

Abstract
PDK1 is crucial for PI3K/AKT/mTOR and Ras/MAPK cancer signaling. It phosphorylates AKT in a PIP3-dependent but S6K, SGK, and RSK kinases in a PIP3-independent manner. Unlike its substrates, its autoinhibited monomeric state has been unclear, likely due to its low population time, and phosphorylation in the absence of PIP3 has been puzzling too. Here, guided by experimental data, we constructed models and performed all-atom molecular dynamics simulations. In the autoinhibited PDK1 conformation that resembles autoinhibited AKT, binding of the linker between the kinase and PH domains to the PIF-binding pocket promotes the formation of the Glu130-Lys111 salt bridge and weakens the association of the kinase domain with the PH domain, shifting the population from the autoinhibited state to states accessible to the membrane and its kinase substrates. The interaction of the substrates’ hydrophobic motif and the PDK1 PIF-binding pocket facilitates the release of the autoinhibition even in the absence of PIP 3 . Phosphorylation of the serine-rich motif within the linker further attenuates the association of the PH domain with the kinase domain. These suggest that while the monomeric autoinhibited state is relatively stable, it can readily shift to its active, catalysis-prone state to phosphorylate its diverse substrates. Our findings reveal the PDK1 activation mechanism and discover the regulatory role of PDK1’s linker, which lead to two innovative linker-based inhibitor strategies: (i) locking the autoinhibited PDK1 through optimization of the interactions of AKT inhibitors with the PH domain of PDK1 and (ii) analogs (small molecules or peptidomimetics) that mimic the linker interactions with the PIF-binding pocket.


Introduction
The monomeric autoinhibited state of 3-phosphoinositide-dependent kinase 1 (PDK1) is still unresolved, and neither is its release in the absence of lipids. PDK1 structurally resembles that of AKT (a.k.a. protein kinase B, PKB), and both can phosphorylate substrates in the presence of signaling phosphoinositide lipids such as phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and phosphatidylinositol 4,5-bisphosphate (PIP2), although PDK1 can also phosphorylate substrates in the absence of PIP3/PIP2. Here we seek to reveal its autoinhibited state and understand how it can be released in the presence and absence of PIP3, and especially, whether its hitherto unknown autoinhibition state could inform an innovative drug strategy for this ultraimportant protein kinase. As we discuss below, we made significant strides in all.
PDK1 serine/threonine kinase is a critical node in the major PI3K/PDK1/AKT/mTOR cell signaling kinase cascade. It phosphorylates AKT and multiple members of the AGC kinase family, activating at least 23 downstream protein kinases. It also acts in the PI3K-dependent, but AKT-independent malignancy route with the PDK1/mTORC2/SGK axis substituting for AKT in survival, migration, and growth, and in homeostasis crosstalk, including Raf/MAPK. It mediates developmental disorders, such as macrocephaly, cardiovascular disease, and acts in insulin resistance and type 2 diabetes, inflammatory and autoimmune disorders. − Its critical role can also be gleaned from the paucity of its mutations, likely since their occurrence, together with its frequent overexpression in cancer, − can elicit oncogene induced senescence (OIS). Altogether, PDK1 is a cardinal therapeutic target, primarily in cancer. − It harnesses PIP3-dependent and independent catalytic mechanisms, raising the tantalizing question of how it is able to readily switch and execute both.
The framework of its mechanism is understood, and the modulation of its substrate specificity also elegantly worked out. Yet, despite efforts, to date no selective drug in the clinics and the vital question of the autoinhibited state, which may help not only in fully elucidating its activation mechanism, but innovative allosteric drug discovery, has not been worked out. Here we uncover the hidden autoinhibited PDK1 conformation, leading us to discover the regulatory role of the linker region, opening a possible new therapeutic window.
PDK1 exists as a dimer in cells, and the one crystal structure of its isolated Pleckstrin homology (PH) domain exhibits a dimer interface. To date, there is no clear evidence for the role of dimerization for AKT. Crystal structures of AKT, where the PH domain interacts with the kinase domain, − raise the question as to whether PDK1 monomeric conformations could also exist in the autoinhibited state. Since the structure of the PH domain of PDK1 resembles that of AKT, it is reasonable to assume that PDK1 monomers may also exhibit such autoinhibited conformations. On the other hand, the linker and C-terminal region of PDK1 are significantly different from those of AKT. If PDK1 can sample such autoinhibited states, it is unclear how it concertedly maintains such conformations through intramolecular interactions between the kinase domain, the PH domain, and the linker region. Such interactions could stabilize the autoinhibited conformations providing a new strategy to allosteric, ATP-noncompetitive kinase inhibitors discovery that would capture the monomeric state. ,
Here, we explore the monomeric conformational states associated with the activation of PDK1. Our results, supported by published experimental data, show that PDK1 can adopt a relatively stable monomeric autoinhibited conformation resembling that of AKT. Unexpectedly, we discover that in this conformation, the linker binds to the PDK1-interacting fragment (PIF)-binding pocket of the kinase domain and simultaneously interacts with the PH domain, promoting the formation of a salt bridge between Glu130 and Lys111, mitigating the association between the two domains. Phosphorylation of the linker Ser393 further weakens the binding of the PH domain to the kinase domain. The shift in the population from the autoinhibited toward the open state suggests a relatively unstable closed state, with a low kinetic barrier, amenable to adopting the active, catalysis-prone state, even in the absence of PIP3. This shift makes PDK1 capable of readily phosphorylating its multiple substrates through PIP3-dependent and independent mechanisms, which could be a reason why in cells PDK1 prefers the dimeric state. Population shift is the paradigm in kinase activation. ,
Our work reveals the “hidden” autoinhibited conformation of PDK1 and determines the role of the linker in regulating PDK1 activation, which then offer an innovative potential drug target. Below, we describe our results and offer a broad mechanistic description of PDK1, its autoinhibition states, activation mechanism, and critically the regulation that we discovered, all in the framework of other kinases and especially, AKT, and leading us to propose original drug discovery approach.
Methods
Construction of the Full-Length PDK1 in Autoinhibition
The full-length PDK1 structure was predicted by AlphaFold (Figure S1A). However, in this model, the kinase domain has only few contacts with the PH domain, and almost no contact with the linker. The radii of gyration of the full-length (1–556) and truncated (71–556) structures are ∼30 Å and ∼26 Å, respectively. To obtain diverse full-length PDK1 structures including those with the kinase domain interacting with the PH domain, we used Rosetta protein–protein docking with the crystal structures of the inactive kinase domain (PDB ID: 3NAX) and the PH domain (PDB ID: 1W1H). We modeled the missing residues in the activation loop of the kinase domain with the cyclic coordinate descent method and refined by the kinematic closure method using the Rosetta loop modeling module. − We randomly placed the initial structure of the PH domain ∼10 Å away from the kinase domain, and applied the Rosetta global docking protocol as follows: (i) we randomly perturbed the PH domain with respect to the kinase domain by 3 Å translation and 8° rotation before the start of each run, (ii) rotated the PH domain around the kinase domain in each run, and (iii) randomized the starting positions of the kinase and PH domains in each run to avoid the effect of the starting positions. We performed three independent Rosetta docking runs, resulting in a total of 30,000 complexes. We sorted these complexes by their binding energy score, which represents the effective binding free energy at the binding interface, selecting the top 8 complexes with the lowest binding energy scores for further modeling of the full-length PDK1. We obtained an additional autoinhibited conformation of PDK1 by superimposing of the crystal structures of the PDK1 kinase domain and PH domain onto the corresponding kinase domain and PH domain of the autoinhibited AKT crystal structure (Figure S1B). At the end, a total of 9 complexes were ready for constructing the full-length PDK1.
Next, we used the same Rosetta loop modeling and refinement methods to model the missing kinase domain loop residues in the linker region (residues 360–408) between the kinase and PH domains in the different models. We selected a linker that satisfies two criteria: (i) the predicted linker features predominantly random coil structures, and (ii) has few contacts with both domains. We added the N-terminal (residues 1–70) and C-terminal tails (residues 550–556) to each model using the same loop modeling protocols. In addition, to meet the two criteria for the linker region and reduce the artifact caused by the interactions of the N-terminal tail with other domains, we selected N-terminal tail models with more compact dimensions. We also included a full-length PDK1 structure predicted by RoseTTAFold, showing a different binding pattern between the kinase and PH domains. In total, we constructed 10 full-length PDK1 models (M1–M10), with the radius of gyration varying between 25 and 27 Å excluding the N-terminal tail from the calculation (Figure S2A). We considered these compact conformations candidate autoinhibited states.
Atomistic Molecular Dynamics (MD) Simulations Protocols
To explore the conformational dynamics of the inactive PDK1, we carried out MD simulations of each model with NAMD 2.14. , The simulation protocols were similar to those used in our previous works. − We represented each PDK1 structure using the updated and modified version of the CHARMM36m force field parameters , and solved in a cubic water box filled with TIP3P water molecules. The minimum distance between protein and the edge of the water box was 12 Å. Each system was neutralized by adding 100 mM NaCl. The Nosé–Hoover Langevin piston pressure control and Langevin temperature control were used to maintain the pressure at 1 atm and the temperature at 310 K. , Long-range electrostatics were treated using the Particle Mesh Ewald (PME) method, with the PME grid spacing of 1.0 Å and PME interpolation order of 6. We calculated the van der Waals interactions using a switching function with a twin cutoff of 10 and 12 Å, and applied the SHAKE algorithm to constrain the motion of bonds involving hydrogen atoms. We used an integration time step of 2 fs. Each system was first energy minimized for 20,000 steps, and then equilibrated in the NVT ensemble (constant volume and temperature at 310 K) for 5 ns with Cα atoms restrained using harmonic restraints. The production run was performed in the NPT ensemble (constant pressure at 1 atm and temperature at 310 K) without any restraints and lasted for at least 1,000 ns. The total simulation systems are summarized in Table S1, including number of replica simulations for each system. Since consistent results were obtained, the following analyses are based on single trajectory for each system.
Results
PDK1 Structure and Substrates
PDK1, a member of the AGC kinase family (PKA, protein kinase A; PKG, protein kinase G; PKC, protein kinase C), phosphorylates a specific threonine or serine residue within the activation loop of at least 23 other AGC kinases, including AKT, PKC isoforms, p70 ribosomal S6 kinase (S6K), serum- and glucocorticoid-induced protein kinase (SGK), and p90 ribosomal protein S6 kinase (RSK). Full-length human PDK1 comprises 556 amino acids, including the N-terminal region (residues 1–70), the kinase domain (71–359), the flexible linker (360–408), the PH domain (409–549), and the C-terminal tail (550–556) (Figure A). The kinase domain shares the conserved structural fold as observed in other protein kinases, consisting of two, N- and C-lobes (Figure B). The small N-lobe contains five β-strands and two α-helices, while the large C-lobe is predominantly α-helical. The ATP-binding site is located between the two lobes, below the glycine-rich loop formed between the β1-strand and β2-strand. The PIF-binding pocket is defined by two α-helices (αB-helix and αC-helix) and two β-strands (β4-strand and β5-strand). , In the active conformation of the kinase domain, the αB-helix and αC-helix are well positioned, and a salt bridge between Glu130 in the αC-helix and Lys111 in the β3-strand is maintained. , Another characteristic feature in the kinase domain is the conversed 223Asp-Phe-Gly225 (DFG) motif, where Asp223 is an important residue for catalysis. , In the active conformation, Asp223 points to the ATP-binding site (DFG-in conformation), but along with Phe224, flips ∼180° in the inactive state (DFG-out conformation). The PH domain is responsible for recruiting PDK1 to the plasma membrane by interacting with the signaling phosphoinositide lipids, PIP3 and PIP2, , and PDK1 monomer–dimer equilibrium. Since crystal structures are only available for individual kinase and PH domains, but not for the full-length PDK1, the role of other regions of PDK1, particularly the linker between the kinase and PH domains, has not been fully elucidated. Full activation requires trans-autophosphorylation of Ser241 on the activation loop. , Autophosphorylation of Thr513 in the PH domain may also contribute. ,, Additional phosphorylation events may further fine-tune PDK1 activity. −
1.

Sequence and crystal structure of PDK1. (A) Sequence of the full-length PDK1. The sequences corresponding to the N-terminal region, kinase domain, linker, PH domain, and C-terminal region are underlined. The nonpolar, basic, acidic, and polar residues are colored black, blue, red, and green, respectively. Two motifs, 375NYD377 and the equivalent hydrophobic motif 383FGCM386 are in the linker region. (B) The crystal structure of the PDK1 kinase domain in the DFG-out conformation (left panel). The PIF-binding pocket is on the kinase surface, formed by αB-helix, αC-helix, β4-strand, and β5-strand. The crystal structure of the isolated PDK1 PH domain (right panel). The N-lobe and C-lobe of the kinase domain are colored in blue and cyan, respectively. The αB-helix, αC-helix, β4-strand, and β5-strand are colored in yellow.
Among the PDK1 substrates, only AKT has a PH domain, which allows it to colocalize with PDK1 at the membrane, bind, get activated by PIP3, and its activation loop to be phosphorylated by PDK1 there. , This unique AKT membrane anchorage requirement among the PDK1 substrates points to its critical role in the PI3K/mTOR pathway. It not only promotes the positioning of AKT in PDK1 physical adjacency, but permits optimal orientation, producing a favorable spatial microcluster organization for efficient cell signaling, suggesting why AKT has been endowed with the PH domain. Other substrates, such as S6K, SGK, and RSK, do not possess PH domains, but can bind to the PIF-binding pocket of PDK1 through the phosphorylated hydrophobic motif (HM) in their C-terminal regions, facilitating PDK1 phosphorylation of their activation segments. , A typical HM contains Phe and Tyr residues that are crucial for the interactions with the PIF-binding pocket. In the crystal structure of PDK1 with ATP and PIFtide (PDB ID: 4RRV), the HM of the bound PIFtide corresponds to the sequence MFRDFDYIA. In contrast to other AGC kinases, PDK1 lacks such HM in its C-terminal region. Instead, a recent study identified an equivalent HM in the linker (383FGCM386) that could bind to the PIF-binding pocket and promote PDK1 trans-autophosphorylation. Another 375NYD377 motif in the linker was also found to promote PDK1 autophosphorylation. Our study reveals the functional role of the linker, how it interacts with the PIF-binding pocket of PDK1 and exactly how it mediates PDK1 activation.
PDK1 Can Adopt Multiple Autoinhibited Conformations
PDK1 can exist in multiple conformational states. It can form an autoinhibitory homodimer in living cells, but dissociates into monomers when Thr513 on the PH domain is phosphorylated. In equilibrium, the monomers are highly dynamic, jumping between the different conformations. Distinct monomer conformations can be stabilized by binding small compounds. However, elucidating the molecular details of the full-length PDK1 conformation is challenging due to the low-resolution data obtained by size exclusion chromatography and small-angle X-ray scattering, and the mechanism by which full-length PDK1 becomes activated in the absence of modulators also remains unclear. To provide the ensembles of the conformations of PDK1 in autoinhibition, we performed MD simulations on the ten independent candidate autoinhibited PDK1 systems (Table S1). During the simulations, they mainly maintained their initial configurations, but some models displayed conformational convergence between different configurations (Figure S2B). To cluster the conformational ensembles, we applied the ensemble clustering method implemented in Chimera , to the conformations generated from the simulations of the 10 models. Conformationally related clusters were generated based on the best pairwise root-mean-square deviations (RMSDs) between different conformations with automatically determined cutoff values in the clustering. The best representative conformations from the top 6 ensemble clusters indicate absence of dominant clusters (Figure ). The populations of the top 6 clusters vary from 15% to 5%, suggesting a highly heterogeneous conformational space of the autoinhibited PDK1. The PH domain can interact with both lobes of the kinase domain (clusters 1 and 3), only the N-lobe (clusters 4 and 5), or the C-lobe (clusters 2 and 6). These interactions may have different outcomes. In cluster 1, the PH domain interacts with the two lobes of the kinase domain, which could impair the binding of ATP and substrate. Depending on the relative position of the PH domain with respect to the kinase domain, the linker displays distinct binding preferences. In cluster 3, the linker region primarily interacts with the N-lobe of the kinase domain. But in cluster 6, the linker region preferentially interacts with the PH domain. Taken together, the clustering results demonstrate that the intramolecular interactions between the kinase domain, the PH domain, and the linker region define the conformations of the autoinhibited PDK1 states.
2.

Clustering of autoinhibited PDK1 conformations. Overlay of all simulated configurations with respect to the kinase domain in a cartoon representation, resulting in clustered locations of the PH domain in a thread representation with different colors (left panel). The best representative conformations from the top 6 ensemble clusters with different populations (right panels). The kinase domain is colored light green, the PH domain in different clusters is colored differently, and the linker region is colored cyan. The population of each cluster is shown in parentheses. Cluster 3 corresponds to the AKT-like autoinhibited conformation.
To further characterize the different binding patterns between the kinase and PH domains, we calculated the relative probabilities of contacting residues at the binding interface for all systems. Because surface charge–charge interactions are important for protein stability, we focused on the analysis of charged interacting residues. We considered charged residues on the surface of the kinase domain as contributing to the PH domain binding, with Lys86 showing the highest probability (Figure A). Lys86 is located on the β1-strand, which forms the glycine-rich loop (G-loop) with the β2-strand. Because of its proximity to the ATP-binding site, Lys86 has been considered as an important contact residue for the development of potent and selective PDK1 inhibitors. Lys86 is also at the binding interface between the kinase and PH domains (Asp433) in the conformation of PDK1(77–549) stabilized by a specific compound. We identified two positively charged motifs 75RKKR78 and 169KYIRK173 on the kinase domain surface, contributing to the association with the PH domains. 75RKKR78 is located at the starting sequence of the kinase domain and connecting to the linker region, and 169KYIRK173 on the αD-helix of the C-lobe. Lys76 is the key residue that forms the phosphate-binding site close to the PIF-binding pocket, and the αD-helix is part of the substrate binding pocket as observed in the crystal structures of PKA (PDB ID: 2QCS) and AKT (PDB ID: 4EKK). , Thus, binding of the PH domain to these regions of the kinase domain would impair the binding of substrates that dock either to the PIF-binding pocket or to the canonical substrate binding site below the ATP-binding site. As expected, we observed charged residues across the surface of the PH domain (Figure B). Lys509 shows the highest contact probability, as it can establish electrostatic interactions with Asp178 or Glu90 of the kinase domain, contributing to the different binding patterns between the PH and kinase domains. Notably, Lys467, Lys495, and Arg521 have been identified as key players that interact with phosphoinositides. Thus, burial of these charged residues by interaction with the kinase domain renders the lipid-binding site in the PH domain inaccessible. Collectively, PDK1 could exist in multiple autoinhibited conformations primarily due to distinct surface electrostatic interactions between the kinase and PH domains.
3.

Key charged residues on the surface of the kinase and PH domains involved in the interdomain interactions of PDK1. Residue–residue contact probabilities for residues on the surface of (A) the kinase domain and (B) the PH domain, obtained by statistical analysis of all simulations. Charged residues with a probability >0.5% are labeled. Mapping of those charged residues with a probability >1.0% on the surface of the kinase domain and the PH domain is also shown. A contact occurs when the Cβ atom of a residue (Cα for Gly) in the kinase domain is within 10 Å of the Cβ atom of a residue (Cα for Gly) in the PH domain. The electrostatic interactions between the kinase and PH domains over the large surface area lead to different autoinhibited conformations.
The Best Representative Model for the Autoinhibited PDK1
We quantitatively assessed the binding preferences of the kinase and PH domains in the autoinhibited PDK1 by their interaction energy, as well as the solvent-accessible surface area (SASA) of the PH domain. As expected, different clusters have different buried surfaces, which are less than ∼8400 Å2 for the free PH domain (PDB ID: 1W1H) (Figure S3A). The interaction energies between the kinase and PH domains also vary among these clusters (Figure S3B). The linker interacts strongly with the kinase domain in clusters 1, 2, and 3, and with the PH domain in clusters 4 and 6. Cluster 3 is of particular interest because (i) it shows the largest SASA and the most favorable domain interactions compared to other clusters, (ii) it originates from the model that adopted an AKT-like autoinhibited conformation, and (iii) the linker binds to the PIF-binding pocket. In the following, we focused on cluster 3 and examined the structural alterations of the inactive kinase domain in response to the interactions with the PH domain and the binding of the linker. To sufficiently sample the conformational space of cluster 3, we extended the corresponding simulation from 1 to 2 μs.
To identify the key residues involved in the interdomain interactions in cluster 3, we calculated the contact maps between different domains. The higher the contact frequency (or probability), the stronger the residue–residue interactions. As shown in Figure , the contact map between the kinase and PH domains reveals that many charged residues in the kinase domain, including Lys120, Glu121, Lys123, Arg129, Lys228, Glu233, Arg238, and Glu256, are involved in the electrostatic interactions with the charged residues in the PH domain, including Glu432, Lys435, Arg436, Glu453, Arg474, Lys495, Glu497, Arg521, and Arg546 (Figure A). The most important electrostatic contacts (probability >50%) are those formed by Glu121-Lys435 (94%), Lys228-Glu453 (84%), Glu233-Lys495 (78%), and Lys120-Glu432 (60%). In addition, less frequent electrostatic contacts (probability <50%, unlabeled) are made by Lys238-Glu497 (35%), Arg129-Glu453 (23%), and Glu233-Arg474 (23%). The involvement of these charged residues in the binding interface defines the high-affinity association of the PH domain with the kinase domain in this AKT-like autoinhibited PDK1. In cluster 3, the binding of the linker to the PIF-binding pocket in the kinase domain is unexpected as it was not observed in other clusters. Clusters 1 and 2 show a relatively strong interaction between the linker and the kinase domain (Figure S3C), but the linker interacts with the C-lobe of the kinase domain (Figure ). This result suggests that the binding of the linker to the PIF-binding pocket depends on the relative positions of the kinase and the PH domains. Since the PIF-binding pocket is in the hydrophobic groove of the N-lobe of the kinase domain, the hydrophobic interactions between Phe383 and Ile118, and the π–π stacking between Phe383 and Tyr126 occur with high probabilities (>90%, Figure B). Electrostatic interactions are also observed between the positively charged residues in the kinase domain (Lys76, Arg131, and Lys144) and the negatively charged residues in the linker region (Asp368, Asp371, and Asp377). The most frequent interactions include Lys76-Asp377 (85%), Arg131-Asp371 (71%), and Lys144-Asp368 (70%). These interactions further stabilize the binding of the linker to the PIF-binding pocket. A previous study showed that Lys144 mutations (K144E and K144A) decrease PIFtide binding. In agreement with the experimental result, our result also highlights Lys144 role in the linker binding. We further identify the four interacting residues Lys76, Arg131, Thr148, and Gln150 forming the phosphate-binding site that interacts with the phosphorylated C-terminal HM of PDK1 substrates. The involvement of the linker Asp residues (Asp368, Asp371, and Asp377) is also consistent with the experimental evidence that some PDK1 substrates such as PKA, PKCζ and PRK2 (protein kinase C-related kinase-2), which possess an acidic (Glu/Asp) residue rather than a Ser/Thr in the C-terminal HM, can also interact with the PIF-binding pocket of PDK1. The linker interacts with the PH domain through its serine-rich region (389SSSSSSHSLSAS400) (Figure C), but no significant specific electrostatic interactions were identified, indicating a relatively weak association of the linker with the PH domain.
4.

Electrostatic interactions contribute to the stability of the AKT-like autoinhibited PDK1 (cluster 3). Contact maps between the kinase and PH domains (A), between the kinase domain and the linker (B), and between the PH domain and the linker (C) calculated for the conformations in the AKT-like autoinhibited PDK1 (cluster 3). Snapshots representing the key residue pairs involved in the interdomain interactions are also shown. The contacting residue pair with a probability >50% is labeled. The underlined residues (Lys76, Arg131, Thr148, and Gln150) correspond to the phosphate-binding site of the kinase domain of PDK1 (PDB ID: 1H1W). S389–S393 denotes the serine-rich motif 389SSSSS393.
Binding of the Linker to the PIF-Binding Pocket Promotes PDK1 Transition to an Active-like Conformation
To confirm the binding site of the linker, we superimposed the conformations of the AKT-like autoinhibited PDK1 with the crystal structure of the PDK1 kinase domain in complex with ATP and PIFtide. The linker binds to the same hydrophobic groove formed by the αB-helix, the αC-helix, the β4-strand, and the β5-strand as the PIFtide in the crystal structure (Figure A). In contrast to the active conformation of the kinase domain, the αB- and αC-helices of inactive PDK1 remain largely unstructured. To further examine the effects of the linker binding, two hallmark features of protein kinases, the Lys111-Glu130 salt bridge and the glycine-rich loop, were compared with the PDK1 active state. In the active conformation of PDK1, the formation of a salt bridge (<4 Å) between Glu130 in the αC-helix and Lys111 in the β3-strand, positions ATP for phosphoryl transfer. The crystal structure of the inactive state shows ∼10 Å distance between the two residues. The AKT-like inactive PDK1 (cluster 3) samples the active-like conformation with high probability salt bridge formation (Figure B). This active-like behavior was observed only in cluster 3. Furthermore, we observed that salt bridge formation is correlated with the binding of the linker to the PIF-binding pocket, as indicated by a decrease in the Arg131-Asp371 distance as the Lys111-Glu130 distance decreases (Figure C).
5.
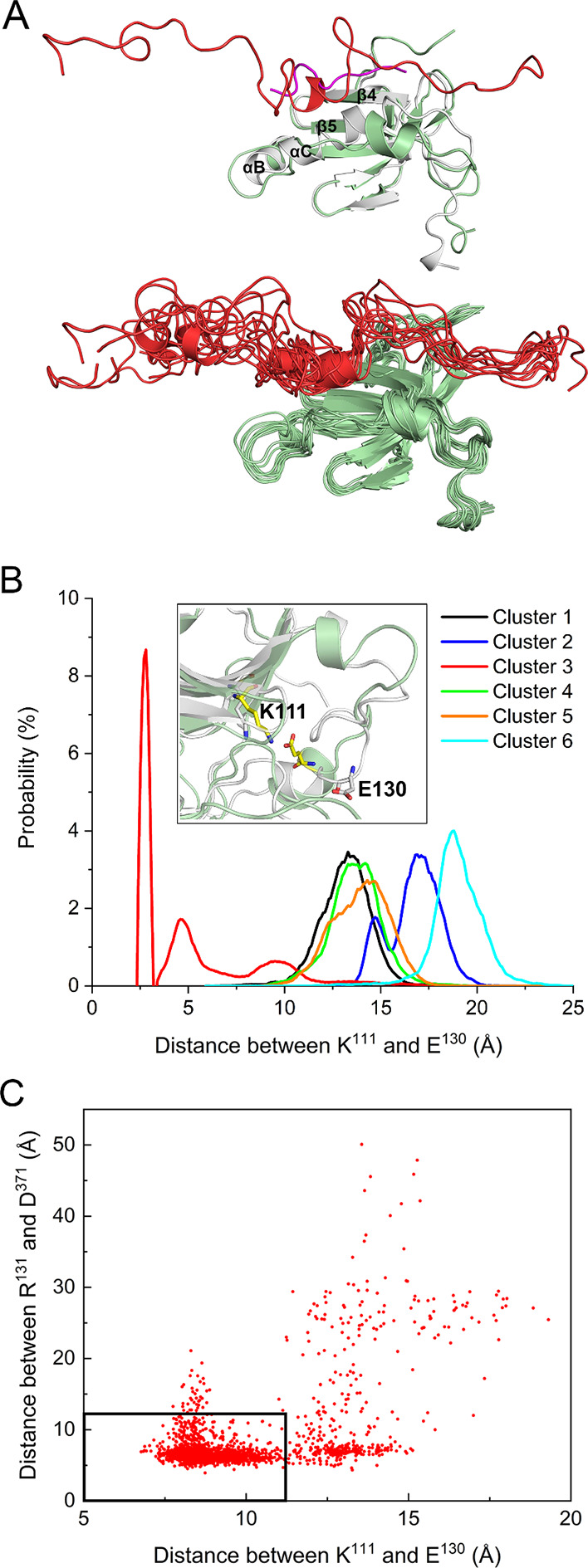
Salt bridge formation in the AKT-like autoinhibited PDK1 (cluster 3). (A) Superimposition of the PIF-binding pocket with the crystal structure of PDK1 kinase domain in complex with PIFtide (purple, PDB ID: 4RRV). The bound linker in the autoinhibited PDK1 is shown in red. The dynamic conformations of the linker in the PIF-binding pocket are also shown. (B) The probability of the Glu130-Lys111 salt bridge formation in different PDK1 clusters. This salt bridge is only formed in cluster 3, and the inset shows an illustration of the formation of this salt bridge. For comparison, the salt bridge in the active kinase domain is also shown (white cartoon, PDB ID: 4RRV). (C) Correlation between the binding of the linker to the PIF-pocket and the formation of the Glu130-Lys111 salt bridge. The center of mass distance between Arg131 and Asp371 is used to characterize the binding of the linker to the PIF-binding pocket. The dense data points in the black rectangle (12 Å × 12 Å) suggest that the decrease in the distance between Arg131 and Asp371 corresponds to the decrease in the distance between Glu130 and Lys111 (formation of the salt bridge).
To further confirm this observation, we performed MD simulations of N-terminal truncated PDK1(71–556) (with the linker), and PDK1(71–556)ΔLinker (without the linker). Excluding the N-terminal tail avoids the effects of its interactions with other regions. The AKT-like autoinhibited conformation taken at 1 μs from the M1 simulation was used as the starting conformation for the new simulations (Figure.S4A). PDK1(71–556) displays the highest probability at the same distance (3 Å) as the full-length PDK1, suggesting formation of the Lys111-Glu130 salt bridge (Figure S4B). However, in PDK1(71–556)ΔLinker, where the kinase domain only interacts with the PH domain, the probability of salt bridge formation is greatly reduced because the peak is located at 8 Å, indicating an increased Lys111-Glu130 distance. This suggests that the binding of the linker to the PIF-binding pocket promotes the formation of the Lys111-Glu130 salt bridge.
The glycine-rich loop is located on top of the ATP-binding site, and mutations of glycine affect the catalytic efficiency of the protein kinase. We used the distance between the glycine-rich loop (residues 90–94) and Asp205 to describe the kinase domain hinge motion between the two lobes, which can adopt open and closed conformations of the active site. For PDK1 in the active state, the average center of mass distances between the glycine-rich loop and Asp205 are 14.7 Å in the presence of ATP (PDB ID: 1H1W) and 15.1 Å in the presence of both ATP and the PIFtide (PDB ID: 4RRV). These active kinase domains states represent the closed state of the active site. For PDK1 in the inactive state, the corresponding distance is 14.8 Å in the presence of an inhibitor (PDB ID: 3NAX). Simulations of inactive PDK1 in the absence of the inhibitor or ATP enabled us to monitor the hinge motion of the kinase domain by calculating the distance between the glycine-rich loop and Asp205 (Figure S5). The inactive apo-kinase domains appear to retain the closed active site conformation (e.g., clusters 1, 2, 3, and 6), but also exhibit the open active site conformation (e.g., clusters 4 and 5). The interaction of the PH domain with the N-lobe kinase domain appears to lead to the open conformation as in clusters 4 and 5 (Figure ). To further check whether those clusters undergo hinge motion, we calculated the distribution of the distance between the glycine-rich loop and Asp205 in each cluster (Figure A). Except for cluster 2, which shows a bimodal distribution but still within 15 Å, the other clusters display unimodal distributions, suggesting the presence of only one dominant conformational state and thus no observable hinge motion in the kinase domain. Cluster 3, the AKT-like autoinhibited PDK1, has the shortest distance of 11.2 ± 0.4 Å compared to other clusters. Superimposition with the crystal structure of the active kinase domain shows that the glycine-rich loop shifts toward the ATP-binding site, resulting in steric clashes with the ATP-binding pocket (Figure B), suggesting that loading of ATP is hindered by the closed active site in autoinhibited PDK1. To explore the effect of linker binding to the PIF-binding pocket, we also calculated the distance between the glycine-rich loop and Asp205 for the two systems, PDK1(71–556) and PDK1(71–556)ΔLinker (Figure C). In the first case, an increase in the distance of 8% (12.1 ± 0.1 Å) is observed, whereas a large increase of 27% (14.2 ± 1.0 Å) is obtained for the second, suggesting that the interaction of the linker with the PIF-binding pocket facilitates the formation of closed active site conformations in autoinhibition.
6.
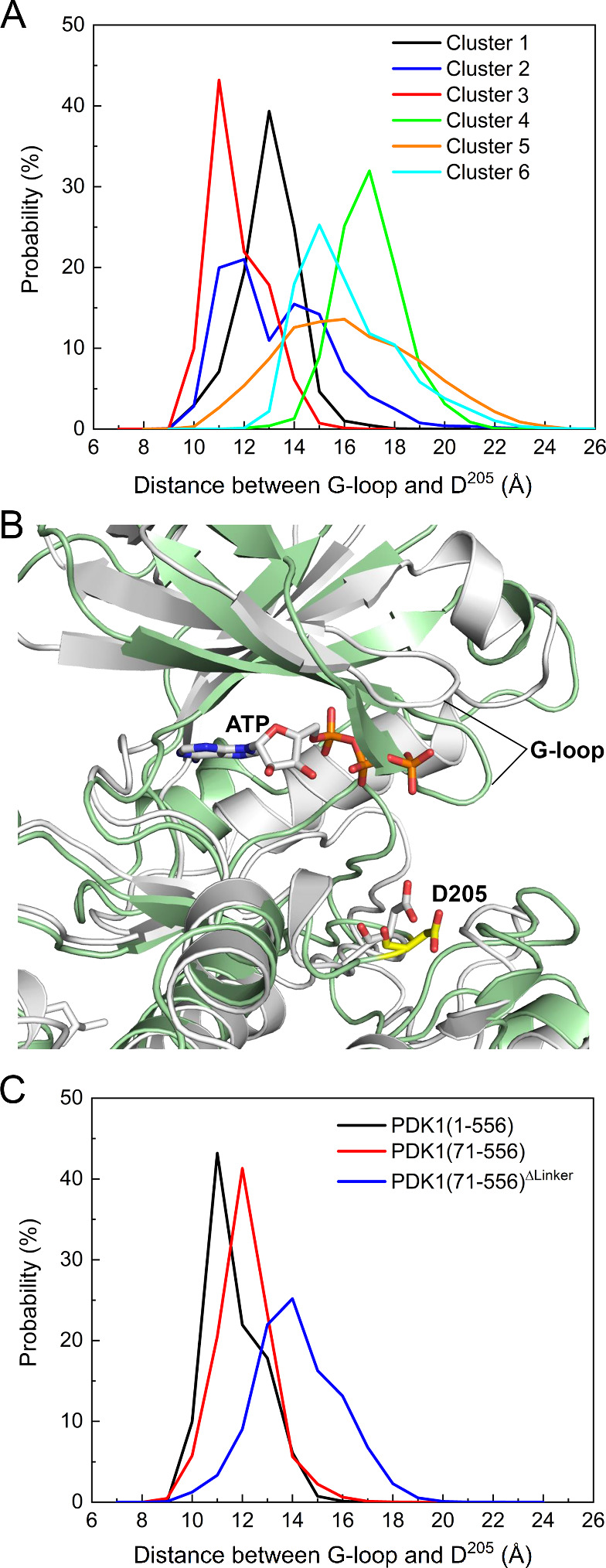
The AKT-like autoinhibited PDK1 shows a closed conformation of the ATP binding site. (A) Distribution of the distance between the center of mass of the glycine-rich loop (G-loop, residues 90–94) and Asp205 for full-length PDK1 in different clusters. Cluster 3 displays the shortest distance between the G-loop and Asp205, resulting in a closed conformation of the active site. (B) Comparison of the positions of the G-loop and Asp205 in the autoinhibited PDK1 (cluster 3) and in the crystal structure after superimposition of the conformation of the autoinhibited PDK1 (green) with the crystal structure of the active kinase domain (white, PDB ID: 4RRV). The conformation of cluster 3 is not accessible to ATP. (C) Distribution of the distance between the G-loop and Asp205 for truncated PDK1. PDK1(71–556) denotes the N-terminal truncated PDK1, and PDK1(71–556)ΔLinker denotes the deletion of both the N-terminal tail and linker. The conformation of the full-length PDK1 at 1 μs from the M1 simulation is used as the starting conformation for both PDK1(71–556) and PDK1(71–556)ΔLinker. All PDK1 refers to the AKT-like autoinhibited conformation (cluster 3).
Effects of the Linker and Phosphorylation on the Autoinhibited PDK1
The structural changes induced by the binding of the linker to the PIF-binding pocket in the small lobe of the kinase domain lead us to ask whether this binding affects the association of the PH domain with the kinase domain. This cardinal question bears on the role of the linker and could have implications for innovative drug discovery. We calculated the interdomain interaction energies for the simulations of the AKT-like PDK1(71–556) and PDK1(71–556)ΔLinker, and compared them with those of full-length PDK1 (Figure ). In the absence of the N-terminal tail and the linker, the interaction energy between the kinase and PH domains becomes more favorable (decreased by 26%), suggesting that the binding of the linker to the PIF-binding pocket actually destabilizes the association of the kinase domain with the PH domain. In the presence of the N-terminal tail, the interaction between the two domains of PDK1(1–556) is more favorable than that of PDK1(71–556), suggesting that the N-terminal tail may stabilize the autoinhibited PDK1. The interaction of the linker with the kinase domain becomes less favorable in PDK1(1–556) than in PDK1(71–556), indicating that the N-terminal tail may also competitively interact with the linker region and consequently reduce the interaction between the kinase domain and the linker region. The PH domain interacts with the serine-rich motif of the linker, including Ser393 (Figure C). Phosphorylation of Ser393 has been shown in vivo, and mutation of Ser393 to Ala did not affect PDK1 activity. To examine the role of the linker in modulating the autoinhibited PDK1, we simulated PDK1(1–556) with phosphorylated Ser393. The phosphorylation results in less favorable interactions between the kinase and PH domains compared to the wild-type PDK1, suggesting that Ser393 phosphorylation tends to destabilize the autoinhibited PDK1 and facilitates its activation, in agreement with previous study. Thus, going back to our question above, binding of the linker to the PIF-binding pocket destabilizes the association of the PH domain with the kinase domain, with PH domain interacting with the linker’s serine-rich motif. Ser393 phosphorylation degrades the interactions between the kinase and PH domains, further shifting the landscape toward the active state. As to the N-terminal tail, its competitive interaction with the linker region reduces the interaction between the kinase domain and the linker region, shifting the ensemble toward the closed autoinhibited state, collectively pointing to the regulatory role of the linker and its hitherto not understood interplay with the N-terminal.
7.
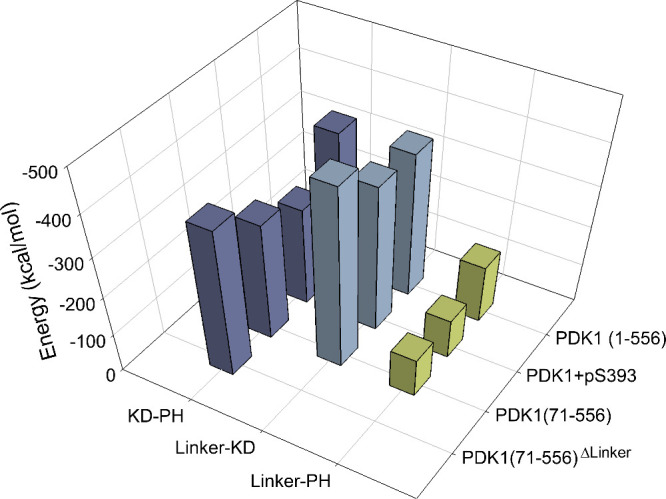
The presence of the linker or phosphorylation of S 393 weakens the interactions between the kinase and the PH domains. Interdomain interaction energies calculated for the full-length PDK1 with S393 phosphorylation, N-terminal truncated PDK1, and N-terminal truncated PDK1 without the linker. For comparison, the interaction energy of the full-length PDK1 is included. All PDK1 refer to the AKT-like conformation (cluster 3). PDK1(1–556)+pS393 denotes the full-length PDK1 with S393 phosphorylation. PDK1(71–556) denotes the N-terminal truncated PDK1, and PDK1(71–556)ΔLinker denotes the N-terminal truncated PDK1 without the linker.
Discussion
AGC Kinase Autoinhibition
Kinase activation requires release of its autoinhibition. , Many AGC kinases, including diverse PDK1 downstream substrates, exhibit autoinhibited conformations in which the substrate binding sites of their kinase domains are blocked by other domains/regions. Release from the autoinhibited states is achieved by distinct mechanisms. In the autoinhibited PKA and PKG, the kinase domains interact with their respective regulatory domains, which block substrate binding. Activation of these kinases is triggered by the binding of cAMP or cGMP to their respective regulatory subunits. For PKC, which is cyclic nucleotide-independent, its regulatory domain contains an autoinhibitory pseudosubstrate domain that resembles the PKC substrate but does not contain a serine/threonine phosphorylation site. In the autoinhibited PKC, the pseudosubstrate domain occupies the catalytic site, thus activation of PKC requires release of this autoinhibitory domain from the kinase core. − Inactive RSK1 and RSK2 are in autoinhibited forms in which the C-terminal inhibitory helix interacts with the kinase core. , In autoinhibited S6K, the C-terminal autoinhibitory domain interacts with the N-terminal kinase domain. Phosphorylation of the autoinhibitory domain disrupts their interactions. − The autoinhibited AKT reveals an interdomain interaction between its N-terminal PH domain and kinase domain, which is relieved by C-tail phosphorylation or PIP3. ,,, In the proposed structural model of the full-length autoinhibited AKT, the unphosphorylated C-terminal tail binds to the PIF-binding pocket of the kinase domain.
PDK1 Autoinhibition Appears to Resemble That of AKT
An autoinhibited state of PDK1 has been suggested by several experiments. ,, If PDK1 could adopt the autoinhibited conformation as its substrates do, it should likely resemble that of AKT, as both contain a PH domain. This work provides structural evidence for the existence of such a conformation. Compared to other possible autoinhibited PDK1 conformations, the one constructed based on the crystal structure of the autoinhibited AKT (cluster 3) shows the most favorable interaction between the kinase and PH domains (Figure S3B). This domain interaction involves multiple strong electrostatic interactions (Figure ). The stable association between the two domains is further supported by our simulation results of the truncated PDK1(71–556)ΔLinker containing only the two domains, which shows an enhanced domain interaction compared to PDK1(1–556) and PDK1(71–556) (Figure ). The finding that the N-terminal tail stabilizes the domain interactions highlights its functional role in the autoinhibited PDK1.
The Regulatory Role of the Linker
Several PDK1 substrates, including S6K, SGK and RSK, require the binding of their respective C-terminal HM to the PIF-binding pocket of PDK1 to be phosphorylated at their activation loops. , A recent study identified an equivalent HM (383FGCM386) as well as the 375NYD377 motif from the linker that can promote PDK1 trans-autophosphorylation by the binding of the linker of one protomer to the PIF-binding pocket of opposing protomer. However, it has been unclear whether the linker itself can bind to the PIF-binding pocket via self-interaction within a PDK1 protomer. The present work provides clear evidence that the linker can bind to the PIF-binding pocket of the full-length autoinhibited PDK1 (Figure A). In addition to hydrophobic interactions, electrostatic interactions further contribute to the binding of the linker to the PIF-binding pocket (Figure ). Electrostatic interactions also played an important role in the binding of PIFtide to the HM of (mouse) Akt. In the autoinhibited PDK1, the binding of the linker to the PIF-binding pocket demonstrates two outcomes. First, it promotes the formation of the salt bridge between Lys111 and Glu130 (Figure B), a characteristic feature of the active kinase that is critical for catalysis. Earlier studies observed the conformational changes in the ATP-binding site induced by binding of small molecules to the PIF-binding pocket of PDK1. , The autoinhibited PDK1 retains the closed conformation of the active site, but also exhibits the open conformation of the active site (Figure A). The latter conformation, which has an exposed active site, refers to a partially autoinhibited PDK1 that is susceptible to ATP loading and activation. Our studies suggest that the binding of the linker to the PIF-binding pocket only partially facilitates the transition from the inactive to the active conformation. Other structural changes, including the disorder-to-order transition of the αC-helix, may be achieved by the binding of the phosphorylated HM of the substrate to the PIF-binding pocket of PDK1. A similar HM phosphorylation-promoted conformational changes have been reported for many PDK1 substrates. ,, The second outcome of the binding of the linker to the PIF-binding pocket is the destabilization of the domain interaction. Our results show an increased interaction between the kinase and PH domains in the absence of the linker (Figure ). Thus, the binding of the linker to the PIF-binding pocket can allosterically modulate the interactions between the kinase and PH domains. Since the PIF-binding pocket is the binding site of the C-terminal HM of substrates, our results suggest that the binding of a substrate’s HM to the PIF-binding pocket of PDK1 may also contribute to the activation of PDK1. And such an effect can be potentiated when the HM is phosphorylated, as electrostatic interactions contribute to its binding (Figure ). Previous studies have shown that phosphorylated S6K1 and SGK1 at their HMs promote their interactions with the PIF-binding pocket of PDK1, and docking of phosphorylated HM of RSK2 to PDK1 activated PDK1. Phosphate-dependent docking site in PDK1, as shown in the crystal structure (PDB ID: 1H1W), involving residues Lys76, Arg131, Thr148, and Gln150, was also identified to contribute to the binding of the linker to the PIF-binding pocket of PDK1, in good agreement with our results (Figure B).
The observation that phosphorylation of the linker Ser393 weakens the interaction between the kinase and PH domains further suggests that the population of the autoinhibited PDK1 is highly context-dependent and could be shifted by multiple factors (Figure ), which may explain why experimental detection of autoinhibited PDK1 remains a challenge. The clustering results (Figure ) provide several representative conformations that the autoinhibited PDK1 could adopt. With the long flexible linker, PDK1 is expected to exist in more autoinhibited states with various populations. The AKT-like autoinhibited PDK1, where the linker binds to the PIF-binding pocket and promotes the activation of PDK1, is consistent with current experimental evidence on how PDK1 becomes active and phosphorylates distinct substrates. Single-molecule studies provided direct evidence for the formation of a membrane-bound PDK1-AKT heterodimer stabilized by PIF interactions. The activation mechanism proposed here highlights the autoinhibited PDK1 state and the regulatory role of the linker region (Figure ). In the cytoplasm, the highly populated autoinhibited PDK1 is in equilibrium with the less populated, PH domain-free partially autoinhibited (or partially activated) states. In the presence of PIP3, the population shifts toward PH domain accessible states, releasing the autoinhibition. In the cytoplasm, PDK1 may interact with scaffold proteins and phosphorylate diverse substrates, independent of PIP3.
8.

Proposed mechanism for the activation of PDK1, highlighting the regulatory role of the linker in PDK1 activation. PDK1 can exit in equilibrium between autoinhibited conformations and other inactive states with free PH domain in the cytoplasm. The autoinhibited states are more populated, with the AKT-like state displaying the high-affinity binding between the kinase and PH domains. Binding of the linker to the PIF-pocket promotes the formation of the Glu130-Lys111 salt bridge and changes the orientation of the αC-helix (from αC-out to αC-in), a hallmark of the active kinase domain. In the presence of PIP3, the population of the autoinhibited PDK1 shifts to the conformation with high affinity for PIP3. PDK1 is then recruited to the membrane and becomes active via trans-autophosphorylation. Once activated, PDK1 phosphorylates the activation loop of AKT. Binding of the hydrophobic motif in the C-terminal region of AKT to the PIF-binding pocket of PDK1 may facilitate phosphorylation. In cytoplasm, the inactive PDK1 may interact with scaffold proteins, become active and phosphorylate other substrate, independent of PIP3, which needs further investigation.
Alternative, Autoinhibition-Based Approaches for the Challenging PDK1 Drug Discovery
Stabilization of AKT in the autoinhibited conformation by allosteric inhibitors has been reported. , However, locking PDK1 in the autoinhibited conformation and impairing membrane recruitment may have been more challenging due to the lack of mechanistic understanding of the autoinhibited state. Complementing efforts to develop PDK1 inhibitors targeting the ATP-binding site, , PIF-binding pocket, − the lipid-binding site, and the DFG-out conformation of PDK1, − the present work proposes two potential strategies for inhibitor design or FDA-approved repurposing (Figure ). The first strategy involves modifying/optimizing inhibitors that lock the AKT in the autoinhibited conformation. We superimposed the conformations of the autoinhibited AKT with inhibitors and found that these inhibitors occupy the same binding site formed by Leu210, Thr211, Leu264, Lys268, Val270–Asp274, Ile290–Asp292, Cys296 and Lys297 residues in the kinase domain, and Leu78–Glu85 in the PH domain (Figure A-C). We performed sequence alignment and identified corresponding residues of PDK1 (Figure C and Figure S6). We then superimposed the conformations of the kinase and PH domains of PDK1 onto the autoinhibited AKT separately (Figure D). We found that these inhibitors interact with similar or identical residues of the kinase domains, but with different residues of the PH domains of AKT and PDK1 (Figures C and D). Optimization of the interactions between the inhibitors and the PH domain of PDK1 by modification of AKT inhibitors appear applicable to the development of inhibitors that stabilize PDK1 in autoinhibited states.
9.

Proposed strategies for the design of PDK1 inhibitors. (A) Inhibitors in the crystal structures of autoinhibited AKT. (B) Superimposition of autoinhibited AKT conformations. The kinase and PH domains of the AKT (PDB ID: 3O96) are shown and colored in orange and light blue, respectively. Residues interacting with inhibitors are shown in surface and colored based on their types (nonpolar: white; polar: green; basic: blue; and acidic: red). (C) Sequence alignment of those interacting residues shown in (B) between AKT and PDK1. For clarity, the nonpolar residues are colored in black. (D) The binding site of those AKT inhibitors in the autoinhibited PDK1. The kinase and PH domains of PDK1 are separately superimposed to the kinase and PH domains of the autoinhibited AKT. The interacting residues based on the sequence alignment in (C) are shown and labeled. The kinase and PH domains of PDK1 are also colored in orange and light blue, respectively. (E) Interaction of the PIFtide 13MFRDFDYIA21 with the PIF-binding pocket (PDB ID: 4RRV). The binding of two inhibitors based on the PIFtide are also shown (PDB IDs: 4RQK and 4RQV). The PIF-binding pocket is shown in surface, and the PIFtide is shown in yellow tube. Residues in the PIFtide are shown as sticks, with Met13 and Ala21 labeled. Residues that define the binding site of the inhibitors are labeled and colored according to their types. The structures of the inhibitors are shown in Table S2. (F) Interaction of the linker segment (368DDEDCYGNYDNLLSQF383) with the PIF-binding pocket. The linker is shown as yellow tube. The binding site of the two inhibitors shown in (E) is obtained by superimposition of the crystal structures (PDB IDs: 4RQK and 4RQV) with the conformation of PDK1. The first inhibitor binding site, as well as the second potential binding site, are indicated as dashed lines.
The second strategy is to develop novel inhibitors that target the PIF-binding pocket. The superposition of available crystal structures of PDK1 with modulators (activators and inhibitors) bound at the PIF-binding pocket revealed that these small molecules occupy the same site, roughly defined by residues Ile118, Arg131, Thr148, Gln150, and Leu155 (Figure S7 and Table S2). The detailed interactions between these modulators and PDK1 are provided in Figure S8. We found that PDK1 activators interact with both Lys76 and Arg131 simultaneously, which resembles the electrostatic interactions between the linker and PIF-pocket observed in our work. In contrast, PDK1 inhibitors interact exclusively with Arg131 (Figure S8). Figure E shows the binding of two of those inhibitors in the PIF-binding pocket, which were designed based on the bound PIFtide (13MFRDFDYIA21), with Phe14 and Phe17 occupying the hydrophobic pocket and Asp16 or Asp18 interacting with Arg131 of PDK1. Correspondingly, the inhibitors contain two aromatic rings and one carboxylic acid moiety (Table S2, PDB IDs: 4RQK and 4RQV). The interactions between the linker and the PIF-binding pocket point to a second site, defined by Lys76, Thr148, Arg131, and Lys144 (Figure F). The interaction of linker residue Asp377 with Arg131 stabilizes the binding of linker in the first site, and interactions of Asp371 with Lys76, and Asp386 with Lys144 stabilize the binding of linker in the second PIF-binding pocket site. Of note, Lys144 plays an important role in the hinge motion of the kinase domain, and Lys144 mutations decreased the binding of PIFtide.
Thus, besides the PIFtide-guided inhibitor design, here we propose alternative approaches for the design of PDK1 inhibitor based on the linker interactions: (1) Introduction of additional carboxylic acid moiety in the structures of existing inhibitors and optimization of the interactions with the second binding site, particularly with Lys144; (2) High-throughput virtual screening of available drug database directly targeting the second binding site in the PIF-binding pocket; and (3) Development of peptidomimetics based on the linker sequence in order to obtain high binding affinity with the PIF-binding pocket. Of note, the newly designed inhibitors should avoid interaction with Lys76, a characteristic interaction with PDK1 activators.
Conclusions
In this work, we elucidate the autoinhibited conformation of PDK1. With the positioning of the PH domain relative to the kinase domain resembling that of AKT, the autoinhibited PDK1 exhibits unique intramolecular interactions between the linker and the PIF-binding pocket of the kinase domain, which promote the characteristic Glu130-Lys111 salt bridge and attenuates the association of the kinase domain with the PH domain. The binding of the two domains is further weakened by the phosphorylation of Ser393 in the linker, shifting the autoinhibited closed state toward conformations with high binding affinity to the substrate, even in the absence of PIP3. Substrate binding relieves the autoinhibition. Different PDK1 substrates such as AKT, PKC, SGK, S6K, and RSK can use their respective hydrophobic motif (HM) to bind to the PIF-binding pocket of PDK1 and promote PDK1 activation.
Taken together, our work suggests that PDK1, like its downstream substrates, could adopt autoinhibited conformations. Our work discovers the regulatory role of the linker in the activation of the full-length PDK1, offers innovative strategies to linker-guided PDK1 inhibitors design. The shift of the autoinhibited population toward the active state even in the absence of PIP 3 suggests how PDK1 can phosphorylate diverse protein kinases.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. All simulations were performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (https://hpc.nih.gov/).
Glossary
ABBREVIATIONS
- PDK1
3-phosphoinositide-dependent kinase 1
- PIF
PDK1-interacting fragment
- PI3K
phosphoinositide 3-kinase
- AKT
protein kinase B
- mTOR
mammalian target of rapamycin
- mTORC2
mTOR complex 2
- S6K
p70 ribosomal S6 kinase
- RSK
p90 ribosomal S6 kinase
- SGK
serum/glucocorticoid regulated kinase
- PIP 3
phosphatidylinositol (3,4,5)-trisphosphate
- PIP 2
phosphatidylinositol 4,5-bisphosphate
- PKA
protein kinase A
- PKG
protein kinase G
- PKC
protein kinase C
- MAPK
mitogen-activated protein kinase
- Raf
rapidly accelerated fibrosarcoma
The crystal structures of proteins are obtained from RCSB: https://www.rcsb.org/. The NAMD (v 2.14) program is available at https://www.ks.uiuc.edu/Research/namd/; the CHARMM program is available at https://academiccharmm.org/; the Chimera program is available at https://www.cgl.ucsf.edu/chimera/; the Rosetta program is available at https://rosettacommons.org/software/; the RoseTTAFold is available at https://robetta.bakerlab.org/; the AlphaFold is available at https://github.com/deepmind/alphafold. The raw data associated with this study are available on request.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.4c01392.
Available structures of PDK1 and AKT (Figure S1); model structures of PDK1 in autoinhibition (Figure S2); solvent-accessible surface area (SASA) and interdomain interaction energy of PDK1 (Figure S3); interaction of the linker with the PIF-binding pocket can promote PDK1 to an active-like structure (Figure S4); the AKT-like autoinhibited PDK1 displays a closed conformation of the ATP binding site (Figure S5); sequence alignment between AKT and PDK1 (Figure S6); small molecule PDK1 modulators bind to the same pocket in the PIF-binding pocket of PDK1 (Figure S7); the interactions of activators and inhibitors with PDK1 (Figure S8); summary of the simulation systems in this work (Table S1); and the crystal structures of PDK1 in complex with activators/inhibitors bound in the PIF-binding pocket (Table S2). (PDF)
L.X., H.J., and R.N. conceived the study. L.X. performed the simulations and data analyses. L.X. wrote the first draft of the manuscript. H.J. and R.N. commented on and revised the manuscript. All authors read and approved the final manuscript.
The authors declare no competing financial interest.
References
- Levina A., Fleming K. D., Burke J. E., Leonard T. A.. Activation of the essential kinase PDK1 by phosphoinositide-driven trans-autophosphorylation. Nat. Commun. 2022;13:1874. doi: 10.1038/s41467-022-29368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordon-Barris L., Pascual-Guiral S., Yang S., Gimenez-Llort L., Lope-Piedrafita S., Niemeyer C., Claro E., Lizcano J. M., Bayascas J. R.. Mutation of the 3-Phosphoinositide-Dependent Protein Kinase 1 (PDK1) Substrate-Docking Site in the Developing Brain Causes Microcephaly with Abnormal Brain Morphogenesis Independently of Akt, Leading to Impaired Cognition and Disruptive Behaviors. Mol. Cell. Biol. 2016;36:2967–2982. doi: 10.1128/MCB.00230-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niba E. T., Nagaya H., Kanno T., Tsuchiya A., Gotoh A., Tabata C., Kuribayashi K., Nakano T., Nishizaki T.. Crosstalk between PI3 kinase/PDK1/Akt/Rac1 and Ras/Raf/MEK/ERK pathways downstream PDGF receptor. Cell Physiol. Biochem. 2013;31:905–913. doi: 10.1159/000350108. [DOI] [PubMed] [Google Scholar]
- Molinaro C., Martoriati A., Lescuyer A., Fliniaux I., Tulasne D., Cailliau K.. 3-phosphoinositide-dependent protein kinase 1 (PDK1) mediates crosstalk between Src and Akt pathways in MET receptor signaling. FEBS Lett. 2021;595:2655–2664. doi: 10.1002/1873-3468.14195. [DOI] [PubMed] [Google Scholar]
- Xia H., Dai X., Yu H., Zhou S., Fan Z., Wei G., Tang Q., Gong Q., Bi F.. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 2018;9:269. doi: 10.1038/s41419-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manne B. K., Munzer P., Badolia R., Walker-Allgaier B., Campbell R. A., Middleton E., Weyrich A. S., Kunapuli S. P., Borst O., Rondina M. T.. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J. Thromb. Haemost. 2018;16:1211–1225. doi: 10.1111/jth.14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E. C., Dibble C. C., Toker A.. PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol. 2017;45:62–71. doi: 10.1016/j.ceb.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A., Newton A. C.. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/S0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- Gagliardi P. A., Puliafito A., Primo L.. PDK1: At the crossroad of cancer signaling pathways. Semin Cancer Biol. 2018;48:27–35. doi: 10.1016/j.semcancer.2017.04.014. [DOI] [PubMed] [Google Scholar]
- Song M., Bode A. M., Dong Z., Lee M. H.. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019;79:1019–1031. doi: 10.1158/0008-5472.CAN-18-2738. [DOI] [PubMed] [Google Scholar]
- Liu P., Cheng H., Roberts T. M., Zhao J. J.. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discovery. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringel M. D., Hayre N., Saito J., Saunier B., Schuppert F., Burch H., Bernet V., Burman K. D., Kohn L. D., Saji M.. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61:6105–6111. [PubMed] [Google Scholar]
- Raimondi C., Falasca M.. Targeting PDK1 in cancer. Curr. Med. Chem. 2011;18:2763–2769. doi: 10.2174/092986711796011238. [DOI] [PubMed] [Google Scholar]
- He Y., Sun M. M., Zhang G. G., Yang J., Chen K. S., Xu W. W., Li B.. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target Ther. 2021;6:425. doi: 10.1038/s41392-021-00828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux A. E., Schulze J. O., Biondi R. M.. AGC kinases, mechanisms of regulation and innovative drug development. Semin. Cancer Biol. 2018;48:1–17. doi: 10.1016/j.semcancer.2017.05.011. [DOI] [PubMed] [Google Scholar]
- Sacerdoti M., Gross L. Z. F., Riley A. M., Zehnder K., Ghode A., Klinke S., Anand G. S., Paris K., Winkel A., Herbrand A. K., Godage H. Y., Cozier G. E., Süß E., Schulze J. O., Pastor-Flores D., Bollini M., Cappellari M. V., Svergun D., Gräwert M. A., Aramendia P. F., Leroux A. E., Potter B. V. L., Camacho C. J., Biondi R. M.. et al. Modulation of the substrate specificity of the kinase PDK1 by distinct conformations of the full-length protein. Sci. Signal. 2023;16:eadd3184. doi: 10.1126/scisignal.add3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidi A., Falasca M.. Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers (Basel) 2017;9:140. doi: 10.3390/cancers9100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R., Tsai C. J., Jang H.. Autoinhibition can identify rare driver mutations and advise pharmacology. FASEB J. 2020;34:16–29. doi: 10.1096/fj.201901341R. [DOI] [PubMed] [Google Scholar]
- Masters T. A., Calleja V., Armoogum D. A., Marsh R. J., Applebee C. J., Laguerre M., Bain A. J., Larijani B.. Regulation of 3-phosphoinositide-dependent protein kinase 1 activity by homodimerization in live cells. Sci. Signal. 2010;3:ra78. doi: 10.1126/scisignal.2000738. [DOI] [PubMed] [Google Scholar]
- Ziemba B. P., Pilling C., Calleja V., Larijani B., Falke J. J.. The PH domain of phosphoinositide-dependent kinase-1 exhibits a novel, phospho-regulated monomer-dimer equilibrium with important implications for kinase domain activation: single-molecule and ensemble studies. Biochemistry. 2013;52:4820–4829. doi: 10.1021/bi400488f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- J. Meuillet E.. Novel inhibitors of AKT: assessment of a different approach targeting the pleckstrin homology domain. Curr. Med. Chem. 2011;18:2727–2742. doi: 10.2174/092986711796011292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W. I., Voegtli W. C., Sturgis H. L., Dizon F. P., Vigers G. P., Brandhuber B. J.. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5:e12913. doi: 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truebestein L., Hornegger H., Anrather D., Hartl M., Fleming K. D., Stariha J. T. B., Pardon E., Steyaert J., Burke J. E., Leonard T. A.. Structure of autoinhibited Akt1 reveals mechanism of PIP(3)-mediated activation. Proc. Natl. Acad. Sci. U. S. A. 2021;118:e2101496118. doi: 10.1073/pnas.2101496118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quambusch L., Landel I., Depta L., Weisner J., Uhlenbrock N., Muller M. P., Glanemann F., Althoff K., Siveke J. T., Rauh D.. Covalent-Allosteric Inhibitors to Achieve Akt Isoform-Selectivity. Angew. Chem., Int. Ed. 2019;58:18823–18829. doi: 10.1002/anie.201909857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae H., Viennet T., Park E., Chu N., Salguero A., Eck M. J., Arthanari H., Cole P. A.. PH domain-mediated autoinhibition and oncogenic activation of Akt. Elife. 2022;11:e80148. doi: 10.7554/eLife.80148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwell M. A., Lapierre J. M., Brassard C., Bresciano K., Bull C., Cornell-Kennon S., Eathiraj S., France D. S., Hall T., Hill J.. et al. Discovery and optimization of a series of 3-(3-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: orally bioavailable, selective, and potent ATP-independent Akt inhibitors. J. Med. Chem. 2012;55:5291–5310. doi: 10.1021/jm300276x. [DOI] [PubMed] [Google Scholar]
- Nussinov R., Jang H., Gursoy A., Keskin O., Gaponenko V.. Inhibition of Nonfunctional Ras. Cell. Chem. Biol. 2021;28:121–133. doi: 10.1016/j.chembiol.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R., Liu Y., Zhang W., Jang H.. Protein conformational ensembles in function: roles and mechanisms. RSC Chem. Biol. 2023;4:850–864. doi: 10.1039/D3CB00114H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A.. et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. J., Moughon S., Wang C., Schueler-Furman O., Kuhlman B., Rohl C. A., Baker D.. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/S0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- Nagashima K., Shumway S. D., Sathyanarayanan S., Chen A. H., Dolinski B., Xu Y., Keilhack H., Nguyen T., Wiznerowicz M., Li L.. et al. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J. Biol. Chem. 2011;286:6433–6448. doi: 10.1074/jbc.M110.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D., Fairservice A., Deak M., Kular G. S., Prescott A. R., Peter Downes C., Safrany S. T., Alessi D. R., van Aalten D. M.. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 2004;23:3918–3928. doi: 10.1038/sj.emboj.7600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canutescu A. A., Dunbrack R. L. Jr.. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Bradley P., Baker D.. Protein-protein docking with backbone flexibility. J. Mol. Biol. 2007;373:503–519. doi: 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- Coutsias E. A., Seok C., Jacobson M. P., Dill K. A.. A kinematic view of loop closure. J. Comput. Chem. 2004;25:510–528. doi: 10.1002/jcc.10416. [DOI] [PubMed] [Google Scholar]
- Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G. R., Wang J., Cong Q., Kinch L. N., Schaeffer R. D.. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kale L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C., Hardy D. J., Maia J. D. C., Stone J. E., Ribeiro J. V., Bernardi R. C., Buch R., Fiorin G., Hénin J., Jiang W., McGreevy R., Melo M. C. R., Radak B. K., Skeel R. D., Singharoy A., Wang Y., Roux B., Aksimentiev A., Luthey-Schulten Z., Kalé L. V., Schulten K., Chipot C., Tajkhorshid E.. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020;153:044130. doi: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Jang H., Li Z., Sacks D. B., Nussinov R.. B-Raf autoinhibition in the presence and absence of 14–3-3. Structure. 2021;29:768–777. doi: 10.1016/j.str.2021.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney R. C., Zhang M., Jang H., Nussinov R.. The mechanism of activation of monomeric B-Raf V600E. Comput. Struct. Biotechnol. J. 2021;19:3349–3363. doi: 10.1016/j.csbj.2021.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Jang H., Nussinov R.. The mechanism of PI3Kα activation at the atomic level. Chem. Sci. 2019;10:3671–3680. doi: 10.1039/C8SC04498H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H., Smith I. N., Eng C., Nussinov R.. The mechanism of full activation of tumor suppressor PTEN at the phosphoinositide-enriched membrane. iScience. 2021;24:102438. doi: 10.1016/j.isci.2021.102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Jang H., Zhang M., Tsai C. J., Maloney R., Nussinov R.. The structural basis of BCR-ABL recruitment of GRB2 in chronic myelogenous leukemia. Biophys. J. 2022;121:2251–2265. doi: 10.1016/j.bpj.2022.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang M., Jang H., Nussinov R.. The allosteric mechanism of mTOR activation can inform bitopic inhibitor optimization. Chem. Sci. 2024;15:1003–1017. doi: 10.1039/D3SC04690G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang M., Jang H., Nussinov R.. Higher-order interactions of Bcr-Abl can broaden chronic myeloid leukemia (CML) drug repertoire. Protein Sci. 2023;32:e4504. doi: 10.1002/pro.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang M., Tsai C. J., Jang H., Nussinov R.. Allosteric regulation of autoinhibition and activation of c-Abl. Comput. Struct. Biotechnol. J. 2022;20:4257–4270. doi: 10.1016/j.csbj.2022.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmuller H., MacKerell A. D. Jr.. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B., Venable R. M., Freites J. A., O’Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr, Pastor R. W.. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga C., Travis K. P.. A configurational temperature Nose-Hoover thermostat. J. Chem. Phys. 2005;123:134101. doi: 10.1063/1.2013227. [DOI] [PubMed] [Google Scholar]
- Martyna G. J., Tobias D. J., Klein M. L.. Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994;101:4177–4189. doi: 10.1063/1.467468. [DOI] [Google Scholar]
- Darden T., York D., Pedersen L.. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- Ciccotti G., Ryckaert J. P.. Molecular-Dynamics Simulation of Rigid Molecules. Comput. Phys. Rep. 1986;4:346–392. doi: 10.1016/0167-7977(86)90022-5. [DOI] [Google Scholar]
- Pearce L. R., Komander D., Alessi D. R.. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Biondi R. M., Komander D., Thomas C. C., Lizcano J. M., Deak M., Alessi D. R., van Aalten D. M.. High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J. 2002;21:4219–4228. doi: 10.1093/emboj/cdf437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi R. M., Kieloch A., Currie R. A., Deak M., Alessi D. R.. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001;20:4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi R. M., Cheung P. C., Casamayor A., Deak M., Currie R. A., Alessi D. R.. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000;19:979–988. doi: 10.1093/emboj/19.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A. P., Haste N. M., Taylor S. S., Ten Eyck L. F.. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U. S. A. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux A. E., Biondi R. M.. The choreography of protein kinase PDK1 and its diverse substrate dance partners. Biochem. J. 2023;480:1503–1532. doi: 10.1042/BCJ20220396. [DOI] [PubMed] [Google Scholar]
- Lucas N., Cho W.. Phosphatidylserine binding is essential for plasma membrane recruitment and signaling function of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2011;286:41265–41272. doi: 10.1074/jbc.M111.300806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamayor A., Morrice N. A., Alessi D. R.. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem. J. 1999;342:287–292. doi: 10.1042/bj3420287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D., Kular G., Deak M., Alessi D. R., van Aalten D. M.. Role of T-loop phosphorylation in PDK1 activation, stability, and substrate binding. J. Biol. Chem. 2005;280:18797–18802. doi: 10.1074/jbc.M500977200. [DOI] [PubMed] [Google Scholar]
- Gao X., Harris T. K.. Role of the PH domain in regulating in vitro autophosphorylation events required for reconstitution of PDK1 catalytic activity. Bioorg. Chem. 2006;34:200–223. doi: 10.1016/j.bioorg.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Heras-Martínez G. d. l., Calleja V., Bailly R., Dessolin J., Larijani B., Requejo-Isidro J.. A Complex Interplay of Anionic Phospholipid Binding Regulates 3′-Phosphoinositide-Dependent-Kinase-1 Homodimer Activation. Sci. Rep. 2019;9:14527. doi: 10.1038/s41598-019-50742-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calleja V., Laguerre M., de Las Heras-Martinez G., Parker P. J., Requejo-Isidro J., Larijani B.. Acute regulation of PDK1 by a complex interplay of molecular switches. Biochem. Soc. Trans. 2014;42:1435–1440. doi: 10.1042/BST20140222. [DOI] [PubMed] [Google Scholar]
- Jiang Q., Zhang X., Dai X., Han S., Wu X., Wang L., Wei W., Zhang N., Xie W., Guo J.. S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat. Commun. 2022;13:1548. doi: 10.1038/s41467-022-28910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riojas R. A., Kikani C. K., Wang C., Mao X., Zhou L., Langlais P. R., Hu D., Roberts J. L., Dong L. Q., Liu F.. Fine tuning PDK1 activity by phosphorylation at Ser163. J. Biol. Chem. 2006;281:21588–21593. doi: 10.1074/jbc.M600393200. [DOI] [PubMed] [Google Scholar]
- Park J., Hill M. M., Hess D., Brazil D. P., Hofsteenge J., Hemmings B. A.. Identification of tyrosine phosphorylation sites on 3-phosphoinositide-dependent protein kinase-1 and their role in regulating kinase activity. J. Biol. Chem. 2001;276:37459–37471. doi: 10.1074/jbc.M105916200. [DOI] [PubMed] [Google Scholar]
- Cheung M., R. Testa J.. Diverse mechanisms of AKT pathway activation in human malignancy. Curr. Cancer Drug Targets. 2013;13:234–244. doi: 10.2174/1568009611313030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lučić I., Rathinaswamy M. K., Truebestein L., Hamelin D. J., Burke J. E., Leonard T. A.. Conformational sampling of membranes by Akt controls its activation and inactivation. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E3940–E3949. doi: 10.1073/pnas.1716109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R.. The spatial structure of cell signaling systems. Phys. Biol. 2013;10:045004. doi: 10.1088/1478-3975/10/4/045004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B. J., Deak M., Arthur J. S., Armit L. J., Alessi D. R.. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003;22:4202–4211. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettenmaier T. J., Sadowsky J. D., Thomsen N. D., Chen S. C., Doak A. K., Arkin M. R., Wells J. A.. A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proc. Natl. Acad. Sci. U. S. A. 2014;111:18590–18595. doi: 10.1073/pnas.1415365112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi R. M.. Phosphoinositide-dependent protein kinase 1, a sensor of protein conformation. Trends Biochem. Sci. 2004;29:136–142. doi: 10.1016/j.tibs.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Kelley L. A., Gardner S. P., Sutcliffe M. J.. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. 1996;9:1063–1065. doi: 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E.. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Strickler S. S., Gribenko A. V., Gribenko A. V., Keiffer T. R., Tomlinson J., Reihle T., Loladze V. V., Makhatadze G. I.. Protein stability and surface electrostatics: a charged relationship. Biochemistry. 2006;45:2761–2766. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- Angiolini M., Banfi P., Casale E., Casuscelli F., Fiorelli C., Saccardo M. B., Silvagni M., Zuccotto F.. Structure-based optimization of potent PDK1 inhibitors. Bioorg. Med. Chem. Lett. 2010;20:4095–4099. doi: 10.1016/j.bmcl.2010.05.070. [DOI] [PubMed] [Google Scholar]
- Lin K., Lin J., Wu W. I., Ballard J., Lee B. B., Gloor S. L., Vigers G. P. A., Morales T. H., Friedman L. S., Skelton N., Brandhuber B. J.. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal. 2012;5:ra37. doi: 10.1126/scisignal.2002618. [DOI] [PubMed] [Google Scholar]
- Kim C., Cheng C. Y., Saldanha S. A., Taylor S. S.. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Schulze J. O., Saladino G., Busschots K., Neimanis S., Suss E., Odadzic D., Zeuzem S., Hindie V., Herbrand A. K., Lisa M. N.. et al. Bidirectional Allosteric Communication between the ATP-Binding Site and the Regulatory PIF Pocket in PDK1 Protein Kinase. Cell Chem. Biol. 2016;23:1193–1205. doi: 10.1016/j.chembiol.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Grant B. D., Hemmer W., Tsigelny I., Adams J. A., Taylor S. S.. Kinetic analyses of mutations in the glycine-rich loop of cAMP-dependent protein kinase. Biochemistry. 1998;37:7708–7715. doi: 10.1021/bi972987w. [DOI] [PubMed] [Google Scholar]
- Nussinov R., Zhang M., Tsai C. J., Liao T. J., Fushman D., Jang H.. Autoinhibition in Ras effectors Raf, PI3Kalpha, and RASSF5: a comprehensive review underscoring the challenges in pharmacological intervention. Biophys. Rev. 2018;10:1263–1282. doi: 10.1007/s12551-018-0461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis S. H.. Mechanisms of autoinhibition in cyclic nucleotide-dependent protein kinases. Front. Biosci. 2002;7:d580–d592. doi: 10.2741/A796. [DOI] [PubMed] [Google Scholar]
- Taylor S. S., Buechler J. A., Yonemoto W.. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- House C., Kemp B. E.. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- Newton A. C.. Protein kinase C: structure, function, and regulation. J. Biol. Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- Steinberg S. F.. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogl G., Alexa A., Kiss B., Katona G., Kovacs M., Bodor A., Remenyi A., Nyitray L.. Structural Basis of Ribosomal S6 Kinase 1 (RSK1) Inhibition by S100B Protein: Modulation of the extracellular signal-regulated kinase (ERK) signaling cascade in a calcium-dependent way. J. Biol. Chem. 2016;291:11–27. doi: 10.1074/jbc.M115.684928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhova M., Tereshko V., Lee S. Y., Yao K., Cho Y. Y., Bode A., Dong Z.. Structural basis for activation of the autoinhibitory C-terminal kinase domain of p90 RSK2. Nat. Struct. Mol. Biol. 2008;15:112–113. doi: 10.1038/nsmb1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis P. B., Pullen N., Pearson R. B., Kozma S. C., Thomas G.. Phosphorylation sites in the autoinhibitory domain participate in p70(s6k) activation loop phosphorylation. J. Biol. Chem. 1998;273:14845–14852. doi: 10.1074/jbc.273.24.14845. [DOI] [PubMed] [Google Scholar]
- Hou Z., He L., Qi R. Z.. Regulation of s6 kinase 1 activation by phosphorylation at Ser-411. J. Biol. Chem. 2007;282:6922–6928. doi: 10.1074/jbc.M607836200. [DOI] [PubMed] [Google Scholar]
- Magnuson B., Ekim B., Fingar D. C.. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- Chu N., Viennet T., Bae H., Salguero A., Boeszoermenyi A., Arthanari H., Cole P. A.. The structural determinants of PH domain-mediated regulation of Akt revealed by segmental labeling. Elife. 2020;9:e59151. doi: 10.7554/eLife.59151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu N., Salguero A. L., Liu A. Z., Chen Z., Dempsey D. R., Ficarro S. B., Alexander W. M., Marto J. A., Li Y., Amzel L. M.. et al. Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis. Cell. 2018;174:897–907. doi: 10.1016/j.cell.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg E. D., Gao T., Newton A. C.. The phosphoinositide-dependent kinase, PDK-1, phosphorylates conventional protein kinase C isozymes by a mechanism that is independent of phosphoinositide 3-kinase. J. Biol. Chem. 2001;276:45289–45297. doi: 10.1074/jbc.M107416200. [DOI] [PubMed] [Google Scholar]
- Yang J., Cron P., Thompson V., Good V. M., Hess D., Hemmings B. A., Barford D.. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell. 2002;9:1227–1240. doi: 10.1016/S1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- Hindie V., Stroba A., Zhang H., Lopez-Garcia L. A., Idrissova L., Zeuzem S., Hirschberg D., Schaeffer F., Jorgensen T. J., Engel M.. et al. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat. Chem. Biol. 2009;5:758–764. doi: 10.1038/nchembio.208. [DOI] [PubMed] [Google Scholar]
- Yang J., Cron P., Good V. M., Thompson V., Hemmings B. A., Barford D.. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 2002;9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- Frodin M., Antal T. L., Dummler B. A., Jensen C. J., Deak M., Gammeltoft S., Biondi R. M.. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 2002;21:5396–5407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M., Jensen C. J., Merienne K., Gammeltoft S.. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon M. T., Ziemba B. P., Falke J. J.. Single-molecule studies reveal regulatory interactions between master kinases PDK1, AKT1, and PKC. Biophys. J. 2021;120:5657–5673. doi: 10.1016/j.bpj.2021.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer C., Alessi D. R.. Small-molecule inhibitors of PDK1. ChemMedChem. 2008;3:1810–1838. doi: 10.1002/cmdc.200800195. [DOI] [PubMed] [Google Scholar]
- Arter C., Trask L., Ward S., Yeoh S., Bayliss R.. Structural features of the protein kinase domain and targeted binding by small-molecule inhibitors. J. Biol. Chem. 2022;298:102247. doi: 10.1016/j.jbc.2022.102247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Chen Y., Fu Q., Ni D., Zhang J., Li X., Lu S.. The chemical diversity and structure-based discovery of allosteric modulators for the PIF-pocket of protein kinase PDK1. J. Enzyme Inhib. Med. Chem. 2019;34:361–374. doi: 10.1080/14756366.2018.1553167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busschots K., Lopez-Garcia L. A., Lammi C., Stroba A., Zeuzem S., Piiper A., Alzari P. M., Neimanis S., Arencibia J. M., Engel M.. et al. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem. Biol. 2012;19:1152–1163. doi: 10.1016/j.chembiol.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Sadowsky J. D., Burlingame M. A., Wolan D. W., McClendon C. L., Jacobson M. P., Wells J. A.. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettenmaier T. J., Fan H., Karpiak J., Doak A., Sali A., Shoichet B. K., Wells J. A.. Small-Molecule Allosteric Modulators of the Protein Kinase PDK1 from Structure-Based Docking. J. Med. Chem. 2015;58:8285–8291. doi: 10.1021/acs.jmedchem.5b01216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina J. R.. Selective 3-phosphoinositide-dependent kinase 1 (PDK1) inhibitors: dissecting the function and pharmacology of PDK1. J. Med. Chem. 2013;56:2726–2737. doi: 10.1021/jm4000227. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A., Arndt J. W., Cancilla M. T., Cao K., Elling R. A., English N., Friedman J., Hansen S. K., Hession C., Joseph I.. et al. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg. Med. Chem. Lett. 2011;21:3078–3083. doi: 10.1016/j.bmcl.2011.03.032. [DOI] [PubMed] [Google Scholar]
- Vijayan R. S., He P., Modi V., Duong-Ly K. C., Ma H., Peterson J. R., Dunbrack R. L. Jr, Levy R. M.. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J. Med. Chem. 2015;58:466–479. doi: 10.1021/jm501603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestito S., Daniele S., Nesi G., Zappelli E., Di Maio D., Marinelli L., Digiacomo M., Lapucci A., Martini C., Novellino E., Rapposelli S.. Locking PDK1 in DFG-out conformation through 2-oxo-indole containing molecules: Another tools to fight glioblastoma. Eur. J. Med. Chem. 2016;118:47–63. doi: 10.1016/j.ejmech.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Vagner J., Qu H., Hruby V. J.. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008;12:292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structures of proteins are obtained from RCSB: https://www.rcsb.org/. The NAMD (v 2.14) program is available at https://www.ks.uiuc.edu/Research/namd/; the CHARMM program is available at https://academiccharmm.org/; the Chimera program is available at https://www.cgl.ucsf.edu/chimera/; the Rosetta program is available at https://rosettacommons.org/software/; the RoseTTAFold is available at https://robetta.bakerlab.org/; the AlphaFold is available at https://github.com/deepmind/alphafold. The raw data associated with this study are available on request.