

Abstract
In an attempt to rationalize the search for new potential Chemotherapeutic agents, a new series of 2-anilinobenzimidazol derivatives with CDK activity have been synthesized. The newly synthesized compounds have been assessed for their cytotoxic effects and CDK activity. These presented compounds showed strong inhibition of cell proliferation in various solid cancer cell lines, suggesting a promising approach for treating malignant tumors. Compounds 4 g, 4j, 4 m, and 4q displayed remarkably strong anticancer potencies against HepG2 cells, with IC50 of 7.59, 8.54, 3.56 and 5.88 µM, respectively, compared to the positive control, DOX (IC50 = 4.50 µM). while compound 4 m, and 4q had the highest anticancer activity against HeLa cells, with an IC50 of 6.39 and 9.71 µM, respectively, compared to the positive control DOX (IC50 = 5.57 µM). On the other hand, comparison of IC50 values against MCF-7 cells revealed that compounds 4 g, 4 m, and 4q showed significant anticancer potency with IC50 of 5.08, 2.18 and 8.19 µM, respectively compared to that of the positive control DOX (IC50 = 4.17 µM). Moreover, compound 4 m and 4q were the most potent CDK9 and CDK12 inhibitors. Furthermore, a molecular docking simulation were performed to explore the ability of compounds 4 m to adopt the common binding pattern of CDK9 and CDK12/T1 inhibitors. In silico ADMET results showed that all compounds have favourable drug-like properties since they met Lipinski’s rule of five criteria. Overall, the synthesized anilinopyrimidine derivatives exhibit significant potential as chemotherapeutic agents.
Supplementary Information
The online version contains supplementary material available at 10.1007/s44446-025-00010-w.
Keywords: Cancer, Benzimidazole, CDK, Antitumor activity, Molecular docking
Introduction
Cancer is a disease of tissue growth regulation. For a normal cell to transform into a cancerous one, the genes that regulate cell growth and differentiation must be altered (Zugazagoitia et al. 2016). It is characterized by hallmarks that was described by Douglas Hanahan, Robert A Weinberg as “sustaining proliferative signalling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis”. Later, another two hallmarks have been added which are: reprogramming of energy metabolism and evading immune destruction (Hanahan and Weinberg 2011). The advancement and variety of available therapeutic approaches over the last few years have led to a notable improvement in the number of cancers survivors (Workman 2005). The discovery of new therapeutic approaches for cancer sustained by the development of structural and chemical cell biology of cancer cells and of the potential therapeutic agents has hastened progression toward clinical trials and served to join the various stages (Yahya and Alqadhi 2021).
Cyclin-dependent kinases (CDKs) are a group of proteins that regulate various cellular processes. CDKs are divided into two subfamilies: cell cycle and transcriptional CDKs. The former, including CDK1, CDK2, CDK4, and CDK6, are directly involved in controlling cell cycle progression (Solary et al. 2002). In contrast, transcriptional CDKs, such as CDK7, CDK8, CDK9, CDK11, CDK12, and CDK13, regulate gene transcription (Protein Modificomics 2019). Transcriptional CDKs, in particular, modulate transcription by phosphorylating the carboxy-terminal domain (CTD) of RNA polymerase II (The RNA Polymerase II CTD Coordinates Transcription and RNA Processing—PMC n.d.). Specifically, CDK9 and CDK12 are key regulators of the elongation phase of transcription, targeting Ser2 of the CTD for phosphorylation (Small Molecule Inhibitors of Cyclin-Dependent Kinase 9 for Cancer Therapy—PMC n.d.; Anshabo et al. 2021a; Chao and Price 2001). Dysregulation in the expression or activity of CDKs is a hallmark of cancer development and progression (Shao et al. 2021; Targeting CDK12 for Cancer Therapy: Function, Mechanism, and Drug Discovery | Cancer Research | American Association for Cancer Research n.d.). Abnormalities in the activity of CDK9 and CDK12 in particular have been linked to various cancers, including leukaemia and solid tumours, such as ovarian and prostate cancers (Kumari et al. 2016).
Over the last several years, the importance of CDK inhibition in cancer has become increasingly apparent, as evidenced by several ongoing clinical trials wherein CDKs inhibitors act as single agents and in combination with other drugs. In addition, the development of small molecules that selectively target CDKs regulating transcriptional programs has only begun (Tadesse et al. 2021; Discovery of N-(4-(3-Isopropyl- 2-Methyl- 2H-Indazol- 5-Yl)Pyrimidin- 2-Yl)− 4-(4-Methylpiperazin- 1-Yl)Quinazolin- 7-Amine as a Novel, Potent, and Oral Cyclin-Dependent Kinase Inhibitor against Haematological Malignancies | Journal of Medicinal Chemistry n.d.). Numerous small molecules were designed and synthesized to target CDKs (either cell cycle or transcriptional). Several molecules, including ribociclib, palbociclib, and abemaciclib, have been approved by the U.S. Food and Drug Administration (FDA) as cyclin-dependent kinase (CDK) inhibitors (Fig. 1) (Zhang et al. 2021). None of these approved drugs are classified as inhibitors of CDK9 or CDK12.
Fig. 1.

The Structures of FDA-approved Cyclin-dependent Kinase Inhibitors
Targeting transcriptional CDK inhibitors, CDK7, 8, 9 and 12 have been reported to be a promising strategy in treating cancer (Eskandrani et al. 2023). Such inhibitors are known to induce apoptosis via inhibition of transcription of several antiapoptotic proteins and oncogenes such as Mcl- 1 and Myc (Anshabo et al. 2021b). There are several inhibitors have been developed to target transcriptional CDKs so far (Baguley and Kerr 2001; Protein Modificomics 2019). Many of them have 2-anilinopyrimidine incorporated in them which is a common scaffold in many protein kinase inhibitors (Fig. 2). Substitution at positions 4 and 5 of the pyrimidine ring is known to play a critical role in modulating the selectivity of the inhibitors against kinases and CDKs in particular (Eskandrani et al. 2023). Therefore, we herein report design, synthesis and biological evaluation of 2-anilino- 4-(benzimidazol- 1-yl)pyrimidine derivatives as potent inhibitors of the transcriptional CDKs 9 and 12. Introducing different groups with different sizes and electronic nature at positions 5 of the pyrimidine and 2 of the benzimidazole rings is expected interact with the gatekeeper regions of both CDK9 and CDK12.
Fig. 2.

Structures of representative CDK9 inhibitors and the designed inhibitors, 3a–h and 4a–t
Materials and methods
Materials
All chemical reagents and solvents used in the synthesis of all target compounds were obtained from commercial suppliers, Apollo Scientific (Manchester, UK), Ark Pharm (Arlington Heights, IL, USA), and Cambridge Isotope Laboratories (Tewksbury, MA, USA), and were used directly without further purification. All reactions were monitored using thin-layer chromatography (TLC) on glass sheets (silica gel F254) and visualized under UV light. An Agilent 6320 Ion Trap mass spectrometer was used to generate the mass spectra (MS). The Melting Point Apparatus Barnstead 9100 Electrothermal was used to record the melting points of the final compounds. A Bruker 700 Ultrashield NMR spectrometer was run at 400 and 175 MHz to generate 1H.
Methods
General procedure for preparation of compounds 4a-t
The general synthetic route for compounds 4a-t is outlined in Scheme 1. Substituted benzimidazoles (1a-e) were prepared by refluxing o-phenylenediamine with the appropriate carboxylic acid in the presence of ammonium chloride as a catalyst. Typically, (1.0 mmol) of o-phenylenediamine and (1.1 mmol) of the appropriate carboxylic acid are dissolved in a minimal amount of a high-boiling solvent such ethanol. Ammonium chloride is added to facilitate the cyclization reaction. The reaction mixture is then heated under reflux for several hours, ensuring complete conversion of the starting materials into the benzimidazole product. Upon completion, the reaction mixture is cooled to room temperature and poured into ice-cold water to precipitate the crude product. Filtration is performed to collect the solid product, which is then purified using recrystallization techniques, with ethanol. The resulting benzimidazoles (1a-e) were subsequently reacted with sodium hydride (NaH) in anhydrous N,N-dimethylformamide (DMF) under an inert atmosphere. Specifically, a mixture of benzimidazole (1.1 equiv) and NaH (2.0 equiv) in dry DMF was stirred at 0 °C for 1 h.
Scheme 1.

Synthesis of 3-((4-(1H-benzimidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamides (4a-t). Reagents and conditions: (a) NaH, dry DMF, 0 °C 1 h, rt overnight; (b) 3-aminobenzenesulfonamide, K2CO3, DMF, 80 °C
Following this, a solution of 2,4-dichloropyrimidine (1.0 equiv) in anhydrous DMF was added dropwise, and the mixture was stirred at room temperature for 24 h. The reaction progress was monitored using thin-layer chromatography (TLC). Upon completion, the mixture was diluted with water and extracted with ethyl acetate (EtOAc). The organic layer was concentrated under vacuum, yielding intermediates 3a-t, which were used in the subsequent step without further purification.
The final products (4a-t) were obtained by refluxing intermediates 3a-t with 3-aminobenzenesulfonamide and potassium carbonate (K₂CO₃) in DMF.
Antiproliferation assay
The MTT assay was performed to evaluate the in vitro antiproliferative activities of the 2-anilino- 4-(benzimidazol- 1-yl)pyrimidines 4a-t according to a previously reported method (Eskandrani et al. 2023).
Kinase assay
The CDK9 and CDK12 inhibition assays were performed as reported previously (Eskandrani et al. 2023).
Results
Chemistry
The findings for compounds 1a-e, 2a-d, and 3a-t have been previously reported (Eskandrani et al. 2023), while the results for 4a-t are presented below.:
3-((4-(1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4a), White solid (35 mg, 12%). M.p. 347—350 °C, 1H NMR (DMSO-d6): δ 5.56 (s, 1H), 6.70 (d, J = 8.0 Hz, 1H), 7.06–7.14 (m, 2H), 7.17 (s, 1H), 7.34–7.42 (m, 2H), 7.62 (s,1H), 7.73 (d, J = 7.6 Hz, 1H), 8.60 (d, J = 5.6 Hz, 1H), 8.66 (d, J = 7.6, Hz, 1H), 9.05 (s, 1H). MS, m/z (%): 367.1 [M + 1]. RP-HPLC, tR = 10.74 min (purity 95%).
3-((4-(1H-benzo[d]imidazol- 1-yl)− 5-fluoropyrimidin- 2-yl)amino)benzenesulfonamide (4b), White solid (78 mg, 47%). M.p. 343—344 °C, 1H NMR (DMSO-d6): δ 4.05 (s, 1H), 6.77 (d, J = 8.0 Hz, 1H), 7.16 (t, J = 8.0 Hz, 1H), 7.28–7.32 (m, 3H), 7.36 (t, J = 7.2 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 8.16 (d, J = 3.2 Hz, 1H), 8.52 (d, J = 3.2 Hz, 1H), 8.64 (s, 1H). MS, m/z (%): 385.1 [M + 1]. RP-HPLC, tR = 13.10 min (purity 99%).
3-((4-(1H-benzo[d]imidazol- 1-yl)− 5-methylpyrimidin- 2-yl)amino)benzenesulfonamide (4c), White solid (53 mg, 35%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 2.30 (s, 3H), 5.55 (s, 1H), 6.69 (d, J = 7.2 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H),7.14 (d, J = 8.0 Hz, 1H), 7.16 (s, 1H), 7.89 (t, J = 7.2 Hz, 1H), 7.32 (t, J = 8.4 Hz, 1H), 7.67 (d, J = 7.6 Hz, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.54 (s, 1H), 8.62 (s, 1H). MS, m/z (%): 381.2 [M + 1]. RP-HPLC, tR = 12.24 min (purity 97.30%).
3-((4-(1H-benzo[d]imidazol- 1-yl)− 5-(trifluoromethyl)pyrimidin- 2-yl)amino)benzenesulfonamide (4 d), White solid (64 mg, 43%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 5.50 (s, 1H, CH = N), 7.01 (s, 1H), 7.03 (s, 1H,),7.07 (s, 1H), 7.19 (s, 1H), 7.29–7.32 (m, 2H), 7.66 (d, J = 7.6 Hz, 1H), 8.35 (s, 1H), 8.65 (d, J = 8.0 Hz, 1H), 8.67 (s, 1H). MS, m/z (%): 435.2 [M + 1]. RP-HPLC, tR = 4.95 (purity 98.80%).
3-((4-(2-methyl- 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4e), White solid (90 mg, 20%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 2.76 (s, 3H), 5.66 (s, 1H), 6.53 (s, 1H), 6.76 (d, J = 6.8 Hz, 1H), 6.99 (d, J = 6.0 Hz, 1H), 7.03 (d, J = 8.0 Hz, 1H), 7.11 (s, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.23–7.27 (m, 2H), 7.56–7.58 (m, 1H), 8.12–8.15 (m, 1H), 8.65 (d, J = 5.6 Hz, 1H). MS, m/z (%): 381.1 [M + 1]. RP-HPLC, tR = 9.38 min (purity 98.45%).
3-((5-fluoro- 4-(2-methyl- 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4f), White solid (127 mg, 60%). M.p. 324—325 °C, 1H NMR (DMSO-d6): δ 2.48 (s, 3H), 6.80 (d, J = 1.6 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H), 7.18 (t, J = 7.6 Hz, 2H), 7.23 (d, J = 5.2 Hz, 1H), 7.28 (s, 1H), 7.60 (d, J = 7.6 Hz, 1H), 8.65 (s, 1H). MS, m/z (%): 399.2 [M + 1]. RP-HPLC, tR = 10.42 min (purity 98.30%).
3-((5-methyl- 4-(2-methyl- 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4 g), White solid (31 mg, 41%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 2.30 (s, 2H), 2.65 (s, 3H), 5.56 (s, 1H), 6.70 (d, J = 8.0 Hz, 1H), 7.01 (d, J = 7.6 Hz, 1H), 7.06 (d, J = 7.4 Hz, 2H), 7.20 (t, J = 8 Hz, 2H), 7.54 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 8.51 (s, 1H). MS, m/z (%): 395.2 [M + 1]. RP-HPLC, tR = 13.31 min (purity 99.20%).
3-((4-(2-methyl- 1H-benzo[d]imidazol- 1-yl)− 5-(trifluoromethyl)pyrimidin- 2-yl)amino)benzenesulfonamide (4 h), White solid (44 mg, 50%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 2.89 (s, 3H), 6.74 (d, 1H), 6.93 (s, 1H), 7.08 (t, 2H), 7.17 (s, 1H), 7.37 (m, 2H), 7.67 (d, 1H), 8.47 (s, 1H). MS, m/z (%): 449.2 [M + 1]. RP-HPLC, tR = 7.13 min (purity 99.20%).
3-((4-(2-(trifluoromethyl)− 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4i), White solid (27 mg, 13%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 5.54 (s, 1H), 6.74 (d, J = 7.6 Hz, 1H), 6.98 (d, J = 7.6 Hz, 1H), 7.10 (t, J = 4.4 Hz, 2H), 7.46–7.50 (m, 3H), 7.93 (d, J = 5.6 Hz, 1H), 8.83 (s, 1H). MS, m/z (%): 335.2 [M + 1]. RP-HPLC, tR = 13.88 min (purity 99.50%).
3-((5-fluoro- 4-(2-(trifluoromethyl)− 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4j), White solid (82 mg, 47%). M.p. 348–349 °C, 1H NMR (DMSO-d6): δ 5.57 (s, 1H), 6.76 (d, J = 8.0 Hz, 1H), 6.98 (d, J = 8.0 Hz, 1H), 7.09–7.13 (m, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.48–7.52 (m, 2H), 7.98 (d, J = 4.8 Hz, 1H), 9.08 (s, 1H). MS, m/z (%): 453.2 [M + 1]. RP-HPLC, tR = 14.88 min (purity 99.20%).
3-((5-methyl- 4-(2-(trifluoromethyl)− 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4k), White solid (18 mg, 11%). M.p. 295–296 °C, 1H NMR (DMSO-d6): δ1.09 (t, J = 6.8 Hz, 1H), 1.86 (s, 3H), 5.48 (s, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 7.04 (t, 1H), 7.09 (s, 1H), 7.45–7.49 (m, 2H), 7.96 (d, J = 6.8 Hz, 1H), 8.75 (s, 1H). MS, m/z (%): 449.1 [M + 1]. RP-HPLC, tR = 3.85 min (purity 99.88%).
3-((5-(trifluoromethyl)− 4-(2-(trifluoromethyl)− 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4 l) White solid (90 mg, 32%). M.p. 290—291 °C, 1H NMR (DMSO-d6): δ 2.55 (s, 2H), 4.05 (s, 1H), 6.67–6.69 (m, 1H), 6.94–7.02 (m, 3H), 7.44–7.48 (m, 2H), 7.85 (d, J = 5.6 Hz, 1H), 7.86 (s, 1H), 8.43 (s, 1H). MS, m/z (%): 503.2 [M + 1]. RP-HPLC, tR = 4.68 min (purity 98.30%).
3-((4-(2-isopropyl- 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4 m), White solid (76 mg, 32%). M.p. 322–323 °C, 1H NMR (DMSO-d6): δ 1.2 (d, J = 6.8 Hz, 6H), 3.98 (septet, J = 6.8 Hz, 1H), 5.66 (s, 1H), 6.78 (d, J = 8.4 Hz, 1H), 7.01 − 7.09 (m, 2H), 7.10 (s, 2H),7.15–7.26 (m, 3H), 7.61 (d, J = 6.8 Hz, 1H), 7.92 (d, J = 7.2 Hz, 1H), 8.70 (s, 1H). MS, m/z (%): 409.2 [M + 1]. RP-HPLC, tR = 11.38 min (purity 98.20%).
3-((5-fluoro- 4-(2-isopropyl- 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4n), White solid (47 mg, 50%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 1.13 (d, J = 6.8 Hz, 6H), 3.09 (septet, J = 6.8 Hz, 1H), 5.59 (s, 1H), 6.78 (d, J = 6.8 Hz, 1H), 6.97 (d, J = 7.6 Hz, 1H), 7.08 (s, 1H),7.12 (t, J = 8.0 Hz, 1H), 7.22 (d, J = 4.8 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 6.8 Hz, 1H), 8.98 (s, 1H). MS, m/z (%): 427.2 [M + 1]. RP-HPLC, tR = 13.32 min (purity 98.40%).
3-((4-(2-isopropyl- 1H-benzo[d]imidazol- 1-yl)− 5-methylpyrimidin- 2-yl)amino)benzenesulfonamide (4o), White solid (27 mg, 40%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 1.07–1.43 (m, 6H), 2.31 (s, 3H), 3.88 (s, 1H), 4.85 (s, 1H), 6.75 (d, J = 8.8 Hz, 1H), 7.02 (t, J = 7.6 Hz, 1H), 7.13 (d, J = 6.7 Hz, 1H), 7.15 (s,1H), 7.26—7.30 (m, 3H), 7.63 (d, J = 7.2 Hz, 1H), 7.77 (d, J = 7.2 Hz, 1H), 8.44 (s, 1H). MS, m/z (%): 423.2 [M + 1]. RP-HPLC, tR = 10.99 min (purity 99.60%).
3-((4-(2-isopropyl- 1H-benzo[d]imidazol- 1-yl)− 5-(trifluoromethyl)pyrimidin- 2-yl)amino)benzenesulfonamide (4p), White solid (23 mg, 51%). M.p. 287–288 °C, 1H NMR (DMSO-d6): δ 0.97 (s, 2H), 1.06 (s, 2H), 2.07 (s, 1H), 6.68 (s, 1H), 7.03 (d, J = 5.2 Hz, 2H), 7.09 (s, 1H), 7.19–7.24 (m, 2H), 7.59 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 8.44 (s, 1H). MS, m/z (%): 477.2 [M + 1]. RP-HPLC, tR = 5.26 min (purity 96.40%).
3-((4-(2-cyclopropyl- 1H-benzo[d]imidazol- 1-yl)pyrimidin- 2-yl)amino)benzenesulfonamide (4q), White solid (76 mg, 51%). M.p. 338–339 °C,1H NMR (DMSO-d6): δ 0.98 (m, 2H), 1.07 (m, 2H), 3.57 (s, 1H), 5.67 (s, 2H), 6.52 (s, 1H), 6.77 (d, J = 1.6 Hz, 1H), 7.06 (t, J = 9.6 Hz, 2H), 7.10 (s, 1H), 7.14–7.22 (m, 3H), 7.52 (d, J = 6.8 Hz, 1H), 7.97 (d, J = 6.8 Hz, 1H), 8.68 (d, J = 5.6 Hz, 1H). MS, m/z (%): 407.2 [M + 1]. RP-HPLC, tR = 11.42 min (purity 95.50%).
3-((4-(2-cyclopropyl- 1H-benzo[d]imidazol- 1-yl)− 5-fluoropyrimidin- 2-yl)amino)benzenesulfonamide (4r), White solid (34 mg, 49%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 0.97 (d, J = 7.6 Hz, 2H), 1.06 (s, 2H), 2.09 (s, 1H), 5.56 (s, 1H), 6.76 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 7.09—7.12 (m, 2H), 7.17 (s,2H), 7.22 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 8.95 (s, 1H), 8.95 (s, 1H). MS, m/z (%): 425.2 [M + 1]. RP-HPLC, tR = 11 min (purity 97.50%).
3-((4-(2-cyclopropyl- 1H-benzo[d]imidazol- 1-yl)− 5-methylpyrimidin- 2-yl)amino)benzenesulfonamide (4 s), White solid (21 mg, 42%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 0.93 (d, J = 8.0 Hz, 2H), 1.03 (s, 2H), 2.31 (s, 3H), 5.59 (s, 1H), 6.71 (d, J = 7.2 Hz, 1H), 7.03—7.11 (m, 4H), 7.14 (d, J = 8.0 Hz, 1H), 7.19 (t, J = 6.8 Hz, 1H),7.49 (d, J = 7.6 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 8.56 (s, 1H). MS, m/z (%): 421.2 [M + 1]. RP-HPLC, tR = 11.10 min (purity 97.50%).
3-((4-(2-cyclopropyl- 1H-benzo[d]imidazol- 1-yl)− 5-(trifluoromethyl)pyrimidin- 2-yl)amino)benzenesulfonamide (4 t), White solid (30 g, 47%). M.p. > 350 °C, 1H NMR (DMSO-d6): δ 0.98 (s, 2H), 1.05 (s, 2H), 6.62 (s, 1H), 6.94 (s, 1H), 7.00—7.041 (m, 2H), 7.07 (s,2H), 7.16 (d, J = 7.6 Hz, 2H), 7.20 (s, 2H), 7.49 (d, J = 7.6 Hz, 1H), 8.07 (d, J = 8.0 Hz, 1H), 8.44 (s, 1H). MS, m/z (%): 475.2 [M + 1]. RP-HPLC, tR = 18.91 min (purity 99.20%).
In vitro antiproliferative activity
In vitro antiproliferative activity assessment serves as a crucial preliminary step in the evaluation of potential anticancer agents. The ability of a compound to inhibit the uncontrolled growth of cancer cells in a controlled laboratory setting provides essential insights into its therapeutic potential. In the pursuit of more effective and targeted cancer treatments, the analysis of antiproliferative activity in vitro has become a fundamental aspect of preclinical research. Therefore, the in vitro antiproliferative activities of the newly synthesized compounds were evaluated using the standard MTT assay (Mosmann 1983; Yahya and Alqadhi 2021) against five human cancer cell lines, namely, HCT116 (colorectal carcinoma), HepG2 (hepatocellular carcinoma), HeLa (cervical epithelioid carcinoma), MCF7 (mammary gland breast cancer), and WI- 38 (embryonic lung fibroblasts) using doxorubicin (DOX) as a positive control as shown in (Table 1).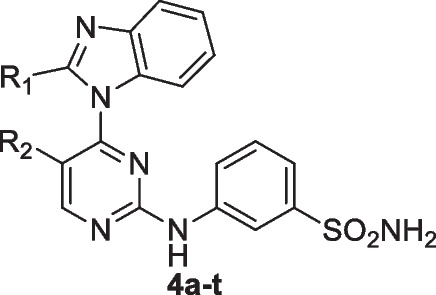
Table 1.
Cytotoxicity (IC50) of the target compounds 4-t toward HCT116, HepG2, HeLa, MCF7, and WI- 38 cells
| Compound | R1 | R2 | IC50 (µM) | ||||
|---|---|---|---|---|---|---|---|
| HCT- 116 | HePG- 2 | Hela | MCF- 7 | WI- 38 | |||
| 4a | H | H | > 50 | > 50 | > 50 | > 50 | 25.39 ± 1.8 |
| 4b | H | F | 24.67 ± 1.7 | 22.26 ± 1.6 | 17.02 ± 1.3 | 14.25 ± 1.2 | 46.71 ± 2.6 |
| 4c | H | CH3 | > 50 | 44.39 ± 2.5 | 49.84 ± 2.7 | > 50 | > 50 |
| 4 d | H | CF3 | > 50 | > 50 | > 50 | > 50 | > 50 |
| 4e | CH3 | H | > 50 | > 50 | > 50 | > 50 | 39.02 ± 2.3 |
| 4f | CH3 | F | 33.48 ± 2.0 | 15.39 ± 1.2 | 23.61 ± 1.7 | 21.65 ± 1.7 | 41.25 ± 2.5 |
| 4 g | CH3 | CH3 | 9.34 ± 0.8 | 7.59 ± 0.6 | 12.68 ± 0.9 | 5.08 ± 0.3 | > 50 |
| 4 h | CH3 | CF3 | > 50 | > 50 | > 50 | > 50 | > 50 |
| 4i | CF3 | H | > 50 | 48.31 ± 2.7 | > 50 | > 50 | > 50 |
| 4j | CF3 | F | 16.83 ± 1.2 | 8.54 ± 0.7 | 19.20 ± 1.5 | 11.71 ± 0.9 | > 50 |
| 4k | CF3 | CH3 | 48.69 ± 2.7 | 34.12 ± 2.1 | 42.82 ± 2.3 | 39.76 ± 2.2 | > 50 |
| 4 l | CF3 | CF3 | > 50 | > 50 | > 50 | > 50 | 18.34 ± 1.3 |
| 4 m | iPr | H | 7.19 ± 0.5 | 3.56 ± 0.2 | 6.39 ± 0.4 | 2.18 ± 0.1 | > 50 |
| 4n | iPr | F | 43.29 ± 2.5 | 19.87 ± 1.4 | 36.41 ± 2.1 | 32.36 ± 2.0 | 76.13 ± 3.9 |
| 4o | iPr | CH3 | > 50 | > 50 | > 50 | > 50 | > 50 |
| 4p | iPr | CF3 | > 50 | > 50 | > 50 | > 50 | 45.29 ± 2.7 |
| 4q | Cyclopropyl | H | 13.72 ± 1.0 | 5.88 ± 0.4 | 9.71 ± 0.7 | 8.19 ± 0.5 | 31.40 ± 2.1 |
| 4r | Cyclopropyl | F | 29.81 ± 1.9 | 11.37 ± 0.9 | 26.53 ± 1.8 | 16.20 ± 1.4 | > 50 |
| 4 s | Cyclopropyl | CH3 | > 50 | 38.06 ± 2.3 | 47.16 ± 2.5 | 45.67 ± 2.4 | 28.01 ± 2.0 |
| 4 t | Cyclopropyl | CF3 | 38.79 ± 2.3 | 29.31 ± 1.9 | 30.29 ± 1.9 | 27.81 ± 1.8 | > 50 |
| Doxorubicin | - | - | 5.23 ± 0.3 | 4.50 ± 0.2 | 5.57 ± 0.4 | 4.17 ± 0.2 | 6.72 ± 0.5 |
IC50 (µM): 1–10 (very strong), 11–20 (strong), 21–50 (moderate), 51–100 (weak), and > 100 (non-cytotoxic). DOX: Doxorubicin
CDK9 and CDK12 inhibitory activities
The ability of compounds 4a–t to inhibit CDK9 and CDK 12 was evaluated in vitro and dinaciclib was used as the positive drug control for comparison. IC50 values (measured in µM) were determined using dose–response curves, and the results are shown in (Table 2).
Table 2.
Inhibitory activities of compounds 4a-t against CDK9 and CDK12
| Compound | IC50 (μM) | |
|---|---|---|
| CDK9 | CDK12 | |
| 4a | 0.396 ± 0.017 | 0.295 ± 0.011 |
| 4b | 1.15 ± 0.05 | 0.566 ± 0.022 |
| 4c | 0.041 ± 0.002 | 0.14 ± 0.005 |
| 4 d | 0.782 ± 0.034 | 1.329 ± 0.052 |
| 4e | 0.409 ± 0.018 | 0.425 ± 0.017 |
| 4f | 0.762 ± 0.033 | 1.143 ± 0.044 |
| 4 g | 0.114 ± 0.005 | 0.071 ± 0.003 |
| 4 h | 0.05 ± 0.002 | 0.153 ± 0.006 |
| 4i | 1.088 ± 0.047 | 0.365 ± 0.014 |
| 4j | 0.055 ± 0.002 | 0.104 ± 0.004 |
| 4k | 5.468 ± 0.236 | 1.211 ± 0.047 |
| 4 l | 1.673 ± 0.072 | 0.899 ± 0.035 |
| 4 m | 0.04 ± 0.002 | 0.06 ± 0.002 |
| 4n | 0.392 ± 0.017 | 0.179 ± 0.007 |
| 4o | 1.045 ± 0.045 | 1.876 ± 0.073 |
| 4p | 0.153 ± 0.007 | 0.664 ± 0.026 |
| 4q | 0.057 ± 0.002 | 0.074 ± 0.003 |
| 4r | 0.242 ± 0.01 | 0.104 ± 0.001 |
| 4 s | 0.408 ± 0.018 | 0.189 ± 0.007 |
| 4 t | 0.121 ± 0.005 | 0.533 ± 0.021 |
| Dinaciclib | 0.032 ± 0.001 | % 1.% 2 ± 0.002 |
In comparison to dinaciclib, a known potent CDK inhibitor, some of the new analogues approach its efficacy but do not surpass it. Compounds 4 m, and 4q stand out for their low IC50 values and present promising candidates for further development. This analysis underscores the importance of small alkyl groups at R1, paired with specific substituents at R2, for maximizing potency against CDK9 and CDK12.
Molecular docking procedure
Initially, we sourced the 3D crystal structures of both CDK9 and CDK12. These were obtained from the renowned RCSB Protein Data Bank, with the reference codes PDB: 6Z45 (Barlaam et al. 2020) for CDK9 and 7nxk (Jiang et al. 2021) for CDK12. Once retrieved, these structures underwent a thorough preparation process within the MOE platform. This procedure ensured that any missing atoms and loops were adequately addressed, steric clashes minimized, and optimal alternative conformations selected. A critical step in this phase was the 3D protonation, ensuring that the proteins mirrored their active physiological states.
Turning our attention to the compounds under study, the MMFF94 force field was employed for the energy minimization step. To provide a more accurate simulation, we incorporated hydrogen atoms, calculated the partial charges, and subsequently saved the compound structures as mol2 files.
The crux of our study was the docking of these compounds into the ATP binding site of the proteins. Leveraging the triangle matcher algorithm ensured precision in this phase. The docking scores, which signify the binding affinity between CDK9, CDK12, and each ligand, were initially evaluated using the London-dG scoring function. To further refine our results and ensure accuracy, we also rescored using the GBVI/WSA-dG function within the MOE software (Ahmed et al. 2022).
Molecular dynamics (MD) simulation overview
Our team initiated the molecular dynamics simulation procedure by deploying the NAMD software, specifically the version Git- 2021–09 - 06 for Linux- × 86 64 multicore. The simulation framework was built upon the renowned AMBER force field. We conducted several 50 ns MD simulations to meticulously evaluate the binding stabilities of compound 4 m.
To generate the configuration files essential for the MD simulations, we utilized the CHARMM-GUI online platform (http://www.charmm-gui.org/), which was accessed on 5 th October 2023. Incorporating physiological conditions, we solvated the poses—those which displayed the most promising binding affinities from our earlier molecular docking experiments—in a 10.0 periodic dodecahedron water box. This was fortified with a salt content of 0.15 M to further enhance the simulation's accuracy.
For parameterizing the ligand, we harnessed the capabilities of the CHARMM General Force Field (CGenFF) online tool (https://cgenff.umaryland.edu/), which was also accessed on 20 th February 2023(Phillips et al. 2005; Lee et al. 2016; Brooks et al. 2009). Ensuring the readiness of our systems for the simulation, a meticulous minimization and equilibration process was undertaken. This spanned 200 ps, encompassing both the NPT (isothermal–isobaric ensemble) and NVT (canonical ensemble) steps.
Our detailed simulation strategy involved running each MD simulation with a precision of a 2 fs time step. To gauge the robustness and stability of each system over the simulation duration, we computed the Root Mean Square Deviation (RMSD). It was imperative that any compound under scrutiny exhibited stable binding throughout the simulation process, characterized by converging RMSD values devoid of significant fluctuations (Ahmed et al. 2022).
Docking study
Compound 4 m with CDK12
The CDK12 kinase structure, as per the provided details by Bösken et al.(Dixon-Clarke et al. 2015), is typified by a canonical kinase fold, which is characterized by the N-terminal lobe and the C-terminal lobe. The CDK12 active site has specific structural features that can crucially interact with ligands, such as the glycine-rich loop, Asp859, and an extended C-terminal segment that meanders towards the ATP-binding cleft.
Compound 4 m with CDK9
CDK9 presents a typical kinase fold with distinct N and C-terminal lobes. While there are similarities with CDK2, the significant distinctions in loop regions and key residues underpin its unique interactions with ligands (Xiao et al. 2023; Zhong et al. 2021). Docking results of compound 4 m with CDK9 reveal intriguing interactions with key residues in the kinase. The nitrogen atom (N27) of the ligand forms a hydrogen bond with the oxygen of CYS 106, exhibiting an interaction energy of − 0.8 kcal/mol. Another nitrogen atom (N35) in the compound forms hydrogen bonds with the OD2 of ASP 109 and the oxygen of ALA 153, showing more pronounced binding energies of − 2.1 kcal/mol and − 2.9 kcal/mol, respectively. Notably, ASP 109 lies at the beginning of the C-terminal lobe, making it crucial for interactions. Additionally, aromatic interactions of compound 4 m's 5-ring and 6-ring with VAL 33, evidenced by the pi-H interactions, further anchor the molecule to the binding site (Table 4 and Fig. 3).
Table 4.
Hydrogen bond interactions of compounds 4 m with CDK9
| Compound | Ligand | Receptor | Interaction | Distance | E (kcal/mol) |
S (kcal/mol) |
|---|---|---|---|---|---|---|
| 4 m | N 27 | O CYS 106 (A) | H-donor | 3.58 | − 0.8 | − 7.25 |
| N 35 | OD2 ASP 109 (A) | H-donor | 2.99 | − 2.1 | ||
| N 35 | O ALA 153 (A) | H-donor | 3.09 | − 2.9 | ||
| 5-ring | CG1 VAL 33 (A) | pi-H | 4.25 | − 0.6 | ||
| 6-ring | CG2 VAL 33 (A) | pi-H | 3.62 | − 0.4 | ||
| Q6E | N13 39 | O CYS 106 (A) | H-donor | 2.82 | − 3.1 | − 7.87 |
| C18 42 | O ASP 104 (A) | H-donor | 3.58 | − 0.3 | ||
| N19 44 | N CYS 106 (A) | H-acceptor | 2.91 | − 5 | ||
| 5-ring | CG1 VAL 33 (A) | pi-H | 3.79 | − 0.4 | ||
| 6-ring | CD1 LEU 156 (A) | pi-H | 3.57 | − 0.3 |
Fig. 3.

A 3D visualization of CDK9 (PDB code: 6Z45) displays the binding points for compound 4 m (shown in green) and the co-crystalline ligand Q6E (represented in brown). The detailed docking configurations emphasize the unique binding directions and areas each molecule interacts with within the kinase's active site
Molecular dynamic simulation
The root-mean-square deviation (RMSD)of CDK12 and CDK9
In the molecular dynamics simulations (MSD) of CDK12 (Fig. 4A), both in its unbound (apo) state and when complexed with compound 4 m, the RMSD values provide insight into structural stability. The slightly lower average RMSD for the CDK12-compound 4 m complex compared to its apo state suggests that the presence of compound 4 m may impart some conformational stability to the CDK12 protein. This observation is further reinforced by the reduced standard deviation in the protein–ligand complex, indicating more consistent structural orientations and potentially hinting at stabilizing interactions between the protein and the compound.
Fig. 4.

RMSD plots for compound 4 m in interaction with (A) CDK12 and (B) CDK9. The apo protein is represented by a block line, while the complex formation is depicted by a red line
The RMSD values provide insights into the structural stability of molecular systems over the course of molecular dynamics simulations. For the CDK9 protein in complex with compound 4 m (PDB code 6Z45), the average RMSD value is 2.33 Å with a standard deviation (SD) of 0.27 Å (Fig. 4B). This suggests that the ligand-bound form of the protein is relatively stable with only minor fluctuations around the mean value. In comparison, the apo form (protein alone) of CDK9 exhibits an average RMSD value of 3.10 Å and a higher SD of 0.62 Å. This indicates greater variability and potentially more pronounced structural rearrangements in the apo form relative to the ligand-bound state. Thus, the binding of compound 4 m appears to confer added stability to the CDK9 protein structure, reducing its conformational flexibility as evidenced by the lower RMSD values and reduced variability in the bound state.
The root mean square fluctuation (RMSF)
The CDK12 kinase structure presents a typical kinase fold delineated between the N-terminal and C-terminal lobes. The kinase's active configuration is emphasized by the orientation of the αC helix, and this specific arrangement enables the substrate's access to the catalytic site. Within this arrangement, several regions, such as the 877DFG motif and the glycine-rich loop, play crucial roles in the kinase's activity. Upon examining the RMSF of compound 4 m within CDK12's active site during a 50 nS MDS, we see distinct changes in the structural flexibility of certain residues when the ligand is present. The subtraction values provided depict the change in fluctuation between the liganded (Pro-LIG) and unliganded (Apo) states for specific residues. A considerable number of residues in the N-terminal lobe (714–719) exhibit decreased flexibility upon ligand binding, suggesting that the compound may stabilize this region. This stabilization is crucial given the proximity of these residues to the glycine-rich loop (734–739) that functions prominently in ATP binding (Fig. 5A).
Fig. 5.

Root Mean Square Fluctuation (RMSF) profiles of compound 4 m when complexed with (A) CDK12 and (B) CDK9. The apo protein is represented by a black line, while the complexed form is depicted in red
Residues 797–805, presumably near the activation segment, show substantial decreases in fluctuation. These values underscore the compound's potential influence on the kinase's activation conformation. The most dramatic decrease is observed in residues 800–805, with residue 800 experiencing a whopping drop of over 6.6 Å. This suggests a profound structural stabilization, implying that compound 4 m could be playing a significant role in modulating the kinase's activation status. Furthermore, residues 1031–1039 in the C-terminal region of the kinase, which meanders around the C-terminal kinase lobe, are particularly interesting. These residues have a direct association with the ATP binding cleft and the bound nucleotide. The increment in RMSF values for residues 1032–1039 signifies increased flexibility upon ligand binding. This dynamic behavior might be due to the ligand altering the usual interactions in the ATP cleft. The most prominent increase is seen in residues 1038 and 1039, with the latter fluctuating by over 4 Å. Considering the pivotal role of this C-terminal stretch in the kinase's ATP binding and its ‘closed conformation’, the ligand's influence here can be critical. The binding of compound 4 m to CDK12 might be causing alterations to the native intra-molecular interactions, potentially impacting the kinase's overall function.
While The CDK9 enzyme, possessing a typical kinase fold, consists of an N-terminal and a C-terminal lobe. This architecture, while sharing a 40% sequence identity with CDK2, has distinct structural variations, especially in the loop regions. Importantly, CDK9's active conformation is typified by the orientation of the αC-helix, the phosphoryl transfer residues'conformation, and the activation segment configuration that makes the catalytic site accessible for substrate binding. In the context of this architecture, the RMSF results provide insights into the dynamic behavior of residues upon binding compound 4 m to the active site of CDK9. The residues with subtraction values greater than 0.5 Å indicate significant conformational changes upon ligand binding. A positive value suggests that the residue is more flexible in the protein–ligand complex than in the apo state, while a negative value indicates that the residue becomes more rigid upon ligand binding. Residues 4, 5, 9, 26, and 88, with positive subtraction values, have their flexibility enhanced upon compound 4 m binding. Specifically, residue 4 shows a substantial increase in flexibility with a value of 3.431839705 Å. Conversely, a multitude of residues, such as 38–41, 52–54, 175–182, 200–208, 233, 234, 249–273, and 276–302, exhibit negative subtraction values, implying that these residues have reduced flexibility in the presence of compound 4 m. Some residues, like 251, 263, and 264, show a stark reduction in flexibility, with values lower than − 2 Å, suggesting a significant structural stabilization upon ligand binding (Fig. 5B).
It's worth noting that residues 175–182 are a part of the activation segment, and their decreased flexibility upon compound 4 m binding might indicate a direct interaction or an indirect stabilization effect. This is consistent with the description of the kinase's active conformation, where residues from the activation segment play pivotal roles in substrate recognition and catalysis. The effect of compound 4 m binding to CDK9 can be further contextualized by considering the spatial orientation of the affected residues in relation to the active site. The residues that demonstrate altered flexibility likely outline the binding pocket or are involved in securing the compound in the active site, ensuring optimal interactions for effective inhibition. Compound 4 m seems to cause substantial conformational changes in CDK9, especially in regions critical for its activity. Such changes might contribute to its inhibitory effect, either by directly obstructing substrate binding or by altering the kinase's conformation to a less active or inactive state.
Compactness
The compactness of a protein–ligand complex can be examined through parameters like the Solvent Accessible Surface Area (SASA) and the Radius of Gyration (Rg) Fig. 4(A-D). Both these parameters offer insights into how a ligand binding affects the protein's overall structure and conformation.
From the provided SASA values for the CDK12 enzyme, it is observed that the mean SASA for the apo form (without the ligand) is 19231.84Å2. Upon binding with compound 4 m (Pro-LIG), there's a slight increase in the mean SASA to 19562.70 Å2. This increase suggests that the protein might adopt a slightly more extended conformation or that certain regions of the protein become more exposed to the solvent upon ligand binding. The standard deviations (SD) for the Apo and Pro-LIG states, 342.93 Å2 and 325.9698348 Å2 respectively, indicate that the fluctuations in SASA are relatively consistent in both states, with the apo form having a slightly higher fluctuation (Fig. 6B).
Fig. 6.

Comparative analysis of Rg and SASA for compound 4 m in complex with CDK12 and CDK9. A Rg plot for compound 4 m with CDK12, B SASA plot for compound 4 m with CDK12, C Rg plot for compound 4 m with CDK9, and D SASA plot for compound 4 m with CDK9. The apo-protein is represented by a black line, while the complex is illustrated with a red line
Turning our attention to the Rg values, the Radius of Gyration offers a measure of the protein's size and overall shape. The mean Rg value for the apo state of CDK12 is 22.01Å, which sees an increase to 22.54769838 Å upon binding with compound 4 m. This indicates that the protein might become slightly less compact or that it adopts a conformation with a broader distribution of its mass around its center when compound 4 m is bound. The standard deviations, 0.195 Å for Apo and 0.229 Å for Pro-LIG, are relatively small, suggesting that the overall conformational fluctuations in the protein's shape are minor and consistent across both states. However, the slightly higher SD in the Pro-LIG state might hint at a bit more variability in the protein's conformation upon ligand binding (Fig. 6A).
When examining the compactness of the complex formed by compound 4 m with the CDK9 enzyme, the SASA values provide a direct indication. The mean SASA for the apo form of CDK9 is 18864.67 Å2, which reduces slightly to 18568.47 Å2 in the Pro-LIG state. This decrease suggests that upon binding compound 4 m, CDK9 becomes marginally more compact, or the binding site becomes less exposed to the solvent. The standard deviation (SD) for the SASA values is 355.11 Å2 for the apo state and slightly higher at 401.12 Å2 for the Pro-LIG state, indicating slightly more variability in the compactness of the protein–ligand complex as compared to the apo state (Fig. 6C) (Antiviral Activity of Some Benzo[g]Quinazolines against Coxsackievirus B4: Biological Screening and Docking Study—PubMed n.d.; Eskandrani et al. 2023).
On the other hand, the Rg provides a measure of the protein's overall shape and the distribution of its mass around its center of gravity. The mean Rg value for the apo form of CDK9 is 21.71 Å, which increases to 21.99 Å when compound 4 m is bound. This rise in Rg suggests that the overall structure of CDK9 expands or becomes slightly less compact upon ligand binding. Such a change could be attributed to local conformational rearrangements caused by the ligand binding. The SD values for Rg are 0.19 Å for the apo state and slightly lesser, 0.16 Å, for the Pro-LIG state, which suggests that the conformational variability or flexibility around the mean shape is slightly reduced upon ligand binding (Fig. 6D).
Hydrogen bond of CDK12 and CDK9
In the complex of compound 4 m with CDK12, the observed mean number of hydrogen bonds during a 50 ns molecular dynamics simulation is 2, signifying a stable interaction. However, the maximum number of hydrogen bonds noted at certain moments reaches up to 6, indicating transient conformations that allow for stronger binding events (Fig. 7A). This dynamic hydrogen bonding profile suggests not only a stable interaction but also the potential for alternative binding modes, providing valuable insights for optimizing ligand–protein interactions.
Fig. 7.

A Patterns of hydrogen bonds in the CDK12–4 m complex during a molecular dynamic simulation over 50 ns. B The hydrogen bond occupancy of several essential CDK12 residues throughout the simulation run in the presence of 5b inhibitor. C Patterns of hydrogen bonds in the CDK9–4 m complex during a molecular dynamic simulation over 50 ns. D The hydrogen bond occupancy of several essential CDK9 residues throughout the simulation run in the presence of 4 m
The CDK12 kinase, with its structure captured in PDB code 7nxk, presents a classical kinase fold with both the N-terminal and C-terminal lobes playing crucial roles in the enzyme's activity. Specifically, the kinase's active conformation, characterized by the orientation of the αC-helix and other motifs, is highly critical for substrate accessibility to the catalytic site. In an evaluation of molecular dynamics simulation over 50 ns for compound 4 m within the CDK12 active site, several notable interactions emerge (Fig. 7 B). Among the most persistent of these interactions is between the side chain of MET816 and the ligand (occupancy of 52.75%). Furthermore, the ligand's side chain also forms a significant hydrogen bond with the main chain of ASP817, having an occupancy of 34%. These interactions indicate the potential anchor points of compound 4 m within the kinase's active site. Other important interactions include the side chain of TYR815 with the ligand (occupancy of 13.6%) and LYS743's side chain interaction with the ligand (occupancy of 9.05%). The ligand's engagement with TYR815, a residue mentioned to have polar interactions with the kinase's extended C-terminal region, further highlights the ligand's potential involvement in modulating the kinase's conformation and activity. Many other residues, such as ILE733, GLU735, HSD818, and SER863, also show interactions with compound 4 m, albeit with lower occupancy percentages. While these interactions are less persistent over the course of the simulation, they still signify the dynamic nature of the ligand within the kinase's active site and suggest various potential points of interaction during the ligand's residence time. The multiple hydrogen bond interactions between the ligand and residues both from the N-terminal and the C-terminal lobes provide insights into the orientation of compound 4 m. The ligand appears to span across the active site, likely interacting with critical regions including the ATP cleft and possibly the extended C-terminal region. Specifically, the interactions with the CYS1039 residue, which is part of the stretch extending after the last helix of the kinase domain, suggest the ligand's potential involvement in modulating ATP binding and substrate accessibility.
However, in the complex of compound 4 m with CDK9, the hydrogen bond dynamics over a 50 ns molecular dynamics simulation reveal an average of 2.4335 hydrogen bonds, indicating a consistent and stable interaction between the ligand and the protein. The peak value, with as many as seven hydrogen bonds, suggests transient moments of enhanced complementarity (Fig. 7C). This consistent hydrogen bonding, combined with moments of higher interaction, underscores the importance of hydrogen bonds in the stability of this complex and might hint at avenues for further ligand optimization.
Within this structural context, compound 4 m exhibits several hydrogen bond interactions with CDK9 during the Molecular Dynamics Simulation (MDS). The interactions between CYS106 and LIG1, with an occupancy of over 50%, suggest a strong binding affinity of the compound near the N-terminal lobe since CYS106 is part of this region (Fig. 7D). Furthermore, the high-occupancy interaction between LIG1 and GLU107, another residue in the vicinity of the N-terminal lobe, underscores this idea. The interaction of LIG1 with ASP104, which boasts an occupancy of 41.65%, adds another layer of stability to the compound's binding. This interaction indicates that compound 4 m is likely nestled firmly within the kinase’s N-terminal region.
Beyond these primary interactions, compound 4 m also interacts with other residues such as ASP109, ILE25, LYS48, and PHE105, albeit less frequently. These interactions still play a role in the compound's overall binding to CDK9. The breadth of interactions suggests that compound 4 m spans a region between the N-terminal and the early C-terminal lobe. This positioning, especially around residues like CYS106 and GLU107, is crucial because it places the compound close to the kinase’s active site, which is vital for its inhibitory activity. Additionally, the bond with ALA166, a part of the activation segment, indicates that compound 4 m might have an influence on the conformation of this segment, which can, in turn, impact the kinase's activity.
Discussion
Chemistry
In this study, a series of 2-anilino- 4-(benzimidazol- 1-yl)pyrimidine derivatives were designed and synthesized as potential CDK9 and CDK12 inhibitors, leveraging key pyrimidine ring modifications at the 4- and 5-positions, which are known to influence kinase selectivity and potency. The compounds were synthesized through a multi-step process involving nucleophilic substitution and condensation reactions and were structurally confirmed using 1H NMR, mass spectrometry, and RP-HPLC. Structure–activity relationship (SAR) analysis revealed that lipophilic groups at R₁, such as isopropyl (iPr) or cyclopropyl, significantly enhanced inhibitory activity against CDK9 and CDK12, likely due to hydrophobic interactions within the kinase active sites. Furthermore, the presence of a hydrogen atom at R₂ resulted in the most potent inhibition, as observed in compounds 4 m and 4q, which displayed notably low IC₅₀ values against both kinases. These findings were further supported by MTT cytotoxicity assays, where 4 m and 4q exhibited strong antiproliferative activity against cancer cell lines while maintaining limited cytotoxicity toward normal WI- 38 fibroblast cells, demonstrating high selectivity and therapeutic potential. This selectivity is particularly significant, as many CDK inhibitors suffer from off-target effects that limit their clinical application. Overall, these results highlight the importance of hydrophobic interactions and steric factors in optimizing kinase inhibition and selectivity. The synthesis of the target compounds was achieved through a multi-step process involving nucleophilic substitution and condensation reactions. The procedure began with the preparation of 4-chloropyrimidine derivatives, which were synthesized by reacting the corresponding aminopyrimidines with phosphorus oxychloride under reflux. The resulting chlorinated intermediates were then coupled with various substituted benzimidazoles in the presence of a base such as potassium carbonate or sodium hydride in anhydrous DMF, leading to the formation anilinobenzimidazole derivatives.
In the subsequent step, the amino group was introduced at the C2 position of the pyrimidine ring through nucleophilic substitution using different amines, including sulfonamides. The reactions were carried out in polar aprotic solvents such as DMF or DMSO, under mild to moderate heating conditions. The crude products were purified by recrystallization or column chromatography using a suitable eluent system.
Compound 4 m exhibits characteristic features in its NMR spectra and HPLC purity. In the 1H NMR spectrum, aromatic protons show shifts influenced by the presence of electronegative substituents. The fluorine or trifluoromethyl groups cause downfield shifts in the aromatic region due to their electron-withdrawing effects. Additionally, 19 F NMR signals, typically observed between − 120 to − 60 ppm, provide insight into the position and environment of the fluorine atoms. The trifluoromethyl group often causes more pronounced downfield shifts in the 1H NMR due to its bulk and electron-withdrawing nature. For compounds with methyl substituents, a singlet around 2.0–2.5 ppm is typically observed in the 1H NMR spectrum, corresponding to the methyl group.
In terms of HPLC purity, Compounds with fluorine or trifluoromethyl groups may show slight variations in retention time on the HPLC column due to the influence of these substituents on the compound’s polarity, but they typically maintain a high level of purity. Methyl-substituted compounds tend to elute faster on reversed-phase columns, due to their increased hydrophobicity, and also exhibit high purity. Overall, the presence of fluorine or trifluoromethyl groups affects the NMR shifts, while HPLC profiles for all these compounds generally show good purity with minor differences in retention times.
The final compounds were characterized using spectroscopic techniques. The 1H NMR spectra displayed characteristic singlets and doublets corresponding to aromatic and heterocyclic protons, along with deshielded signals indicative of sulfonamide or trifluoromethyl groups. Additionally, mass spectrometry confirmed the molecular weights of the synthesized compounds, while RP-HPLC analysis ensured their purity, with most derivatives exhibiting a high degree of purity (above 95%).
This synthetic strategy successfully yielded a diverse library of anilinobenzimidazole derivatives with varying substituents, demonstrating the effectiveness of the employed methodology.
In vitro antiproliferative activity
Compounds 4 g, 4j, 4 m, and 4q displayed remarkably strong anticancer potencies against HepG2 cells, with IC50 of 7.59, 8.54, 3.56 and 5.88 µM, respectively. Compounds 4f, 4n and 4r showed strong activity with IC50 of 15.39, 19.78 and 11.37 µM, respectively, compared to the positive control, DOX (IC50 = 4.50 µM).nCompound 4 m, and 4q had the highest anticancer activity against HeLa cells, with an IC50 of 6.39 and 9.71 µM, respectively. Compounds 4b, 4 g, and 4j showed higher inhibitory activity, with IC50 values of 17.02, 12.63, and 19.39 µM, respectively. compared to the positive control DOX (IC50 = 5.57 µM). Comparison of IC50 values against MCF- 7 cells revealed that compounds 4 g, 4 m, and 4q showed significant anticancer potency with IC50 of 5.08, 2.18 and 8.19 µM, respectively compared to that of the positive control DOX (IC50 = 4.17 µM). All the tested compounds displayed different antitumor activities, ranging from weak to moderate, against HCT116 cells, except for compounds 4 g, 4j, 4 m and 4q with strong anticancer activity against HCT116 cells IC50 9.34, 16.83, 7.19 and 13.72 µM respectively against the positive control DOX (IC50 = 5.23 µM).
The Structure–Activity Relationship (SAR) of the series of compounds was analyzed, revealing distinct patterns based on the substitutions at the R1 and R2 positions. When R1 is a hydrogen atom, the IC50 values against CDK9 and CDK12 exhibited a significant range, with the lowest value for CDK9 observed at 0.041 µM in compound 4c, where R2 is a methyl group. This finding suggests that the combination of hydrogen at R1 and a methyl group at R2 are particularly potent against CDK9. For CDK12, the most favourable outcome with hydrogen at R1 occurred when R2 is also hydrogen, as seen in compound 4a. Substitution of R1 with a methyl group generally enhances the potency of the compounds against CDK9, particularly when R2 is trifluoromethyl (CF3), as evidenced by compound 4 h, which demonstrated a low IC50 value of 0.05 µM. A similar trend is observed for CDK12, where the combination of a methyl group at R1 and a methyl group at R2 in compound 4 g yielded a potent IC50 of 0.071 µM. Conversely, replacement of R1 with CF3 generally resulted in reduced potency for both CDK9 and CDK12, with the exception of compound 4j, where fluorine is present at R2, leading to lower IC50 values. When R1 is substituted with isopropyl (iPr), a notable improvement in the compounds'efficacy is observed, particularly for CDK9. Compound 4 m, with an iPr group at R1 and a hydrogen atom at R2, exhibits one of the lowest IC50 values for both CDK9 (0.04 µM) and CDK12 (0.06 µM), indicating that this combination is highly favourable. The cyclopropyl group at R1 also contributes to consistent potency across the series, with compound 4q (R2 = H) showing low IC50 values, particularly for CDK9.
However, when it comes to R2 substitutions, the presence of fluorine at R2 with various R1 groups yields varying results, with some combinations, such as iPr at R1 and F at R2 (compound 4n), showing enhanced activity against both CDK9 and CDK12. The presence of a methyl group at R2 generally leads to moderate potency, though a significant drop in efficacy is observed when paired with a CF3 group at R1 (compound 4k). CF3 at R2 generally enhances activity when combined with smaller R1 groups, though bulkier combinations lead to decreased potency.
CDK9 and CDK12 inhibitory activities
The transcriptional cyclin-dependent kinases (CDKs), particularly CDK9 and CDK12, play a crucial role in regulating gene expression by phosphorylating the carboxy-terminal domain (CTD) of RNA polymerase II, specifically at Ser2, during the elongation phase of transcription. Dysregulation of these kinases has been linked to various malignancies, including leukemia and solid tumors such as ovarian and prostate cancers, making them attractive therapeutic targets. In comparison to dinaciclib, a known potent CDK inhibitor, some of the new analogues approach its efficacy but do not surpass it. Compounds 4 m, and 4q stand out for their low IC50 values and present promising candidates for further development. This analysis underscores the importance of small alkyl groups at R1, paired with specific substituents at R2, for maximizing potency against CDK9 and CDK12.
Molecular docking studies
Compound 4 m with CDK12
Compound 4 m exhibits significant interactions within the CDK12 active site. Notably, the hydrogen bond it forms with MET 816 (with a distance of 2.79 Å and an energy of − 3.7 kcal/mol) indicates a key interaction within the N-terminal lobe. Additionally, the compound interacts with CYS 1039 and HIS 1040, residues found in the extended C-terminal segment that approaches the ATP cleft. The interaction with HIS 1040 is particularly strong, with an energy of − 4.5 kcal/mol, highlighting the potential critical role of the C-terminal lobe in mediating the binding of compound 4 m. Furthermore, an aromatic interaction with VAL 741, denoted as a pi-H interaction, suggests that compound 4 m might engage residues near the P-loop, another critical region in kinase function (Table 3 & Fig. 8).
Table 3.
Hydrogen bond interactions of compounds 4 m with CDK12
| Compound | Ligand | Receptor | Interaction | Distance | E (kcal/mol) |
S (kcal/mol) |
|---|---|---|---|---|---|---|
| 4 m | N 27 | O MET 816 (A) | H-donor | 2.79 | − 3.7 | − 6.61 |
| N 35 | SG CYS 1039 (A) | H-donor | 3.6 | − 0.9 | ||
| N 12 | NZ LYS 756 (A) | H-acceptor | 2.96 | − 3.8 | ||
| O 34 | NE2 HIS 1040 (A) | H-acceptor | 2.85 | − 4.5 | ||
| N 45 | N MET 816 (A) | H-acceptor | 3.46 | − 1.4 | ||
| 6-ring | CG1 VAL 741 (A) | pi-H | 3.73 | − 0.3 | ||
| UUB | C19 12 | O ASP 817 (A) | H-donor | 3.11 | − 0.3 | − 8.26 |
| C22 15 | OD1 ASP 819 (A) | H-donor | 3.72 | − 0.3 | ||
| N7 32 | OD1 ASP 877 (A) | H-donor | 3.1 | − 0.5 | ||
| N13 29 | N MET 816 (A) | H-acceptor | 3 | − 1 | ||
| 6-ring | CG1 VAL 741 (A) | pi-H | 3.84 | − 0.3 | ||
| 6-ring | CD1 LEU 866 (A) | pi-H | 3.63 | − 0.4 |
Fig. 8.

Three-dimensional representation of CDK12 (PDB code: 7nxk) showing the interaction sites with compound 4 m (depicted in green) and co-crystalline ligand (UUB) (depicted in yellow). The intricate docking patterns highlight the distinct binding orientations and regions targeted by each molecule within the kinase's active site
While the UUB, the co-crystalline ligand, has a more favourable binding energy of − 8.26 kcal/mol, suggesting a stronger affinity to CDK12 compared to compound 4 m. It forms interactions with residues such as ASP 817 and ASP 819, which are located near the ATP cleft. The interaction with MET 816 further underscores the significance of the N-terminal lobe in ligand binding. Notably, UUB also has an aromatic interaction with VAL 741, much like compound 4 m, emphasizing the potential importance of this residue in ligand anchoring (Table 3 & Fig. 8).
In comparing the binding of compound 4 m and UUB, both ligands seem to target regions that are pivotal for the kinase's catalytic function. While compound 4 m demonstrates a strong interaction with residues on both the N-terminal and the C-terminal lobe, UUB seems to have a slight preference for the N-terminal region, particularly with its interactions around the ATP-binding cleft. The stronger binding energy for UUB suggests that it might occupy the active site of CDK12 more effectively than compound 4 m. This could be due to a better alignment with the kinase's structural features or due to more favourable molecular interactions.
Compound 4 m with CDK9
In contrast, the co-crystalline ligand Q6E shows an even stronger affinity to CDK9. N13 of Q6E forms a strong hydrogen bond with CYS 106, exhibiting an interaction energy of − 3.1 kcal/mol. Additionally, N19 of the ligand engages in a robust hydrogen bonding with the nitrogen of CYS 106, displaying an interaction energy of − 5 kcal/mol. The pi-H interactions of Q6E with VAL 33 and LEU 156 further suggest its close proximity and alignment with the kinase's key residues. While both molecules engage with CYS 106, indicative of its importance in ligand binding, Q6E's interactions are notably stronger, as evidenced by the lower interaction energy of − 7.87 kcal/mol compared to compound 4 m's − 7.25 kcal/mol. This difference in binding energy, along with an RMSD of 2.19 for Q6E, suggests that Q6E may present a closer alignment to the original binding mode of the native ligand in CDK9 (Table 4 and Fig. 2). Considering the orientation and location within the active site, while both molecules appear to target the region around CYS 106 and VAL 33, the specific interactions with other residues, such as ASP 109 for compound 4 m and ASP 104 and LEU 156 for Q6E, suggest slightly different orientations and perhaps binding modes. This could have implications in their mechanism of action, potential off-target effects, and selectivity.
Conclusion
In conclusion, the newly synthesized series of 2-anilinobenzimidazole derivatives exhibit strong potential as chemotherapeutic agents, as demonstrated by their significant cytotoxic effects and CDK inhibitory activity across various solid tumour cell lines. Compounds 4 g, 4j, 4 m, and 4q showed particularly robust anticancer efficacy, with notable activity against HepG2, HeLa, and MCF- 7 cells. Among these, compounds 4 m and 4q emerged as the most effective inhibitors of CDK9 and CDK12, suggesting their promise as targeted cancer therapies. Molecular docking simulations and in silico ADMET analysis further support the drug-like properties of these compounds, indicating their potential for further development in cancer treatment.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are thankful to the Researchers Supporting Project number (RSPD2025R930), King Saud University, Riyadh, Saudi Arabia.
Author contribution
L.S.A.-R., A.S.A and S.A.A. carried out the experiments and contributed to interpreting and validating the results. A.H.B., H.H.A and A.A.A. contributed to the performance of molecular docking and dynamic simulation and writing the manuscript. H.M.A. conceived, planned, and contributed to interpreting and validating the results and writing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Researchers Supporting Project number (RSPD2025R930), King Saud University, Riyadh, Saudi Arabia.
Data availability
Not Applicable.
Declarations
Ethics
Not Applicable.
Competing interest
Not Applicable.
Footnotes
In the Acknowledgment and Funding Sections of this article the project number was incorrectly given as RSPD2024R930 but should have been RSPD2025R930. The original article has been corrected.
References
- Ahmed AF, Wen Z-H, Bakheit AH, Basudan OA, Ghabbour HA, Al-Ahmari A, Feng C-W (2022) A major Diplotaxis harra-derived bioflavonoid glycoside as a protective agent against chemically induced neurotoxicity and Parkinson’s Models; in silico target prediction; and biphasic HPTLC-based quantification. Plants 11(5):5. 10.3390/plants11050648 [DOI] [PMC free article] [PubMed]
- Anshabo AT, Milne R, Wang S, Albrecht H (2021a) CDK9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front Oncol 11:678559. 10.3389/fonc.2021.678559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anshabo AT, Milne R, Wang S, Albrecht H (2021b) CDK9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front Oncol 11. 10.3389/fonc.2021.678559 [DOI] [PMC free article] [PubMed]
- Antiviral activity of some benzo[g]quinazolines against coxsackievirus B4: biological screening and docking study—PubMed (n.d.). Retrieved 24 August 2024, from https://pubmed.ncbi.nlm.nih.gov/37210695/ [DOI] [PMC free article] [PubMed]
- Baguley BC, Kerr DJ (2001) Anticancer drug development. Elsevier
- Barlaam B, Casella R, Cidado J, Cook C, De Savi C, Dishington A, Donald CS, Drew L, Ferguson AD, Ferguson D, Glossop S, Grebe T, Gu C, Hande S, Hawkins J, Hird AW, Holmes J, Horstick J, Jiang Y, … Yao T (2020) Discovery of AZD4573, a potent and selective inhibitor of CDK9 that enables short duration of target engagement for the treatment of hematological malignancies. J Med Chem 63(24):15564–15590. 10.1021/acs.jmedchem.0c01754 [DOI] [PubMed]
- Brooks BR, Brooks CL, MacKerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, … Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30(10):1545–1614. 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed]
- Chao SH, Price DH (2001) Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276(34):31793–31799. 10.1074/jbc.M102306200 [DOI] [PubMed] [Google Scholar]
- Discovery of N-(4-(3-isopropyl-2-methyl-2H-indazol-5-yl)pyrimidin-2-yl)-4-(4-methylpiperazin-1-yl)quinazolin-7-amine as a Novel, Potent, and Oral Cyclin-Dependent Kinase Inhibitor against Haematological Malignancies | Journal of Medicinal Chemistry (n.d.) Retrieved 3 September 2022, from https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.1c00271 [DOI] [PubMed]
- Dixon-Clarke SE, Elkins JM, Cheng S-WG, Morin GB, Bullock AN (2015) Structures of the CDK12/CycK complex with AMP-PNP reveal a flexible C-terminal kinase extension important for ATP binding. Sci Rep 5:17122. 10.1038/srep17122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandrani R, Al-Rasheed LS, Ansari SA, Bakheit AH, Almehizia AA, Almutairi M, Alkahtani HM (2023) Targeting transcriptional CDKs 7, 8, and 9 with anilinopyrimidine derivatives as anticancer agents: design, synthesis, biological evaluation and in silico studies. Molecules 28(11):11. 10.3390/molecules28114271 [DOI] [PMC free article] [PubMed]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Jiang B, Jiang J, Kaltheuner IH, Iniguez AB, Anand K, Ferguson FM, Ficarro SB, Seong BKA, Greifenberg AK, Dust S, Kwiatkowski NP, Marto JA, Stegmaier K, Zhang T, Geyer M, Gray NS (2021) Structure-activity relationship study of THZ531 derivatives enables the discovery of BSJ-01-175 as a dual CDK12/13 covalent inhibitor with efficacy in Ewing sarcoma. Eur J Med Chem 221:113481. 10.1016/j.ejmech.2021.113481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari N, Dwarakanath BS, Das A, Bhatt AN (2016) Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol 37(9):11553–11572. 10.1007/s13277-016-5098-7 [DOI] [PubMed] [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, Im W (2016) CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 12(1):405–413. 10.1021/acs.jctc.5b00935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1–2):55–63. 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26(16):1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protein Modificomics (2019) https://shop.elsevier.com/books/protein-modificomics/dar/978-0-12-811913-6
- Shao H, Foley DW, Huang S, Abbas AY, Lam F, Gershkovich P, Bradshaw TD, Pepper C, Fischer PM, Wang S (2021) Structure-based design of highly selective 2,4,5-trisubstituted pyrimidine CDK9 inhibitors as anti-cancer agents. Eur J Med Chem 214:113244. 10.1016/j.ejmech.2021.113244 [DOI] [PubMed] [Google Scholar]
- Small molecule inhibitors of cyclin-dependent kinase 9 for cancer therapy—PMC (n.d.) Retrieved 21 September 2024, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7919902/ [DOI] [PMC free article] [PubMed]
- Solary E, Droin N, Sordet O, Rébé C, Filomenko R, Wotawa A, Plenchette S, Ducoroy P (2002) Cell death pathways as targets for anticancer drugs. pp 55–76. 10.1016/B978-012072651-6/50005-X
- Tadesse S, Duckett DR, Monastyrskyi A (2021) The promise and current status of CDK12/13 inhibition for the treatment of cancer. Future Med Chem 13(2):117–141. 10.4155/fmc-2020-0240 [DOI] [PubMed] [Google Scholar]
- Targeting CDK12 for Cancer Therapy: Function, Mechanism, and Drug Discovery | Cancer Research | American Association for Cancer Research (n.d.) Retrieved 4 September 2023, from https://aacrjournals.org/cancerres/article/81/1/18/647808/Targeting-CDK12-for-Cancer-Therapy-Function
- The RNA polymerase II CTD coordinates transcription and RNA processing—PMC (n.d.) Retrieved 21 September 2024, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3465734/
- Workman P (2005) Genomics and the second golden era of cancer drug development. Mol bioSyst 1(1):1. 10.1039/b501751n [DOI] [PubMed]
- Xiao L, Liu Y, Chen H, Shen L (2023) Targeting CDK9 with selective inhibitors or degraders in tumor therapy: an overview of recent developments. Cancer Biol Ther 24(1):2219470. 10.1080/15384047.2023.2219470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahya EB, Alqadhi AM (2021) Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci 269:119087. 10.1016/j.lfs.2021.119087 [DOI] [PubMed] [Google Scholar]
- Zhang M, Zhang, L, Hei R, Li X, Cai H, Wu X, Zheng Q, Cai C (2021) CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res 11(5):1913–1935. Retrieved September 04, 2023, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8167670/ [PMC free article] [PubMed]
- Zhong L, Li Y, Xiong L, Wang W, Wu M, Yuan T, Yang W, Tian C, Miao Z, Wang T, Yang S (2021) Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Sig Transduct Target Ther 6(1):201. 10.1038/s41392-021-00572-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L (2016) Current challenges in cancer treatment. Clin Ther 38(7):1551–1566. 10.1016/j.clinthera.2016.03.026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not Applicable.