

Abstract
The mouse 3D8 anti‐DNA antibody can enter cells and localize in the cytoplasm, primarily facilitated by the complementarity‐determining region 1 of the variable light chain (CDR L1) domain. In this study, we grafted the CDR L1 loop from 3D8 onto non‐cell‐penetrating IgG antibodies to investigate whether these IgGs could acquire cytoplasmic localization ability while retaining antigen‐binding activity. One of three IgGs was successfully delivered into the cytoplasm while maintaining antigen‐binding activity. In silico protein modeling suggests that this capability is linked to structural similarity between CDR L1 in the grafted Ab and that in 3D8. This study proposes a strategy to confer cell‐penetrating capability by incorporating a specific CDR loop into an antibody backbone while retaining affinity.
Keywords: anti‐DNA antibody, CDR grafting, cell penetration, cytoplasmic delivery, IgG
We achieved cytoplasmic delivery of non‐cell‐penetrating IgGs by grafting a single functional complementarity‐determining region 1 (CDR1) from the light chain variable region (VL) of the cell‐internalizable 3D8 antibody. The engineered IgG acquired cell‐penetrating ability while maintaining antigen affinity, highlighting CDR1 grafting as a promising strategy for intracellular antibody delivery.
Abbreviations
BLI, bio‐layer interferometry
CDR, complementarity‐determining
C L , constant light chain
CPPs, cell‐penetrating peptides
CSPGs, chondroitin sulfate protein glycans
DMEM, dulbecco Modified Eagle Medium
DR‐5, death receptor 5
ELISA, enzyme‐linked immunosorbent assay
ENT2, equilibrative nucleoside transporter 2
FR, framework region
HSPGs, heparan sulfate proteoglycans
IMGT, ImMunoGeneTics
K D , equilibrium dissociation constant
KIFC1, kinesin family member C1
mAbs, monoclonal antibodies
MD, molecular dynamics
MFI, mean fluorescence intensity
PBS, phosphate‐buffered saline
PEI, polyethyleneimine
s, seconds
scFv, single‐chain variable fragments
SEC, size‐exclusion chromatography
SSA, super streptavidin
TAR, trans‐activation responsive region
T m , melting temperature
TSA, thermal shift assay
V L , variable light chain
V H , variable heavy chain
Monoclonal antibodies (mAbs), which primarily target extracellular or cell surface molecules, have been widely used to treat various diseases [1]. Meanwhile, research is increasingly focused on developing mAbs that target intracellular proteins for applications in medicine, biotechnology, and cell biology, either to modulate the functions of target molecules or to visualize intracellular targets in living cells [2, 3]. However, full‐sized antibodies (Abs) and their fragments, such as Fab and single‐chain variable fragments (scFv), cannot naturally cross cell membranes, which limits their use against intracellular targets. Several strategies have been studied to target intracellular molecules with Abs: (1) direct physical methods such as electroporation and microinjection of Ab proteins, (2) intracellular expression of the Ab gene to allow the Ab to function as an “intrabody,” (3) Ab protein delivery using various nanoparticle carriers, and (4) engineering Abs into ‘transbodies (transMabs)’ capable of cellular entry using cell‐penetrating peptides (CPPs) or functional parts of internalizing autoAbs [2, 4].
Only a limited number of naturally occurring anti‐DNA autoAbs can penetrate live cell membranes. Mouse mAbs such as 2C10, 3E10, and 3D8 derived from MRL‐lpr mice, a model of human systemic lupus erythematosus, exhibit different mechanisms of cellular entry and vary in terms of their intracellular localization. The 2C10 anti‐DNA mAb enters the cell by forming a complex with DNA via micropinocytosis and then traffics to the nucleus [5]. 3E10 is proposed to enter cells directly in a manner dependent on equilibrative nucleoside transporter 2 (ENT2), bypassing the endocytosis mechanism, and further traffics to the nucleus [6, 7]. 3D8 enters cells through endocytosis by attaching to cell surface proteoglycans such as heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate protein glycans (CSPGs), undergoes conformational changes at the endosomal acidic pH, and is then released into the cytosol without further trafficking to the nucleus [8, 9, 10, 11]. The factors that determine mAb movements remain unidentified.
The cell‐penetrating ability of the 3D8 antibody—including its IgG, scFv, and variable light chain (VL) forms—is mediated by the VL domain, particularly the CDR L1 [12, 13, 14]. Replacing an IgG VL molecule with 3D8 VL enables cytoplasmic internalization [13, 14]. Furthermore, grafting the 3D8 CDR L1 loop onto a human scFv Ab confers internalization capability, although it abrogates its antigen‐binding activity. By contrast, the synthetic peptide corresponding to CDR L1 cannot independently enter cells, suggesting that the cell‐penetrating activity is determined by the conformation of CDR L1 rather than by its linear amino acid sequence [12].
Building upon these findings, this study aimed to explore the potential of grafting 3D8 CDR L1 onto IgG Abs as a strategy to create Abs that retain antigen‐binding activity while gaining cellular internalization capability. We generated two versions of three non‐cell‐penetrating IgGs (adalimumab/Humira®, omalizumab/Xolair®, and house‐made 6C407 IgG) grafted with 3D8 CDR L1, defined based on the Kabat and ImMunoGeneTics (IMGT) numbering systems [15], and analyzed their antigen‐binding and cellular internalization capabilities. The 6C407 IgG variant (6CD1‐IMGT) grafted with 3D8 CDR L1 successfully acquired internalization capabilities while maintaining full antigen‐binding activity. The underlying mechanism was investigated using computer‐aided protein modeling.
Materials and methods
Plasmid construction
Genes encoding the variable heavy chain (VH) and VL regions of Abs were synthesized and cloned into the KV10‐IgCw‐γκ plasmid to express the IgG1 format. The KV10 vector contains human Ab constant genes controlled by two CMV promoters, enabling simultaneous expression of heavy and light chains [16]. The VH and VL genes were cloned into specific cloning sites downstream of the leader sequences. The VH and VL sequences of adalimumab were obtained from GenBank (IDs: 2449538014 and OP892521.1), and those of omalizumab were obtained from PDB (ID: CPX‐213192). The VH and VL sequences of 6C407 are available in a previous study [16] and GenBank (MH638366 and MH638367), while those of 3D8 are available in a previous study [17] and GenBank (AAF79128 and AAF79129). The plasmid vector to express the 6C407 IgE isotype was constructed by replacing the Cγ gene with the Cε gene in the KV10‐6C407 IgG plasmid. The CDR L1 loops of Abs were defined using both the Kabat (https://www.novoprolabs.com/tools/cdr) and IMGT (https://www.imgt.org/IMGT_vquest/analysis) numbering schemes. When designing the VLs grafted with 3D8 CDR L1, the Ab's CDR L1 sequence defined by the Kabat scheme was replaced with the 3D8 CDR L1 sequence defined by the Kabat scheme. Similarly, the Ab's CDR L1 defined by the IMGT scheme was replaced with the 3D8 CDR L1 defined by the IMGT scheme.
Cell culture
HeLa (RRID:CVCL_0030) cells were maintained in Dulbecco modified Eagle medium (DMEM) (Welgene; cat# LM001‐05) supplemented with 10% fetal bovine serum, 100 U·mL−1 penicillin, and 100 μg·mL−1 streptomycin. Cells were incubated in a humidified incubator in 5% CO2 at 37 °C. HEK293F cells (RRID:CVCL_6642) were cultured using Freestyle 293 Expression Medium (Thermo Fisher Scientific, Waltham, MA, USA; cat# 12338018) in a humidified incubator in 8% CO2 at 37 °C. All cell lines were cultured at low passage (<20) in mycoplasma‐free conditions.
Ab preparation
HEK293F cells were seeded at a density of 1 × 106 cells·mL−1 in a 500 mL flask (SPL Life Sciences, Pocheon‐si, Gyeonggi‐do, South Korea; cat# 73500) using Freestyle 293 Expression Medium. When the cell density reached 2 × 106 cells·mL−1, KV10 plasmids encoding each IgG were transiently transfected into HEK293F cells using polyethyleneimine (PEI) reagent (Polysciences, Warrington, PA, USA; cat# 23966). For transfection, 400 μg of PEI was incubated with 200 μg of plasmid DNA at room temperature for 10 min and then added to 100 mL of cells to achieve a final PEI concentration of 4 μg·mL−1. After 7 days of culture at 37 °C in 8% CO2 with 130 rpm shaking, the supernatant was harvested by centrifugation and filtered through a 0.45 μm cellulose acetate filter. IgGs were purified using Protein A‐Sepharose (Cytiva, Marlborough, MA, USA; cat# 17‐1279‐01), and 6C407 IgE was purified using Protein L‐Sepharose (Cytiva; cat# 17‐5478‐01) according to the manufacturer's instructions. All purified Abs were sterilized using a 0.22 μm cellulose acetate filter.
Confocal microscopy
To assess the cell‐penetrating ability of IgG Abs, HeLa cells were seeded at 3 × 104 cells per 12‐mm coverslip in 24‐well plates and treated with 3 μm antibodies for 6 h at 37 °C. After five washes with phosphate‐buffered saline (PBS), cells were fixed with 4% paraformaldehyde for 10 min at room temperature and permeabilized with 0.1% Triton X‐100 for 20 min at 4 °C. Cells were then incubated with goat anti‐human IgG Fc (Thermo Fisher Scientific; cat# 31125) followed by Alexa Fluor 488‐conjugated donkey anti‐goat IgG (Invitrogen, Carlsbad, CA, USA; cat# 11055) for 1 h at 4 °C. Each incubation step was followed by three washes with PBS. Nuclei were stained with Hoechst 33342 (Invitrogen; cat# C10329) at room temperature. The coverslips were mounted on glass slides using PermaFluor (Thermo Fisher Scientific; cat# TA‐030‐FM), and images were captured with a 40× objective on a Carl Zeiss LSM710 confocal microscope.
To analyze the colocalization of 6CD1 IgG and kinesin family member C1 (KIFC1), HeLa cells stably expressing GFP‐tagged KIFC1 were synchronized in M phase [18, 19]. Cells were treated with 3 mm thymidine (Sigma, St. Louis, MO, USA; cat# T1895) for 18 h and released into complete medium for 7 h, and this process was repeated to fully arrest cells in G1/S phase. After washing with PBS, cells were treated with 9 μm RO‐3306 (Sigma; cat# SML0569), a CDK1 inhibitor, for 2 h to induce M phase entry followed by 6CD1 IgG (2 μm) for 2 h at 37 °C. Floating cells were gently dislodged, seeded onto poly‐L‐lysine‐coated coverslips in 24‐well plates, and briefly incubated. After adhesion, cells were fixed with 4% paraformaldehyde and incubated with rabbit anti‐human IgG Fc (Thermo Fisher Scientific; cat# 31142) followed by Alexa Fluor 647‐conjugated goat anti‐rabbit IgG (Invitrogen; cat# A21244).
Flow cytometry
HeLa cells were seeded in six‐well plates at a density of 5 × 105 cells·well−1 and incubated with 3 μm IgG Abs for 6 h at 37 °C. The cells were washed five times with ice‐cold PBS and fixed with 4% paraformaldehyde diluted in PBS for 10 min at 4 °C. After washing with PBS, the cell membranes were either permeabilized with PERM buffer (PBS containing 0.1% saponin, 0.1% sodium azide, and 1% bovine serum albumin) for 30 min at 4 °C or the permeabilization step was omitted. The cells were then incubated with rabbit anti‐human IgG Fc (Thermo Fisher Scientific; cat# 31142) followed by Alexa Fluor 488‐conjugated donkey anti‐rabbit IgG (Invitrogen; cat# A21206) for 1 h at 4 °C. Each incubation step was followed by three washes with PBST. Finally, the cells were analyzed using a flow cytometer (BD, FACSAria™ III).
Enzyme‐linked immunosorbent assay (ELISA)
To assess the antigen‐binding activity of IgG Abs, wells of 96‐well polystyrene ELISA plates were coated overnight at 4 °C with 2 μg·mL−1 specific antigens diluted in PBS. The antigens used were the synthesized 6C peptide (EDGLEPEKKRTR) for 6C407 and 6CD1s, active trimeric human TNF‐α (Acrobiosystems, Newark, DE, USA; cat# TNA‐H5228) for adalimumab and AdaD1s, and in‐house prepared 6C407 IgE for omalizumab and OmaD1s. Wells were blocked with 3% bovine serum albumin for 1 h at room temperature and subsequently incubated with 10–160 ng·mL−1 IgG Abs and alkaline phosphatase‐conjugated goat anti‐human Fc IgG (Sigma‐Aldrich; cat# A9544). Each incubation step was followed by three washes with TBST (TRIS‐buffered saline containing 0.05% Tween‐20). Finally, p‐nitrophenyl phosphate (Sigma‐Aldrich; cat# N2765) solution (1 mg·mL−1 prepared in 0.1 m glycine, 1 mm ZnCl2, and 1 mm MgCl2, pH 10.3) was added to each well, and absorbance at 405 nm was measured using a microplate reader (BioTek, Epoch2).
To assess the binding of 6CD1‐IMGT to full‐length KIFC1, wells coated with KIFC1 protein (Abcam, Cambridge, Cambridgeshire, UK; cat# ab152492) were incubated with IgG Abs (7.5 μg·mL−1) followed by alkaline phosphatase‐conjugated goat anti‐human Fc IgG. To assess antigen specificity, wells were coated with 2 μg·mL−1 peptides corresponding to the N terminus of KIFC1: 2C (PSLTTVPQTQGQ), 6C (EDGLEPEKKRTR), and 10C (IATGLKNQKPVP). To evaluate heparin‐binding activity, wells were coated with 10 μg·mL−1 heparin (Sigma‐Aldrich; cat# H3149). To assess the binding of 6CD1 Ab to intracellularly expressed KIFC1, a sandwich ELISA was performed. Wells were coated with 0.5 μg·mL−1 rabbit anti‐KIFC1 IgG (Cell Signaling Technology; cat# 12313) and incubated with a mixture of 6CD1‐IMGT protein (50 μg·mL−1) and lysates of HeLa cells stably expressing GFP‐tagged KIFC1 (1 mg·mL−1). Detection was carried out using goat anti‐human IgG heavy chain (Invitrogen; cat# 31118) followed by alkaline phosphatase‐conjugated rabbit anti‐goat IgG (Pierce, Rockford, IL, USA; cat# 31300).
Immunoblotting analysis
HeLa cells were seeded in 60‐mm dishes at a density of 1 × 106 cells·dish−1 24 h prior to being treated with the IgG Abs at a final concentration of 3 μm for 6 h. After five washes with PBS, cells were lysed with cytoplasm isolation buffer (Cell Signaling Technology; cat #9038) or whole cell lysis buffer (0.025 m Tris, 0.15 m NaCl, 0.001 m EDTA, 1% NP‐40, and 5% glycerol, pH 7.4). Supernatants were obtained by centrifugation at 16 000 g at 4 °C for 10 min. The protein concentration in the cell lysate was measured using a BCA protein assay kit (Thermo Fisher Scientific; cat# 23227). SDS/PAGE of cytosolic fractions and whole cell lysates was performed under reducing conditions using 4–20% gradient gels, and resolved proteins were transferred to polyvinylidene fluoride membranes. The membranes were probed with the following primary Abs: mouse anti‐GAPDH (Santacruz, Dallas, TX, USA; cat# sc‐365 062), rabbit anti‐BiP (Cell Signaling; cat# 3183), goat anti‐human IgG Fc (Abcam; cat# ab97221), and mouse anti‐Lamin A/C (Santacruz; cat# sc‐7292). Membranes were subsequently reacted with corresponding secondary Abs: horse anti‐mouse IgG‐HRP (Cell Signaling; cat# 7076) for mouse Abs, goat anti‐rabbit IgG‐HRP (Jackson ImmunoResearch, West Grove, PA, USA; cat# 111‐035‐003) for rabbit Abs, and rabbit anti‐goat IgG‐HRP (Invitrogen; cat# 81‐1620) for goat Abs.
Preparation of Fab and molecular docking
Structural modeling of 6C407 Fab fragments was performed to obtain a three‐dimensional structure using the Ab modeler embedded in Biovia Discovery Studio 2020 (Modeler ver. 9.22). The crystal structure of M2177 (PDB ID: 5TL5) was used as a template for the overall structures of 6C407 Fab. The light chain structures of Abs 14.1 (PDB ID: 5FB8) were used as templates for the light chain portion of Fab, while the heavy chain structure of Ab 48G7 (PDB ID: 1AJ7) served as the template for generating heavy chain structures. The CDR L1, L2, and L3 loops of Ab A13‐D6.3 (PDB ID: 2XQY) were used as templates for the CDR L loops of 6C407 Fab, while the CDR1 of the heavy chain (CDR H1), H2, H3 structures of Abs 2A12 (PDB ID: 4WEB), 1F9 (PDB ID: 1DZB), and 12E (PDB ID: 5UEA), respectively, served as templates for generating the CDR H loops of 6C407 Fab. The 6C407 Fab model with the lowest total potential energy, identified through probability density function total energy analysis, was validated using Ramachandran plots and underwent explicit‐water molecular dynamics (MD) simulation.
Subsequently, molecular docking was performed on the predicted 6C407 structure to anticipate interactions with its peptide antigen (6C peptide), employing ZDOCK and RDOCK protocols in Discovery Studio 2020. Initially, 6C407 Fab was docked to the 6C peptide, specifically targeting binding residues in the Ab's variable region. The most promising cluster of binding sites was selected, and the pose was refined and optimized using the RDOCK protocol to enhance the accuracy of peptide binding predictions. Finally, MD simulation was conducted on this optimized structure to verify its stability.
Structure prediction of light chains
To analyze the configuration of the VL‐CDR loops, light chain structures were generated using AlphaFold. These structures were further refined through MD simulations [20].
Size‐exclusion chromatography (SEC)
Purified 6C407 IgG, 6CD1‐IMGT IgG, and 6CD1‐Kabat IgG were analyzed by size‐exclusion chromatography using a Shimadzu UFLC system (DGU‐20A3) equipped with a TSK G3000SWXL column (7.8 × 300 mm; Tosoh Bioscience, Grove City, OH, USA). The mobile phase consisted of 100 mm HEPES and 85 mm HNaSO4 (pH 6.8), with a flow rate of 1 mL·min−1. Chromatograms were recorded by monitoring absorbance at 280 nm.
Thermal shift assay (TSA)
Thermal stabilities of 6C407 and 6CD1 IgGs were evaluated using a Protein Thermal Shift Dye Kit (Thermo Fisher Scientific; cat# 4461146), which contains a fluorescent dye that specifically binds to hydrophobic residues exposed during protein unfolding. Proteins (1 mg·mL−1) were prepared in a total volume of 20 μL per well in a 96‐well PCR plate (Thermo Fisher Scientific; cat# 4346907). The melting temperature (Tm) was determined using a QuantStudio™ 3 Real‐Time PCR System by applying a temperature ramp from 25 °C to 99 °C at a rate of 0.05 °C·s−1. Fluorescence was detected using an optical filter set at excitation/emission wavelengths of 580/623 nm (Ex/Em).
Bio‐layer interferometry (BLI)
Binding kinetics and affinity of Abs against biotin‐conjugated KIFC1 (residues 1–50; biotin‐MDPQRSPLLEVKGNIELKRPLIKAPSQLPLSGSRLKRRPDQMEDGLEPEK, synthesized by Apeptide Co., Ltd.) were analyzed using an Octet® R8 system (Sartorius). The antigen was immobilized onto Super Streptavidin (SSA) biosensors (Sartorius; cat# 18‐5057) at 50 μg·mL−1 for 300 s (s). Abs were serially diluted twofold starting from 50 nm in PBS containing 1 mg·mL−1 BSA. Association and dissociation phases were measured for 300 s and 600 s, respectively. The equilibrium dissociation constant (K D) was calculated using the equation K D = k off/k on, where k off and k on are the dissociation and association rate constants, respectively.
Results
Generation of IgG Abs grafted with 3D8‐CDR L1
We investigated the effect of grafting the 3D8‐CDR L1 loop onto three IgGs that do not naturally enter cells (Fig. 1A). The three donor IgGs were our in‐house 6C407 (a chimeric IgG targeting KIFC1) and two well‐known Abs: Humira® (adalimumab, targeting TNF‐α) and Xolair® (omalizumab, targeting IgE) [16]. For each Ab, we constructed two versions of the 3D8‐CDR L1 graft, based on the Kabat and IMGT numbering systems, resulting in six Abs in total (Fig. 1B). We collectively named the Abs grafted with 3D8 CDR L1 as D1 Abs. D1 Abs with CDR L1 defined by the Kabat scheme were named ‘X’D1‐Kabat, while those with CDR L1 defined by the IMGT scheme were called ‘X’D1‐IMGT. Here, ‘X’ represents the prefix of the Ab name, such that CDR L1‐grafted 6C407, adalimumab, and omalizumab are referred to as 6CD1, AdaD1, and OmaD1, respectively. 3D8 CDR L1 defined by the Kabat scheme consists of 17 amino acid residues, longer than the 12 amino acids defined by the IMGT scheme. IgG proteins were obtained with a high degree of purity and in their intact form through transient expression of Ab genes in HEK293F cells (Fig. 1C). The production yields of each parental Ab and its CDR L1‐grafted counterparts did not significantly differ. Production yield can reflect Ab stability; therefore, the lack of variation suggests that the grafted CDR L1 did not negatively impact the stability of these Abs (Table 1).
Fig. 1.

Generation of 3D8‐CDR L1‐grafted Abs. (A) Schematic representation of D1 Ab generation by replacing the CDR L1 region with the 3D8‐CDR L1 sequence. (B) Amino acid sequences of the light chain FR1‐CDR1‐FR2 region, aligned according to the Kabat and IMGT numbering systems. The 3D8 CDR1 sequence is highlighted in yellow, while the grafted CDR L1 sequences in D1 Abs are shown in gray. (C) SDS/PAGE analysis of purified parental and D1 Abs (2 μg) under reducing and non‐reducing conditions using a 4–20% gradient polyacrylamide gel, with proteins visualized by Coomassie brilliant blue staining.
Table 1.
Production yield of Abs obtained in transfected HEK293F cells.
| Antibody | Production yield (mg·L−1) |
|---|---|
| 6C407 | 18.4 ± 1.0 |
| 6CD1‐Kabat | 22.7 ± 0.9 |
| 6CD1‐IMGT | 19.3 ± 0.6 |
| Adalimumab | 44.6 ± 0.9 |
| AdaD1‐Kabat | 45.1 ± 0.3 |
| AdaD1‐IMGT | 44.8 ± 0.6 |
| Omalizumab | 40.7 ± 0.5 |
| OmaD1‐Kabat | 38.7 ± 0.5 |
| OmaD1‐IMGT | 37.2 ± 0.2 |
Certain D1 Abs retain antigen‐binding activity
We examined the antigen‐binding activity of D1 Abs by an ELISA. 6CD1‐IMGT retained the antigen‐binding capacity of its parental 6C407, while 6CD1‐Kabat showed less than 50% of that activity (Fig. 2A). However, neither AdaD1 Abs (Kabat and IMGT) nor OmaD1 Abs (Kabat and IMGT) bound to their antigens. This observation may not be surprising given the findings of studies on the contribution of CDRs to antigen binding in adalimumab and omalizumab [21, 22]. A study of the crystal structure of TNF‐α in complex with adalimumab Fab showed that all interactions with TNF‐α arise from the CDRs; CDR L2 and CDR H2 contribute the most, with additional input from CDRs L1, L3, H1, and H3 [21]. Although CDR L1 plays a minor role in antigen binding, replacing it with 3D8 CDR L1 likely caused the loss of antigen‐binding ability. In omalizumab, five CDRs contact its antigen IgE‐Fc, except for the CDR L3 loop [22]. CDR L1 of omalizumab is involved in antigen binding; therefore, OmaD1 (generated by both the Kabat and IMGT schemes) likely lost its binding ability. Molecular docking simulations were conducted to investigate the contribution of each CDR in 6C407 to antigen binding (Fig. 2B). The results, which predicted the most favorable binding disposition of the 6C peptide antigen within 6C407 Fab, revealed that CDR H3, CDR H1, CDR L2, and CDR L3 directly contact the peptide antigen and likely play a crucial role in binding. By contrast, CDR H2 and CDR L1 are located considerably farther from the antigen‐binding site. These findings support the conclusion that substituting CDR L1 in 6C407 allowed 6CD1 Abs to retain their antigen‐binding ability.
Fig. 2.

Evaluation of antigen binding of D1 Abs and structural docking of 6C407 Fab with 6C peptide. (A) Direct ELISA to assess the antigen‐binding activity of D1 Abs. ELISA plates were coated with 6C peptide (derived from KIFC1 protein), TNF‐α, and 6C407 IgE, and then incubated with the indicated Abs at various concentrations, followed by alkaline phosphatase‐conjugated goat anti‐human Fc IgG detection. (B) Molecular docking of 6C407 Fab and 6C peptide performed using Discovery Studio 2020 software (modeler v.9.22). The Fd (VH–CH1) region of 6C407 Fab is shown in dark blue, the light chain is shown in light blue, and the docked 6C peptide is shown in pink. The left panel shows the side view of the docked molecule, while the right panel displays the top view.
We evaluated the structural stability of three Abs (6C407, 6CD1‐IMGT, and 6CD1‐Kabat) using size‐exclusion chromatography (SEC) and thermal shift assay (TSA) after storage at 4 °C for 2 weeks (Fig. 3A,B). SEC analysis revealed a single peak at approximately 150 kDa for each Ab, confirming that all Abs were predominantly intact monomers with over 97% purity and no apparent aggregation propensity (Fig. 3A). The thermal stabilities of these Abs were also similar, with melting temperatures (Tm) of 69.9 °C (6C407), 69.7 °C (6CD1‐IMGT), and 70.0 °C (6CD1‐Kabat), indicating that CDR L1 grafting did not negatively impact structural stability (Fig. 3B). Because 6CD1‐IMGT and 6C407 exhibited similar antigen‐binding activity by ELISA (Fig. 2A), their binding kinetics and affinities were further characterized using bio‐layer interferometry (Fig. 3C). Both Abs displayed comparable equilibrium dissociation constants (K D), with values of 10.57 nm (6C407) and 9.31 nm (6CD1‐IMGT), indicating similar overall binding strength. However, 6CD1‐IMGT showed approximately 1.78‐fold slower association (k on = 7.31 × 104 m −1 s−1) and 2.01‐fold slower dissociation (k off = 6.82 × 10−4 s−1) rates compared to 6C407 (k on = 1.30 × 105 m −1 s−1; k off = 1.37 × 10−3 s−1). These results indicate that the binding kinetics of a CDR L1‐grafted Ab can be altered even if its overall antigen‐binding affinity is maintained.
Fig. 3.

Comparison of stability and binding characteristics of 6C407 IgG and 6CD1 IgGs. (A) SEC analysis of Abs to assess structural stability. The purity of each antibody is indicated at the top right of the respective peak. (B) Thermal stability analysis of the Abs using a Protein Thermal Shift Dye Kit. The melting curves were measured with a QuantStudio™ 3 Real‐Time PCR System, and the data were plotted using prism software. (C) Bio‐Layer Interferometry (BLI) sensorgram showing the kinetic profile of Abs binding to biotin‐conjugated KIFC1 peptide (50 amino acids). Binding measurements were performed at six different antibody concentrations, starting at 50 nm. The binding parameters (k on, k off, and K D) were presented on the right.
Certain D1 Abs gain the ability to enter cells
We examined the cell‐penetrating ability of D1 Abs by incubating HeLa cells with these Abs for 6 h at 37 °C and assessing penetration using flow cytometry and confocal microscopy. Flow cytometric analysis, which included a membrane permeabilization step, showed significant cell‐penetrating ability of 6CD1 and AdaD1 Abs. By contrast, both OmaD1‐IMGT and OmaD1‐Kabat demonstrated only marginal capabilities compared with the parental Ab omalizumab (Fig. 4A). Cell‐penetrating ability was quantified by subtracting the mean fluorescence intensity (MFI) of nonpermeabilized cells from that of permeabilized cells. These values were then expressed as a ratio of the MFI in Ab‐treated cells to that in PBS‐treated cells. Using this method, 6CD1‐IMGT achieved approximately 67% of the cell‐penetrating ability observed in the positive control, 3D8 IgG, while 6CD1‐Kabat showed around 40%. Similarly, AdaD1‐IMGT reached about 47% of the cell‐penetrating activity of 3D8 IgG, while AdaD1‐Kabat showed around 40% (Fig. 4B). The confocal microscopy results were consistent with the flow cytometric findings, confirming that 6CD1‐IMGT has the highest cell‐penetrating ability among D1 Abs (Fig. 4C).
Fig. 4.

Internalization of D1 Abs into living cells. (A, B) Flow cytometric analysis of D1 Ab internalization. HeLa cells were treated with 3 μm Abs for 6 h at 37 °C, and internalized IgGs were detected using rabbit anti‐human IgG Fc followed by Alexa Fluor 488‐conjugated donkey anti‐rabbit IgG. Internalization of D1 Abs was assessed relative to PBS‐treated controls, with 3D8 IgG serving as a positive control. Representative cytometry profiles are shown in (A). Data are presented as MFI ± standard deviation from three independent experiments. (C) Confocal microscopy. HeLa cells were treated with 3 μm Abs for 6 h at 37 °C, and internalized Abs were visualized by staining with goat anti‐human IgG Fc followed by Alexa Fluor 488‐conjugated donkey anti‐goat IgG. Nuclei were stained with Hoechst 33342. Scale bars: 10 and 20 μm.
Cell‐penetrating D1 Abs inherit cytoplasmic localization characteristics from 3D8 Abs
We examined whether the cellular localization of cell‐penetrating D1 Abs is restricted to the cytoplasm without being transported to the nucleus, as in the case of 3D8 IgG. Cytosolic fractions from HeLa cells that had been incubated with D1 Abs for 6 h at 37 °C were subjected to western blot analysis (Fig. 5). The purity of the cytosolic fractions was confirmed using specific markers: GAPDH, a cytosolic marker and loading control, was detected, while the membrane/organelle marker BiP and the nuclear marker Lamin A/C were not. 6CD1 and AdaD1 (‐Kabat and ‐IMGT) were detected as strong protein bands in the cytosolic fraction but not in the nuclear fraction. Among OmaD1 Abs with very low cell‐penetrating efficiency, OmaD1‐IMGT was barely detectable at a subvisible level, while OmaD1‐Kabat was not detected at all. These results demonstrate that cell‐penetrating D1 Abs are released into the cytosol and none are transported to the nucleus. Thus, 6CD1 and AdaD1 appear to have inherited the cellular localization characteristics of 3D8, which are provided by 3D8 CDR L1.
Fig. 5.

Cytosolic release of internalized D1 Abs. (A–C) Immunoblot analysis of cytosolic release of Abs. HeLa cells were treated with purified Abs (3 μm) for 6 h at 37 °C, followed by cell lysis and fractionation. Fractionation was validated using specific markers: GAPDH (cytosolic), BiP (membrane/organelle), and Lamin A/C (nuclear). GAPDH also served as a loading control. 3D8 IgG was used as a positive control.
6CD1‐IMGT IgG binds to KIFC1 protein in living cells
6CD1‐IMGT retained its antigen‐binding activity while gaining high cell‐penetrating ability, unlike adalimumab and AdaD1 Abs, which lost their antigen‐binding activity in exchange for cell‐penetrating ability. Given the potential of 6CD1‐IMGT, we explored its binding to heparin, a soluble analog of heparan sulfate because 3D8 Ab enters cells by binding to sugar chains on HSPGs and CSPGs. The ELISA confirmed that 6CD1‐IMGT gained heparin‐binding ability (Fig. 6A), suggesting it follows the endocytosis mechanism of 3D8 Ab. Furthermore, 6CD1‐IMGT maintained antigen specificity, binding exclusively to the 6C peptide (Fig. 6B), and also bound to purified KIFC1 protein (Fig. 6C).
Fig. 6.

Intracellular KIFC1‐targeting of 6CD1‐IMGT in living cells. (A) Heparin‐binding activity. Heparin‐coated wells were incubated with IgG Abs, and bound IgGs were detected using alkaline phosphatase‐conjugated goat anti‐human IgG. 3D8 IgG served as a positive control. (B) Direct ELISA assessing the antigen specificity of 6CD1‐IMGT against plate‐coated 6C, 2C, and 10C peptides derived from KIFC1. (C) Direct ELISA evaluating the antigen‐binding activity of 6CD1‐IMGT to purified full‐length KIFC1‐coated plates. (D) Sandwich ELISA evaluating binding activity to intracellularly expressed KIFC1. A schematic representation is shown in the left panel. Wells coated with rabbit anti‐KIFC1 Ab were incubated with a mixture of IgG Ab (6CD1‐IMGT or AdaD1‐IMGT) and lysate of HeLa cells that stably expressed GFP‐tagged KIFC1. Bound IgG protein was detected using alkaline phosphatase‐conjugated rabbit anti‐goat IgG (right panel). (E) Confocal microscopy of co‐localization of cytosol‐released 6CD1‐IMGT with KIFC1 protein. HeLa cells that stably expressed GFP‐tagged KIFC1 were treated with 2 μm IgG Ab (6CD1‐IMGT or 3D8) for 2 h at 37 °C, followed by rabbit anti‐human IgG Fc and then Alexa Fluor 647‐conjugated goat anti‐rabbit IgG. Images show KIFC1 (green), Abs (red), and Hoechst 33342‐stained nuclei (blue). Scale bar: 10 μm. Data are presented as the mean ± standard deviation from three independent experiments.
Next, we assessed whether 6CD1‐IMGT IgG penetrates the cytoplasm and recognizes KIFC1, a C‐terminal kinesin motor that primarily localizes to the nucleus but is exposed to the cytoplasm during cell division. HeLa cells stably expressing GFP‐tagged KIFC1 were treated with 6CD1‐IMGT, and then a sandwich ELISA of the cell lysates was performed. 6CD1‐IMGT bound to intracellularly expressed KIFC1 protein (Fig. 6D). Confocal microscopy of cells synchronized in M phase or anaphase to observe KIFC1 on the spindle showed significant colocalization of 6CD1‐IMGT (red) and KIFC1 (green) on mitotic spindles. These results demonstrate that 6CD1‐IMGT enters cells, escapes into the cytoplasm, and successfully targets its specific KIFC1 antigen (Fig. 6E).
D1 Abs with cell‐penetrating ability retain a CDR L1 loop configuration similar to 3D8 CDR L1
To understand why some CDR L1‐grafted Abs acquire cell‐penetrating ability while others do not, we compared the CDR L1 loop structure of D1 Abs and 3D8 Ab using in silico modeling. Three‐dimensional structures of the light chains (VL–CL) of each D1 Ab were generated using AlphaFold, and then MD simulation was carried out for structure optimization using the CHARMm forcefield embedded in the Discovery Studio 2020 software. The CDR L1 shapes of parental and D1 Abs are distinctly different. The CDR L1 loops of parental Abs are relatively flat, while those of all D1 Abs protrude upward, similar to the protruding 3D8 CDR L1 loop (Fig. 7A,B). OmaD1‐Kabat and OmaD1‐IMGT, which barely enter cells, did not superimpose well with the 3D8 CDR L1 loop despite the protruding CDR L1 shapes due to a 5.9 angstrom distance from the 3D8 CDR L1 loop (Fig. 7C). The CDR L1 loops of 6CD1 and AdaD1, which exhibited high cell‐penetrating ability, superimposed well with the 3D8 CDR L1 loop. This superimposition was more pronounced in the CDR L1 loops defined by IMGT for 6CD1 and AdaD1 than in those defined by Kabat (Fig. 7D). This suggests that mimicking the spatial configuration and protruding shape of 3D8 CDR L1 is critical to confer cell‐penetrating ability.
Fig. 7.
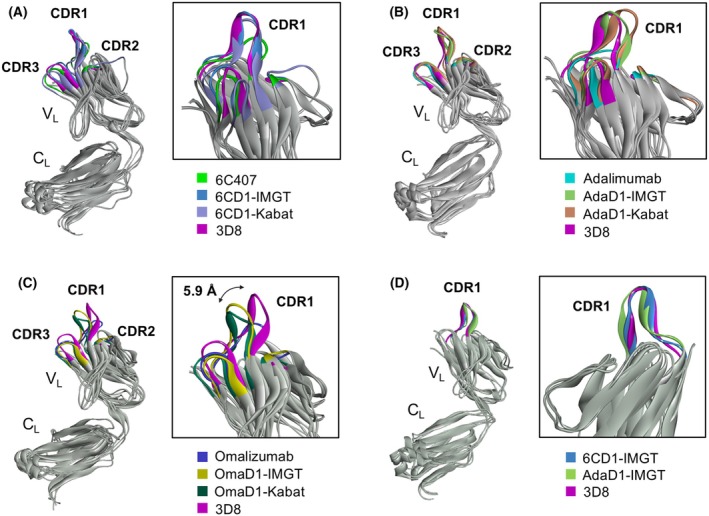
Superpositions of Ab VL structures. (A–C) Superimposition of the light chains of the specified Abs. The VL domains are magnified in boxes to highlight the CDR1 loop. In (C), the double‐headed arrow indicates the distance between two Cα atoms of Arg33 at the tip of the CDR1 loop in 3D8 VL and OmaD1 VL. (D) Superimposition of the CDR L1 loop from 6CD1‐IMGT and AdaD1‐IMGT onto that of 3D8. Structures were generated using AlphaFold and optimized with MD simulations in the Discovery Studio 2020 software. All images were rendered using Discovery Studio 2020 software.
Discussion
The entire 3D8 VL domain has been used to generate a transbody capable of cytoplasmic entry by replacing the original VL of an IgG molecule with 3D8 VL, enabling cytoplasmic internalization. However, this approach is only applicable to Abs that rely exclusively on their VH domain for antigen binding [13, 23]. In this study, we developed a novel approach to create a cell‐penetrating Ab by grafting a single CDR L1 loop from 3D8 VL onto other Abs. Although tested on a limited set of Abs, the observation that 6CD1 Abs retained the KIFC1 antigen‐binding ability of parental 6C407, while gaining cell‐penetrating capability, supports the feasibility of this approach to create cell‐penetrating Abs. This feasibility could be further enhanced when the single CDR domain‐grafting strategy is combined with computation‐aided antibody structure prediction.
Only 20–33% of amino acids in CDRs directly contact the antigen. Noncontact residues are proposed to shape the CDR loops, optimizing the positioning of contact residues for efficient and specific antigen binding [24, 25]. Although residues from grafted CDRs can affect the overall conformation of the Ab by interacting with adjacent CDR loops, 6CD1 successfully retained its antigen‐binding ability and acquired cell‐penetrating capability. This can be attributed to three key factors. First, CDR L1 of the parental Ab 6C407 does not significantly contribute to antigen binding. Second, the reshaped 6CD1 preserves the overall VL domain structure. Third, the VH–VL interface, which is important to form the antigen‐binding site, remains compatible.
Similarly, our previous work showed that replacing the 3D8 CDR L1 sequence, as defined by Kabat, with the HW6 scFv targeting death receptor 5 (DR‐5) allows the grafted scFv (HW6/3D8L1) to gain cellular entry ability at the expense of losing antigen‐binding activity [12]. Therefore, the ability of 3D8 CDR L1 grafting to confer cellular entry does not appear uncommon, and further research is needed to develop strategies for creating cell‐penetrating Abs without sacrificing their intrinsic antigen‐binding capabilities. Additionally, we previously found that grafting the Tat48–60 peptide (GRKKRRQRRRPPQ) onto CDR H3 of the 3D8 scFv retains its DNA‐binding ability and adds Tat functions, such as nuclear translocation and enhanced trans‐activation responsive region (TAR) RNA binding [26]. Examples from 3D8 and 6C407 Abs suggest that some Abs can tolerate CDR sequence replacements, as long as the CDR to be replaced is not essential to contact the antigen. It would be interesting to explore whether grafting a functional peptide (even if it is not necessarily a CDR‐derived sequence) onto other CDRs that are predicted not to significantly contribute to antigen binding can add new functions while retaining affinity. For example, grafting a functional sequence such as 3D8 CDR L1 onto CDR L3 of omalizumab or CDR H2 of 6C407 IgG, neither of which directly contribute to antigen binding, could be explored as potential strategies [22].
Various Ab numbering systems, such as the Kabat, the Chothia or Martin (Enhanced Chothia), Honegger's (AHo's), and IMGT have evolved to enable easy comparison of structural or functional similarities between Ab variable domains. These systems differ in how they assign numbers to amino acid residues, consequently affecting the definition of CDR lengths and boundaries. [27]. 6CD1‐IMGT was superior to 6CD1‐Kabat in terms of both antigen‐binding and cell‐penetrating abilities. AdaD1 Abs gained cellular entry ability at the cost of losing antigen‐binding activity, with AdaD1‐IMGT showing superior cellular entry compared with AdaD1‐Kabat (Figs 2 and 4). These findings suggest that CDR L1 (12 amino acids) defined by the IMGT numbering system is sufficient as a functional region and aligns well with the actual structure of the variable domain.
Unlike other anti‐DNA Abs that can enter the nucleus, 3D8 remains exclusively in the cytoplasm when expressed as a VL domain, scFv, or full‐size IgG, but does not internalize as a VH domain [11, 12, 13, 28]. Interestingly, 6CD1 and AdaD1 Abs exhibited behavior similar to that of 3D8, remaining in the cytoplasm without translocating to the nucleus (Fig. 5). 6CD1‐IMGT acquired heparin‐binding activity, potentially enabling it to bind to cell surface HSPGs, which are endocytic receptors for cellular internalization of 3D8 [10]. Consequently, 6CD1 likely enters cells through HSPG binding, similar to 3D8. This behavior underscores the role of the 3D8 CDR L1 loop in governing not just cell‐penetrating ability but also cytoplasmic localization. This point is particularly interesting given that a previous study with humanized 3D8 IgG demonstrated that Ab internalization and endosomal escape are mediated independently by CDR L1 and CDR L3, respectively. Specifically, the YYH motif within CDR L3 responds to acidic pH, undergoing conformational changes that facilitate interaction with endosomal membranes, leading to pore formation [8]. However, in our study, 6CD1 and AdaD1 Abs grafted with only 3D8 CDR L1—without CDR L3—achieved efficient internalization and subsequent cytoplasmic localization via endosomal escape. Considering that the YYH motif is absent in the CDR L3 of 6CD1 and AdaD1, it may not be strictly necessary for endosomal escape. Further studies are required to identify alternative functional residues or motifs to replace the YYH motif of 3D8, which may help clarify the mechanism underlying endosomal escape by 6CD1 and AdaD1. While CPPs [29] and synthetic peptides derived from the CDR H2 and CDR H3 sequences of certain anti‐DNA Abs enter cells [30], the peptide corresponding to 3D8 CDR L1 does not. This indicates that 3D8 CDR L1, which requires structural support from framework region 1 (FR1) and FR2 for cellular entry, behaves differently from these peptides.
Various methods for delivering Ab proteins into living cells have been attempted to either functionally block or induce E3 ligase‐mediated degradation of target molecules. These methods include electroporation [31, 32], microinjection [33, 34], Tat peptide fusion to IgG or scFv Abs [26, 35, 36] nanoparticle carriers [37], and fusion Abs of antigen‐targeting and cell‐penetrating Abs [38]. These methods neither provide cell specificity nor strictly ensure cytoplasmic localization. To target cytoplasmic molecules in specific cells, three moieties are required: one that recognizes specific cell surface molecules, one that enables cell penetration, and one that targets cytoplasmic molecules. By employing the single CDR grafting strategy to develop an Ab with cell‐penetrating and cytoplasmic‐targeting capabilities, the addition of a moiety for cell specificity is the only remaining step, which presents a major advantage in Ab engineering.
Author contributions
YJ conducted the investigation and drafted the paper. JC provided guidance on computer modeling. Y R analyzed the data and provided resources. EL and LV purified proteins. MK supervised the conceptualization and edited the manuscript.
Peer review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1002/1873‐3468.70058.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) under Grant NRF‐22020R1A2C2008258.
Edited by Wilfried Ellmeier
Data accessibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Wang Z, Wang G, Lu H, Li H, Tang M and Tong A (2022) Development of therapeutic antibodies for the treatment of diseases. Mol Biomed 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang C, Otjengerdes RM, Roewe J, Mejias R and Marschall ALJ (2020) Applying antibodies inside cells: principles and recent advances in neurobiology, virology and oncology. BioDrugs 34, 435–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ueda H, Dai Y and Ghadessy F (2023) Visualizing intracellular target antigens in live cells. Trends Cell Biol 33, 277–279. [DOI] [PubMed] [Google Scholar]
- 4. Slastnikova TA, Ulasov AV, Rosenkranz AA and Sobolev AS (2018) Targeted intracellular delivery of antibodies: the state of the art. Front Pharmacol 9, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inoue K, Ishizawa M and Kubota T (2020) Monoclonal anti‐dsDNA antibody 2C10 escorts DNA to intracellular DNA sensors in normal mononuclear cells and stimulates secretion of multiple cytokines implicated in lupus pathogenesis. Clin Exp Immunol 199, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zack DJ, Stempniak M, Wong AL, Taylor C and Weisbart RH (1996) Mechanisms of cellular penetration and nuclear localization of an anti‐double strand DNA autoantibody. J Immunol 157, 2082–2088. [PubMed] [Google Scholar]
- 7. Hansen JE, Tse CM, Chan G, Heinze ER, Nishimura RN and Weisbart RH (2007) Intranuclear protein transduction through a nucleoside salvage pathway. J Biol Chem 282, 20790–20793. [DOI] [PubMed] [Google Scholar]
- 8. Kim JS, Choi DK, Shin JY, Shin SM, Park SW, Cho HS and Kim YS (2016) Endosomal acidic pH‐induced conformational changes of a cytosol‐penetrating antibody mediate endosomal escape. J Control Release 235, 165–175. [DOI] [PubMed] [Google Scholar]
- 9. Jang JY, Jeong JG, Jun HR, Lee SC, Kim JS, Kim YS and Kwon MH (2009) A nucleic acid‐hydrolyzing antibody penetrates into cells via caveolae‐mediated endocytosis, localizes in the cytosol and exhibits cytotoxicity. Cell Mol Life Sci 66, 1985–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park H, Kim M, Kim HJ, Lee Y, Seo Y, Pham CD, Lee J, Byun SJ and Kwon MH (2017) Heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs) function as endocytic receptors for an internalizing anti‐nucleic acid antibody. Sci Rep 7, 14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park H, Kim M, Seo Y, Ham Y, Cho MY and Kwon MH (2018) Cytosolic internalization of anti‐DNA antibodies by human monocytes induces production of pro‐inflammatory cytokines independently of the tripartite motif‐containing 21 (TRIM21)‐mediated pathway. Front Immunol 9, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee J, Kim HJ, Roh J, Seo Y, Kim M, Jun HR, Pham CD and Kwon MH (2013) Functional consequences of complementarity‐determining region deactivation in a multifunctional anti‐nucleic acid antibody. J Biol Chem 288, 35877–35885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi DK, Bae J, Shin SM, Shin JY, Kim S and Kim YS (2014) A general strategy for generating intact, full‐length IgG antibodies that penetrate into the cytosol of living cells. MAbs 6, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee WR, Jang JY, Kim JS, Kwon MH and Kim YS (2010) Gene silencing by cell‐penetrating, sequence‐selective and nucleic‐acid hydrolyzing antibodies. Nucleic Acids Res 38, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiu ML, Goulet DR, Teplyakov A and Gilliland GL (2019) Antibody structure and function: the basis for engineering therapeutics. Antibodies 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seo Y, Lee Y, Kim M, Park H and Kwon MH (2020) Assembly and folding properties of cytosolic IgG Intrabodies. Sci Rep 10, 2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim DS, Lee SH, Kim JS, Lee SC, Kwon MH and Kim YS (2009) Generation of humanized anti‐DNA hydrolyzing catalytic antibodies by complementarity determining region grafting. Biochem Biophys Res Commun 379, 314–318. [DOI] [PubMed] [Google Scholar]
- 18. Chen G and Deng X (2018) Cell synchronization by double thymidine block. Bio Protoc 8, e2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amin MA and Varma D (2017) Combining mitotic cell synchronization and high resolution confocal microscopy to study the role of multifunctional cell cycle proteins during mitosis. J Vis Exp 56513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi J, Jeon Y, Roh Y, Jang J, Lee E, Villamante L, Kim M and Kwon MH (2024) The dispensability of V(H)‐V(L) pairing and the indispensability of V(L) domain integrity in the IgG1 secretion process. Front Mol Biosci 11, 1346259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu S, Liang S, Guo H, Zhang D, Li H, Wang X et al. (2013) Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor alpha‐associated diseases from a molecular view. J Biol Chem 288, 27059–27067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pennington LF, Tarchevskaya S, Brigger D, Sathiyamoorthy K, Graham MT, Nadeau KC, Eggel A and Jardetzky TS (2016) Structural basis of omalizumab therapy and omalizumab‐mediated IgE exchange. Nat Commun 7, 11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shin SM, Choi DK, Jung K, Bae J, Kim JS, Park SW, Song KH and Kim YS (2017) Antibody targeting intracellular oncogenic Ras mutants exerts anti‐tumour effects after systemic administration. Nat Commun 8, 15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Padlan EA (1994) Anatomy of the antibody molecule. Mol Immunol 31, 169–217. [DOI] [PubMed] [Google Scholar]
- 25. MacCallum RM, Martin AC and Thornton JM (1996) Antibody‐antigen interactions: contact analysis and binding site topography. J Mol Biol 262, 732–745. [DOI] [PubMed] [Google Scholar]
- 26. Jeong JG, Kim DS, Kim YS and Kwon MH (2011) A tat‐grafted anti‐nucleic acid antibody acquires nuclear‐localization property and a preference for TAR RNA. Biochem Biophys Res Commun 406, 403–407. [DOI] [PubMed] [Google Scholar]
- 27. Dondelinger M, Filee P, Sauvage E, Quinting B, Muyldermans S, Galleni M et al. (2018) Understanding the significance and implications of antibody numbering and antigen‐binding surface/residue definition. Front Immunol 9, 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pham CD, Woo MY, Kim YS, Park S and Kwon MH (2012) An anti‐nucleic acid antibody delivers antigen to the cross‐presentation pathway in dendritic cells and potentiates therapeutic antitumor effects. J Immunol 189, 5755–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gori A, Lodigiani G, Colombarolli SG, Bergamaschi G and Vitali A (2023) Cell penetrating peptides: classification, mechanisms, methods of study, and applications. ChemMedChem 18, e202300236. [DOI] [PubMed] [Google Scholar]
- 30. Ternynck T, Avrameas A, Ragimbeau J, Buttin G and Avrameas S (1998) Immunochemical, structural and translocating properties of anti‐DNA antibodies from (NZBxNZW)F1 mice. J Autoimmun 11, 511–521. [DOI] [PubMed] [Google Scholar]
- 31. Marschall AL, Zhang C, Frenzel A, Schirrmann T, Hust M, Perez F and Dübel S (2014) Delivery of antibodies to the cytosol: debunking the myths. MAbs 6, 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Freund G, Sibler AP, Desplancq D, Oulad‐Abdelghani M, Vigneron M, Gannon J, van Regenmortel MH and Weiss E (2013) Targeting endogenous nuclear antigens by electrotransfer of monoclonal antibodies in living cells. MAbs 5, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC et al. (2017) A method for the acute and rapid degradation of endogenous proteins. Cell 171, 1692–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mercer WE, Nelson D, DeLeo AB, Old LJ and Baserga R (1982) Microinjection of monoclonal antibody to protein p53 inhibits serum‐induced DNA synthesis in 3T3 cells. Proc Natl Acad Sci USA 79, 6309–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang JF, Xiong HL, Cao JL, Wang SJ, Guo XR, Lin BY, Zhang Y, Zhao JH, Wang YB, Zhang TY et al. (2022) Erratum: a cell‐penetrating whole molecule antibody targeting intracellular HBx suppresses hepatitis B virus via TRIM21‐dependent pathway: erratum. Theranostics 12, 3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramezani A, Asgari A, Kaviani E, Hosseini A and Ghaderi A (2021) Tatibody, a recombinant antibody with higher internalization potency. Mol Immunol 135, 320–328. [DOI] [PubMed] [Google Scholar]
- 37. Hershman RL, Li Y, Ma F, Xu Q and Van Deventer JA (2022) Intracellular delivery of antibodies for selective cell signaling interference. ChemMedChem 17, e202100678. [DOI] [PubMed] [Google Scholar]
- 38. Weisbart RH, Gera JF, Chan G, Hansen JE, Li E, Cloninger C, Levine AJ and Nishimura RN (2012) A cell‐penetrating bispecific antibody for therapeutic regulation of intracellular targets. Mol Cancer Ther 11, 2169–2173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.