷kel M. S.; Iversen B. B. Magnetic Structure of Ni(DCOO)2(D2O)2. Inorg. Chem. 2011, 50 (4), 1441–1446. 10.1021/ic102008a. [DOI] [PubMed] [Google Scholar]
- Collings I. E.; Manna R. S.; Tsirlin A. A.; Bykov M.; Bykova E.; Hanfland M.; Gegenwart P.; Van Smaalen S.; Dubrovinsky L.; Dubrovinskaia N. Pressure Dependence of Spin Canting in Ammonium Metal Formate Antiferromagnets. Phys. Chem. Chem. Phys. 2018, 20 (37), 24465–24476. 10.1039/C8CP03761B. [DOI] [PubMed] [Google Scholar]
- Sánchez-Andújar M.; Presedo S.; Yáñez-Vilar S.; Castro-García S.; Shamir J.; Señarís-Rodríguez M. A. Characterization of the Order-Disorder Dielectric Transition in the Hybrid Organic-Inorganic Perovskite-like Formate Mn(HCOO)3[(CH 3)2NH2]. Inorg. Chem. 2010, 49 (4), 1510–1516. 10.1021/ic901872g. [DOI] [PubMed] [Google Scholar]
- Xu G. C.; Ma X. M.; Zhang L.; Wang Z. M.; Gao S. Disorder-Order Ferroelectric Transition in the Metal Formate Framework of [NH4][Zn(HCOO)3]. J. Am. Chem. Soc. 2010, 132 (28), 9588–9590. 10.1021/ja104263m. [DOI] [PubMed] [Google Scholar]
- Mullangi D.; Evans H. A.; Yildirim T.; Wang Y.; Deng Z.; Zhang Z.; Mai T. T.; Wei F.; Wang J.; Hight Walker A. R.; Brown C. M.; Zhao D.; Canepa P.; Cheetham A. K. Noncryogenic Air Separation Using Aluminum Formate Al(HCOO)3 (ALF). J. Am. Chem. Soc. 2023, 145 (17), 9850–9856. 10.1021/jacs.3c02100. [DOI] [PubMed] [Google Scholar]
- Dybtsev D. N.; Chun H.; Yoon S. H.; Kim D.; Kim K. Microporous Manganese Formate: A Simple Metal-Organic Porous Material with High Framework Stability and Highly Selective Gas Sorption Properties. J. Am. Chem. Soc. 2004, 126 (1), 32–33. 10.1021/ja038678c. [DOI] [PubMed] [Google Scholar]
- Shang R.; Chen S.; Wang B. W.; Wang Z. M.; Gao S. Temperature-Induced Irreversible Phase Transition from Perovskite to Diamond but Pressure-Driven Back-Transition in an Ammonium Copper Formate. Angew. Chemie - Int. Ed. 2016, 55 (6), 2097–2100. 10.1002/anie.201510024. [DOI] [PubMed] [Google Scholar]
- Duan Z.; Wang Z.; Gao S. Irreversible Transformation of Chiral to Achiral Polymorph of K[Co(HCOO)3]: Synthesis, Structures, and Magnetic Properties. Dalt. Trans. 2011, 40 (17), 4465–4473. 10.1039/c0dt01701a. [DOI] [PubMed] [Google Scholar]
- Bovill S. M.; Saines P. J. Structure and Magnetic Properties of the AB(HCO2)3 (A = Rb+ or Cs+, B = Mn2+, Co2+ or Ni2+) Frameworks: Probing the Effect of Size on the Phase Evolution of the Ternary Formates. CrystEngComm 2015, 17 (43), 8319–8326. 10.1039/C5CE01628B. [DOI] [Google Scholar]
- Wang Z.; Zhang B.; Inoue K.; Fujiwara H.; Otsuka T.; Kobayashi H.; Kurmoo M. Occurrence of a Rare 49·66 Structural Topology, Chirality, and Weak Ferromagnetism in the [NH4][M II(HCOO)3] (M = Mn, Co, Ni) Frameworks. Inorg. Chem. 2007, 46 (2), 437–445. 10.1021/ic0610031. [DOI] [PubMed] [Google Scholar]
- Ichiraku Y.; Takeda R.; Shimono S.; Mito M.; Kubota Y.; Inoue K.; Kato Y. Magnetic Phase Diagram and Chiral Soliton Phase of Chiral Antiferromagnet [NH4][Mn(HCOO)3]. J. Phys. Soc. Jpn. 2019, 88 (9), 094710 10.7566/JPSJ.88.094710. [DOI] [Google Scholar]
- Mazzuca L.; Cañadillas-Delgado L.; Fabelo O.; Rodríguez-Velamazán J. A.; Luzón J.; Vallcorba O.; Simonet V.; Colin C. V.; Rodríguez-Carvajal J. Microscopic Insights on the Multiferroic Perovskite-Like [CH3NH3][Co(COOH)3] Compound. Chem. - A Eur. J. 2018, 24 (2), 388–399. 10.1002/chem.201703140. [DOI] [PubMed] [Google Scholar]
- Canãdillas-Delgado L.; Mazzuca L.; Fabelo O.; Rodríguez-Carvajal J.; Petricek V. Experimental Evidence of the Coexistence of Proper Magnetic and Structural Incommensurability on the [CH3NH3][Ni(COOH)3] Compound. Inorg. Chem. 2020, 59 (24), 17896–17905. 10.1021/acs.inorgchem.0c01722. [DOI] [PubMed] [Google Scholar]
- Greenfield J. T.; Unger C. D.; Chen M.; Izquierdo N.; Woo K. E.; Garlea V. O.; Kamali S.; Kovnir K. A Series of Chiral, Polar, Homospin Topological Ferrimagnets: M3(OOCH)5Cl(OH2) (M = Fe, Co, Ni). Chem. Mater. 2017, 29 (18), 7716–7724. 10.1021/acs.chemmater.7b01755. [DOI] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42 (2), 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71 (1), 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C. Struct. Chem. 2015, 71 (Md), 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington W. T. DIAMOND - Visual Crystal Structure Information System. J. Appl. Crystallogr. 1999, 32, 1028–1029. 10.1107/S0021889899011486. [DOI] [Google Scholar]
- Bain G. A.; Berry J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85 (4), 532–536. 10.1021/ed085p532. [DOI] [Google Scholar]
- Duisenberg A. J. M. Indexing in Single-Crystal Diffractometry with an Obstinate List of Reflections. J. Appl. Crystallogr. 1992, 25 (pt 2), 92–96. 10.1107/S0021889891010634. [DOI] [Google Scholar]
- Katcho N. A.; Cañadillas-Delgado L.; Fabelo O.; Fernández-Díaz M. T.; Rodríguez-Carvajal J. Int3d: A Data Reduction Software for Single Crystal Neutron Diffraction. Crystals 2021, 11 (8), 897. 10.3390/cryst11080897. [DOI] [Google Scholar]
- Wilkinson C.; Khamis H. W.; Stansfield R. F. D.; McIntyre G. J. Integration of Single-Crystal Reflections Using Area Multidetectors. J. Appl. Crystallogr. 1988, 21 (5), 471–478. 10.1107/S0021889888005400. [DOI] [Google Scholar]
- Matthewman J. C.; Thompson P.; Brown P. J. The Cambridge Crystallography Subroutine Library. J. Appl. Crystallogr. 1982, 15 (2), 167–173. 10.1107/S0021889882011728. [DOI] [Google Scholar]
- Schefer J.; Fischer P.; Heer H.; Isacson A.; Koch M.; Thut R. A Versatile Double-Axis Multicounter Neutron Powder Diffractometer. Nucl. Inst. Methods Phys. Res. A 1990, 288 (2–3), 477–485. 10.1016/0168-9002(90)90141-R. [DOI] [Google Scholar]
- Fischer P.; Frey G.; Koch M.; Konnecke M.; Pomjakushin V.; Schefer J.; Thut R.; Schlumpf N.; Burge R.; Greuter U.; Bondt S.; Berruyer E. High-Resolution Powder Diffractometer HRPT for Thermal Neutrons at SINQ. Phys. B Condens. Matter 2000, 276–278, 146–147. 10.1016/S0921-4526(99)01399-X. [DOI] [Google Scholar]
- Huq A.; Kirkham M.; Peterson P. F.; Hodges J. P.; Whitfield P. S.; Page K.; Hűgle T.; Iverson E. B.; Parizzi A.; Rennich G. POWGEN: Rebuild of a Third-Generation Powder Diffractometer at the Spallation Neutron Source. J. Appl. Crystallogr. 2019, 52, 1189–1201. 10.1107/S160057671901121X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.; Fischer H. E.; Henry P. F.; Convert P.; Brunelli M.; Torregrossa J. The D20 Instrument at the ILL: A Versatile High- Intensity Two-Axis Neutron Diffractometer. Recent Developments and Future Prospective. Acta Crystallogr., Sect. A: Found. Crystallogr. 2009, 65, s325. 10.1107/S0108767309093064. [DOI] [Google Scholar]
- Rodríguez-Carvajal J.; Roisnel T. Line Broadening Analysis Using Fullprof: Determination of Microstructural Properties. Mater. Sci. Forum 2004, 443-444, 123–126. 10.4028/www.scientific.net/MSF.443-444.123. [DOI] [Google Scholar]
- Roisnel T.; Rodríguez-Carvajal J. WinPLOTR: A Windows Tool for Powder Diffraction Pattern Analysis. Mater. Sci. Forum 2001, 378-381 (I), 118–123. 10.4028/www.scientific.net/MSF.378-381.118. [DOI] [Google Scholar]
- Bertaut E. F. Representation Analysis of Magnetic Structures. Acta Crystallogr., Sect. A 1968, 24 (1), 217–231. 10.1107/S0567739468000306. [DOI] [Google Scholar]
- Aroyo M. I.; Perez-Mato J. M.; Orobengoa D.; Tasci E.; De La Flor G.; Kirov A. Crystallography Online: Bilbao Crystallographic Server. Bulg. Chem. Commun. 2011, 43 (2), 183–197. [Google Scholar]
- Perez-Mato J. M.; Gallego S. V.; Tasci E. S.; Elcoro L.; De La Flor G.; Aroyo M. I. Symmetry-Based Computational Tools for Magnetic Crystallography. Annu. Rev. Mater. Res. 2015, 45, 217–248. 10.1146/annurev-matsci-070214-021008. [DOI] [Google Scholar]
- Aroyo M. I.; Kirov A.; Capillas C.; Perez-Mato J. M.; Wondratschek H. Bilbao Crystallographic Server. II. Representations of Crystallographic Point Groups and Space Groups. Acta Crystallogr. Sect. A Found. Crystallogr. 2006, 62 (2), 115–128. 10.1107/S0108767305040286. [DOI] [PubMed] [Google Scholar]
- Aroyo M. I.; Perez-Mato J. M.; Capillas C.; Kroumova E.; Ivantchev S.; Madariaga G.; Kirov A.; Wondratschek H. Bilbao Crystallographic Server: I. Databases and Crystallographic Computing Programs. Zeitschrift fur Krist. 2006, 221 (1), 15–27. 10.1524/zkri.2006.221.1.15. [DOI] [Google Scholar]
- Brese N. E.; O’Keeffe M. Bond-valence Parameters for Solids. Acta Crystallogr. Sect. B 1991, 47 (2), 192–197. 10.1107/S0108768190011041. [DOI] [Google Scholar]
- Eikeland E.; Lock N.; Filsø M.; Stingaciu M.; Shen Y.; Overgaard J.; Iversen B. B. Alkali Metal Ion Templated Transition Metal Formate Framework Materials: Synthesis, Crystal Structures, Ion Migration, and Magnetism. Inorg. Chem. 2014, 53 (19), 10178–10188. 10.1021/ic501152j. [DOI] [PubMed] [Google Scholar]
- Wöhlert S.; Wriedt M.; Jess I.; Näther C. Polymeric Potassium Triformatocobalt(II). Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67 (4), m422. 10.1107/S1600536811008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.